
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAerobic oxidation and oxidative esterification of alcohols through cooperative catalysis under metal-free conditions†
Babak
Karimi
 *ab,
Mina
Ghahremani
a,
Hojatollah
Vali
c,
Rosaria
Ciriminna
d and
Mario
Pagliaro
*d
*ab,
Mina
Ghahremani
a,
Hojatollah
Vali
c,
Rosaria
Ciriminna
d and
Mario
Pagliaro
*d
aFaculty of Chemistry, Institute for Advanced Studies in Basic Sciences (IASBS), Gava Zang, PO Box 45195-1159, Zanjan 45137-66731, Iran. E-mail: karimi@iasbs.ac.ir
bResearch Center for Basic Sciences & Modern Technologies (RBST), Institute for Advanced Studies in Basic Sciences (IASBS), Zanjan 45137-66731, Iran
cDepartment of Anatomy and Cell Biology and Facility for Electron Microscopy Research McGill University, Montreal, Quebec, H3A 2A7, Canada
dIstituto per lo Studio dei Materiali Nanostrutturati, CNR via U. La Malfa 153, Palermo 90146, Italy. E-mail: mario.pagliaro@cnr.it
First published on 9th August 2021
Abstract
The ABNO@PMO-IL-Br material obtained by anchoring 9-azabicyclo[3.3.1]nonane-3-one N-oxyl (keto-ABNO) within the mesopores of periodic mesoporous organosilica with bridged imidazolium groups is a robust bifunctional catalyst for the metal-free aerobic oxidation of numerous primary and secondary alcohols under oxygen balloon reaction conditions. The catalyst, furthermore, can be successfully employed in the first metal-free self-esterification of primary aliphatic alcohols affording valued esters.
Thanks to their high selectivity and pronounced versatility, stable nitroxyl radicals of the TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) family are now well established as environmentally benign catalysts for the selective oxidation of a wide variety of alcohols to carbonyl compounds (and to carboxylic acids when reaction is carried out in water).1 Primary oxidants such as bleach (NaOCl), hypervalent iodine reagents, and O2 in combination with NOx co-catalysts such as tert-butylnitrite (TBN)2 and sodium nitrite3 are generally employed to promote catalytic turnover. Since the first transition-metal-free TEMPO-catalyzed aerobic oxidation of alcohols using molecular oxygen as a terminal oxidant, the catalytic method has replaced most alcohol oxidation stoichiometric routes, especially those based on highly toxic chromium(VI) reagents and other metal oxidants that until a decade ago were still widely employed to oxidise alcohols in the fine chemical and pharmaceutical industries.4
Sterically hindered by the four methyl groups, the oxoammonium group of TEMPO ensures the chemoselective oxidation of primary alcohols in the presence of unprotected secondary alcohol groups, but the applicability of TEMPO-based catalysts in the oxidation of non-activated aliphatic, secondary and sterically hindered primary alcohols is often limited by the same steric constraints.5 To expand the scope of nitroxyl radical mediated-oxidations, the use of less hindered bicyclic nitroxyl radicals such as 2-azaadamantan-N-oxyl (AZADO) and 9-azabicyclo[3.3.1]nonane-N-oxyl (ABNO) has become increasingly common.6
Obtained through a multi-step (i.e. up to four steps to synthesis 1-Me-AZADO) synthetic process,7 AZADO derivatives are relatively difficult to access. Prepared through a three-step synthetic route, the radical ABNO is an economically viable nitroxyl radical bearing hydrogen atoms on the α-carbons (avoiding to violate Bredt's rule) alternative to AZADO.8 Known since the mid-1960s,9 such less-hindered nitroxyl radicals mediate the oxidation of sterically hindered alcohols for which TEMPO is not a suitable catalyst. Applications include the efficient oxidation of chiral alcohols using chirally modified AZADOs,10 as well as 9-azanoradamantane N-oxyl (Nor-AZADO) exhibiting higher catalytic activity than AZADO, ABNO and TEMPO.11 Furthermore, combined with HNO3, NaNO2 (or both) as co-catalysts and O2 as primary oxidant, ABNO and 9-azabicyclo[3.3.1]nonane-3-one N-oxyl (keto-ABNO) effectively mediate the oxidation of secondary alcohols along with imines bearing diverse functional groups.12 Similarly, in combination with a catalytic amount of Cu(I), keto-ABNO successfully catalyzes the selective oxidation of secondary alcohols, including unactivated aliphatic substrates.13 The first metal- and halogen-free keto-ABNO catalyst for the aerobic oxidation of alcohols under mild reaction conditions was reported in 2014.14a Denoted as SABNO, the latter is an heterogeneous catalytic system comprised of SBA-15 functionalized with keto-ABNO showing superior catalytic activity over SBA-15-supported TEMPO in the aerobic oxidation of a wide range of alcohols bearing diverse functional groups under conditions similar to those used with homogenous catalysts.14b We now report a newly supported ABNO heterogeneous catalyst using a periodic mesoporous silica functionalized with an imidazolium framework (PMO-IL) as support in the metal-free aerobic oxidation of alcohols, in analogy to the catalyst system comprising a robust PMO with bridged imidazolium network (PMO-IL) further functionalized with TEMPO (Fig. 1).15
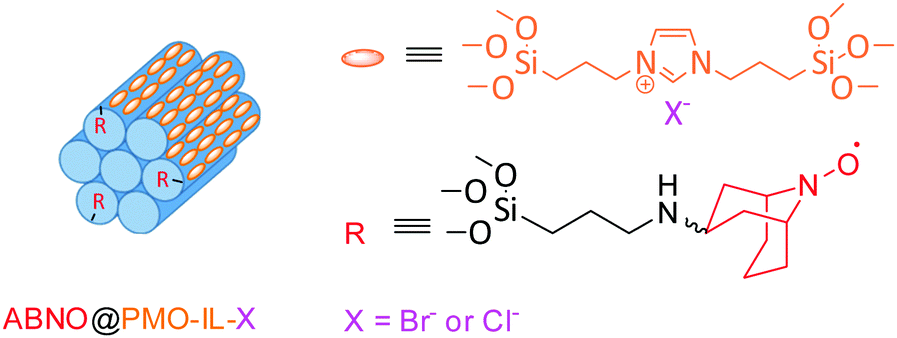 | ||
| Fig. 1 Schematic representation of ABNO@PMO-IL; for details of the preparation, see ESI.† | ||
The new catalyst, ABNO@PMO-IL, displays enhanced catalytic activity in comparison to the latter PMO-IL system as well to IL-free analogues due to the imidazolium bromide units in close proximity to the ABNO moieties. A robust PMO with bridged imidazolium network (PMO-IL) was initially prepared according to previously reported protocol, wherein 1,3-bis(3-trimethoxysilylpropyl)imidazolium chloride and Si(OMe)4 are hydrolyzed and co-condensed in the presence of Pluronic P123 as a template under acidic conditions.16 The ABNO radical was then anchored to the inner mesoporous network by mixing ABNO-functionalized silane, obtained via reductive amination of keto-ABNO with 3-(amino propyl) trimethoxysilane in the presence of NaBH3CN. The textural properties of the resulting ABNO@PMO-IL-Cl catalyst and PMO-IL are shown in Table 1 and Fig. 2a (wherein for clarity isotherms are shifted along the y-axis) illustrate.
| Material | S BET (m2 g−1) | r P (nm) | V P (cm3 g−1) |
|---|---|---|---|
| PMO-IL | 526.65 | 5.29 | 1.002 |
| ABNO@PMO-IL | 231.10 | 3.53 | 0.43 |
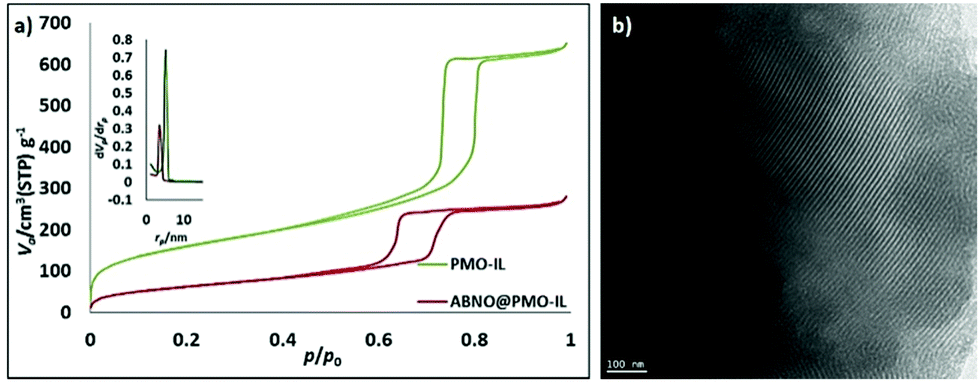 | ||
| Fig. 2 (a) N2 adsorption–desorption isotherms of all materials including PMO-IL and ABNO@PMO-IL-Cl and inset BJH pore size distribution. Va refers to the volume of adsorbed nitrogen in the pores of the materials. (b) Side view TEM images of ABNO@PMO-IL-Cl. (Scale bar: 100 nm). | ||
The type IV N2 adsorption–desorption isotherms with sharp hysteresis loop (Fig. 2a) are characteristic of highly ordered mesoporous materials with a regular array of 2D hexagonal pore structure. The specific surface area, total pore volume and pore diameter of ABNO@PMO-IL-Cl lower than those of pristine PMO-IL, indicate that the organic moieties are grafted in the inner surface of the mesochannels. This outcome is further supported by TEM images of ABNO@PMO-IL, showing highly ordered cylindrical pores of uniform size (Fig. 2b). The X-ray photoelectron spectroscopy (XPS) survey of ABNO@PMO-IL-Cl (Fig. 3) confirms the presence of C, Si, O, Cl and N as the only elements comprising this material, with chlorine present in the form of chloride ions balancing the positive charge of the IL framework. A closer look at the photoemission spectrum of the N (1s) line reveals (inset in Fig. 3) two main contributions at 399.26 eV (with a full width at half maximum, FWHM, of 1.48 eV) and 401.17 eV (FWHM = 2.02) binding energy, respectively. According to literature data,17 the peak at 401.17 is attributed to two nitrogen atoms in the imidazolium ring, whereas the first signal at 399.26 is attributed to the nitrogen of the nitroxide group NO,18 part of the ABNO molecular moieties attached to the surface of PMO-IL through the C–N covalent bond. This finding is further corroborated by the results of the thermogravimetric analysis (TGA) pointing to a loading of ABNO and IL groups at 0.26 and 0.97 mmol g−1, respectively (ESI,† Fig. S1).
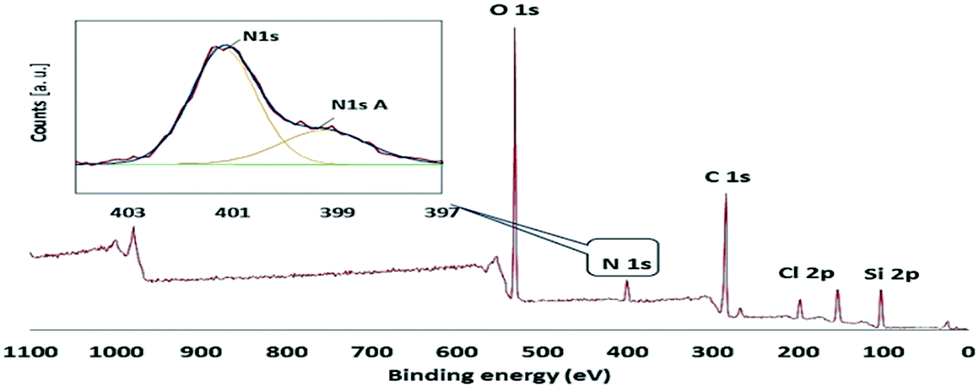 | ||
| Fig. 3 XPS survey spectrum of ABNO@PMO-IL, and (inset) XPS spectrum of the N (1s) electrons of ABNO@PMO-IL. | ||
Evidence of successful anchoring of ABNO on the inner surface of PMO-IL was further obtained by Fourier-transform infrared spectroscopy (FT-IR). Besides the signals characteristic of alkylimidazolium groups, the FTIR spectrum of ABNO@PMO-IL-Cl displays a new band at 1351 cm−1 related to the N–O free radicals (Fig. S2, ESI†) in good agreement with previous results concerning PMO-IL functionalized with TEMPO.15
Tested in the aerobic oxidation of benzyl alcohol (BnOH) as a model substrate under relatively mild conditions with TBN as nitrite source (1 mol% catalyst, 0.5 mL toluene, 10 mol% HCl, 8 mol% TBN) at 50 °C, the ABNO@PMO-IL-Cl showed more activity than SABNO (Table 2, entries 1 and 2). Therefore, we decided to replace the chloride counter ions with bromide ions by stirring the ABNO@PMO-IL-Cl catalyst in a saturated solution of NaBr for 24 h. To our delight, employed in the aerobic oxidation of BnOH under the aforementioned conditions, the new ABNO@PMO-IL-Br catalyst exhibited excellent catalytic activity not only in comparison to ABNO@PMO-IL-Cl but also to IL-free SABNO, PMO-IL functionalized with TEMPO, and even to homogeneous keto-ABNO (Table 2). The use PMO-IL as a catalyst (Table 2; entry 6) resulted only in 15% yields of benzaldehyde even after 12 h, clearly showing the crucial importance of co-supported ABNO and imidazolium bromide in obtaining high catalytic activity. We thus tested the new catalyst and the catalytic protocol in the selective oxidation of different alcohol substrates. Results in Table 3 show that good to excellent yields were obtained in relatively short time with all substrates.
| Entry | Material | t (min) | Conv. (%) |
|---|---|---|---|
| a Conditions: catalyst (1 mol%) in the presence of O2 (oxygen balloon, 621–622 torr) HCl (10 mol%) as additive and t-BuONO (TBN, 8 mol%) in toluene (0.5 mL) as a solvent in the presence of molecular oxygen. b Using homogeneous Keto-ABNO (1 mol%). c Using PMO-IL at the corresponding IL loading (10%) (40 mg) prolonged time to 12 h. All yields obtained via GC analysis using a standard method. | |||
| 1 | ABNO@PMO-IL-Cl | 120 | 97a |
| 2 | ABNO@SBA-15 (SABNO) | 120 | 90a |
| 3 | ABNO@PMO-IL-Br | 30 | >99 |
| 4 | TEMPO@PMO-IL-Br | 120 | 98 |
| 5 | Keto-ABNO | 30 | 90b |
| 6 | PMO-IL | 12 h | 15c |
|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | t (h) | Yielda (%) |
| Conditions.a Catalyst (1 mol%) in the presence of O2 (oxygen balloon, 621–622 torr), 10 mol% HCl, 8 mol% TBN, 0.3 mL toluene.b 5 mol% HCl.c 1.5–2 mol% catalyst, 16 mol% TBN, 5 mol% HCl, 0.3 mL toluene.d Catalyst-free conditions. All yields obtained via GC analysis using an internal standard FID calibration method (Fig. S4–S33). | ||||
| 1 | C6H5 | H | 0.5 | >99 |
| 2 | 4-Me–C6H5 | H | 0.5 | >99 |
| 3 | 3-Me–C6H5 | H | 0.5 | >99 |
| 4 | 2-Me–C6H5 | H | 0.66 | >99 |
| 5 | 4-iPr-C6H5 | H | 0.66 | >99 |
| 6 | 4-Cl–C6H5 | H | 1 | >99 |
| 7 | 3-Cl–C6H5 | H | 1 | 97 |
| 8 | 2-Cl–C6H5 | H | 1.25 | 97 |
| 9 | 4-Br–C6H5 | H | 1 | >98 |
| 10 | 2-Br–C6H5 | H | 1.5 | 95 |
| 11 | 2,4-DiCl–C6H5 | H | 2 | 87 |
| 12 | 4-NO2–C6H5 | H | 1.5 | 94 |
| 13 | 3-NO2–C6H5 | H | 2 | 86 |
| 14 | 2-NO2–C6H5 | H | 5 | 67 |
| 15 | 4-MeO–C6H5 | H | 1.5 | 94 |
| 16 | 2-MeO–C6H5 | H | 2 | 95 |
| 17 | 4-MeS–C6H5 | H | 1.5 | 95 |
| 18 | 1-Naphtyl | H | 1 | >98 |
| 19 | C6H5 | Me | 1 | 98b |
| 20 | 4-Ph–C6H5 | Me | 12 | >99b |
| 21 | C6H5 | Et | 1 | 97b |
| 22 | C6H5 | Ph | 4 | 96b |
| 23 | C6H5 | Cyclopentyl | 3 | 95b |
| 24 | α-Tetralol | — | 4 | 97b |
| 25 | CH3(CH2)4 | Me | 7 | 92c |
| 26 | CH3(CH2)5 | Me | 7 | 90c |
| 27 | Cyclohexyl | Me | 8 | 95c |
| 28 | Cycloheptyl | — | 9 | 99c |
| 29 | Cyclooctyl | — | 10 | 63c |
| 30 | Norbornyl | — | 10 | 86c |
| 31 | 2-Admantyl | — | 8 | >99c |
| 32 | C6H5 | H | 12 | 7d |
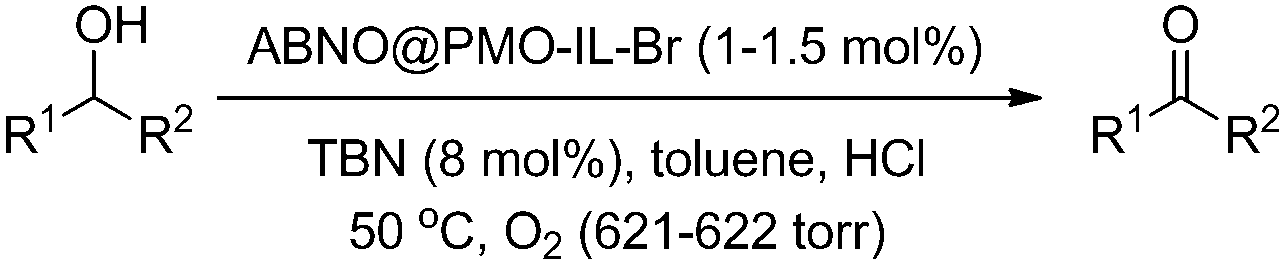
Even deactivated 2-nitrobenzyl alcohol was converted into the corresponding 2-nitrobenzaldehyde (an important precursor of indigo carmine) in 67% yield (entry 14 in Table 3). The oxidation of sterically hindered α-tetralol smoothly proceeded, affording in 4 h a 96% yield in 1-tetralone (entry 24). For comparison, the state-of-the art oxidation at room temperature of the same substrate affording 89% yield in 1-tetralone mediated by (Ph4P)[RuO2Cl3] dissolved in dichloromethane requires a three-fold excess of N-methylmorpholine-N-oxide (NMO) a primary oxidant in the presence of 4 Å molecular sieves.19 The lowest yield in selectively oxidised product was recorded for cyclooctanol affording 63% yield in cyclooctanone (entry 29). For comparison, the oxidative dehydrogenation of cyclooctanol to cyclooctanone over Pd/MgO under O2 at 70–80 °C affords a 58% yield.20 Not involving catalytic metal species, the ABNO@PMO-IL-Br catalyst is ideally suited for the conversion of sulfur-containing substrates such as 4-(methylthio)benzyl alcohol which is quickly converted into 4-(methylthio) benzaldehyde in 98% yield (entry 17). The aldehyde is the valued raw material of nonsteroidal anti-inflammatory drug “sulindac”. For comparison the best current catalyst using air as primary oxidant, MnCo2O4, catalyzes the selective oxidation of 4-(methylthio)benzyl alcohol to the 4-(methylthio) benzaldehyde in 96.5% yield.21 Finally, it is remarkable how the new catalyst successfully mediates the oxidation of secondary alcohols (entries 19–23 and 25–27) affording excellent (>95%) yields under the same conditions employed for converting primary and secondary alcohols to aldehydes and to ketones, respectively. The latter alcohols are generally considered to be unaffected under the conditions employed for the conversion of primary and secondary alcohol substrates.22 The suitability of the catalysis for acid-sensitive silyl ether and acetal groups remains to be investigated.
The direct oxidative esterification of alcohols to esters over metal catalysts such as Ru,23 Pd,24 Ir,25 Au,26 Co (and Co3O4)27 has been widely investigated as an important and highly atom-economic process. Although being a highly desirable process for the industrial production of esters, the environmental and economic concerns associated with the high temperature and pressure involved in state-of-the-art catalytic processes using precious metal catalysts are important issues that remain to be addressed.28 We thus investigated the activity of ABNO@PMO-IL-Br in the direct oxidative esterification of alcohols, carrying out the aerobic oxidation of BnOH under the optimized conditions of entry 1 in Table 3. Prolonging the reaction time to 24 h showed no self-esterification product. Similarly, increasing the amount of catalyst to 2 mol% and using 16 mol% of TBN nitrite source and 5 mol% of HCl, did not lead to the formation of benzyl benzoate. However, using the new catalyst in the aerobic oxidation of primary aliphatic alcohols efficiently affords self-esterified products (Table 4). To the best of our knowledge, this is the first report on the self-esterification of primary aliphatic alcohols using a nitroxyl radical catalyst system under metal-free conditions (for proposed mechanism of self-esterification see Scheme S1 in the ESI†). Attempts to synthesis self-esterification products either using homogenous keto-ABNO or SABNO under optimized conditions were unsuccessful (aldehydes are the sole products). This result highlights the importance of near molecular proximity of supported ABNO and imidazolium bromide in the nanoscale cavities of ABNO@PMO-IL-Br that could synergistically contribute to the unique selective oxidative self-esterification of primary aliphatic alcohols.
|
|
|||
|---|---|---|---|
| Entry | R | t (h) | Conv.a (%) |
| Conditions.a 1.5–2 mol% of catalyst in the presence of molecular oxygen, 5 mol% HCl as an additive and 16 mol% TBN in 0.3 mL toluene as a solvent. All products were detected using NMR spectroscopy. The progress of the reactions was monitored by GC.b Using Keto-ABNO (2 mol%), yields in parenthesis refer to the corresponding aldehyde.c Using SABNO (2 mol%), yield refer to the corresponding aldehyde. | |||
| 1 | PhCH2CH2 | 20 | 75 |
| 2 | CH3(CH2)2 | 15 | 91 |
| 3 | CH3(CH2)3 | 15 | 80 |
| 4 | CH3(CH2)6 | 15 | 87 |
| 5 | CH3(CH2)7 | 15 | 85 |
| 6 | CH3(CH2)8 | 20 | 83 |
| 7 | PhCH2CH2 | 20 | (95)b |
| 8 | PhCH2CH2 | 20 | 94c |

In summary, we have discovered that anchoring of ABNO moieties within the nanochannels of a PMO functionalized with imidazolium bromide network affords a catalyst showing enhanced catalytic activity in the metal-free aerobic oxidation of numerous primary benzylic and secondary alcohols. Activity is much higher than that of IL-free SABNO and PMO-IL functionalized with TEMPO excellent catalysts, and even slightly higher than homogenous keto-ABNO. The same ABNO@PMO-IL-Br solid catalyst allows to perform the one-pot oxidative esterification of non-activated aliphatic alcohols under mild reaction conditions. The catalyst could be recycled in three consecutive reactions run in the oxidation of benzyl alcohol with only a slight decrease in catalytic activity. Besides a few interesting metal-free protocols for aerobic oxidation of alcohols,28 these results open the route to new effective and versatile approach for carrying out alcohol selective oxidations and selective oxidative self-esterification of primary aliphatic alcohols under mild metal-free conditions (oxygen balloon, 50 °C).
The authors acknowledge IASBS Research Councils, and Iranian National Science Foundation (INSF) to grant the research (No. 99022450), the Alexander von Humboldt Foundation (B. K.), NanoQuebec and NSER Council of Canada (H. V.) for support of this work.
Conflicts of interest
The authors have no conflicts of interest to declare.Notes and references
- R. A. Sheldon, Catal. Today, 2015, 247, 4 CrossRef CAS
.
-
(a) Y. Xie, W. M. Mo, D. Xu, Z. L. Shen, N. Sun, B. X. Hu and X. Q. Hu, J. Org. Chem., 2007, 72, 4288 CrossRef CAS PubMed
-
(a) X. Wang, R. Liu, Y. Jin and X. Liang, Chem. – Eur. J., 2008, 14, 2679 CrossRef CAS PubMed
-
(a) R. H. Liu, X. M. Liang, C. Y. X. Dong and Q. Hu, J. Am. Chem. Soc., 2004, 126, 4112 CrossRef CAS PubMed
- H. A. Beejapur, Q. Zhang, K. Hu, L. Zhu, J. Wang and Z. Ye, ACS Catal., 2019, 9(4), 2777 CrossRef CAS
- S. Wertz and A. Studer, Green Chem., 2013, 15, 3116 RSC
-
(a) M. Shibuya, Y. Osada, Y. Sasano, M. Tomizawa and Y. Iwabuchi, J. Am. Chem. Soc., 2011, 133, 6497 CrossRef CAS PubMed
- M. Shibuya, M. Tomizawa, Y. Sasano and Y. Iwabuchi, J. Org. Chem., 2009, 74, 4619 CrossRef CAS PubMed
- R. M. Dupeyre and A. Rassat, J. Am. Chem. Soc., 1966, 88, 3180 CrossRef CAS
- M. Tomizawa, M. Shibuya and Y. Iwabuchi, Org. Lett., 2009, 11, 1829 CrossRef CAS PubMed
-
(a) M. Hayashi, Y. Sasano, S. Nagasawa, M. Shibuya and Y. Iwabuchi, Chem. Pharm. Bull., 2011, 59, 1570 CrossRef CAS PubMed
-
(a) M. B. Lauber and S. S. Stahl, ACS Catal., 2013, 3, 2612 CrossRef CAS
- L. Rogan, N. L. Hughes, Q. Cao, L. M. Dornan and M. J. Muldoon, Catal. Sci. Technol., 2014, 4, 1720 RSC
-
(a) B. Karimi, E. Farhangi, H. Vali and S. Vahdati, ChemSusChem, 2014, 7, 2735 CrossRef CAS PubMed
- B. Karimi, S. Vahdati and H. Vali, RSC Adv., 2016, 6, 63717 RSC
- B. Karimi, D. Elhamifar, J. H. Clark and A. J. Hunt, Chem. – Eur. J., 2010, 16, 8047 CrossRef CAS PubMed
-
(a) J. Tan, D. Fang, Y. Liu and L. Hu, New J. Chem., 2019, 43, 2583 RSC
-
(a) O. Swiech, R. Bilewicz and E. Megiel, RSC Adv., 2013, 3, 5979 RSC
- U. R. Pillai and E. Sahle-Demessie, Green Chem., 2004, 6, 161 RSC
- D. Li, F. Ruan, Y. Jin, Q. Ke, Y. Cao, H. Wang, T. Wang, Y. Song and P. Cui, Catal. Sci. Technol., 2019, 9, 418 RSC
- D. Chen, Y. Zhang, X. Pan, F. Wang and S. Huang, Adv. Synth. Catal., 2018, 360, 3607 CrossRef CAS
-
(a) J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840 CrossRef CAS PubMed
-
(a) S. Gowrisankar, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5139 CrossRef CAS PubMed
- N. Yamamoto, Y. Obora and Y. Ishii, J. Org. Chem., 2011, 76, 2937 CrossRef CAS PubMed
- H. Miyamura, T. Yasukawa and S. Kobayashi, Green Chem., 2010, 12, 776 RSC
-
(a) R. V. Jagadeesh, H. Junge, M.-M. Pohl, J. Radnik, A. Brückner and M. Beller, J. Am. Chem. Soc., 2013, 135, 10776 CrossRef CAS PubMed
-
J. Otera and J. Nishikido, Esterification: Methods, Reactions, and Applications, John Wiley & Sons, New York, 2nd edn, 2010 Search PubMed
-
(a) Z. Wang, J. Shi, D. Wang, Y. Pu, J. X. Wang and J. F. Chen, React. Chem. Eng., 2019, 4, 507–515 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc02937a |
| This journal is © The Royal Society of Chemistry 2021 |