
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effects of overhang placement and multivalency on cell labeling by DNA origami†
Ying
Liu‡
 a,
Piyumi
Wijesekara‡
b,
Sriram
Kumar
a,
Weitao
Wang
a,
Xi
Ren
*ab and
Rebecca E.
Taylor
*abc
a,
Piyumi
Wijesekara‡
b,
Sriram
Kumar
a,
Weitao
Wang
a,
Xi
Ren
*ab and
Rebecca E.
Taylor
*abc
aDepartment of Mechanical Engineering, Carnegie Mellon University, 5000 Forbes Avenue, Pittsburgh, USA. E-mail: bex@andrew.cmu.edu; Fax: +1-412-268-3348; Tel: +1-412-268-2500
bDepartment of Biomedical Engineering, Carnegie Mellon University, 5000 Forbes Avenue, Pittsburgh, USA. E-mail: xiren@cmu.edu
cDepartment of Electrical and Computer Engineering, Carnegie Mellon University, 5000 Forbes Avenue, Pittsburgh, USA
First published on 2nd April 2021
Abstract
Through targeted binding to the cell membrane, structural DNA nanotechnology has the potential to guide and affix biomolecules such as drugs, growth factors and nanobiosensors to the surfaces of cells. In this study, we investigated the targeted binding efficiency of three distinct DNA origami shapes to cultured endothelial cells via cholesterol anchors. Our results showed that the labeling efficiency is highly dependent on the shape of the origami as well as the number and the location of the binding overhangs. With a uniform surface spacing of binding overhangs, 3D isotropic nanospheres and 1D anisotropic nanorods labeled cells effectively, and the isotropic nanosphere labeling fit well with an independent binding model. Face-decoration and edge-decoration of the anisotropic nanotile were performed to investigate the effects of binding overhang location on cell labeling, and only the edge-decorated nanotiles were successful at labeling cells. Edge proximity studies demonstrated that the labeling efficiency can be modulated in both nanotiles and nanorods by moving the binding overhangs towards the edges and vertices, respectively. Furthermore, we demonstrated that while double-stranded DNA (dsDNA) bridge tethers can rescue the labeling efficiency of the face-decorated rectangular plate, this effect is also dependent on the proximity of bridge tethers to the edges or vertices of the nanostructures. A final comparison of all three nanoshapes revealed that the end-labeled nanorod and the nanosphere achieved the highest absolute labeling intensities, but the highest signal-to-noise ratio, calculated as the ratio of overall labeling to initiator-free background labeling, was achieved by the end-labeled nanorod, with the edge-labeled nanotile coming in second place slightly ahead of the nanosphere. The findings from this study can help us further understand the factors that affect membrane attachment using cholesterol anchors, thus providing guidelines for the rational design of future functional DNA nanostructures.
 Rebecca Taylor | Rebecca Taylor is an Assistant Professor of Mechanical Engineering, and, by courtesy, of Biomedical Engineering and Electrical and Computer Engineering at Carnegie Mellon University (CMU). Her degrees are in Mechanical Engineering with a B.S.E in 2001 from Princeton University and a Ph.D. in 2013 with Prof. Beth Pruitt at Stanford University. During her postdoctoral training she worked in the laboratory of Prof. James Spudich in Biochemistry at the Stanford University School of Medicine. She now combines both microfabrication and nanofabrication to create hybrid top-down and bottom-up fabricated sensors and actuators for nanobiosensing, robotics, and advanced manufacturing applications. |
Introduction
DNA origami is a self-assembly technique for folding long single-stranded DNA (ssDNA) “scaffolds” into one-, two-, or three-dimensional nanoshapes typically using around 200 unique short ssDNA “staples”.1,2These nanostructures can be decorated like breadboards with nanometer-scale control over the placement of biological molecules, fluorophores, and other nanoparticles.3,4 Incorporation of flexible and responsive elements further allows numerous applications in drug delivery, presentation of growth factors, and the creation and delivery of nucleic acid-based nanobiosensors and nanorobots.5,6
The aforementioned capabilities have enabled both qualitative and quantitative studies with emerging applications that are difficult to achieve through other means, one of which is the interface with cellular membranes.7 Cellular membranes are crucial for cell functionalities like cell communications, mobility, proliferation and differentiation.8–11 For applications that target cellular membranes, such as force sensing12 and delivery of drugs that target membrane-bound proteins,13 it is critical to attach functional DNA nanostructures to the exterior surface of the membrane. Utilizing hydrophobic molecules like cholesterol tags as anchors is one of the most commonly used methods.14–19 Among which, a two-step approach was usually applied when introducing DNA nanostructures to the cell surface: first, the lipophilic cholesterol molecules are covalently conjugated to small stretches of hydrophilic ssDNA, which allows insertion of these amphiphilic initiators into the lipid bilayer's hydrophobic core. Then DNA nanostructures carrying complementary ssDNA hybridize with the cholesterol-ssDNA initiators, leading to the attachment to the membrane.20,21 This approach helps avoid aggregation of DNA nanostructures otherwise caused by conjugated hydrophobic cholesterol groups.22,23 Researchers have also been investigating factors that affect the membrane attachment, such as the number and nanoscale spatial distribution of anchors on DNA nanostructures, using artificial membrane like giant unilamellar vesicles (GUVs) as model system.24,25 Furthermore, using spacers, ssDNA or dsDNA that elongate the distance of anchors to DNA nanostructures, has been shown to provide the conformational flexibility through attachment chemistry, enhancing the accessibility to the initiators and thus increasing the attachment efficiency.26
However, translating attachment strategies on GUVs to the cellular plasma membrane is a more complex challenge and thus different design rules may be necessary for targeting DNA nanostructures to cells. The cell membrane offers numerous receptors for targeted attachment,27 but navigating DNA nanostructures through the crowded cell surface environment is a critical challenge, since the surfaces of many prokaryotic and eukaryotic cells, including endothelial cells, are coated with a glycocalyx layer composed of glycoproteins and glycolipids.28 This glycocalyx has diverse biological roles as a regulator of inflammation,29 a modulator of capillary red blood cell filling,30 and a protective barrier. For this reason, we hypothesized that the shape and placement of binding ligands on DNA origami would be critical to cellular attachment. Further, in this work we sought understand the role of targeted versus non-targeted adhesion, because there can be substantial membrane interaction and resulting cellular uptake of non-target DNA origami.31,32 Live cells constantly uptake nanostructures around the membrane nonspecifically. This uptake, however, has been shown to be highly dependent on the cell types as well as the size, shape, and decoration of DNA nanostructures with no universal behavior pattern reported. For example, Bastings et al. studied the endocytosis of 11 distinct DNA origami shapes that were coated with oligolysine(K10)-PEG(5K) for enhanced stability, finding that DNA origami with greater compactness were preferentially internalized compared with elongated, high-aspect-ratio ones.33 In contrast, Wang et al.34 investigated uptake of four distinct undecorated DNA origami shapes by multiple human cancer lines and found nearly the opposite trend, that larger and elongated structures were taken up more efficiently.
Membrane embedding has also emerged as a powerful tool for programming cell–cell interactions.35,36 Akbari et al. have demonstrated that cholesterol-embedding and attachment of DNA origami breadbroads via a tether can be used as a universal method for targeting cellular membranes,21 but to date, design rules for tether-free binding of DNA origami via cholesterol-embedding have not been elucidated.
The high level of nanoscale geometric control within DNA origami makes it possible to systematically investigate the role of local shape and ligand placement on targeted origami binding to cells.37–42 Studies on cellular and artificial membranes using receptor binding suggest that copy number, position and spacing of ligands are critical to enhance binding efficiency.43–46 Therefore, in this work, we systematically investigated the nanostructure shape, as well as the origami-based multivalency and positioning of binding overhangs to modulate binding efficiency through cholesterol-mediated membrane embedding. All studies were performed on human umbilical vein endothelial cells (HUVECs), a cell type that is particularly relevant for future vascular studies.
Results
Here we designed three DNA origami shapes: a rectangular nanotile, a nanorod and a wire-framed nanosphere. Each of the three shapes was folded from a M13mp18 scaffold with a set of approximately 200 unique staple sequences.47 Therefore, the three nanostructures with distinct dimensions and shapes have similar molecular weights. Atomic force microscopy (AFM) images indicated successful formation of these shapes (Fig. 1). The detailed design and sequence information of each origami is provided in Fig. S1–S7.† | ||
| Fig. 1 The design of the three DNA origami shapes. We have designed and synthesized three distinct DNA origami shapes including a nanotile, nanorod, and a nanosphere with similar molecular weights. The AFM images indicated successful formation of the origami structures. Scale bars: 100 nm. | ||
Using a two-step cholesterol anchoring technique,21 we targeted the DNA origami to the plasma membrane of our model cell type, cultured HUVECs. In the first step of this process, cholesterol-conjugated ssDNA oligonucleotides or “initiators” were introduced into cell culture with the hydrophobic moiety cholesterol on the initiators inserted into the middle plane of the lipid bilayer via hydrophobic interaction.48,49 Subsequently, the DNA origami with complementary ssDNA could hybridize to the cell surface-tethered initiator oligonucleotides (Fig. 2a). To avoid potential hydrophobic effects of fluorophore decoration during self-assembly,50,51 each DNA origami nanostructure was pre-annealed with 35 biotin tags to allow post-assembly fluorescence decoration. After cell fixation, origami was stained by introducing Alexa Fluor 647-labeled streptavidin (SA647) that binds to biotin. Thus the labeling efficiency of DNA origami to the cell membrane can be quantified by measuring the fluorescence intensity of SA647 binding. In this study we utilized both fluorescence microscopy and spectrophotometer microplate reader studies to assess labeling efficiency (Fig. 2). Spectrophotometer microplate reader measurement was used to quantify the fluorescence intensity of the whole well area, providing a time-efficient approach for the assessment of labeling effectiveness. In addition, using a plate reader allows a direct readout from the cultured endothelial cells. The fluorescence intensity read from the spectrophotometer microplate reader represents the labeling efficiency of DNA origami to the cell surfaces and the microscope images were used to observe and confirm the cell morphology. In each experiment, we also carried a condition without the cholesterol-ssDNA initiators as a control signal, wherein the DNA origami were not targeted to the cell surface and only attached to the cell surface in a non-targeted manner. We define effective targeted labeling (the signal) as that which provides greater than a twofold increase beyond the non-targeted initiator-free condition (the noise).
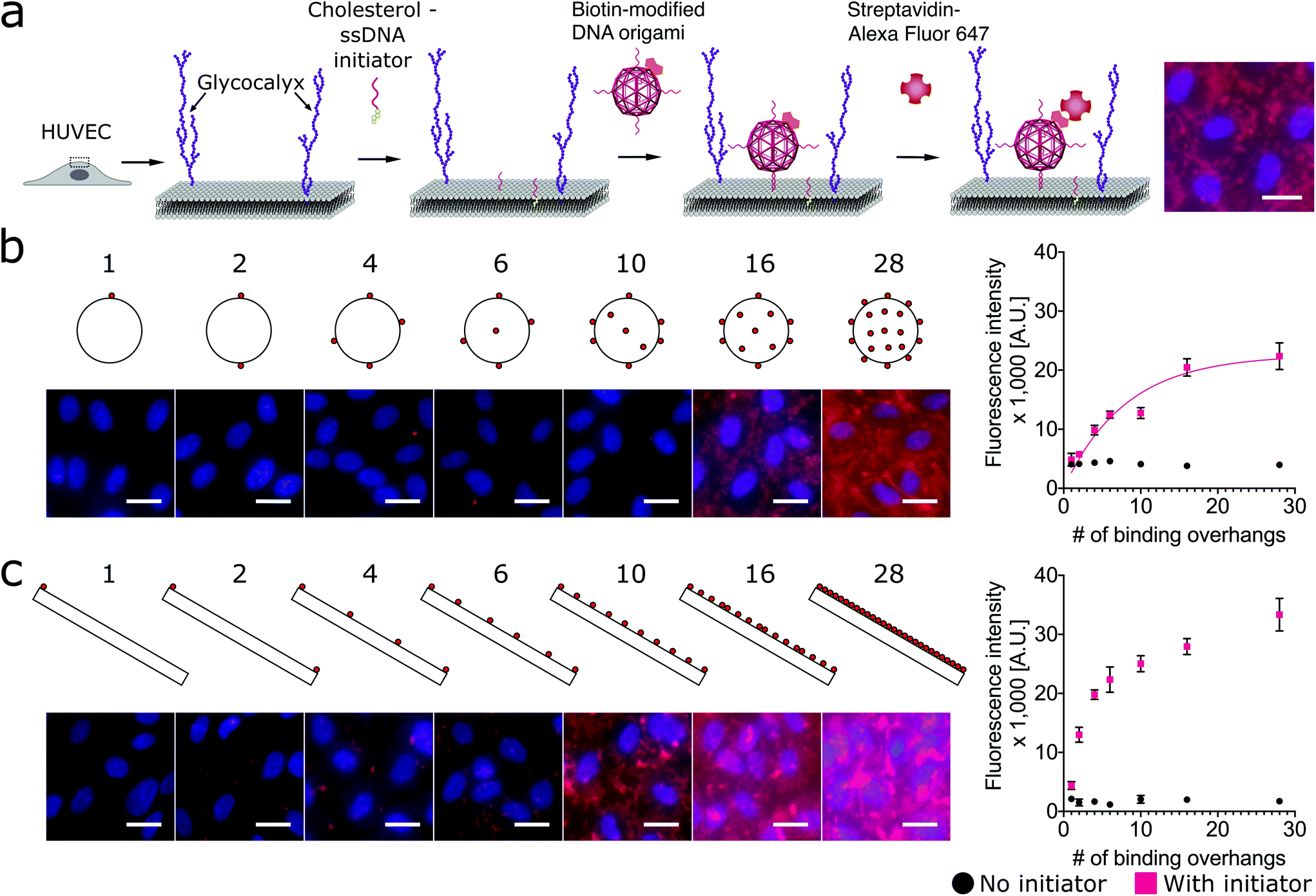 | ||
Fig. 2 Targeting DNA origami to cells through a two-step targeting via cholesterol anchors: increasing the number of binding overhangs on the nanosphere and nanorod improves the cell labeling efficiency in a nonlinear fashion. (a) The cartoon shows the two-step approach to target the DNA origami with binding overhangs to the surface of human umbilical vein endothelial cells (HUVECs). Cholesterol-modified ssDNA were first introduced to anchor within the lipid bilayer of the membrane. Next, DNA origami nanostructures with biotin tags and complementary ssDNA binding overhangs were introduced to hybridize with the anchor strands and label the membrane. To assess binding efficiency, the cells were fixed and stained with Alexa Fluor 647-labeled streptavidin (SA647). (b) DNA origami nanospheres and (c) nanorods were prepared with up to 28 approximately uniformally-spaced ssDNA binding overhangs. Fluorescence intensity studies quantified by spectrophotometer microplate reader indicated that for both shapes labeling efficiency increased nonlinearly with increasing multivalency. The nanorod and nanosphere both provided strong labeling, requiring as few as 2 or 4 binding overhangs, respectively, to achieve an initiator![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :initiator-free (signal:noise) ratio greater than two. On the fluorescence microscope images, red: SA647 channel that indicated DNA origami labeling of cells; blue: Hoechst channel that showed the nuclei of the cells. Error bars were calculated from the standard deviation from the triplicate data. Scale bars: 20 μm. :initiator-free (signal:noise) ratio greater than two. On the fluorescence microscope images, red: SA647 channel that indicated DNA origami labeling of cells; blue: Hoechst channel that showed the nuclei of the cells. Error bars were calculated from the standard deviation from the triplicate data. Scale bars: 20 μm. | ||
To investigate how geometries and multivalency regulate cell surface targeting, we engineered an isotropic nanosphere and an anisotropic nanorod with an approximately uniform distribution of binding overhangs, from 1 to 28. The agarose gel and AFM results confirmed the origami nanostructures were intact before being introduced to cells, and the actual number of binding sites correspond to the design, see Fig. S8–S15.† For the isotropic nanosphere, if we assume that the binding probability of each overhang is independent, we can model the relationship of binding intensity versus the number of binding overhangs with eqn (1). This equation states that the labeling intensity I is proportional to the probability of at least one overhang binding.
| I = Imax·[1 − (1 − p)n] | (1) |
I max is the maximum labeling intensity assuming all origami structures were captured and attached to the cell surface, p is the probability of membrane binding for a single binding overhang, and n is the number of binding overhangs on each DNA origami nanoshape.
As shown in Fig. 2b, we found that increasing the number of binding overhangs on the DNA origami nanospheres could effectively increase their affinity to the cell surface resulting in more than a twofold change above the initiator-free control signal. Given the nature of this model, it is clear that there are diminishing returns for increasing labeling intensity using large numbers of overhangs. To identify a critical number of overhangs for effective labeling, we define a parameter that represents the number of overhangs needed for 50% of maximal labeling (L50). This parameter can serve as a guideline for DNA nanostructure designers. Extracting the L50 from a nonlinear regression analysis of the nanosphere data fit to eqn (1), we find that the L50 for the nanosphere is 6 binding overhangs.
While the nanorod labeling trend was qualitatively similar to the three-dimensional (3D) nanosphere, the assumption of independent and equal binding probabilities can not be applied to the 1-dimensional (1D) nanorod due to its anisotropy. That is, we would expect the location of the binding overhangs to impact their binding probabilities. To understand the role of anisotropy, we next investigated a two-dimensional (2D) plate-like nanotile which has sharp edge regions and flat face regions. We make the assumption that overhangs on edges have uniform binding probabilities that are distinct from the uniform binding probabilities of overhangs on flat face regions. Accordingly, we hypothesized that increasing numbers of binding overhangs would likely result in higher fluorescence, but that face decoration and edge decoration might result in different levels of labeling efficiency.
To assess the relative labeling efficiency of overhangs on edges versus faces, two populations of nanotiles were prepared: face-decorated nanotiles with up to 28 binding overhangs (Fig. 3a) and edge-decorated nanotiles with up to 12 binding overhangs (number limited due to the total number of staple sequences available on the edge of the nanotile). For these studies the face-decorated nanotiles performed notably worse than the edge-decorated nanotiles. For all numbers of face-decorated binding overhangs, the fluorescence signal intensity did not exceed twice than that of the initiator-free control group and therefore did not effectively label the cell membranes (Fig. 3a). In contrast, edge-decorated nanotiles were able to effectively label the cell surface with as few as four binding overhangs resulting in labeling more than four times that of the initiator-free control. The L50 of the edge-decorated nanotiles was found to be 4 binding overhangs.
 | ||
| Fig. 3 Increasing the number of binding overhangs on the edges of the nanotiles is more effective than face decoration for cell surface targeting. DNA origami nanotiles were prepared with (a) up to 28 ssDNA binding overhangs on one face and (b) up to 12 ssDNA binding overhangs on two opposite edges. As with the nanorods and nanospheres, fluorescence intensity studies indicated that labeling efficiency increased with increasing multivalency. However, the labeling of face-decorated nanotile were ineffective with no more than twice the initiator-free control for all numbers of binding overhangs. Edge-decoration of nanotiles, however, did result in effective cell labeling with as few as four binding overhangs. This indicates that edge-decoration with binding overhangs is a tether-free approach for rescuing the labeling efficiency of nanotiles. On the fluorescence microscope images, red: SA647 channel; blue: Hoechst channel. Error bars were calculated from the standard deviation from the triplicate data. Scale bars: 20 μm. | ||
This result is consistent with a “corner attack” approach, as has been described previously.34,52 For this model of approach, the sharp or “pointy” portions of a DNA nanostructure like the vertices of the nanorod or edges of the 2D nanotile interact more directly with the membrane. Furthermore, this result may simply reflect the practical challenges of approaching the cell membrane that is crowded with glycoproteins of the glycocalyx. Our results suggest that the crowded membrane inhibits binding at the face of nanotiles, and only binding overhangs near the edge of nanotiles can access the initiators on the membrane.
To test the hypothesis that proximity to the edge or end of DNA nanostructures modulates labeling efficiency, we must relax the previous assumption that all face-decorated binding overhangs have the same binding probabilites. We synthesized both anisotropic shapes, 1D nanorods with sharp ends and 2D nanotiles with sharp edges, each with a group of binding overhangs at increasing distances from one end or edge, respectively (Fig. 4a and b). The nanorods were decorated with three binding overhangs with the closest distance to the end ranging from 0 nm to 175 nm (Fig. 4a). Unlike our previous valence study on the nanorod (Fig. 2b), which always had at least one binding overhang at the end, this end proximity test only demonstrated effective labeling greater than twice the initator-free control signal for the binding overhangs located 0 nm from the end. This suggests that valence is not enough to predict labeling efficiency and proximity to the end is also critical to ensure the nanorod binding. The data was analyzed with one-way ANOVA analysis with post hoc Tukey's test and the P values between neighbor conditions was shown in Fig. 2. The full table of the P value can be found in Fig. S17.†
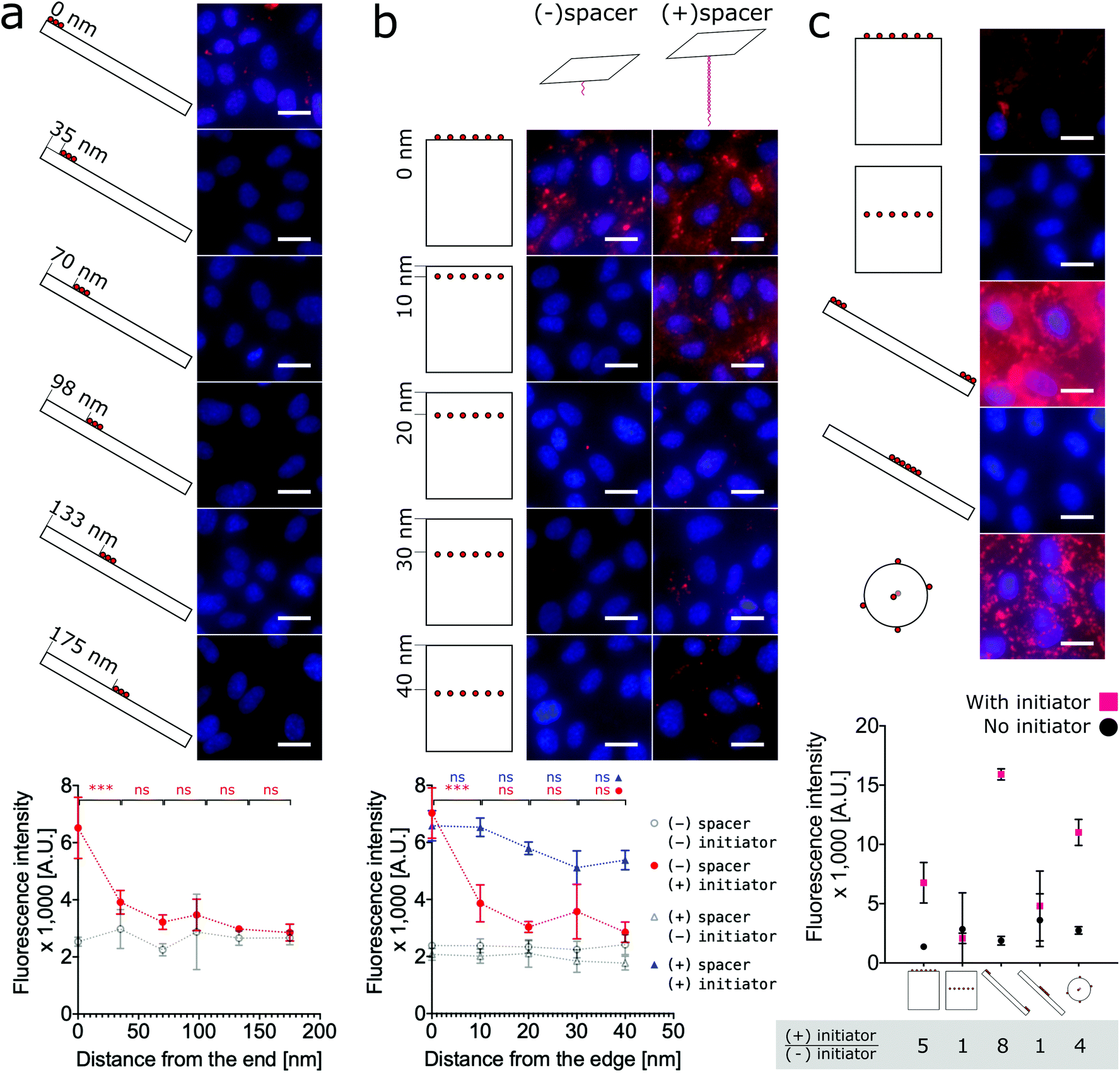 | ||
| Fig. 4 Cell labeling is dependent on the proximity of binding overhangs to the edge or end of the nanostructure. (a) Nanorods were decorated with groups of three binding overhangs located with increasing distance from the end. The labeling was effective when the binding overhands were at the end of the nanorod. For cases with 35 nm and farther from the end, the labeling efficiency was not twice the initiator-free control signal. (b) For an analogous 2D study, nanotiles were prepared with rows of six binding overhangs located from 0 nm to 40 nm from the edge. Labeling with these nanotiles was only effective at greater than twice the initiator-free control signal when the row of binding overhangs was located at the edge. When binding overhangs were extended from the nanotile with 80 base pair long bridge tethers, labeling efficiency for all cases was effective. However, labeling efficiency did decrease with increasing distance from the edge even for the rescued bridge tether conditions. To test the dependence of labeling efficiency on proximity to the edge, one-way ANOVA analysis with post hoc Tukey's test was run with Prism Graphpad on the neighboring data for those conditions with initiator. ns P > 0.05, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. (c) A direct comparison the labeling efficiency of the three shapes and extreme labeling locations is provided. For consistency each DNA origami shape was decorated with six binding overhangs, and the resulting labeling efficiencies show that end-labeled nanorods, nanospheres, and edge-labeled nanotiles efficiently labeled the cells. Centrally-located overhangs on anisotropic shapes did not result in strong labeling. The highest signal:noise ratio from the initiator:initiator-free condition was achieved by the end-labeled nanorod, with the edge-labeled nanotile coming in second place slightly ahead of the nanosphere. On the fluorescence microscope images, red: SA647 channel; blue: Hoechst channel. Scale bars: 20 μm. | ||
The analogous investigation for our 2D nanotile was performed by decorating the tile with rows of six overhangs at distances ranging from 0 nm to 40 nm from the edge. This study likewise demonstrated that valence is not the only factor in predicting labeling efficiency by the nanotile. Effective labeling greater than twice the initiator-free control signal was similarly only observed for the nanotiles with a row of binding overhangs right on the edge (Fig. 4b). These findings demonstrate that nanorods and nanotiles with a small number of binding overhangs can effectively label the cell membrane, but labeling efficiency is dependent on proximity to the end or edge of the nanostructure.
Previous studies have demonstrated that long bridge tethers could improve binding, providing a universal method for membrane labeling.26 To confirm the utility of this approach and test its dependence of edge proximity, nanotiles were decorated with rows tethering “bridges” at positions 0 nm to 40 nm from the edge. These bridge tethers consist of 80 base-pair dsDNA or approximately 27 nm in length. As shown in Fig. 4b, the presence of both the initiator and the bridge tether resulted in efficient labeling that gradually decreased with distance from the edge. Surprisingly, the labeling was effective even when the distance from the edge was greater than the length of the tether. In fact, all bridge tether conditions resulted in effective labeling greater than twice the initiator-free controls. This result indicates that effective tether-mediated labeling is dependent on both the presence of and the location of the bridge tethers. This work finds, however, that bridge tethers are not necessary for efficient cell labeling by nanotiles with edge-located binding overhangs. Further, bridge tethers can rescue labeling efficiency on nanotiles, but labeling is greatest for edge-located bridge tethers.
Finally, to provide a direct comparison of all three shapes and the location-specific labeling approach in a single experiment, we decorated cells with representative conditions of each nanoshape. As shown in Fig. 4c, for consistency we applied six binding overhangs in each of the conditions, and for the anisotropic nanotiles and nanorods, we tested two extreme conditions with the binding sites at the end/edge and in the middle. This ensemble study confirms that edge-labeled nanotiles and end-labeled nanorods can efficiently label cells, while centrally-labeled nanotiles and nanorods are unsuccessful. Maximum labeling intensities were achieved by the end-labeled nanorod and the nanosphere. However, the highest signal:noise ratios calculated as the ratio of overall labeling to initiator-free background labeling, was achieved by the end-labeled nanorod, with edge-labeled nanotile coming in second place slightly ahead of the nanosphere.
Discussion
In this work, we labeled HUVECs with three distinct DNA origami shapes, a nanosphere, a nanorod, and a nanotile using a two-step cholesterol anchoring process. This approach has numerous applications in the presentation and delivery of drug molecules, growth factors, and nanobiosensors to the surfaces of cells. In order to elucidate the design rules for nanostructure decoration to maximize labeling efficiency we investigated the effects of both geometry and valence on the efficiency of cell labeling.First, we demonstrated for the nanospheres and nanorods that increasing the number of approximately uniformally-distributed binding overhangs effectively increased the labeling efficiency in a non-linear fashion. The trend for the isotropic nanosphere agreed with a model wherein the binding probability of each overhang is independent. Our L50 metric provides a design guideline for future researchers aiming to achieve efficient labeling with a small number of overhangs. The L50 of 6 for the nanosphere indicates that only 6 binding overhangs are needed to achieve maximal labeling intensity.
Simply increasing the number of binding overhangs on a 2D nanotile, however, did not universally increase labeling efficiency. None of the face-labeled nanotiles with up to 28 binding overhangs were able to efficiently label the cell membrane. Whereas, nanotiles with as few as four edge-located binding overhangs were able to effectively label the cell membranes. More careful investigation of this edge-proximity effect revealed that labeling efficiency with both nanorods and nanotiles is highly dependent on the spatial placement of binding overhangs: a small number of binding overhangs at the end or edge of the nanostructures resulted in effective labeling, while labeling was not effective for other distances from the end or edge. This finding suggests that the labeling efficiency is highly dependent on the location of binding overhangs, and that we can potentially rescue the labelling performance of certain shapes like the nanotile by moving the binding overhangs to the pointy regions. Efficient labeling strategies for anisotropic nanorod and nanotile are particularly important because, when end- and edge-labeled, these shapes achieved higher signal:noise ratios than the isotropic sphere. Additional labeling improvements for nanotiles can be realized by utilizing bridge tethers. However, even though a bridge tether was able to rescue the labeling efficiency of the binding overhangs far away from the edge of the nanotile, the labeling efficiency did decrease with the proximity to edge.
Future studies of DNA origami on cell membranes may require the nanostructures to make direct contact with and through the cell membrane, and for those studies bridge tethers or tethers would not be effective. It is therefore an important finding that bridge tethers may not be necessary for labeling cell membranes if binding overhangs are located near sharp nanostructural features like ends, corners and edges.
Previous simulation work52 by Ding et al. and TEM investigations34 by Wang et al. that studied the process of a non-specific cell uptake of DNA origami nanorods and tetrahedra found that these nanostructures demonstrate a “corner attack mode” whereby nanostructures enter membranes corner-first. Our result extends their findings for targeted binding to cellular membranes and suggests that locating binding overhangs at the “pointy” regions of DNA origami nanostructures can increase labeling efficiency.
We speculate that the end- and edge-preferred interaction mode between the DNA origami and lipid membrane may be due to a combination of glycocalyx repulsion and the geometric hindrance. Negatively charged nucleic acids experience electrostatic repulsion from the glycocalyx on the cell surface.53 Additionally, from the literature, the glycocalyx on cultured HUVECs is reported to have a zone of interaction around 20–30 nm,54 which is comparable to the dimension of the tether and the DNA origami we used. Approaching the cell membrane with the end or the edge may therefore reduce the electrostatic repulsive force. Similarly, nanostructures with tether attachments may further be able to circumvent geometric and charge-based barriers.
Our study further reveals the requirements for an efficient membrane attachment of DNA nanostructures through a two-step cholesterol anchoring technique, and future studies should investigate whether these findings hold for other hydrophobic and membrane-bound protein anchors. The geometry of the structure, the valency and spatial positioning of binding overhangs, and the usage of bridge spacer or tether must all be taken into consideration for the rational design of membrane-targeting nucleic acid-based nanomaterials and nanobiosensors.
Methods
Preparation of DNA origami
The DNA origami nanotile and nanorod were designed using CaDNAno.55 For the nanosphere design, we used the vHelix-generated structure described by Benson et al. in 2015.47 The detailed staple sequences are listed in the Fig. S4–S7.† Each DNA origami shape was folded from a M13mp18 scaffold (Bayou Biolabs) together with a set of about 200 distinct and carefully designed short ssDNA sequences (purchased from Integrated DNA Technologies, Inc.). The scaffold and staples were mixed following the conditions described in Table 1 in 1× TAE buffer. For the DNA origami nanostructures with the dsDNA bridge spacer, 1 μM of bridge sequences were added to the annealing mixture.| Name | Scaffold (nM) | Staples (nM) | MgCl2 (mM) |
|---|---|---|---|
| Nanotile | 20 | 100 | 12.5 |
| Nanorod | 10 | 100 | 12.5 |
| Nanosphere | 10 | 100 | 20 |
The annealing ramps to form the DNA origami were performed in a MiniAmp™ Plus Thermal Cycler. The details of the ramps are as follows.
For the origami nanotile:
90 °C for 5 min
90 °C–70 °C, decrease 0.1 °C per 6 seconds
70 °C–45 °C, decrease 0.1 °C per 30 seconds
45 °C–30 °C, decrease 0.1 °C per 6 seconds
30 °C–4 °C, decrease 0.1 °C per 3 seconds
Hold the sample at 4 °C.
For the DNA origami nanorod and nanosphere:
80 °C for 5 min
80 °C–65 °C, decrease 0.1 °C per 24 seconds
65 °C–24 °C, decrease 0.1 °C per 6 minutes 18 seconds
24 °C–4 °C, decrease 0.1 °C per 18 seconds
Hold the sample at 4 °C.
The annealed origami was precipitated by centrifugation at 10500g for 25 minutes in a buffer containing 7.5% PEG 8000 (Sigma-Aldrich), 10 mM MgCl2, 255 mM NaCl, 22.5 mM Tris, 10 mM acetic acid, and 1 mM EDTA mixture. The supernatant was removed and the pellet was reconstituted to 15 nM using 1× PBS buffer with 12.5 mM MgCl2. The purified DNA origami was stored at 4 °C for no more than 24 hours before application to cells.
Agarose gel electrophoresis
15 μl of 15 nM purified DNA origami samples were analyzed by electrophoresis in 2% agarose gel in 1× TBE with 12.5 mM MgCl2 at 100 V for 1.5–2 hours in a cold room, stained with 1× SYBR Safe DNA gel stain (Invitrogen), and imaged with Bio-rad ChemiDoc Imaging System.Atomic force microscopy
DNA origami samples was diluted to 2 nM (0.5 nM for the nanorod) in 1× PBS with 12.5 mM MgCl2 for purified samples. For unpurified samples, see Table 1. 10 μL diluted DNA origami structures was deposited onto a freshly cleaved mica surface, incubated at room temperature in a humid chamber for 5 minutes, and blow-dried with nitrogen. The sample was washed twice with 20 μL DI water and thoroughly blow-dried with nitrogen before imaging. The AFM scan was performed on a MFP-3D-BIO AFM (Asylum Research) with a 5 nm AFM tip (NanoAndMore) in tapping mode.Labeling HUVECs with DNA origami through cholesterol anchoring
15 nM DNA origami in 1× PBS with 12.5 mM MgCl2 was diluted into 3 volumes with endothelial cell growth medium-2 (EGM2, Lonza) medium right before application to cells at 5 nM. HUVECs (purchased from Lonza, tested negative for mycoplasma contamination) were maintained in EGM2 medium in gelatin-coated tissue-culture flask. 50000 cells of passage 4 HUVECs were plated in each well of a 96-well plate and cultured for 6–9 hours before introduction of the origami. Three replicated wells were analyzed for each experimental condition. Cells were incubated with 0.5 μM cholesterol-ssDNA (IDT) initiator in EGM2 media for one hour at 37 °C. For the control wells, we added the EGM2 medium without the initiator instead. Subsequently, all wells including the test and control wells were then incubated in 5 nM diluted DNA origami at 37 °C for half hour and then fixed with 4% paraformaldehyde (PFA) in 1× PBS. The fixed cells was then passivated with 1% bovine serum albumin (BSA, Fisher BioReagents) in 1× PBS with Ca2+ & Mg2+ for half an hour and stained with 4 μg ml−1 streptavidin-Alexa Fluor 647 (Invitrogen) and 1% BSA in 1× PBS with Ca2+ & Mg2+ for half an hour. The fluorescence signal was read in a Molecular Devices SpectraMax i3X microplate reader. The microscope images of the cells were imaged with a Nikon Ti2 system with a 20× objective, a Cy5 filter cube and a Prime 95B Photometrics camera.
Statistical analysis
All the cell experiments were performed with triplicate wells and the result was averaged from the data with error bars as the standard deviation. All cell experiments and gel results was performed at least twice in different days with the similar trend. And the data shown in this paper was a representative one. The data analysis was performed with Prism Graphpad. The P value was obtained from one-way ANOVA analysis with post hoc Tukey's test with Prism Graphpad.Author contributions
Conceptualization, Y.L., P.W, S.K, W.W, X.R, and R.E.T; data curation, Y.L.; formal analysis, Y.L., and P.W.; funding acquisition, R.E.T., and X.R.; investigation, Y.L., P.W, S.K, and W.W; methodology, Y.L., P.W, S.K, W.W, X.R, and R.E.T; project administration, R.E.T., and X.R.; supervision, R.E.T., and X.R.; validation, Y.L.; visualization, Y.L., P.W, S.K, W.W, X.R, and R.E.T; writing – review & editing, Y.L., P.W, S.K, W.W, X.R, and R.E.T.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Department of Biomedical Engineering (X. R.) and the Department of Mechanical Engineering (R. E. T.) at Carnegie Mellon University, a T32 pre-doctoral training grant (Biomechanics in Regenerative Medicine, BiRM) from the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health (NIH) (P. W.), a 2018 Dowd Fellowship (Y. L.), the Samuel and Emma Winters Foundation (R. E. T.), the National Heart Lung and Blood Institute of the NIH 1R21HL152147 (R. E. T.), and the Berkman Faculty Development Fund (R. E. T.). We are grateful to Drs Adam Feinberg, Philip LeDuc and Rachelle Palchesko for sharing the AFM, and to Misti West for laboratory management.References
- P. W. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS.
- C. E. Castro, F. Kilchherr, D.-N. Kim, E. L. Shiao, T. Wauer, P. Wortmann, M. Bathe and H. Dietz, Nat. Methods, 2011, 8, 221–229 CrossRef CAS PubMed.
- A. Kuzuya and M. Komiyama, Nanoscale, 2010, 2, 309–321 RSC.
- A. Samanta and I. L. Medintz, Nanoscale, 2016, 8, 9037–9095 RSC.
- H. Ijäs, S. Nummelin, B. Shen, M. A. Kostiainen and V. Linko, Int. J. Mol. Sci., 2018, 19, 2114 CrossRef PubMed.
- Y. Liu, S. Kumar and R. E. Taylor, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2018, 10, e1518 Search PubMed.
- W. Wang, D. S. Arias, M. Deserno, X. Ren and R. E. Taylor, APL Bioeng., 2020, 4, 041507 CrossRef CAS PubMed.
- H. Lodish, A. Berk, S. L. Zipursky, P. Matsudaira, D. Baltimore and J. Darnell, Molecular Cell Biology, WH Freeman, 4th edn, 2000 Search PubMed.
- W. R. Loewenstein, Physiol. Rev., 1981, 61, 829–913 CrossRef CAS PubMed.
- J. Kwik, S. Boyle, D. Fooksman, L. Margolis, M. P. Sheetz and M. Edidin, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13964–13969 CrossRef CAS PubMed.
- G. Nikolova, N. Jabs, I. Konstantinova, A. Domogatskaya, K. Tryggvason, L. Sorokin, R. Fässler, G. Gu, H.-P. Gerber and N. Ferrara, et al. , Dev. Cell, 2006, 10, 397–405 CrossRef CAS PubMed.
- P. K. Dutta, Y. Zhang, A. T. Blanchard, C. Ge, M. Rushdi, K. Weiss, C. Zhu, Y. Ke and K. Salaita, Nano Lett., 2018, 18, 4803–4811 CrossRef CAS.
- S. M. Douglas, I. Bachelet and G. M. Church, Science, 2012, 335, 831–834 CrossRef CAS.
- A. Kurz, A. Bunge, A.-K. Windeck, M. Rost, W. Flasche, A. Arbuzova, D. Strohbach, S. Müller, J. Liebscher and D. Huster, et al. , Angew. Chem., Int. Ed., 2006, 45, 4440–4444 CrossRef CAS PubMed.
- J. R. Burns, N. Al-Juffali, S. M. Janes and S. Howorka, Angew. Chem., Int. Ed., 2014, 53, 12466–12470 CrossRef CAS PubMed.
- P. A. Beales and T. K. Vanderlick, J. Phys. Chem. A, 2007, 111, 12372–12380 CrossRef CAS.
- A. Czogalla, D. J. Kauert, R. Seidel, P. Schwille and E. P. Petrov, Nano Lett., 2015, 15, 649–655 CrossRef CAS PubMed.
- A. Rodríguez-Pulido, A. I. Kondrachuk, D. K. Prusty, J. Gao, M. A. Loi and A. Herrmann, Angew. Chem., Int. Ed., 2013, 52, 1008–1012 CrossRef PubMed.
- J. R. Burns, K. Göpfrich, J. W. Wood, V. V. Thacker, E. Stulz, U. F. Keyser and S. Howorka, Angew. Chem., Int. Ed., 2013, 52, 12069–12072 CrossRef CAS PubMed.
- S. Kocabey, S. Kempter, J. List, Y. Xing, W. Bae, D. Schiffels, M. Shih, F. C. Simmel and T. Liedl, ACS Nano, 2015, 9(4), 3530–3539 CrossRef CAS PubMed.
- E. Akbari, M. Y. Mollica, C. R. Lucas, S. M. Bushman, R. A. Patton, M. Shahhosseini, J. W. Song and C. E. Castro, Adv. Mater., 2017, 29, 1703632 CrossRef.
- J. List, M. Weber and F. C. Simmel, Angew. Chem., Int. Ed., 2014, 53, 4236–4239 CrossRef CAS PubMed.
- S. Krishnan, D. Ziegler, V. Arnaut, T. G. Martin, K. Kapsner, K. Henneberg, A. R. Bausch, H. Dietz and F. C. Simmel, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- A. Khmelinskaia, H. G. Franquelim, E. P. Petrov and P. Schwille, J. Phys. D: Appl. Phys., 2016, 49, 194001 CrossRef.
- P. Chidchob, D. Offenbartl-Stiegert, D. McCarthy, X. Luo, J. Li, S. Howorka and H. F. Sleiman, J. Am. Chem. Soc., 2019, 141, 1100–1108 CrossRef CAS PubMed.
- A. Khmelinskaia, J. Mücksch, E. P. Petrov, H. G. Franquelim and P. Schwille, Langmuir, 2018, 34, 14921–14931 CrossRef CAS PubMed.
- L. Song, Q. Jiang, J. Liu, N. Li, Q. Liu, L. Dai, Y. Gao, W. Liu, D. Liu and B. Ding, Nanoscale, 2017, 9, 7750–7754 RSC.
- M. Pavelka and J. Roth, Functional ultrastructure: atlas of tissue biology and pathology, Springer, 2015 Search PubMed.
- M. L. Smith, D. S. Long, E. R. Damiano and K. Ley, Biophys. J., 2003, 85, 637–645 CrossRef CAS.
- S. Reitsma, D. W. Slaaf, H. Vink, M. A. Van Zandvoort and M. G. oude Egbrink, Pflügers Arch., 2007, 454, 345–359 CrossRef CAS PubMed.
- P. D. Halley, C. R. Lucas, E. M. McWilliams, M. J. Webber, R. A. Patton, C. Kural, D. M. Lucas, J. C. Byrd and C. E. Castro, Small, 2016, 12, 308–320 CrossRef CAS.
- Y. Zhang, C. Ge, C. Zhu and K. Salaita, Nat. Commun., 2014, 5, 1–10 CAS.
- M. M. Bastings, F. M. Anastassacos, N. Ponnuswamy, F. G. Leifer, G. Cuneo, C. Lin, D. E. Ingber, J. H. Ryu and W. M. Shih, Nano Lett., 2018, 18, 3557–3564 CrossRef CAS PubMed.
- P. Wang, M. A. Rahman, Z. Zhao, K. Weiss, C. Zhang, Z. Chen, S. J. Hurwitz, Z. G. Chen, D. M. Shin and Y. Ke, J. Am. Chem. Soc., 2018, 140, 2478–2484 CrossRef CAS.
- J. Li, K. Xun, K. Pei, X. Liu, X. Peng, Y. Du, L. Qiu and W. Tan, J. Am. Chem. Soc., 2019, 141, 18013–18020 CrossRef CAS.
- Z. Ge, J. Liu, L. Guo, G. Yao, Q. Li, L. Wang, J. Li and C. Fan, J. Am. Chem. Soc., 2020, 142, 8800–8808 CrossRef PubMed.
- C. Scheibe, S. Wedepohl, S. B. Riese, J. Dernedde and O. Seitz, ChemBioChem, 2013, 14, 236–250 CrossRef CAS PubMed.
- K. Gorska, K.-T. Huang, O. Chaloin and N. Winssinger, Angew. Chem., Int. Ed., 2009, 48, 7695–7700 CrossRef CAS PubMed.
- M. Yamabe, K. Kaihatsu and Y. Ebara, Bioorg. Med. Chem. Lett., 2019, 29, 744–748 CrossRef CAS PubMed.
- A. Angelin, S. Weigel, R. Garrecht, R. Meyer, J. Bauer, R. K. Kumar, M. Hirtz and C. M. Niemeyer, Angew. Chem., Int. Ed., 2015, 54, 15813–15817 CrossRef CAS PubMed.
- D. Huang, K. Patel, S. Perez-Garrido, J. F. Marshall and M. Palma, ACS Nano, 2018, 13, 728–736 CrossRef PubMed.
- A. Suma, A. Stopar, A. W. Nicholson, M. Castronovo and V. Carnevale, Nucleic Acids Res., 2020, 48, 4672–4680 CrossRef CAS PubMed.
- R. Veneziano, T. J. Moyer, M. B. Stone, E.-C. Wamhoff, B. J. Read, S. Mukherjee, T. R. Shepherd, J. Das, W. R. Schief and D. J. Irvine, et al. , Nat. Nanotechnol., 2020, 15, 716–723 CrossRef CAS PubMed.
- W. Hawkes, D. Huang, P. Reynolds, L. Hammond, M. Ward, N. Gadegaard, J. F. Marshall, T. Iskratsch and M. Palma, Faraday Discuss., 2019, 219, 203–219 RSC.
- A. Shaw, V. Lundin, E. Petrova, F. Fördős, E. Benson, A. Al-Amin, A. Herland, A. Blokzijl, B. Högberg and A. I. Teixeira, Nat. Methods, 2014, 11, 841 CrossRef CAS PubMed.
- K. Liu, C. Xu and J. Liu, J. Mater. Chem. B, 2020, 8, 6802–6809 RSC.
- E. Benson, A. Mohammed, J. Gardell, S. Masich, E. Czeizler, P. Orponen and B. Högberg, Nature, 2015, 523, 441–444 CrossRef CAS PubMed.
- M. Langecker, V. Arnaut, T. G. Martin, J. List, S. Renner, M. Mayer, H. Dietz and F. C. Simmel, Science, 2012, 338, 932–936 CrossRef CAS PubMed.
- A. Johnson-Buck, S. Jiang, H. Yan and N. G. Walter, ACS Nano, 2014, 8, 5641–5649 CrossRef CAS.
- T. Schroder, M. B. Scheible, F. Steiner, J. Vogelsang and P. Tinnefeld, Nano Lett., 2019, 19, 1275–1281 CrossRef PubMed.
- F. Nicoli, M. K. Roos, E. A. Hemmig, M. Di Antonio, R. de Vivie-Riedle and T. Liedl, J. Phys. Chem. A, 2016, 120, 9941–9947 CrossRef CAS PubMed.
- H. Ding, J. Li, N. Chen, X. Hu, X. Yang, L. Guo, Q. Li, X. Zuo, L. Wang and Y. Ma, et al. , ACS Cent. Sci., 2018, 4, 1344–1351 CrossRef CAS PubMed.
- M. J. Palte and R. T. Raines, J. Am. Chem. Soc., 2012, 134, 6218–6223 CrossRef CAS.
- D. Chappell, M. Jacob, O. Paul, M. Rehm, U. Welsch, M. Stoeckelhuber, P. Conzen and B. F. Becker, Circ. Res., 2009, 104, 1313–1317 CrossRef CAS PubMed.
- S. M. Douglas, H. Dietz, T. Liedl, B. Högberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/D0NR09212F |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |