
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrdered polymer composite materials: challenges and opportunities
Yuping
Wang
 ,
Griffen J.
Desroches
and
Robert J.
Macfarlane
*
,
Griffen J.
Desroches
and
Robert J.
Macfarlane
*
Department of Materials Science and Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA. E-mail: rmacfarl@mit.edu
First published on 14th December 2020
Abstract
Polymer nanocomposites containing nanoscale fillers are an important class of materials due to their ability to access a wide variety of properties as a function of their composition. In order to take full advantage of these properties, it is critical to control the distribution of nanofillers within the parent polymer matrix, as this structural organization affects how the two constituent components interact with one another. In particular, new methods for generating ordered arrays of nanofillers represent a key underexplored research area, as emergent properties arising from nanoscale ordering can be used to introduce novel functionality currently inaccessible in random composites. The knowledge gained from developing such methods will provide important insight into the thermodynamics and kinetics associated with nanomaterial and polymer assembly. These insights will not only benefit researchers working on new composite materials, but will also deepen our understanding of soft matter systems in general. In this review, we summarize contemporary research efforts in manipulating nanofiller organization in polymer nanocomposites and highlight future challenges and opportunities for constructing ordered nanocomposite materials.
 Robert J. Macfarlane | Rob Macfarlane received his Ph.D. in chemistry from Northwestern University in 2013, and subsequently was a Kavli Nanoscience Institute postdoctoral fellow at Caltech from 2013–2015. In 2015, he joined the Department of Materials Science and Engineering at the Massachusetts Institute of Technology, where he is currently the Paul M. Cook Associate professor. His research interests focus on the development of methods to synthesize hierarchically ordered polymer, nanoparticle, and biomaterial composite architectures via supramolecular-chemistry directed self-assembly. |
1. Introduction
Composites are materials composed of two or more constituent components with different physicochemical characteristics that exhibit hybrid properties unseen in either constituent individually. The development of composite materials can be traced back thousands of years, when people discovered that mixing dried mud and straw resulted in a new material that resisted both squeezing and tearing, allowing bricks of this composite to serve as excellent construction materials. Modern scientists have discovered that such effects also exist at the microscopic level, where the incorporation of nanoscale filler materials into polymer matrices can impart unique physical behaviors that arise due to the interactions between the matrix and filler. For example, mixing glass fibers into a polymer medium generates light weight fiberglass with outstanding mechanical properties, and mixing carbon black and silica with tire rubber improves wear resistance, leading to a longer service life. Significant research effort has therefore been put forth to study how the compositions of different nanofillers (e.g. their sizes, shapes, chemical make-up, and relative concentration in the matrix) affect the final properties of the nanocomposites. While these studies have given rise to greater fundamental knowledge as well as multiple industrially relevant technologies, a key area of structural control remains underinvestigated, specifically examination of how the relative organization of nanofiller materials throughout the polymer matrix affects material properties and behavior. The majority of polymer nanocomposites investigated in research laboratories and used in commercial applications consist primarily of nanofillers that are randomly dispersed throughout the polymer. While our ability to manipulate nanoscale ordering within a composite remains underdeveloped compared to other aspects of compositional control in these materials, there is ample evidence exhibited in natural nanocomposite materials that precise and hierarchical organization of the filler material is critical for achieving new materials and enabling new applications. For example, it is believed that nacre (mother of pearl) is strong and resilient because its mechanical properties are dictated by the lamellar alignment of aragonite platelets connected by thin layers of elastic biopolymers. The resulting nanocomposite possesses significantly higher fracture toughness than both the individual parent components by themselves and a random mixture of the two. It is also true that organized arrays of nanoparticles (NPs) have been observed to exhibit complex emergent and metamaterial phenomena as a result of structural ordering, such as photonic band gaps, controlled electrical transport, and anisotropic mechanical behavior. Incorporating these phenomena into composites by dictating nanoparticle organization would further provide a means to tune the physical properties of a composite for various applications including tissue engineering,1–3 sensing,4–7 energy storage,8,9conductors,10 and catalysis.11–13The development of methods for generating nanoscale organization of filler material is therefore critical for further progress in the field of composite materials science. Nevertheless, this research field is still in its infancy, and many important fundamental research questions are yet to be answered, such as (i) how nanofiller shape, size, and surface chemistry affect structural ordering and material organization at the nano, micro, and macroscopic length scales; (ii) how the formation of ordered arrays of nanofiller affect the behavior and properties of the composite, and (iii) how the properties of these arrays can be harnessed to develop materials with novel applications. This review will therefore highlight some of the state-of-the-art efforts that have been made for controlling nanoscale organization of individual components within polymer matrices, and provide a road map for elevating the field of nanocomposite synthesis by taking advantage of these different types of structural organization. Furthermore, we will highlight key opportunities that this structural control brings in terms of understanding structure–property relationships, as well as developing next-generation nanocomposite materials with novel properties in the near future. This summary of the current field should therefore serve as inspiration for other researchers in the field to explore different possibilities for making and studying ordered nanocomposite materials.
2. Approaches, achievements and opportunities
The toolbox for manipulating the structures of nanofillers within polymer matrices has been greatly enriched during the past several decades by advances in the fields of organic, polymer, and nanomaterial synthesis. Several methods have been developed to control the distributions of nanomaterial and polymers within the composite materials (Scheme 1). These methods can be classified into two categories based on the nature of the composite materials: (1) The nanofillers are addressed independently, and the polymer chains simply act as a medium to occupy space between the nanoobjects within the nanocomposite. In these types of methods, the alignment of nanoobjects is typically controlled by external forces based on their intrinsic properties (e.g., using magnetic fields to regulate the organization of magnetically responsive NPs). (2) The formation of the final composite materials relies on the interplay between the polymer chains and nanofillers, and supramolecular interactions guide the formation of complex particle arrangements. This second category can be further subdivided as a function of the in situ assembly mechanism: (i) an ordered polymer superstructure (e.g. a block copolymer array) serves as a template to dictate the distribution of the nanofillers throughout the composite by segregating the nanoparticles to different volumes within the material; (ii) the nanofillers and polymers are functionalized with chemical groups that have complementary interactions, and chemical bonding forces the particles to be heterogeneously distributed throughout a material (e.g. layer-by-layer deposition); (iii) polymer chains are directly tethered onto the surface of NPs, such that each building block is inherently a nanoscale composite, and the resulting composite material's structure is dictated by the assembly of these building blocks into higher ordered structures. We will expand our discussion in the following sections by highlighting the progress, advantages, challenges, and opportunities for each type of strategy. | ||
| Scheme 1 (a) Polymer nanocomposites with nanoscale structural ordering have been achieved via three principal means: (i) bare nanofiller and bulk polymer, (ii) surface-compatibilized nanofiller and bulk polymer, and (iii) nanofillers modified with complementary recognition pairs. The materials design principles used to create these ordered nanocomposites have thus far included (b) external field application,14,15 adapted with permission from ref. 14 (copyright 2016, American Chemical Society), ref. 15 (copyright 2019, Wiley-VCH), (c) spatial confinement of fillers,16,17 adapted with permission from ref. 16 (copyright 2012, American Chemical Society), ref. 17 (copyright 2013, American Chemical Society), (d) layer-by-layer deposition,18,19 adapted with permission from ref. 18 (copyright 2011, American Chemical Society), ref. 19 (copyright 2015, The Authors), and (e) self-assembly,20 adapted with permission from ref. 20 (copyright 2011, American Chemical Society). | ||
2.1. Field-assisted alignment of nanocomponents
A straightforward approach for manipulating nanofiller positions within a composite is to use applied fields to direct the assembly of particles into desired arrangements. In this type of approach, the polymer component typically serves as simply a space-filling medium that occupies the volume between organized nanofillers within the final material. It should be noted that many methods exist which use external fields to organize NPs in other media besides polymers, but this review will focus solely on controlling nanoparticle organization in composites; we direct the reader to other reviews that have covered particle assembly for more information on non-composite structures.21–27 The main advantage of using applied fields to control nanoscale organization is that they can be controlled remotely and independently from intrinsic nanomaterial design parameters like particle size or shape. For example, magnetic fields can regulate the alignment of magnetically-responsive nanofillers,28–30 such as paramagnetic nanoparticles,31–33 metal–oxide nanosheets,34 magnetically active nanofibers,35–38 and carbon nanotubes,39–42 resulting in the formation of nanofiller arrays with internally organized fillers. When incorporated into polymer matrixes, these well-ordered nanofillers can be fixed in position upon crosslinking of the polymer network, giving rise to the formation of a nanocomposite material with permanent ordered nanostructure. Wang and coworkers demonstrated this concept using very weak magnetic fields to drive the alignment of magnetite-carbon nanofibers in an epoxy nanocomposite.38 This process is likely difficult to scale, as nanofiber alignment must be achieved before gelation of the epoxy occurs, but at the scale demonstrated in this example, the nanofiber alignment proceeded rapidly even at a field strength of 50 mT. Within the final nanocomposites, the nanofiber fillers ran parallel to the direction of the applied magnetic field, resulting in anisotropic electrical and mechanical properties that differ significantly from those with randomly oriented nanofibers (Fig. 1a). In their example, electrical conductivity measured parallel with the aligned nanofibers was consistently an order of magnitude higher than that measured for randomly oriented nanofiber composites, while no such conductivity difference was observed when measuring normal to the aligned fibers (Fig. 1b). For mechanical properties, significant improvements in fracture toughness were observed when the crack surface was normal to the aligned fibers. In addition, when compared to the disorganized structures present in conventional magnetically responsive polymer composites, composites containing ordered arrays of magnetically active nanofillers exhibit more controlled and uniform thermogenesis behavior. All these observations demonstrate how controlling structural organization within the composite materials can modify material properties. Furthermore, the well-ordered nanomaterial arrays can be further utilized as a template to regulate the alignment of other small molecules within the composite network in such a way that the properties of the small molecules (e.g., absorption and emission bands) can be controlled (Fig. 1c).41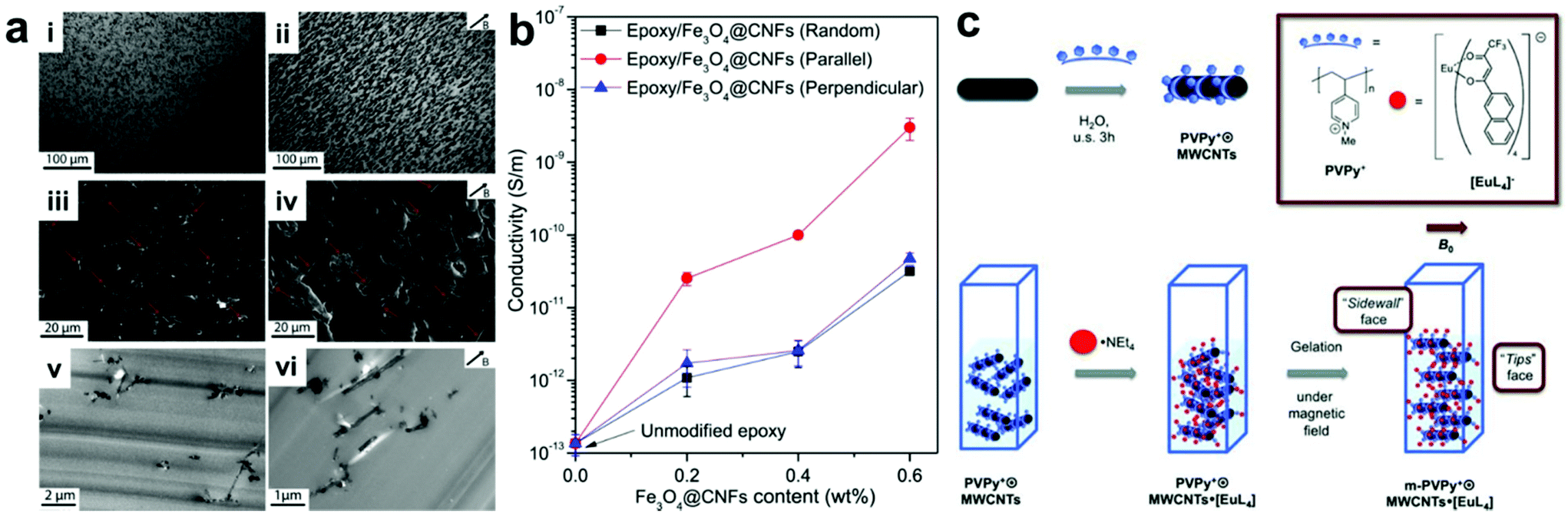 | ||
| Fig. 1 (a) Optical micrographs (top row), SEM (middle row) and TEM (bottom row) images of epoxy nanocomposites containing Fe3O4–C nanofibers in random orientation (left column) and aligned by applied magnetic field during curing (right column). (b) Electrical conductivity as a function of Fe3O4–C nanofiber content for randomly-oriented and field-aligned nanocomposites (measurement direction given in inset), demonstrating significant anisotropic enhancement of electrical properties owing to the uniaxial nanofiber alignment,38 adapted with permission from ref. 38 (copyright 1969, Elsevier). (c) Preparation of magnetic field-aligned nanocomposite hydrogels with rare earth-containing carbon nanotubes,41 adapted with permission from ref. 41 (copyright 2013, Wiley-VCH). | ||
Applied electric fields14,43–62 have also been used to control the organization of nanocomponents with permanent or induced dipole moments by means of dielectrophoretic forces (DEP),14,63–65 where the structure of the assembled nanocomponents can be manipulated by varying electrode geometry to create non-homogenous electric fields. As with magnetic field assembly, in situ polymerization is usually carried out after forming ordered nanofiller structures to fix their positions within the polymer matrix (Fig. 2). For example, Velev and coworkers reported the directed assembly of latex microspheres within a soft thermos-responsive hydrogel by manipulating DEP, and these field-oriented NP assemblies acted as endoskeletal structures within the polymer network which guide the macroscopic bending of the hydrogel (Fig. 2b).66
 | ||
| Fig. 2 (a) Optical micrograph (OM, left) and SEM (right) images of DEP-patterned nanocomposite hydrogels, demonstrating the ability of non-homogenous electric fields to drive the assembly of organized nanoparticle array,63 adapted with permission from ref. 63 (copyright 2007, American Chemical Society). (b) Schematic illustrations, OM images, and fluorescence micrographs demonstrating the effect of NP structural arrangement on hydrogel equilibrium bending. DEP-organized NPs act as an endoskeleton which guides hydrogel bending along a preferred axis perpendicular to that of the NP chain alignment,66 adapted with permission from ref. 66 (copyright 2016, Wiley-VCH). | ||
Mechanical force is another simple way for fabricating ordered structures of nanocomposite materials.67–69 Upon applying shear force, anisotropic nanoobjects undergo rotation and align in the direction parallel to the shear force;15,70–76 when tensile strength is applied to a polymer network, 1D and 2D nanofillers such as nanorods and nanoplatelets can be forced to align with the direction of deformation (Fig. 3a and b).77–80 A secondary step (i.e. crosslinking or in situ polymerization) can then be employed to fix the nanofillers in their “strain-induced” orientation,81,82 leading to anisotropic nanocomposite materials with physical properties that diverge significantly from their unaligned precursors. For example, Shigehara and coworkers demonstrated this concept with an imigolite-filled nanocomposite hydrogel, in which inorganic imigolite nanotubes were brought into alignment with the tensile axis under strain and then fixed in position via secondary in situ polymerization (Fig. 3c).81 The resulting material exhibited significant mechanical and optical anisotropy; the maximum tensile stress and strain were both significantly higher when measured perpendicular to the aligned imigolite nanotubes than when measured in the parallel direction. These materials also exhibited a degree of anisotropic birefringence that was only possible with fixed uniaxial nanotube alignment and that could only otherwise be achieved by placing uncrosslinked hydrogels under active tensile load. In another example, Yan and coworkers prepared a strain-aligned polyvinyl alcohol/silicon carbide nanowire composite without the use of a secondary crosslinking step; instead, the strain-induced uniaxial alignment of SiC nanowires was preserved in a kinetically-trapped state via film drying under tension.82 Compared to their unaligned counterparts, the strain-aligned SiC nanocomposites exhibited marked improvements in tensile strength, elastic modulus, and thermal conductivity, demonstrating the importance of nanomaterial alignment for the dissipation of both strain energy and heat.
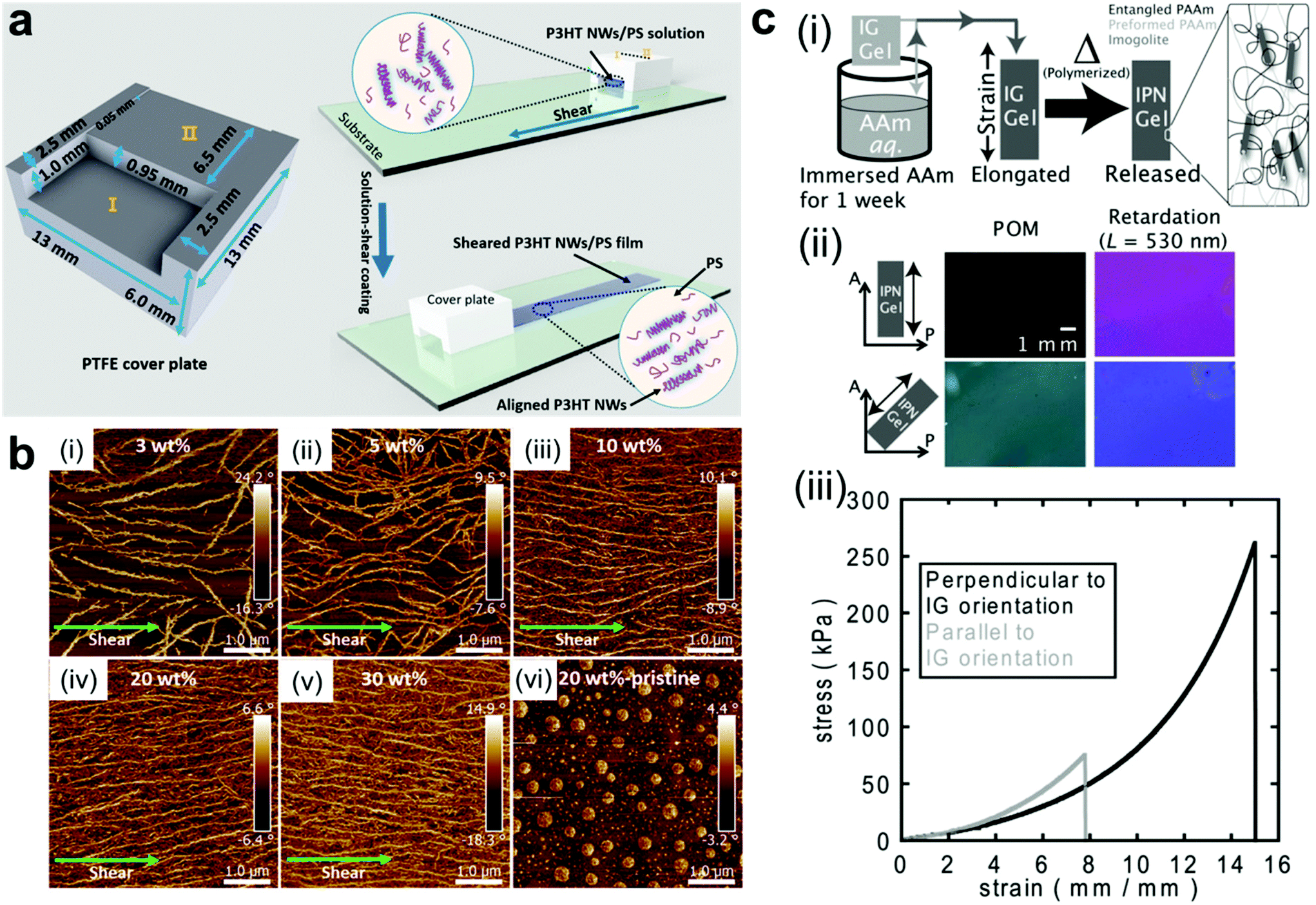 | ||
| Fig. 3 (a and b) Fabrication of uniaxially-aligned polymer nanowires in a nanocomposite thin film by solution shear coating, in which nanowire fillers align parallel to the shear axis.69 As the number density of nanowire fillers increases, the alignment of the nanowires along the shearing direction improves. Adapted with permission from ref. 69 (copyright 2016, American Chemical Society). (c-i) Preparation of strain-oriented imigolite nanocomposite hydrogels. As-synthesized hydrogels were placed under tension to bring imigolite fillers into parallel alignment, and the strain-induced alignment was then fixed with a secondary in situ polymerization. (c-ii) Polarized OM images of strain-oriented imigolite gels at different angles with respect to the polarizer, demonstrating strong anisotropic birefringence. (c-iii) Tensile stress–strain curves for strain-oriented imigolite gels measured both parallel and perpendicular to filler orientation, demonstrating clear mechanical anisotropy.81 Adapted with permission from ref. 81 (copyright 2014, Wiley). | ||
The use of applied fields presents a unique strategy for preparing ordered nanofiller arrays (especially in the case of anisotropic structures) due to the directional, noncontact nature of the applied field as well as the independence of this method from nanofiller geometry. However, it should be noted that this strategy can only be applied to nanofillers that are responsive to the applied field, and the methods outlined above each face their own challenges. Strong magnetic fields require bulky equipment to be in close proximity to the composite material, which increases the difficulty of material processing. Electric fields have difficulty in fabricating large-scale nanocomposites, as the applied voltage is limited by the electrochemical stability of the nanocomponents. Mechanical force has limited ability to control the alignment of nanoobjects with low aspect ratio and thus is typically used to align rod- or plate-like particles. In addition, while the alignment of the nanofillers in one direction (parallel or perpendicular to the applied field) can be well controlled, the distribution of these filler materials in the plane perpendicular to the applied field is generally either random (i.e., the composite has a nematic phase) or possesses only transient ordering which requires a continuous input of external stimuli. In other words, applied fields can be used to control the alignment of individual nanofillers but not their positions and distances relative to one-another. These intrinsic drawbacks have provided inspiration for researchers to develop additional approaches for controlling nanofiller distribution in a more precise fashion, where it is crucial to take the interactions between NPs, polymers and environment into account.
2.2. Strategies involving interplay between nanofillers and polymers
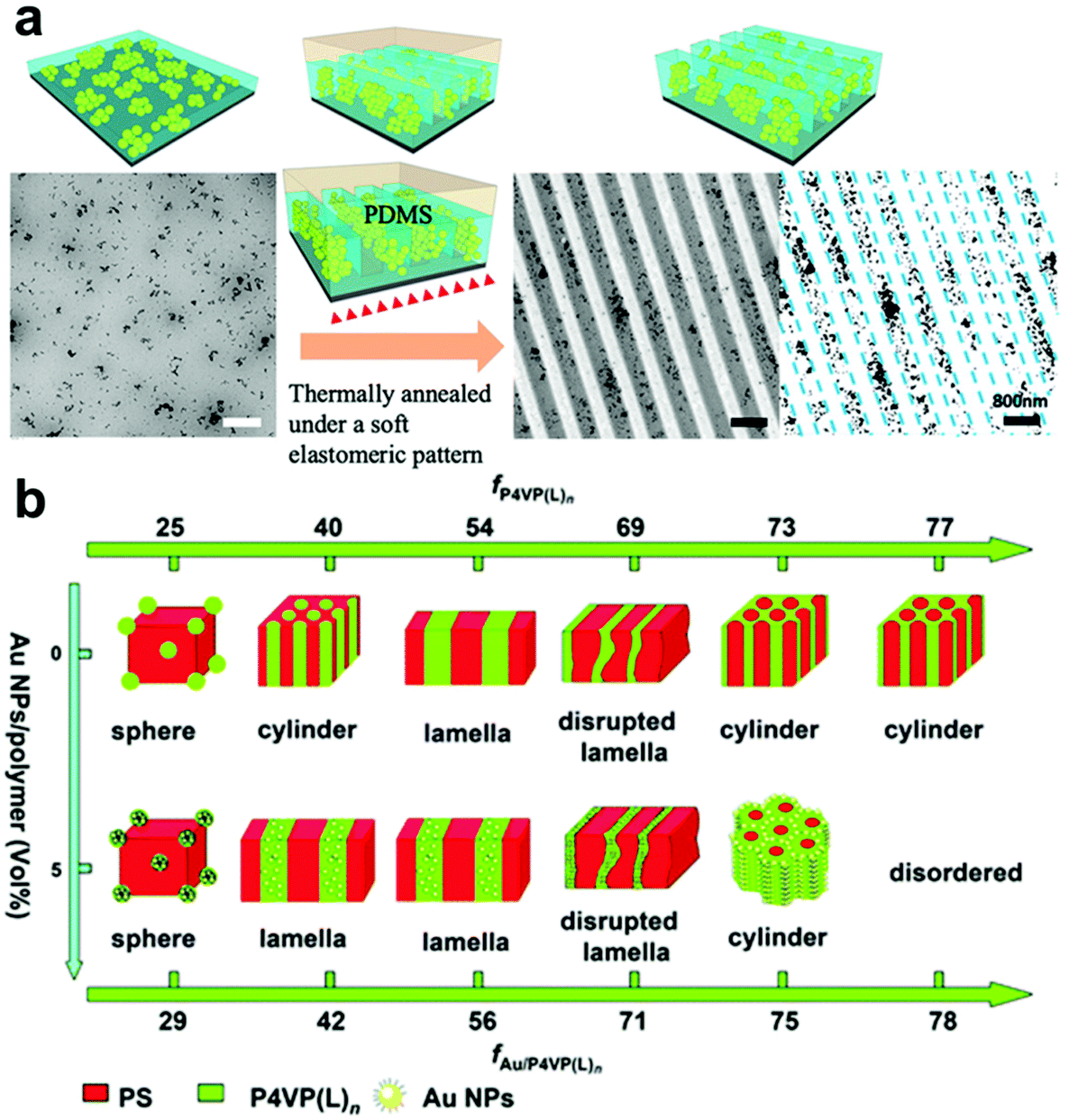 | ||
| Fig. 4 (a) Nanoimprint patterning of PS-grafted TiO2 nanoparticles in a PS thin film in which entropic localization effects confine the PS-TiO2 NPs to the mesa regions of the patterned film.84 Adapted with permission from ref. 84 (copyright 2019, American Chemical Society). (b) Phase diagram of self-assembled Au NP/BCP hybrid structures, demonstrating that nanofiller content and BCP composition can be tuned to produce a wide range of nanocomposite morphologies.91 Adapted with permission from ref. 91 (copyright 2013, Royal Society of Chemistry). | ||
A more advanced level of structural organization can be achieved by using block-copolymers (BCPs) that can self-assemble into various types of periodic nanostructures, including spheres, lamella, and cylinders due to the microphase separation of their constituent polymer blocks (Fig. 4b).88–93 BCP-based methods for confining NPs within a matrix have attracted significant attention as templates to dictate the spatial distribution of NPs due to their ability to generate complex hierarchical architectures. Enthalpy-driven control over particle organization can be achieved by functionalizing NPs with molecules that have programmed interactions with complementary functional groups on one block of the BCP structures, causing them to be preferentially sequestered in one of the blocks at equilibrium.94–97 Alternatively, periodic BCP superstructures can also control the distribution of NPs in an entropic fashion, where the introduction of NPs leads to deformation of polymer chains and introduces an entropic penalty that affect polymer organization.98–102 This entropic penalty is dictated by the size of NPs relative to the BCP domains, drastically increasing concomitantly with nanoparticle size. As a result, larger particles the particles typically fill “softer” regions in the BCP superstructures (i.e. at the center each BCP domain) to minimize the entropy loss of the polymer chains, whereas smaller NPs tend to occupy the interface of the BCP domains to gain translational entropy.95,97 BCPs are particularly useful for the preparation of anisotropic polymer-NP composite materials, which can be achieved by either (i) assembling anisotropic NPs in controlled orientation and positions,101,103 or (ii) assembling isotropic NPs in an anisotropic manner by tuning the alignment of BCP periodic structures and NP surface functionalization.17 Loading of NPs usually occurs when the polymer is in a swollen state, in which polymer occupies a fairly low volume percentage relative to solvent guest molecules. It is noteworthy that NPs can be introduced into the polymer network by the addition of either pre-synthesized NPs or chemical precursors that can yield NPs in situ.89,104–107
The wide variety of available methods for controlling the conformation of BCPs, such as changes in graft length,91,108 selection of solvent,109 and addition of stimuli-responsive small molecules that bind to polymer chains,2,110,111 have made BCPs a versatile tool for spatial confinement of NPs. In particular, the use of small molecule additives to bridge BCPs and NPs can help mitigate differences in BCP and NP surface chemistry, which expands the range of NP chemical compositions that can be solubilized in a given polymer. For example, Xu and coworkers developed a general strategy to prepare stimuli-responsive nanocomposites with controlled spatial distribution of NPs without requiring chemical modification of either NPs or BCPs; various nanoparticle compositions, including PbS, CdSe, Au, and CoFe2O4, of varying geometries were selectively incorporated into BCP microdomains in the presence of small molecule bridging moieties (Fig. 5a–e).112 BCP nanocomposites with ordered NP arrays have been successfully prepared not only as 3D materials in bulk solution, but also as 2D materials formed at a solution interface (Fig. 5f).103,105 In these composite materials, the mechanical properties of the BCP network were reinforced by NP infiltration, and the controllable swelling and deswelling of the ordered BCP superstructure allowed for reversible changes in interparticle distances.104,113–115 This feature makes the BCP mediated strategy a unique one for the preparation of ordered polymer composite materials with dynamic, NP-interaction dependent (e.g. photonic) properties.104
 | ||
| Fig. 5 (a–e) TEM images of various BCP/inorganic NP hybrid structures prepared by small-molecule-directed assembly where the BCP microdomain morphologies dictate the regions in which the NPs are spatially confined. NPs in lamellar BCP structures were confined to certain phase domains to maximize favorable interactions.112 Adapted with permission from ref. 112 (copyright 2009, Springer Nature). (f) Preparation (i–iv, top), SEM image (bottom left) and TEM image (bottom right) of conductive Ag NP/PEG gel elastomer nanocomposites, in which Ag NPs were generated in situ during gel formation by the simultaneous reduction of silver trifluoroacetate.105 Adapted with permission from ref. 105 (copyright 2011, Wiley). | ||
Nanoscale confinement has also been achieved by using liquid droplets, in which surface-functionalized nanofillers are driven to the air–liquid interface to form a monolayer.16,116,117 For example, water droplets have been utilized to encapsulate patchy nanofillers, where the steric hinderance resulting from the bulkiness of nanofillers and the enhanced hydrophilic forces induced by the water droplet can drive the clustering and assembling of the nanofillers with specific morphology.16 Yu and coworkers made use of these phenomena to assemble montmorillonite (MTM) clay nanoplatelets with chitosan into a 2D lamellar biomimetic material: polymer-associated nanoplatelets aqueous suspension were first prepared, and as water molecules were removed by evaporation or vacuum filtration, molecular crowding forced these nanoplatelets to assemble into a well-aligned lamellar structure reminiscent of nacre, bone, and other similar biomaterials.116 These lamellar nanocomposite materials exhibited improved mechanical properties compared to randomly dispersed nanocomposites prepared by simple mixing, as evidenced by the significantly increased Young's modulus and ultimate tensile strength as well as decreased ultimate strain. The ability to collect single monolayers of nanofillers for incorporation into a growing nanocomposite by simple tuning of surface chemistries have made this interfacial confinement-based strategy a convenient one for assembling 2D nanocomposite materials.
While spatial confinement-based strategies have already made considerable achievement in controlling the spatial distribution of nanofillers, further improvements are still possible in a number of difference aspects. First, BCP assembly for many contemporary systems is not only limited by the timescale of the process itself (assembly times ranging from a few hours to several days are common),96,104,112 but is also sensitive to the very presence of nanofiller materials; the excluded volume occupied by NPs can impede the movement of BCP chains, which can seriously disrupt or even prevent entirely the formation of an ordered phase.91,118 Some examples have been developed that have significantly decreased the time needed for BCP ordering and NP segregation to be achieved,119 but more effort is needed to make such approaches both generally applicable and readily adoptable. Secondly, the types of ordered BCP structures that can be prepared are restricted to those that are thermodynamically favorable (e.g. lamellae, gyroids, etc.), and the phase space of possible structures can be complicated when using large amounts of nanoparticle fillers. Thus, BCP confinement methods would greatly benefit from the development of new materials and techniques for both rapid formatting periodic superstructures and robust expansion of the types of superstructures that can be achieved with these methods, enabling both a wider array of materials for further investigation and more facile scale-up for industrial applications.101,120 Thirdly, fundamental questions on how NP morphology, concentration, and surface chemistry affect the BCP mesophase structure must be answered;118,121–124 these NP parameters are not simply “passive scalars” existing in isolation from the surrounding BCP matrix, and can potentially alter the resulting BCP structure by modulating polymer-polymer forces of interaction or altering preferred polymer chain conformations.108,125 Finally, while NP distribution within the nanocomposite can be governed by the confined nanoscale environment, controlling the spatial organization of each NP within a polymer block remains a significant challenge. Methods of generating ordered arrays of NPs within these confined spaces are limited, and most composites have disordered clusters or amorphous arrangements of particles (as opposed to, for example, crystalline superlattices).126 This could potentially be achieved with further NP surface functionalization with moieties that could engage in a secondary self-assembly process independent of BCP phase segregation.
LBL assembly is commonly driven by electrostatic interactions, where a substrate is coated via sequential rounds of deposition with nanofiller and polymers functionalized with oppositely-charged moieties.21,128,131,135–142 Other types of interactions besides electrostatic attraction can also be employed, such as hydrophobic forces, hydrogen-bonding and host–guest interactions, where the overall process for generating the composites remains similar (complementary binding interactions allow sequential, alternating rounds of particle and polymer deposition).140,142,143 For example, Gauckler and coworkers reported a method of preparing multilayer 2D biomimetic materials where hydrophobic surface groups and sonication were used to direct the formation of a well-oriented aluminum platelet monolayer at the air–water interface. The monolayers were then transferred to a flat substrate by dip-coating and subsequently spin-coated with a layer of chitosan polymer, and a lamellar nanocomposite was readily fabricated simply by repeating this procedure through multiple cycles (Fig. 6a).144 Notably, this method avoids the undesirable aggregation and poor orientation control that has frustrated previous attempts to produce such materials by dip-coating.145,146 It should also be noted that materials selection was determined only by design principles of the intended final material and was not limited by the LBL strategy itself. By selecting organic and inorganic phases with certain inherent tensile strengths, mutual binding affinities, and (in the case of platelets) aspect ratios, several ordered lamellar nanocomposites with different chemical compositions were prepared which possessed tensile strengths and ductilities far superior to that of any naturally occurring substance (Fig. 6b and c). The combination of ab initio materials selection with precise nanofiller orientation therefore demonstrates that LBL assembly is a very advantageous approach for composite thin films fabrication from a materials design standpoint.
 | ||
| Fig. 6 (a) Layer-by-layer fabrication of nacre-like films where hydrophobically-modified Al2O3 nanoplatelets form a well-oriented monolayer at the air–water interface and transferred to a substrate by dip coating to await subsequent spin-coating of a desired polymer. Repetition of this process over multiple cycles leads to the formation of nacre-like nanocomposite thin films. (b) Tensile stress/strain curves for chitosan films with different platelet volume fractions (Vp). Mechanical strength rapidly increases with Vp, demonstrating the benefit of these layered structures for property enhancement. (c) Tensile stress–strain curves of naturally occurring layered biomaterials compared to the artificial Al2O3–chitosan hybrid films.144 Adapted with permission from ref. 144 (copyright 2008, AAAS). | ||
Another advantage of LBL assembly is that it can be applied to a wide array of filler material compositions and morphologies such as nanorods, nanotubes, nanosheets and nanoplates.147,148 Surface modification required for LBL incorporation is typically facile and dictated by the chemistry of the selected filler materials. For example, SWNTs can be readily functionalized with carboxylic acid groups,149 while clay nanosheets can be functionalized with ammonium groups,146,150 and these functionalized fillers can then be assembled with oppositely-charged polymers via electrostatically-driven LBL. The simplicity and versatility of LBL assembly has garnered much research attention, and numerous studies have focused on further improving the methods’ ability to dictate structural organization.10,151 For example, spray-coating protocols have been developed to shorten deposition time for each layer,152 and nanofabrication techniques such as lithography and printing, which can pre-modify the substrate surface into arbitrary patterns, have been used in conjunction with LBL assembly to enable the formation of composite materials with more complicated nanostructures.1,153–156
In addition, LBL techniques allow for a number of input parameters beyond simple materials selection when designing protocols. A number of physicochemical parameters (i.e. pH,157 ionic strength,158 temperature,159 and solvent selection;160 the exact parameters vary depending on the type of intermolecular forces governing interactions between material components) have been varied to tune properties such as growth rate for each deposition step, thermodynamic stability of the final nanocomposite, and many others.128,161 Modulation of these structures and thus the material properties can even be modified post-fabrication via the introduction of stimuli-responsive moieties.19,162–168 Hammond and coworkers, for example, reported the electrostatic LBL assembly of cationic linear poly(ethyleneimine) (LPEI) and anionic prussian blue (PB) NPs into lamellar films with the ability to undergo redox-driven film swelling (Fig. 7).169 Film swelling was driven by electrochemical reduction of the redox-active PB NPs, resulting in migration of water and counterions into the film (Fig. 7b). Apart from changes in interlayer spacings caused by swelling, the electrochemically reduced swollen films were found to have significantly diminished load-depth response and elastic modulus compared to the oxidized, deswelled state.
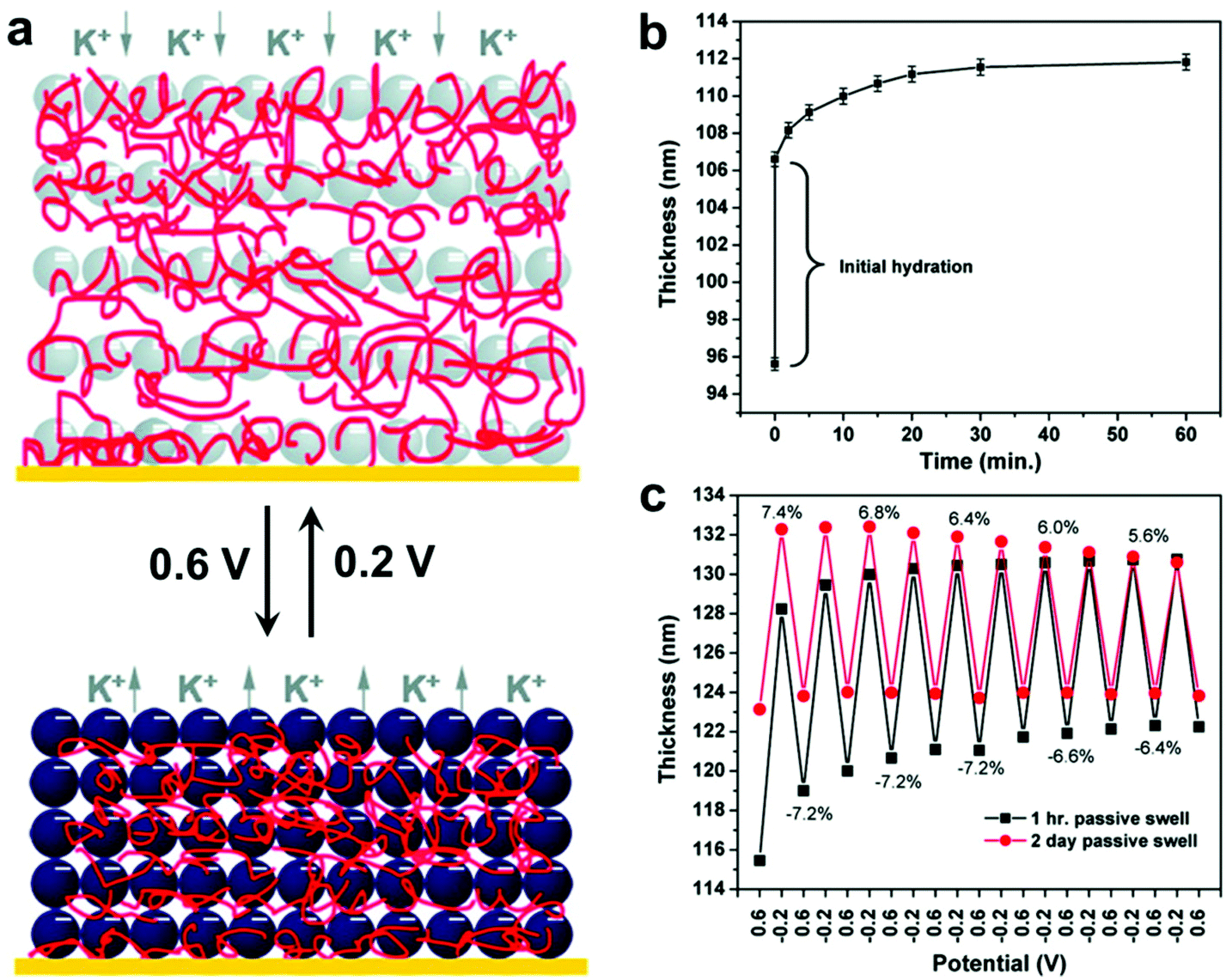 | ||
| Fig. 7 (a) Schematic depiction of the reversible electrochemically-induced swelling of LBL-produced LPEI-PB NP composite films. Reduction of PB NPs causes cation-bearing electrolyte solution to migrate into the film to maintain charge balance, resulting in significant film swelling. The opposite process happens upon PB NP oxidation. (b) Time-dependent passive swelling of LPEI-PB films in electrolyte solution as measured by ellipsometry. Film thickness sees a 17% increase from the dry state after 1 h and a 30% increase from the dry state after 2 days. (c) Electrochemically-induced active swelling of LPEI-PB films across multiple redox cycles as measured by ellipsometry. Films that were passively swollen for two days beforehand exhibited highly reversible swelling behavior, whereas those that were passively swollen for only 1 h exhibited somewhat irreversible swelling initially, eventually achieving reversibility once their thickness values converged with those of the 2-day preswollen films after repeated cycling.169 Adapted with permission from ref. 169 (copyright 2009, American Chemical Society). | ||
While the control of ordering along the layer stacking direction has been achieved by tuning many types of complementary interactions between adjacent layers, methods for control the intra-layer organization of nanofillers remain rare and warrant further research efforts in the future. Furthermore, LBL-produced nanocomposite films typically experience defect formation and loss of ordering as the number of layers increases,170 making the development of post-synthetic annealing or reorganization techniques as well as higher-fidelity NP-NP interactions highly desirable. In addition, the majority of nanocomposite thin films developed so far are binary. The use of multiple orthogonal recognition pairs (e.g., hydrogen bonding and coordination interactions) would enable the fabrication of thin films containing multiple types of nanoparticles or polymers into a single structure, significantly increasing the compositional versatility and this programmability of material properties. More complex synthetic protocols would also allow for the exploration of how NPs of unlike composition interact in an ordered nanoscale array to impart emergent phenomena as a result of interparticle or particle-polymer interactions.171–173
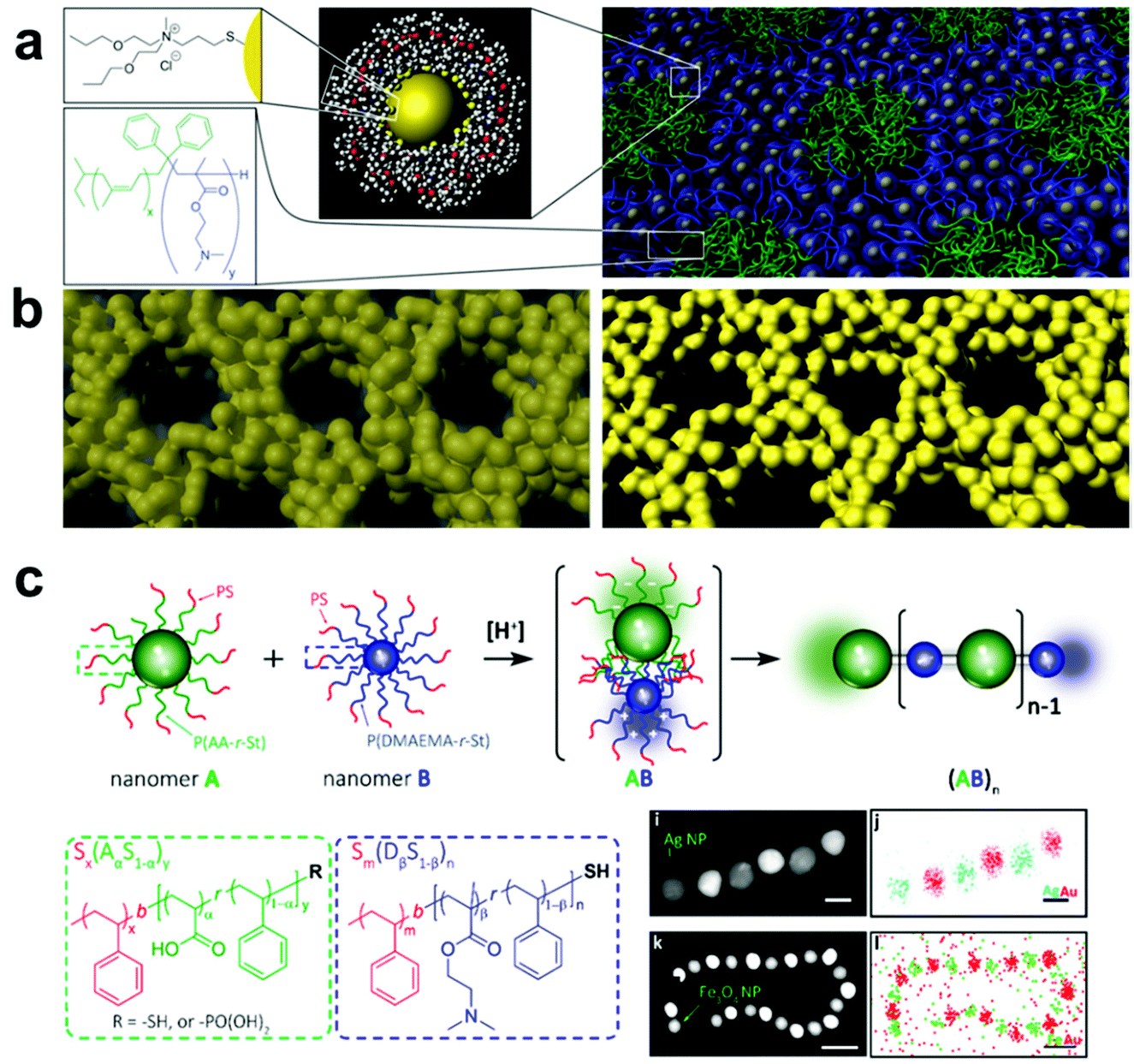 | ||
| Fig. 8 (a) Schematic representation of the assembly of ligand-capped Pt NPs into organized structures with BCPs. The hydrophilic surface ligands drive assembly of NPs with similarly hydrophilic BCP domains, creating an inverse hexagonal morphology. (b) Pyrolysis of the as-assembled NP-BCP hybrids, followed by plasma or acid etch treatment, yields well-ordered mesoporous Pt structures.180 Adapted with permission from ref. 180 (copyright 2008, AAAS). (c) Schematic representation of alternating assembly of polymer-grafted NPs into linear polymeric chains. Here, NPs functionalized with either poly(acrylic acid-r-styrene) or poly(N,N-dimethylaminoethyl methacrylate-r-styrene) assemble in alternating sequence by electrostatic attraction in a manner mimicking Nylon 66 polymerization.200 Adapted with permission from ref. 200 (copyright 2019, American Chemical Society). | ||
A more viable strategy to form ordered polymer and nanoparticle composite arrays is to directly tether polymer chains to the nanofiller surface, thereby making each nanomaterial building block inherently a composite structure.183,184 Compared with systems that employ small molecules as surface ligands, grafting a brush of polymer chains to the surfaces of particles allows the hydrodynamic diameter of each building block to be much more widely tuned by changing polymer length, polymer composition, and choice of solvent.,185–187 When these polymer-grafted particles are then assembled, this diversity in material design parameters leads to much greater control over interparticle spacings and particle organization. As an additional benefit, tethering polymer chains to nanofillers can help avoid phase segregation between the two in the final composite. Because the steric bulk of the polymer brush prevents particle aggregation, a much larger range of nanofiller weight percentages in the final material can be achieved compared with other methods of incorporating nanoparticles in a polymer matrix.
A major area of study in this type of composite synthesis has been the use of hydrophobic interactions as a means to direct nanofiller self-assembly.188–190 By functionalizing nanofillers with both hydrophobic (such as polystyrene, PMMA) and hydrophilic polymers (such as PEO), NP surface anisotropy can be readily induced due to the lateral phase-separation of immiscible polymers.191–194 As a result, these NPs can aggregate into nanowires,195 bundles,196 lamellae,17,197 and vesicles20,198 upon varying the solvent conditions to change the interactions between polymer domains. As the hydrophobicity of polymer chains can be tuned by temperature, the formation of these hierarchical structures exhibits reversibility upon the treatment of external stimuli, such as heat or light, by taking advantage of the photothermal effect.198,199 In addition, by coupling composite formation with other processes that disturb the system's hydrophobicity, other types of stimuli-responsiveness can be achieved. For example, by incorporating 4-vinylpyridine groups as comonomers in the PMMA chains grafted on NPs, the hydrophobicity of the resulting polymer grafts and consequently the assembled structure can respond to changes in pH via protonation/deprotonation of the pyridine moieties.20 Electrostatic interactions can also be employed to direct the assembly of nanocomposite architectures by grafting binary sets of nanoparticles with oppositely charged polymers. Inspired by the synthesis of Nylon-66, Nie and coworkers reported an interesting approach for synthesizing 1D nanowires with copolymer grafted NPs in which NP assembly was driven by electrostatic attraction between poly(acrylic acid-r-styrene)- and poly(N,N-dimethylaminoethyl methacrylate-r-styrene) blocks functionalized on complementary NPs, where the final structure produced linear chains of particles resembling the namesake copolymer system (Fig. 8c).200 It should be noted, however, that because neither hydrophobic forces nor electrostatic interactions are stoichiometrically accurate, structures prepared from these strategies have limited control over the interactions between each individual component. Thus, the prepared structures usually lack long-rang ordering. More importantly, since these two types of interactions are determined primarily by the polymer composition, any alteration of the polymer component may require a redesign of the entire assembly system and conditions, meaning that expansion of these strategies to different types of composites may require significant additional design and investigation.
To circumvent the abovementioned drawbacks, polymer grafts containing end-tethered supramolecular recognition groups with precise binding behavior have been employed in regulating the superstructures of NP assemblies.197,201–204 When compared with hydrophobic forces, supramolecular interactions with chemically accurate stoichiometry have several key advantages: (1) the binding strength between complementary recognition group pairs can be determined using small molecules as model systems; (2) the supramolecular recognition motifs can be designed independently from the polymer component, thereby significantly increasing the modularity of the nanocomposite building blocks; (3) the versatile nature of supramolecular binding groups can introduce multi-stimuli responsiveness into the assembly.
A new class of nanoscale building block has been developed based on these design parameters, termed “nanocomposite tectons” (NCTs), which consist of an inorganic NP core grafted with a dense monolayer of polymer chains whose termini are functionalized with supramolecular recognition groups (Fig. 9a).205 Upon mixing solutions of NCTs that possess complementary recognition groups, these particles rapidly assemble into larger structures, which subsequently form ordered 3D superlattices upon annealing (Fig. 9c and d).201 These NCT structures possess a number of different design variables that can be used to tune the assembly behavior and final structures of the superlattices, such as particle size and shape, particle composition, polymer length, polymer composition, brush grafting density, and supramolecular binding group identity.196,201,202,204,206 As a result, different types of ordered arrays can be achieved with unique particle organizations, and significant insight has been gained on how NCT architecture affects the supramolecular chemistry-driven assembly process.
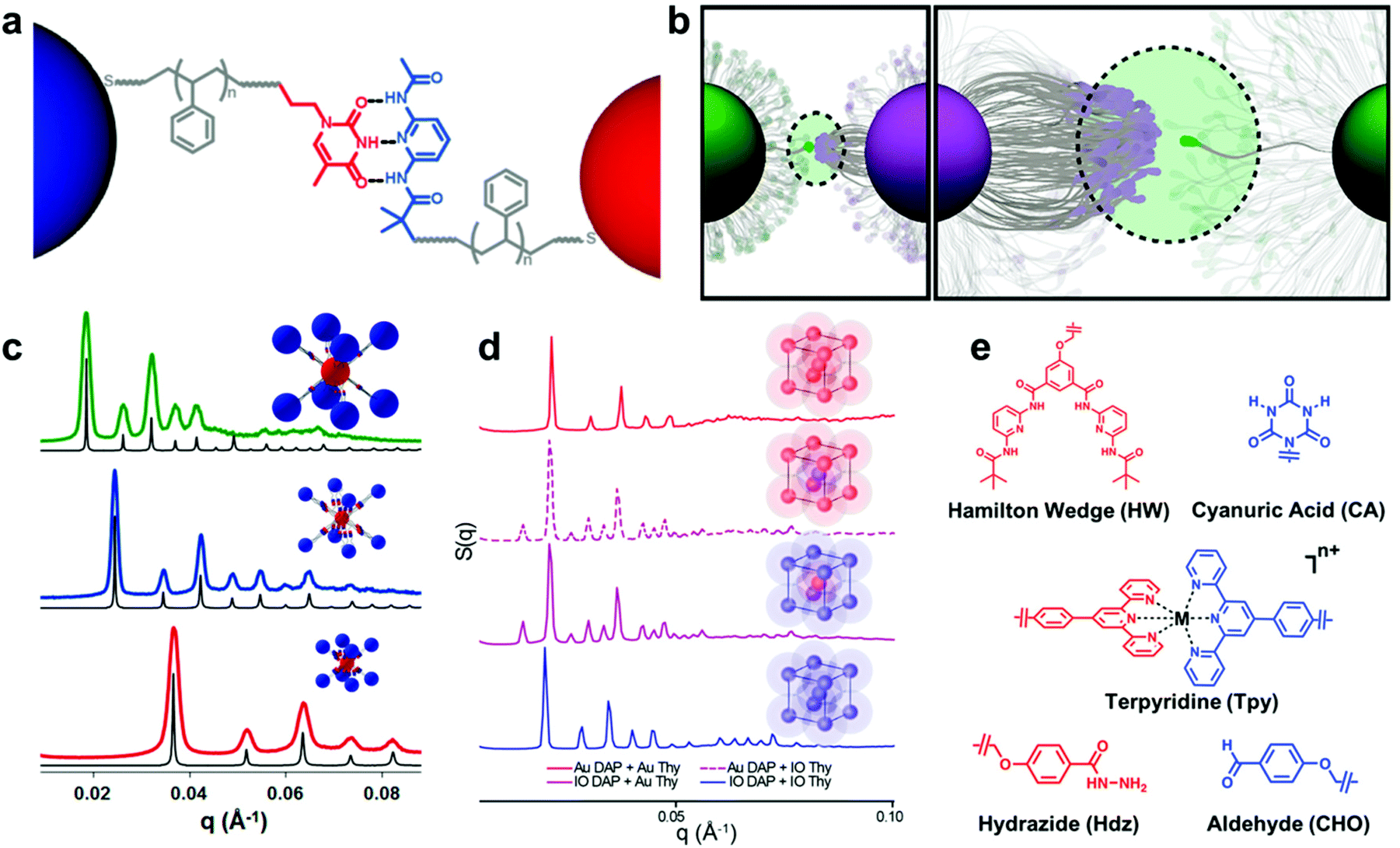 | ||
| Fig. 9 (a) Schematic illustration of bonding interactions between complementary NCTs. Au NPs are grafted with PS chains capped with supramolecular binding moieties to direct their assembly.205 Adapted with permission from ref. 205 (copyright 2019, American Chemical Society). (b) The thermodynamics of the NCT–NCT bonds driven by complementary binding groups is dictated by the multivalent interactions of many binding moieties acting together.206 Adapted with permission from ref. 206 (copyright 2019, American Chemical Society). (c) Small-angle X-ray scattering (SAXS) data of assembled NCTs exhibiting body-centered cubic (bcc) lattice structures. Shifts in peak positions are a consequence of different Au NP core sizes and PS graft lengths.201 Adapted with permission from ref. 201 (copyright 2016, American Chemical Society). (d) SAXS data of NCT assemblies with both Au and iron oxide (IO) cores. Assembled lattices of just IO or Au NCTs exhibited bcc symmetry, while combinations of IO and Au NCTs yielded cesium chloride-type structures. The preservation interparticle spacings between patterns demonstrates that NCT superlattice symmetry can be controlled independently from core NP composition.204 Adapted with permission from ref. 204 (copyright 2020, American Chemical Society). (e) Chemical structures of the alternative binding moieties that have been used to drive NCT assembly, demonstrating the flexibility of the NCT concept across multiple types of bonding interactions (hydrogen bonding, metal ion complexation, and dynamic covalent bond formation).202 Adapted with permission from ref. 202 (copyright 2019, American Chemical Society). | ||
For instance, the entropic penalty arising from the restricted conformation of the NCT-tethered polymer chains allows for significant tuning of the characteristic dissociation temperature of the composite arrays (Fig. 9b), and changes to supramolecular recognition group (Fig. 9e), polymer length, and density of binding groups can all be used independently to manipulate this process in a rational manner.202 An additional benefit of the conformational flexibility of the polymer brush is that NCT superlattice formation is highly tolerant to dispersity in both NP radius and polymer molecular weight, which is a significant advantage when compared to other top-down nanocomposite ordering systems that require highly uniform building blocks.205 In comparison to the other strategies introduced so far, employing non-covalent interactions between the polymer grafted nanocomponents allows one to control the interactions between the nanoscale building blocks in a more precise manner and facilitate the formation of highly ordered nanocomposite structures. Preliminary evidence even suggests that such structures may possess unique and tunable optical, magnetic, or mechanical properties as a function of their structural organization.203,204
In order to further develop the potential of NCTs for forming ordered nanocomposites, it is necessary to expand their viability to a wider array of both NCT chemical compositions (i.e., different polymers, NP cores, and supramolecular recognition groups) and assembly conditions. This will allow the preparation of different model systems for ordered nanocomposites, enabling the further study of structure–property relationships and the establishment of design rules for ordered nanocomposite materials with programmable properties. Moreover, combining NCTs functionalized with different types of supramolecular groups of different binding strength will enable the construction of hierarchical NCT superlattices in a more complex manner.
3. Conclusions and perspectives
In general, controlling both chemical composition and nanoscale organization in polymer composite materials allows for two different means to tune the physical properties of the composites. First, composite materials can be synthesized take advantage of the emergent properties that arise from ordered arrays of nanoparticles, such as electromagnetic coupling, controlled transport, or metamaterial behaviors.11,116,144,155,171–173,207–215 In these materials, the polymers primarily serve as fillers, spacers, and crosslinking agents to occupy the void space between NPs and hold the particle arrays in place.112,137,146 However, more advanced materials can also be developed where the polymers serves to regulate the connectivity or coupling between particles as a means of modulating their collective behavior (e.g. altering the refractive index, or thermal/electrical conductivity of the material); with appropriate polymer selection, the medium surrounding the particles can even be modulated in a dynamic manner to serve as an actuating component of the composite material.207,214 With the advent of increasingly sophisticated particle arrays within these composites, another important view point is beginning to emerge, specifically an understanding of how ordered arrays of NPs can potentially affect the properties of polymer matrix. Depending on the type of NP alignment within the polymer, the distribution and conformation of matrix polymer chains should theoretically be regulated by the periodic structures of the NPs.81,118,216,217 Consequently, one would expect that a nanocomposite material will exhibit different mechanical, transport, or other chain-dynamics-driven properties upon varying the organization of particles within the polymer network. Further research effort to determine what types of structures may be beneficial and to develop a comprehensive understanding of these phenomena is required, but significant potential exists for examining this unique strategy to control composite properties.In this review, we have covered different types of assembly strategies for the preparation of nanocomposite materials with structural control at the nanoscale, and presented a number of current challenges and future opportunities facing each type of composite synthetic strategy. In addition to the challenges discussed above, several additional potential barriers are common to multiple strategies; identifying potential solutions for these challenges would greatly advance the development of ordered nanocomposite materials.
(1) While bottom-up approaches have achieved great success in preparing superlattices of nanofillers inside a polymer matrix, the grain sizes of these arrays typically remains small. Given that grain boundaries in such materials are likely to impede beneficial properties like uniform optical response or transport, manipulating the polycrystalline nature of such structures needs to be a key focus for future efforts. Novel strategies that control not only nanoscale organization but also material microstructure are necessary to develop composites with more complicated structural hierarchy that may be beneficial in future device architectures.58,218–221
(2) Development of modular chemistries for both particle functionalization, self-assembly, and particle-polymer interactions would significantly broaden the types of composites that could be fabricated without the need to develop many different particle functionalization or assembly strategies. In addition, developing versatile techniques for dynamically modifying particle-polymer interactions (e.g. via photo-, redox-, temperature, or pH-sensitive motif) would allow for stimuli responsive materials that alter both nanoscale organization and material properties on demand.20,26,112,162,164,165,167,168,202
(3) It is generally accepted that hybrid nanocomposite materials can exhibit novel properties unseen in individual components, but exactly how the arrangement of the component phases translates into bulk-scale properties is not as thoroughly understood for all types of composite architectures; fundamental structure property investigations on composites with controlled organization of particles are significantly less numerous than their disordered counterparts. Moreover, some functions of nanocomposite materials rely simply on “clustering” of the of the nanofillers rather than true ordering (e.g., magnetothermal effects for therapeutic applications33). In order to fully exploit the benefit of structural ordering, a quantitative correlation between physicochemical (especially mechanical) properties and the degree of structural ordering must be established.222,223 It is therefore imperative to first understand the interplay between polymers, nanomaterials and solvents in the system.224
(4) The future development of ordered nanocomposite materials will also be aided by future breakthroughs in instrumentation. For example, while SERS has proven useful in investigating the interactions between small molecules tethered to the NP surface, and NMR and ITC techniques can be used to determine the interactions between polymers in solution, there is currently not an effective way to directly probe the interactions between polymers anchored on the NP surface quantitatively.
Development of novel strategies for making ordered polymer composites not only will grant us access to polymer composite materials with different types of ordering nanostructures to explore their unprecedented properties, but will also afford a better understanding of structure–property relationships for the design of next-generation composite materials. We hope this review can inspire other researchers in the related field to explore different possibilities for making and studying ordered nanocomposite materials in a wide variety of different composite systems.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was primarily supported by an NSF CAREER Grant, award number CHE-1653289.References
- A. N. Leberfinger, D. J. Ravnic, A. Dhawan and I. T. Ozbolat, Stem Cells Transl. Med., 2017, 6, 1940–1948 CrossRef.
- K. R. Zhang, H. L. Gao, X. F. Pan, P. Zhou, X. Xing, R. Xu, Z. Pan, S. Wang, Y. M. Zhu, B. Hu, D. H. Zou and S. H. Yu, Matter, 2019, 1, 770–781 CrossRef.
- E. Masaeli, V. Forster, S. Picaud, F. Karamali, M. H. Nasr-Esfahani and C. Marquette, Biofabrication, 2020, 12, 025006 CrossRef CAS.
- A. M. Yu, Z. J. Liang, J. Cho and F. Caruso, Nano Lett., 2003, 3, 1203–1207 CrossRef CAS.
- M. H. Yang, Y. Yang, H. F. Yang, G. L. Shen and R. Q. Yu, Biomaterials, 2006, 27, 246–255 CrossRef CAS.
- A. Afzal, S. Feroz, N. Iqbal, A. Mujahid and A. Rehman, Sens. Actuators, B, 2016, 231, 431–439 CrossRef CAS.
- S. Mariani, A. Paghi, A. A. La Mattina, A. Debrassi, L. Dahne and G. Barillaro, ACS Appl. Mater. Interfaces, 2019, 11, 43731–43740 CrossRef CAS.
- S. Wang, X. Y. Huang, G. Y. Wang, Y. Wang, J. L. He and P. K. Jiang, J. Phys. Chem. C, 2015, 119, 25307–25318 CrossRef CAS.
- P. H. Hu, Z. Y. Jia, Z. H. Shen, P. Wang and X. R. Liu, Appl. Surf. Sci., 2018, 441, 824–831 CrossRef CAS.
- Y. Kim, J. Zhu, B. Yeom, M. Di Prima, X. L. Su, J. G. Kim, S. J. Yoo, C. Uher and N. A. Kotov, Nature, 2013, 500, 59–63 CrossRef CAS.
- E. H. Hill, C. Hanske, A. Johnson, L. Yate, H. Jelitto, G. A. Schneider and L. M. Liz-Marzan, Langmuir, 2017, 33, 8774–8783 CrossRef CAS.
- Q. Zhang, Y. H. Zhang, K. Xiao, Z. L. Meng, W. S. Tong, H. W. Huang and Q. An, Chem. Eng. J., 2019, 358, 389–397 CrossRef CAS.
- Z. B. Shifrina, V. G. Matveeva and L. M. Bronstein, Chem. Rev., 2020, 120, 1350–1396 CrossRef CAS.
- K. Wang, S. M. Jin, J. P. Xu, R. J. Liang, K. Shezad, Z. G. Xue, X. L. Xie, E. Lee and J. T. Zhu, ACS Nano, 2016, 10, 4954–4960 CrossRef CAS.
- X. J. Zhao, C. R. Zhou, Y. Lvov and M. X. Liu, Small, 2019, 15, 1900357 CrossRef.
- M. Grzelczak, A. Sanchez-Iglesias, H. H. Mezerji, S. Bals, J. Perez-Juste and L. M. Liz-Marzan, Nano Lett., 2012, 12, 4380–4384 CrossRef CAS.
- T. Nakano, D. Kawaguchi and Y. Matsushita, J. Am. Chem. Soc., 2013, 135, 6798–6801 CrossRef CAS.
- X. C. Ye, C. H. Zhu, P. Ercius, S. N. Raja, B. He, M. R. Jones, M. R. Hauwiller, Y. Liu, T. Xu and A. P. Alivisatos, Nat. Commun., 2015, 6, 10052 CrossRef.
- A. Zhuk, R. Mirza and S. Sukhishvili, ACS Nano, 2011, 5, 8790–8799 CrossRef CAS.
- J. B. Song, L. Cheng, A. P. Liu, J. Yin, M. Kuang and H. W. Duan, J. Am. Chem. Soc., 2011, 133, 10760–10763 CrossRef CAS.
- J. W. Liu, H. W. Liang and S. H. Yu, Chem. Rev., 2012, 112, 4770–4799 CrossRef CAS.
- L. B. Wang, L. G. Xu, H. Kuang, C. L. Xu and N. A. Kotov, Acc. Chem. Res., 2012, 45, 1916–1926 CrossRef CAS.
- Y. Z. Long, M. Yu, B. Sun, C. Z. Gu and Z. Y. Fan, Chem. Soc. Rev., 2012, 41, 4560–4580 RSC.
- S. Y. Zhang, M. D. Regulacio and M. Y. Han, Chem. Soc. Rev., 2014, 43, 2301–2323 RSC.
- Z. Liu, J. Xu, D. Chen and G. Z. Shen, Chem. Soc. Rev., 2015, 44, 161–192 RSC.
- M. Grzelczak, L. M. Liz-Marzan and R. Klajn, Chem. Soc. Rev., 2019, 48, 1342–1361 RSC.
- H. B. Hu, S. C. Wang, X. L. Feng, M. Pauly, G. Decher and Y. Long, Chem. Soc. Rev., 2020, 49, 509–553 RSC.
- D. Lorenzo, D. Fragouli, G. Bertoni, C. Innocenti, G. C. Anyfantis, P. D. Cozzoli, R. Cingolani and A. Athanassiou, J. Appl. Phys., 2012, 112, 083927 CrossRef.
- A. Stopin, F. Pineux, R. Marega and D. Bonifazi, Chem. – Eur. J., 2015, 21, 9288–9301 CrossRef CAS.
- Y. S. Kim, M. J. Liu, Y. Ishida, Y. Ebina, M. Osada, T. Sasaki, T. Hikima, M. Takata and T. Aida, Nat. Mater., 2015, 14, 1002–1007 CrossRef CAS.
- J. Nunes, K. P. Herlihy, L. Mair, R. Superfine and J. M. DeSimone, Nano Lett., 2010, 10, 1113–1119 CrossRef CAS.
- K. E. Roskov, J. E. Atkinson, L. M. Bronstein and R. J. Spontak, RSC Adv., 2012, 2, 4603–4607 RSC.
- K. Hu, J. F. Sun, Z. B. Guo, P. Wang, Q. Chen, M. Ma and N. Gu, Adv. Mater., 2015, 27, 2507–2514 CrossRef CAS.
- M. J. Liu, Y. Ishida, Y. Ebina, T. Sasaki, T. Hikima, M. Takata and T. Aida, Nature, 2015, 517, 68–72 CrossRef CAS.
- D. L. Shi, P. He, J. Lian, X. Chaud, S. L. Bud'ko, E. Beaugnon, L. M. Wang, R. C. Ewing and R. Tournier, J. Appl. Phys., 2005, 97, 064312 CrossRef.
- D. Fragouli, R. Buonsanti, G. Bertoni, C. Sangregorio, C. Innocenti, A. Falqui, D. Gatteschi, P. D. Cozzoli, A. Athanassiou and R. Cingolani, ACS Nano, 2010, 4, 1873–1878 CrossRef CAS.
- T. Nagai, N. Aoki, Y. Ochiai and K. Hoshino, ACS Appl. Mater. Interfaces, 2011, 3, 2341–2348 CrossRef CAS.
- S. Y. Wu, R. B. Ladani, J. Zhang, A. J. Kinloch, Z. H. Zhao, J. Ma, X. H. Zhang, A. P. Mouritz, K. Ghorbani and C. H. Wang, Polymer, 2015, 68, 25–34 CrossRef CAS.
- M. S. Mauter, M. Elimelech and C. O. Osuji, ACS Nano, 2010, 4, 6651–6658 CrossRef CAS.
- B. Wang, Y. F. Ma, N. Li, Y. P. Wu, F. F. Li and Y. S. Chen, Adv. Mater., 2010, 22, 3067–3070 CrossRef CAS.
- L. Maggini, M. J. Liu, Y. Ishida and D. Bonifazi, Adv. Mater., 2013, 25, 2462–2467 CrossRef CAS.
- M. R. Liu, H. Younes, H. P. Hong and G. P. Peterson, Polymer, 2019, 166, 81–87 CrossRef CAS.
- K. M. Ryan, A. Mastroianni, K. A. Stancil, H. T. Liu and A. P. Alivisatos, Nano Lett., 2006, 6, 1479–1482 CrossRef CAS.
- R. Wakabayashi, K. Kaneko, M. Takeuchi and S. Shinkai, New J. Chem., 2007, 31, 790–799 RSC.
- K. P. Herlihy, J. Nunes and J. M. DeSimone, Langmuir, 2008, 24, 8421–8426 CrossRef CAS.
- M. Mittal and E. M. Furst, Adv. Funct. Mater., 2009, 19, 3271–3278 CrossRef CAS.
- Y. F. Zhu, C. Ma, W. Zhang, R. P. Zhang, N. Koratkar and J. Liang, J. Appl. Phys., 2009, 105, 054319 CrossRef.
- L. T. Yan, H. G. Schoberth and A. Boker, Macromol. Chem. Phys., 2009, 210, 1003–1010 CrossRef CAS.
- L. T. Yan, H. G. Schoberth and A. Boker, Soft Matter, 2010, 6, 5956–5964 RSC.
- M. Osada and T. Sasaki, Adv. Mater., 2012, 24, 210–228 CrossRef CAS.
- K. Hayashida, RSC Adv., 2013, 3, 221–227 RSC.
- O. Karatay, M. Dogan, T. Uyar, D. Cokeliler and I. C. Kocum, IEEE Trans. Nanotechnol., 2014, 13, 101–108 CAS.
- M. Arguin, F. Sirois and D. Therriault, AMPCS, 2015, 1, 16–25 Search PubMed.
- M. Mohammadimasoudi, Z. Hens and K. Neyts, RSC Adv., 2016, 6, 55736–55744 RSC.
- X. H. Li, J. Cai, Y. Y. Shi, Y. Yue and D. Y. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 1593–1601 CrossRef CAS.
- Y. Thakur, T. Zhang, C. Iacob, T. N. Yang, J. Bernholc, L. Q. Chen, J. Runt and Q. M. Zhang, Nanoscale, 2017, 9, 10992–10997 RSC.
- E. C. Sengezer, G. D. Seidel and R. J. Bodnar, Smart Mater. Struct., 2017, 26, 095027 CrossRef.
- S. Kaur, G. Murali, R. Manda, Y. C. Chae, M. Yun, J. H. Lee and S. H. Lee, Adv. Opt. Mater., 2018, 6, 1800235 CrossRef.
- J. S. Gao, Y. He and X. B. Gong, Results Phys., 2018, 9, 493–499 CrossRef.
- Y. J. Zhang, X. W. Zheng, W. T. Jiang, J. Han, J. T. Pu, L. L. Wang, B. Lei, B. D. Chen, Y. S. Shi, L. Yin, H. Z. Liu, F. Luo, X. K. Liu and J. J. Chen, RSC Adv., 2019, 9, 15238–15245 RSC.
- J. Kim, H. Ryu, J. H. Lee, U. Khan, S. S. Kwak, H. J. Yoon and S. W. Kim, Adv. Energy Mater., 2020, 10, 1903524 CrossRef CAS.
- F. Besharat, M. Manteghian, G. Gallone and A. Lazzeri, Nanotechnology, 2020, 31, 155701 CrossRef CAS.
- M. Suzuki, T. Yasukawa, H. Shiku and T. Matsue, Langmuir, 2007, 23, 4088–4094 CrossRef CAS.
- T. A. Asoh, M. Matsusaki, T. Kaneko and M. Akashi, Adv. Mater., 2008, 20, 2080–2083 CrossRef CAS.
- Q. Lu, S. M. Bai, Z. Z. Ding, H. Guo, Z. Z. Shao, H. S. Zhu and D. L. Kaplan, Adv. Mater. Interfaces, 2016, 3, 1500687 CrossRef.
- D. Morales, B. Bharti, M. D. Dickey and O. D. Velev, Small, 2016, 12, 2283–2290 CrossRef CAS.
- W. A. Deheer, W. S. Bacsa, A. Chatelain, T. Gerfin, R. Humphreybaker, L. Forro and D. Ugarte, Science, 1995, 268, 845–847 CrossRef CAS.
- X. J. Zan, S. Feng, E. Balizan, Y. Lin and Q. Wang, ACS Nano, 2013, 7, 8385–8396 CrossRef CAS.
- M. Chang, Z. Su and E. Egap, Macromolecules, 2016, 49, 9449–9456 CrossRef CAS.
- M. Shibayama, T. Karino, S. Miyazaki, S. Okabe, T. Takehisa and K. Haraguchi, Macromolecules, 2005, 38, 10772–10781 CrossRef CAS.
- G. Schmidt, A. I. Nakatani, P. D. Butler, A. Karim and C. C. Han, Macromolecules, 2000, 33, 7219–7222 CrossRef CAS.
- M. M. Malwitz, S. Lin-Gibson, E. K. Hobbie, P. D. Butler and G. Schmidt, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 3237–3248 CrossRef CAS.
- A. Sulong and J. Park, J. Compos. Mater., 2011, 45, 931–941 CrossRef CAS.
- A. E. Eken, E. J. Tozzi, D. J. Klingenberg and W. Bauhofer, Polymer, 2012, 53, 4493–4500 CrossRef CAS.
- W. S. Tong, Y. H. Zhang, L. Yu, X. L. Luan, Q. An, Q. Zhang, F. Z. Lv, P. K. Chu, B. Shen and Z. L. Zhang, J. Phys. Chem. C, 2014, 118, 10567–10573 CrossRef CAS.
- S. Vowinkel, G. Schafer, G. Cherkashinin, C. Fasel, F. Roth, N. Liu, C. Dietz, E. Ionescu and M. Gallei, J. Mater. Chem. C, 2016, 4, 3976–3986 RSC.
- K. Murata and K. Haraguchi, J. Mater. Chem., 2007, 17, 3385–3388 RSC.
- H. Endo, S. Miyazaki, K. Haraguchi and M. Shibayama, Macromolecules, 2008, 41, 5406–5411 CrossRef CAS.
- K. Shikinaka, Y. Koizumi, K. Kaneda, Y. Osada, H. Masunaga and K. Shigehara, Polymer, 2013, 54, 2489–2492 CrossRef CAS.
- J. S. Yang, R. Downes, A. Schrand, J. G. Park, R. Liang and C. Y. Xu, Scr. Mater., 2016, 124, 21–25 CrossRef CAS.
- K. Shikinaka, Y. Koizumi and K. Shigehara, J. Appl. Polym. Sci., 2015, 132, 41691 Search PubMed.
- J. Y. Yan, J. S. Yang, S. M. Dong, K. Huang, J. Ruan, J. B. Hu, H. J. Zhou, Q. Zhu, X. Y. Zhang and Y. S. Ding, Nanotechnology, 2019, 30, 275602 CrossRef CAS.
- R. J. Liang, J. P. Xu, R. H. Deng, K. Wang, S. Q. Liu, J. Y. Li and J. T. Zhu, ACS Macro Lett., 2014, 3, 486–490 CrossRef CAS.
- X. T. Wang, S. Bhadauriya, R. Zhang, P. Pitliya, D. Raghavan, J. N. Zhang, M. R. Bockstaller, J. F. Douglas and A. Karim, ACS Appl. Polym. Mater., 2019, 1, 3242–3252 CrossRef CAS.
- Y. Zare, Composites, Part A, 2016, 84, 158–164 CrossRef CAS.
- J. Liu, Z. X. Wang, Z. Y. Zhang, J. X. Shen, Y. L. Chen, Z. J. Zheng, L. Q. Zhang and A. V. Lyulin, J. Phys. Chem. B, 2017, 121, 9311–9318 CrossRef CAS.
- X. Y. Ma, Y. Zare and K. Y. Rhee, Nanoscale Res. Lett., 2017, 12, 621 CrossRef.
- R. Shenhar, T. B. Norsten and V. M. Rotello, Adv. Mater., 2005, 17, 657–669 CrossRef CAS.
- M. R. Bockstaller, R. A. Mickiewicz and E. L. Thomas, Adv. Mater., 2005, 17, 1331–1349 CrossRef CAS.
- Y. Lin, A. Boker, J. B. He, K. Sill, H. Q. Xiang, C. Abetz, X. F. Li, J. Wang, T. Emrick, S. Long, Q. Wang, A. Balazs and T. P. Russell, Nature, 2005, 434, 55–59 CrossRef CAS.
- T. Y. Ye, X. F. Chen, X. H. Fan and Z. H. Shen, Soft Matter, 2013, 9, 4715–4724 RSC.
- P. Bai, S. Yang, W. Bao, J. Kao, K. Thorkelsson, M. Salmeron, X. Zhang and T. Xu, Nano Lett., 2017, 17, 6847–6854 CrossRef CAS.
- D. Zhao, V. Gimenez-Pinto, A. M. Jimenez, L. X. Zhao, J. Jestin, S. K. Kumar, B. Kuei, E. D. Gomez, A. S. Prasad, L. S. Schadler, M. M. Khani and B. C. Benicewicz, ACS Cent. Sci., 2017, 3, 751–758 CrossRef CAS.
- W. A. Lopes and H. M. Jaeger, Nature, 2001, 414, 735–738 CrossRef CAS.
- M. R. Bockstaller, Y. Lapetnikov, S. Margel and E. L. Thomas, J. Am. Chem. Soc., 2003, 125, 5276–5277 CrossRef CAS.
- J. J. Chiu, B. J. Kim, E. J. Kramer and D. J. Pine, J. Am. Chem. Soc., 2005, 127, 5036–5037 CrossRef CAS.
- M. E. Mackay, A. Tuteja, P. M. Duxbury, C. J. Hawker, B. Van Horn, Z. B. Guan, G. H. Chen and R. S. Krishnan, Science, 2006, 311, 1740–1743 CrossRef CAS.
- T. L. Morkved, P. Wiltzius, H. M. Jaeger, D. G. Grier and T. A. Witten, Appl. Phys. Lett., 1994, 64, 422–424 CrossRef CAS.
- Y. Liu, M. H. Rafailovich, J. Sokolov, S. A. Schwarz, X. Zhong, A. Eisenberg, E. J. Kramer, B. B. Sauer and S. Satija, Phys. Rev. Lett., 1994, 73, 440–443 CrossRef CAS.
- A. M. Higgins and R. A. L. Jones, Nature, 2000, 404, 476–478 CrossRef CAS.
- W. K. Li, P. Zhang, M. Dai, J. He, T. Babu, Y. L. Xu, R. H. Deng, R. J. Liang, M. H. Lu, Z. H. Nie and J. T. Zhu, Macromolecules, 2013, 46, 2241–2248 CrossRef CAS.
- M. J. A. Hore and R. J. Composto, Macromolecules, 2014, 47, 875–887 CrossRef CAS.
- D. Nepal, M. S. Onses, K. Park, M. Jespersen, C. J. Thode, P. F. Nealey and R. A. Vaia, ACS Nano, 2012, 6, 5693–5701 CrossRef CAS.
- C. Kang, E. Kim, H. Baek, K. Hwang, D. Kwak, Y. Kang and E. L. Thomas, J. Am. Chem. Soc., 2009, 131, 7538–7539 CrossRef CAS.
- D. C. Hyun, M. Park, C. Park, B. Kim, Y. Xia, J. H. Hur, J. M. Kim, J. J. Park and U. Jeong, Adv. Mater., 2011, 23, 2946–2950 CrossRef CAS.
- H. Xu, Y. C. Xu, X. C. Pang, Y. J. He, J. H. Jung, H. P. Xia and Z. Q. Lin, Sci. Adv., 2015, 1, e1500025 Search PubMed.
- X. C. Pang, Y. J. He, J. H. Jung and Z. Q. Lin, Science, 2016, 353, 1268–1272 CrossRef CAS.
- R. B. Thompson, V. V. Ginzburg, M. W. Matsen and A. C. Balazs, Science, 2001, 292, 2469–2472 CrossRef CAS.
- R. J. Spontak, R. Shankar, M. K. Bowman, A. S. Krishnan, M. W. Hamersky, J. Samseth, M. R. Bockstaller and K. O. Rasmussen, Nano Lett., 2006, 6, 2115–2120 CrossRef CAS.
- T. Kato and J. M. J. Frechet, Macromolecules, 1989, 22, 3818–3819 CrossRef CAS.
- J. Ruokolainen, R. Makinen, M. Torkkeli, T. Makela, R. Serimaa, G. ten Brinke and O. Ikkala, Science, 1998, 280, 557–560 CrossRef CAS.
- Y. Zhao, K. Thorkelsson, A. J. Mastroianni, T. Schilling, J. M. Luther, B. J. Rancatore, K. Matsunaga, H. Jinnai, Y. Wu, D. Poulsen, J. M. J. Frechet, A. P. Alivisatos and T. Xu, Nat. Mater., 2009, 8, 979–985 CrossRef CAS.
- A. Jayaraman, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 524–534 CrossRef CAS.
- J. Lee, S. Yoo, M. Shin, A. Choe, S. Park and H. Ko, J. Mater. Chem. A, 2015, 3, 11730–11735 RSC.
- M. Benkovicova, A. Holos, P. Nadazdy, Y. Halahovets, M. Kotlar, J. Kollar, P. Siffalovic, M. Jergel, E. Majkova, J. Mosnacek and J. Ivanco, Phys. Chem. Chem. Phys., 2019, 21, 9553–9563 RSC.
- H. B. Yao, Z. H. Tan, H. Y. Fang and S. H. Yu, Angew. Chem., Int. Ed., 2010, 49, 10127–10131 CrossRef CAS.
- H. L. Gao, S. M. Chen, L. B. Mao, Z. Q. Song, H. B. Yao, H. Colfen, X. S. Luo, F. Zhang, Z. Pan, Y. F. Meng, Y. Ni and S. H. Yu, Nat. Commun., 2017, 8, 287 CrossRef.
- J. Kao and T. Xu, J. Am. Chem. Soc., 2015, 137, 6356–6365 CrossRef CAS.
- J. Kao, K. Thorkelsson, P. Bai, Z. Zhang, C. Sun and T. Xu, Nat. Commun., 2014, 5, 4053 CrossRef CAS.
- J. Kao, S. J. Jeong, Z. Jiang, D. H. Lee, K. Aissou, C. A. Ross, T. P. Russell and T. Xu, Adv. Mater., 2014, 26, 2777–2781 CrossRef CAS.
- S. W. Yeh, K. H. Wei, Y. S. Sun, U. S. Jeng and K. S. Liang, Macromolecules, 2003, 36, 7903–7907 CrossRef CAS.
- H. Chung, K. Ohno, T. Fukuda and R. J. Composto, Nano Lett., 2005, 5, 1878–1882 CrossRef CAS.
- S. W. Yeh, K. H. Wei, Y. S. Sun, U. S. Jeng and K. S. Liang, Macromolecules, 2005, 38, 6559–6565 CrossRef CAS.
- J. Y. Huang, Y. W. Qian, K. Evans and T. Xu, Macromolecules, 2019, 52, 5801–5810 CrossRef CAS.
- J. Y. Lee, R. B. Thompson, D. Jasnow and A. C. Balazs, Macromolecules, 2002, 35, 4855–4858 CrossRef CAS.
- V. Kalra, J. Lee, J. H. Lee, S. G. Lee, M. Marquez, U. Wiesner and Y. L. Joo, Small, 2008, 4, 2067–2073 CrossRef CAS.
- B. V. Lotsch, Annu. Rev. Mater. Res., 2015, 45, 85–109 CrossRef CAS.
- J. J. Richardson, J. W. Cui, M. Bjornmalm, J. A. Braunger, H. Ejima and F. Caruso, Chem. Rev., 2016, 116, 14828–14867 CrossRef CAS.
- X. F. Pan, H. L. Gao, Y. Lu, C. Y. Wu, Y. D. Wu, X. Y. Wang, Z. Q. Pan, L. Dong, Y. H. Song, H. P. Cong and S. H. Yu, Nat. Commun., 2018, 9, 2974 CrossRef.
- J. Z. Xue, H. L. Gao, X. Y. Wang, K. Y. Qian, Y. Yang, T. He, C. X. He, Y. Lu and S. H. Yu, Angew. Chem., Int. Ed., 2019, 58, 14152–14156 CrossRef CAS.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- J. Chen, A. Fasoli, J. D. Cushen, L. Wang and R. Ruiz, Macromolecules, 2017, 50, 9636–9646 CrossRef CAS.
- Y. H. Song, K. J. Wu, T. W. Zhang, L. L. Lu, Y. Guan, F. Zhou, X. X. Wang, Y. C. Yin, Y. H. Tan, F. Li, T. Tian, Y. Ni, H. B. Yao and S. H. Yu, Adv. Mater., 2019, 31, 1905711 CrossRef CAS.
- J. F. Wang, L. Lin, Q. F. Cheng and L. Jiang, Angew. Chem., Int. Ed., 2012, 51, 4676–4680 CrossRef CAS.
- X. L. Zhang, Y. Xu, X. Zhang, H. Wu, J. B. Shen, R. Chen, Y. Xiong, J. Li and S. Y. Guo, Prog. Polym. Sci., 2019, 89, 76–107 CrossRef CAS.
- E. Donath, G. B. Sukhorukov, F. Caruso, S. A. Davis and H. Mohwald, Angew. Chem., Int. Ed., 1998, 37, 2202–2205 CrossRef CAS.
- R. A. Caruso, A. Susha and F. Caruso, Chem. Mater., 2001, 13, 400–409 CrossRef CAS.
- S. H. Sun, S. Anders, H. F. Hamann, J. U. Thiele, J. E. E. Baglin, T. Thomson, E. E. Fullerton, C. B. Murray and B. D. Terris, J. Am. Chem. Soc., 2002, 124, 2884–2885 CrossRef CAS.
- Y. Wang, Z. Y. Tang, P. Podsiadlo, Y. Elkasabi, J. Lahann and N. A. Kotov, Adv. Mater., 2006, 18, 518–522 CrossRef CAS.
- X. Zhang, H. Chen and H. Y. Zhang, Chem. Commun., 2007, 1395–1405 RSC.
- S. Srivastava and N. A. Kotov, Acc. Chem. Res., 2008, 41, 1831–1841 CrossRef CAS.
- J. Borges and J. F. Mano, Chem. Rev., 2014, 114, 8883–8942 CrossRef CAS.
- B. L. Zhu, N. Jasinski, A. Benitez, M. Noack, D. Park, A. S. Goldmann, C. Barner-Kowollik and A. Walther, Angew. Chem., Int. Ed., 2015, 54, 8653–8657 CrossRef CAS.
- L. J. Bonderer, A. R. Studart and L. J. Gauckler, Science, 2008, 319, 1069–1073 CrossRef CAS.
- N. Sheng, M. C. Boyce, D. M. Parks, G. C. Rutledge, J. I. Abes and R. E. Cohen, Polymer, 2004, 45, 487–506 CrossRef CAS.
- P. Podsiadlo, A. K. Kaushik, E. M. Arruda, A. M. Waas, B. S. Shim, J. D. Xu, H. Nandivada, B. G. Pumplin, J. Lahann, A. Ramamoorthy and N. A. Kotov, Science, 2007, 318, 80–83 CrossRef CAS.
- H. B. Yao, L. B. Mao, Y. X. Yan, H. P. Cong, X. Lei and S. H. Yu, ACS Nano, 2012, 6, 8250–8260 CrossRef CAS.
- G. F. Wang, H. L. Qin, X. Gao, Y. Cao, W. Wang, F. C. Wang, H. A. Wu, H. P. Cong and S. H. Yu, Chem, 2018, 4, 896–910 CrossRef CAS.
- A. A. Mamedov, N. A. Kotov, M. Prato, D. M. Guldi, J. P. Wicksted and A. Hirsch, Nat. Mater., 2002, 1, 190–194 CrossRef CAS.
- A. Walther, I. Bjurhager, J. M. Malho, J. Ruokolainen, L. Berglund and O. Ikkala, Angew. Chem., Int. Ed., 2010, 49, 6448–6453 CrossRef CAS.
- G. Bantchev, Z. H. Lu and Y. Lvov, J. Nanosci. Nanotechnol., 2009, 9, 396–403 CrossRef CAS.
- A. Izquierdo, S. S. Ono, J. C. Voegel, P. Schaaf and G. Decher, Langmuir, 2005, 21, 7558–7567 CrossRef CAS.
- W. S. Liao, S. Cheunkar, H. H. Cao, H. R. Bednar, P. S. Weiss and A. M. Andrews, Science, 2012, 337, 1517–1521 CrossRef CAS.
- M. R. Beaulieu, N. R. Hendricks and J. J. Watkins, ACS Photonics, 2014, 1, 799–805 CrossRef CAS.
- H. Bai, F. Walsh, B. Gludovatz, B. Delattre, C. L. Huang, Y. Chen, A. P. Tomsia and R. O. Ritchie, Adv. Mater., 2016, 28, 50–56 CrossRef CAS.
- D. N. Minh, S. Eom, L. A. T. Nguyen, J. Kim, J. H. Sim, C. Seo, J. Nam, S. Lee, S. Suk, J. Kim and Y. Kang, Adv. Mater., 2018, 30, 1802555 CrossRef.
- S. Srivastava, V. Ball, P. Podsiadlo, J. Lee, P. Ho and N. A. Kotovlt, J. Am. Chem. Soc., 2008, 130, 3748–3749 CrossRef CAS.
- V. Selin, J. F. Ankner and S. A. Sukhishvili, Macromolecules, 2015, 48, 3983–3990 CrossRef CAS.
- J. J. Richardson, B. L. Tardy, H. Ejima, J. L. Guo, J. W. Cui, K. Liang, G. H. Choi, P. J. Yoo, B. G. De Geest and F. Caruso, ACS Appl. Mater. Interfaces, 2016, 8, 7449–7455 CrossRef CAS.
- R. Gill, M. Mazhar, O. Felix and G. Decher, Angew. Chem., Int. Ed., 2010, 49, 6116–6119 CrossRef CAS.
- B. G. De Geest, N. N. Sanders, G. B. Sukhorukov, J. Demeester and S. C. De Smedt, Chem. Soc. Rev., 2007, 36, 636–649 RSC.
- M. Delcea, H. Mohwald and A. G. Skirtach, Adv. Drug Delivery Rev., 2011, 63, 730–747 CrossRef CAS.
- P. Kekicheff, G. F. Schneider and G. Decher, Langmuir, 2013, 29, 10713–10726 CrossRef CAS.
- A. Zhuk and S. A. Sukhishvili, Soft Matter, 2013, 9, 5149–5154 RSC.
- W. F. Zhao, R. W. N. Nugroho, K. Odelius, U. Edlund, C. S. Zhao and A. C. Albertsson, ACS Appl. Mater. Interfaces, 2015, 7, 4202–4215 CrossRef CAS.
- Y. Q. Gu, E. K. Weinheimer, X. Ji, C. G. Wiener and N. S. Zacharia, Langmuir, 2016, 32, 6020–6027 CrossRef CAS.
- F. Q. Fan, L. Wang, F. Li, Y. Fu and H. P. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 17004–17010 CrossRef CAS.
- X. Fu, L. Hosta-Rigau, R. Chandrawati and J. W. Cui, Chem, 2018, 4, 2084–2107 CAS.
- D. J. Schmidt, F. C. Cebeci, Z. I. Kalcioglu, S. G. Wyman, C. Ortiz, K. J. Van Vliet and P. T. Hammond, ACS Nano, 2009, 3, 2207–2216 CrossRef CAS.
- K. J. Si, D. Sikdar, Y. Chen, F. Eftekhari, Z. Q. Xu, Y. Tang, W. Xiong, P. Z. Guo, S. Zhang, Y. R. Lu, Q. L. Bao, W. R. Zhu, M. Premaratne and W. L. Cheng, ACS Nano, 2014, 8, 11086–11093 CrossRef CAS.
- S. Jahani and Z. Jacob, Nat. Nanotechnol., 2016, 11, 23–36 CrossRef CAS.
- N. I. Zheludev and E. Plum, Nat. Nanotechnol., 2016, 11, 16–22 CrossRef CAS.
- J. D. Caldwell, I. Vurgaftman, J. G. Tischler, O. J. Glembocki, J. C. Owrutsky and T. L. Reinecke, Nat. Nanotechnol., 2016, 11, 9–15 CrossRef CAS.
- A. Boker, J. He, T. Emrick and T. P. Russell, Soft Matter, 2007, 3, 1231–1248 RSC.
- S. K. Ghosh and A. Boker, Macromol. Chem. Phys., 2019, 220, 1900196 CrossRef.
- H. Y. Li, J. D. Carter and T. H. LaBean, Mater. Today, 2009, 12, 24–32 CrossRef CAS.
- G. A. Ozin, K. Hou, B. V. Lotsch, L. Cademartiri, D. P. Puzzo, F. Scotognella, A. Ghadimi and J. Thomson, Mater. Today, 2009, 12, 12–23 CrossRef CAS.
- Z. H. Nie, A. Petukhova and E. Kumacheva, Nat. Nanotechnol., 2010, 5, 15–25 CrossRef CAS.
- D. P. Song, C. Li, W. H. Li and J. J. Watkins, ACS Nano, 2016, 10, 1216–1223 CrossRef CAS.
- S. C. Warren, L. C. Messina, L. S. Slaughter, M. Kamperman, Q. Zhou, S. M. Gruner, F. J. DiSalvo and U. Wiesner, Science, 2008, 320, 1748–1752 CrossRef CAS.
- Z. H. Li, K. Hur, H. Sai, T. Higuchi, A. Takahara, H. Jinnai, S. M. Gruner and U. Wiesner, Nat. Commun., 2014, 5, 3247 CrossRef.
- J. M. Ha, S. H. Lim, J. Dey, S. J. Lee, M. J. Lee, S. H. Kang, K. S. Jin and S. M. Choi, Nano Lett., 2019, 19, 2313–2321 CrossRef CAS.
- Z. H. Nie, D. Fava, E. Kumacheva, S. Zou, G. C. Walker and M. Rubinstein, Nat. Mater., 2007, 6, 609–614 CrossRef CAS.
- C. R. Iacovella, M. A. Horsch and S. C. Glotzer, J. Chem. Phys., 2008, 129, 044902 CrossRef.
- J. S. Xia, N. Horst, H. X. Guo and A. Travesset, Macromolecules, 2019, 52, 8056–8066 CrossRef CAS.
- R. A. LaCour, C. S. Adorf, J. Dshemuchadse and S. C. Glotzer, ACS Nano, 2019, 13, 13829–13842 CrossRef CAS.
- E. J. Bailey and K. I. Winey, Prog. Polym. Sci., 2020, 105, 101242 CrossRef CAS.
- B. J. Kim, G. H. Fredrickson, C. J. Hawker and E. J. Kramer, Langmuir, 2007, 23, 7804–7809 CrossRef CAS.
- B. J. Kim, G. H. Fredrickson, J. Bang, C. J. Hawker and E. J. Kramer, Macromolecules, 2009, 42, 6193–6201 CrossRef CAS.
- S. G. Jang, D. J. Audus, D. Klinger, D. V. Krogstad, B. J. Kim, A. Cameron, S. W. Kim, K. T. Delaney, S. M. Hur, K. L. Killops, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, J. Am. Chem. Soc., 2013, 135, 6649–6657 CrossRef CAS.
- E. R. Zubarev, J. Xu, A. Sayyad and J. D. Gibson, J. Am. Chem. Soc., 2006, 128, 15098–15099 CrossRef CAS.
- B. Zhao and L. Zhu, Macromolecules, 2009, 42, 9369–9383 CrossRef CAS.
- L. Chen and H. A. Klok, Soft Matter, 2013, 9, 10678–10688 RSC.
- A. M. Percebom, J. J. Giner-Casares, N. Claes, S. Bals, W. Loh and L. M. Liz-Marzan, Chem. Commun., 2016, 52, 4278–4281 RSC.
- R. Gunawidjaja, S. Peleshanko, H. Ko and V. V. Tsukruk, Adv. Mater., 2008, 20, 1544–1549 CrossRef CAS.
- A. Abdilla, N. D. Dolinski, P. de Roos, J. M. Ren, E. van der Woude, S. E. Seo, M. S. Zayas, J. Lawrence, J. R. de Alaniz and C. J. Hawker, J. Am. Chem. Soc., 2020, 142, 1667–1672 CrossRef CAS.
- S. G. Jang, E. J. Kramer and C. J. Hawker, J. Am. Chem. Soc., 2011, 133, 16986–16996 CrossRef CAS.
- K. C. Wei, J. Li, J. H. Liu, G. S. Chen and M. Jiang, Soft Matter, 2012, 8, 3300–3303 RSC.
- C. Durand-Gasselin, N. Sanson and N. Lequeux, Langmuir, 2011, 27, 12329–12335 CrossRef CAS.
- C. L. Yi, Y. Q. Yang and Z. H. Nie, J. Am. Chem. Soc., 2019, 141, 7917–7925 CrossRef CAS.
- J. Y. Zhang, P. J. Santos, P. A. Gabrys, S. Lee, C. Liu and R. J. Macfarlane, J. Am. Chem. Soc., 2016, 138, 16228–16231 CrossRef CAS.
- Y. P. Wang, P. J. Santos, J. M. Kubiak, X. H. Guo, M. S. Lee and R. J. Macfarlane, J. Am. Chem. Soc., 2019, 141, 13234–13243 CrossRef CAS.
- J. M. Kubiak and R. J. Macfarlane, Adv. Funct. Mater., 2019, 29, 1905168 CrossRef CAS.
- P. J. Santos and R. J. Macfarlane, J. Am. Chem. Soc., 2020, 142, 1170–1174 CrossRef CAS.
- P. J. Santos, T. C. Cheung and R. J. Macfarlane, Nano Lett., 2019, 19, 5774–5780 CrossRef CAS.
- P. J. Santos, Z. Cao, J. Y. Zhang, A. Alexander-Katz and R. J. Macfarlane, J. Am. Chem. Soc., 2019, 141, 14624–14632 CrossRef CAS.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- L. M. Liz-Marzan, Langmuir, 2006, 22, 32–41 CrossRef CAS.
- P. C. Ma, M. Y. Liu, H. Zhang, S. Q. Wang, R. Wang, K. Wang, Y. K. Wong, B. Z. Tang, S. H. Hong, K. W. Paik and J. K. Kim, ACS Appl. Mater. Interfaces, 2009, 1, 1090–1096 CrossRef CAS.
- F. A. Rahim and K. Dong-Hwan, Nano Today, 2016, 11, 415–434 CrossRef.
- M. A. Boles, M. Engel and D. V. Talapin, Chem. Rev., 2016, 116, 11220–11289 CrossRef CAS.
- J. T. Hu, Y. Huang, Y. M. Yao, G. R. Pan, J. J. Sun, X. L. Zeng, R. Sun, J. B. Xu, B. Song and C. P. Wong, ACS Appl. Mater. Interfaces, 2017, 9, 13544–13553 CrossRef CAS.
- A. Akelah, A. Rehab, M. Abdelwahab and M. A. Betiha, Nanocomposites, 2017, 3, 20–29 CrossRef CAS.
- S. Homaeigohar and M. Elbahri, Adv. Opt. Mater., 2019, 7, 1970004 CrossRef.
- K. R. Deng, Z. S. Luo, L. Tan and Z. W. Quan, Chem. Soc. Rev., 2020, 49, 6002–6038 RSC.
- J. Huh, V. V. Ginzburg and A. C. Balazs, Macromolecules, 2000, 33, 8085–8096 CrossRef CAS.
- X. W. Gu, X. C. Ye, D. M. Koshy, S. Vachhani, P. Hosemann and A. P. Alivisatos, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2836–2841 CrossRef CAS.
- S. J. Wan, J. S. Peng, L. Jiang and Q. F. Cheng, Adv. Mater., 2016, 28, 7862–7898 CrossRef CAS.
- G. P. Wu, X. Y. Liu, X. X. Chen, H. S. Suh, X. Li, J. X. Ren, C. G. Arges, F. X. Li, Z. Jiang and P. F. Nealey, Adv. Mater. Interfaces, 2016, 3, 1600048 CrossRef.
- A. K. Singh, A. Parhi, B. P. Panda, S. Mohanty, S. K. Nayak and M. K. Gupta, J. Mater. Sci.: Mater. Electron., 2017, 28, 17655–17674 CrossRef CAS.
- M. R. Begley, D. S. Gianola and T. R. Ray, Science, 2019, 364, 1250–1257 CrossRef.
- A. Maiti, J. Wescott and P. Kung, Mol. Simul., 2005, 31, 143–149 CrossRef CAS.
- L. R. Dalton, Organic Photonic Materials and Devices Xv, 2013, p. 8622 Search PubMed.
- C. L. Yi, Y. Q. Yang, B. Liu, J. He and Z. H. Nie, Chem. Soc. Rev., 2020, 49, 465–508 RSC.
| This journal is © The Royal Society of Chemistry 2021 |