
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Standardisation needs for organ on chip devices†
Monica
Piergiovanni
 *,
Sofia B.
Leite
,
Raffaella
Corvi
and
Maurice
Whelan
*,
Sofia B.
Leite
,
Raffaella
Corvi
and
Maurice
Whelan
European Commission, Joint Research Centre (JRC), Ispra, Italy. E-mail: monica.piergiovanni@ec.europa.eu
First published on 12th July 2021
Abstract
Organ on chip (OoC) devices represent the cutting edge of biotechnologies, combining advanced cell and tissue culture with microengineering. OoC is accelerating innovation in the life sciences and has the potential to revolutionise many fields including biomedical research, drug development and chemical risk assessment. In order to gain acceptance by end-users of OoC based methods and the data derived from them, and to establish OoC approaches as credible alternatives to animal testing, OoC devices need to go through an extensive qualification process. In this context, standardisation can play a key role in ensuring proper characterisation of individual devices, benchmarking against appropriate reference elements and aiding efficient communication among stakeholders. The development of standards for OoC will address several important issues such as basic terminology, device classification, and technical and biological performance. An analysis of technical and biological aspects related to OoC is presented here to identify standardisation areas specific for OoC, focusing on needs and opportunities. About 90 standards are already available from related fields including microtechnologies, medical devices and in vitro cell culture, laying the basis for future work in the OoC domain. Finally, two priority areas for OoC are identified that could be addressed with standards, namely, characterisation of small molecule absorption and measurement of microfluidic parameters.
Introduction
According to the International Organization for Standardization (ISO), a standard is a document, established by consensus and approved by a recognized body that provides, for common and repeated use, rules, guidelines or characteristics for activities or their results, aimed at the achievement of the optimum degree of order in a given context. In relation to disruptive innovation, standards can play a key role in advancing new technologies from R&D to commercial products. For organ on chip (OoC) devices, proper qualification is required to demonstrate their technological and biological relevance in order to increase their uptake and implementation by end-users, and to contribute to their acceptance in regulatory contexts of use. The development and use of standards should facilitate, for example, systematic characterisation of different devices by describing their structure and functionality, and the specification of performance requirements and test methods to verify them. In addition, such standards will provide a means for better comparison and benchmarking of OoC devices, by ensuring a consistent use of predefined sets of parameters and measurement units. Standards dealing with proper terminology and reporting can also prove essential for effective and efficient communication and ensure a proper understanding between R&D, end-user, and stakeholder communities. Here we first review the current status of standardisation in the OoC field to understand what is already available and to identify emerging needs. Standards already existing in different but related technological domains are then presented as potential starting points for future standards development work specific to OoC. Finally, molecule adsorption and microfluidic control are described as two priority areas for OoC standardisation activities.Current status
Various consortia have developed position papers that describe the collective vision of many stakeholders regarding standardisation needs to advance the OoC field. For example, the ORCHID project identified standardisation as a fundamental pillar for the advancement of OoC technologies at the European level.1 The t4 (transatlantic think tank for toxicology) 2019 workshop report2 summarises the view of 46 international stakeholders on the challenges for the OoC community and identifies standards as tools to support qualification and reach regulatory acceptance.Only recently, however, the OoC community started to actively involve Standards Development Organisations (SDOs) in their work and to discuss actions that could be taken together. The PSIS (Putting Science into Standards) 2021 workshop,‡ organised by the European Commission's Joint Research Centre (JRC) and CEN-CENELEC is an important step in this direction, bringing together stakeholders from academia, industry and regulatory agencies. The Standards Coordinating Body in the USA is also targeting SDOs to steer their standards work in the OoC area. In particular, the Microphysiological System working group is coordinating activities together with ASTM international within the Standards Advancement Project.§
The analysis presented here builds on these pre-normative initiatives and complements their findings. It is divided in sub-sections, each one focusing on a specific aspect of OoC technology, providing an overview of the current situation and suggesting future needs. Table 1 summarises the analysis of the current scenario, together with needs and recommendations in standardisation for OoC and a proposal on priorities.
| Current scenario | Future needs | Priority | |
|---|---|---|---|
| a TRL = Technology Readiness Level. | |||
| Definition | No uniform definition of MPS/OoC and related vocabulary | A consensus on terminology is necessary to start the qualification process | +++ |
| Classification | Some categories can be identified (OoC focusing on barrier, parenchyma, multi organs) but there is no consensus | Identification of categories will facilitate the qualification process | +++ |
| Functional requirements | Functional requirements are not uniformly agreed upon. Many developers perform an internal technical validation, but it is usually only partial and not fully reported | Identification of requirements and performance indicators (relevant parameters, units, measurement method, acceptability range...) | +++ |
| Device material | There is a wide use of PDMS but also other plastics. The issue of molecule absorption is not uniformly addressed | Identify suitable test methods for molecule adsorption quantification | +++ |
| Production process | Low TRLa devices are produced with soft lithography and rapid prototyping, no standardisation is usually needed | Standards for plastic materials can be used for high TRLa devices | ++ |
| Compatibility | Standardisation effort to create common interfaces among different OoCs and with laboratory equipment | Promote the development of standards for OoC integration | ++ |
| Sterilization and packaging | Low TRLa devices are sterilized with non-standard methods (UV light under biological hood or autoclave) | Standards for sterilization and packaging can be used for high TRLa devices | + |
| Quality | Some developers of commercialized OoC already perform quality control | Promote the use of GMP | + |
| Ancillary devices | Many standards are applicable in this field. Many products offered by major companies are already CE marked | Monitoring and updating of existing standards | + |
| Assays/endpoints | Relevant endpoints are organ-specific and application-specific | Standardized lists for specific context of uses have to be agreed upon and used as a basis for qualification | +++ |
| Test compounds/drugs | Building lists of reference compounds is a widely accepted validation method in regulatory sciences | Standardized lists have to be agreed upon and used as a basis for qualification | +++ |
| Cell source | Primary cells, iPSC and cell lines are all widely used. Standardisation in the field is rather poor | Promote the standardisation of protocols for cell culture and maintenance, sharing of best practices | ++ |
| Practical use | GLP, GIVIMP and GCCP are applicable | Disseminate and increase the use of best practices among developers | ++ |
| Other material | Some standards exist for materials used as matrixes or scaffold. Issue of molecule absorption is not uniformly addressed | Identify test methods for molecule adsorption on matrixes (measurement methods, units, ranges) | + |
Definition and classification
Since OoC first appeared in the scientific field, researchers and developers all over the world have been trying to find the best terminology to describe their innovations. Generally speaking, both Organ on Chip and Micro Physiological System (MPS) are currently used, often interchangeably, although for many, MPS is taken to be technically broader in scope, also including in vitro models such as 3D cultures, spheroids and organoids which usually lack the engineered fluidics component specific to OoC. Recently, the FDA Alternative Methods Working Group proposed some draft definitions3 but there is still no agreement on the meaning or relevance of terms such as tissue-on-chip, body-on-chip, and so on. Future work should focus on the identification of the specifics that constitute an OoC device and use this as a basis to compile a list of relevant terms.Classification aims to group together devices that have technically similar features. This is particularly challenging for OoC since there are many different types of devices based on various technologies and design concepts, developed to represent specific aspects of biology and physiology. However, it should be possible to identify common technical characteristics that can be used as a basis to set up a classification scheme using suitable classification criteria. For instance, some devices are designed to represent the combination of multiple organs, providing two or more connected chambers where nutrients and signalling molecules are shared among different cell types. Other devices reproduce barrier functions, for example by culturing endothelial and epithelial cells of the same organ on opposite sides of a membrane. Some others focus on the co-culture of various cell types by recreating a relevant microenvironment. The use of fluid flow can also be used as a classification criterion, since many devices use a flow of continuously fresh medium, while others prefer to recirculate the medium for a certain amount of time. The use of various pumping systems also influences the performance of a device, determining the stability of the flow-rate and thus of the shear stress on the cells and medium renewal.
Technical performance of OoC devices
When selecting a primary material for an OoC device, the issue of absorption of molecules circulating in the liquid medium is a significant concern since it can compromise the functioning of the device and the accuracy of the results obtained. The absorption of small molecules by PDMS has been extensively studied through both experimental testing and mathematical modelling. However, no standards exist in terms of test methods, suitable measurement units and performance criteria (e.g. acceptability range). Such standards would greatly benefit the whole community by providing a sound basis for reliable characterisation and comparison of materials in different OoC setups and assays.
OoC devices are rarely completely standalone and thus need to be compatible with laboratory equipment. Devices are designed to be operated inside an incubator in order to provide the right conditions for air, CO2, temperature and humidity required for maintaining cells and tissues. OoC devices that require continuous fluid flow inside the microfluidic channels need a connection with its pumping and control system. A lot of work has been done to minimise pump dimensions so that they can fit in the incubator. In addition, the number of tubes that connect a pumping system located outside the incubator with the OoC devices inside it have been minimised. Some commercial providers have implemented platforms to easily manage multiple devices in order to facilitate parallel experiments to increase flexibility and throughput.
Compatibility issues also arise in the analysis phase of an OoC method or assay. Many OoC devices provide optical access to the culture chambers through a transparent bottom but in many cases there are no standard solutions for mounting the device on microscopes or imaging platforms. Devices typically need to be physically accessed to retrieve the biological material for further analysis (e.g. immunostaining, gene-expression, etc.). To address these needs, OoC developers are opting to use standard dimensions and tolerances already being used for common lab ware, such as multi-well plates, glass slides, coverslips and petri dishes.
Biological model performance
The recent Resource Identification Initiative (RII) has built a freely accessible database that lists well-characterised biological resources and gives them unique identifiers that can be reported in research papers.14 A wide variety of biological materials is considered, such as micro-organisms and antibodies and the RII approach is gaining popularity among scientific journals. Standards covering requirements and test methods related to scaffold and matrix materials have already been developed for tissue engineered medical products (TEMPs). These standards can be directly applied or used as a basis to develop new standards for materials specifically used in OoC. As discussed more below, a high priority for OoC is the development and use of standards to address the issue of molecule absorption by biological materials since this can influence the effective concentration of a test compound that the target cells or tissues are exposed to.
Existing standards relevant to OoC
OoC devices were developed by combining micro-process engineering and cell culture with the goal of improving the relevance of in vitro methods for basic and applied research. Thus OoC integrates and exploits scientific and technical knowledge from a variety of fields. Our preliminary analysis showed that about ninety published standards, originally developed in different fields, could be related to specific aspects of OoC. In particular, several existing standardised test methods and requirements refer to sterilization, pumping system safety, and materials characterization. Many standards on components widely used in design/prototyping phase (such as needles, connections, syringes) and standards on compatibility (like microplate geometry, pitch-spacing) are already available from the medical device, plastics and in vitro diagnostic fields. These standards are listed in the ESI† section, together with the number and title of the standard, the specific technical committee (TC) that developed it, the standard type (definition, test method, requirement, reference material, best practice) and the OoC aspect the standard relates to. The TCs that are more involved are ISO/TC 276 (Biotechnology), ASTM – F04.41–44 (focused on TEMPs), ISO/TC 210 (quality management and general aspects for medical devices) and CEN/TC 140 (in vitro diagnostic medical devices). The standards presented here cannot be immediately applied in the OoC domain, but they provide a practical starting point to identify what could be adapted for OoC and what significant OoC-specific gaps there might be. In addition, this list of relevant standards helps identify the most relevant TC that could address a particular OoC standardisation need.General standards
Many OoC devices are built as laboratory prototypes by university groups and research institutions. Several standard requirements and test methods related to consumables used in the healthcare sector are typically used, such as needles, tubing and connectors. Even if most current applications of OoC do not involve contact with a human subject (patient), these standards still guarantee that the components used in OoC devices meet such specifications.
For devices produced at an industry level, the batch control standard21 deals with the production process of any product on the market and is thus available to be used also for OoC devices. CEN/TC 102 published a series of standard requirements for sterilization for medical purposes including those using ethylene oxide and radiation, and provides chemical and biological indicators to verify the sterilization outcome. These standards can be easily adjusted to be applicable with OoC devices.
The US Pharmacopoeia (USP) identified about 10 reference materials commonly used for OoC, including collagenase, foetal bovine serum (FBS), growth factors and cytokines. The use of reference materials is generally needed to develop accurate test methods and to ensure that perform as intended. These materials can also be used during inter-laboratory comparisons to increase reproducibility.
A group of seven standards developed by the American Society of Mechanical Engineering focuses on the measurement of flow rates in pipes, covering definition of key terminology, device requirements, and test methods for certain flow meters. These standards are quite general but can be applied also to flow meters for microfluidic applications. Awareness of these standards can help when assessing the flow rate in OoC devices setups.
| Chapter | Content |
|---|---|
| 1. Roles and responsibilities | The in vitro method life cycle from development to the use for safety assessment purposes has a variety of key actors and the guidance identifies clearly their responsibilities, both individually and collectively |
| 2. Quality considerations | To realise fully the potential of in vitro methods and allowing them to become a key tool for a new way of doing toxicology, they need to be developed and applied in a way that scientific integrity and quality is assured |
| 3. Facilities | In vitro cell and tissue culture facilities should be fit for purpose and a detailed understanding of the workflow for the in vitro method related processes is essential. The separation of specific laboratory functions and elements that can adversely affect in vitro method work need to be understood |
| 4. Apparatus, materials and reagents | Apparatus, including computerised systems, should be regularly maintained, calibrated and validated (if required). Material and reagents should be purchased from well-established sources to ensure the integrity and reliability of the in vitro method results |
| 5. Test systems | With the advances in science and technology a variety of different cell and tissue culture-based test systems have been developed, but only few have been used in regulatory-approved test guideline methods due to reliability issues caused by a variety of elements described in this chapter |
| 6. Test and reference/control items | The preparation and characterisation of test, reference and control items and their interaction with the in vitro environment should be well understood, to ensure the acquisition of reliable and relevant results |
| 7. Standard operating procedures | Standard operating procedures (SOPs) and the accompanying forms, templates or worksheets should be written and prepared in a way that they will form the tools to simplify the work of the user when carrying out an in vitro method study |
| 8. Performance of the method | In vitro method developers need to ensure that in vitro methods they design will produce good quality data, i.e. fit for purpose, thanks to a stringent assessment of the performance of the method |
| 9. Reporting of results | Good reporting of in vitro methods can only be achieved when all important details are recorded in a way that allows others to reproduce the work or reconstruct fully the in vitro method study |
| 10. Storage and retention of records and materials | Before collecting data from in vitro methods it is important to assess the format of collection, the complexity involved and requirements for traceability, storage, verification and transmission of data |
These efforts are crucial to standardise the test methods and thus ensure an adequate biological performance and improve reproducibility, especially for methods that are not formally validated. The OECD Guidance Document 21132 presents a harmonised approach to describing non-guideline in vitro test methods, which could be applied to OoC. The guidance document is addressed to developers and end-users for reporting fundamental information that a data-user would need to know such as: details of the experimental protocol; the reference chemicals used; the stage of development and validation of the method; assay acceptance criteria; data interpretation procedures; and performance metrics including reproducibility and accuracy. A test method reporting template was also developed by the toxicological research community, setting out specific questions to be addressed and providing comments and additional notes to help the user complete the template.33 Something similar was developed for in vitro methods intended for developmental neurotoxicity testing, where ‘readiness criteria’ were also proposed to evaluate methods with a view to their application in regulatory safety assessment.34
TEMPs
Tissue Engineered Medical Products (as per ASTM terminology) or cellular therapeutic products (as per ISO terminology) are medical products that may achieve a therapeutic potential from cells, biomolecules, scaffolds, and other materials, and processed tissues and derivatives used in various combinations or alone.41 Standards to regulate these new products were developed between 2011 and 2019 by ASTM and ISO using a strategy that could be replicated for OoC. Firstly, standard terminology and classification of TEMPs was proposed, followed by a series of standards setting out critical requirements and associated test methods for materials and matrices used in scaffolds, e.g. alginate, chitosan salts and hyaluronan. A standard guide for evaluation of in vitro release of biomolecules from biomaterial-scaffolds recognises the need to measure molecules released by TEMPs materials,42 as opposed to the issue of absorption of molecules in OoC devices mentioned earlier.Medical devices
Standards for medical devices are developed by CEN/ISO and are referred to by the European Commission in the list of harmonised standards that can be used to prove compliance with the medical device regulation.43 There are more than 200 standards included in this list; some of these can be used as a model for the OoC field. The standards related to quality management in a risk assessment framework and those on biological evaluation of medical devices are fundamental pillars of the regulatory acceptance for medical devices. Moreover, standards on the various sterilization alternatives (radiation, ethylene oxide, moist heat methods but also biological, chemical and microbiological indicators to assess reliable sterilization) and packaging requirements complement the list of relevant standards that can be used as a reference to address similar issues in the OoC field.In vitro diagnostics
Standards on in vitro diagnostics are developed by CEN/ISO and are indicated by the European Commission in the list of harmonised standards that can be used to prove compliance with the EU Regulation on in vitro diagnostic medical devices.44 Those of interest for the OoC field are mainly related to performance evaluation of these devices, stability-in-time of the various components (e.g. reagents and culture medium) and statistical aspects to be considered for acceptability of results.Critical aspects of OoC devices that need standardisation
In addition to standardising OoC terminology and classification, there are two areas where standards could play a valuable and immediate role: small molecule absorption on OoC materials and the evaluation of fluidic shear stress. These two widely acknowledged issues in OoC applications are, however, still addressed in a very heterogeneous way, with every end-user using a different approach. The development and use of test methods and reporting standards with adequate information would greatly increase the reliability, relevance, and utility of the results obtained.Methods to determine small molecule adsorption
As already discussed, some polymeric materials used in many OoC devices can raise doubts about their suitability, due to absorption of small molecules (i.e. molecular weight lower than 1 kDa) from the fluid medium. This material property can be a fundamental issue for many OoC applications, but it becomes specifically relevant in toxicity or efficacy studies, where knowing and controlling the effective concentration of a test compound acting on the cells is crucial to obtain accurate results on the biological response.45 PDMS is the material most used for OoC devices, yet it has significant absorption issues. A number of studies were conducted to investigate this phenomenon and it is now widely known that the absorption rate depends on the chemical properties of the molecule itself. For example, hydrophobic molecules (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P > 1.85) with no H-bond donor groups are the molecules most easily sequestered into PDMS.46
P > 1.85) with no H-bond donor groups are the molecules most easily sequestered into PDMS.46
Researchers and companies started to use various coatings on the inner walls of microfluidic chambers to avoid direct contact between PDMS and the small molecules present in the medium, with the goal of reducing or preventing absorption (see ESI†). Coatings are produced using various types of biological matrixes (most commonly: collagen, fibronectin, polylisine) or by growing a layer of organ-specific endothelial cells. However, small molecules can also be sequestered by coatings, bind to proteins and lipids dissolved in the medium, or can even evaporate, decreasing the amount of compound that the cells are effectively exposed to, as demonstrated for collagen I with ibuprofen.47 Thus, while the use of a coating can solve the issues posed by PDMS, it can actually cause others.
To gain a better understanding of the problem, computational models such as the VCBA (virtual cell based assay) can be used to estimate the amount of compound that the cells are exposed to and the factors influencing it.48–50 These models are able to simulate the kinetics and dynamics of a chemical compound in cell-based in vitro assays, by integrating a transport model, a cell partitioning model, a cell growth and division model, and an effects model, together with many important characteristics of the experimental setup. In order to be adapted to OoC devices, these models will need to be modified to account for OoC specific design features. In particular, the PDMS needs to be implemented as a new material with its specific physical and chemical properties, as well as the materials used for the coatings. Moreover, current VCBA models do not include fluid flow, which is key to represent the transport of molecules in the devices. VCBA models have been built to represent typical in vitro assays using multi-well plates and thus they are based on a simple cylindrical geometry. This is not applicable to the majority of OoC devices, which usually employ rectangular chambers connected by microfluidic channels.
The relationship between a material and the biological entity it is in contact with is of great relevance. This certainly the case in the medical device domain, especially for permanent implants and prostheses. Two standard test methods were developed to comply with regulatory safety assessment for biocompatibility (ISO 1099351 and ASTM F748 – 1625). A standard test method52 describing an experimental procedure to extract plastic material from a medical device, to simulate its release in a biological environment, was developed by ASTM to demonstrate compliance with regulations. A similar approach could be used to define a suitable test method that allows a correct and reproducible evaluation of the compound actually binding to the OoC system used, accounting for coatings, matrices, scaffolds and other ancillary materials involved.
Shear stress estimation
One of the major characteristics of OoC technologies is the possibility to include fluid flow, thus exposing the cell culture to a physiologically relevant biomechanical solicitation and continuous exchange of biomolecules. This is largely possible due to the integration of microfluidic circuits that, with specific pumping systems and sometimes with the help of hydraulic valves, can modulate the flow in various chambers to achieve different fluid dynamic conditions. To evaluate the effect of the flow on the outcome, it is important to correctly understand the fluid dynamics at the microscale, how the fluid flow can be controlled with different pumps and how the device resistance can influence the flow rate.In some OoC devices, for instance, the fluid is not flowing through empty channels, but rather through a matrix that embeds the cells in a 3D structure. In this specific situation, it is possible to estimate the hydraulic resistance with the Darcy law, which accounts for the permeability k of a porous material through which a fluid is flowing (an example from the biomedical field is cartilage):
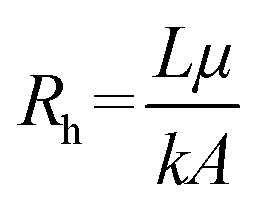
The cross-section of the channel greatly influences its resistance. In particular, the relationship between fluidic resistance and channel radius (or the channel height and width for rectangular cross-section) follows a 4th order power law.53 Thus, even a small variation in channel geometry will greatly influence the channel resistance and thus the fluid flow. For commercialised products, proper production control can ensure design specifications and tolerances are respected and each batch is controlled. However, this dimensional issue becomes relevant for OoC devices which are in the early phases of design and prototyping. With soft lithography, where moulds can easily have varying heights, and 3D printing, where every device is printed as a single entity or in very small batches, the geometrical features can vary from one device (or batch) to another. This uncertainty can influence the hydraulic resistance, leading to non-uniform flow rates, especially if the pumping system is not used properly (see ESI†).
Geometry is also fundamental to estimate the shear stress. For very small channels, the presence of a cell layer can significantly reduce the cross-section space, thus considerably increasing the shear stress acting on the cells.
A correct understanding and use of microfluidic systems in OoC is necessary to ensure reliability of the results, while accurate reporting of fluidic variables and protocols will ensure reproducibility. Standard methods to measure, derive and report fluidic variables are thus of crucial importance to ensure reliability of results, clearly demonstrate the added value of OoC approaches and ultimately build end-user confidence.
Conclusions
There is a clear consensus in the OoC community on the need for standardisation to advance the field, with several initiatives now in progress worldwide. It is important to emphasise that the development and use of standards in no way impedes innovation. On the contrary, standards are an important enabler of innovation. Researchers and developers need to keep thinking ‘outside the box’ to push boundaries and drive innovation. On the contrary, standards should be considered as valuable tools that can be readily used to demonstrate the reliability and relevance of their novel devices and pave the way for their application and regulatory acceptance.The analysis presented here builds on those efforts and has identified specific needs and priorities. Clearly, a focus on terminology and device classification are the first aspects to be tackled and in this regard, with ISO-IWA 23 (2016) and ISO 10991 (2009) providing a good basis. However, specific features and peculiarities of OoC need to be properly addressed. A second priority is the technical assessment of OoC devices, covering various aspects including materials, production processes and test methods. Some existing standards related to medical devices and IVD can be used as a starting point to develop specific standards for OoC, considering their intended use in various fields. To really boost the implementation of OoC for regulatory and biomedical sciences, it is crucial to understand their biological performance, intended as the capacity to recapitulate relevant and specific organ functions. The development of standards to facilitate the assessment of biological performance for specific contexts of use would not only advance the OoC field, but complex in vitro methods in general.
Even though OoC has emerged as a potentially disruptive technology in its own right, the OoC community should acknowledge the many existing standards in related fields and evaluate how best they could be exploited and expanded for the OoC domain. As proposed here, material absorption and microfluidic flow are two specific issues common across all OoC devices where a considerable amount of work has already been done and where there is a real need for standardisation to progress further. Irrespective of which priority areas are pursued first however, it is imperative that the OoC community joins forces with SDOs (e.g. CEN-CENELEC, ISO, ASTM) and with experts from related fields to devise a roadmap for bringing standards into the OoC domain.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Ms. Adelaide Dura for her support in preparing the graphical abstract.References
- M. Mastrangeli, S. Millet, C. Mummery, P. Loskill, D. Braeken, W. Eberle, M. Cipriano, L. Fernandez, M. Graef, X. Gidrol, N. Picollet-D'Hahan, B. Van Meer, I. Ochoa, M. Schutte and J. Van den Eijnden-van Raaij, Altex, 2019, 36, 481–492 CrossRef PubMed.
- U. Marx, T. Akabane, T. B. Andersson, E. Baker, M. Beilmann, S. Beken, S. Brendler-Schwaab, M. Cirit, R. David, E. M. Dehne, I. Durieux, L. Ewart, S. C. Fitzpatrick, O. Frey, F. Fuchs, L. G. Griffith, G. A. Hamilton, T. Hartung, J. Hoeng, H. Hogberg, D. J. Hughes, D. E. Ingber, A. Iskandar, T. Kanamori, H. Kojima, J. Kuehnl, M. Leist, B. Li, P. Loskill, D. L. Mendrick, T. Neumann, G. Pallocca, I. Rusyn, L. Smirnova, T. Steger-Hartmann, D. A. Tagle, A. Tonevitsky, S. Tsyb, M. Trapecar, B. Van de Water, J. Van den Eijnden-van Raaij, P. Vulto, K. Watanabe, A. Wolf, X. Zhou and A. Roth, Altex, 2020, 37, 365–394 Search PubMed.
- Food and Drug Administration (FDA), https://www.fda.gov/science-research/about-science-research-fda/advancing-alternative-methods-fda#:~:text=Objectives of FDA's Alternative Methods Working Group Discuss,development of draft performance criteria for such assays.
- S. Batista Leite, M. Zincke Dos Reis Fernandes Cipriano, D. Carpi, S. Coecke, M. Holloway, R. Corvi, A. Worth, J. F. Viegas Barroso and M. Whelan, Establishing the scientific validity of complex in vitro models, EUR 30556 EN, Publications Office of the European Union, Luxembourg, 2021, ISBN 978-92-76-28410-9, DOI:10.2760/376171, JRC122394.
- International Organization for Standardization, Interoperability of microfluidic devices — Guidelines for pitch spacing dimensions and initial device classification, 2016, ISO-IWA 23, 2016 Search PubMed.
- K. Fabre, B. Berridge, W. R. Proctor, S. Ralston, Y. Will, S. W. Baran, G. Yoder and T. R. Van Vleet, Lab Chip, 2020, 20, 1049–1057 RSC.
- A. R. Baudy, M. A. Otieno, P. Hewitt, J. Gan, A. Roth, D. Keller, R. Sura, T. R. Van Vleet and W. R. Proctor, Lab Chip, 2020, 20, 215–225 RSC.
- S. Casati, P. Aeby and I. Kimber, et al. , ATLA, Altern. Lab. Anim., 2009, 37(3), 305–312 CrossRef CAS PubMed.
- D. Kirkland, P. Kasper, H. J. Martus, L. Müller, J. van Benthem, F. Madia and R. Corvi, Mutat. Res., 2016, 795, 7–30 CAS.
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), ICH S5 (R3) guideline on reproductive toxicology: detection of toxicity to reproduction for medicinal products including toxicity to male fertility, 2020, ICH S5 (R3).
- F. Madia and R. Corvi, EURL ECVAM Genotoxicity and Carcinogenicity Consolidated Database of Ames Positive Chemicals, https://ec.europa.eu/jrc/en/scientific-tool/eurl-ecvam-genotoxicity-and-carcinogenicity-consolidated-database-ames-positive-chemicals).
- P. Jennings, M. Schwarz, B. Landesmann, S. Maggioni, M. Goumenou, D. Bower, M. O. Leonard and J. S. Wiseman, Arch. Toxicol., 2014, 88, 2099–2133 CrossRef CAS.
- D. E. Ingber, Adv. Sci., 2020, 7, 2002030 CrossRef CAS.
- A. Bandrowski, M. Brush, J. S. Grethe, M. A. Haendel, D. N. Kennedy, S. Hill, P. R. Hof, M. E. Martone, M. Pols, S. C. Tan, N. Washington, E. Zudilova-Seinstra and N. Vasilevsky, Brain Behav., 2016, 6, e00417 Search PubMed.
- International Organization for Standardization (ISO), Micro process engineering — Vocabulary, 2009, ISO 10991, 2009 Search PubMed.
- ANSI/SLAS, Microplates: Footprint Dimensions, 2004, ANSI SLAS 1–2004.
- ANSI/SLAS, Microplates: Height Dimensions, 2004, ANSI SLAS 2–2004.
- ANSI/SLAS, Microplates: Bottom Outside Flange Dimensions, 2004, ANSI SLAS 3–2004.
- ANSI/SLAS, Microplates: Well Positions, 2004, ANSI SLAS 4–2004.
- ANSI/SLAS, Microplates: Well Bottom Elevation, 2012, ANSI SLAS 6–2012.
- International Society for Automation (ISA), Batch Control, 2010, ANSI/ISA-88.
- ASTM International, Standard Guide for Silicone Elastomers, Gels, and Foams Used in Medical Applications Part I—Formulations and Uncured Materials, 2018, ASTM F2038-18.
- ASTM International, Standard Guide for Silicone Elastomers, Gels, and Foams Used in Medical Applications Part II—Crosslinking and Fabrication, 2018, ASTM F2042-18.
- VDI - the Association of German Engineers, Medical Grade Plastics, 2017, VDI2017.
- ASTM International, Standard Practice for Selecting Generic Biological Test Methods for Materials and Devices, 2016, ASTM F748 - 16.
- S. Coecke, M. Balls, G. Bowe, J. Davis, G. Gstraunthaler, T. Hartung, R. Hay, O. W. Merten, A. Price, L. Schechtman, G. Stacey and W. Stokes, ATLA, Altern. Lab. Anim., 2005, 33, 261–287 CrossRef CAS PubMed.
- D. Pamies, A. Bal-Price, C. Chesné, S. Coecke, A. Dinnyes, C. Eskes, R. Grillari, G. Gstraunthaler, T. Hartung, P. Jennings, M. Leist, U. Martin, R. Passier, J. C. Schwamborn, G. N. Stacey, H. Ellinger-Ziegelbauer and M. Daneshian, Altex, 2018, 35, 353–378 CrossRef.
- D. Pamies, A. Bal-Price, A. Simeonov, D. Tagle, D. Allen, D. Gerhold, D. Yin, F. Pistollato, T. Inutsuka, K. Sullivan, G. Stacey, H. Salem, M. Leist, M. Daneshian, M. C. Vemuri, R. McFarland, S. Coecke, S. C. Fitzpatrick, U. Lakshmipathy, A. Mack, W. B. Wang, D. Yamazaki, Y. Sekino, Y. Kanda, L. Smirnova and T. Hartung, Altex, 2017, 34, 95–132 Search PubMed.
- D. Pamies, M. Leist, S. Coecke, G. Bowe, D. Allen, G. Gstraunthaler, A. Bal-Price, F. Pistollato, R. DeVries, T. Hartung and G. Stacey, Altex, 2020, 37, 490–492 CrossRef.
- Organisation for Economic Co-operation and Development (OECD), Guidance Document on Good In Vitro Method Practices (GIVIMP) - Series on Testing and Assessment No. 286, 2018 Search PubMed.
- M. Baker, Nature, 2016, 533, 452–454 CrossRef CAS PubMed.
- Organisation for Economic Co-operation and Development (OECD), Guidance document for describing non-guideline in vitro test methods - Series on Testing and Assessment No. 211, 2014 Search PubMed.
- A. Krebs, T. Waldmann, M. F. Wilks, B. M. A. Van Vugt-Lussenburg, B. Van der Burg, A. Terron, T. Steger-Hartmann, J. Ruegg, C. Rovida, E. Pedersen, G. Pallocca, M. Luijten, S. B. Leite, S. Kustermann, H. Kamp, J. Hoeng, P. Hewitt, M. Herzler, J. G. Hengstler, T. Heinonen, T. Hartung, B. Hardy, F. Gantner, E. Fritsche, K. Fant, J. Ezendam, T. Exner, T. Dunkern, D. R. Dietrich, S. Coecke, F. Busquet, A. Braeuning, O. Bondarenko, S. H. Bennekou, M. Beilmann and M. Leist, Altex, 2019, 36, 682–699 CrossRef.
- A. Bal-Price, H. T. Hogberg, K. M. Crofton, M. Daneshian, R. E. FitzGerald, E. Fritsche, T. Heinonen, S. Hougaard Bennekou, S. Klima, A. H. Piersma, M. Sachana, T. J. Shafer, A. Terron, F. Monnet-Tschudi, B. Viviani, T. Waldmann, R. H. S. Westerink, M. F. Wilks, H. Witters, M. G. Zurich and M. Leist, Altex, 2018, 35, 306–352 CrossRef PubMed.
- S. Brunak, C. Bjerre Collin, K. Eva Ó Cathaoir, M. Golebiewski, M. Kirschner, I. Kockum, H. Moser and D. Waltemath, J. Integr. Bioinform., 2020, 17(2–3), 20200006 CrossRef.
- Elsevier, MethodsX, https://www.journals.elsevier.com/methodsx.
- E. Marcus, Cell, 2016, 166, 1059–1060 CrossRef CAS.
- L. Molander, M. Ågerstrand, A. Beronius, A. Hanberg and C. Rudén, Hum. Ecol. Risk Assess., 2015, 21(3), 753–762 CrossRef CAS.
- T. Hartung, S. Bremer, S. Casati, S. Coecke, R. Corvi, S. Fortaner, L. Gribaldo, M. Halder, S. Hoffmann, A. J. Roi, P. Prieto, E. Sabbioni, L. Scott, A. Worth and V. Zuang, ATLA, Altern. Lab. Anim., 2004, 32, 467–472 CrossRef CAS PubMed.
- Organisation for Economic Co-operation and Development (OECD), OHT 201 Intermediate effects - mechanistic information, 2020 Search PubMed.
- ASTM International, Standard Classification for Tissue Engineered Medical Products (TEMPs), 2013, ASTM F2211 - 13.
- ASTM International, Standard Guide for Evaluation of in vitro Release of Biomolecules from Biomaterials Scaffolds for TEMPs, 2016, ASTM F3142 - 16.
- European Parliament and Council of the European Union, Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC, 2017.
- European Parliament and Council of the European Union, Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU, 2017.
- B. J. van Meer, H. de Vries, K. S. A. Firth, J. van Weerd, L. G. J. Tertoolen, H. B. J. Karperien, P. Jonkheijm, C. Denning, A. P. IJzerman and C. L. Mummery, Biochem. Biophys. Res. Commun., 2017, 482(2), 323–328 CrossRef CAS.
- A. W. Auner, K. M. Tasneem, D. A. Markov, L. J. McCawley and M. S. Hutson, Lab Chip, 2019, 19, 864–874 RSC.
- G. L. Truisi, E. D. Consiglio, C. Parmentier, C. C. Savary, G. Pomponio, F. Bois, B. Lauer, R. Jossé, P. G. Hewitt, S. O. Mueller, L. Richert, A. Guillouzo and E. Testai, Toxicol. Lett., 2015, 233, 172–186 CrossRef CAS PubMed.
- J. M. Z. Comenges, E. Joossens, J. V. S. Benito, A. Worth and A. Paini, Toxicol. In Vitro, 2017, 45, 209–221 CrossRef CAS PubMed.
- R. Graepel, L. Lamon, D. Asturiol, E. Berggren, E. Joossens, A. Paini, P. Prieto, M. Whelan and A. Worth, Toxicol. In Vitro, 2017, 45, 258–267 CrossRef CAS.
- A. Paini, M. Mennecozzi, T. Horvat, K. Gerloff, T. Palosaari, J. V. Sala Benito and A. Worth, Toxicol. In Vitro, 2017, 45, 233–240 CrossRef CAS.
- International Organization for Standardization, Biological evaluation of medical devices — Part 1: Evaluation and testing within a risk management process, 2018, ISO 10993-1, 2018.
- ASTM International, Standard Practice for Extraction of Materials Used in Medical Devices, 2020, ASTM F619 - 20.
- H. Bruus, Theoretical Microfluidics, OUP Oxford, 2008 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1lc00241d |
| ‡ https://www.cencenelec.eu/news/events/Pages/EV-2021-20.aspx |
| § https://www.standardscoordinatingbody.org/project-organonachip-standards-landscape-assessment |
| This journal is © The Royal Society of Chemistry 2021 |