
Recent advances in the heteroatom doping of perovskite oxides for efficient electrocatalytic reactions
Yifan
Liu†
,
Honglan
Huang†
,
Liang
Xue
,
Jingwen
Sun
,
Xin
Wang
 ,
Pan
Xiong
* and
Junwu
Zhu
*
,
Pan
Xiong
* and
Junwu
Zhu
*
Key Laboratory for Soft Chemistry and Functional Materials of Ministry Education, School of Chemistry and Chemical Engineering, Nanjing University of Science and Technology, Nanjing, 210094, China. E-mail: pan.xiong@njust.edu.cn; zhujw@njust.edu.cn
First published on 10th November 2021
Abstract
Perovskite-type transition metal oxides have emerged as promising electrocatalysts for various electrocatalytic reactions owing to their low cost, compositional tunability and high stability. However, insufficient electrocatalytic activities of pristine perovskite oxides hinder their pathway towards real-world applications. The incorporation of heteroatoms into perovskite oxide structures has been regarded as an efficient way to improve the electrocatalytic performance. This minireview summarizes the recent advances in the heteroatom doping of perovskite oxides as efficient electrocatalysts for the hydrogen evolution reaction (HER), oxygen evolution reaction (OER) and oxygen reduction reaction (ORR). These heteroatom doping strategies are classified based on various types of doping sites. The mechanisms of improved electrocatalytic activities are discussed in detail within different doping sites and various kinds of dopants. Finally, the remaining challenges and perspectives are outlined for future developments of perovskite oxide-based catalysts.
 Yifan Liu | Yifan Liu received his BSc degree in Chemical Engineering from Nanjing Tech University, China, in 2019. He is now pursuing his Ph.D. degree under the supervision of Professor Junwu Zhu in the School of Chemistry and Chemical Engineering at Nanjing University of Science and Technology, China. His research focuses on perovskite oxide nanomaterials for electrocatalytic applications. |
 Honglan Huang | Honglan Huang received his BSc degree in Chemistry both from Nanjing Tech University and the University of Sheffield in 2018, and MRes degree in Nanomaterials in Imperial College London under the supervision of Dr James E. M. Lewis. Currently, he is pursuing his Ph.D. degree under the supervision of Professor Junwu Zhu in the School of Chemistry and Chemical Engineering at Nanjing University of Science and Technology. His current research focuses on the design of solid-state electrolytes for lithium metal batteries. |
 Xin Wang | Xin Wang received his PhD degree in inorganic chemistry at the Department of Chemistry, Nanjing University in 1985. After that, he joined Nanjing University of Science and Technology and was promoted to full professor in 1988. His research interests are focused on the design and synthesis of nanostructured materials for use in energy storage and conversion. |
 Pan Xiong | Pan Xiong is a Professor at the Nanjing University of Science and Technology, China. He received his PhD degree in Materials Science and Engineering from the same university in 2015. He was a visiting PhD student at the University of Texas at Austin followed by working as a postdoctoral researcher at the National Institute for Materials Science (NIMS), Japan and then a research associate at the University of Technology Sydney, Australia. His research focuses on two-dimensional nanomaterials and their heterostructures/superlattices for energy conversion and storage applications. |
 Junwu Zhu | Junwu Zhu received his PhD degree in materials chemistry from the Nanjing University of Science and Technology in 2005. He is presently working as a professor at the same university. His main research interests are focused on the preparation and application of functional materials based on carbon and nanostructured composites. |
1. Introduction
With the accelerated depletion of fossil fuels and environmental issues, advanced energy conversion technologies have attracted considerable attention in academia and industry.1–9 Among them, energy conversion based on electrocatalytic reactions is a promising strategy, which can convert energies efficiently and flexibly through chemical bond transitions.10–13 Although noble metal (e.g., Pt, Ir, Ru, Pd, and Rh) based catalysts have been widely used for electrochemical and renewable energy technologies,14–17 their prohibitive cost and extreme scarcity hindered their large-scale applications in the energy conversion industry. Therefore, it is imperative to seek low-cost and Earth-abundant alternative catalysts with comparable electrocatalytic activities for commercial demands.4,18–24Perovskite-type transition metal oxides with a general formula of ABO3 show high compositional tunability, where the A-site is generally occupied by a rare-earth/alkaline metal with 12-fold coordination and the B-site is occupied by a transition metal with 6-fold coordination with O atoms.25 Due to their low cost, environmental friendliness, high catalytic stability and large potential window, perovskite oxides have been intensively studied as promising catalysts for multiple electrocatalytic reactions, such as the hydrogen evolution reaction (HER), oxygen evolution reaction (OER), oxygen reduction reaction (ORR), carbon dioxide reduction reaction (CO2RR), and nitrogen reduction reaction (NRR).26–30 However, in most instances, the catalytic activities of perovskite oxides are incomparable to those of conventional noble metal catalysts. This is because disunited 3d orbitals of surface transition metal cations usually result in either too weak or too strong surface adsorption/desorption strength of reactants.31 Meanwhile, the relatively low electrical conductivities of many perovskite oxides lead to a sluggish charge-transfer process and slow the electrocatalytic kinetics.32 To curb these adverse factors, many strategies have been attempted to improve the electrocatalytic performance of perovskite oxide catalysts, including heteroatom doping,33,34 interfacial engineering,35 strain modulation,36,37 and introduction of defects.38,39
Among these strategies, heteroatom doping is one of the most facile and effective methods to optimize the catalytic properties of perovskite oxides. By affecting the interaction between oxygen and B-site atoms, the doped heteroatoms can improve the coupling efficiency of active centers and intermediate reactants, leading to facilitated electrocatalytic reactions.40 In terms of the elemental composition of perovskite oxides, in principle, more than 30 and 50 kinds of elements in the periodic table have been found at the A- and B-sites in ABO3 perovskite oxides, respectively.25 The great flexibility in composition determines the diversity of doped heteroatoms, which could be doped into different sites of the ABO3 perovskite oxides, namely, the A-site, B-site, O-site, and dual-sites. This results in various optimizing concepts, such as A-site doping can impact indirectly via activating the transition metal catalytic sites,41,42 B-site doping can optimize the B-site catalytic activities via electron exchange between the heteroatoms and the intrinsic B-site cations of perovskite oxides,43,44 O-site doping can create abundant oxygen vacancies to improve the electrical conductivity,45,46 and dual-site doping can promote the multi-active sites synergistically.47 Recently, various types of dopants have been studied, including heteroatoms doped into the A site (e.g., Na, K, Sr, Ca, Yb, Ce and La),41,42,48–52 B site (e.g., Ru, Ir, Fe, Co, Ni, Mn, W, P, S and Si),34,53–59 O site (e.g., F, S, N and Cl),46,60–62 and dual sites (e.g., Sr&Fe and Fe&Sn).47,63,64 Although some work on heteroatom-doped strategies for electrocatalysis has been outlined briefly as a branch of optimizing approaches,45,65,66 a few reports are systematically and comprehensively concerned with reviewing perovskite oxide electrocatalysts from the aspect of different doping sites. An up-to-date summarization of the latest discoveries and achievements is indispensable.
Herein, this minireview focuses on the recent advances in heteroatom doping strategies for the design of perovskite oxides towards efficient electrocatalytic reactions. These heteroatom doping strategies are classified based on the various types of doping sites, namely, A site, B site, O site and dual sites in perovskite oxides. The mechanisms of improved electrocatalytic activity are discussed in detail within different doping sites for three important electrocatalytic reactions, namely, the HER, OER, and ORR (Fig. 1). The current challenges and future perspectives are also outlined. This minireview aims to provide general guidelines and timely viewpoints for the design of highly efficient perovskite oxide catalysts based on heteroatom doping approaches.
 | ||
| Fig. 1 Schematic illustration of the A-site, B-site, O-site and dual-site heteroatom doping of ABO3-type perovskite oxides for efficient electrocatalytic reactions including the HER, OER and ORR. | ||
2. A-site cation doping
Generally, the A-site cations in the pristine perovskite oxide structures are considered not to participate in the electrocatalytic process.45 However, it will indirectly affect the electrocatalytic performance via activating the transition metal catalytic sites. In previous works, many heteroatoms have been doped into A sites to tailor the electronic structures, and thereby to enhance the electrocatalytic activities of perovskite oxides. Accordingly, the effects of A-site doping in perovskite oxides on the electrocatalytic activities will be summarized based on the categories of doped heteroatoms, including alkali metals (e.g., Na+ and K+),48,49 alkaline-earth metals (e.g., Sr2+ and Ca2+),50,67 and rare-earth metals (e.g., Yb3+, Ce3+ and La3+).41,42,522.1 A-site doping with alkali metals
Acting as p-type dopants, alkali metals usually have a lower valence of +1 and smaller size than the A-site cations of perovskite oxides. Among them, Na+ and K+ are widely adopted to modify the A-site and tailor the properties of perovskite oxides both physically and chemically. For example, the significant volumetric mismatch between Na+/K+ dopants and A-site metal cations of perovskite oxides will result in lattice rearrangements.48 Besides, the alkali metal dopants with a lower valence than the A-site metal cations in perovskite oxides will increase oxygen vacancies or change the oxidation state of B-site active centers owing to charge compensation effects.67 All of these structural changes can effectively manipulate the electrocatalytic activity and durability.Pavone et al. proposed K-doped Sr2Fe1.5Mo0.5O6 (KSFMO) (Fig. 2a) as an excellent bifunctional catalyst for both the OER and ORR via first principles calculations.48 They analyzed the structural and electronic features of both Sr1.5Fe1.5Mo1.5O6 (SFMO) and KSFMO. In the SFMO/KSFMO topmost surface layer, the oxygen vacancies (VO) have two different environments due to different adjacent atoms, i.e., Fe–VO–Fe (Fig. 2b) and Mo–VO–Fe (Fig. 2c). When K atoms occupy A-site lattices, the VO formation energies of Fe–VO–Fe and Mo–VO–Fe in KSFMO are lower than those in SFMO because of charge compensation effects.68 Interestingly, KSFMO will suffer a peculiar surface reconstruction upon VO formation along with Mo–VO–Fe. The Mo atom and surrounding O atoms undergo a structural rearrangement from an unsaturated octahedron to a tetrahedron MoO4 moiety, and Fe atoms become highly under-coordinated (Fig. 2c). H-Bonds are formed during the reconstruction of the MoO4 tetrahedron to stabilize *OOH intermediates (Fig. 2d), which can accelerate the rate-determining step of the OER process. Meanwhile, the 3d orbital of the under-coordinated Fe cation, with a mixed Fe(III)/Fe(IV) character, has a strong overlap with the O 2p orbital, which is also beneficial for the ORR process.
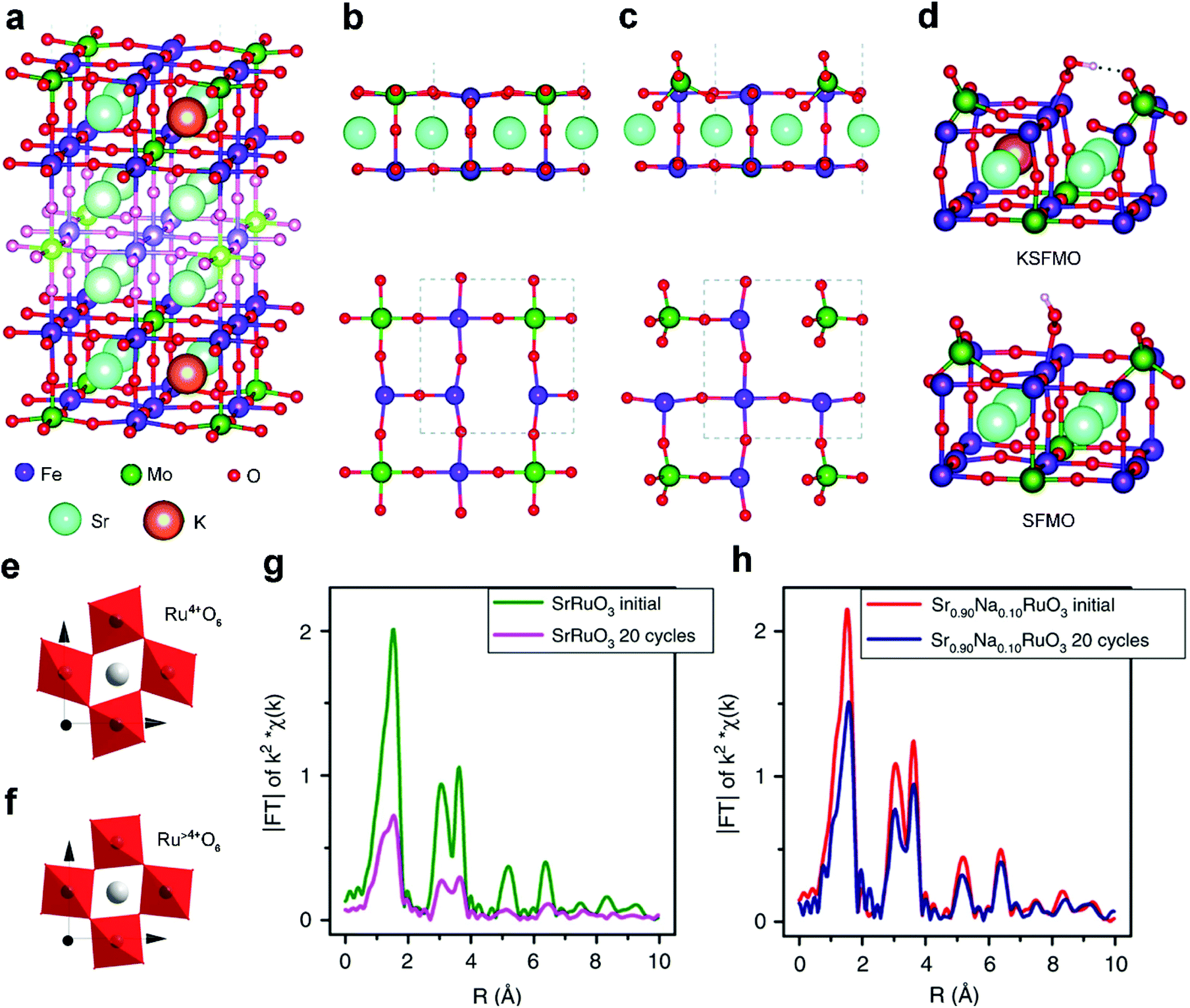 | ||
| Fig. 2 (a) Structure of the 9-layer slab of KSFMO. Lateral and top views of the outermost layers after the formation of (b) Fe–VO–Fe and (c) Mo–VO–Fe. (d) Lateral view of the structures of the *OOH intermediates at Fe sites in KSFMO and in SFMO. Reproduced with permission from ref. 48. Copyright 2017, Royal Society of Chemistry. Schematic representation of octahedral distortion in (e) SrRuO3 and (f) Sr0.9Na0.1RuO3. Fourier transform from Ru K-edge EXAFS signals of (g) SrRuO3 and (h) Sr0.9Na0.1RuO3 before and after OER cycles. Reproduced with permission from ref. 49. Copyright 2019, Nature Publishing Group. | ||
The A-site doping of perovskite oxides with alkali metals also induces excellent cycling durability.49 Pristine SrRuO3 exhibits high specific OER activity but usually suffers from unsatisfactory durability, which is related to the distortion of Ru4+O6 octahedra in SrRuO3 (Fig. 2e). Upon Na+ doping, Ru4+ is slightly over-oxidized to Ru>4+ because of charge compensation effects. Accordingly, the Ru>4+O6 octahedra are more regular in Sr0.9Na0.1RuO3 (Fig. 2f) due to decreased Jahn–Teller effects. Thus, a severely decreased Ru–O signal is observed for the cycled SrRuO3 (Fig. 2g), indicating severe structural degradation. Conversely, similar signals are displayed in fresh and cycled Sr0.9Na0.1RuO3 (Fig. 2h), confirming the high structural stability. Na locally increases the oxidation state of Ru from +4 to +5 and weakens the too strong adsorption energies of the OER intermediates. If Ru5+ sites are instrumental for the development of SrRuO3 perovskite oxide catalysts, other monovalent cations such as small radius Li+ or large radius Cs+ can be tested to ascertain whether they can also enhance the activity or the stability. This interesting hypothesis would contribute to us for exploring other A-site alkali metal dopants.
2.2 A-site doping with alkaline-earth metals
In addition to alkali metal cations, bivalent alkaline-earth metal cations, such as Sr2+ and Ca2+, have also been doped into the A-site of perovskite oxides to improve the electrocatalytic efficiency. The substitution of bivalent Sr2+ for trivalent A-site cations usually causes a charge imbalance throughout the perovskite oxides.69 This imbalance leads to the generation of oxygen vacancies, the modulation of oxidation states of the B-site transition metals, and the regulation of surface morphology.67,70,71 Ca2+ doping can suppress surface segregation or induce a reformation into a stable structure, thereby yielding enhanced durability.50,72In a recent study, aliovalent Sr2+ cations were used to substitute La3+ in LaMO3 (M = Co, Fe, Ni, Mn), forming La1−xSrxCoO3−δ,67,71 La1−xSrxFeO3−δ,73 La1−xSrxNiO3−δ,51 and La1−xSrxMnO3−δ,74 as efficient OER and ORR catalysts. For example, Stevenson and co-workers reported that the oxidation states of Co and the band overlap of Co 3d/O 2p are increased through Sr2+ doping.67 The formed ligand holes induce the release of lattice oxygen (Fig. 3a); therefore more oxygen vacancies will be created with the increased concentration of Sr2+ in the La1−xSrxCoO3 system. Compared with the crystal structures of low-level Sr2+ doped La0.8Sr0.2CoO3 (Fig. 3b and d), obvious ordering of oxygen vacancies was observed in SrCoO3 (Fig. 3c and e). The resulting layered ordered oxygen vacancies and atomic columns were directly observed in the high-level Sr2+ doped La0.2Sr0.8CoO3 (Fig. 3f) and SrCoO3 (Fig. 3g), due to the decreased formation energy of oxygen vacancies as the Sr2+ concentration increases (Fig. 3h). In addition, the oxygen evolution mechanism will transfer from the adsorbate evolution mechanism (AEM) to the lattice oxygen oxidation mechanism (LOM) with the increased Sr2+ concentration. The OER occurs via the AEM on LaCoO3−δ and La0.75Sr0.25CoO3−δ, whereas on La0.5Sr0.5CoO3−δ and SrCoO3−δ, it is mainly following the LOM mechanism (Fig. 3i).
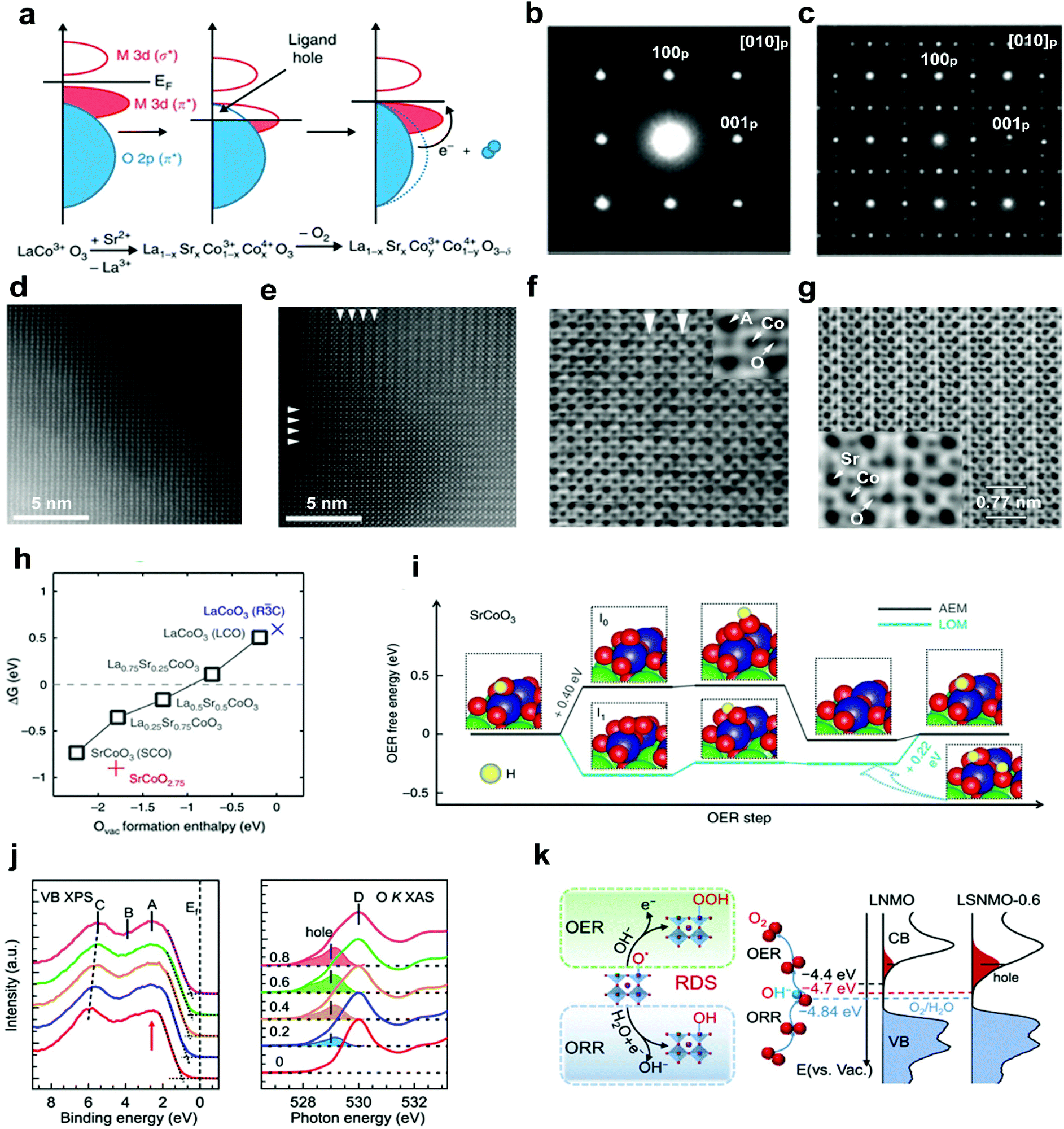 | ||
| Fig. 3 (a) Schematic representation of the relationship between the oxygen vacancy concentration and Co–O bond covalency through Sr2+ doping. SAED patterns of (b) La0.8Sr0.2CoO3 and (c) SrCoO3. HAADF-STEM images of (d) La0.8Sr0.2CoO3 and (e) SrCoO3. ABF-STEM imaging of oxygen vacancy ordering in (f) La0.2Sr0.8CoO3 and (g) SrCoO3. (h) ΔG (free energy difference between the LOM and AEM) versus the oxygen vacancy formation energy in perovskite oxides with different Sr2+ concentrations. (i) The OER free energy changes of the LOM and AEM on SrCoO3. Reproduced with permission from ref. 67. Copyright 2014, Nature Publishing Group. (j) X-ray photoemission spectroscopy valence band and O K-edge XAS spectra of LSNMO with different Sr2+concentration. (k) Left panel: rate-determining step of the OER and the ORR. Right panel: schematics diagram for the electronic structure near the Fermi level. Reproduced with permission from ref. 70. Copyright 2021, American Chemical Society. | ||
The electronic structures of perovskite oxides can also be optimized by Sr2+ doping. It was found that when Sr2+ was doped into A sites, a shift of the whole valence band spectra towards the Fermi level (Ef) and excess hole states were observed in La2−xSrxNiMnO6 (LSNMO) (Fig. 3j).70 Such electronic modulation enhances the hybridization of O 2p with Ni/Mn 3d, further reducing the charge transfer barrier and improving the intrinsic ORR and OER activities (Fig. 3k). The Sr2+ doping can also change the surface morphology due to the charge imbalance between Sr2+ and trivalent A-site cations. The unbalanced charge distribution may cause the local structural disorder in the [BO6] octahedron, resulting in an increased specific surface area of La1−xSrxMnO3−δ and a newly formed amorphous layer in La1−xSrxCoO3−δ with electrocatalytic activities.71,75 A series of grain boundaries would form as an internal interface when amorphous layers and crystal layers are in direct contact with each other. The grain boundaries are considered as distinctive platforms to exhibit new physical and chemical properties.35 This would be interesting to explore the effect of dopant-induced grain boundaries on perovskite oxide electrocatalytic performance.
The durability of perovskite oxides can also be significantly enhanced after doping with alkaline-earth cations. Perovskite oxide catalysts usually suffer from degradations during long-term operations because of the adverse changes in their crystal-structures and unstable surface-properties.76 For instance, a decrease in the activity of Ba-containing perovskite oxides is related to the surface degradation and Ba leaching.77 Luo et al. stabilized the crystal structure of PrBa0.85Ca0.15MnFeO5+δ (PBCMF) perovskite oxides by doping the A-site with Ca2+.50 Its pristine Ca-free counterpart, PrBaMnFeO5+δ (PBMF), suffered from an obvious degradation after a 1000-cycle test in the water oxidation reactions. A ∼10 nm Ba-rich shell was observed on the surface of degraded PBMF. In contrast, all the elements were uniformly distributed across the entire region in PBCMF. The calculation results reveal that a small amount of Ca-doping can increase the corrosion resistance in perovskite oxides, preventing surface degradation and Ba leaching. In addition, the Ca doping was reported to induce a structural transformation from layered perovskite oxides to cubic perovskite oxides with improved structural stability, which can deliver a durable OER performance.77
2.3 A-site doping with rare-earth metals
The A-site doping with rare-earth metals will result in a large cation-size mismatch and charge imbalance between rare-earth metal dopants and A-site metal cations, which can improve the conductivity, optimize the B-site electron states, and construct an active secondary phase for improved activities of perovskite oxides.41,52Xie et al.42 reported a combined method of Yb3+ doping and hydrogen treatment to improve the conductivity of CaMnO3. The Yb3+ dopants with a higher valence compared to Ca2+ can help to inject electrons to the perovskite oxides,78 introducing suitable oxygen vacancies for improved conductivity (Fig. 4a). Meanwhile, a similar radius between Yb3+ dopants and Ca2+ maintains the orthorhombic phase crystal structure of Ca0.9Yb0.1MnO3−δ (Fig. 4b). The hydrogen treatment further improves the conductivity of CaMnO3 by optimizing the eg electron filling status (Fig. 4c), showing a linear increasing relationship (Fig. 4d). The introduction of Yb also caused the degeneration of Mn eg orbitals into two sub-levels. The degeneration caused the elongation of Mn–O bonds owing to the Jahn–Teller effects (Fig. 4d). Such an elongation weakened the Mn–O covalent bond and facilitated the adsorption of oxygen intermediates. Therefore, significantly enhanced electrocatalytic activities were exhibited by the Yb-doped Ca0.9Yb0.1MnO3−δ.
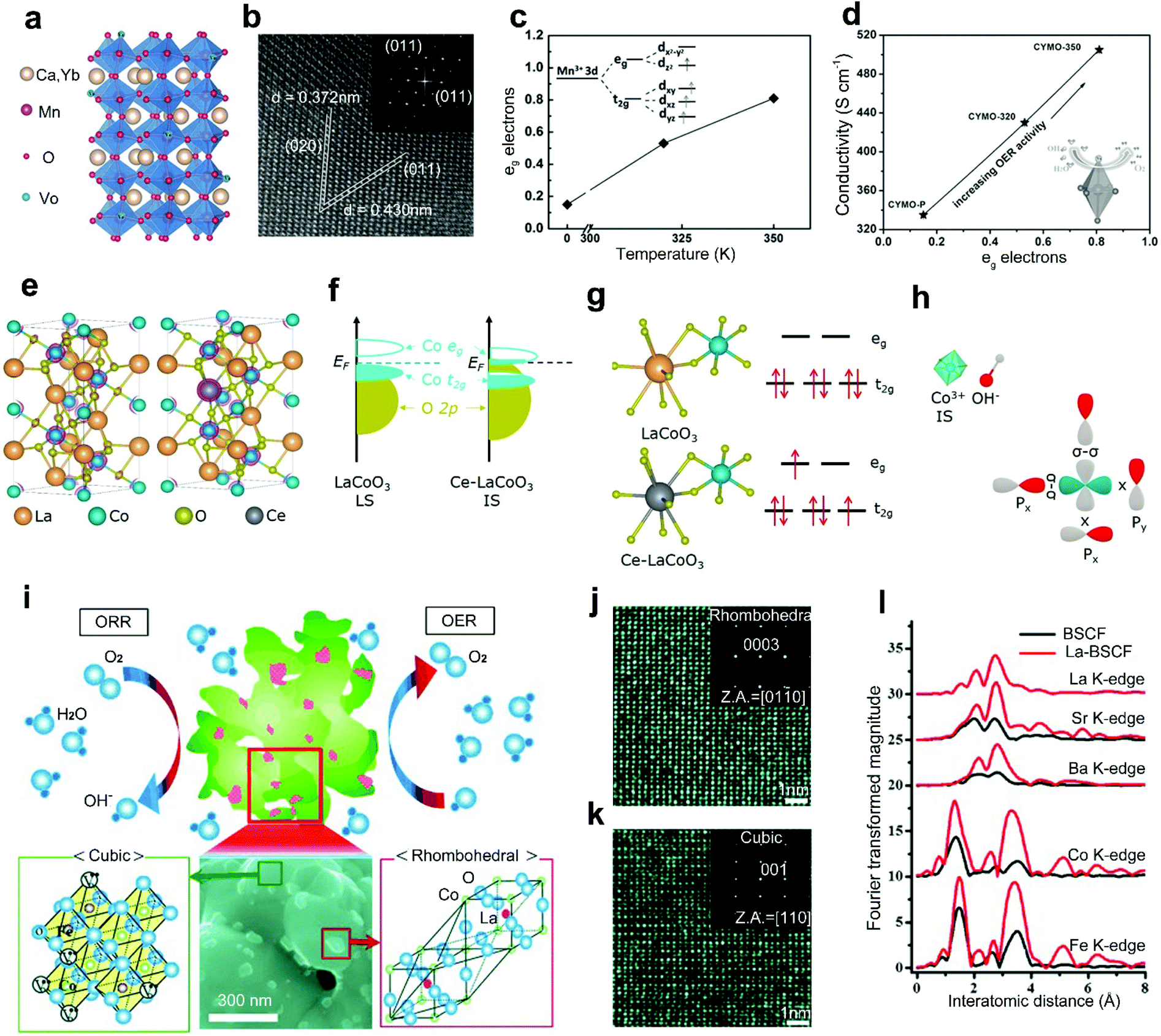 | ||
| Fig. 4 (a) Crystal structure of Ca0.9Yb0.1MnO3−δ with oxygen vacancies. (b) High-resolution TEM (HRTEM) image of Ca0.9Yb0.1MnO3−δ. (c) Hydrogen treatment temperature versus average eg electrons. (d) The average eg electrons versus electrical conductivity. Reproduced with permission from ref. 42. Copyright 2015, Wiley-VCH. (e) The crystal structure of LaCoO3 and Ce-LaCoO3. (f) Schematic representation of Co 3d–O 2p overlap for LaCoO3 and Ce-LaCoO3. (g) Schematic representation of the orbital splitting of Co 3d in LaCoO3 and Ce-LaCoO3. (h) Schematic representation of the interaction between the Co3+ and OH− in Ce-LaCoO3. Reproduced with permission from ref. 41. Copyright 2020, Elsevier B.V. (i) Schematic representation of La-BSCF perovskite oxide catalysts. HRTEM images and corresponding SAED patterns of (j) rhombohedral LaCoO3 particles on the (k) cubic BSCF surface. (l) Radial distribution function for the all-atomic EXAFS spectra for BSCF and La-BSCF. Reproduced with permission from ref. 52. Copyright 2014, Wiley-VCH. | ||
In addition, rare-earth metal doping of the A-site in perovskite oxides can also contribute to the optimization of the electronic structure, leading to improved catalytic performance. Gao and co-workers reported an A-site doped LaCoO3 with Ce3+ as a robust OER and ORR catalyst (Fig. 4e).41 In pristine LaCoO3, Co3+ is stabilized in the low-spin state (LS: t62ge0g) and no obvious overlap is shown between the eg band of Co and O 2p states near the Fermi level (Fig. 4f). Differently, in the Ce-LaCoO3, the spin state of Co3+ transitions from the LS to the steady intermediate-spin state (IS: t52ge1g). The downshifted eg band toward the Fermi level enlarged the Co 3d/O 2p band overlap, resulting in efficient acceleration of electron transfer between active sites and oxygen intermediates (Fig. 4f). Accordingly, the electrons of Co3+ in 3d orbital transition from t2g to eg (Fig. 4g). Then, improved OER activities under alkaline conditions can be achieved through a steady σ interaction between the activated eg orbitals of Co3+ and the O 2p orbitals of OH− (Fig. 4h).79
The surface morphology evolution, which would be induced by rare-earth metal doping in perovskite oxides, can also effectively affect the catalytic activities. Cho et al. reported a La-doped La0.3(Ba0.5Sr0.5)0.7Co0.8Fe0.2O3−δ (La-BSCF) as a promising bifunctional perovskite catalyst for the ORR and OER.52 Compared with the conventional powder morphology of BSCF, La-BSCF shows a flower-shaped morphology with particles on surfaces (Fig. 4i). The dynamic microstructure phenomenon is attributed to the charge imbalance by aliovalent La3+ doping, forming rhombohedral LaCoO3 (Fig. 4j) on the cubic BSCF surface (Fig. 4k). Additionally, the original Co–O bond in pristine BSCF is separated into two size-mismatched Co–O bonds (Fig. 4l). When compared with the BSCF, the La-BSCF with an excess active secondary phase provides more active sites for both the ORR and OER. For rhombohedral LaCoO3 and cubic BSCF perovskite oxides, these two crystalline grains may form a grain boundary as an internal interface. This distinctive work provides an interesting idea to realize in situ formation of grain boundaries by heteroatom doping strategies.
3. B-site cation doping
In an ideal perovskite structure, transition metals occupying the [BO6] octahedron can directly affect the electrocatalytic performance of perovskite oxides.80 To design high-performance perovskite oxide catalysts, it is rational to introduce heteroatoms into B-site lattices of perovskite oxides. To comprehensively elucidate the types of B-site dopants in perovskite oxides, noble metals (e.g., Ir, Ru, Pt, Ag),34,81–83 non-noble metals (e.g., Mn, Fe, Co, Ni),59,84–86 and non-metals (e.g., P, Si)58,87 will be introduced in this section.3.1 B-site doping with noble metals
Doping highly active noble metals into the B-site of perovskite oxides is a typical strategy used to design highly efficient electrocatalysts, such as Ag-BSCF,83 La0.9Fe0.92Ru0.08O3−δ,88 Ba0.9Sr0.1Co0.8Fe0.1Ir0.1O3−δ,89 and LaCo1−xPtxO3−δ.82 According to the work of Singhal et al.,90 Ru-doped perovskite oxides show higher current densities and more capacitive behaviors than their Ru-free counterparts and RuO2. This originates from the excellent ionic activities of Ru in doped perovskite oxides, which are even higher than that of ionic Ru in the benchmark RuO2. Hereafter, the positive effects of B-site doping with noble metals mainly stem from modulated electronic structures of original catalytic sites and generated new heteroatom catalytic sites.Zou and co-workers reported Ir doped SrTiO3 (Ir-STO) with a remarkable OER activity in acidic media (Fig. 5a).81 Pristine STO exhibits a negligible OER activity because the Ti 3d band is far from the Fermi level of STO (Fig. 5b). After the incorporation of Ir dopants, the electron density around Ti sites increases and the electrons are redistributed (Fig. 5c).91 The density of states (DOS) of Ir-STO (Fig. 5b) also reveals an additional Ir electronic state crossing the Fermi level, and the Ti 3d band and O 2p band are closer to the Fermi level compared with STO. In this regard, the adsorption of oxygen intermediates on the Ti site is strengthened in the potential-determining step (Fig. 5d). This Ir doping strategy enables the possibility of designing high catalytic activity electrocatalysts for the OER under acid conditions, with a smaller amount of precious metals than their IrO2 counterpart.
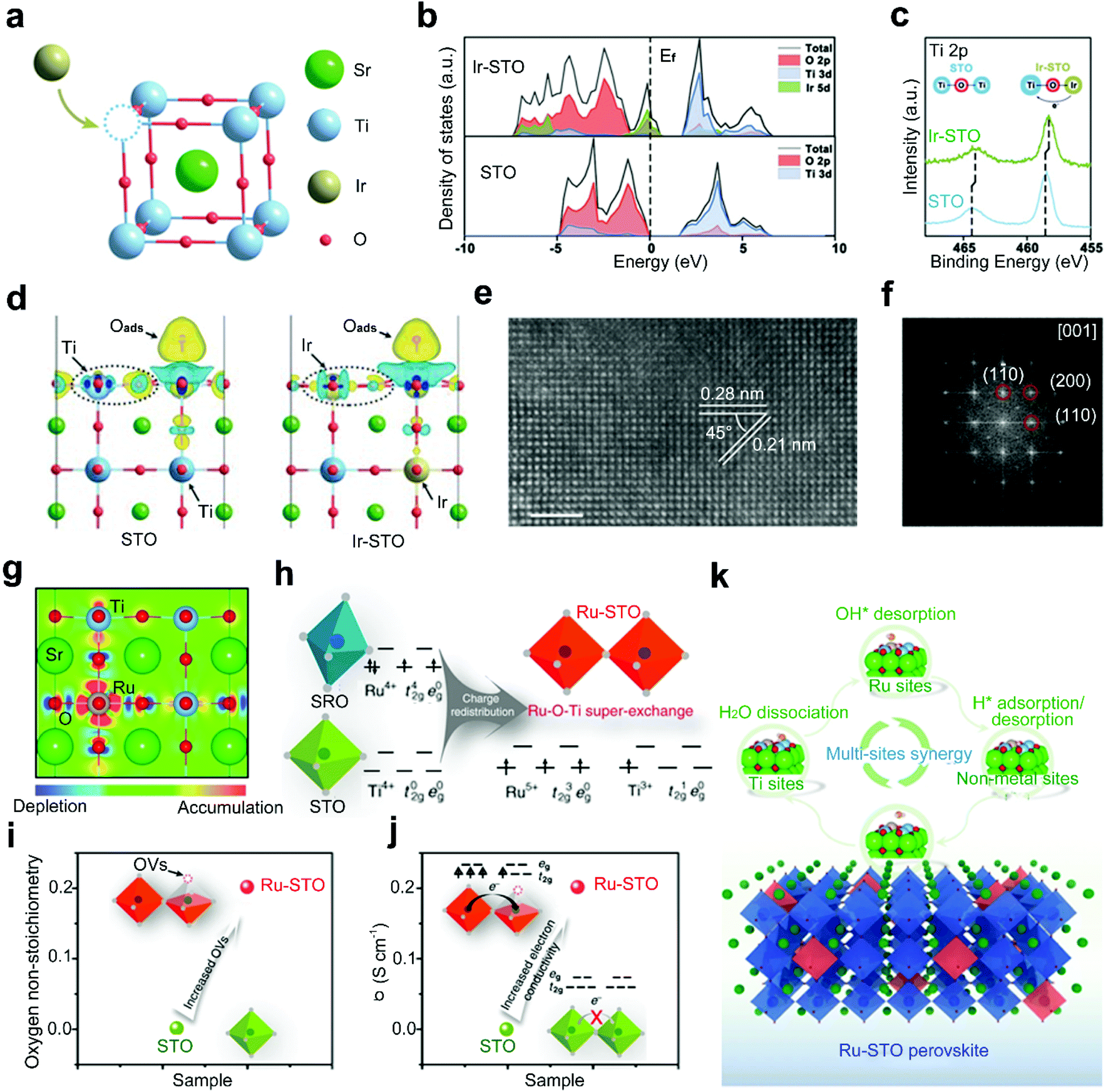 | ||
| Fig. 5 (a) Schematic representation for the incorporation of Ir dopants in STO matrix. (b) DOS of Ir-STO and STO. (c) Ti 2p XPS spectra of Ir-STO and STO. (d) The charge density difference induced by oxygen adsorption on the Ti site in STO and Ir-STO. Reproduced with permission from ref. 81. Copyright 2019, Wiley-VCH. (e) HRTEM image and (f) corresponding fast Fourier transform (FFT) pattern of Ru-STO. (g) Schematic representation for top view of the charge distribution in Ru-STO. (h) Schematic representation of the super-exchange interaction in Ru-STO. (i) The oxygen non-stoichiometry and (j) electronic conductivity of STO and Ru-STO. (k) Schematic representation of the synergistic catalysis mechanism for an alkaline HER on Ru-STO. Reproduced with permission from ref. 34. Copyright 2020, Nature Publishing Group. | ||
Dai and co-workers introduced a Ru-doped single-phase SrTiO3, SrTi0.7Ru0.3O3−δ (Ru-STO), with ultrafast HER performance in alkaline media (Fig. 5e and f).34 The incorporation of Ru dopants into STO leads to a charge redistribution between the neighbouring Ti and Ru ions (Ti4+ + Ru4+ → Ti3+ + Ru5+) (Fig. 5g), resulting in an unusual super-exchange interaction between adjacent Ti3+ (with a 3d1 configuration) and doped Ru5+ (with a 4d3 configuration) (Fig. 5h).43 In addition, oxygen vacancies are generated in the Ru-STO (Fig. 5i), which enhances the electrical conductivity (Fig. 5j) and contributes to exceptional catalytic activities. Density functional theory (DFT) calculations further reveal that the excellent activities are primarily attributed to a unique synergistic effect among the activated Ti sites, doped Ru sites and increased oxygen sites, which facilitate the water dissociation, enhance the OH* desorption, and promote the optimal H* adsorption and H2 desorption, respectively (Fig. 5k).
Though B-site doping of perovskite oxides with noble metals is promising for achieving fast kinetics and high current density with a low overpotential, the large-scale commercial applications are restricted by the high cost and resource scarcity of the noble metals.54 Besides, drastic structural instability during long-term operations has been observed, due to the dissolution of the formed noble metal oxides (RuOx and IrOx) on the surfaces of doped perovskite oxides.49
3.2 B-site doping with non-noble metals
Non-noble transition metals (e.g., Fe, Co, Ni, Mn) have been intensively investigated as B-site dopants because of their common merits of low cost and Earth-abundance.84–86 Some non-noble transition metals also have their specific merits. Mn-incorporated materials usually work well in the ORR,52,92 Co/Ni-doped materials show high OER activities,93–95 and Fe-based materials are relatively environmentally friendly.96,97 Therefore, it is highly desirable to dope these non-noble transition metals into perovskite oxides for the development of large-scale energy production.Perovskite oxides are found to be more active toward the OER when a relatively small amount of Fe is doped into the B-site.98,99 Xu and co-workers analysed the role of Fe-doping in perovskite oxide catalysts in the OER and found that the structure of LaCoO3 is changed with the incorporated content of Fe. The LaCo1−xFexO3 with a low concentration of doped Fe corresponds to a rhombohedral structure (Fig. 6a).100 When the concentration of doped Fe increases, the LaCo1−xFexO3 transitions from the rhombohedral phase to an orthorhombic phase (Fig. 6b). The phase change leads to the spin state transition of Co3+ from LS to HS and results in enhanced hybridization between Co 3d states and O 2p states (Fig. 6c). At an optimized concentration of doped Fe, remarkably enhanced OER activity is achieved (Fig. 6d). The doped Fe can also extend the potential range of thermodynamic metastability and improve the stability of the perovskite oxide structures through the formation of an oxy(hydroxide) layer on surfaces under OER conditions,101 which is the actual active layer for the OER.102
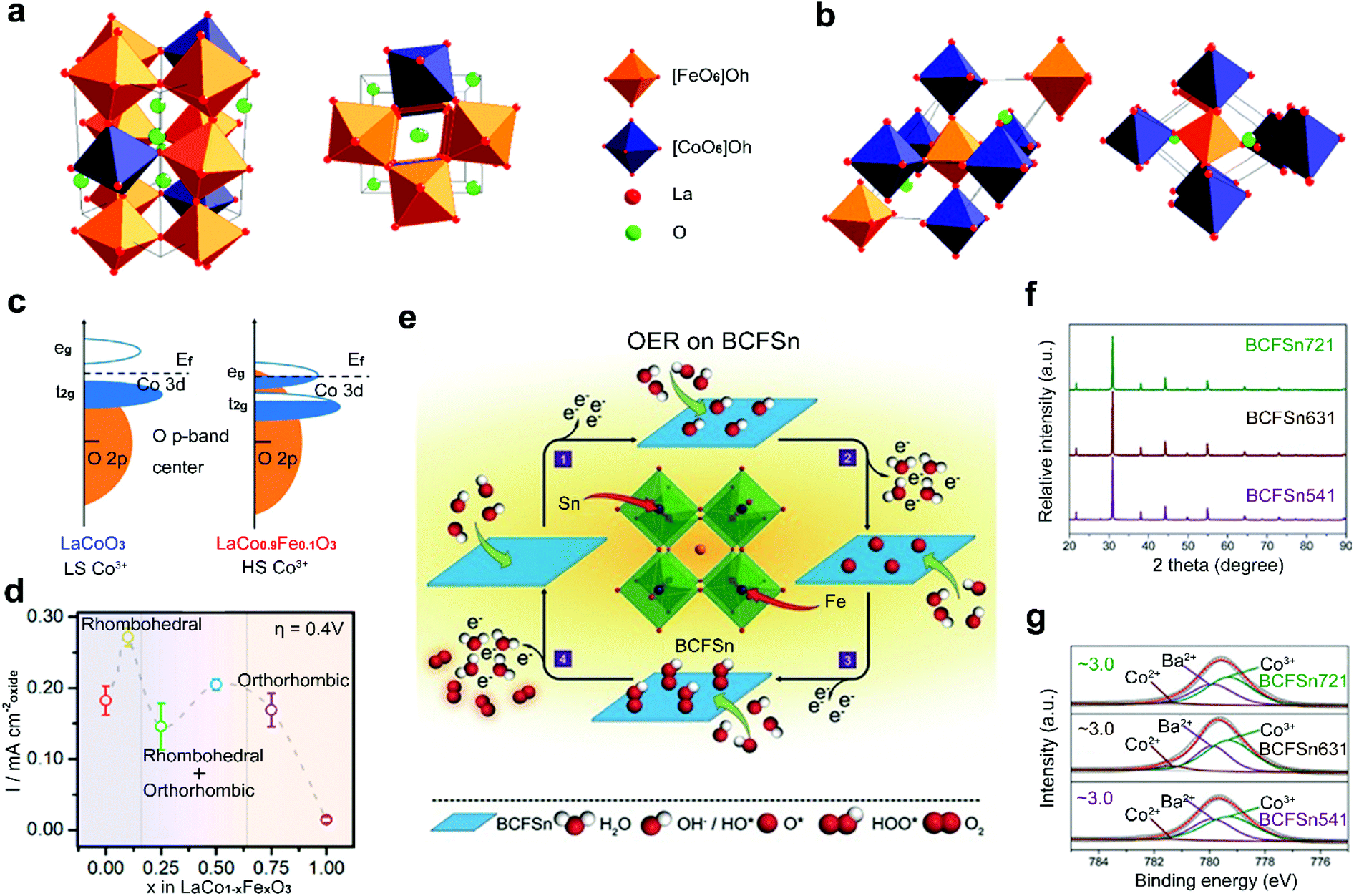 | ||
| Fig. 6 Schematic representation for perovskite oxide structures with (a) distorted rhombohedral structure and (b) orthorhombic structure. (c) Schematic representation of Co 3d–O 2p overlap for LaCoO3 and LaCo0.9Fe0.1O3. (d) Specific current density and corresponding structure for LaCo1−xFexO3 at an overpotential of 0.4 V versus RHE. Reproduced with permission from ref. 85. Copyright 2017, American Chemical Society. (e) Schematic representation of OER process of BCFSn perovskite oxides. (f) XRD patterns of BCFSn perovskite oxides. (g) High-resolution XPS spectra of Co 2p3/2 for BCFSn perovskite oxides. Reproduced with permission from ref. 103. Copyright 2016, Wiley-VCH. | ||
Two kinds of transition metals can also be co-doped into the B-site of perovskite oxides. Shao et al. reported a co-doping strategy by introducing Fe and Sn into the B-site of inert BaCoO3−δ to design highly active BaCo0.9−xFexSn0.1O3−δ (BCFSn) as OER catalysts (Fig. 6e).103 The introduction of both Fe and Sn elements can not only optimize the OH− adsorption capability, O2 desorption capability, average metal–oxygen bond energy and electrical conductivity, but also stabilize the cubic-phase structure (Fig. 6f) and the oxidation state of surface Co atoms (Fig. 6g). Thus, a four-electron transfer pathway for the OER is greatly boosted in BCFSn. Some other co-doped perovskite oxides have also been developed, such as Co/Ni co-doped Sr2FeMoO6−δ,104 Cu/Co co-doped LaMnO3−δ,105 and Cu/Fe co-doped LaNiO3−δ.106 The optimized electronic structure of active sites and increased amount of oxygen vacancies are found in these co-doped perovskite oxides with improved catalytic activities.
3.3 B-site doping with non-metals
B-site doping of perovskite oxides with non-metal elements provides another attractive way to improve the electrocatalytic activities. Non-metal elemental dopants are inert by themselves towards electrocatalysis, such as silicon (Si) and phosphorus (P).57,58,87,107 However, the non-metal elemental doping of perovskite oxides usually results in additional reaction pathways, favorable structural transitions, boosting electrical conductivities and optimized active sites, which are beneficial to efficient electrocatalysis.Si doping can influence the catalytic performance of perovskite oxides through diverse ways, such as activation of additional reaction pathways, optimization of surface structures, and creation of oxygen vacancies. Shao's group introduced Si to the B-site of SrCoO3 (Fig. 7a) to form a tetragonal-phase Si-SrCoO3 (Si-SCO) (Fig. 7b).58 The pH-dependence of the OER kinetics and almost unchanged eg filling of Co in Si-SCO prove that the boosted electrocatalytic activities should be attributed to an activated LOM mechanism (Fig. 7c) instead of the AEM.108 Besides, the increased pseudocapacitive behaviors of Si-SCO also indicate the involvement of lattice oxygen redox during the OER. The OER enhancement can also result from structural transition upon non-metal doping of perovskite oxides.58 A Si-doped SrFeO3−δ (Si-SFO) is designed and shows an approximately three-fold increase in OER activity relative to SrFeO3−δ.107 This obviously enhanced OER activity can be ascribed to the structural transition from tetragonal to cubic phase originating from Si doping, accompanying the generation of abundant oxygen vacancies and further acceleration of charge transfer.
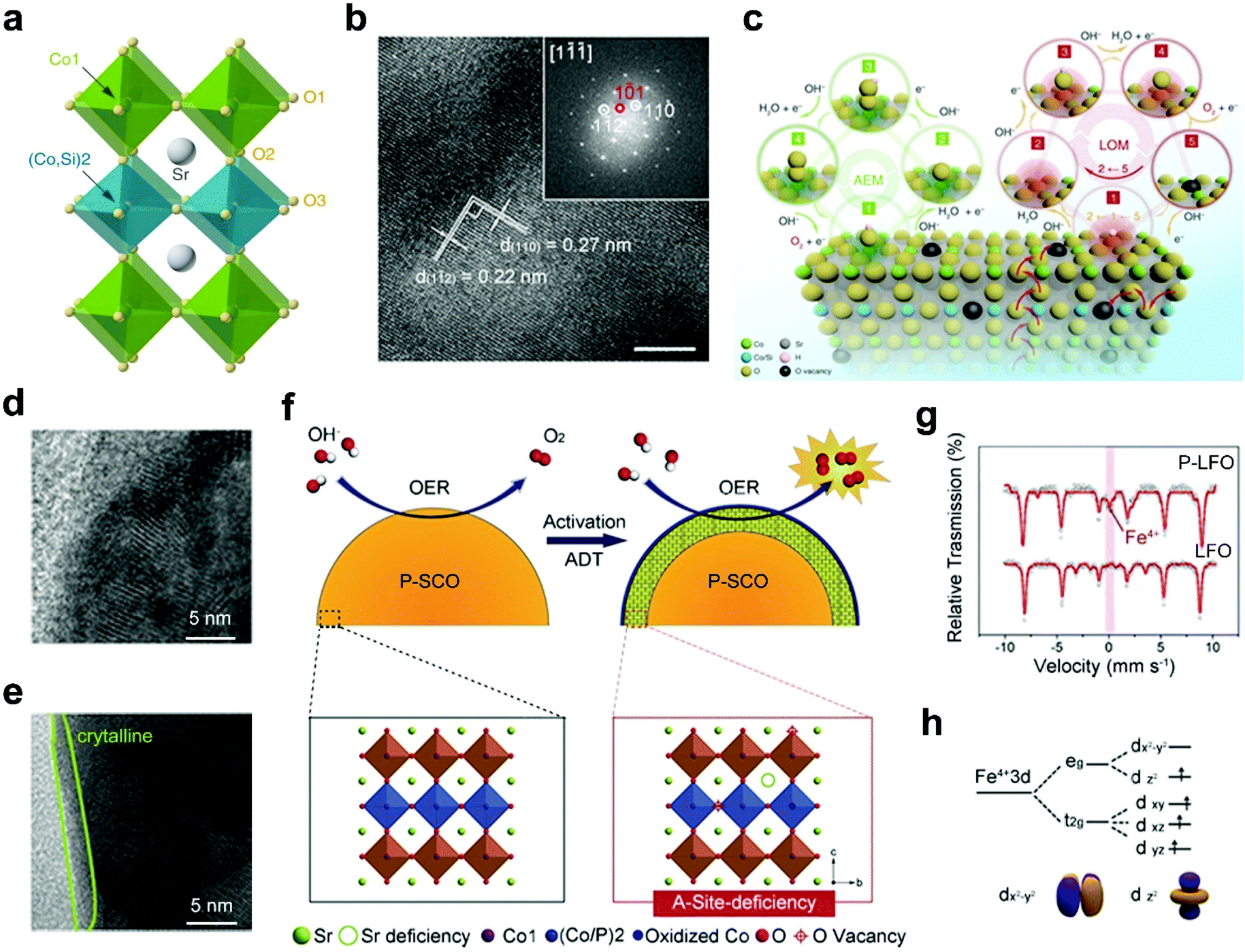 | ||
| Fig. 7 (a) Schematic representation of the crystal structure of Si-SCO. (b) HRTEM image of Si-SCO and the corresponding FFT pattern. (c) Schematic representation of the AEM and LOM reaction pathways on Si-SCO. Reproduced with permission from ref. 58. Copyright 2020, Nature Publishing Group. (d) HRTEM images of surface regions of P-SCO (d) before and (e) after 1000 cycles. (f) Schematic representation of the main origin of the OER activation in the SCP sample. Reproduced with permission from ref. 57. Copyright 2016, Wiley-VCH. (g) Mössbauer spectrum of LFO and P-LFO. (h) Schematic representation of Fe 3d orbit degeneration. Reproduced with permission from ref. 87. Copyright 2018, Elsevier B.V. | ||
The introduction of P elements into perovskite oxides can affect the electrochemical surface areas and B-site electronic structures of perovskite oxides. For example, an increase in OER activities is observed for P-doped SrCoO3 (P-SCO) after a 1000-cycle test.57 This increase is due to the formation of an amorphous layer with a thickness of 1–2 nm on the surfaces of P-SCO during the durability tests (Fig. 7d and e). The surface amorphization, associated with the leaching of A-site Sr2+, can lead to the exposure of active B sites and oxygen vacancies on the perovskite surface (Fig. 7f) and thus increase the electrochemical surface areas. Afterwards, Liu et al. found that compared with the pristine LaFeO3−δ (LFO), P-doped LaFeO3−δ (P-LFO) shows almost double times higher ORR activities under alkaline conditions.87 Upon the incorporation of P, the valence states of partial Fe-sites increase from Fe3+ to Fe4+ (Fig. 7g). In particular, the generation of Fe4+ results in the degeneration of the Fe 3d orbital (Fig. 7h). With the coexistence of Fe4+/Fe3+/Fe2+, the eg electron filling is optimized close to 1. What's more, the incorporation of P cations can also generate rich oxygen vacancies. With the optimized eg orbital filling and abundant oxygen vacancies, P-LFO possesses enhanced ORR catalytic activity.
4. O-site anion doping
In addition to the above cation doping, replacement of partial oxygen anions in the perovskite oxides by other non-metal heteroatom anions has also been explored to improve the activities of perovskite oxides, such as F,33,60 S,61,109 N,62 and Cl.110 The introduction of anions with different electronegativities or valences into the perovskite matrix can decrease the formation energy of oxygen vacancies, modify the electronic state and induce surface structure transformation of perovskite, and therefore contribute to the manipulation of catalytic performance.F is a widely used dopant to improve the electrocatalytic performance of perovskite oxides. Due to a less negative valence of −1 than −2 of O anions, the substitution of O with F induces a transformation from the [CoO6] octahedral structure to [CoO5F] and [CoO5] in cubic F-doped La0.5Ba0.25Sr0.25CoO3−δ and La0.5Ba0.25Sr0.25CoO3−δF0.1 (LBSCOF) (Fig. 8a).60 The formed octahedral [CoO5F] symmetry and square pyramidal [CoO5] symmetry lead to a reconfiguration of Co in the eg and t2g orbitals (Fig. 8b). Besides, owing to the larger electronegativity of F− than that of O2−, the coulombic force between B-site cations and O can be weakened upon F doping. Thus, a lower formation energy of the oxygen vacancy is needed in LBSCOF than that in pristine La0.5Ba0.25Sr0.25CoO3−δ (LBSCO). More content of active oxygen species is observed in LBSCOF than LBSCO (Fig. 8c). The optimized electronic orbital of Co and formed active oxygen species greatly promote the water-splitting reactions. As an element of the halogen group, Cl anions have the same −1 valence but lower electronegativity than F anions. Wang et al. experimentally investigated the role of Cl dopants. The enhanced OER activity of LaFeO2.9−δCl0.1 was ascribed to the fact that the Cl dopants enriched abundant oxygen vacancies, increased the electrical conductivity and enhanced the Fe–O covalency.110
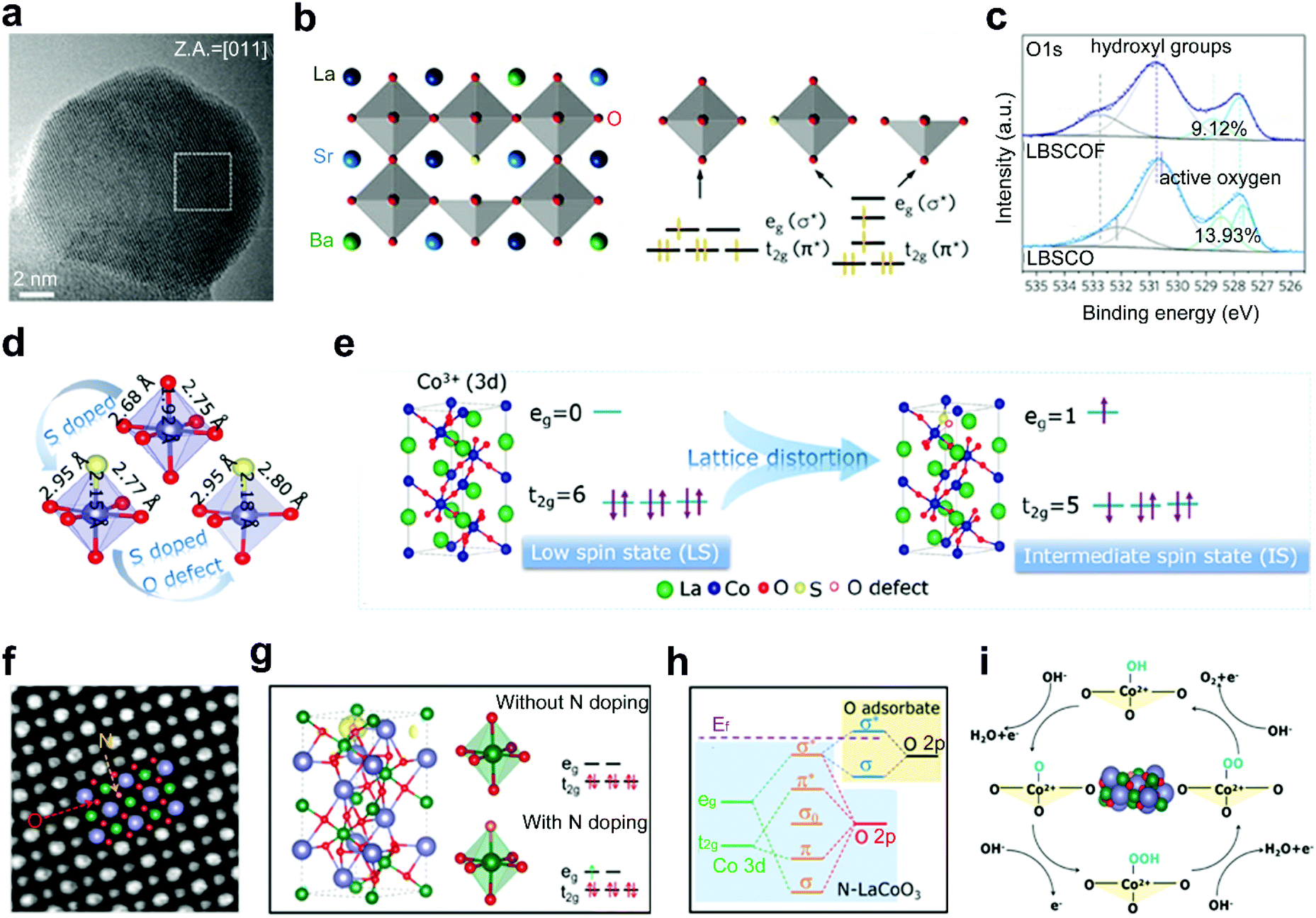 | ||
| Fig. 8 (a) HRTEM image of LBSCOF. (b) Schematic representation of the perovskite oxide structure and featured [CoO6−δ] polyhedron. (c) XPS spectra of O 1s for LBSCOF and LBSCO. Reproduced with permission from ref. 60. Copyright 2018, Elsevier Inc. (d) Schematic representation for optimized atomic structures. (e) Schematic representation of the evolution of the IS state and the transition of electrons from t2g to eg orbital. Reproduced with permission from ref. 109. Copyright 2020, American Chemical Society. (f) ABF-STEM image of N-LCO. (g) Spin density distribution and electronic configuration of LCO and N-LCO. (h) Schematic representation of orbital bonding. (i) Illustration diagram of the four-step process of the OER. Reproduced with permission from ref. 62. Copyright 2021, American Chemical Society. | ||
S is another efficient dopant for the O-site doping of perovskite oxides. S anions have the same −2 valence as O anions, but larger ionic radius than O anions. Therefore, the B–O bond lengths of [BO6] octahedra are changed and oxygen vacancies are formed after doping S into the O-site. These phenomena are observed in a recently reported S-doped LaCoO3 (S-LCO).109 During the in situ vapor–solid reaction process, the sulfur atoms react with LaCoO3 perovskite oxides and are reduced into sulfur anions. The sulfur anions are anchored on the O sites and a distorted [BO6] octahedral structure is formed (Fig. 8d). The distorted structure induces the spin state transition of Co from low spin to intermediate spin in S-LCO (Fig. 8e). Compared with pristine LCO, S-LCO shows greatly improved activities for both the OER and ORR under alkaline conditions. The influences of S dopants during the electrocatalytic process are also investigated by DFT calculations. Theoretical simulation reveals an elongated O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond in S doped perovskite oxides during the ORR process.61 The easier O2 cleavage indicates that S doping can endow perovskite oxides with an optimal electronic structure and surface adsorption feature for ORR catalysis.
O double bond in S doped perovskite oxides during the ORR process.61 The easier O2 cleavage indicates that S doping can endow perovskite oxides with an optimal electronic structure and surface adsorption feature for ORR catalysis.
With a low electronegativity and a high level of the 2p orbital, N atoms can exchange electrons in 2p orbitals with 3d orbitals of B-site active cations.111 Recently, Gao and co-workers reported an N-doped LaCoO3 (N-LCO) with enhanced oxygen catalytic activities (Fig. 8f).62 In typical LaCoO3, the Co cations show a low spin state with eg occupancy of ∼0. In contrast, in the N-LCO, N provides Co with an additional electron through the N–Co bond, resulting in a moderate eg occupancy of ∼1 for the Co cations (Fig. 8g). Then, the interaction between the 3d orbital of Co3+ and the 2p orbital of surface-adsorbed O intermediates is promoted (Fig. 8h). Eventually, a four-step process of the OER is greatly accelerated (Fig. 8i).
5. Dual-site doping
Although single-site doping of heteroatoms into ABO3 perovskite oxides has been intensively studied, the optimized performance is still somehow far from satisfaction. Recently, a dual-site doping strategy has been explored by doping diverse heteroatom dopants into A and B sites or B and O sites of perovskite oxides. A synergistic promotion is supposed to be achieved among multi-active sites induced by dual-site doping.47,63,64Zhang et al. doped Sr and Fe heteroatoms into A and B sites of LaNiO3 (LNO), respectively (Fig. 9a).64 Single-site doping of LNO with Fe (Fe-LNO) only shows a Fe3+ oxidation state with a shift of the whole valence band (VB) photoemission spectrum of Fe-LNO away from Ef (Fig. 9b). However, the Sr and Fe dual-site doped LNO (Sr&Fe-LNO) shows a promoted Fe state from +3 to +4. The activated Fe4+ states induce an inverse shift of the whole VB spectrum back close to Ef (Fig. 9b). The presence of Fe4+ can also reduce the electron–electron repulsion from bridging O (Fig. 9c) and facilitate the generation of active Ni4+ states. Besides, a reduced energy barrier of electron transfer is achieved for the Sr&Fe-LNO compared with LNO (Fig. 9d). Consequently, a six times higher current density is obtained for the Sr&Fe-LNO than that of LNO and Fe-LNO. In another report, by regulating the proportion of Sr and Fe nitrate precursors, an optimal eg-filling value of 1.2 is achieved through the dual-site doping with Sr and Fe (Fig. 9e).63
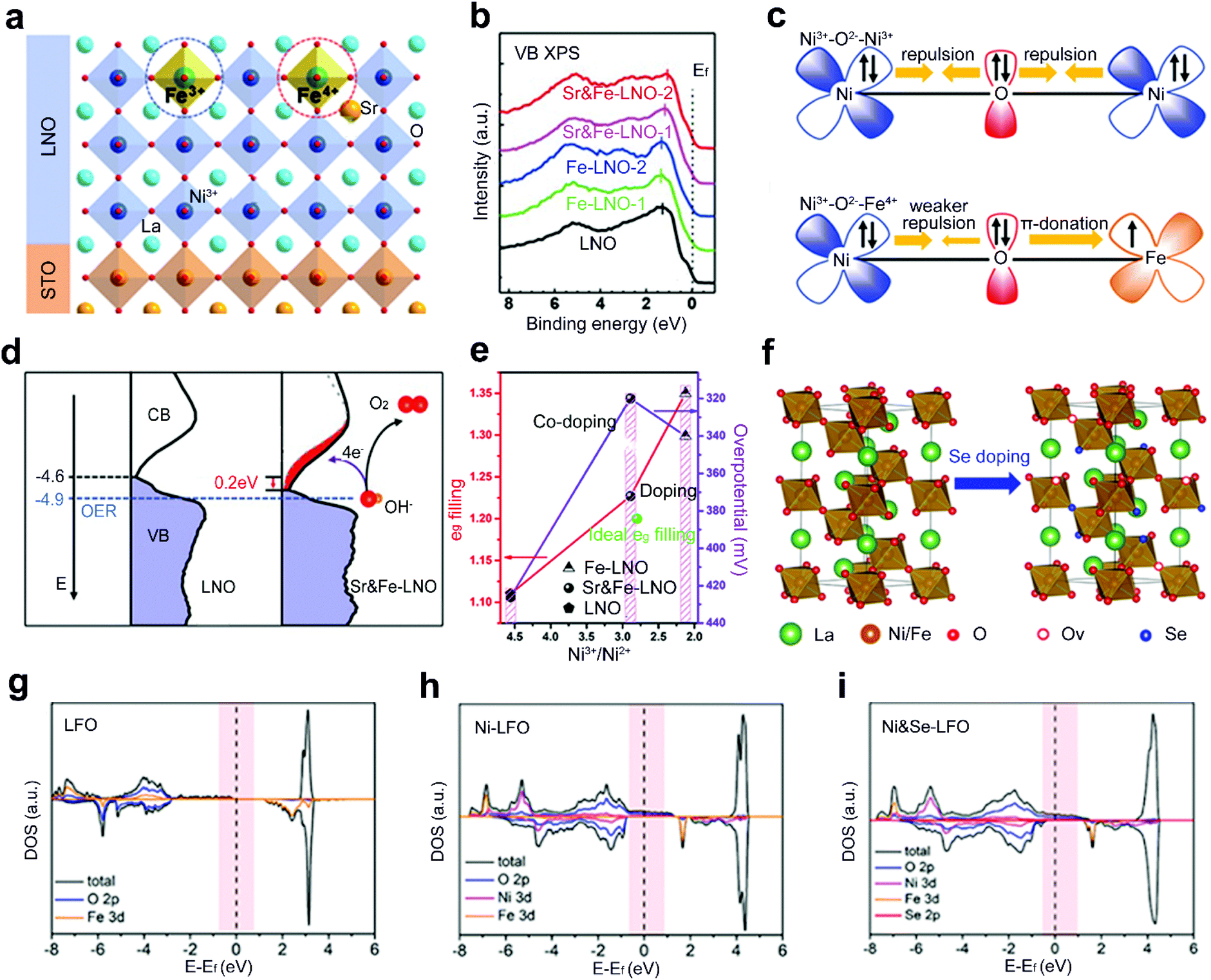 | ||
| Fig. 9 (a) Schematic representation of epitaxial growth of Sr&Fe-LNO perovskite oxide thin films on a SrTiO3 substrate. (b) XPS valence band spectrum and (c) schematic representation of the electronic coupling between the Ni3+ and Fe3+ in LNO and between the Ni3+ and Fe4+ in Sr&Fe-LNO, respectively. (d) The comparison of the occupied DOS (VB) and unoccupied DOS (CB) near Ef for LNO and Sr&Fe-LNO on an absolute energy scale. Reproduced with permission from ref. 64. Copyright 2021, Wiley-VCH. (e) The eg orbital filling of different perovskite oxides and OER activity as a function of Ni3+/Ni2+ rate. Reproduced with permission from ref. 63. Copyright 2019, Frontiers Media S.A. (f) Schematic representation of crystal structure of Ni-LFO and Ni&Se-LFO. DOS of (g) LFO, (h) Ni-LFO, and (i) Ni&Se-LFO, respectively. Reproduced with permission from ref. 47. Copyright 2020, American Chemical Society. | ||
Dual-site doped LaFeO3 with Ni and Se into B and O sites, respectively, is explored as a robust OER catalyst (Fig. 9f).47 The adsorption energies of oxygenated species on Fe sites are stronger than those of Ni sites.112 The substitution of the B-site Fe by adding different amounts of nickel salts into precursor solutions can optimize the adsorption of oxygen intermediates during the OER process. Furthermore, a mixture of Se powder and Ni single-site doped LFO (Ni-LFO) is annealed under an argon atmosphere. Se atoms are doped into O sites and lead to the formation of tremendous oxygen vacancies. The electron-occupied states crossing the Fermi level are increased in Ni and Se dual-site doped LFO (Ni&Se-LFO) compared to Ni-LFO and LFO (Fig. 9g–i). Finally, both experimental results and theoretical calculations demonstrate that Ni&Se-LFO shows superior OER catalytic activities to single-doped Ni-LFO and LFO.
6. Conclusion and perspective
This review summarizes the recent advances in the heteroatom doping of perovskite oxides. The catalytic performances of current doped perovskite oxide catalysts are listed in Table 1. The types of doping were classified in terms of the categories of the doping sites in ABO3-type perovskite oxide structures, including A-site doping, B-site doping, O-site doping, and dual-site doping. Based on these doping strategies, the electrocatalytic activities of perovskite oxides can be effectively enhanced by the optimization of active site states, introduction of oxygen vacancies, regulation of surface morphologies, and surface reconstruction. Nevertheless, there are also many challenges that deserve further investigation.| Composition engineering | Dopants | Catalysts | Catalytic performance | Ref. | ||
|---|---|---|---|---|---|---|
| Doped/pristine materials | HERa | OERb | ORRc | |||
| a The overpotential at a current density of 10 mA cm−2 (V vs. RHE). b The overpotential at a current density of 10 mA cm−2 (V vs. RHE). c Half-wave potential (V vs. RHE). | ||||||
| A site doping | Na | Sr0.95Na0.05RuO3/SrRuO3 | 0.16/0.19 | 49 | ||
| Sr | La0.8Sr0.2CoO3/LaCoO3 | 0.41/0.44 | 67 | |||
| Ce | Ce-LaCoO3/LaCoO3 | 0.41/0.65 | 0.68/0.45 | 41 | ||
| La | La0.3(Ba0.5Sr0.5)0.7Co0.8Fe0.2O3/Ba0.5Sr0.5Co0.8Fe0.2O3 | 0.32/0.44 | 52 | |||
| Sr | La0.2Sr0.8Co0.8Fe0.2O3−δ/LaCo0.8Fe0.2O3−δ | 0.45/0.56 | 71 | |||
| Sr | La0.5Sr0.5FeO3−δ/LaFeO3 | 0.24/0.35 | 73 | |||
| B site doping | Ti | SrTi0.7Ru0.3O3−δ/SrRuO3−δ | 0.05/0.09 | 33 | ||
| Ir | SrTi0.67Ir0.33O3/SrTiO3 | 0.24/0.32 | 81 | |||
| Pt | LaCo0.94Pt0.06O3−δ/LaCoO3−δ | 0.29/0.44 | 0.454/0.541 | 82 | ||
| Ag | Ag-Ba0.5Sr0.5Co0.8Fe0.2O3−δ/Ba0.5Sr0.5Co0.8Fe0.2O3−δ | 0.44/0.51 | 83 | |||
| Si | Si-SrCoO3−δ/SrCoO3−δ | 0.417/0.488 | 58 | |||
| Co | LaFe0.8Co0.2O3/LaFeO3 | 0.34/0.36 | 59 | |||
| Fe | La0.58Sr0.4Co0.8Fe0.2O3/La0.58Sr0.4CoO3 | 0.422/0.490 | 92 | |||
| Ru | SrCo0.9Ru0.1O3−δ/SrCoO3−δ | 0.36/0.44 | 43 | |||
| Sn/Fe | BaCo0.7Fe0.2Sn0.1O3−δ/BaCoO3−δ | 0.42/0.45 | 103 | |||
| O site doping | S | S doped LaCoO3/LaCoO3 | 0.364/0.473 | 109 | ||
| Cl | LaFeO2.9−δCl0.1/LaFeO3 | 0.47/0.51 | 110 | |||
| F | F-Ba0.5Sr0.5Co0.8Fe0.2O3−δ/Ba0.5Sr0.5Co0.8Fe0.2O3−δ | 0.28/0.34 | 33 | |||
| F | La0.5Ba0.25Sr0.25CoO2.9−δF0.1/La0.5Ba0.25Sr0.25CoO3−δ | 0.17/0.22 | 0.39/0.43 | 60 | ||
| S | S doped CaMnO3/CaMnO3 | 0.47/0.62 | 0.72/0.68 | 61 | ||
| N | N doped LaCoO3/LaCoO3 | 0.37/0.57 | 0.63/0.62 | 62 | ||
| Dual site doping | Se, Ni | Se, Ni doped LaFeO3/LaFeO3 | 0.307/0.330 | 47 | ||
| Sr, Fe | La0.4Sr0.6Ni0.5Fe0.5O3/LaNiO3 | 0.320/0.473 | 63 | |||
| Sr, Fe | Sr, Fe doped LaNiO3/LaNiO3 | 0.33/0.49 | 64 | |||
Most of the present research studies focus on single-site doping of perovskite oxides with heteroatoms. Compared with single-site doping, dual-site doping is supposed to show higher energy-efficiency for perovskite oxides.47,63,64 Available examples mentioned above have demonstrated that dual-site doping could introduce more remarkable influences on the electrocatalytic behaviors of perovskite oxides, thus worthy of our in-depth study. Current research studies have focused on doping different types of heteroatoms into multiple sites. However, there are enormous possible combinations of heteroatom dopants in perovskite oxide materials, which intensively hinder our efficient screening in the search for a combination with optimal catalytic properties. Thus, for a dual-site doping strategy, how to select the optimal heteroatom dopants remains a huge challenge. Recently, S has been doped into both B sites and O sites of perovskite oxides simultaneously, due to its relatively moderate electronegativity.57,61 It is highly possible that some other non-metal elements with similar electronegativity could also function as both an electron acceptor and an electron donor, showing great potential of multi-site dopants for next-generation doping strategies.
While searching for optimal doping strategies, advanced in situ techniques should be considered to clarify the dynamic variation of dopant precursors during the doping process and real-time interaction between the doped heteroatoms and active sites during catalytic reactions. For example, in situ scanning transmission electron microscopy (STEM) characterization can facilitate our accurate determination of the locations of heteroatoms and record of atomic displacements of active sites.35 Operando X-ray absorption spectral (XAS) measurements can help to understand the functional role of doped heteroatoms in perovskite oxides during the catalytic reactions.101In situ Raman technology can capture the dynamic surface reconstruction and facilitate our understanding of the surface catalytic mechanism during the evolution of the active phase.113 Currently, Bao's group discovered the reversible exsolution and dissolution of metal nanoparticles in Co-doped Sr2Fe1.5Mo0.5O6−δ (Co-SFMC) perovskite oxides using advanced in situ techniques.114In situ XRD and STEM revealed that the doped Co was exsolved from the parent perovskite and facilitated the generation of CoFe alloy nanoparticles on the surface of Co-SFMC, leading to enhanced CO2 electrolysis performance. These operando techniques visually demonstrate the structural evolution of electrocatalysts and provide us with more detailed research to understand the specific behavior of heteroatoms for catalyst synthesis and reaction processes.
The current research studies of doped perovskite oxide electrocatalysts are mainly focused on the HER, OER, and ORR. Currently, perovskite oxides have been demonstrated with activities toward the NRR under ambient conditions.30,115 The activities are mainly attributed to the favourable merits produced by the oxygen vacancies, which could have originated from heteroatom doping. Indeed, Bi's group improved the NRR catalytic performance of heteroatom-doped perovskite oxides.116 They found that A-site Sr doping can create abundant oxygen vacancies in Sr-doped LFO (La0.5Sr0.5FeO3−δ). The enriched oxygen vacancies weaken the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bonds and accelerate the formation of N–H bonds. Considering the high composition tunability of perovskite oxides and various types of heteroatom dopants, we anticipate that perovskite oxide catalysts doped with specific and optimized heteroatoms may be applied to other catalytic fields.
N triple bonds and accelerate the formation of N–H bonds. Considering the high composition tunability of perovskite oxides and various types of heteroatom dopants, we anticipate that perovskite oxide catalysts doped with specific and optimized heteroatoms may be applied to other catalytic fields.
Overall, perovskite oxide-based electrocatalysts will indeed have a promising outlook in the electrocatalytic field, owing to their multiple advantages of Earth abundance, low cost, structural diversity and high efficiency. With the gradually optimized synthetic strategies and advanced in situ techniques, the role of heteroatom dopants will be further understood at the atomic level. The heteroatom doping strategy could pave the way for the development of practical perovskite oxide catalysts for widespread applications in energy-related catalysis fields.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52125202, 22179062, 51902161, U2004209, and 21908110). J. S. thanks the Natural Science Foundation of Jiangsu Province (BK20190479). L. X. acknowledges the support from the China Postdoctoral Science Foundation (2020M681613 and 2021T140326).References
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- H. N. Nong, L. J. Falling, A. Bergmann, M. Klingenhof, H. P. Tran, C. Spori, R. Mom, J. Timoshenko, G. Zichittella, A. Knop-Gericke, S. Piccinin, J. Perez-Ramirez, B. R. Cuenya, R. Schlogl, P. Strasser, D. Teschner and T. E. Jones, Nature, 2020, 587, 408–413 CrossRef CAS PubMed.
- F. Pelayo García de Arquer, C. T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D. H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed.
- B. Sun, P. Xiong, U. Maitra, D. Langsdorf, K. Yan, C. Wang, J. Janek, D. Schroder and G. Wang, Adv. Mater., 2020, 32, 1903891 CrossRef CAS PubMed.
- P. Xiong, R. Ma, G. Wang and T. Sasaki, Energy Storage Mater., 2019, 19, 281–298 CrossRef.
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Science, 2017, 358, 751–756 CrossRef CAS PubMed.
- C. Guo, P. He, R. Cui, Q. Shen, N. Yang and G. Zhao, Adv. Energy Mater., 2019, 9, 1900364 CrossRef.
- X. Huang, Q. Shen, J. Liu, N. Yang and G. Zhao, Energy Environ. Sci., 2016, 9, 3161 RSC.
- Z. Zhou, Z. Wu, Q. Xu and G. Zhao, J. Mater. Chem. A, 2017, 5, 25450 RSC.
- D. J. Kubicki, S. D. Stranks, C. P. Grey and L. Emsley, Nat. Rev. Chem., 2021, 5, 624 CrossRef CAS.
- P. Xiong, Y. Wu, Y. Liu, R. Ma, T. Sasaki, X. Wang and J. Zhu, Energy Environ. Sci., 2020, 13, 4834–4853 RSC.
- Z. Zhou, Y.-N. Xie, W. Zhu, H. Zhao, N. Yang and G. Zhao, Appl. Catal., B, 2021, 286, 119868 CrossRef CAS.
- M. Zhang, G. Jeerh, P. Zou, R. Lan, M. Wang, H. Wang and S. Tao, Mater. Today, 2021 DOI:10.1016/j.mattod.2021.05.004.
- Y. R. Zheng, P. Wu, M. R. Gao, X. L. Zhang, F. Y. Gao, H. X. Ju, R. Wu, Q. Gao, R. You, W. X. Huang, S. J. Liu, S. W. Hu, J. Zhu, Z. Li and S. H. Yu, Nat. Commun., 2018, 9, 2533 CrossRef PubMed.
- H. Huang, M. Yan, C. Yang, H. He, Q. Jiang, L. Yang, Z. Lu, Z. Sun, X. Xu, Y. Bando and Y. Yamauchi, Adv. Mater., 2019, 31, 1903415 CrossRef CAS PubMed.
- Q. Yao, B. Huang, Y. Xu, L. Li, Q. Shao and X. Huang, Nano Energy, 2021, 84, 105909 CrossRef CAS.
- C. Cui, R. Cheng, C. Zhang and X. Wang, Chin. Chem. Lett., 2020, 31, 988–991 CrossRef CAS.
- P. Xiong, F. Zhang, X. Zhang, S. Wang, H. Liu, B. Sun, J. Zhang, Y. Sun, R. Ma, Y. Bando, C. Zhou, Z. Liu, T. Sasaki and G. Wang, Nat. Commun., 2020, 11, 3297 CrossRef CAS PubMed.
- P. Xiong, R. Ma, N. Sakai and T. Sasaki, ACS Nano, 2018, 12, 1768–1777 CrossRef CAS PubMed.
- P. Xiong, X. Zhang, F. Zhang, D. Yi, J. Zhang, B. Sun, H. Tian, D. Shanmukaraj, T. Rojo, M. Armand, R. Ma, T. Sasaki and G. Wang, ACS Nano, 2018, 12, 12337–12346 CrossRef CAS PubMed.
- S. Wang, P. Xiong, X. Guo, J. Zhang, X. Gao, F. Zhang, X. Tang, P. H. L. Notten and G. Wang, Adv. Funct. Mater., 2020, 30, 2001588 CrossRef CAS.
- S. Wang, P. Xiong, J. Zhang and G. Wang, Energy Storage Mater., 2020, 29, 310–331 CrossRef.
- L. Peng, P. Xiong, L. Ma, Y. Yuan, Y. Zhu, D. Chen, X. Luo, J. Lu, K. Amine and G. Yu, Nat. Commun., 2017, 8, 15139 CrossRef PubMed.
- K. Jiang, P. Xiong, J. Ji, J. Zhu, R. Ma, T. Sasaki and F. Geng, Acc. Chem. Res., 2020, 53, 2443–2455 CrossRef CAS PubMed.
- W.-J. Yin, B. Weng, J. Ge, Q. Sun, Z. Li and Y. Yan, Energy Environ. Sci., 2019, 12, 442–462 RSC.
- H. J. Song, H. Yoon, B. Ju and D. W. Kim, Adv. Energy Mater., 2020, 11, 2002428 CrossRef.
- C. Sun, J. A. Alonso and J. Bian, Adv. Energy Mater., 2020, 11, 2000459 CrossRef.
- L. Wang, K. A. Stoerzinger, L. Chang, J. Zhao, Y. Li, C. S. Tang, X. Yin, M. E. Bowden, Z. Yang, H. Guo, L. You, R. Guo, J. Wang, K. Ibrahim, J. Chen, A. Rusydi, J. Wang, S. A. Chambers and Y. Du, Adv. Funct. Mater., 2018, 28, 1803712 CrossRef.
- R. P. Singh, P. Arora, S. Nellaiappan, C. Shivakumara, S. Irusta, M. Paliwal and S. Sharma, Electrochim. Acta, 2019, 326, 134952 CrossRef CAS.
- S. Zhang, G. Duan, L. Qiao, Y. Tang, Y. Chen, Y. Sun, P. Wan and S. Zhang, Ind. Eng. Chem. Res., 2019, 58, 8935–8939 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 27, 1606459 CrossRef PubMed.
- J. Xiong, H. Zhong, J. Li, X. Zhang, J. Shi, W. Cai, K. Qu, C. Zhu, Z. Yang, S. P. Beckman and H. Cheng, Appl. Catal., B, 2019, 256, 117817 CrossRef CAS.
- J. Dai, Y. Zhu, H. A. Tahini, Q. Lin, Y. Chen, D. Guan, C. Zhou, Z. Hu, H. J. Lin, T. S. Chan, C. T. Chen, S. C. Smith, H. Wang, W. Zhou and Z. Shao, Nat. Commun., 2020, 11, 5657 CrossRef CAS PubMed.
- Y. Heo, S. Choi, J. Bak, H. S. Kim, H. B. Bae and S. Y. Chung, Adv. Energy Mater., 2018, 8, 1802481 CrossRef.
- J. R. Petrie, H. Jeen, S. C. Barron, T. L. Meyer and H. N. Lee, J. Am. Chem. Soc., 2016, 138, 7252–7255 CrossRef CAS PubMed.
- M. J. Choi, T. L. Kim, J. K. Kim, T. H. Lee, S. A. Lee, C. Kim, K. Hong, C. W. Bark, K. T. Ko and H. W. Jang, Nano Lett., 2020, 20, 8040–8045 CrossRef CAS PubMed.
- Y. Zhu, W. Zhou, J. Yu, Y. Chen, M. Liu and Z. Shao, Chem. Mater., 2016, 28, 1691–1697 CrossRef CAS.
- J. I. Jung, H. Y. Jeong, M. G. Kim, G. Nam, J. Park and J. Cho, Adv. Mater., 2015, 27, 266–271 CrossRef CAS PubMed.
- Y. Chen, H. Li, J. Wang, Y. Du, S. Xi, Y. Sun, M. Sherburne, J. W. Ager III, A. C. Fisher and Z. J. Xu, Nat. Commun., 2019, 10, 572 CrossRef CAS PubMed.
- J. Qian, T. Wang, Z. Zhang, Y. Liu, J. Li and D. Gao, Nano Energy, 2020, 74, 104948 CrossRef CAS.
- Y. Guo, Y. Tong, P. Chen, K. Xu, J. Zhao, Y. Lin, W. Chu, Z. Peng, C. Wu and Y. Xie, Adv. Mater., 2015, 27, 5989–5994 CrossRef CAS PubMed.
- J. Dai, Y. Zhu, Y. Yin, H. A. Tahini, D. Guan, F. Dong, Q. Lu, S. C. Smith, X. Zhang, H. Wang, W. Zhou and Z. Shao, Small, 2019, 15, 1903120 CrossRef PubMed.
- Y. R. Sun, X. Zhang, L. G. Wang, Z. K. Liu, N. Kang, N. Zhou, W. L. You, J. Li and X. F. Yu, Chem. Eng. J., 2021, 421, 129698 CrossRef CAS.
- Y. Zhu, X. Liu, S. Jin, H. Chen, W. Lee, M. Liu and Y. Chen, J. Mater. Chem. A, 2019, 7, 5875–5897 RSC.
- J. Zhang, Y. Cui, L. Jia, B. He, K. Zhang and L. Zhao, Int. J. Hydrogen Energy, 2019, 44, 24077–24085 CrossRef CAS.
- Z. Li, K. H. Xue, J. Wang, J. G. Li, X. Ao, H. Sun, X. Song, W. Lei, Y. Cao and C. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 41259–41268 CrossRef CAS PubMed.
- A. B. Muñoz-García and M. Pavone, J. Mater. Chem. A, 2017, 5, 12735–12739 RSC.
- M. Retuerto, L. Pascual, F. Calle-Vallejo, P. Ferrer, D. Gianolio, A. G. Pereira, A. Garcia, J. Torrero, M. T. Fernandez-Diaz, P. Bencok, M. A. Pena, J. L. G. Fierro and S. Rojas, Nat. Commun., 2019, 10, 2041 CrossRef PubMed.
- B. Hua, Y. F. Sun, M. Li, N. Yan, J. Chen, Y. Q. Zhang, Y. Zeng, B. S. Amirkhiz and J. L. Luo, Chem. Mater., 2017, 29, 6228–6237 CrossRef CAS.
- J. Liu, E. Jia, L. Wang, K. A. Stoerzinger, H. Zhou, C. S. Tang, X. Yin, X. He, E. Bousquet, M. E. Bowden, A. T. S. Wee, S. A. Chambers and Y. Du, Adv. Sci., 2019, 6, 1901073 CrossRef CAS PubMed.
- J. I. Jung, H. Y. Jeong, J. S. Lee, M. G. Kim and J. Cho, Angew. Chem., Int. Ed., 2014, 53, 4582–4586 CrossRef CAS PubMed.
- D. Zhang, Y. Song, Z. Du, L. Wang, Y. Li and J. B. Goodenough, J. Mater. Chem. A, 2015, 3, 9421–9426 RSC.
- G. Chen, Z. Hu, Y. Zhu, Z. G. Chen, Y. Zhong, H. J. Lin, C. T. Chen, L. H. Tjeng, W. Zhou and Z. Shao, J. Mater. Chem. A, 2018, 6, 9854–9859 RSC.
- Q. Wang, Y. Xue, S. Sun, S. Li, H. Miao and Z. Liu, Electrochim. Acta, 2017, 254, 14–24 CrossRef CAS.
- D. Mierwaldt, S. Mildner, R. Arrigo, A. Knop-Gericke, E. Franke, A. Blumenstein, J. Hoffmann and C. Jooss, Catalysts, 2014, 4, 129–145 CrossRef.
- Y. Zhu, W. Zhou, J. Sunarso, Y. Zhong and Z. Shao, Adv. Funct. Mater., 2016, 26, 5862–5872 CrossRef CAS.
- Y. Pan, X. Xu, Y. Zhong, L. Ge, Y. Chen, J. M. Veder, D. Guan, R. O'Hayre, M. Li, G. Wang, H. Wang, W. Zhou and Z. Shao, Nat. Commun., 2020, 11, 2002 CrossRef CAS PubMed.
- J. Dai, Y. Zhu, Y. Zhong, J. Miao, B. Lin, W. Zhou and Z. Shao, Adv. Mater. Interfaces, 2019, 6, 1801317 CrossRef.
- B. Hua, M. Li, W. Pang, W. Tang, S. Zhao, Z. Jin, Y. Zeng, B. S. Amirkhiz and J. L. Luo, Chem, 2018, 4, 2902–2916 CAS.
- S. Peng, X. Han, L. Li, S. Chou, D. Ji, H. Huang, Y. Du, J. Liu and S. Ramakrishna, Adv. Energy Mater., 2018, 8, 1800612 CrossRef.
- B. Xia, T. Wang, J. Ran, S. Jiang, X. Gao and D. Gao, ACS Appl. Mater. Interfaces, 2021, 13, 2447–2454 CrossRef CAS PubMed.
- Q. Guo, X. Li, H. Wei, Y. Liu, L. Li, X. Yang, X. Zhang, H. Liu and Z. Lu, Front. Chem., 2019, 7, 224 CrossRef CAS PubMed.
- G. Fu, W. Li, J. Y. Zhang, M. Li, C. Li, N. Li, Q. He, S. Xi, D. Qi, J. L. MacManus-Driscoll, J. Cheng and K. H. Zhang, Small, 2021, 17, 2006930 CrossRef CAS PubMed.
- Y. Dai, J. Yu, C. Cheng, P. Tan and M. Ni, Chem. Eng. J., 2020, 397, 125516 CrossRef CAS.
- D. Liu, P. Zhou, H. Bai, H. Ai, X. Du, M. Chen, D. Liu, W. F. Ip, K. H. Lo, C. T. Kwok, S. Chen, S. Wang, G. Xing, X. Wang and H. Pan, Small, 2021, 2101605 CrossRef CAS PubMed.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- M. Pavone, A. B. Muñoz-García, A. M. Ritzmann and E. A. Carter, J. Phys. Chem. C, 2014, 118, 13346–13356 CrossRef CAS.
- Z. Shen, Y. Zhuang, W. Li, X. Huang, F. E. Oropeza, E. J. M. Hensen, J. P. Hofmann, M. Cui, A. Tadich, D. Qi, J. Cheng, J. Li and K. H. L. Zhang, J. Mater. Chem. A, 2020, 8, 4407–4415 RSC.
- M. Qu, X. Ding, Z. Shen, M. Cui, F. E. Oropeza, G. Gorni, V. A. de la Peña O’Shea, W. Li, D.-C. Qi and K. H. L. Zhang, Chem. Mater., 2021, 33, 2062–2071 CrossRef CAS.
- S. Song, J. Zhou, J. Sun, S. Zhang, X. Lin, Z. Hu, J. Hu, L. Zhang and J. Q. Wang, Chin. J. Catal., 2020, 41, 592–597 CrossRef CAS.
- R. Liu, F. Liang, W. Zhou, Y. Yang and Z. Zhu, Nano Energy, 2015, 12, 115–122 CrossRef CAS.
- S. She, J. Yu, W. Tang, Y. Zhu, Y. Chen, J. Sunarso, W. Zhou and Z. Shao, ACS Appl. Mater. Interfaces, 2018, 10, 11715–11721 CrossRef CAS PubMed.
- V. Celorrio, L. Calvillo, E. Dann, G. Granozzi, A. Aguadero, D. Kramer, A. E. Russell and D. J. Fermín, Catal. Sci. Technol., 2016, 6, 7231–7238 RSC.
- Y. Zhao, Y. Hang, Y. Zhang, Z. Wang, Y. Yao, X. He, C. Zhang and D. Zhang, Electrochim. Acta, 2017, 232, 296–302 CrossRef CAS.
- N. Tsvetkov, Q. Lu, L. Sun, E. J. Crumlin and B. Yildiz, Nat. Mater., 2016, 15, 1010–1016 CrossRef CAS PubMed.
- D. He, G. He, H. Jiang, Z. Chen and M. Huang, Chem. Commun., 2017, 53, 5132–5135 RSC.
- J. Reszczyńska, T. Grzyb, J. W. Sobczak, W. Lisowski, M. Gazda, B. Ohtani and A. Zaleska, Appl. Catal., B, 2015, 163, 40–49 CrossRef.
- Y. Sun, Z. Liu, W. Zhang, X. Chu, Y. Cong, K. Huang and S. Feng, Small, 2019, 15, 1803513 CrossRef PubMed.
- D. Neagu, G. Tsekouras, D. N. Miller, H. Menard and J. T. Irvine, Nat. Chem., 2013, 5, 916–923 CrossRef CAS PubMed.
- X. Liang, L. Shi, Y. Liu, H. Chen, R. Si, W. Yan, Q. Zhang, G. D. Li, L. Yang and X. Zou, Angew. Chem., Int. Ed., 2019, 58, 7631–7635 CrossRef CAS PubMed.
- C. Wang, L. Zeng, W. Guo, C. Gong and J. Yang, RSC Adv., 2019, 9, 35646–35654 RSC.
- E. Y. Göl, A. Aytekin, E. E. Özkahraman and E. Karabudak, J. Appl. Electrochem., 2020, 50, 1037–1043 CrossRef.
- N. Sun, H. Liu, Z. Yu, Z. Zheng and C. Shao, RSC Adv., 2016, 6, 13522–13530 RSC.
- Y. Duan, S. Sun, S. Xi, X. Ren, Y. Zhou, G. Zhang, H. Yang, Y. Du and Z. J. Xu, Chem. Mater., 2017, 29, 10534–10541 CrossRef CAS.
- J. Sun, L. Du, B. Sun, G. Han, Y. Ma, J. Wang, H. Huo, P. Zuo, C. Du and G. Yin, J. Energy Chem., 2021, 54, 217–224 CrossRef.
- Z. Li, L. Lv, J. Wang, X. Ao, Y. Ruan, D. Zha, G. Hong, Q. Wu, Y. Lan, C. Wang, J. Jiang and M. Liu, Nano Energy, 2018, 47, 199–209 CrossRef CAS.
- Y. Jiang, Z. Geng, L. Yuan, Y. Sun, Y. Cong, K. Huang, L. Wang and W. Zhang, ACS Sustainable Chem. Eng., 2018, 6, 11999–12005 CrossRef CAS.
- Q. Luo, D. Lin, W. Zhan, W. Zhang, L. Tang, J. Luo, Z. Gao, P. Jiang, M. Wang, L. Hao and K. Tang, ACS Appl. Energy Mater., 2020, 3, 7149–7158 CrossRef CAS.
- V. Vats and A. Singhal, Electroanalysis, 2020, 33, 618–626 CrossRef.
- M. N. Grisolia, J. Varignon, G. Sanchez-Santolino, A. Arora, S. Valencia, M. Varela, R. Abrudan, E. Weschke, E. Schierle, J. E. Rault, J. P. Rueff, A. Barthelemy, J. Santamaria and M. Bibes, Nat. Phys., 2016, 12, 484–492 Search PubMed.
- K. Elumeeva, J. Masa, F. Tietz, F. Yang, W. Xia, M. Muhler and W. Schuhmann, ChemElectroChem, 2016, 3, 138–143 Search PubMed.
- J. X. Flores-Lasluisa, F. Huerta, D. Cazorla-Amoros and E. Morallon, J. Colloid Interface Sci., 2019, 556, 658–666 CrossRef CAS PubMed.
- Y. Han, Z. Zhu, L. Huang, Y. Guo, Y. Zhai and S. Dong, Nanoscale, 2019, 11, 19579–19585 RSC.
- X. Liu, H. Gong, T. Wang, H. Guo, L. Song, W. Xia, B. Gao, Z. Jiang, L. Feng and J. He, Chem. – Asian J., 2018, 13, 528–535 CrossRef CAS PubMed.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- B. Han, A. Grimaud, L. Giordano, W. T. Hong, O. Diaz-Morales, L. Yueh-Lin, J. Hwang, N. Charles, K. A. Stoerzinger, W. Yang, M. T. M. Koper and Y. Shao-Horn, J. Phys. Chem. C, 2018, 122, 8445–8454 CrossRef CAS.
- S.-L. Zhang, D. Cox, H. Yang, B.-K. Park, C.-X. Li, C.-J. Li and S. A. Barnett, J. Mater. Chem. A, 2019, 7, 21447–21458 RSC.
- P. Kolla, G. Nasymov, R. Rajappagowda and A. Smirnova, J. Power Sources, 2020, 446, 227234 CrossRef CAS.
- Y. Liu, X. Kong, X. Guo, Q. Li, J. Ke, R. Wang, Q. Li, Z. Geng and J. Zeng, ACS Catal., 2019, 10, 1077–1085 CrossRef.
- B. J. Kim, E. Fabbri, D. F. Abbott, X. Cheng, A. H. Clark, M. Nachtegaal, M. Borlaf, I. E. Castelli, T. Graule and T. J. Schmidt, J. Am. Chem. Soc., 2019, 141, 5231–5240 CrossRef CAS PubMed.
- M. S. Burke, S. Zou, L. J. Enman, J. E. Kellon, C. A. Gabor, E. Pledger and S. W. Boettcher, J. Phys. Chem. Lett., 2015, 6, 3737–3742 CrossRef CAS PubMed.
- X. Xu, C. Su, W. Zhou, Y. Zhu, Y. Chen and Z. Shao, Adv. Sci., 2016, 3, 1500187 CrossRef PubMed.
- H. Sun, X. Xu, Z. Hu, L. H. Tjeng, J. Zhao, Q. Zhang, H.-J. Lin, C.-T. Chen, T.-S. Chan, W. Zhou and Z. Shao, J. Mater. Chem. A, 2019, 7, 9924–9932 RSC.
- Y. Lv, Z. Li, Y. Yu, J. Yin, K. Song, B. Yang, L. Yuan and X. Hu, J. Alloys Compd., 2019, 801, 19–26 CrossRef CAS.
- B. Hou, C. C. Wang, R. Tang, Q. Zhang, Z. Tan, X. Fan and X. Cui, Nano, 2020, 15, 2050077 CrossRef CAS.
- X. Xu, Y. Chen, W. Zhou, Y. Zhong, D. Guan and Z. Shao, Adv. Mater. Interfaces, 2018, 5, 1701693 CrossRef.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y. L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- J. Ran, T. Wang, J. Zhang, Y. Liu, C. Xu, S. Xi and D. Gao, Chem. Mater., 2020, 32, 3439–3446 CrossRef CAS.
- Y. Zhu, Q. Lin, Z. Wang, D. Qi, Y. Yin, Y. Liu, X. Zhang, Z. Shao and H. Wang, J. Energy Chem., 2021, 52, 115–120 CrossRef.
- B. Xia, T. Wang, X. Jiang, J. Li, T. Zhang, P. Xi, D. Gao and D. Xue, J. Mater. Chem. A, 2019, 7, 4729–4733 RSC.
- M. Li, L. Tao, X. Xiao, X. Lv, X. Jiang, M. Wang, Z. Peng and Y. Shen, ChemCatChem, 2018, 10, 4119–4125 CrossRef CAS.
- Y. Sun, R. Li, X. Chen, J. Wu, Y. Xie, X. Wang, K. Ma, L. Wang, Z. Zhang, Q. Liao, Z. Kang and Y. Zhang, Adv. Energy Mater., 2021, 11, 2003755 CrossRef CAS.
- H. Lv, L. Lin, X. Zhang, Y. Song, H. Matsumoto, C. Zeng, N. Ta, W. Liu, D. Gao, G. Wang and X. Bao, Adv. Mater., 2020, 32, 1906193 CrossRef CAS PubMed.
- K. Chu, F. Liu, J. Zhu, H. Fu, H. Zhu, Y. Zhu, Y. Zhang, F. Lai and T. Liu, Adv. Energy Mater., 2021, 11, 2003799 CrossRef CAS.
- Y. Xu, X. Xu, N. Cao, X. Wang, X. Liu, M. Fronzi and L. Bi, Int. J. Hydrogen Energy, 2021, 46, 10293–10302 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |