
Synergy between dynamic covalent boronic ester and boron–nitrogen coordination: strategy for self-healing polyurethane elastomers at room temperature with unprecedented mechanical properties†
Kai
Song
a,
Wujin
Ye
a,
Xingchen
Gao
a,
Huagao
Fang
 *ab,
Yaqiong
Zhang
c,
Qi
Zhang
d,
Xueliang
Li
e,
Shanzhong
Yang
ab,
Haibing
Wei
*ab and
Yunsheng
Ding
*ab
*ab,
Yaqiong
Zhang
c,
Qi
Zhang
d,
Xueliang
Li
e,
Shanzhong
Yang
ab,
Haibing
Wei
*ab and
Yunsheng
Ding
*ab
aDepartment of Polymer Science and Engineering, School of Chemistry and Chemical Engineering, Hefei University of Technology, Hefei, Anhui 230009, China. E-mail: fanghg@hfut.edu.cn; dingys@hfut.edu.cn; hbwei@hfut.edu.cn
bAnhui Province Key Laboratory of Advanced Functional Materials and Devices, Hefei, Anhui 230009, China
cBiomass Molecular Engineering Center, School of Forestry and Landscape Architecture, Anhui Agricultural University, Hefei, Anhui 230036, China
dAnhui Province Key Lab of Aerospace Structural Parts Forming Technology and Equipment, Institute of Industry & Equipment Technology, Hefei University of Technology, Hefei, Anhui 230009, China
eAnhui Province Key Laboratory of Advanced Catalytic Materials and Reaction Engineering, School of Chemistry and Chemical Engineering, Hefei University of Technology, Hefei, Anhui 230009, China
First published on 5th November 2020
Abstract
Achieving mechanical robustness and highly efficient self-healing simultaneously at room temperature is always a formidable challenge for polymeric materials. Herein, a series of novel supramolecular polyurethane elastomers (SPUEs) are developed by incorporating dynamic covalent boronic ester and boron–nitrogen (B–N) coordination. The SPUEs demonstrate the highest tensile toughness (∼182.2 MJ m−3) to date for room-temperature self-healable polymers, as well as an excellent ultimate tensile strength (∼10.5 MPa) and ultra-high fracture energy (∼72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 J m−2), respectively, owing to a synergetic quadruple dynamic mechanism. It is revealed that the B–N coordination not only facilitates the formation and dissociation of boronic ester at room temperature but also dramatically enhances the mechanical properties by the intermolecular coordinated chain crosslinking and intramolecular coordinated chain folding. Meanwhile, the B–N coordination and urethane hydrogen interaction also serve as sacrificial bonds, which rupture during stretching to dissipate energy and recover after release, leading to superior notch insensitiveness and recoverability. The SPUEs restore their mechanical robustness after self-healing at room temperature and the self-healing efficiency can be dramatically accelerated by surface wetting.
100 J m−2), respectively, owing to a synergetic quadruple dynamic mechanism. It is revealed that the B–N coordination not only facilitates the formation and dissociation of boronic ester at room temperature but also dramatically enhances the mechanical properties by the intermolecular coordinated chain crosslinking and intramolecular coordinated chain folding. Meanwhile, the B–N coordination and urethane hydrogen interaction also serve as sacrificial bonds, which rupture during stretching to dissipate energy and recover after release, leading to superior notch insensitiveness and recoverability. The SPUEs restore their mechanical robustness after self-healing at room temperature and the self-healing efficiency can be dramatically accelerated by surface wetting.
New conceptsAchieving mechanical robustness and healing efficiency simultaneously at room temperature is a great challenge in self-healing materials. Previously, the dynamic boronic ester and boron–nitrogen (B–N) coordination were often used to fabricate self-healing polymers, in which the boronic ester-containing polymer was coordinated with the ortho-site nitrogen donor or that from the exotic small molecules. This coordination was reported to be effective to accelerate the dynamics of transesterification of boronic ester and beneficial to the self-healing performance, but has no contributions to the mechanical enhancement. The current work highlights the synergy between the boronic ester and B–N coordination to improve the mechanical properties and the self-healing efficiency in polyurethanes. Two types of B–N coordination are formed when the boron- and nitrogen-containing chain extenders are incorporated into the backbone of polyurethane. The resultant supramolecular polyurethane elastomers (SPUEs) demonstrate the unprecedented mechanical properties because of the coordinated intermolecular crosslinking and intramolecular folding. The coordination also serves as a sacrificial bond to dissipate energy during stretching, leading to superior toughness and notch insensitiveness. Moreover, the SPUEs show high self-healing efficiency because of the accelerated transesterification of the boronic ester, especially when healed with the aid of water. |
1 Introduction
The ability to autonomously repair the damage of self-healable polymers, not only extends the lifetime and improves the safety of service, but also decreases the material consumption and reduces maintenance costs, making them a class of promising materials with numerous potential applications in the field of wearable and implantable electronics, smart coatings, and durable sensors.1–5According to the different mechanisms, these materials can be categorized into two types: extrinsic and intrinsic.6,7 Extrinsic self-healing is based on an elaborate design in which reparable monomers and initiators are entrapped in capsules,1 vascular networks8 or their hybrids,9 which are dispersed beforehand in the polymer matrix. Once damaged, the reaction of the monomers starts with the aid of initiators, and then the crack is repaired. Even though the method is reliable, the limited number of healing times seriously inhibits its further development. In contrast, the intrinsic-type mechanism enables polymeric materials to repeatedly repair, which depends on the polymers themselves and is originated from the reconstruction of dynamic noncovalent interactions10–13 or dynamic covalent bonds.2,14–16 Intrinsic type polymeric materials lack the need for healing reagents, but external stimuli input usually is required, e.g., light,17 heat,2,15 pressure or others.3,18,19
Polymeric materials that can autonomously repair at room temperature are highly desired because most materials are used at ambient conditions under which it is inconvenient or inhibited to input external energy. To realize autonomous self-healability at room temperature, the following two criteria must be met in polymeric materials: (1) dynamic noncovalent interactions or dynamic covalent bonds that are readily operable at room temperature. Such dynamic noncovalent interactions include hydrogen bonds,3,12,20,21 metal–ligand bonds,10,22 host–guest interactions,23 ionic interactions,11 and van der Waals forces,24 and dynamic covalent bonds include urea chemistry,16 disulfide metathesis,25 oxime chemistry,26 diselenide metathesis,27 boronic ester,15,19,28,29 and boroxine.18 (2) Sufficient molecular mobility that facilitates reconstruction of dynamic bonding at room temperature. These two criteria ensure that polymeric materials self-heal within a reasonable timescale. But the mechanical robustness and efficient self-healing of the polymeric materials are contradictory properties, and the improvement of the mechanical properties is usually achieved at the expense of the self-healing efficiency. To reconcile this contradiction, scientists have proposed different strategies. Guan and co-workers proposed a multiphase design strategy, in which the covalent connectivity in the soft segments of traditional TPEs was replaced by non-covalent hydrogen bonds.20 You and co-workers proposed a dynamic covalent bond and dynamic non-covalent bond cooperative strategy in which the copper ion–oxime coordination played a crucial role. The resultant polymer possessed a tensile strength of up to 14.8 MPa and a significantly higher toughness (87.0 MJ m−3) than all the previously reported room-temperature self-healing elastomers.26
Boronic acids, which are known to form a variety of dynamic covalent bonds and dynamic noncovalent bonds, have been widely employed to fabricate covalent organic frameworks (COFs),30 molecular networks31 and self-healing polymeric materials.15,18,19,32,33 The direction of boroxines/boronic acids and boronic esters/boronic acids can be simply adjusted by temperature, the addition of water, or the addition of Lewis bases. Previous researchers have demonstrated that a nitrogen-donor, such as small molecular additives pyridine and amine29 or ortho-amino,18,28 imine,34 can coordinate with the boron atom of boroxines or boronic esters, leading to the acceleration of the equilibrium of boroxines and boronic esters at room temperature and the improved self-healing efficiency of the resultant polymeric materials.18,28 However, the coordination does not contribute to the strength of the material because it hardly enhances the interaction between the molecular chains. Herein, we present a dynamic covalent boronic ester and boron–nitrogen coordination synergy strategy to design and prepare supramolecular polyurethane elastomers (SPUEs). For this purpose, a synthetic dihydroxyl-bearing boronic ester and commercialized tertiary amine-bearing diol are used as the chain extenders to incorporate boronic esters and nitrogen-donors into the backbone of polyurethane, separately. Boron–nitrogen coordination bonds form between boronic esters and nitrogen-donors, which not only facilitates the formation and dissociation of boronic ester at room temperature but also dramatically enhances the mechanical properties by the intermolecular coordinated chain crosslinking and intramolecular coordinated chain folding. Meanwhile, the B–N coordination and urethane hydrogen interaction also serve as sacrificial bonds, which rupture during stretching to dissipate energy and recover after releasing, leading to superior notch insensitiveness and recoverability.
2 Results and discussion
Material design and characterizations
SPUEs were readily synthesized by two-step polymerization from commercially available polytetramethylene ether glycol (PTMEG), isophorone diisocyanate (IPDI), N-(3-dimethylaminopropyl)-N,N-diisopropanolamine (DPA) and the customized 2,2′-(1,4-phenylene)-bis[4-(4-hydroxybutyl)-1,3,2-dioxaborolane] (HDB) (Fig. S1, ESI†) in the presence of dibutyltin dilaurate (DBTDL) as the catalyst. The detailed reaction procedure is described in the ESI† (Fig. S2) and the resultant polymers are denoted as PU–BNx, in which x indicates the weight content of HDB. HDB and DPA are the keys to the design of the SPUEs. HDB acts as the chain extender to introduce reversible boronic esters, in which the flexible hexyl segments enhance the mobility of the boronic esters. DPA acts as the chain extender to incorporate nitrogen-donors to facilitate the formation of boron–nitrogen (B–N) coordination with the boronic esters (Fig. 1).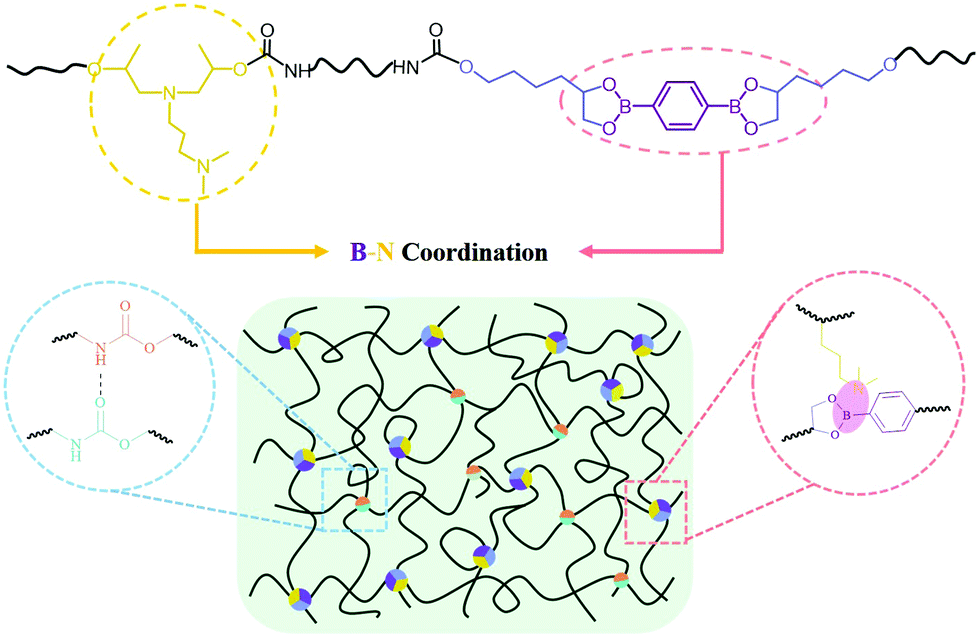 | ||
| Fig. 1 The proposed ideal supramolecular structure of SPUEs. | ||
To verify the hydrolytic reversibility of boronic esters in the present study, HDB was evaluated in a model experiment by 1H NMR spectroscopy under both dry and wet conditions to show the extreme shifts in equilibrium. The dry sample demonstrated a clear downfield shift of the protons near the oxygen atom of the boronic esters. After adding a small amount of water and waiting for 30 min, all of the boronic esters were completely hydrolyzed to their corresponding boronic acids and triol components (Fig. 2a), clearly showing the hydrolytic reversibility. The configuration of B–N coordination between HDB and DPA was investigated by density functional theory (DFT) calculation. The phenyl group of HDB is quite coplanar with the boronic ester. However, in the tertiary amine-coordinated boronic ester, the phenyl group is noncoplanar with the boronic ester ring due to the formation of a B–N coordination bond (Fig. 2b). Two possible configurations existed in the tertiary amine-coordinated boronic ester, which is a B–N1 and B–N2 adduct with the coordination bond length of 1.707 Å and 1.754 Å, respectively. The electronic energy of eqn (S1) (B–N1) and eqn (S2) (B–N2) (ESI†) was −13.98 and 1.16 kcal mol−1, respectively (Table S2, ESI†), revealing that the B–N1 adduct is favorable compared to the B–N2 adduct. The 11B NMR spectrum of HDB exhibited a peak at around 30 ppm, which was consistent with the trigonal sp2 boron center in boronic esters. The addition of DPA led to the upfield shift of the boron center to 11 ppm, which was consistent with the tetrahedral sp3 boron center in boronic esters involving B–N coordination.29 As the molar ratio of DPA:HDB increased, the intensity of the sp3 boron center increased and the intensity of the sp2 boron center decreased. When the molar ratio reached 1:2, the sp2 boron center completely disappeared (Fig. 2c). According to the literature, the B–N coordination facilitated the formation of boronic esters by accelerating the equilibrium of esterification at room temperature and was beneficial to the self-healing of polymeric materials.29 Meanwhile, DFT calculations indicated that the length of B–O bonds near the B–N coordination bond in B–N1 and B–N2 adducts was 1.451 Å, which was longer than those in boronic esters (1.369 Å), thereby expediting the dissociation of boronic esters (Table S2, ESI†).18 Therefore, it can be inferred that the formation and dissociation of nitrogen-coordinated boronic esters is highly reversible and can be beneficial to improve self-healing efficiency. The ATR-FTIR spectroscopy of the PU–BNx showed the complete consumption of the NCO group reflected by the absence of the peak at 2266 cm−1. The peaks at 3318, 1720 and 1698 cm−1 were ascribed to N–H, free and hydrogen-bonded C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations, respectively, indicating the formation of urethane groups. The negligible peak at 1745 cm−1, which corresponds to DMAc, was confirmed to exclude solvent-mediated chain mobility for self-healing. The peaks at around 1367 and 1312 cm−1 corresponded to B–C stretching vibration, and those at 1025 and 661 cm−1 corresponded to B–O stretching vibration and out-of-plane displacement of a boron atom diagnostic of the formation of boronic esters, respectively (Fig. S3, ESI†).3511B NMR spectroscopy of the PU–BNx showed only one peak around 11 ppm, which was consistent with a tetrahedral sp3 boron center involved in B–N coordination. PU–B9 without the nitrogen-donor showed one peak around 31 ppm, which was consistent with a trigonal sp2 boron center in boronic esters (Fig. 2d). The above results indicated that the boronic ester was incorporated in the polyurethane backbone and B–N coordination formed between the boronic ester and nitrogen-donor of DPA.
O stretching vibrations, respectively, indicating the formation of urethane groups. The negligible peak at 1745 cm−1, which corresponds to DMAc, was confirmed to exclude solvent-mediated chain mobility for self-healing. The peaks at around 1367 and 1312 cm−1 corresponded to B–C stretching vibration, and those at 1025 and 661 cm−1 corresponded to B–O stretching vibration and out-of-plane displacement of a boron atom diagnostic of the formation of boronic esters, respectively (Fig. S3, ESI†).3511B NMR spectroscopy of the PU–BNx showed only one peak around 11 ppm, which was consistent with a tetrahedral sp3 boron center involved in B–N coordination. PU–B9 without the nitrogen-donor showed one peak around 31 ppm, which was consistent with a trigonal sp2 boron center in boronic esters (Fig. 2d). The above results indicated that the boronic ester was incorporated in the polyurethane backbone and B–N coordination formed between the boronic ester and nitrogen-donor of DPA.
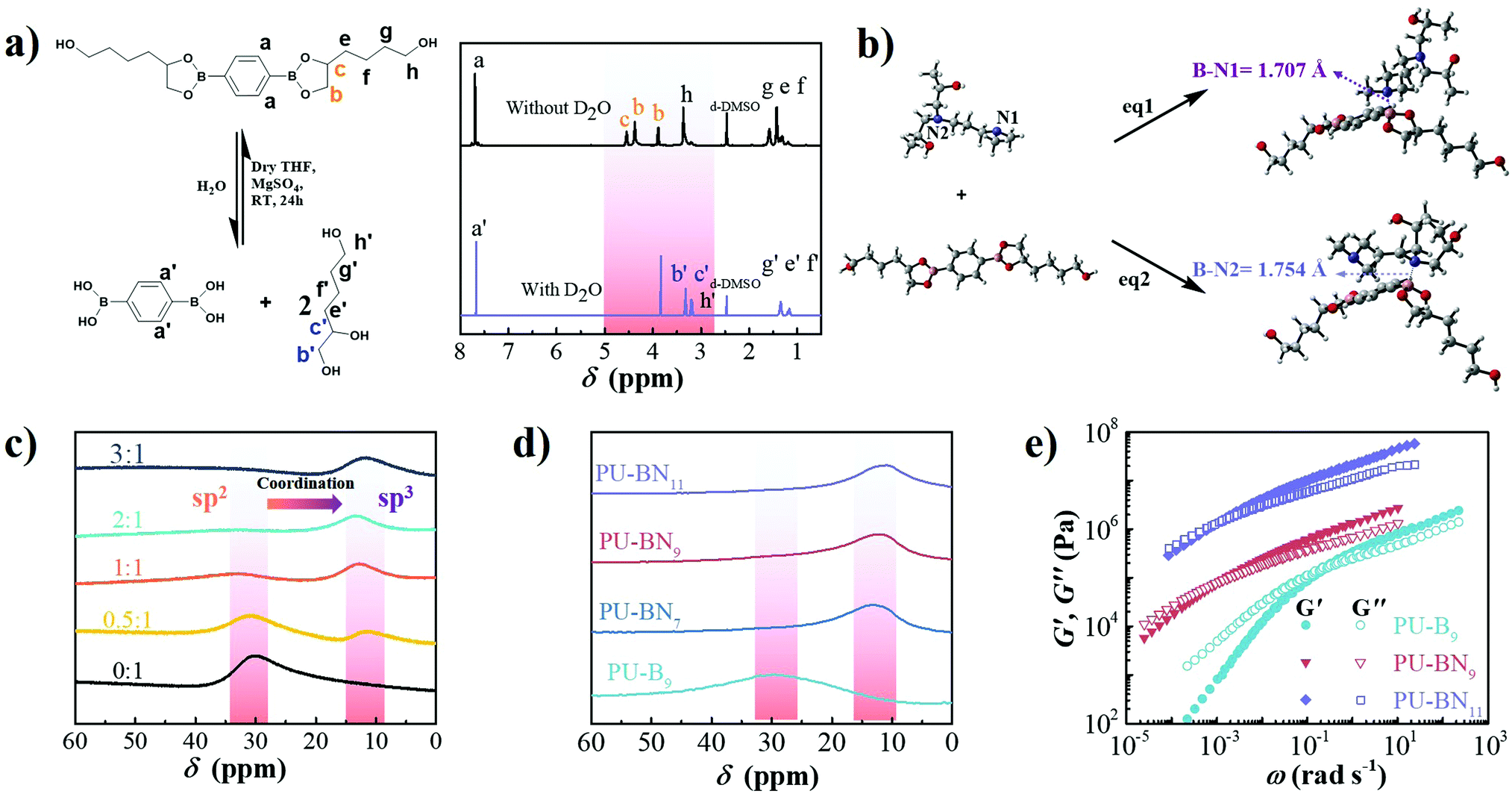 | ||
| Fig. 2 (a) 1H NMR spectra of HDB in d-DMSO before and after the addition of D2O. (b) DFT-calculated configurations of HDB, DPA, B–N1, and B–N2 adduct. (c) 11B NMR spectra of DPA and HDB with the different molar ratios in CDCl3. (d) 11B NMR spectra of PU–B9, and PU–BNx in CDCl3. (e) Master curves of PU–B9, PU–BN9, and PU–BN11 at Tref of 25 °C. | ||
Thermal, rheological, and mechanical properties
PU–BNx and PU–B9 have similar initial decomposition temperatures at around 220 °C, thus suggesting sufficient thermal stability in applications (Fig. S4, ESI†). All one-dimensional curves featured a broad amorphous halo around 19° and a tiny crystalline peak at 24.7° as compared with the pattern of HDB (Fig. S5 and S6, ESI†), suggesting that the crystallization of HDB was restricted after incorporating it in the polyurethane backbone. The small-angle X-ray scattering (SAXS) pattern of PU–B9 showed a broad peak of scattering vector, indicating weak microphase separation. The separation was restricted due to the B–N coordination and the peak moved to low vector, reflecting the inter-domain spacing and phase size increase with the content of B–N coordination (Fig. S7, ESI†). Small-amplitude oscillatory shear (SAOS) experiments were performed to gain more insights into the effect of B–N coordination on the bulk properties of PU–BNx. In the master curves scaled by time–temperature superposition (TTS) (eqn (S1), ESI†), PU–B9 demonstrated a typical response for viscous materials, showing the slope of G′ and G′′ in the low-frequency region close to the Maxwell model (approximate G′ ∝ ω2 and G′′ ∝ ω). Due to its simplicity, the reciprocal of crossover of G′ and G′′(1/ω0) was usually utilized to give the terminal relaxation time (τd = 1/ω0), which represented the lifetime of transient bonds and correlated to the healing efficiency of dynamic materials. The crossover point moved to the lower frequency and the modulus increased when the B–N coordination was incorporated, indicating the retarded terminal relaxation process and improved mechanical robustness (Fig. 2e). This is expected since the crosslinking effect was introduced with B–N coordination. Interestingly, as compared with PU–BN9, the increase of B–N coordination content in PU–BN11 only led to an obviously higher rubber plateau while it possessed a weak effect on terminal relaxation time, showing roughly identical frequency at the crossover point in PU–BN9 and PU–BN11 (Table S3, ESI†).The mechanical properties are shown in Fig. 3 and some parameters including the yield strength, Young's modulus, ultimate tensile strength, elongation at break, and tensile toughness are collected and listed in Table S4 (ESI†). PU–BN7 demonstrated impressively ultra-high extensibility of ∼14000% strain and relatively low yield strength (∼0.6 MPa) (Fig. 3a and Movie S1, ESI†). The strength of the material improved dramatically with the increase of the B–N coordination. The ultimate tensile strength increased to 4.2 MPa for PU–BN9 and 10.5 MPa for PU–BN11, while the elongation at break dropped remarkably to 4960% and 3120%, respectively (Fig. 3b). In contrast, the reference samples (PU–B0 and PU–B9) only possessed a low yield strength of 0.35 MPa and 0.3 MPa, respectively, and showed the typical characteristic of a viscoelastic liquid after passing the yield point (Fig. S8 and Table S4, ESI†). However, PU–BN9 and PU–BN11 showed superior strength and extensibility as compared to most of the self-healing materials and tough hydrogels. It should be emphasized that the strength and toughness of PU–BN11 reached 10.5 MPa and 182.2 MJ m−3 respectively, and the toughness is significantly higher than other room temperature self-healing polymers (Fig. 3c and Table S5, ESI†). The high strength and stretchability of PU–BNx may originate from the B–N coordination between HDB and DPA which were incorporated into the PU backbone. In the previous work, Bao and coworkers fabricated a series of metal–ligand crosslinked poly(dimethylsiloxane) (PDMS), in which the ligands were incorporated into the polymer backbone. The materials showed high stretchability, due to the reversible formation of intermolecular coordination between different PDMS chains and intramolecular coordination within the same PDMS chain.36 A similar behavior can also be found in supramolecular polyurethane elastomers with 2-amino-4-hydroxy-6-methylpyrimidine (UPy) as the quadruple H-bonding motif.12 In our materials, a similar conformation was possibly formed via the B–N coordination. In the unstretched state, the intramolecular coordination results in the folding of the PU backbone, which enables a very high chain extension under deformation. Meanwhile, the intermolecular coordination contributes to the formation of the transient network, which dramatically enhances the mechanical properties. Because of the dynamic reversible nature of the B–N coordination, the opening of the inter- or intramolecular connection by progressively disrupting the B–N coordination bonds upon stretching should bring about the unfolding and sliding of the polymeric networks, thereby enhancing the stretchability and toughness of PU–BNx (Fig. 3d).
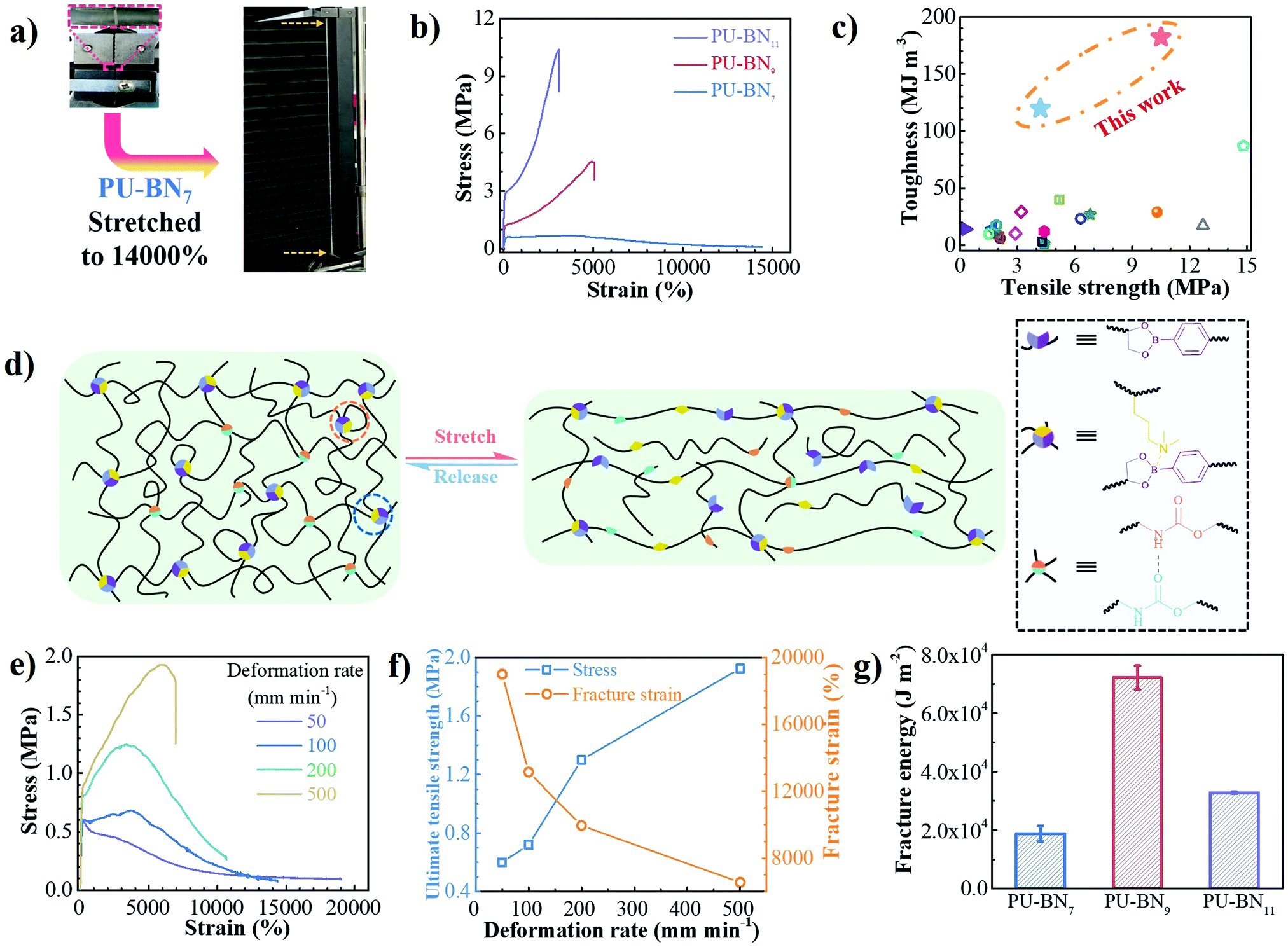 | ||
| Fig. 3 (a) Photographs of a PU–BN7 specimen before and after stretching to 14000%. (b) Stress–strain curves of PU–BNx. (c) Ashby plot of “toughness” versus “tensile strength” of PU–BN11, PU–BN9 and other room-temperature self-healable polymers reported in the literature. PU–BN11 exhibited the highest toughness. (d) The proposed mechanism of bond rupture and reformation in PU–BNx during stretching and releasing. Orange dotted circle: intramolecular B–N coordination; blue dotted circle: intermolecular B–N coordination. (e) Stress–strain curves of PU–BN7 at the deformation rates in the range of 50 to 500 mm min−1. (f) Deformation rate dependence of PU–BN7 on the ultimate tensile strength and fracture strain. (g) Fracture energy of PU–BNx. | ||
The deformation-rate-dependent tensile behaviors of the PU–BNx were consistent with the proposed chain dynamics. The tensile behavior of PU–BN7 demonstrated an obvious rate dependence, showing that the strain gradually decreased but the yield and ultimate tensile strength increased with increase of the deformation rate. The ultimate tensile strength increased by a factor of 3.2x, but the strain at break experienced a 2.7× decrease, along with the deformation rate increasing from 50 to 500 mm min−1 (Fig. 3e and f). PU–BN9 (Fig. S9, ESI†) and PU–BN11 (Fig. S10, ESI†) showed similar behaviors but the dependence became weak. It should be noted that PU–BN11 can be stretched to over 3000% of its original length at the high deformation rate of 500 mm min−1 with the yield and ultimate tensile strength of 4.2 and 10.7 MPa, respectively. This ability to maintain high extensibility and strength at the high deformation rate was rarely reported in other materials. The above results also indicated the dynamic characteristics of B–N coordination bonds which can undergo rapid dissociation and reconstruction under strain. Then, we performed a crack propagation experiment on single-edged notch samples. Using PU–BN9 as an example, the notch area became blunt dramatically during the stretching process and remained stable up to an average elongation of 1400% (Fig. S11 and Movie 2, ESI†). The fracture energies of PU–BN7, PU–BN9, and PU–BN11 were estimated to be 18800, 72100, and 32800 J m−2 using the Rivlin and Thomas method37 (Fig. 3g and Fig. S12, ESI†), respectively. This fracture energy (72100 J m−2) far exceeded the value of bulk elastomers reported by Bao and coworkers (12000 J m−2, published in 2018)38 and Sun and coworkers (22900 J m−2, published in 2017)39 which were calculated using the same method (Fig. S13 and S14, ESI†). In our SPUEs, the intermolecular B–N coordination and crystalline hard domains endowed the mechanical strength while the dynamic characteristic of inter- and intramolecular coordination, as well as the hydrogen bonds undoubtedly contributed to the notch-insensitivity with ultra-high fracture energy. The sufficient intermolecular physical bonding transferred the fracture stress concentrated at the notch front to the entire polymeric network via the possible synergy of the two toughening mechanisms: (1) crack bridging, which is enabled by the strong and sufficient B–N coordination bonds to postpone the transverse advancing crack front and (2) background hysteresis which is resulted from the dissociation of weak H-bonds to dissipate energy under strain.
To demonstrate the recoverability of the SPUEs, the repeated cyclic tensile test at a large strain of 500% for PU–BN11 as an example was shown in Fig. S15, ESI.† The large hysteresis circle in the first cycle indicated a significant energy dissipation by rupturing the dynamic crosslinks. An obvious reduction in the hysteresis circle area during the second loading–unloading cycle can be observed when the second cycle was conducted immediately after the first one. This was because the ruptured sacrificial bonds during the first loading did not have enough time to recover to their original state. The hysteresis circle slightly decreased with the increase of cycle in sequential tests, suggesting the balance of rupturing and reorganization of sacrificial bonds. When the sample was allowed to relax for 4 h at room temperature and subjected to the 6th cyclic tensile, it showed a similar curve to the first cycle, indicating the full recovery of dynamic crosslinks. The good recoverability of the PU–BN11 primarily originated from the highly dynamic B–N coordination within the networks that can be readily broken when the polymers are under stress to dissipate energy and reconstructed when the applied stress is released to allow the extended polymer chains to recover to their original state. The dynamic characteristics were also reflected by the temperature-dependent creep and recovery, in which irreversible deformation was prominent even at 30 °C (Fig. S16, ESI†). Although the toughness of the material is very high, the combination with irreversible deformation should be taken into account when applications are foreseen.
Self-healing properties
The dynamic characteristics of B–N coordination and hydrogen bonds, as well as the incorporation of hydrolytically reversible HDB units in the backbone, would endow the PU–BNx with self-healing capability at room temperature, especially with the aid of water. Surface scratch recovery tests were first performed by scratching the films with a razor blade. The X-shaped scratch on the film of PU–BN7 (Fig. S17, ESI†) almost completely disappeared within 0.5 h, suggesting autonomous self-healing behavior. The increase of B–N content resulted in a longer time needed for recovery, i.e., at least 2 h for PU–BN9 (Fig. S18, ESI†) and 20 h for PU–BN11 (Fig. S19, ESI†). PU–BN7 had good chain mobility, which contributed to its fast recovery. The high content of hard segments and B–N coordination of PU–BN9 and PU–BN11 largely increased its strength, but dramatically decreased its chain mobility, leading to the reduced self-healing ability. The bulky self-healing behavior of PU–BN9 (Fig. S20, ESI†) and PU–BN11 (Fig. 4a) was also evaluated. With the increase of healing time, improved self-healing efficiency was observed in both samples. For example, the self-healing efficiency of PU–BN9 was 59.3% after 12 h and climbed to 84.3% after 36 h, along with the large elongation at break of 4850%. While for PU–BN11, no further healing effect was observed after 24 h, and only 31.1% of efficiency was achieved in the process (Fig. 4b and c). The weakening of the self-healing efficiency was attributed to the increase of mechanical strength for PU–BN11, which slowed down the migration of the segments and reduced the availability of healing components on the damaged surfaces. It should also be stressed that despite the low self-healing efficiency, the healed PU–BN11 showed an ultimate tensile strength of 5.2 MPa, which is already among the highest strengths for room temperature self-healing materials (Table S5, ESI†). The self-healing behaviors of PU–BN9 and PU–BN11 can be dramatically improved with the aid of water. The X-shaped scratches of PU–BN9 (Fig. S21, ESI†) and PU–BN11 (Fig. 4d) almost completely recovered within 0.5 h and 1 h, respectively. It was observed that PU–BN11 self-healed for 24 h with the aid of water can hold a weight of 500 g which was about 4000 times the weight of the sheet itself and can be readily stretched to about 17 times of its original length (Fig. S22, ESI†). Fig. S23 (ESI†) and Fig. 4e depicted the representative stress–strain curves for PU–BN9 and PU–BN11 that were healed for different lengths of time with the aid of water, respectively. The strain can be completely recovered in 36 h and the calculated self-healing efficiency reached 91.7% for PU–BN9 (Fig. 4b). PU–BN11 also showed an improved self-healing performance with the aid of water and an efficiency of 67.8% was achieved after 36 h (Fig. 4c). It could be argued that the healing efficiency is not high enough, but the healed sample showed an ultimate tensile strength of 8.6 MPa, which is already among the highest strengths for the room temperature self-healing materials (Table S5, ESI†). We believe that the absolute restored strength could be a much more indicative parameter in self-healing materials than the healing efficiency itself. The self-healing mechanism of PU–BNx can be schematically described in Fig. S24 (ESI†). Without water, the B–N coordination bonds, the hydrogen bonds, and boronic ester metathesis contribute to the self-healing,15,29 but the limited chain mobility decreases its self-healing efficiency, especially for the stiffer polymer of PU–BN11. When the damaged surface is dipped in water, the B–N coordinated boronic esters on the surface are hydrolyzed into boronic acids and at the same time, the hydrogen bonds of urethane and B–N coordination bonds are also partially broken. The dissociation of boronic ester bonds, B–N coordination bonds, and hydrogen bonds dramatically enhanced the mobility of the polymer chains. When the two damaged surfaces are brought into contact, the reformation of boronic ester bonds, B–N coordination bonds, and hydrogen bonds takes place at room temperature upon drying. Thereby, the damaged PU–BNx are healed and their original mechanical properties are restored to a large extent.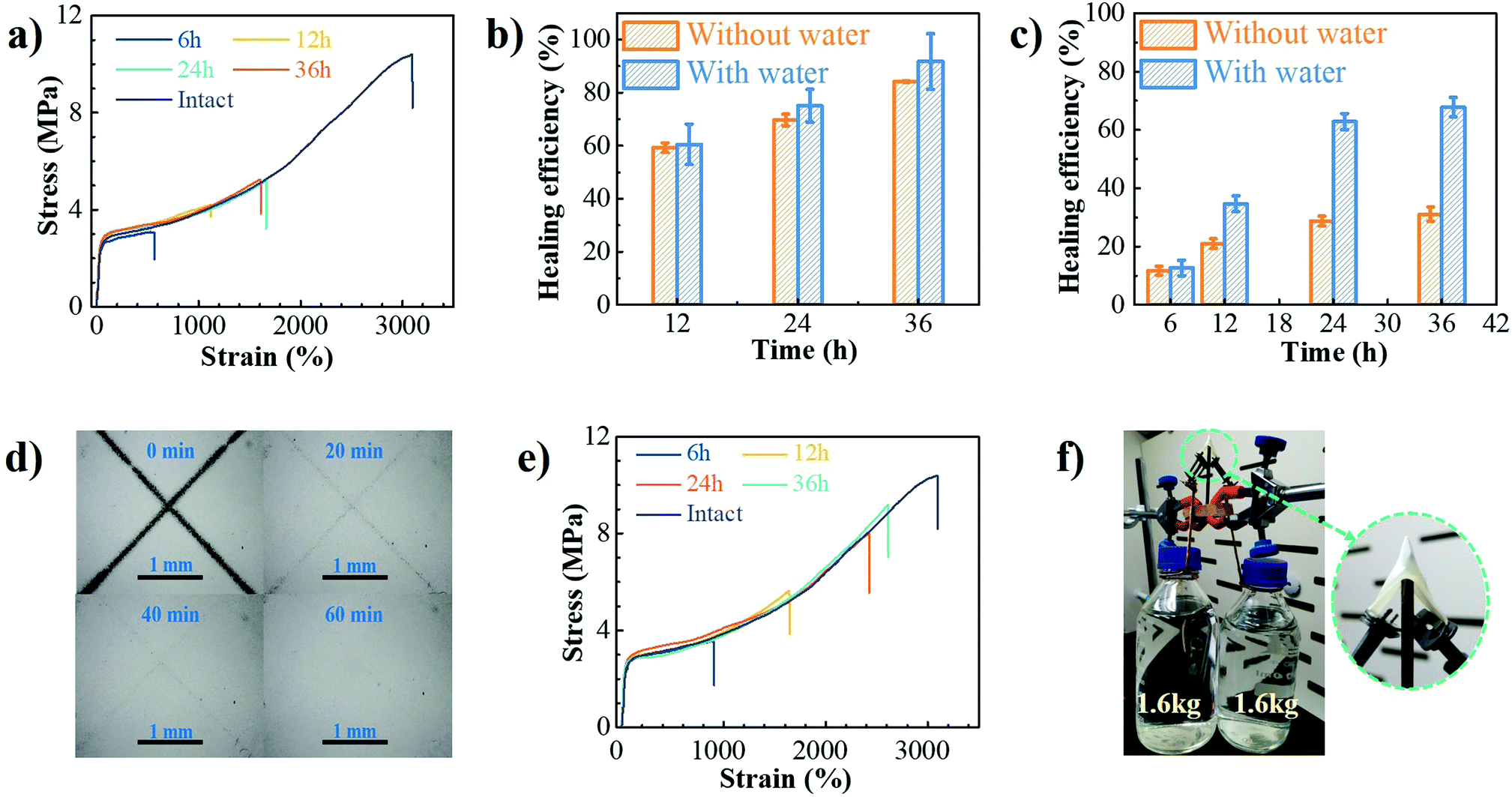 | ||
| Fig. 4 (a) Stress–strain curves of original and self-healed PU–BN11 at room temperature for different times without the aid of water. Self-healing efficiency of (b) PU–BN9 and (c) PU–BN11 with and without the aid of water for a different time at room-temperature. (d) Optical microscopy images of the X-shaped scratch on PU–BN11 films with the aid of water. (e) Stress–strain curves of original and self-healed PU–BN11 at room temperature for different time with the aid of water. (f) Puncture resistant property of PU–BN11. | ||
Applications
Because of their excellent mechanical properties, the SPUEs have promising applications in soft protective armor and wearable flexible electronics that can heal after routine damage. As described in Fig. 4f and Movie S3 (ESI†), the PU–BN11 can bear about 3.2 kg of weight with the puncture of a sharp nail, showing excellent puncture resistance. The exemplary application in flexible electronics of these self-healing PUs is tentatively demonstrated by fabricating a self-healable electrical conductor, which was constructed by directly brushing silver paste on the surface of PU–BN11. A circuit with this conductor and 3 V voltage batteries efficiently illuminated a connected lamp (Fig. S25, ESI†). When the composite conductor was cut into two separate pieces, the lamp was off. The damaged ends of the conductor were dipped into water for 1 min, then brought into contact for 2 min and the residual water was removed by drying the samples at room temperature for different time durations to heal the damage. Within 120 min, the two pieces readily self-healed, the lamp turned back on and the healed conductor can hold a weight of 50 g. It should be noted that the presence of water also has a dramatic effect on the mechanical properties of PU–BNx. After exposing in humidity at RH55, PU–BN11 could easily absorb water and demonstrated a dramatic decrease in the ultimate tensile strength with increasing exposure time. Meanwhile, the elongation at break exhibited an opposite change trend and achieved remarkable increase. Consequently, the tensile toughness that accounts both stress and strain exhibited a similar slower decline with increasing water content (Fig. S26 and S27, ESI†). As discussed above, the water absorbed in the sample could lead to the hydrolysis of boronic esters and breaking of the hydrogen bonds and B–N coordination, which deteriorated the mechanical properties seriously. Further works are needed to improve water resistance before their applications in broader areas.3 Conclusion
We designed and synthesized a series of novel room-temperature self-healing supramolecular polyurethane elastomers with outstanding mechanical properties. PU–BN11 exhibited excellent ultimate tensile strength (∼10.5 MPa) and superior stretchability (∼3120%), resulting in ultra-high tensile toughness (∼182.2 MJ m−3), which is twice the highest tensile toughness in the previously reported room-temperature self-healing polymeric materials. The material also demonstrated notch-insensitivity with the fracture energy of ∼72100 J m−2. Meanwhile, the material showed outstanding room temperature self-healing performance and the tensile strength reached 5.2 MPa after being healed for 24 h and the healing efficiency can be improved with the aid of water. The integration of mechanical robustness and self-healing was based on the synergistic effect of quadruple dynamic mechanisms including metathesis and hydrolysis-reesterification of boronic ester bonds, B–N coordination bonds, and hydrogen bonds. The incorporation of B–N coordination not only benefited the self-healing of polymeric materials but also dramatically enhanced the mechanical properties. Based on the demonstration of an auto-repairing electrical conductor and punch resistant film, we suggest that these mechanically robust, notch-insensitive, and self-healable SPUEs are promising materials in many advanced applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 51673056, 51503055 and 21702041), the National Key R&D Program of China (Grant No. 2017YFB0903800) and the Fundamental Research Funds for the Central Universities (Grant No. PA2020GDKC0009). The authors acknowledge Prof. Zhongkai Wang (Anhui Agricultural University) and Prof. Xiaoliang Wang (Nanjing University) for their assistance in experiments and insightful discussions.Notes and references
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS.
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Science, 2018, 359, 72–76 CrossRef CAS.
- J. Kang, J. B. H. Tok and Z. Bao, Nat. Electron., 2019, 2, 144–150 CrossRef.
- S. H. Cho, S. R. White and P. V. Braun, Adv. Mater., 2009, 21, 645–649 CrossRef CAS.
- D. Y. Wu, S. Meure and D. Solomon, Prog. Polym. Sci., 2008, 33, 479–522 CrossRef CAS.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- K. S. Toohey, N. R. Sottos, J. A. Lewis, J. S. Moore and S. R. White, Nat. Mater., 2007, 6, 581–585 CrossRef CAS.
- R. C. R. Gergely, W. A. S. Cruz, B. P. Krull, E. L. Pruitt, J. Wang, N. R. Sottos and S. R. White, Adv. Funct. Mater., 2017, 28, 1704197 CrossRef.
- D. Mozhdehi, S. Ayala, O. R. Cromwell and Z. Guan, J. Am. Chem. Soc., 2014, 136, 16128–16131 CrossRef CAS.
- Y. Miwa, J. Kurachi, Y. Kohbara and S. Kutsumizu, Chem. Commun., 2018, 1, 5 CrossRef.
- X. Yan, Z. Liu, Q. Zhang, J. Lopez, H. Wang, H. C. Wu, S. Niu, H. Yan, S. Wang, T. Lei, J. Li, D. Qi, P. Huang, J. Huang, Y. Zhang, Y. Wang, G. Li, J. B. Tok, X. Chen and Z. Bao, J. Am. Chem. Soc., 2018, 140, 5280–5289 CrossRef CAS.
- C. H. Li and J. L. Zuo, Adv. Mater., 2019, 1903762, DOI:10.1002/adma.201903762,.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS.
- M. Rottger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolay and L. Leibler, Science, 2017, 356, 62–65 CrossRef.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS.
- C. Bao, Y.-J. Jiang, H. Zhang, X. Lu and J. Sun, Adv. Funct. Mater., 2018, 28, 1800560 CrossRef.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098–2106 CrossRef CAS.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS.
- D. Wang, J. Xu, J. Chen, P. Hu, Y. Wang, W. Jiang and J. Fu, Adv. Funct. Mater., 2019, 30, 1907109 CrossRef.
- J. C. Lai, X. Y. Jia, D. P. Wang, Y. B. Deng, P. Zheng, C. H. Li, J. L. Zuo and Z. Bao, Nat. Commun., 2019, 10, 1164 CrossRef.
- J. Liu, C. S. Y. Tan, Z. Yu, N. Li, C. Abell and O. A. Scherman, Adv. Mater., 2017, 29, 1605325 CrossRef.
- M. W. Urban, D. Davydovich, Y. Yang, T. Demir, Y. Zhang and L. Casabianca, Science, 2018, 362, 220–225 CrossRef CAS.
- S. M. Kim, H. Jeon, S. H. Shin, S. A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 CrossRef.
- L. Zhang, Z. Liu, X. Wu, Q. Guan, S. Chen, L. Sun, Y. Guo, S. Wang, J. Song, E. M. Jeffries, C. He, F. L. Qing, X. Bao and Z. You, Adv. Mater., 2019, 31, 1901402 CrossRef.
- X. An, R. H. Aguirresarobe, L. Irusta, F. Ruipérez, J. M. Matxain, X. Pan, N. Aramburu, D. Mecerreyes, H. Sardon and J. Zhu, Polym. Chem., 2017, 8, 3641–3646 RSC.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS.
- C. Kim, H. Ejima and N. Yoshie, J. Mater. Chem. A, 2018, 6, 19643–19652 RSC.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O’Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef.
- E. Sheepwash, V. Krampl, R. Scopelliti, O. Sereda, A. Neels and K. Severin, Angew. Chem., Int. Ed., 2011, 50, 3034–3037 CrossRef CAS.
- F. Vidal, J. Gomezcoello, R. A. Lalancette and F. Jakle, J. Am. Chem. Soc., 2019, 141, 15963–15971 CrossRef CAS.
- A. P. Bapat, B. S. Sumerlin and A. Sutti, Mater. Horiz., 2020, 7, 694–714 RSC.
- S. Delpierre, B. Willocq, J. De Winter, P. Dubois, P. Gerbaux and J. M. Raquez, Chem. – Eur. J., 2017, 23, 6730–6735 CrossRef CAS.
- A. Breuillac, A. Kassalias and R. Nicolaÿ, Macromolecules, 2019, 52, 7102–7113 CrossRef CAS.
- C. H. Li, C. Wang, C. Keplinger, J. L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X. Z. You and Z. Bao, Nat. Chem., 2016, 8, 618–624 CrossRef CAS.
- R. S. Rivlin and A. G. Thomas, J. Polym. Sci., 1953, 10, 291–318 CrossRef CAS.
- J. Kang, D. Son, G. N. Wang, Y. Liu, J. Lopez, Y. Kim, J. Y. Oh, T. Katsumata, J. Mun, Y. Lee, L. Jin, J. B. Tok and Z. Bao, Adv. Mater., 2018, 30, e1706846 CrossRef.
- Y. Wang, X. Liu, S. Li, T. Li, Y. Song, Z. Li, W. Zhang and J. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 29120–29129 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0mh01142h |
| This journal is © The Royal Society of Chemistry 2021 |