
Rational design of metal oxide catalysts for electrocatalytic water splitting
Yiming
Xu†
a,
Kaicai
Fan†
a,
Yu
Zou
a,
Huaiqin
Fu
a,
Mengyang
Dong
a,
Yuhai
Dou
a,
Yun
Wang
 a,
Shan
Chen
a,
Huajie
Yin
ab,
Mohammad
Al-Mamun
*a,
Porun
Liu
*a and
Huijun
Zhao
*a
a,
Shan
Chen
a,
Huajie
Yin
ab,
Mohammad
Al-Mamun
*a,
Porun
Liu
*a and
Huijun
Zhao
*a
aCentre for Catalysis and Clean Energy, School of Environment and Science, Griffith University, Gold Coast Campus, Queensland, 4222, Australia. E-mail: m.al-mamun@griffith.edu.au; p.liu@griffith.edu.au; h.zhao@griffith.edu.au
bKey Laboratory of Materials Physics, Centre for Environmental and Energy Nanomaterials, Anhui Key Laboratory of Nanomaterials and Nanotechnology, CAS Center for Excellence in Nanoscience Institute of Solid State Physics, HFIPS, Chinese Academy of Sciences, Hefei 230031, P. R. China
First published on 23rd November 2021
Abstract
Electrocatalytic energy conversion between electricity and chemical bonding energy is realized through redox reactions with multiple charge transfer steps at the electrode–electrolyte interface. The surface atomic structure of the electrode materials, if appropriately designed, will provide an energetically affordable pathway with individual reaction intermediates that not only reduce the thermodynamic energy barrier but also allow an acceptably fast kinetic rate of the overall redox reaction. As one of the most abundant and stable forms, oxides of transitional metals demonstrated promising electrocatalytic activities towards multiple important chemical reactions. In this topical review, we attempt to discuss the possible avenues to construct the electrocatalytic active surface for this important class of materials for two essential chemical reactions for water splitting. A general introduction of the electrochemical water splitting process on the electrocatalyst surface with applied potential will be provided, followed by a discussion on the fundamental charge transfers and the mechanism. As the generally perceived active sites are chemical reaction dependent, we offer a general overview of the possible approaches to construct or create electrocatalytically active sites in the context of surface atomic structure engineering. The review concludes with perspectives that summarize challenges and opportunities in electrocatalysis and how these can be addressed to unlock the electrocatalytic potentials of the metal oxide materials.
 Mohammad Al-Mamun | Dr Mohammad Al-Mamun studied transition metal based electrocatalysts and obtained his PhD degree in materials chemistry in 2016 from Griffith University, Australia. Prior to his PhD, he received research training at Korea National University of Transportation (KNUT) and Shah Jalal University of Science and Technology (SUST). Currently, he is a Research Fellow in Centre for Catalysis and Clean Energy (CCCE), Griffith University. His main research interests include one-step synthesis of earth-abundant functional nanomaterials for electrocatalytic reactions involved in various energy conversion applications. |
 Porun Liu | Dr Porun Liu received his Ph.D. in Chemical Science in 2011 from Griffith University. He subsequently worked as a research fellow in Centre for Catalysis and Clean Energy (CCCE). He was awarded a Griffith University Postdoctoral Research Fellowship in 2015 and an Australian Research Council Discovery Early Career Researcher Award in 2017. He is currently an Associate Professor at School of Environment and Science, focusing on synthesis, in situ characterizations and electrocatalytic applications of nanostructured catalysts in energy conversion and storage devices. |
 Huijun Zhao | Professor Huijun Zhao is the Director of CCCE at Griffith University, the elected Fellow of the Royal Society of Chemistry (FRSC) and the Royal Australian Chemical Institute (FRACI). He has extensive expertise in energy and environmental nanomaterials, water source control & management, field-based sensing technologies and aquatic environmental quality assessment. One of his current pursuits is to unlock the catalytic powers of nonprecious materials as high-performance catalysts for important catalysis reactions. He has published over 500 refereed journal papers, gained 68 international patents within 8 world-wide patent families and has been a Clarivate Analytics Highly Cited Researcher since 2018. |
1. Introduction
With the increase of global demand for energy, rapid depletion of fossil fuels and intensification of environmental concerns, exploitation of clean and sustainable energy carrier alternatives to fossil fuel with industrial-scale has significantly attracted universal interests.1,2 Among the various alternatives, hydrogen has emerged as a promising energy carrier to fulfil the increasing energy demand due to its large energy density per unit mass and eco-friendly production possibilities.1 However, hydrogen does not exist in molecular structures in nature; thus, efficient and sustainable H2 production, conversion technologies and storage technologies are indispensable for the clean hydrogen cycle. Unlike fossil fuels, hydrogen in molecular or ionic forms needs to be generated for further usage/storage. Currently, large-scale H2 production involves steam reforming of fossil fuels with CO2 as a by-product.1 One promising approach to accomplish clean and sustainable hydrogen production is splitting water molecules into hydrogen and oxygen molecules driven by renewable energy, such as wind power and solar energy.3,4 However, the practical applications of water electrolysis are partly hindered by the large overpotential and unsatisfactory cost-effectiveness of electrode materials for anodic oxygen evolution reaction (OER) and cathodic hydrogen evolution reaction (HER). Implementation of efficient electrocatalysts is an effective strategy to lower the large HER, OER overpotentials. Ideal water splitting electrocatalysts are featured with high activities with low overpotential to achieve sizeable current density, remarkable robustness and long-term stability. Nowadays, electrocatalysts based on platinum (Pt), ruthenium (Ru) or iridium (Ir) have been the benchmark materials for HER and OER in electrolyzers.4 However, the scarcity and high cost of these noble metals greatly limit their large scale applications in water electrolysis. Thus, rational design and scalable preparation of highly active and stable noble-metal-free electrocatalysts for efficient hydrogen production have been one of the keys to an industry-scale and rapid deployment of hydrogen technology for sustainable clean energy consumption.Transition metal oxides are a group of materials abundantly available in Earth's crust. Several features such as mechanical and chemical stability, relatively high charge transfer property even in the amorphous state, and particularly tuneable electronic structure, make metal oxides one of the most promising candidatures for water splitting.5 Moreover, even though metal phosphides, sulfides, carbides, nitrides are more active materials towards HER, when considering overall water splitting, metal oxides are generally more stable, especially under OER conditions.6 Also, the fabrication approaches toward metal oxides can be facile and free from harmful chemicals or harsh reaction conditions. Based on these unique properties, metal oxides have been identified as crucial candidates in practical applications, such as thin-film transistors, solar cells, catalysis, energy storage. It is expected that the versatility and functionality of the metal oxides could provide unique opportunities for water splitting. The unexplored potentials of metal oxide as catalysts have inspired and driven many recently published research articles on these fundamentally essential groups of materials as electrocatalysts. Part of the key findings has been summarized in a number of insightful reviews on HER,7–9 OER10–14 and overall water splitting.15–17 A comprehensive review summarizing the active sites, in situ characterization techniques, and material engineering strategies for earth-abundant metal oxides for the application of water splitting (HER and OER) has not been published to date. The information provided in the proposed review will allow a timely overview of the useful approaches and also inspirations towards the design of high-performance electrocatalysts for water splitting applications.
In this topical review, we provide an overview of the construction of electrocatalytic active sites (primarily for HER and OER) on metal oxide surfaces. In section 1, we introduce the nature of electrocatalytic active sites in general and their expected features for heterogeneous electrocatalytic water splitting. This is followed by a summary of the characterization techniques for the metal oxides in section 2. The aim of section 3 is to summarize current approaches that could achieve the targeted features of metal oxides. The review is concluded in section 4 with perspectives and outlooks on the future development of high-performance metal oxide electrocatalysts. The review aims to provide colleagues in nanoscience and nanotechnology with a well-established baseline by summarising recent knowledge advance on structure–activity relationship and developments in material engineering strategies for high-performance metal oxide electrocatalysts. We believe the included advancements from theories, characterizations, and strategies surveyed in the review will inspire ideas and innovations in real-world electrolysis. The information included herein has broader implications for the fundamental researches on nanoscale functional material, surface chemistry, electrochemistry that extendable to the aforementioned widespread applications of metal oxides.
1.1 Water splitting mechanisms
Nowadays, more than 95% of the global hydrogen is produced by steam reforming natural gas or other fossil fuels.18 However, the industrial processes produce low purity hydrogen with a large proportion of carbonaceous species such as carbon monoxide. In contrast, water electrolysis is an effective process to produce high-quality hydrogen through the electrochemical conversion of water to hydrogen and oxygen.19 The electrochemical water splitting provides a clean and sustainable way to convert abundant and renewable water sources into high purity hydrogen.20 Overall electrochemical water-splitting are composed of two essential reactions, i.e., cathodic HER and anodic OER.Water splitting occur in two different pathways, in which the reactions are expressed as:
In acidic solution:
| Cathode: 2H+ + 2e− ↔ H2 | (1) |
| Anode: H2O ↔ 2H+ + 1/2O2 + 2e− | (2) |
In neutral and alkaline solutions:
| Cathode: 2H2O + 2e− ↔ H2 + 2OH− | (3) |
| Anode: 2OH− ↔ H2O + 1/2O2 + 2e− | (4) |
The thermodynamic voltage of water splitting is 1.23 V at 25 °C, 1 atm regardless the water media yet it decreases with increase of temperature. In practise, nevertheless, to achieve the electrochemical water splitting, the applied voltages are higher than the thermodynamic potential value. The overpotential (η) is primarily applied to overcome the intrinsic activation barriers on both anode (ηa), cathode (ηc) and some other resistances, e.g., ohmic drop (ηother). The practical operational voltage (EOP) for water splitting can be described as:2
| EOP = 1.23 V + ηa + ηc + ηother | (5) |
(1) Electrochemical adsorption (Volmer or discharge reaction)
| H+ + M* + e− ↔ M–H* (acidic solution) | (6) |
| H2O + M* + e− ↔ M–H* + OH− (alkaline solution) | (7) |
(2) Electrochemical desorption (Heyrovsky or ion + atom reaction)
| M–H* + H+ + e− ↔ M* + H2 (acidic solution) | (8) |
| M–H* + H2O + e− ↔ M* + OH− + H2 (alkaline solution) | (9) |
(3) Chemical desorption (Tafel or combination reaction)
| 2M–H* ↔ 2M* + H2 (acid and alkaline solutions) | (10) |
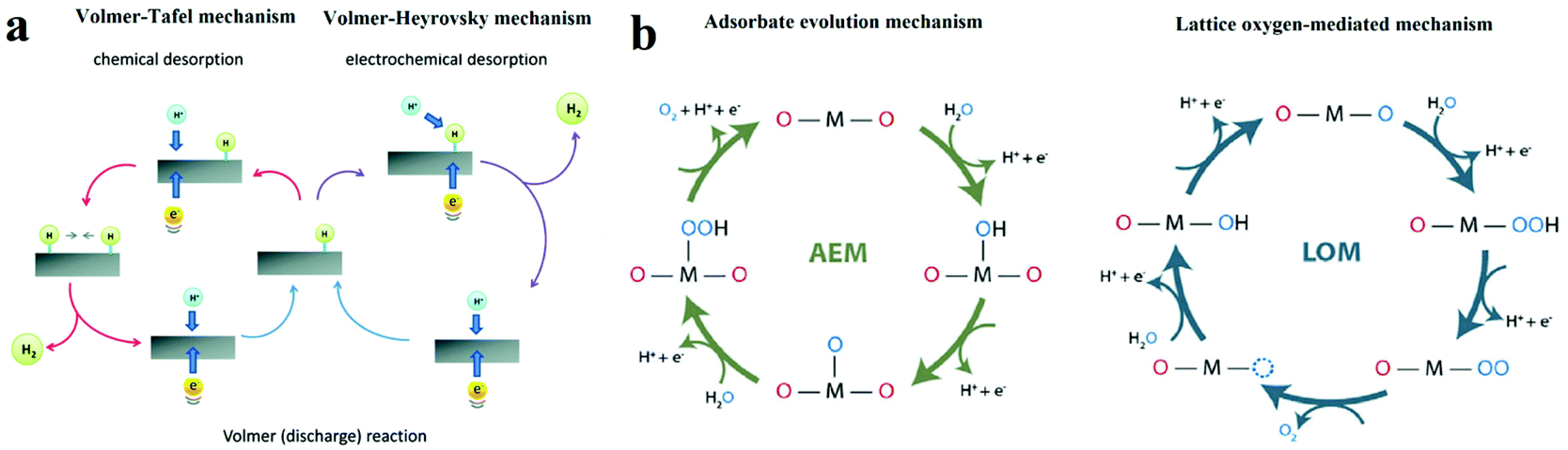 | ||
| Fig. 1 (a) The schematic of electrocatalytic turn-over of hydrogen evolution on the surface of an electrode.21 (b) The adsorbate evolution mechanism (AEM) and lattice oxygen mediated mechanism (LOM) of oxygen evolution turnover on the surface of catalyst.23 | ||
Although the two mechanisms are applicable in both acid and alkaline solutions, it should be noticed that reactants and products are different in various pH values. For example, in acid, only the reduction of a proton into an H* is involved in the Volmer step. While in alkaline solution, besides the creation of the H*, water dissociation is also a critical step for the entire HER process, which requires a breaking of the strong H–O–H covalent bond.
In acidic solution:
| H2O + M* ↔ M–OH* + H+ + e− | (11) |
| M–OH* ↔ M–O* + H+ + e− | (12) |
| M–O* + H2O ↔ M–OOH* + H+ + e− | (13) |
| M–OOH* ↔ M + O2 + H+ + e− | (14) |
In alkaline solution:
| OH− + M* ↔ M–OH* + e− | (15) |
| M–OH* + OH− ↔ M–O* + H2O + e− | (16) |
| M–O* + OH− ↔ M–OOH* + e− | (17) |
| M–OOH* + OH− ↔ M* + O2 + H2O + e− | (18) |
The single-site adsorption mechanism of OER is schematically shown in Fig. 1b.23 It is generally acknowledged that the OER activity of an electrocatalyst depends strongly on the bonding strength of the absorbents on the M active site (M–OH*, M–O* and M–OOH*), of which the adsorption energies are linearly correlated.
Moreover, another OER mechanism (lattice oxygen mediated mechanism, LOM, Fig. 1b) that involves the formation of O–O bonds on adjacent metal ions have also been proven.23,24 At first, two HO* on the two neighbouring metal ions deprotonate and form two M–O*. Then, the two adjacent O* couple directly with each other, resulting in the formation of O–O bond. O2 is eventually released, and two vacant metal sites are occupied by OH−. In contrast to AEM, intermediate HOO* is not involved in LOM.
1.2 Kinetics and volcano plots
As the mechanisms shown above, a heterogeneous catalytic reaction consists of a number of coupled reaction steps, i.e., the adsorption of reactant, surface-mediated activation of the adsorbed reactant, and surface-mediated recombination and the eventual desorption of the products from the surface. Based on the Brønsted–Evans–Polanyi relationship, the reaction kinetics of two consecutive dissociative chemisorption steps, the activation energies for each step are estimated to be linear to the dissociative chemisorption energy:25| Ea1 = α1 ΔE + β1 | (19) |
| Ea2 = α2 ΔE + β2 | (20) |
As the perfectors α1 is positive and α2 is negative, one can see the plots of the curves for overall reaction rate (by converting the Ea and reaction rate r with Arrhenius equation) exhibits a Sabatier volcano shape, i.e., with a maximal rate curve at the top of the full solution to the microkinetic model.
Based on the Sabatier principle, for an efficient HER catalyst, its surface ought to allow an interaction strong enough to absorb H* for facilitating the proton–electron-transfer process, while also weak enough to promote the bond breakage and gaseous H2 release.26 Nevertheless, it is difficult to establish the quantitative relationship between the energetics of the H* intermediate and the reaction rate. From the perspectives of physical chemistry, the Gibbs free energy change for H* adsorption on a catalyst surface (ΔGH*) is a widely accepted descriptor for the catalysts. According to the Sabatier principle (a full microkinetic solution for a simple generalized heterogeneous catalytic reaction25), if ΔGH* is zero, the overall reaction has the maximum rate. Furthermore, the relationship between the experimentally measured exchange current density (j0) and the quantum chemistry-derived ΔGH* form a volcano curve for a wide range of electrode surfaces.27 Following this trend, the relationship between the intrinsic electrochemical nature and HER kinetics could be built. Pt-based metals are at the summit of the volcano, as shown in Fig. 2a,27 which means they have the best activities, and the hydrogen adsorption energy is close to zero. Furthermore, the metals to the left of Pt have strong adsorption to the hydrogen atoms, which blocks the active site and hinders hydrogen generation. While metals to the right of Pt adsorb the hydrogen too weakly, which will not effectively stabilize the intermediate state, thus hindering the occurrence of following hydrogen generation.25,27
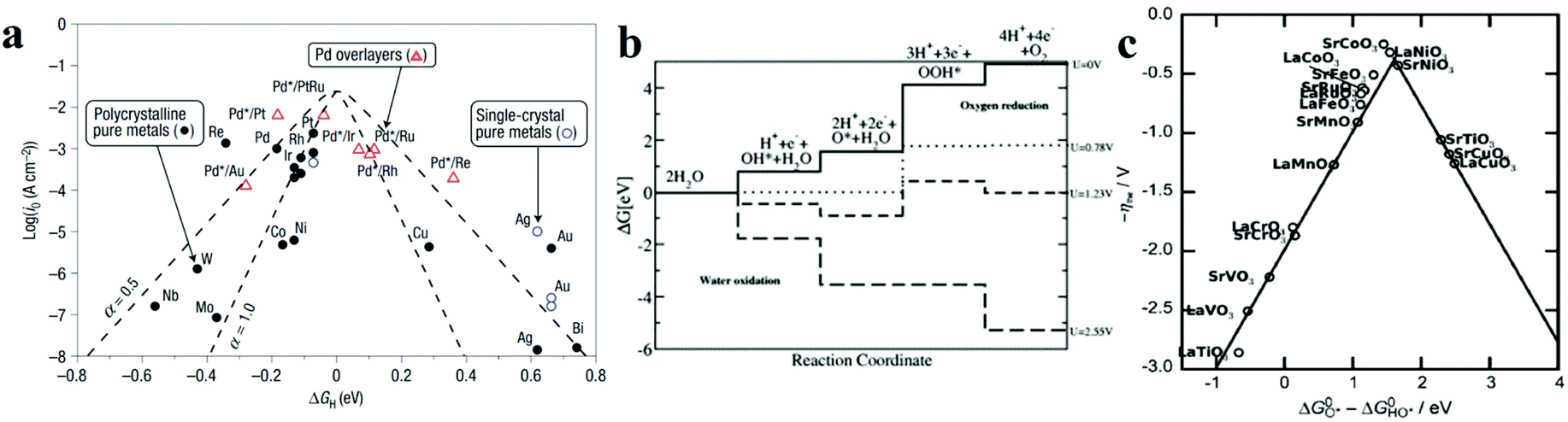 | ||
Fig. 2 (a) A volcano plot of experimentally measured exchange current density as a function of the calculated ΔGH.27 (b) The free energy diagram for OER steps (from left to right) at different applied potentials (U). Due to the scaling relation between the free energy of the intermediates (OH*, O*, OOH*), they are correlated and thus moved in a concerted manner. At U = 2.55 V, all the OER steps are exothermic (downhill from left to right).34 (c) Activity trends of the perovskite OER electrocatalysts. The negative theoretical overpotential was plotted against the difference of standard free energy of the 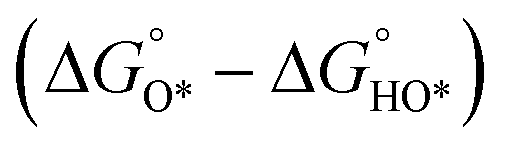 step.22 step.22 | ||
Even though the trends in activity for the HER in alkaline media have never been established independently from those results for the HER in acidic media, differences between the descriptors for alkaline and acid HER have been identified. In acid solutions, the reaction is mainly controlled by the hydrogen recombination (the Tafel step), indicating the strong relationship between the activity and the ΔGH*. However, for HER that occurs in alkaline solutions, the kinetics are determined by the balance between the water dissociation (Volmer step) and the accompanying interaction of the water dissociation product (OH*) on the catalysts.28–30 In this regard, the optimal ΔGH* as the sole descriptor is insufficient to describe the HER activity (exchange current in alkaline media is 2–3 orders of magnitude smaller). Thus, other parameters should be further involved and optimized to explore the highly active catalysts. Gong et al. suggested that the water adsorption energy and the hydroxide anion adsorption energy can also influence the activity.31 Low water adsorption decreases the number of reactants, and high hydroxide adsorption energy may result in active site poisoning. Alonso-Vante and co-workers suggested that the kinetics of HER in alkaline is determined by OH adsorption.32 Thereby, modification of Pt by inducing oxophilic metal centres or sp2 carbon sites favours the adsorption of OH at low potentials, removes Hads intermediate produced in Volmer step, and thus improves the HER kinetics. Markovic et al., established the relationship overall catalytic activities for HER in alkaline media as a function of OH–M2+δ bond strength using 3d-M hydro(oxy)oxides and determined that the activity for the established systems follows the order Ni > Co > Fe > Mn.29 Moreover, Markovic's group identified three parameters based on recent researches, (i) the proton donor, (ii) the energy of formation of the activated complex from proton donor, (iii) the availability of active sites.33 They believed these parameters could be applied in any aqueous system. These results provided a foundation for the rational design of catalysts for practical HER in alkaline electrolytes.
In contrast, for the AEM of OER, free energy (ΔG) diagrams have been used to investigate the rate-determining steps based on Density-functional theory (DFT) calculations (Fig. 2b).34 The ΔG values of the intermediates at different steps have a scaling relation and thus change correlatedly.34 In other words, one free parameter can determine both the free energy diagram and the activity. Later, the scaling relation between the adsorption energies of OOH* and OH* was found to be universal for most of the investigated metal oxide electrocatalysts (Fig. 2c).22 When the theoretical thermodynamic overpotentials were plotted against the difference of standard free energy of the O* and OH* 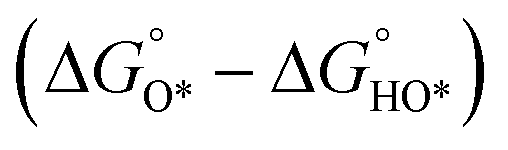 , a volcano relation has been obtained. In line with the Sabatier principle, adsorbate binds to the electrocatalysts on the left branch of the plot too strongly and binds to the counterparts on the right branch too weakly. Also, the
, a volcano relation has been obtained. In line with the Sabatier principle, adsorbate binds to the electrocatalysts on the left branch of the plot too strongly and binds to the counterparts on the right branch too weakly. Also, the 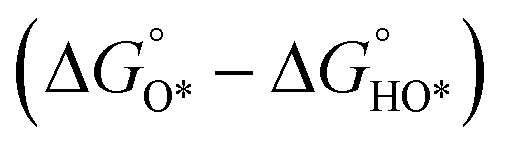 values of all the good OER catalysts are constantly about 3.2 eV, compared to the perfect value of 2.46 eV, indicating a minimum overpotential of 0.4–0.2 V. This explains the relatively large overpotential of OER compared to the HER. To tune the
values of all the good OER catalysts are constantly about 3.2 eV, compared to the perfect value of 2.46 eV, indicating a minimum overpotential of 0.4–0.2 V. This explains the relatively large overpotential of OER compared to the HER. To tune the 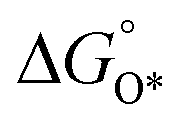 and
and 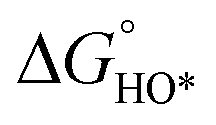 values through varying the oxide surface or electrochemical interface may help to improve the OER activities.
values through varying the oxide surface or electrochemical interface may help to improve the OER activities.
On the other hand, in OER pathway following LOM, the intermediate HOO* is not involved (Fig. 1b).23 Instead, the direct coupling of the lattice oxygen occurs. It is generally believed that oxygen vacancy participation plays an important role in activating the LOM mechanism.35 Thereby, OER following LOM breaks the scaling relation of HOO* and HO*, and surpassing the maximal activities predicted in the volcano plot in AEM become possible. However, there are still some limitations in the LOM. The process of OER following LOM still involves oxygen intermediates, HO* and O*. Thereby the activity in the LOM is affected by the binding energy between oxygen intermediates and active sites,36 which means the optimization of adsorption energies of intermediates is important for LOM. For example, Shao-Horn's group performed DFT calculations and galvanostatic oxidation test and proposed that the deprotonation of the surface OH* group was the rate-limiting step.37 Therefore, a rational-designed OER catalyst following LOM with optimal activities requires both breaking the scaling relation between HOO* and HO* and optimizing the adsorption energies of intermediates.
1.3 Activity descriptors
With the established Sabatier volcano plot and surface scaling relation, one could quantitatively compare the catalytic activity of different surfaces by assessing the bonding strength between the active site and the H adsorbate in HER with DFT calculations. To allow rational design of efficient catalyst, identification of few microscopic surface properties (descriptors) that could link directly with the bonding strength with accuracy is essential. As the d-band model is useful for the understanding of bond formation and prediction of reactivity among transition metals,38 DFT calculation of the d band centre (relative to the Fermi level) of the transition state has been used as one of the useful descriptors for HER. The higher the d band centre, the more active the metal is (the lower the transition state energy and strong absorption).39 Also, it is note that other relevant properties such as the filling of the d bands and the width of d bands are strongly coupled and could be used as well. Based on a high-throughput DFT screening scheme, more than 700 binary surface alloys have been assessed theoretically. BiPt has been identified to have high HER activity and this has been experimentally verified.27A number of reaction descriptors have been proposed for the oxygen catalysis (OER and oxygen reduction reaction, ORR), such as the energy of the metal–OH bond (metals),40 enthalpy of transition from lower oxide to nominal oxide transition 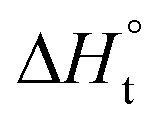 (metal oxides),41 the delocalization and filling (occupancy) of the antibonding σ* (eg) states (metal oxides),42–46 the metal–oxygen covalency (metal oxides),45,46
(metal oxides),41 the delocalization and filling (occupancy) of the antibonding σ* (eg) states (metal oxides),42–46 the metal–oxygen covalency (metal oxides),45,46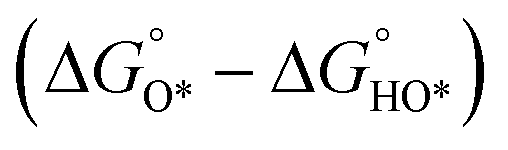 (DFT, metal oxides),34 the number of outer electrons (DFT, metals and oxides).47 For metal oxide catalyst in solid oxide fuel cell, the descriptor of O p-band centre relative to the Fermi level (DFT)48 and oxygen and hydroxyl adsorption energies22 have been proposed. A statistical machine learning and analysis of the descriptors has done by Shao-Horn et al., who found the number of d electrons, charge-transfer energy and optimality of eg are important in correlating the OER activity.49 It should be noted that the utilization of the above descriptors is limited by the structural type of catalysts, and it is vital to find the appropriate descriptor to predict and explain the OER activity. For example, the eg orbital filling and number of d electrons can be the effective descriptors for perovskite materials, while the eg occupancy of the cations at octahedral sites can be used for spinel materials.9,46,50
(DFT, metal oxides),34 the number of outer electrons (DFT, metals and oxides).47 For metal oxide catalyst in solid oxide fuel cell, the descriptor of O p-band centre relative to the Fermi level (DFT)48 and oxygen and hydroxyl adsorption energies22 have been proposed. A statistical machine learning and analysis of the descriptors has done by Shao-Horn et al., who found the number of d electrons, charge-transfer energy and optimality of eg are important in correlating the OER activity.49 It should be noted that the utilization of the above descriptors is limited by the structural type of catalysts, and it is vital to find the appropriate descriptor to predict and explain the OER activity. For example, the eg orbital filling and number of d electrons can be the effective descriptors for perovskite materials, while the eg occupancy of the cations at octahedral sites can be used for spinel materials.9,46,50
These as-mentioned descriptors will always provide the theoretical foundations to improve the electrocatalytic activity towards a specific reaction. Important parameters have been used to describe the performance of an electrocatalyst, such as overpotential (η), Tafel slope, areal activity, mass activity, faradaic efficiency (FE) etc.12 However, when comparing the electrochemical activity, the overpotentials under the same current density are always influenced by the mass loading. Compared with other parameters, turnover frequency (TOF) is the kinetic parameter indicating the intrinsic activity of each active site. TOF is defined as the number of reactant molecules that a catalyst can convert to the desired product per active site in a unit of time. It can be calculated by the equation: TOF = (j × NA)/(n × F × Γ). j stands for the current density, NA is the Avogadro number, n is the number of transferred electrons, F is the Faraday constant, and Γ represents the number of active sites. From this equation, it can be deduced that at a given TOF, the more active sites the catalyst own, the larger the current density becomes, representing faster kinetics of the specific reaction. Therefore, it is expected that efficient catalysts possess well-exposed abundant active sites.
These analyses have shown the possibility of fine-tuning of both intrinsic activity and the number of active sites on metal/metal oxides. Thus, in the coming section, we will summarize characterization techniques that have been used for such purposes to allow better constructing the active catalytic sites.
2. In situ characterization methods
As exchange current provides useful yet limited information, appropriate characterization methodologies should be involved to study both the electrocatalytic activity and its origin. Two metrics are usually applied to evaluate the electrocatalytic performance of a catalyst. One is the apparent total electrode activity and another is the intrinsic activity of each active site. The relationship between the intrinsic activity and the apparent total electrode activity is vital to the insight of the faradaic charge transfer processes on the electrode surface. Electrochemical approaches, such as voltammetry, electrochemical impedance spectroscopy (EIS) and electrochemical active surface area (ECSA) are widely applied to investigate the relationships and processes towards a specific electrochemical reaction. Other characterizations, including optical spectroscopy and microscopy techniques, are also employed to investigate the composition and structure of electrocatalysts. In addition to these experimental measurements, Density Functional Theory (DFT) calculations are intensively applied to identify the active sites and build the relationship between the structure and the electrochemical activity of the electrocatalysts. Therefore, DFT studies could pave the way in guiding the construction of the electrocatalyst with high activity and stability.Even though these conventional methods have been proved to be adequate to gain details of the electrocatalyst, it is noteworthy that these details are strongly influenced by environmental conditions, such as atmosphere, temperature, and pressure. Further information is still needed to detect the actual active sites and reaction mechanism towards a specific electrochemical reaction those may guide towards the activity enhancement. Therefore, characterizations at specific voltages are required, and in situ experiments are designed for this purpose. Spectroscopic characterizations include Raman spectroscopy, X-ray absorption spectroscopy (XAS), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and UV/visible spectroscopy are useful to in situ capture the structural transformation of the electrode and oxidation state change of the catalytic species during a specific electrochemical reaction. While microscopic techniques such as atomic force microscope (AFM) and scanning tunnelling microscope (STM) provide direct morphological snap shoot of the evolving electrocatalysts.
The selection of the characterization technique from the tool box is also subject dependent. Currently, material scientists mainly focus on two aspects to improve the performance of electrocatalysts. One aspect is exposing or building more active sites. In this regard, strategies including nanoscale confinement, phase engineering, and facet engineering are utilized. Since some new active phases or facets may form during the electrochemical test, in situ Raman spectroscopy, XRD, AFM, and STM are effective tools to detect the newly formed active part and investigate the underlying mechanism. The other aspect is improving the intrinsic activity of each active site. Commonly used methods involve heteroatom doping, defect and strain engineering, etc. These methods often result in the changes in electronic structure, such as metal d band and oxygen p band. Therefore, in situ XAS, XPS, and UV/visible spectroscopy can be useful techniques to study the properties of metal oxides. The following part illustrates the specific applications of the aforementioned in situ techniques.
2.1 In situ Raman spectroscopy
Raman spectroscopy is one of the routinely used spectroscopic techniques to observe the vibration, rotation, and other low-frequency modes. Since M–OH, M–OH2, and M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrations appear in low-frequency range, Raman spectroscopy is a powerful tool to provide information about structure of metal oxides and adsorbates on the surface of metal oxides. This feature endorses it as a useful in situ method for the direct investigation of structural changes and intermediates under electrochemical reaction conditions. Edvinsson's group synthesized NiFe-layered double hydroxides as the efficient electrocatalyst towards alkaline water splitting.51 For HER, combining in situ Raman spectroscopy data and other ex situ characterization results, the author proposed that Ni2+ centre adsorbed Had intermediate (Had–NiO) and Fe3+ centre adsorbed OHad intermediate (OHad–FeO) (Fig. 3a–c).51 A second H2O would associate with NiO and produce another adsorbed H atom. The two adsorbed H combined to form H2. The presence of FeOOH facilitated the formation of the second hydrogen with optimal bonding energy and improved the electrocatalytic activity towards HER. For OER, the active phase was identified as γ-NiOOH. The existence of Fe3+ introduced high-valent Ni into γ-NiOOH, which was believed to be the reason for the improvement in OER activity (Fig. 3d–f).51 Wang's group developed Pt–WO3 as an efficient electrocatalyst towards HER and utilized in situ Raman spectroscopy to confirm the real active sites in Pt–WO3.52 The characteristic peaks of WO3 at 268, 710, and 806 cm−1 disappeared and re-appeared during CV cycles, which proved the phase transition from WO3 to HxWO3. EIS studies further illustrated that the HxWO3 phase facilitated fast electron and hydrogen transfer and thus acted as the actual active sites towards HER.
O vibrations appear in low-frequency range, Raman spectroscopy is a powerful tool to provide information about structure of metal oxides and adsorbates on the surface of metal oxides. This feature endorses it as a useful in situ method for the direct investigation of structural changes and intermediates under electrochemical reaction conditions. Edvinsson's group synthesized NiFe-layered double hydroxides as the efficient electrocatalyst towards alkaline water splitting.51 For HER, combining in situ Raman spectroscopy data and other ex situ characterization results, the author proposed that Ni2+ centre adsorbed Had intermediate (Had–NiO) and Fe3+ centre adsorbed OHad intermediate (OHad–FeO) (Fig. 3a–c).51 A second H2O would associate with NiO and produce another adsorbed H atom. The two adsorbed H combined to form H2. The presence of FeOOH facilitated the formation of the second hydrogen with optimal bonding energy and improved the electrocatalytic activity towards HER. For OER, the active phase was identified as γ-NiOOH. The existence of Fe3+ introduced high-valent Ni into γ-NiOOH, which was believed to be the reason for the improvement in OER activity (Fig. 3d–f).51 Wang's group developed Pt–WO3 as an efficient electrocatalyst towards HER and utilized in situ Raman spectroscopy to confirm the real active sites in Pt–WO3.52 The characteristic peaks of WO3 at 268, 710, and 806 cm−1 disappeared and re-appeared during CV cycles, which proved the phase transition from WO3 to HxWO3. EIS studies further illustrated that the HxWO3 phase facilitated fast electron and hydrogen transfer and thus acted as the actual active sites towards HER.
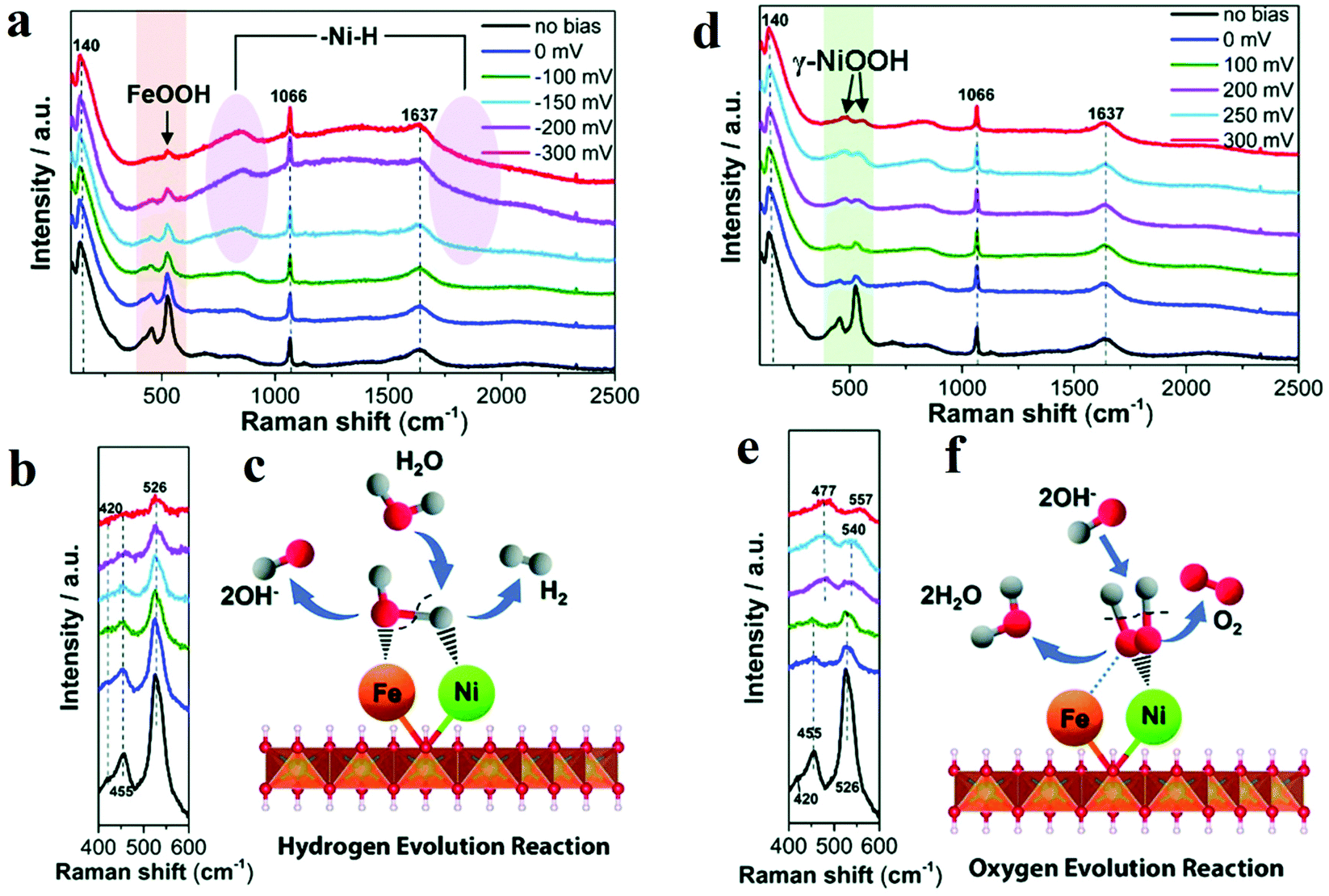 | ||
| Fig. 3 (a) In situ Raman spectra collected in a large wavenumber region from NiFe LDH during HER process in 1 M KOH at various overpotentials vs. RHE. (b) Magnification of the corresponding orange wavelength region of (a). (c) Schematic illustration. (d–f) In situ Raman spectra of NiFe LDH with 532 nm excitation under OER condition. (d) In situ Raman spectra collected in a large wavenumber region from NiFe LDH during OER process in 1 M KOH at various overpotentials vs. RHE. (e) Magnification of the corresponding green wavelength region of (d). (f) Schematic picture.51 | ||
Yeo prepared different OER electrochemical catalysts by depositing Co3O4 on various metals and found that substrate metal with larger electronegativity owned better performance towards OER.53In situ Raman spectroscopy results indicated that at an OER active anodic potential, Co species were oxidized to the valence which was higher than 3 and acted as the active species for OER. By performing the in situ surface-enhanced Raman spectroscopy, Smith's group studied the electrocatalytic performance towards OER of two Ni-based catalysts (Ni(Fe)–Bi and Ni(Fe)OOH).54 The authors concluded from the region ca. 900–1150 cm−1 that at the potential before the actual OER occurred, some “active” oxygen sites in Ni(Fe)–Bi and Ni(Fe)OOH got negatively charged and acted as OER precursors. Bron's group also investigated Ni and Ni/Fe thin-film electrodes by in situ Raman spectroscopy.55 At OER potentials, the peaks at 475 and 557 cm−1 in the Raman spectroscopy results showed the formation of γ-NiOOH. With the increasing Fe contents, the peaks of γ-NiOOH deceased, and I475/I557 band ratio also decreased, which meant disorder was introduced into γ-NiOOH. The authors believed that certain amount of Fe can maintain the presence of γ-NiOOH and induce the occurrence of some other structural disorders, which were vital to the improved OER activity. 15% Fe content was determined to be optimum to obtain the best performance.
2.2 In situ X-ray absorption spectroscopy
XAS is based on the electronic excitations from the core level to an empty electronic state induced by X-ray photon for the element being investigated. By combining X-ray absorption near edge structure (XANES) and the extended X-ray absorption fine structure (EXAFS), this technique can be used to monitor the changes of the electronic structure and the local geometries of the atoms of interest on the catalyst. Herein, it has been applied to study the redox process of metal oxides and identify the active sites during the electrochemical reaction. For the metal oxides, K-edge XAS spectra (with edge positions above 4000 eV) are usually probed, and the ultrahigh vacuum (UHV) condition is not necessary for the “hard” X-ray. Therefore, in situ spectrochemical XAS has become a powerful characterization in electrolyte with applied potentials. Brodsky et al. prepared oxidic cobalt OER catalysts, and they built the active site as Co4O4 cubane.56 Through electrochemical studies and in situ XAS, they found the successive oxidation of Co(III)4 to Co(III)2(IV)2 (Fig. 4a and b).56 Combined with DFT calculations, it could be concluded that the antiferromagnetically exchange-coupled Co(IV) sites catalysed OER. Similarly, Bergmann and his group studied the spinel Co3O4 as an OER catalyst.57 The in situ cobalt L-edge XAS and in situ XRD results showed in Fig. 4c–e indicated that at an OER potential (+1.62 V), the Co species were oxidized to higher valence, which could be proved by the large amount of Co3+ and Co4+, and the crystalline phase transformed into an amorphous phase which provided the structure flexibility.57 The authors believed that the higher valent Co species at the surface, as well as the amorphization, were responsible for catalytic activity. Friebel et al. utilized operando XAS techniques to observe the changes in the oxidation states and M–O bond lengths of Ni–Fe oxyhydroxides under applied OER potential.58 The in situ EXAFS data suggested that when the Fe contents below 25%, the Fe atoms doped into oxyhydroxide structure and the oxidized catalysts were noted as γ-Ni1−xFexOOH. While the Fe levels were above 25%, the Fe atoms tend to nuclear and form γ-FeOOH. Based on the data of changes in oxidation states and M–O bond lengths, DFT results showed that Fe3+ cations in Ni–Fe-based oxides lowed the overpotential for OER apparently and were the real active sites for OER.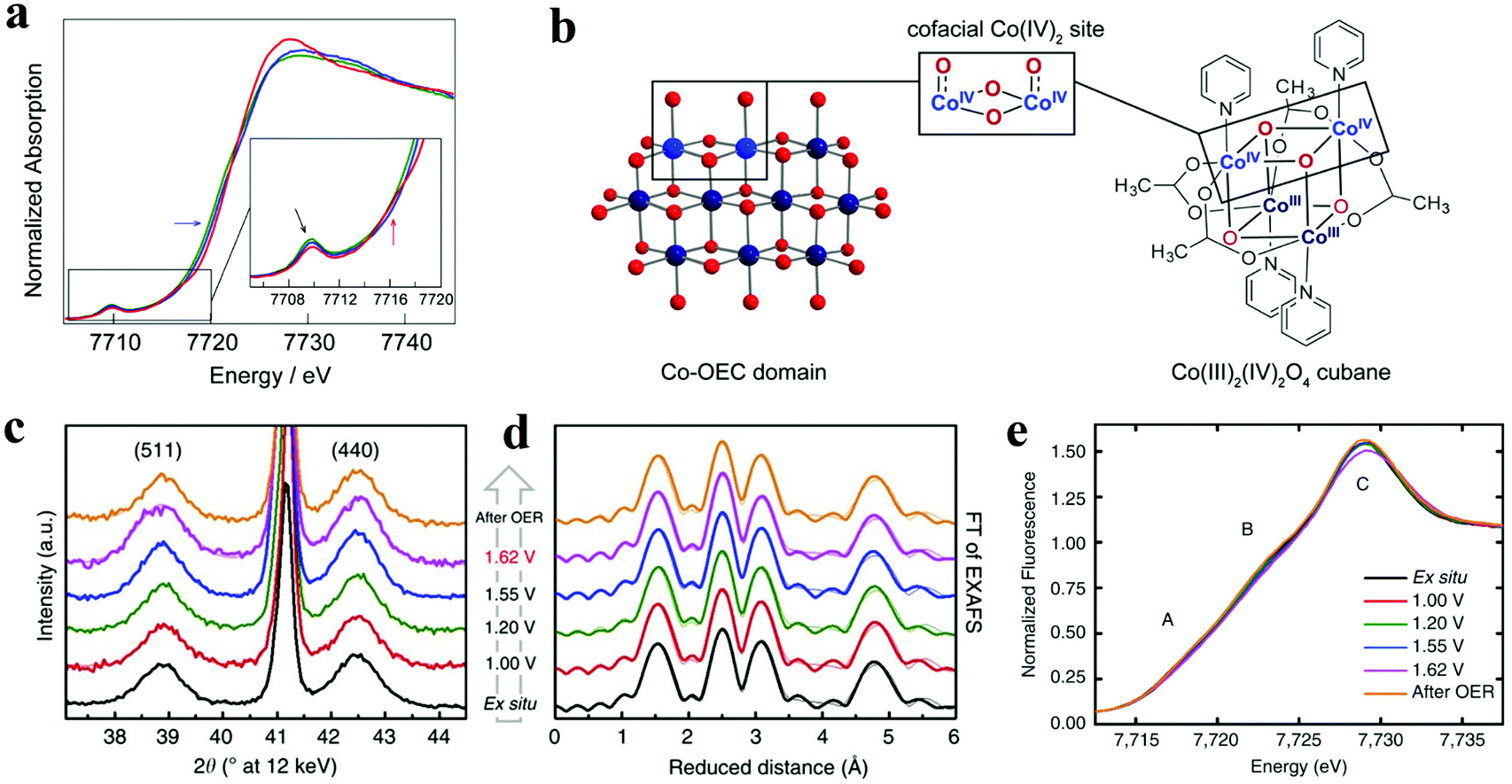 | ||
| Fig. 4 (a and b) X-ray absorption near edge structure spectra of the three accessible oxidation states of cubane, performed in MeCN + 0.1 M n-Bu4NPF6.56 (a) Co K-edge X-ray absorption data for the Co(III)4 (green lines), Co(III)3(IV) (blue lines), and Co(III)2(IV)2 (red lines) states, obtained by applying no potential, 0.80, and 1.8 V vs. Fc+/0, respectively. (b) Co-facial Co(IV)2 sites in Co oxygen-evolving catalyst (Co-OEC) are modelled by a Co4O4 cubane. (c–e) In situ structural characterization of Co3O4 films.57 (c) In situ X-ray diffraction patterns. (d) In situ Fourier-transforms (FT) of quasi-in situ EXAFS spectra collected at the Co K-edge. (e) In situ XANES spectra. | ||
2.3 In situ X-ray diffraction
Another widely used X-ray characterization technique is XRD for investigating the crystal structure of the electrocatalysts. When the incident X-ray beam passes through a crystal, constructive interference and specific diffraction patterns will occur. Different crystalline structures own distinct diffraction patterns. Therefore, in situ XRD can offer researchers structure evolution information about crystalline structure, such as crystal phase, crystallinity, lattice parameters (strains), and average grain size, etc. during the electrochemical reaction. Combined with in situ Raman spectroscopy, it is effective to investigate the active phase changes. Chen's group electrodeposited nanocrystalline cerium dioxide (CeO2) and amorphous nickel hydroxide (Ni(OH)2) on graphite.59 For OER, the catalyst showed low overpotential (177 mV@10 mA cm−2) and good durability (over 300 h) at 1000 mA cm−2. The authors compared the in situ XRD results of Ni(OH)2 with and without Ce intercalation and they ound intercalation of cerium facilitated the formation of γ-NiOOH exposing (003) facets, which was considered to be the active phase for OER (Fig. 5).59 As mentioned before, Bergmann investigated the spinel Co3O4 at OER potentials.57 At OER potentials, the crystalline Co3O4 structure transformed into amorphous state. While after OER, the amorphous structure returned into the thermodynamically stable spinel crystal. Lattice oxygen was believed to participate OER, leading to the formation of oxygen vacancies. These vacancies ultimately resulted in amorphization.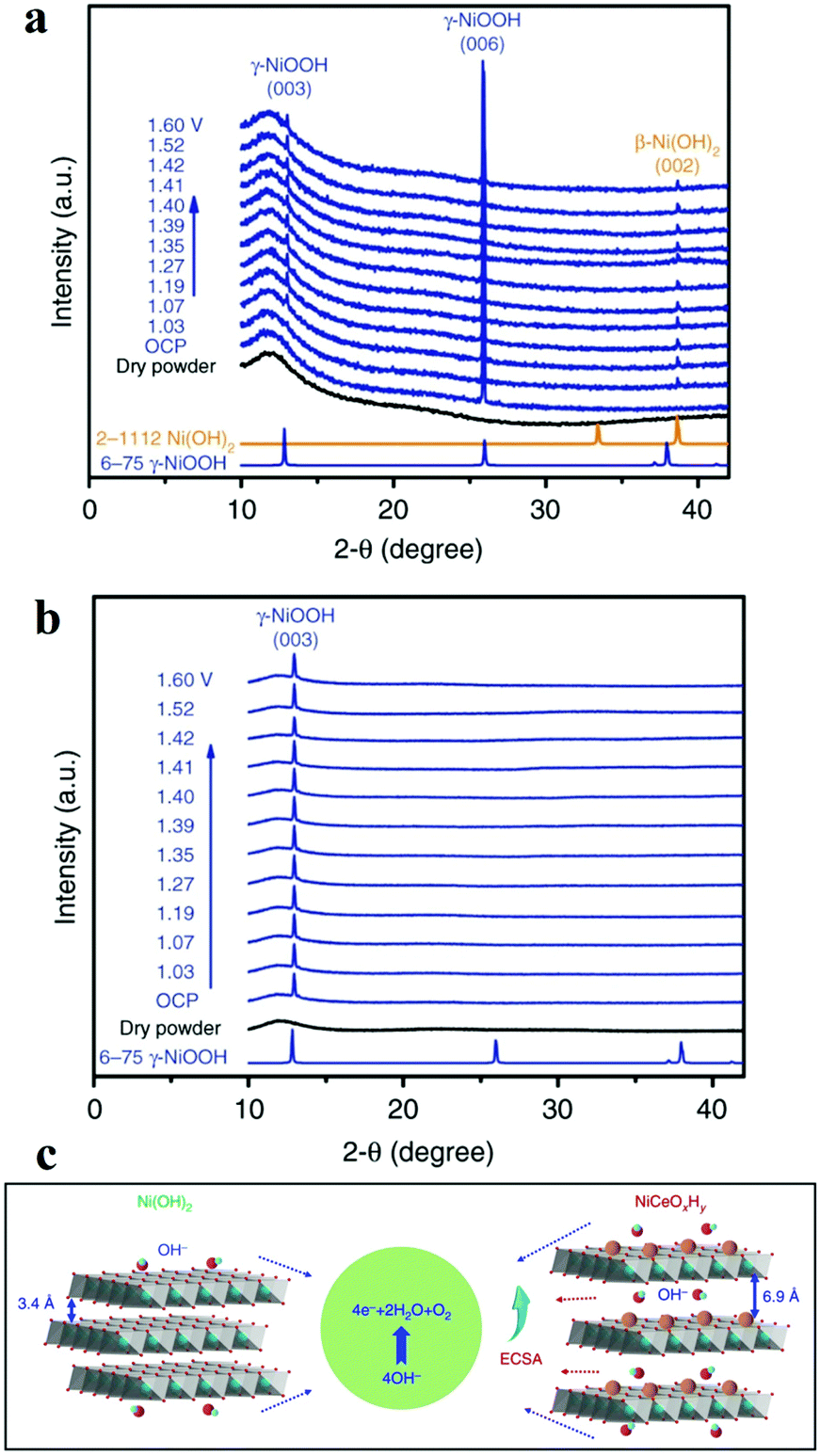 | ||
| Fig. 5 (a and b) Operando XRD patterns collected on (a) Ni(OH)2/NF and (b) NiCeOxHy /NF electrodes at different potentials. (c) The OER proceeds more favourably on NiCeOxHy with larger interlamellar spacing and higher ECSA relative to Ni(OH)2.59 | ||
2.4 In situ X-ray photoelectron spectroscopy
XPS is a photoemission spectroscopic technique to characterize surface chemistry, bonding structure and composition of surfaces/interfaces by measuring the binding energies of core-level electron generated with soft X-rays based on a photoelectric effect. Since the energy of photoelectron is determined by the energy of the photon and the binding energy of the atomic orbital, one can easily investigate the valence of metal sites and the charge transfer between active sites and coordination atoms. In situ XPS therefore enables researchers to obtain bonding information about of different elements in the catalysts under operando conditions. Nenning and co-workers employed in situ XPS to study SrTi0.7Fe0.3O3−x (STF) thin film.60 Fe 2p spectra illustrated that at open-circuit voltage, the valence of Fe was between 2 and 3, and the valence band edge was below the Fermi level, resulting in semiconductor-like electronic structure. When cathodic bias was applied, the valence of Fe was reduced to 0. The metallic Fe(0) also extended the orbitals in the valence band towards Fermi level, leading to enhanced HER activity. Friebel's group reported the use of X-ray ambient pressure XPS to investigate a thin Ni–Fe oxyhydroxide film electrodeposited on Au surface as an OER catalyst.61 When the applied potential increased from 0 to 0.3 V vs. Ag/AgCl, the increase of O/OH was detected (Fig. 6a–c),61 which they believed was due to the oxidation of Ni(OH)2 to NiOOH. Combined with Ni and Fe 2p in situ XPS spectra (Fig. 6d and e),61 the authors concluded that oxidation of both Ni and Fe in the film occurred during OER process. Nilsson and co-workers utilized ambient-pressure XPS to in situ probe the iridium oxides under OER conditions.62 The results indicated that under OER conditions, the surface Ir4+ was oxidized to Ir5+. This in situ observation suggested that Ir5+ played an indispensable role in the OER and provided evidence for the OOH-mediated mechanism.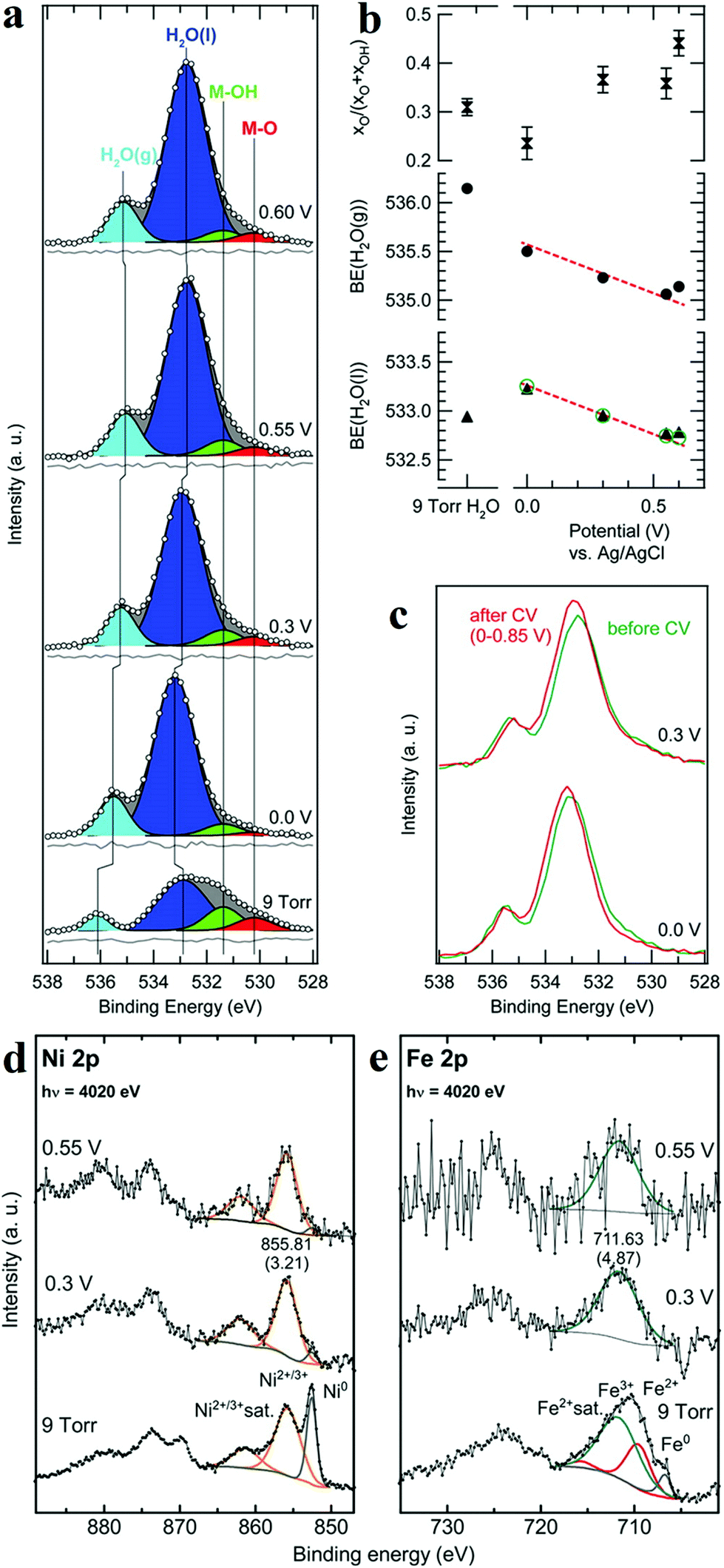 | ||
| Fig. 6 (a) Normalized and background-subtracted O 1s XPS spectra of Ni–Fe electrocatalyst. (b) Fit results for spectra in (a): relative intensity of the M–O component in the MOx(OH)y catalyst, and measured H2O peak positions (black symbols). (c) Comparison of XPS measurements taken at identical applied potentials before (green) and after (red) the second electrochemical conditioning. Normalized XPS spectra of (d) Ni 2p and (e) Fe 2p transitions for a Ni–Fe electrocatalyst.61 | ||
2.5 In situ UV/visible spectroscopy
Electron transitions between different energy levels will lead to the absorption of specific wavelengths of ultraviolet or visible (UV/Vis) light. Thereby, one can easily determine the metal redox states by identifying the wavelength of the absorbed UV/Vis light. In situ UV/Visible Spectroscopy (UV/Vis) is commonly used to track the change of metal redox states. Görlin et al. studied the influence of pH value on the performance of Ni–Fe oxyhydroxide.63 By using in situ UV/vis, they monitored the amount of oxidized Ni3+/Ni4+ and found that there was an earlier onset of the redox process with a higher pH value. It was also demonstrated that low-valence Ni was favourable for OER process to occur. Yoshida and co-workers prepared the nickel oxide electrocatalyst by electrodeposition, and they found that during the electrodeposition, the addition of diamines could improve the OER activity of the nickel oxide.64Operando UV/Vis absorption spectroscopy indicated that 1,2-ethylenediamine combined with Ni2+ and formed complexes, which finally facilitated the nickel oxide electrodeposition and increased the number of Ni sites. The improved OER activity mainly derived from the increased number of Ni, which served as active sites.2.6 In situ electrochemical atomic probe microscopy
Integrated with a classical electrochemical cell, Electrochemical Atomic Force Microscopy (EC-AFM) can in situ observe the surface morphology or measure the height of catalysts on flat substrates under reaction conditions. Boettcher and co-workers utilized in situ EC-AFM to investigate dynamic process of the incorporation of Fe into Ni(OH)2 nanosheets.65 When applied with high anodic potentials, Fe ions quickly deposited on the top of NiOxHy nanosheets and formed FeOxHy nanoparticles. While at the open circuit potential, Fe incorporated into and expanded NiOxHy, which can be detected by measuring the mean height of the nanosheet. The incorporation of Fe had been further verified by anodic shifts in Ni redox wave. Scanning Tunnelling Microscopy (STM), combined with electrochemical cells, can be employed to in situ probe the change of morphology, such as dissolution, degradation, under varying potentials. Moreover, EC-STM can achieve fine measurement of the morphological change with atomic resolution. Paoli et al. used EC-STM to observe the dissolution of RuO2 catalyst under OER potentials directly.66 The STM images demonstrated that RuO2 exhibited better stability than deposited Ru, since Ru disappeared from view while RuO2 showed no corrosion with the same potential scan from 1.3 V to 1.5 V.2.7 Other techniques determining active sites
Besides the forementioned in situ techniques, other electrochemical analyses are also used to determine the concentration of active sites on surface if the current responses are well understood. Since most published work on OER agree that the OER active metal sites undergo in situ oxidation and reduction upon voltametric scans. A relatively reliable method is to calculate OER active sites from the redox features of the metal in the CV.67 The surface concentration of metal sites that participate in OER can be calculated with the integrated area associated with the metal oxidation or reduction (the associated charge) and the number of the transferred electron.As for determining the concentration of active sites towards HER, since it is difficult to differentiate current associated with the metal redox reactions (if any) superimposed in the cathodic faradaic current, the as-mentioned method is no longer suitable. For the determination of electrochemical active surface area, other electrochemical methods, such as carbon monoxide (CO) stripping, hydrogen underpotential deposition (H-UPD) and peroxide oxidation, have also been used.68,69 One can also assume that all the metal atoms are active towards HER or OER, and then calculate the active site number and TOF. However, it is noteworthy that this method produced numerous errors.
DFT is often coupled with characterizations to identify the real active sites. After characterizing the geometrical and electronic structure of electrocatalysts, one can build an appropriate model on the basis of the structural information acquired from the characterizations. The essential information such as Gibbs free energy diagram, density of states (DOS), band structure, and charge distribution, etc. are accessible with the help of DFT methods.70–72 These results will finally reflect in descriptors such as d band centre, M–O covalency, and O p-band centre, which regulate the reaction pathways by governing the adsorption energy of intermediates and potential-determining step (PDS). Therefore, the activity of active sites can be predicted with the calculated descriptors and compared with the experimental results.72 Notably, some electrocatalysts will undergo surface reconstruction and phase transformation, especially when working under OER conditions.23 Thus it is more accurate to call these catalysts used in the published work “pre-catalysts”. The calculation results based on the ex situ characterizations without surface reconstruction/activation could be misleading. Thereby, truthful structural information from in situ observations under operando conditions is more suitable for DFT calculation. Despite some challenges in combinatorial research, such as the lack of DFT tool to effectively simulate the reaction interface under operando conditions with affordable computational cost, or the structural complexity at the interface in atomic scale, computation modelling coupled with in situ characterizations have been the most effective approach to date and will accelerate exploring high-performance electrocatalysts.
To summarize, in situ characterization methods have been briefly summarized as the toolbox for electrocatalyst development that provide researchers with precise information under working conditions as well as guidance to deeper mechanism. The next section of the review is to illustrate that with the help of the characterization techniques, how specific material engineering strategies influence geometric and electronic structures of electrocatalysts and further construct the active catalytic sites on metal oxides.
3. Strategies for activity enhancement
3.1 Nanoscale confinement
Nowadays, the performance of heterogeneous electrocatalysts is still limited by the numbers and intrinsic reactivity of active sites. Moreover, most metal oxides possess limited electronic conductivity and charge transport to the active sites. Compared with the bulk materials, nanostructured materials, such as nanoparticles and nanosheets, typically own higher specific surface areas and thus are endowed with a larger number of surface-active sites. When the diameter of a particulate catalyst is smaller than a specific size (3–5 nm), the significantly reduced coordination number of atoms in outer shells induces dramatic changes in the electronic structure and may improve the catalytic performance. In addition, modulation of the nanostructure, mainly reducing the size of electrocatalysts to the nanoscale, provides an efficient solution to facilitate charge transfer by shortening the charge transport distance. Recently, significant improvements have been achieved by designing the nanostructure of metal oxide catalysts.The top-down creation of nanoparticles from the parent particles with larger sizes is a common strategy to create a large active area or abundant active sites. Wang et al. introduced lithium ions into transition metal oxides and fabricated nanoparticles with an ultrasmall diameter (2–5 nm).73 It was demonstrated that the ultrasmall NiFeOx and CoO nanoparticles exhibited superior catalytic activity towards HER and OER compared to nanoparticles with large diameters (∼20 nm). Moreover, the obtained catalyst achieved excellent stability of 200 h at 10 mA cm−2 without any decay. Different from the catalysts prepared by other traditional chemical synthetic methods,74 the nanoparticles in situ formed via this lithium-induced conversion reaction still maintain excellent electrical interconnection while introducing large surface areas and exposed active sites (Fig. 7a–e).73 From transmission electron microscopy (TEM) images, the interconnected ultrasmall nanoparticles were manifested to have abundant grain boundaries, which acted as the origin of increased surface area and active centres. The rich boundaries were further verified by calculating electrochemical double layer capacities from cyclic voltammetry results. The authors attributed the excellent stability to the in situ preparation method involving delithiation reaction process, which endowed excellent mechanical and electrical contacts between the catalyst and the substrate. On the other hand, the coordination environment of atoms in outer shells is significantly affected by the size. The electronic structure is also primarily altered. Zeng et al. managed to obtain optimal eg orbital occupation by controlling the particle size of perovskite LaCoO3.75 The authors found that when the size was down to 80 nm, LaCoO3 showed the best performance for OER with an overpotential of only 320 mV in 0.1 M KOH electrolyte. The authors explained that the cobalt ions have an optimal electron configuration of eg ∼1.2 according to Shao-Horn’ principle46 when the particle size was ∼80 nm which was beneficial to improve the OER activity. By conducting electron energy loss spectroscopy (EELS) spectra, the enhancement stemmed from the spin-state transition from the low-spin state to a high-spin state of cobalt ions at the surface.
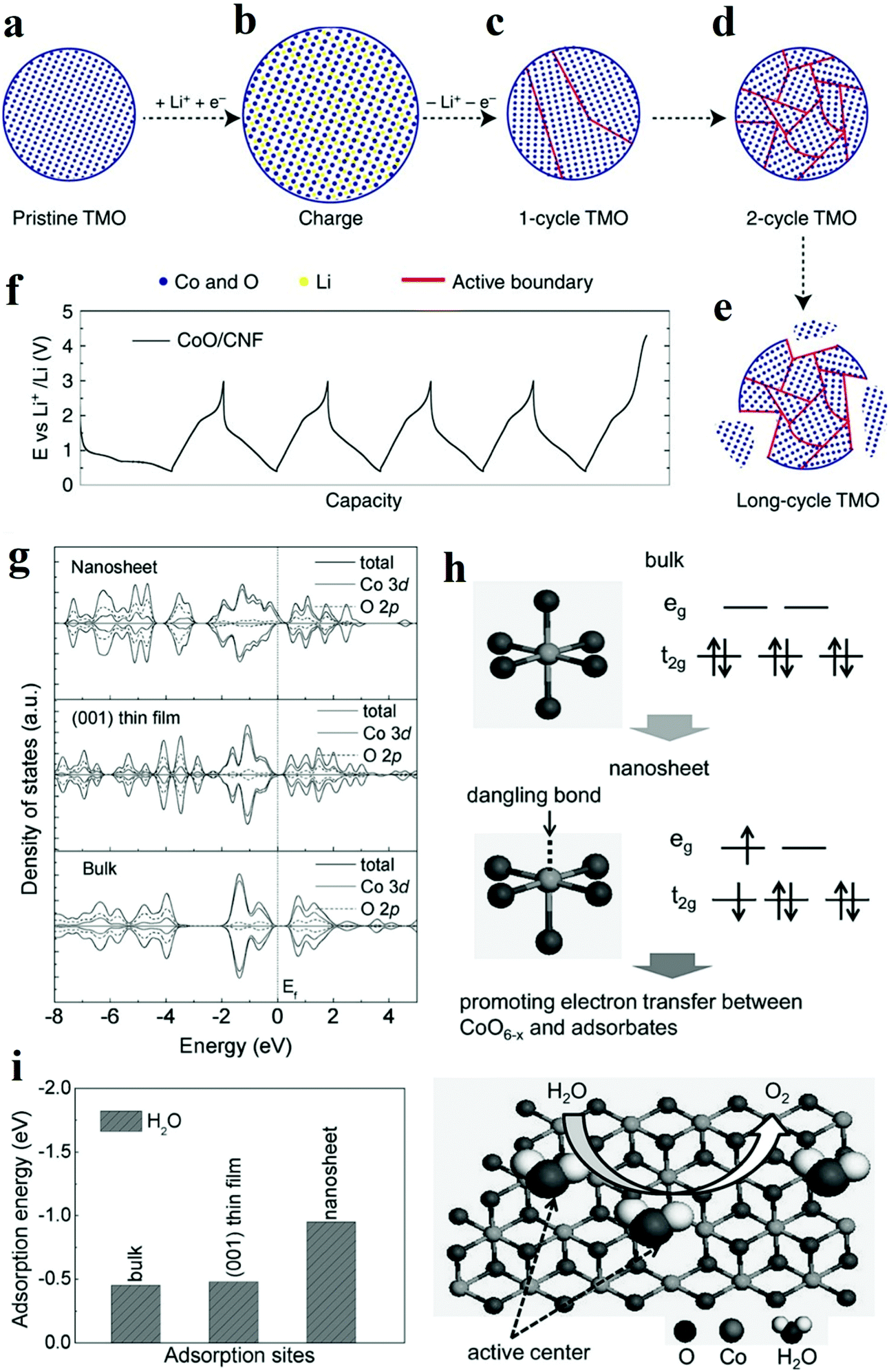 | ||
| Fig. 7 (a–f) Schematic of transition metal oxides (TMO) morphology evolution under galvanostatic cycles.73 (a–e) TMO particles gradually change from single crystalline to ultra-small interconnected crystalline NPs. Long-term battery cycling may result in the break-up of the particle. (f) The galvanostatic cycling profile of CoO/CNF galvanostatic cycling. (g–i) The XAS results and theoritical results of γ-CoOOH with different size.77 (g) Calculated density of states (DOS) for γ-CoOOH nanosheet, γ-CoOOH (001) thin film and bulk γ-CoOOH. (h) Electronic structure transformation of ultrathin γ-CoOOH nanosheet and model of water oxidation on the nanosheet surface. (i) Calculated adsorption energy for ultrathin γ-CoOOH nanosheet, γ-CoOOH (001) thin film and bulk γ-CoOOH. | ||
Two-dimensional materials, such as nanosheets and thin films, are also widely explored due to their large surface areas, good electrical conductivity, and abundant low-coordinated surface atoms which are essential to accelerate electrochemical kinetics. Sun et al. prepared atomically thin cobalt oxide porous sheets and carefully investigated the impact of the nanostructure on OER both theoretically and experimentally.76 The authors synthesized porous Co3O4 ultrathin sheets by heating CoO sheets. TEM and AFM results showed that the porous Co3O4 sheets possessed a thickness of only 0.43 nm, which was about half of a unit cell. This ultrathin thickness induced a larger surface area and the expose of all Co3+ of Co3O4, which was regarded as the active site for OER. More importantly, the coordination number of Co3+ atoms decreased from 6 and 5 to 4 or even 3 because of the ultrasmall thickness. According to DFT calculations, the adsorption energy of H2O molecules on lower coordinated Co3+ atoms was larger (0.45 eV), leading to a higher catalytic activity. Furthermore, the density of states at the edge of the valence band and conduction band increased due to the structure disorder on the surface of atomically thick sheets. Therefore, the electron transfer along the conducting channel was promoted, and thus the sluggish OER kinetics was significantly accelerated. Besides, the OER performance for the Co3O4 nanosheet was retained even after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 CV cycles, while the bulk counterpart experienced a dramatic drop in OER performance. Through the interfacial charge-transfer resistance test, the outstanding stability was attributed to the unique 2D configuration, which provided intimate contact between the glassy carbon (GC) electrode and the catalyst, and enabled a feasible release of evolved gas bubbles. In another work, ultrathin γ-CoOOH nanosheets were fabricated by atomic-scale phase transformation strategy and manifested superior performance for OER than the state-of-art catalyst.77 The ultrathin nanosheets were found to be half-metallic, and the electric conductivity was 52 times better than the bulk CoOOH. EXAFS and XPS results in Fig. 7g–i showed that the size confinement led to the existence of surface structural distortion of CoO6−x and rearrangement of Co 3d electrons population.77 This rearrangement resulted in forming an electron configuration of t2g5eg1.2, which ultimately improved the electron transfer between surface cations and adsorbed intermediates. The thickness effect of metal oxides on their electrical conductivity was also investigated. Viswanathan et al. investigated the relationship between the critical thickness of TiO2 and the location of valence band maximum relative to the limiting potential for OER to study the charge transport mechanism.78 With the help of atomic layer deposition (ALD), the authors managed to control the thickness of TiO2. Through theoretical analysis, it was predicted that the thermodynamic limiting potential for OER was in the band gap of TiO2, located close to the valence band, and a bias potential was needed to maintain the electrochemical current when the thickness of TiO2 surpassed a certain value. The experimental results also proved that the overpotential occurred when the thickness exceeded 4 nm.
000 CV cycles, while the bulk counterpart experienced a dramatic drop in OER performance. Through the interfacial charge-transfer resistance test, the outstanding stability was attributed to the unique 2D configuration, which provided intimate contact between the glassy carbon (GC) electrode and the catalyst, and enabled a feasible release of evolved gas bubbles. In another work, ultrathin γ-CoOOH nanosheets were fabricated by atomic-scale phase transformation strategy and manifested superior performance for OER than the state-of-art catalyst.77 The ultrathin nanosheets were found to be half-metallic, and the electric conductivity was 52 times better than the bulk CoOOH. EXAFS and XPS results in Fig. 7g–i showed that the size confinement led to the existence of surface structural distortion of CoO6−x and rearrangement of Co 3d electrons population.77 This rearrangement resulted in forming an electron configuration of t2g5eg1.2, which ultimately improved the electron transfer between surface cations and adsorbed intermediates. The thickness effect of metal oxides on their electrical conductivity was also investigated. Viswanathan et al. investigated the relationship between the critical thickness of TiO2 and the location of valence band maximum relative to the limiting potential for OER to study the charge transport mechanism.78 With the help of atomic layer deposition (ALD), the authors managed to control the thickness of TiO2. Through theoretical analysis, it was predicted that the thermodynamic limiting potential for OER was in the band gap of TiO2, located close to the valence band, and a bias potential was needed to maintain the electrochemical current when the thickness of TiO2 surpassed a certain value. The experimental results also proved that the overpotential occurred when the thickness exceeded 4 nm.
Since the hierarchical structure provides highly exposed active sites and facilitates the charge and electron transfer of active species,79,80 electrocatalysts with different hierarchical structures have been fabricated and presented high electrocatalytic performance. Hierarchical NiCo2O4 hollow microcuboids with 1D porous nanowires subunits were fabricated by Gao and co-workers.81 The electrode exhibited excellent activity for overall water splitting, with a small applied potential (1.65 V) to reach a current density of 10 mA cm−2 towards overall water splitting as well as excellent stability of 36 h at 20 mA cm−2. Zhu et al. prepared a class of novel nickel cobalt oxide hollow nanosponges, which showed excellent catalytic activity towards OER due to their porous and hollow nanostructures.82
Overall, the nanoscale confinement strategy, aiming to control the morphology and size of the metal oxide, benefit the electrocatalytic activity by the following four ways. Firstly, the engineering of morphology in nanoscales, such as nanoparticles and nanosheets, often improves the surface specific area and exposes more active sites. Secondly, downsizing into a scale less than a unit cell can dramatically influence the coordination environment of the surface atoms and further impact the intrinsic activity of metal sites. Thirdly, the modulation of size brings a substantial change in the energy band. Then, the modified metal oxides may transform from insulator or semi-conductor to conductor, and the electron transfer pathway is facilitated. Finally, confinement strategies could facilitate more contact between catalysts and substrates and optimize the release of gas bubble, which significantly improve the durability. Thus, nanoscale confinement can be an effective measure to improve catalytic performance.
3.2 Crystal phase and facet engineering
Since most heterogeneous catalysis processes occur on the catalysts’ surface, the composition and arrangement of surface atoms are decisive to the activity of catalysts.83,84 The anisotropic atomic structures such as the coordination environment could be finely tuned by controlled crystal growth. Moreover, distinctive facets and phases usually expose different number of active sites. Therefore, crystal phase and facet can influence the overall activity of electrodes. Accordingly, various surface phase and facet control strategies have been developed to manipulate the catalytic activity and/or create more active sites.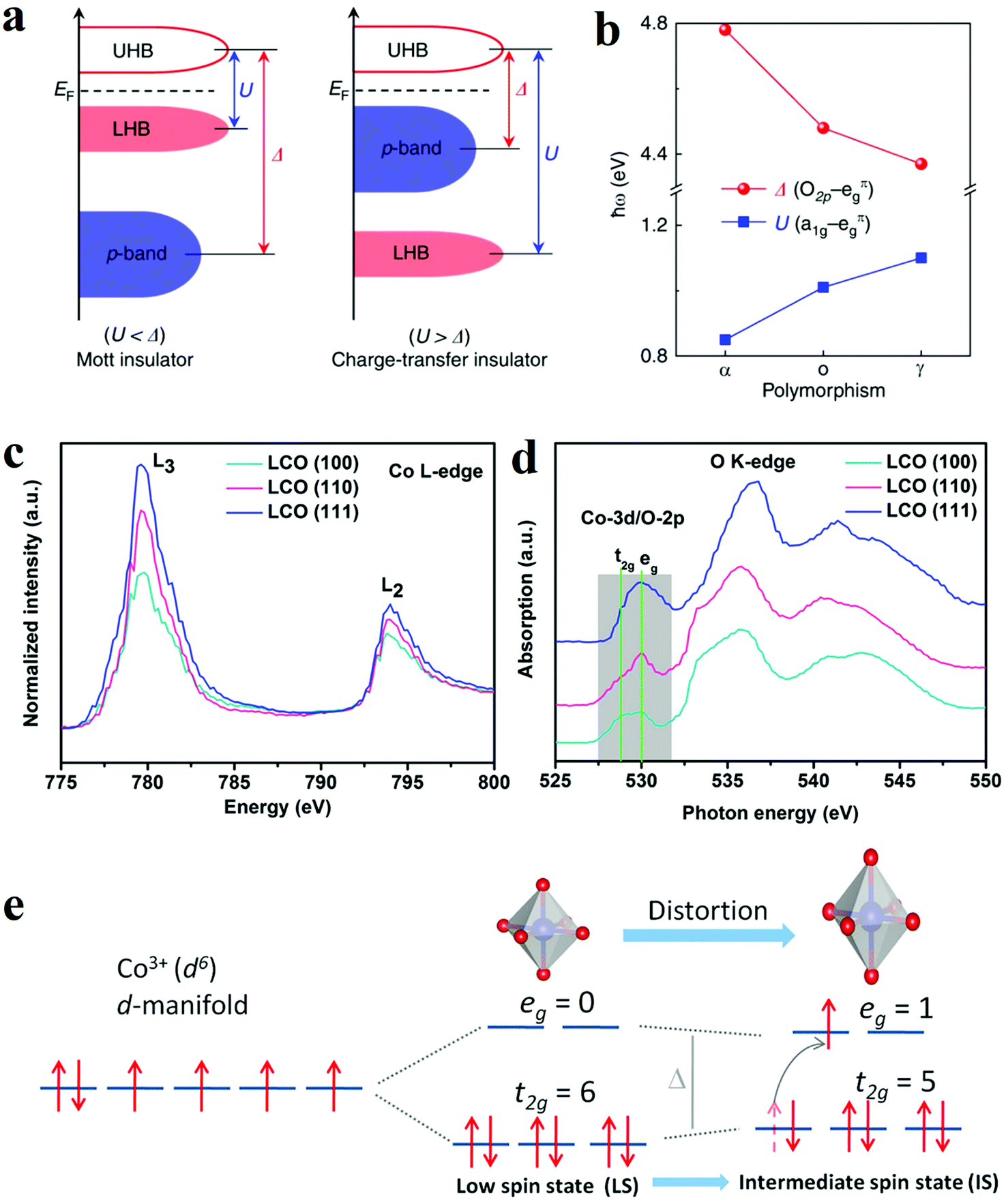 | ||
| Fig. 8 (a and b) The influence of phase engineering on the energy band of Ti2O3 polymorphs.85 (a) Schematic energy band diagram for the Mott insulator and charge-transfer insulator. (b) Evolution of the U and Δ in Ti2O3 polymorphs. (c–e) XANES spectra and schematic illustration of intermediate spin state transition.93 (c and d) Co L-edge (c) and O K-edge (d) XANES spectra for LCO (100), (110), and (111) films. (e) Schematic illustration of the transition of electrons from t2g to eg orbital and the evolution of intermediate spin state. | ||
O elongation and optimize the adsorption, dissociation and desorption of oxygen species. Hence, the Co3O4-NP/N-rGO exposing (112) facets with more abundant Co3+Oh sites presented better catalytic performance than the counterparts with other exposing crystal facets. Wu et al. also compared the OER electrocatalytic activities of α-Fe2O3 nanocrystals exposing different facets, namely (012)-O, (012), (104) and (110).92 The hematite exposing (012)-O and (012) showed advantageous catalytic activity for OER, with a lower overpotential of 305 mV and 317 mV at 10 mA cm−2. Theoretical results through DFT calculations indicated the (012) facet owned the active sites with the highest activity since they possessed a relatively low free energy for all intermediates from OER. As a result, the process of adsorption/desorption was optimized, and the kinetics barrier became lower. The facet engineering have also been used to improve perovskite materials. Wu and co-workers optimized the OER performance of LaCoO3 through spin-state regulation by controlling facet orientation.93 Three different facet orientations, basically (111), (110), and (100), brought a varied degree of distortion of CoO6 octahedron, which corresponded to the transformation of the spin state from the low spin state (t2g6eg0) to the intermediate spin state (t2g5eg1) (Fig. 8e).93 The change in spin state was confirmed by XANES spectra (Fig. 8c and d).93 Benefit from the intermediate spin state, the film with (100) orientation obtained optimal conductivity, eg electron filling states (0.87), as well as adsorption free energy of O species. These characters collectively resulted in an outstanding performance for OER with an overpotential of 470 mV at 10 mA cm−2.
To sum up briefly, surface atomic arrangements of a bulk crystal are readily controllable with crystal phase and facet engineering. This provides scientists with an essential “knob” to fine tune the electronic structure and electrocatalytic nature even with the similar elemental composition. Firstly, engineering of phase and facet endow changes in energy band of metal oxides, which might change insulating characteristics into half-metallic or metallic one and modulate electronic transportation. Secondly, the engineering in phase and facet often changes the coordination environment of metal site and the M–O electronic structure, and thereby modulates the intrinsic activity, especially for OER. Finally, even assuming that active sites are with equal activity, crystalline catalysts with specific phases and facets expose more active sites, which certainly improve the overall electrochemical performance.
3.3 Heterostructure
It is scarce that overall water splitting in practice undergo electrochemical turnover on electrocatalysts with high homogeneity. Recently, construction of heterostructure or creation of interfaces with the remarkable synergistic effect has become a focused research topic. According to the experimental and theoretical studies, the strong coupling interactions and interface reconstruction between composites can not only promote electrochemical reaction activity but also expose more active sites. The strategy of fabricating the rationally designed heterostructures can be adopted to prepare catalysts with excellent performance.It has been proven that HER in alkaline solution significantly depends on activating and cleaving O–H bonds of water molecules and facilitating the adsorption and desorption of the formed H atoms. In this regard, the novel concept of composite materials facilitating these two key aspects of HER process in alkaline solution has been reported. A typical example is a heterostructure composed of oxide and metal, in which the oxide acts as the active site for the dissociation of water, while the metal facilitates the adsorption and desorption of hydrogen atoms. Subbaraman et al. prepared nanometre-scale Ni(OH)2 clusters with high water dissociation properties on platinum electrode surfaces.30 With a dual effect, the edges of the Ni(OH)2 clusters facilitated the dissociation of water, and the produced hydrogen intermediates were then adsorbed on the nearby Pt surfaces and recombined into molecular hydrogen. Inspired by this encouraging work, various metal/metal oxide heterostructures have been fabricated. In 2014, Gong et al. prepared a nanoscale NiO/Ni heterostructure attached to a carbon nanotube (CNT) network, which exhibited excellent HER activity in a wide range of pH values as shown in Fig. 9a.94 The NiO/Ni was partially reduced from Ni(OH)2 through thermal decomposition. It was proposed that the exposed NiO/Ni nano-interfaces might be the synergistically active sites towards HER. Specifically, the OH− generated by H2O splitting could preferentially adsorb to a NiO site at the interface, while a nearby Ni site would facilitate H adsorption, boosting synergistic HER catalytic activity to NiO/Ni. In another work, Cr2O3 blended with NiO/Ni was later fabricated, in which the chemically stable Cr2O3 was essential to prevent oxidation of the Ni core and to preserve the catalytically active sites in the heterostructure, and the catalyst with heterostructure obtained great stability of more than 80 h at 200 mA cm−2.95 To date, a number of metal/metal oxide interface nanostructures have been successfully also explored, collectively showcasing that the strategy can be extended to boost the HER performance in alkaline media.96–99
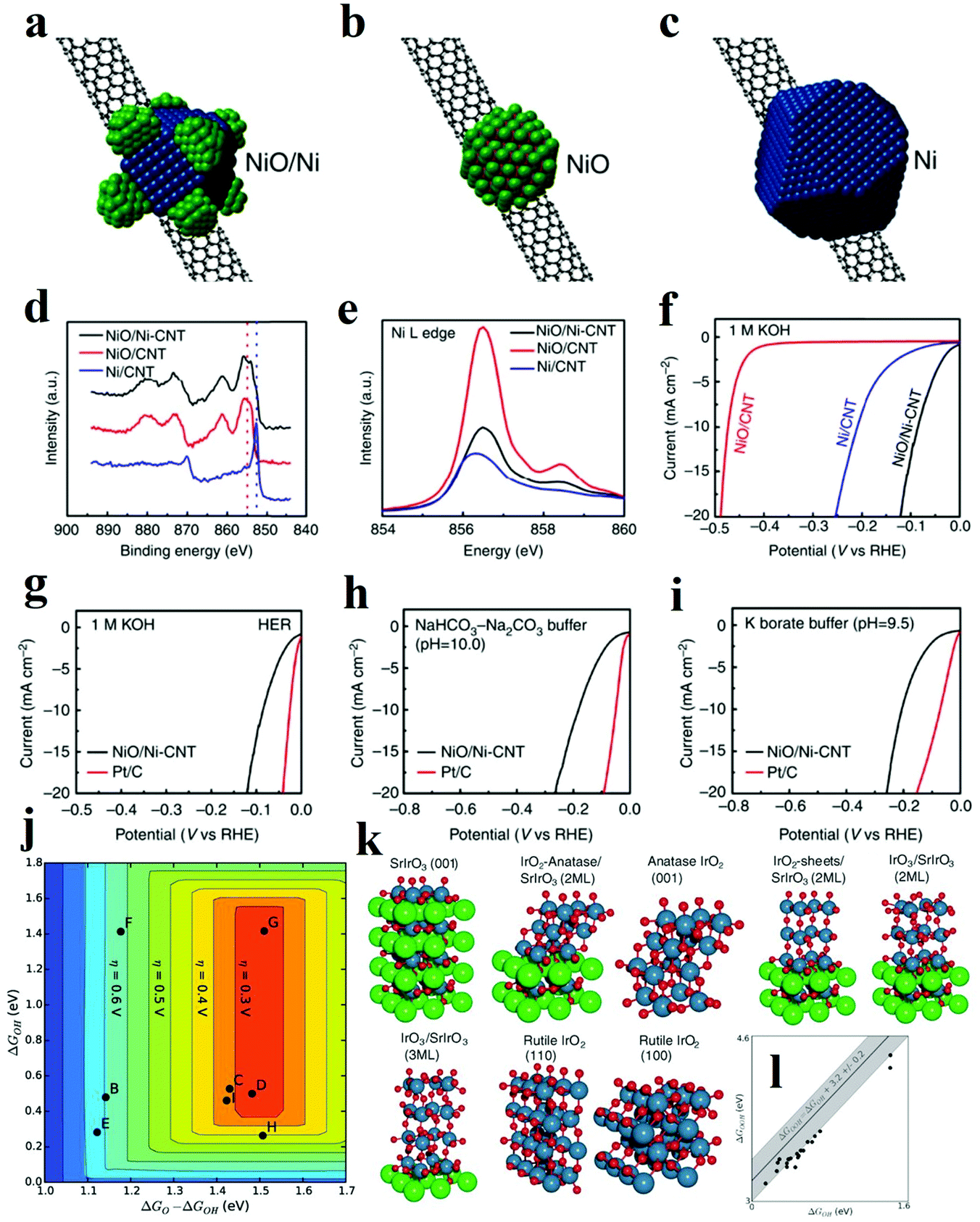 | ||
| Fig. 9 (a–c) Schematic structure of (a) NiO/Ni-CNT nano-hybrid, (b) NiO/CNT, (c) Ni/CNT structure. (d) High-resolution Ni XPS spectra of the three hybrid materials. (e) Ni L edge XANES spectra of the three hybrid materials. (f) Linear sweep voltametry of the three hybrid materials in 1 M KOH. (g–i) Linear sweep voltammetry of NiO/Ni-CNT and Pt/C in (g) 1 M KOH (h) NaHCO3-Na2CO3 buffer (pH = 10.0) and (i) potassium borate buffer (pH = 9.5).94 (j–l) OER theoretical analysis of IrOx/SrIrO3 heterostructure. (j) Theoretical overpotential volcano plot with O* and OH* binding energies as descriptors, using the scaling relationship between OH and OOH. (k) Visual representation of IrOx and SrIrO3 surfaces used for the DFT calculations. (l) OH* and OOH* binding energies (black circles) overlaid on the universal scaling relationship (gray line with shaded uncertainty) that has been found for many transition metals and transition metal oxides.109 | ||
Fabricating heterostructures based on metal oxides is also an efficient strategy to obtain remarkable OER electrocatalysts. Specifically, the metallic compounds with good conductivity and large surface area are intensively utilized as the support to improve the electron transfer process and expose active sites. For example, metallic CoSe2 nanobelts were integrated with other metal oxides as a highly active and stable electrocatalyst for OER.100,101 In addition, the OER activity of pristine metal oxide is often hampered by its poor OER kinetics and mass-transfer ability while fabricating heterostructure with other metal oxides/hydroxides is proved to be an efficient strategy to optimize the energy barriers of intermediates and thus boost the catalytic activities.102–104 Cerium(IV) oxide (CeO2) has reversible surface oxygen ion exchange, good electronic/ion conductivity and oxygen storage capacity, and these properties are expected to enhance the catalytic performance of the electrocatalysts by altering the electrochemical reaction kinetics. For instance, electrodeposited FeOOH/CeO2 hybrids were explored as efficient electrocatalysts for OER, exhibiting the synergistic effects between two compounds.105 According to DFT calculations, the free energy changes of intermediates and products were lower than those free energy for CeO2 and FeOOH. The adsorption energy of OH− on the heterostructure was lower than those on individual FeOOH and CeO2 surfaces, which indicated that the OH− anions tended to be adsorbed on FeOOH/CeO2 more efficiently, and this trait endowed the FeOOH/CeO2 with superior catalytic activity for OER. The authors also believed that the strong electronic interaction improved durability since this heterostructure owned great stability of 50 h under 80 mA cm−2. Additionally, CeO2/Ni-TMO hybrid and CeO2/CoSe2 hybrid have also been recently fabricated, exemplifying the efficacious heterostructure methodology for high-performance electrocatalyst development.106,107
The heterostructure with abundant interfaces is also illustrated to improve charge transfer and to facilitate electrochemical turnover. For instance, ultrafine NiO nanosheets stabilized by TiO2 with abundant interfaces were fabricated by annealing a monolayer layered NiTi LDH precursor.108 It has been previously demonstrated that NiO nanosheets with a high proportion of exposed (110) facets presented efficient catalytic performance. The overall enhanced OER activity is originated from three contributing aspects. Firstly, for 3d transition metal-based electrocatalysts, the one with a surface cation eg orbital occupancy approaching unity, shows the best performance for OER. Different from Ni2+ atoms in the hexagonal ultrathin bulk NiO with t2g6eg2 electron configuration, the Ni2+ atoms on the ultrafine and ultrathin NiO nanosheet were partially oxidized to Ni3+ with t2g6eg1 electronic configuration. The Ni3+ atoms highly exposed on (110) facets were stabilized by TiO2 and acted as active sites with higher OER activity. Secondly, according to the DOS and the partial DOS (PDOS) results, the Ni/TiO2 heterostructure is featured with a continuous DOS around the Fermi level, strongly indicating a high carrier concentration and good electrical conductivity. Thirdly, the adsorption energy of H2O for this kind of heterostructure experienced a dramatic increase compared with other materials leading to the significantly enhanced OER activity. It is noteworthy that the heterostructure might form during the electrochemical OER. Thus extra attention is required when investigating the reaction mechanism and identifying the possible active sites. Seitz et al. thoroughly studied the formation of IrOx/SrIrO3 heterostructure in the OER test, during which Sr ions leached from the surface and minor surface rearrangement occurred.109 The authors utilized DFT calculations to examine the activity and stability of catalyst with possible heterostructures, such as IrO2-anatase/SrIrO3, IrO4/SrIrO3, IrO3/SrIrO3 with different layer thicknesses (Fig. 9k).109 Theoretical results indicated that IrO2-anatase/SrIrO3 and IrO3/SrIrO3 with larger IrO3 layer thickness owned both good activity and stability. The OER performance of IrOx/SrIrO3 heterostructure is outstanding with a small overpotential of 270 to 290 mV (at 10 mA cm−2), which was very near to the peak of the OER volcano. Further AFM and XPS characterizations also proved that Sr leached during OER test and thus IrOx film in situ formed as active sites. Besides the metal compound hybrids, integrating the metal oxide catalysts with carbon-based materials can also enhance electrochemical reaction kinetics and dramatically boost activity through tailoring the adsorption energy of intermediates on catalysts. Li et al. fabricated ultrathin edge-rich FeOOH@carbon nanotubes for efficient OER.110 The carbon nanotubes not only improved the electron transfer, but also promoted the electrochemical oxidation of oxygen species. Specifically, due to the p, π-conjugation effect of carbon atoms in graphene, oxygen atoms in FeOOH decreased the density of electron clouds around the Fe atoms. Herein, hybrid to carbon materials will enhance the adsorption of reaction intermediate on central Fe atoms, and their good electronic conductivity can also contribute to improved OER performance. However, since oxidation of carbon may occur under the OER condition, it is important not to mistake the oxidation of carbon as the OER activity.
To sum up, the synergetic effects of heterostructure can be categorized into three aspects. Firstly, the electronic structures of each composite can be efficiently modulated upon heterostructure formation. Thereby, the disassociation of water, the adsorption and desorption of the intermediates and the overall intrinsic electrochemical reaction kinetic process on the active sites are tuned and facilitated. Secondly, the heterostructures combine the unique advantages from each component and provide dual active sites at the interface, which has contributed to the excellent electrochemical performance. The support materials, especially the ones with high conductivity, can also facilitate charge transfer. Finally, some supports may mediate the growth of loaded catalytically active materials and hinder agglomerations of the nanostructured catalysts, which will contribute to higher durability. Because of the aforementioned multiple merits, heterostructure engineering has been one of the primary strategies toward the development of catalysts with high activity and excellent stability.
3.4 Heteroatom doping
With the development of nanostructure engineering in recent decades, controllable introduction of exotic atoms into a pristine atomic structure (doping) has been proven to be significantly effective in boosting the catalytic activity of active site. Depending on the type of foreign atoms, the elemental doping can be divided into cation atom doping and anion atom doping. When metal oxides are doped with foreign ions, these foreign atoms are injected into the lattice and interacted with metal oxides. The strong interactions between these dopants and the metal oxides matrix efficaciously alter the electronic structure of the oxides, which also greatly influence the electrocatalytic performance. Moreover, introduction of proton acceptors has been proved to be effective method to produce OER electrocatalysts beyond the top of volcano in AEM.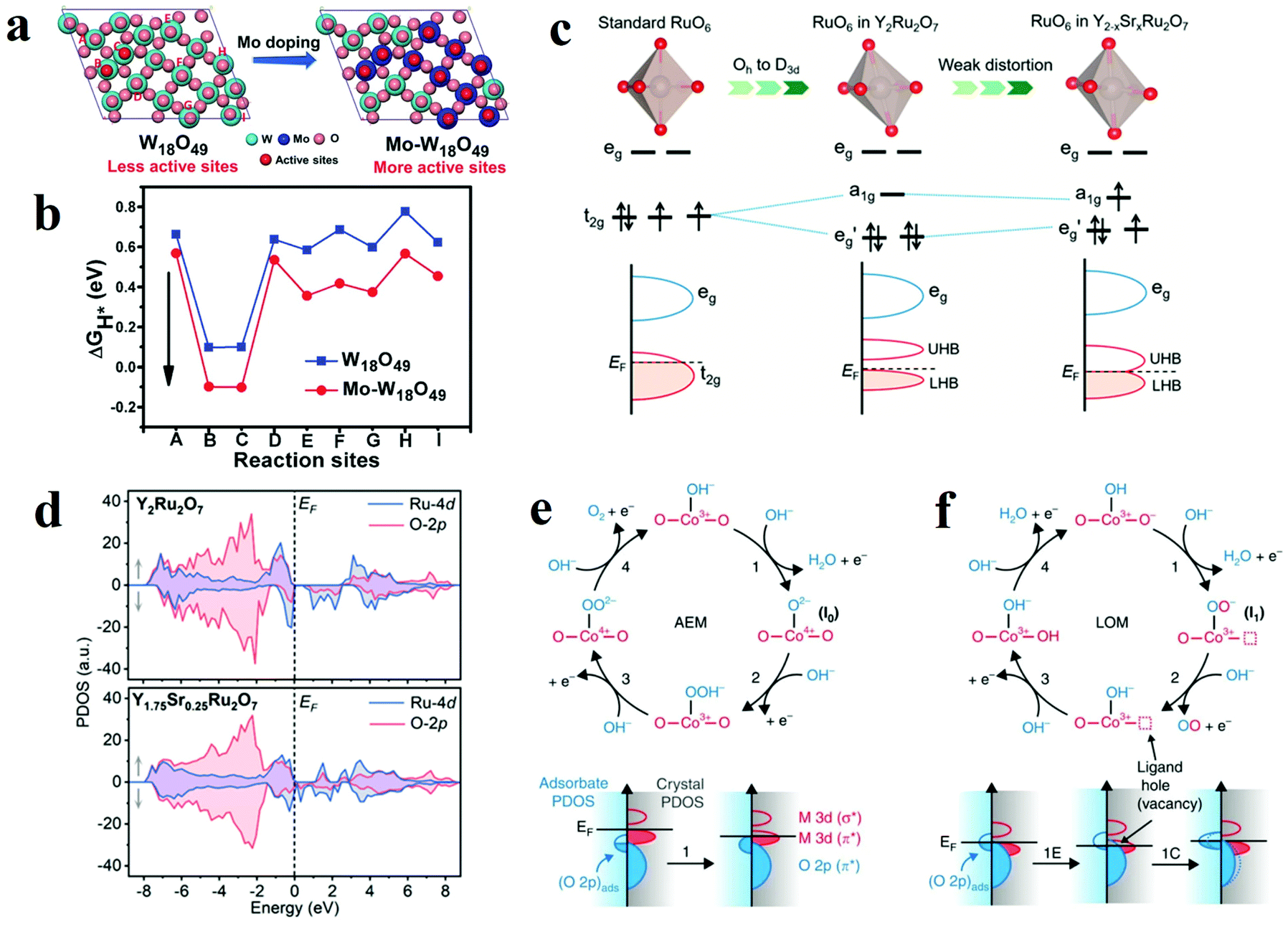 | ||
| Fig. 10 (a) Different oxygen sites on W18O49 (010) and Mo–W18O49 (010). (b) The calculated Gibbs free energy of HER on different oxygen sites of W18O49 (010) and Mo–W18O49 (010).113 (c) Schematic illustrations of different RuO6 along with the splitting of Ru 4d orbitals. (d) Schematic evolution of band alignments through Sr2+ substitution for Y3+ in Y2Ru2O7.127 (e and f) Relationship between oxygen vacancy concentration and Co–O bond.115 | ||
For OER, although the role of cation dopant is still unclear in improving the electrochemical reaction, cation atoms doping has been indicated to efficiently lower the kinetic energy barrier of the water dissociation step and desorption of the formed OH− from the surface.114–116 Recently, researches about cation doped metal oxides, such as ZnxCo3−xO4 nanoarrays, Fe doped BaCo0.9Sn0.1O3−δ and Na1−xNiyFe1−yO2, demonstrated the importance of foreign cations in enhancing OER performance of metal oxides.117–119 Fominykh et al. compared the OER activity of the NiO samples with different Fe dopant concentrations.120 Fe0.1Ni0.9O was found to demonstrate the highest electrocatalytic performance towards OER with an overpotential of 297 mV at a current density of 10 mA cm−2. The Fe3+ doped nickel oxide for significantly enhanced OER has been well studied, and theoretical calculation demonstrated that Fe3+ sites provided optimal binding energy for the OH* and OOH* intermediates and worked as the active site other than Ni3+ sites. Meanwhile, through a in situ XAS experiment, Mukerjee et al. confirmed Fe substitutes stabilized Ni in +2 oxidation state and acted as the active sites for OER.121 However, Li et al. utilized the Lewis acid–base theory to study the role of Fe dopant and they observed that Fe3+ can also function as a Lewis acid and promoted the formation of Ni4+ resulting to an improved catalytic performance.122 The Fe dopant influences the Ni valency, and the formal oxidation state of Ni(IV)-oxo had a character of Ni(III)–O* resonance with an increased covalency. The oxyl radical character played a key role as oxygen radicals in O–O bond formation according to the proton-coupled electron transfer (PCET) mechanism. The research highlighted the role of Fe dopant in boosting OER activity in metal oxide catalyst. Huang and co-worker used Fe dopant to improve the OER performance of NiCo2O4 spinel materials.123 XAS studies showed that Fe ions mainly occupied tetrahedral site of spinel lattice and induced more Ni3+ and Co2+. The increased ratio of Ni3+/Ni2+ and decreased ratio of Co3+/Co2+ made the eg occupancies of Ni and Co closer to unity, respectively, which was the optimal eg filling for OER. Cation doping is also effective in promoting OER performance of perovskite materials. Shao-Horn's group found that the optimum eg filling should be ∼1.2 to achieve the top of the volcano plot.46 Cation doping can finely tune the eg filling of perovskites to achieve better performance towards OER. Based on this guidance, SrNb0.1Co0.7Fe0.2O3, CaCu3Fe4O12 perovskites were explored as catalysts with good activity and stability towards OER, which were comparable or even better than state-of-art OER catalysts.124,125 Xu's group also investigated the influence of Fe dopant on the OER performance of LaCoO3.126 With 10% Fe substitution, the spin state of Co3+ was transformed from a low spin state (LS: t2g6eg0) to a higher spin state, altering the catalyst characteristics from insulator to half-metal. Moreover, the change also increased overlap between Co 3d and O 2p state. Thus, M–O covalency was enhanced, which finally facilitated the OER activity. Zhang et al. improved the OER performance of pyrochlore ruthenate Y2Ru2O7 in acidic medium by substituting Y3+ with Sr2+.127 The mass activity was 1018 A gRu−1 at an overpotential of 300 mV in 0.5 M H2SO4 electrolyte. Compared to standard RuO6 octahedra, the RuO6 octahedra in Y2Ru2O7 are severely distorted, and the 4d electrons of Ru in Y2Ru2O7 are distributed into a lower Hubbard band (LHB), resulting in the upper Hubbard band (UHB) empty. This Mott–Hubbard splitting phenomenon impedes the charge transfer between active sites and intermediates, resulting in an inferior OER performance. For the Y1.7Sr0.3Ru2O7 catalyst, the exotic Sr2+ cations on one hand can regulate coordination geometry and charge redistribution. From the results of XRD Rietveld refinement and Raman spectra, the bond angle of Ru–O–Ru increased from 128.2° to 129.8° after Sr substitution, which alleviated the distortion of RuO6 moieties and improved the overlap of Ru 4d and O 2p orbitals (Fig. 10c).127 On the other hand, the Sr substitution also increased the Ru valence and facilitated the charge transfer from oxygen ligand to Ru centre, which also resulted in enhanced orbital hybridization and bond covalency (Fig. 10d).127
It should be noticed that substitution is the effective method to produce OER electrocatalysts beyond the top of the volcano in AEM. For example, substitution by metals with higher electronegativity can act as proton acceptors. Halck et al. utilized the incorporation of Ni and Co into RuO2 surface to improve the OER performance.128 According to the reaction pathway and scaling relationship in AEM, the smallest overpotential for OER would be 0.4 V. However, after substitution of Ru by Ni and Co, the oxygen atoms on the bridge positions were activated and acted as a proton acceptor. Unlike the traditional scaling relation of HOO* and HO*, the adsorption free energy difference between HOO* and HO* was changed. As a result, the overpotentials for Co- and Ni-substituted RuO2 were only ca. 0.1 V and 0.25 V, respectively. In addition, cation substitutions can also promote OER with the LOM mechanism. Stevenson et al. first proposed the LOM mechanism by using Sr substitution into La1−xSrxCoO3−δ.115 The covalency of the Co–O bond and the concentration of oxygen vacancies can be regulated by controlling Sr substitution (Fig. 10e and f).115 It was demonstrated that the transition from AEM to LOM occurred when x ≥ 0.4. With increasing Sr substitution, the oxidation state of Co was also increased, which led to a more significant overlap between Co 3d and O 2p orbitals and the formation of π* and σ* bands. When the overlap was great enough, ligand holes (oxygen vacancies) were formed, and the Co 3d π* band cannot be treated as an energy level isolated from the oxygen 2p π* band. Therefore, the Fermi energy can be modulated by controlling Co–O covalency, and thus Sr substitution provided the opportunity for lattice oxygen redox activity. The formation of superoxide-like –OO via the coupling of lattice oxygen and adsorption oxygen finally resulted in fast OER kinetics.
In 2017, Xiao et al. developed a facile strategy to fill the oxygen vacancies in the Co3O4 with phosphorus by thermal coupled plasma modification approach.129 The electrocatalytic performance of P-Co3O4 for HER and OER was much better than these of pristine Co3O4 and VO-Co3O4 (VO = oxygen vacancies). The overpotential of overall water splitting was only 420 mV at 100 mA cm−2 in 5 M KOH electrolyte. The electronic properties of the P-Co3O4 have been investigated through both theoretical calculation and experiments. By comparing XAS results of P-Co3O4 and VO-Co3O4, P dopants were confirmed to fill into the O-vacancies. When VO was formed, electrons transferred into Co 3d orbitals, preferably forming Co3+Oh, instead of Co2+Td. While with P dopants in the O-vacancy site, electrons transferred out of Co 3d states, and thereby the number of Co2+Td sites (with better HER and OER catalytic activity) increased. More importantly, P-filling can also significantly improve the intermediate binding, hence remarkably boost the HER and OER activity. According to the calculated free energy diagram for HER, P-Co3O4 has a favourable ΔGH* of −0.08 eV. The free energy profiles along the OER process also demonstrated enhanced binding strength of O* and OOH* intermediates on P-Co3O4. Co3O4 with P-doping showed superior OER performance to pristine Co3O4. Nitrogen doped molybdenum trioxide (N-doped MoO3) was fabricated by heating commercial MoO3 at NH3 atmosphere and exhibited 6 times higher activity towards HER than intact MoO3.130 Positron annihilation spectrometry (PAS) confirmed that the N-doping induced increased concentration of VOVO divacancies, which might be the active sites for HER. Further DFT calculations verified that the existence of VOVO divacancies improved the state density near the valence band edge and thereby boosted the catalyst's electronic conductivity.
Anion doping can be facile to improve electrocatalytic performance of metal oxides in terms of activity and durability of OER. Through solid-state reaction, P doped SrCoO3−δ (SCP) perovskite has been fabricated and showed high OER performance benefiting from its high electrical conductivity and large amount of OO2−/O− species.131 The doping of high-valence-state P5+ in SCP resulted in charge compensation and boosted the electrical conductivities. Importantly, compared to SrCoO3, P doped SrCoO3−δ showed better durability which can be attributed to the stable tetragonal structure after P-doping. Controllable nitrogen doped cobalt oxides (N-Co3O4 and N-CoO) were synthesized from cobalt-alanine complexes by calcination at different atmosphere.132 The authors highlighted the OER performance of N-Co3O4, which required overpotential of only 190 mV at 10 mA cm−2 and showed the smallest Tafel slope of 29.8 mV dec−1 in 1.0 M KOH. The authors, through DFT calculations, claimed that N-doping can not only improve the OH-adsorption capability, but also facilitate OH-cleavage. Moreover, N-doping also reduced the bandgap from 1.80 eV to 1.42 eV, which made the excitation of charge carriers to the conduction band easier and improve the electronic conductivity. Sulphur doped cobalt oxide (CoO0.87S0.13/GN) was also investigated to explore the benefit of anion doping to the electrochemical reaction performance.133
It is acknowledged that the heterogeneous electrocatalytic process only occurred on the surface of the electrocatalysts, and the activities of the electrocatalysts are determined by their size and the accessible active sites. Therefore, preparing electrocatalyst in atomic scale will contribute to the enlarged exposure of the active sites and thus enhance electrochemical activity. Recently, Dou et al. prepared atomic-scale CoOx species through on-site transformation of the atomically distributed Co2+ in ZIF-67 by O2 plasma treatment.134 Benefiting from the large surface area and abundant exposed active species, the electrocatalyst exhibited remarkable OER performance.
In addition, metal oxide is usually utilized as the support to promote the distribution of the electrocatalytic active species. Especially used for selective activation of methane or CO oxidation, various single atom noble metals supported on metal oxide are intensively explored with remarkably enhanced catalytic performance.135,136 More recently, Nong et al. prepared Ru doped TiO2, in which the penta-valence Ru atom was homogeneously distributed in the lattice.137 The experiment and PDOS calculation confirmed that the Ru5+ existed in a row of –RuO3 in the surface of TiO2, which was defined as TiO2:Rusurf. According to the DFT calculation, the adsorption of hydrogen is preferred on the TiO2:Rusurf structure with increase of hydrogen coverage and the calculated ΔGH on these sites was −0.28 eV, suitable for HER.
To sum up, elemental doping can be a facile and effective method in boosting the electrochemical performance of metal oxides. Dopants can regulate the charge transfer properties and intermediate adsorption energy by modifying the bulk electronic structure, spin state of metal site, and M–O covalency. Besides, some dopants, especially the metals with higher electronegativity, can serve as proton acceptor. These dopants can introduce lattice oxygen, and thus produce efficient OER catalyst following LOM instead of AEM, providing opportunities to break the scaling relation. Finally, some dopants are the efficient active sites by themselves, especially for cases of cation doping and single atom doping.
3.5 Defect and strain engineering
Atomic discontinuity in nanoscale in metal oxides, metal sulphides and metal selenides have demonstrated as active sites. Thus defect, and strains enriched metal related compounds have been applied to various electrochemical reaction.138–140 | ||
| Fig. 11 (a and b) Plausible reaction mechanism and DFT calculation results of electrocatalytic H2 evolution on WO2.9.143 (c) The projected density of states (PDOS) on pristine CoO and (d) CoO with O-vacancies.144 (d–f) Computational predictions for the strain effect on the HER activity of CoO.146 (d) Schematic illustration of H2O adsorption and dissociation on the CoO (111) surface with O-vacancies (e) hydrogen adsorption free energy, ΔGH*, vs. tensile strain for the CoO (111)-Ov surface. (f) Schematic illustration of the effect of strain on the electronic structure of (111)-Ov surface of CoO. | ||
In general, defect engineering in metal oxides mainly focus on the control over oxygen vacancies and metal site vacancies. By tuning vacancies, the charge transfer ability and adsorption energy of reactants and intermediates are optimized, thus, the intrinsic catalytic activity of active site is improved. In addition, when the defects accumulate to an exact extent, amorphization may be introduced, and more active sites can be exposed. Strain engineering can also control the vacancies and achieve similar results of anion defect engineering. Also, expansion and compression of catalyst lattice allows modulation in electronic structure towards optimal catalytic activity.
3.6 Amorphization
Amorphization has been proved to be an effective and general strategy to engineer the surface structure of catalysts. The strategy has been widely used in facilitating a variety of catalysts, such as transition metal oxides, sulfides and phosphides.151–153 Partial and complete loss of long-range crystalline structure in to disordered phases will expose more catalytic active site and modulate the electron structure.Indra et al. compared the electrochemical catalytic activity of amorphous and crystalline cobalt iron mixed metal oxides for OER and ORR.154 It was found that the amorphous CoFe2O4 can achieve a current density of 10 mA cm−2 with a small overpotential of 490 mV for OER, which was 70 mV lower than that of crystalline one. The authors considered that the better activity was from amorphous surface which exposed more Co3+ active sites and owned higher specific surface area. In 2014, Geng's group reported an aerosol-spray-assisted fabrication of amorphous metal oxides with precise control over compositions and structures.155 Among all the Fe–Ni–Ox prepared, Fe6Ni10Ox showed the best performance for electrochemical water oxidation, with a low overpotential of 0.286 V at 10 mA cm−2 under alkaline conditions. The effect of crystalline phase on electrochemical water oxidation by calcining the samples in different temperatures was further investigated. The results showed that the superior electrochemical catalytic activity of amorphous phase derived from its larger capacitance, which indicated the amorphous phase possessed a larger ECSA. Other authors also drew the similar conclusion that the large surface specific area of amorphous phase was the origin of improvement in electrochemical catalytic performance for OER.156,157
In general, compared with the crystalline phase, the amorphous phase obtains a larger surface specific area and thereby exposes more active sites.156 Besides, the amorphous surface possesses a high concentration of active sites, which play an important role in enhancing the adsorption of reactants and optimizing the coverage of intermediates.158
3.7 In situ transformation
Recently, metal phosphides, sulfides, and selenides etc. are reported as extraordinary catalysts towards water-splitting.159–161 However, one should be very careful when determining the real active phase especially under OER conditions, since these electrocatalysts often experience complete or partial oxidation, and could be transformed into inhomogeneous materials when OER occurs.162 Therefore, composition and structure studies on these catalysts via ex situ method may not accurate when identifying the active phases, and it should be more appropriate to name these catalysts as pre-catalyst. The active phases are usually composed of oxides, which are formed under OER conditions and lead to an enhancement in catalytic performance. Overall, in situ transformation is a useful method to obtain metal oxides from phosphides, nitrides, sulfides, and selenides etc.Hu's group reported Ni2P/NiOx catalysts with the core–shell structure derived from Ni2P particles towards overall water-splitting.163 The Ni2P/NiOx delivered a current density of 10 mA cm−2 with an overpotential of only 290 mV in alkaline electrolyte. The partial oxidation of Ni2P generated NiOx shell, which served as the active part towards OER. The Ni2P core acted as conducting support and provided favourable electron transfer pathway to the NiOx. The author declared there might be synergistic effects between these two parts, which needed further study. In another work, NiFeOx nanosheet was in situ transformed from NiFeSe2 nanoplate by implying a galvanostatic scan of 5 mA cm−2.164 The as-prepared catalyst exhibited a small overpotential of 195 mV at 10 mA cm−2 and a good stability of 24 h under 10 mA cm−2. From TEM images and EDS mapping results, the precursor nanoplate NiFeSe2 was entirely converted into NiFeOx, and the obtained oxide maintained the nanosheet morphology with only 1–2 nm thickness. Benefiting from the ultrathin nanosheet morphology and Ni–Fe synergic effect, the NiFeOx outperformed NiFe LDH and even other benchmark catalysts. In addition, Cui et al. utilized in situ oxidation to develop binary, ternary, and quaternary transition metal oxide from sulfides.165 Impressively, CoNiFeOx grown on carbon fibre exhibited a small overpotential of 232 mV at reach 10 mA cm−2 and extraordinary durability of more than 100 h, outperforming most of other non-noble OER catalysts.
Overall, in situ transformation is an effective method to obtain metal–oxide-based catalysts with good performance. Firstly, the metal oxides normally possess analogous morphology to their pre-catalyst counterparts, and the specific morphology would substantially boost the electrocatalytic activity. Secondly, partial in situ oxidation may produce specific core–shell heterostructures, and the synergic effect between these two different parts may be beneficial to the enhanced overall electrocatalytic performance. In addition, the anion elements of the pre-catalysts inevitably undergo leaching into the electrolytes. Thus, the resultant true electrocatalysts are endowed with higher specific areas and more exposed active sites which benefit the electrocatalytic turnovers.
Taken together, this part of the review has surveyed strategies towards modulations of the morphology, composition, and structure of metal oxide electrocatalysts. A summary of the high-performance metal oxide electrocatalysts towards water splitting developed by these strategies is given in Table 1. Generally, by tuning the electronic structures and surface properties, these strategies can boost the intrinsic activity of each active site, improve the number of active sites, and optimize the charge transfer process.
| Strategy | Electrocatalyst/substratea | Mass loading (mg cm−2) | Reaction | Electrolyte | η 10 (mV) | Tafel slope (mV dec−1) | Stability | Ref. |
|---|---|---|---|---|---|---|---|---|
| a CFP = carbon fibre paper; GC = glassy carbon; FTO = fluoride doped tin oxide; CNT = carbon nanotube; NC = nitrogen-doped carbon. | ||||||||
| Nanoscale confinement | NiFeOx nanoparticles/CFP | 3 | HER | 1 M KOH | 88 | 150 | 100 h (@10 mA cm−2) | 73 |
| OER | 230 | 31.6 | ||||||
| Water splitting | 280 | N.A. | ||||||
| LiCo0.33Ni0.33Fe0.33O2 nanoparticles/CFP | ∼0.1 | OER | 0.1 M KOH | 340 | 73.6 | 1000 cycles | 74 | |
| 80 nm-LaCoO3/GC | 0.25 | OER | 0.1 M KOH | 490 | 69 | 3 h (@10 A g−1) | 75 | |
| Co3O4 nanosheets/GC | 0.34 | OER | 1.0 M KOH | 790 (@341.7 mA cm−2) | 25 | 10000 cycles |
76 | |
| γ-CoOOH nanosheets/GC | ∼0.15 | OER | 1.0 M KOH | 300 | 38 | 13 h (@10 mA cm−2) | 77 | |
| Co3O4 microtubes/Ni foam | N.A. | HER | 1.0 M KOH | 170 | 98 | 12 h (@20 mA cm−2) | 79 | |
| OER | 260 (@150 mA cm−2) | 84 | ||||||
| Water splitting | 400 | N.A. | ||||||
| Co3O4 nanotubes/GC | 0.208 | OER | 0.1 M KOH | 390 | 76 | 2000 cycles/40 h (@20 mA cm−2) | 80 | |
| NiCo2O4 hollow microcuboids/Ni foam | ∼1 | HER | 1.0 M NaOH | −110 | 49.7 | 36 h (@20 mA cm−2) | 81 | |
| OER | 290 | 53 | ||||||
| Water splitting | 420 | N.A. | ||||||
| Ni–Co2–O hollow nanosponges/GC | 0.2 | OER | 0.1 M KOH | 362 | 64.4 | 500 cycles | 82 | |
| Crystal phase and facet engineering | γ-Ti2O3 film/(001)SrTiO3 | N.A. | HER | 0.5 M H2SO4 | 271 | 199 | N.A. | 85 |
| MnOx film/FTO or Au | N.A. | OER | 1.0 M NaOH | N.A. | 63 | N.A. | 86 | |
| Co3O4@CoO nanocubes/GC | ∼0.025 | OER | 0.5 M KOH | 430 | 89 | 1000 h (@8 mA cm−2)/6000 cycles | 88 | |
| 0.5 M Na2SO4 | 851 | 375 | ||||||
| BaNi0.83O2.5/GC | 0.295 | OER | 0.1 M KOH | ∼380 | N.A. | 1000 cycles | 89 | |
| (110) NiCo2O4 nanosheets/Ni foam | 1.12 | HER | 1.0 M KOH | 157 (@5 mA cm−2) | 71.2 | 50 h (@5 mA cm−2) | 90 | |
| OER | 330 (@5 mA cm−2) | 59.2 | ||||||
| Water splitting | 360 | N.A. | ||||||
| (112) Co3O4 nanoparticles/GC | 0.15 | OER | 0.1 M KOH | 380 | 62 | ∼7 h(@1 mA cm−2) | 91 | |
| (012)-O α-Fe2O3/CFP | 0.014 | OER | 1.0 M NaOH | 317 | 58.5 | 30 h (@10 mA cm−2) | 92 | |
| LaCoO3 (100) film/GC | N.A. | OER | 1.0 M KOH | 470 | 180 | 20000 s |
93 | |
| Heterostructure | Ni(OH)2-Pt/GC | N.A. | HER | 0.1 M KOH | 100 | 100 | N.A. | 30 |
| NiO-Ni/CNT | 0.28 | HER | 1.0 M KOH | <100 | 82 | 20 h (@−20 mA cm−2) | 94 | |
| Ni@Cr2O3-NiO/Ni foam | 8 | HER | 1.0 M KOH | 115 (@100 mA cm−2) | N.A. | 80 h (@−200 mA cm−2) | 95 | |
| CoOx@CN/GC | 1 | HER | 1.0 M KOH | 232 | 115 | 9000 s (@10 mA cm−2) | 96 | |
| OER | 260 | N.A. | ||||||
| Water splitting | 320(@20 mA cm−2) | N.A. | ||||||
| Ni(OH)2-Pt/GC | N.A. | HER | 0.1 M KOH | 100 | 100 | N.A. | 30 | |
| NiO-Ni/CNT | 0.28 | HER | 1.0 M KOH | <100 | 82 | 20 h (@−20 mA cm−2) | 94 | |
| Ni@Cr2O3-NiO/Ni foam | 8 | HER | 1.0 M KOH | 115 (@100 mA cm−2) | N.A. | 80 h (@−200 mA cm−2) | 95 | |
| CoOx@CN/GC | 1 | HER | 1.0 M KOH | 232 | 115 | 9000 s (@10 mA cm−2) | 96 | |
| OER | 260 | N.A. | ||||||
| Water splitting | 320(@20 mA cm−2) | N.A. | ||||||
| Ni/NiO-NSAs/Ni foam | 0.59 | HER | 0.1 M KOH | ∼230 | 114 | 10 h | 97 | |
| Ni-CeO2/CNT | 0.14 | HER | 1.0 M KOH | <100 | N.A. | 10 h(@−0.153 V) | 98 | |
| Co-Co3O4/Ni foam | 0.85 | HER | 1.0 M KOH | 90 | 44 | 6000 s (@−0.120 V) | 99 | |
| Mn3O4-CoSe2/GC | ∼0.2 | OER | 0.1 M KOH | ∼350 | 49 | 12000 cycles |
100 | |
| CoO-CoSe2/Ti | ∼2 | HER | 0.5 M PBS | 337 | 131 | 10 h | 101 | |
| OER | 510 | 137 | ||||||
| Water splitting | 950 | |||||||
| Mn3O4@MnxCo3−xO4/Ni foam | 0.3 | OER | 1.0 M KOH | 246 | 46 | 40 h | 102 | |
| Co3O4-NiCo2O4/Ni foam | 1 | OER | 1.0 M KOH | 340 | 88 | 12000 cycles |
103 | |
| NiO-Co3O4/NC | 0.2 | OER | 1.0 M KOH | 240 | 73 | 48 h | 104 | |
| FeOOH-CeO2/Ni foam | N.A. | OER | 1.0 M NaOH | ∼240 | N.A. | 50 h (@80 mA cm−2) | 105 | |
| CeO2-CoSe/GC | 0.2 | OER | 0.1 M KOH | 288 | 44 | 10 h (@10 mA cm−2) | 106 | |
| CeO2-TMO/Ni | N.A. | HER | 1.0 M KOH | 93 | 69 | 30 h (@10 mA cm−2) | 107 | |
| OER | 220 | 38 | ||||||
| Water splitting | 350 | N.A. | ||||||
| NiO/TiO2 | 0.34 | OER | 1.0 M KOH | 320 | 52 | 10 h | 108 | |
| IrOx/SrIrO3 film | N.A. | OER | 0.5 M H2SO4 | 270 | N.A. | 30 h (@10 mA cm−2) | 109 | |
| FeOOH/CNTs | 0.05 | OER | 1.0 M KOH | 206 | 31 | 10 h (@10 mA cm−2) | 110 | |
| Heteroatom doping | Pr0.5(Ba0.5Sr0.5)0.5Co0.8, Fe0.2O3−δ/GC | 0.232 | HER | 1.0 M KOH | 237 | 45 | 25 h (@−50 mA cm−2) | 111 |
| Ni,Zn-CoO/CFP | N.A. | HER | 1.0 M KOH | 53 | 47 | 24 h (@−10 mA cm−2, 6 M KOH) | 112 | |
| Mo–W18O49/GC | 0.16 | HER | 0.5 M H2SO4 | 45 | 54 | 10 h (@−0.14 V) | 113 | |
| Sr0.90Na0.10RuO3/GC | 0.1 | OER | 0.1 M HClO4 | 170 | 40 | N.A. | 114 | |
| La1−xSrxCoO3−δ/GC | 0.051 | OER | 0.1 M KOH | ∼370 | 31 | 10 h (@10A g−1) | 115 | |
| LiCo0.8Fe0.2O2/GC | 0.232 | OER | 0.1 M KOH | 340 | 50 | 6 h (@10 mA cm−2) | 116 | |
| Na1−xNiyFe1−yO2/GC | 0.13 | OER | 1.0 M KOH | 260 | 44 | 30 h (@10 mA cm−2) | 117 | |
| ZnxCo3−xO4/Ti | N.A. | OER | 1.0 M KOH | 320 | 51 | ∼2 h (@1.63 V) | 118 | |
| BaCo0.9−xFexSn0.1O3−δ/GC | 0.232 | OER | 0.1 M KOH | ∼390 | 69 | 2 h (@5 mA cm−2) | 119 | |
| Fe0.1Ni0.9O/Au | N.A. | OER | 0.5 M KOH | 297 | 37 | 10 h(@10 mA cm−2) | 120 | |
| Ni0.8Fe0.1Co0.1O/GC | 0.25 | OER | 0.1 M KOH | ∼410 | N.A. | N.A. | 121 | |
| Fe-NiCo2O4/FTO | N.A. | OER | 1.0 M NaOH | 201 | 39 | N.A. | 122 | |
| SrNb0.1Co0.7Fe0.2O3/GC | 0.232 | OER | 0.1 M KOH | 420 | 76 | 1000 cycles | 124 | |
| CaCu3Fe4O12/GC | 0.25 | OER | 0.1 M KOH | 310 | 51 | 100 cycles | 125 | |
| Y2−xSrxRu2O7/GC | 0.071 | OER | 0.5 M H2SO4 | 264 | 44.8 | 28 h (@−10 mA cm−2) | 127 | |
| P-Co3O4/Ti mesh | N.A. | HER | 1.0 M KOH | 120 | 52 | ∼2 h (@1.64 V) | 129 | |
| OER | 280 | 51.6 | ||||||
| N-MoO3/GC | 0.694 | HER | 0.5 M H2SO4 | 210 | 101 | 1000 cycles | 130 | |
| SrCo0.95P0.05O3−δ/GC | 0.232 | OER | 0.1 M KOH | 480 | 84 | 1000 cycles | 131 | |
| N-Co3O4/Ni foam | 0.25 | OER | 1.0 M KOH | 190 | 30 | 1.11 h | 132 | |
| CoO0.87S0.13/graphene nanomesh | 0.36 | OER | 0.1 M KOH | 360 | N.A. | 3000 cycles | 133 | |
| CoOx-ZIF/GC | ∼0.2 | OER | 1.0 M KOH | 318 | 70.3 | 2000 cycles | 134 | |
| TiO2:Ru/GC | ∼0.2 | HER | 0.1 M KOH | 150 | 97 | 1000 cycles | 137 | |
| Defect and strain engineering | Co3−xO4/GC | N.A. | OER | 1.0 M KOH | 268 | 38.2 | 2000 cycles | 141 |
| Sn1−xCo0.9Fe0.1(OH)6/GC | ∼0.1 | OER | 1.0 M KOH | 270 | 42.3 | 2000 cycles | 142 | |
| WO2.9/GC | 0.285 | HER | 0.5 M H2SO4 | 70 | 50 | ∼4 h (@−1.64 V) | 143 | |
| CoO/CFP | ∼0.19 | OER | 1.0 M KOH | 330 | 44 | N.A. | 144 | |
| 3.0% S-CoO/CFP | ∼0.46 | HER | 1.0 M KOH | 73 | 83 | 28 h (@−0.073 V) | 146 | |
| 4.2% S-SrCoO3−δ/GC | N.A. | OER | 0.1 M KOH | ∼330 | 40 | ∼3 h (@5 μA) | 147 | |
| S-LaNiO3/GC | N.A. | OER | 0.1 M KOH | ∼350 | N.A. | N.A. | 150 | |
| Amorphization | IrOx/FTO | 0.0001 | OER | 1.0 M H2SO4 | 190 | 34 | 24 h (@1 mA cm−2) | 151 |
| CoFeOx/GC | 0.051 | OER | 0.1 M KOH | 490 | N.A. | N.A. | 154 | |
| Fe6Ni10Ox/GC | 0.1 | OER | 1.0 M KOH | 286 | 48 | N.A. | 155 | |
| VFeCoOx/GC | 0.28 | OER | 1.0 M KOH | 307 | 36 | 12 h (@−1.55 V) | 156 | |
| In situ transformation | Ni2P/NiOx/GC | 0.14 | OER | 1.0 M H2SO4 | 290 | 47 | 10 h (@10 mA cm−2) | 163 |
| NiFeOx nanosheet/Ni foam | N.A. | OER | 0.1 M KOH | 195 | 28 | 24 h (@10 mA cm−2) | 164 | |
| CoNiFeOx/carbon fibre cloth | N.A. | OER | 1.0 M KOH | 232 | 38 | 100 h (@20 mA cm−2) | 165 | |
4. Concluding remarks and perspectives
In recent years, various metal oxide materials have been successfully fabricated as high-performance electrocatalysts for water splitting. In this review, different characterization techniques and fabrication strategies for HER and OER have been systematically summarized. We highlight that the abundance, diversity, and versatility of the transition metal oxides foresees a successful application in energy conversion devices. However, limited knowledge of the actual active sites in atomic scale during the electrochemistry still pose enormous challenges in further improvement in performance. We are in belief that metal oxide electrocatalysts will fulfil their potentials by addressing, at least if not all, the following fundamentally important barriers.Firstly, although a variety of high-performance electrocatalysts have been developed, a mechanistic understanding of the surface charge transfer and catalytic sites during the electrochemical reaction is still highly demanding. Recently, some descriptors different from conventional ΔGH* and (ΔGO* − ΔGHO*) for HER and OER have been reported to evaluate the catalytic performance as well as rationally design materials. For example, the descriptor Φ considering the topological, bonding, and electronic structures of catalytic centre, the eg-filling descriptor in perovskite oxides, and the one involved d band position are applied to predict the electrocatalytic activities towards HER and OER.46,166,167 Additionally, advance in mechanistically understanding fundamental reaction steps has been realized by theoretical calculation, along with well-recognized principles providing useful avenues to study various electrochemistry towards other applications. However, mechanistic studies lack the understanding of reaction kinetics, which may need delicate experimental design. Therefore, combining with theoretical elucidations and experimental exploration, robust, high-performance electrocatalysts will be developed through effective catalyst activation strategies, such as single-atom or dual-atom sites creation, crystal phase engineering, heteroatoms doping, and interface manipulation.168–171
Secondly, the poor stability of the metal oxides electrocatalysts severely blocks their industrial application, compared with noble metal counterparts. Under long-term electrochemical test, the surface of metal oxides could undergo surface reconstruction, such as elemental enrichment or depletion.172 Meanwhile, the dissolution or deposition of electrode materials/impurities at the electrocatalyst/substrate interface may hamper the physical contact or restrict the reaction. For instance, Ru-based oxides are regarded as the most active OER materials, while they suffer from severe performance degradation due to Ru dissolution experimentally.66 Therefore, improvement in structural robustness is essential to drive HER or OER for long-term operation. Moreover, in practical application, the current density required is usually higher than 1000 mA cm−2 for proton-exchange membrane (PEM) electrolyser, requiring catalysts with robust stability up to 1000 h.173 To improve the long-term durability, incorporating molecular catalyst with carbon materials has been reported, significantly accelerating water oxidation half-reaction.174 Although endowing with many advantages, the biggest challenge of PEM is that only noble metal-based materials can be used as working electrodes so far, wherein Pt-group metals (PGM) serve as cathode and Ir or Ru-based oxides as the anode. Recently, the development of alkaline anion exchange membrane (AEM) electrolysers provides possibility for the use of PGM-free catalysts, while the limited durability remains a challenge to be solved.175 Over the last decade, alkaline membrane fuel cells (AMFCs) have been achieved with improved current densities and stability.176 An AMFC operated continuously for over 1000 h at 600 mA cm−2 has been reported by developing gas diffusion electrodes (GDL) with hydrophobic PTFE in both the GDL and catalyst layers.177 Thus, the structural integrity of the metal oxides should be assessed with a detailed investigation on the structural degradation or mechanical breakdown.178
Thirdly, accompanying with the development of theoretical methods, complementary operando characterization techniques are also highly desirable to precisely detect structures vibration and identify active sites during electrocatalysis process. Therefore, a rational design of well-defined electrochemically active components needs to comprehensively be tested with various parameters combining with in situ probing techniques for the metal oxides (as shown in Part 2). These technologies are also of paramount importance for understanding the reaction mechanism more profoundly. For example, accurate in situ detection of the atomic or electron structure could be realized by employing well-defined atomically thin electrode materials or adopting surface-sensitive probing techniques.65,179 Additionally, reactant isotope labelling experiments that utilize the kinetic isotope effects are also useful to probe the forming mechanisms of intermediates as well as rate-limiting step.180
In summary, the future industrial application of robust metal oxides for energy conversion and storage demands comprehensive advancement from techniques, theories, characterizations, and devices. We believe with a great development of these key elements, impactful breakthroughs in real-world large-scale water electrolysis are becoming indispensable in the expectable future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from Australian Research Council (Discovery Project and Discovery Early Career Researcher Award).References
- J. A. Turner, Science, 2004, 305, 972 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474 CrossRef CAS PubMed.
- J. K. Norskov and C. H. Christensen, Science, 2006, 312, 1322 CrossRef CAS PubMed.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2016, 16, 57 CrossRef PubMed.
- W. Chen, X. Hou, X. Shi and H. Pan, ACS Appl. Mater. Interfaces, 2018, 10, 35289 CrossRef CAS PubMed.
- M. P. Browne, C. Domínguez and P. E. Colavita, Curr. Opin. Electrochem., 2017, 7, 208 CrossRef.
- G. Ming, D. Y. Wang, C. C. Chen, B. J. Hwang and H. Dai, Nano Res., 2016, 9, 28 CrossRef.
- G. Zhao, K. Rui, S. X. Dou and W. Sun, Adv. Funct. Mater., 2018, 28, 180329 Search PubMed.
- Y. Zhu, Q. Lin, Y. Zhong, H. A. Tahini, Z. Shao and H. Wang, Energy Environ. Sci., 2020, 12, 3361 RSC.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404 RSC.
- M. Tahir, L. Pan, F. Idrees, X. Zhang, L. Wang, J. J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136 CrossRef CAS.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337 RSC.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748 CrossRef CAS PubMed.
- J. Song, C. Wei, Z. F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196 RSC.
- C. Hu, L. Zhang and J. Gong, Energy Environ. Sci., 2019, 12, 2620 RSC.
- L. Li, P. Wang, Q. Shao and X. Huang, Chem. Soc. Rev., 2020, 49, 3072 RSC.
- J. Wang, Y. Gao, H. Kong, J. Kim, S. Choi, F. Ciucci, Y. Hao, S. Yang, Z. Shao and J. Lim, Chem. Soc. Rev., 2020, 49, 9154 RSC.
- C. Acar and I. Dincer, Int. J. Hydrogen Energy, 2014, 39, 1 CrossRef CAS.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148 RSC.
- P. C. Vesborg, B. Seger and I. Chorkendorff, J. Phys. Chem. Lett., 2015, 6, 951 CrossRef CAS PubMed.
- C. G. Morales-Guio, L. A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555 RSC.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159 CrossRef CAS.
- F. Y. Chen, Z. Y. Wu, Z. Adler and H. Wang, Joule, 2021, 5, 1 CrossRef.
- T. Maiyalagan, K. A. Jarvis, S. Therese, P. J. Ferreira and A. Manthiram, Nat. Commun., 2014, 5, 3949 CrossRef CAS PubMed.
- J. K. Norskov, T. Bligaard, B. Hvolbaek, F. Abild-Pedersen, I. Chorkendorff and C. H. Christensen, Chem. Soc. Rev., 2008, 37, 2163 RSC.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. B. Chorkendorff and J. K. Norskov, Nat. Mater., 2006, 5, 909 CrossRef CAS PubMed.
- J. Staszak-Jirkovsky, C. D. Malliakas, P. P. Lopes, N. Danilovic, S. S. Kota, K. C. Chang, B. Genorio, D. Strmcnik, V. R. Stamenkovic, M. G. Kanatzidis and N. M. Markovic, Nat. Mater., 2016, 15, 197 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, K. C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K. C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256 CrossRef CAS PubMed.
- C. Hu, L. Zhang and J. J. Gong, Energy Environ. Sci., 2019, 12, 2620 RSC.
- C. Campos-Roldán, R. González-Huerta and N. Alonso-Vante, Electrochim. Acta, 2018, 283, 1829 CrossRef.
- D. Strmcnik, P. P. Lopes, B. Genorio, V. R. Stamenkovic and N. M. Markovic, Nano Energy, 2016, 29, 29 CrossRef CAS.
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Chem. Phys., 2005, 319, 178 CrossRef CAS.
- Z. Shi, X. Wang, J. Ge, C. Liu and W. Xing, Nanoscale, 2020, 12, 13249 RSC.
- X. Rong, J. Parolin and A. M. Kolpak, ACS Catal., 2016, 6, 1153 CrossRef CAS.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y. L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457 CrossRef CAS PubMed.
- B. Hammer and J. K. Norskov, Nature, 1995, 376, 238 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71 CAS.
- P. Rüetschi and P. Delahay, J. Chem. Phys., 1955, 23, 556 CrossRef.
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1980, 111, 125 CrossRef CAS.
- Y. Matsumoto, H. Yoneyama and H. Tamura, J. Electroanal. Chem. Interfacial Electrochem., 1977, 83, 167 CAS.
- Y. Matsumoto and E. Sato, Mater. Chem. Phys., 1986, 14, 397 CrossRef CAS.
- J. O. M. Bockris, J. Electrochem. Soc., 1984, 131, 290 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383 CrossRef CAS PubMed.
- F. Calle-Vallejo, N. G. Inoglu, H.-Y. Su, J. I. Martínez, I. C. Man, M. T. M. Koper, J. R. Kitchin and J. Rossmeisl, Chem. Sci., 2013, 4, 1245 RSC.
- Y. L. Lee, J. Kleis, J. Rossmeisl, Y. Shao-Horn and D. Morgan, Energy Environ. Sci., 2011, 4, 3966 RSC.
- W. T. Hong, R. E. Welsch and Y. Shao-Horn, J. Phys. Chem. C, 2015, 120, 78 CrossRef.
- C. Wei, Z. Feng, G. G. Scherer, J. Barber, Y. Shao-Horn and Z. J. Xu, Adv. Mater., 2017, 29, 1606800 CrossRef PubMed.
- Z. Qiu, C. W. Tai, G. A. Niklasson and T. Edvinsson, Energy Environ. Sci., 2019, 12, 572 RSC.
- C. Xie, W. Chen, S. Du, D. Yan, Y. Zhang, J. Chen, B. Liu and S. Wang, Nano Energy, 2020, 71, 104653 CrossRef CAS.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587 CrossRef CAS PubMed.
- B. J. Trześniewski, O. Diaz-Morales, D. A. Vermaas, A. Longo, W. Bras, M. T. Koper and W. A. Smith, J. Am. Chem. Soc., 2015, 137, 15112 CrossRef PubMed.
- M. Steimecke, G. Seiffarth and M. Bron, Anal. Chem., 2017, 89, 10679 CrossRef CAS PubMed.
- C. N. Brodsky, R. G. Hadt, D. Hayes, B. J. Reinhart, N. Li, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3855 CrossRef CAS PubMed.
- A. Bergmann, E. Martinez-Moreno, D. Teschner, P. Chernev, M. Gliech, J. F. De Araújo, T. Reier, H. Dau and P. Strasser, Nat. Commun., 2015, 6, 8625 CrossRef CAS PubMed.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M. Cheng, D. Sokaras, T. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305 CrossRef CAS PubMed.
- Z. Yan, H. Sun, X. Chen, H. Liu, Y. Zhao, H. Li, W. Xie, F. Cheng and J. Chen, Nat. Commun., 2018, 9, 1 CrossRef PubMed.
- A. Nenning, A. K. Opitz, C. Rameshan, R. Rameshan, R. Blume, M. Havecker, A. Knop-Gericke, G. Rupprechter, B. Klotzer and J. Fleig, J. Phys. Chem. C, 2016, 120, 1461 CrossRef CAS PubMed.
- H. Ali-Löytty, M. W. Louie, M. R. Singh, L. Li, H. G. Sanchez Casalongue, H. Ogasawara, E. J. Crumlin, Z. Liu, A. T. Bell, A. Nilsson and D. Friebel, J. Phys. Chem. C, 2016, 120, 2247 CrossRef.
- H. G. Sanchez Casalongue, M. L. Ng, S. Kaya, D. Friebel, H. Ogasawara and A. Nilsson, Angew. Chem., Int. Ed., 2014, 53, 7169 CrossRef CAS PubMed.
- M. Görlin, J. Ferreira de Araújo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2017, 139, 2070 CrossRef PubMed.
- T. Miura, S. Tsunekawa, S. Onishi, T. Ina, K. Wang, G. Watanbe, C. Hu, H. Kondoh, T. Kawai and M. Yoshida, Phys. Chem. Chem. Phys., 2021, 23, 23280 RSC.
- J. Deng, M. R. Nellist, M. B. Stevens, C. Dette, Y. Wang and S. W. Boettcher, Nano Lett., 2017, 17, 6922 CrossRef CAS PubMed.
- E. A. Paoli, F. Masini, R. Frydendal, D. Deiana, C. Schlaup, M. Malizia, T. W. Hansen, S. Horch, I. E. Stephens and I. Chorkendorff, Chem. Sci., 2015, 6, 190 RSC.
- K. Karthick, S. Anantharaj, P. E. Karthik, B. Subramanian and S. Kundu, Inorg. Chem., 2017, 56, 6734 CrossRef CAS PubMed.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069 CrossRef CAS.
- S. Anantharaj, P. E. Karthik, B. Subramanian and S. Kundu, ACS Catal., 2016, 6, 4660 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886 CrossRef.
- C. Zhu, A. L. Wang, W. Xiao, D. Chao, X. Zhang, N. H. Tiep, S. Chen, J. Kang, X. Wang and J. Ding, Adv. Mater., 2018, 30, 1705516 CrossRef PubMed.
- L. Li, P. Wang, Q. Shao and X. Huang, Adv. Mater., 2021, 2004243 CrossRef PubMed.
- H. Wang, H. W. Lee, Y. Deng, Z. Lu, P. C. Hsu, Y. Liu, D. Lin and Y. Cui, Nat. Commun., 2015, 6, 7261 CrossRef CAS PubMed.
- Z. Lu, H. Wang, D. Kong, K. Yan, P. C. Hsu, G. Zheng, H. Yao, Z. Liang, X. Sun and Y. Cui, Nat. Commun., 2014, 5, 4345 CrossRef CAS PubMed.
- S. Zhou, X. Miao, X. Zhao, C. Ma, Y. Qiu, Z. Hu, J. Zhao, L. Shi and J. Zeng, Nat. Commun., 2016, 7, 11510 CrossRef CAS PubMed.
- Y. Sun, S. Gao, F. Lei, J. Liu, L. Liang and Y. Xie, Chem. Sci., 2014, 5, 3976 RSC.
- J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu, Y. Jiang, Z. Pan and S. Wei, Angew. Chem., Int. Ed., 2015, 54, 8722 CrossRef CAS PubMed.
- V. Viswanathan, K. L. Pickrahn, A. C. Luntz, S. F. Bent and J. K. Norskov, Nano Lett., 2014, 14, 5853 CrossRef CAS PubMed.
- Y. P. Zhu, T. Y. Ma, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 1324 CrossRef CAS PubMed.
- H. Wang, S. Zhuo, Y. Liang, X. Han and B. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9055 CrossRef CAS PubMed.
- X. Gao, H. Zhang, Q. Li, X. Yu, Z. Hong, X. Zhang, C. Liang and Z. Lin, Angew. Chem., Int. Ed., 2016, 55, 6290 CrossRef CAS PubMed.
- C. Zhu, D. Wen, S. Leubner, M. Oschatz, W. Liu, M. Holzschuh, F. Simon, S. Kaskel and A. Eychmuller, Chem. Commun., 2015, 51, 7851 RSC.
- Y. Wang, F. Silveri, M. K. Bayazit, Q. Ruan, Y. Li, J. Xie, C. R. A. Catlow and J. Tang, Adv. Energy Mater., 2018, 8, 1801084 CrossRef.
- K. D. Gilroy, X. Yang, S. Xie, M. Zhao, D. Qin and Y. Xia, Adv. Mater., 2018, 30, e1706312 CrossRef PubMed.
- Y. Li, Z. G. Yu, L. Wang, Y. Weng, C. S. Tang, X. Yin, K. Han, H. Wu, X. Yu, L. M. Wong, D. Wan, X. R. Wang, J. Chai, Y. W. Zhang, S. Wang, J. Wang, A. T. S. Wee, M. B. H. Breese, S. J. Pennycook, T. Venkatesan, S. Dong, J. M. Xue and J. Chen, Nat. Commun., 2019, 10, 3149 CrossRef PubMed.
- F. Zhou, A. Izgorodin, R. K. Hocking, L. Spiccia and D. R. MacFarlane, Adv. Energy Mater., 2012, 2, 1013 CrossRef CAS.
- H. Y. Su, Y. Gorlin, I. C. Man, F. Calle-Vallejo, J. K. Norskov, T. F. Jaramillo and J. Rossmeisl, Phys. Chem. Chem. Phys., 2012, 14, 14010 RSC.
- C. W. Tung, Y. Y. Hsu, Y. P. Shen, Y. Zheng, T. S. Chan, H. S. Sheu, Y. C. Cheng and H. M. Chen, Nat. Commun., 2015, 6, 8106 CrossRef CAS PubMed.
- J. G. Lee, J. Hwang, H. J. Hwang, O. S. Jeon, J. Jang, O. Kwon, Y. Lee, B. Han and Y. G. Shul, J. Am. Chem. Soc., 2016, 138, 3541 CrossRef CAS PubMed.
- L. Fang, Z. Jiang, H. Xu, L. Liu, Y. guan, X. Gu and Y. Wang, J. Catal., 2018, 357, 238 CrossRef.
- X. Han, G. He, Y. He, J. Zhang, X. Zheng, L. Li, C. Zhong, W. Hu, Y. Deng and T. Ma, Adv. Energy Mater., 2018, 10, e1702222 CrossRef.
- H. Wu, T. Yang, Y. Du, L. Shen and G. W. Ho, Adv. Mater., 2018, 30, e1804341 CrossRef PubMed.
- Y. Tong, Y. Guo, P. Chen, H. Liu, M. Zhang, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Chem, 2017, 3, 812 CAS.
- M. Gong, W. Zhou, M. C. Tsai, J. Zhou, M. Guan, M. C. Lin, B. Zhang, Y. Hu, D. Y. Wang, J. Yang, S. J. Pennycook, B. J. Hwang and H. Dai, Nat. Commun., 2014, 5, 4695 CrossRef CAS PubMed.
- M. Gong, W. Zhou, M. J. Kenney, R. Kapusta, S. Cowley, Y. Wu, B. Lu, M. C. Lin, D. Y. Wang, J. Yang, B. J. Hwang and H. Dai, Angew. Chem., Int. Ed., 2015, 54, 11989 CrossRef CAS PubMed.
- H. Jin, J. Wang, D. Su, Z. Wei, Z. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688 CrossRef CAS PubMed.
- Y. Kuang, G. Feng, P. Li, Y. Bi, Y. Li and X. Sun, Angew. Chem., Int. Ed., 2016, 55, 693 CrossRef CAS PubMed.
- Z. Weng, W. Liu, L. C. Yin, R. Fang, M. Li, E. I. Altman, Q. Fan, F. Li, H. M. Cheng and H. Wang, Nano Lett., 2015, 15, 7704 CrossRef CAS PubMed.
- X. Yan, L. Tian, M. He and X. Chen, Nano Lett., 2015, 15, 6015 CrossRef CAS PubMed.
- M. R. Gao, Y. F. Xu, J. Jiang, Y. R. Zheng and S. H. Yu, J. Am. Chem. Soc., 2012, 134, 2930 CrossRef CAS PubMed.
- K. Li, J. Zhang, R. Wu, Y. Yu and B. Zhang, Adv. Sci., 2016, 3, 1500426 CrossRef PubMed.
- C. Hu, L. Zhang, Z. J. Zhao, J. Luo, J. Shi, Z. Huang and J. Gong, Adv. Mater., 2017, 29, 1701820 CrossRef PubMed.
- H. Hu, B. Guan, B. Xia and X. W. Lou, J. Am. Chem. Soc., 2015, 137, 5590 CrossRef CAS PubMed.
- M. Tahir, L. Pan, R. Zhang, Y. C. Wang, G. Shen, I. Aslam, M. A. Qadeer, N. Mahmood, W. Xu, L. Wang, X. Zhang and J. J. Zou, ACS Energy Lett., 2017, 2, 2177 CrossRef CAS.
- J. X. Feng, S. H. Ye, H. Xu, Y. X. Tong and G. R. Li, Adv. Mater., 2016, 28, 4698 CrossRef CAS PubMed.
- Y. R. Zheng, M. R. Gao, Q. Gao, H. H. Li, J. Xu, Z. Y. Wu and S. H. Yu, Small, 2015, 11, 182 CrossRef CAS PubMed.
- X. Long, H. Lin, D. Zhou, Y. An and S. Yang, ACS Energy Lett., 2018, 3, 290 CrossRef CAS.
- Y. Zhao, X. Jia, G. Chen, L. Shang, G. I. Waterhouse, L. Z. Wu, C. H. Tung, D. O'Hare and T. Zhang, J. Am. Chem. Soc., 2016, 138, 6517 CrossRef CAS PubMed.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011 CrossRef CAS PubMed.
- H. Li, Q. Zhou, F. Liu, W. Zhang, Z. Tan, H. Zhou, Z. Huang, S. Jiao and Y. Kuang, Appl. Catal., B, 2019, 255, 117755 CrossRef CAS.
- X. Xu, Y. Chen, W. Zhou, Z. Zhu, C. Su, M. Liu and Z. Shao, Adv. Mater., 2016, 28, 6442 CrossRef CAS PubMed.
- T. Ling, T. Zhang, B. Ge, L. Han, L. Zheng, F. Lin, Z. Xu, W. B. Hu, X. W. Du, K. Davey and S. Z. Qiao, Adv. Mater., 2019, 31, e1807771 CrossRef PubMed.
- X. Zhong, Y. Sun, X. Chen, G. Zhuang, X. Li and J. G. Wang, Adv. Funct. Mater., 2016, 26, 5778 CrossRef CAS.
- M. Retuerto, L. Pascual, F. Calle-Vallejo, P. Ferrer, D. Gianolio, A. G. Pereira, A. Garcia, J. Torrero, M. T. Fernandez-Diaz, P. Bencok, M. A. Pena, J. L. G. Fierro and S. Rojas, Nat. Commun., 2019, 10, 2041 CrossRef PubMed.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- Y. Zhu, W. Zhou, Y. Chen, J. Yu, M. Liu and Z. Shao, Adv. Mater., 2015, 27, 7150 CrossRef CAS PubMed.
- B. Weng, F. Xu, C. Wang, W. Meng, C. R. Grice and Y. Yan, Energy Environ. Sci., 2017, 10, 121 RSC.
- X. Liu, Z. Chang, L. Luo, T. Xu, X. Lei, J. Liu and X. Sun, Chem. Mater., 2014, 26, 1889 CrossRef CAS.
- X. Xu, C. Su, W. Zhou, Y. Zhu, Y. Chen and Z. Shao, Adv. Sci., 2016, 3, 1500187 CrossRef PubMed.
- K. Fominykh, P. Chernev, I. Zaharieva, J. Sicklinger, G. Stefanic, M. Doblinger, A. Muller, A. Pokharel, S. Bocklein, C. Scheu, T. Bein and D. Fattakhova-Rohlfing, ACS Nano, 2015, 9, 5180 CrossRef CAS PubMed.
- M. K. Bates, Q. Jia, H. Doan, W. Liang and S. Mukerjee, ACS Catal., 2015, 6, 155 CrossRef.
- N. Li, D. K. Bediako, R. G. Hadt, D. Hayes, T. J. Kempa, F. von Cube, D. C. Bell, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1486 CrossRef CAS PubMed.
- Y. Liu, Y. Ying, L. Fei, Y. Liu, Q. Hu, G. Zhang, S. Y. Pang, W. Lu, C. L. Mak, X. Luo, L. Zhou, M. Deng and H. Huang, J. Am. Chem. Soc., 2019, 141, 8136 CrossRef CAS PubMed.
- Y. Zhu, W. Zhou, Z. G. Chen, Y. Chen, C. Su, M. O. Tadé and Z. Shao, Angew. Chem., 2015, 127, 3969 CrossRef.
- S. Yagi, I. Yamada, H. Tsukasaki, A. Seno, M. Murakami, H. Fujii, H. Chen, N. Umezawa, H. Abe, N. Nishiyama and S. Mori, Nat. Commun., 2015, 6, 8249 CrossRef PubMed.
- Y. Duan, S. Sun, S. Xi, X. Ren, Y. Zhou, G. Zhang, H. Yang, Y. Du and Z. J. Xu, Chem. Mater., 2017, 29, 10534 CrossRef CAS.
- N. Zhang, C. Wang, J. Chen, C. Hu, J. Ma, X. Deng, B. Qiu, L. Cai, Y. Xiong and Y. Chai, ACS Nano, 2021, 15, 8537 CrossRef CAS PubMed.
- N. B. Halck, V. Petrykin, P. Krtil and J. Rossmeisl, Phys. Chem. Chem. Phys., 2014, 16, 13682 RSC.
- Z. Xiao, Y. Wang, Y. C. Huang, Z. Wei, C. L. Dong, J. Ma, S. Shen, Y. Li and S. Wang, Energy Environ. Sci., 2017, 10, 2563 RSC.
- K. Liu, W. Zhang, F. Lei, L. Liang, B. Gu, Y. Sun, B. Ye, W. Ni and Y. Xie, Nano Energy, 2016, 30, 810 CrossRef CAS.
- Y. Zhu, W. Zhou, J. Sunarso, Y. Zhong and Z. Shao, Adv. Funct. Mater., 2016, 26, 5862 CrossRef CAS.
- X. Li, J. Wei, Q. Li, S. Zheng, Y. Xu, P. Du, C. Chen, J. Zhao, H. Xue and Q. Xu, Adv. Funct. Mater., 2018, 28, 1800886 CrossRef.
- J. Fu, F. M. Hassan, C. Zhong, J. Lu, H. Liu, A. Yu and Z. Chen, Adv. Mater., 2017, 29, 1702526 CrossRef PubMed.
- S. Dou, C. L. Dong, Z. Hu, Y. C. Huang, J. l. Chen, L. Tao, D. Yan, D. Chen, S. Shen, S. Chou and S. Wang, Adv. Funct. Mater., 2017, 27, 1702546 CrossRef.
- Y. Kwon, T. Y. Kim, G. Kwon, J. Yi and H. Lee, J. Am. Chem. Soc., 2017, 139, 17694 CrossRef CAS PubMed.
- J. C. Liu, Y. G. Wang and J. Li, J. Am. Chem. Soc., 2017, 139, 6190 CrossRef CAS PubMed.
- S. Nong, W. Dong, J. Yin, B. Dong, Y. Lu, X. Yuan, X. Wang, K. Bu, M. Chen, S. Jiang, L. M. Liu, M. Sui and F. Huang, J. Am. Chem. Soc., 2018, 140, 5719 CrossRef CAS PubMed.
- F. Cheng, J. Shen, B. Peng, Y. Pan, Z. Tao and J. Chen, Nat. Chem., 2011, 3, 79 CrossRef CAS PubMed.
- J. Hong, Z. Hu, M. Probert, K. Li, D. Lv, X. Yang, L. Gu, N. Mao, Q. Feng, L. Xie, J. Zhang, D. Wu, Z. Zhang, C. Jin, W. Ji, X. Zhang, J. Yuan and Z. Zhang, Nat. Commun., 2015, 6, 6293 CrossRef CAS PubMed.
- Y. Liu, H. Cheng, M. Lyu, S. Fan, Q. Liu, W. Zhang, Y. Zhi, C. Wang, C. Xiao, S. Wei, B. Ye and Y. Xie, J. Am. Chem. Soc., 2014, 136, 15670 CrossRef CAS PubMed.
- R. Zhang, Y. C. Zhang, L. Pan, G. Q. Shen, N. Mahmood, Y. H. Ma, Y. Shi, W. Jia, L. Wang, X. Zhang, W. Xu and J. J. Zou, ACS Catal., 2018, 8, 3803 CrossRef CAS.
- D. Chen, M. Qiao, Y. R. Lu, L. Hao, D. Liu, C. L. Dong, Y. Li and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 8691 CrossRef CAS PubMed.
- Y. H. Li, P. F. Liu, L. F. Pan, H. F. Wang, Z. Z. Yang, L. R. Zheng, P. Hu, H. J. Zhao, L. Gu and H. G. Yang, Nat. Commun., 2015, 6, 8064 CrossRef CAS PubMed.
- T. Ling, D. Y. Yan, Y. Jiao, H. Wang, Y. Zheng, X. Zheng, J. Mao, X. W. Du, Z. Hu, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2016, 7, 12876 CrossRef CAS PubMed.
- Q. Feng, S. Zhao, D. He, S. Tian, L. Gu, X. Wen, C. Chen, Q. Peng, D. Wang and Y. Li, J. Am. Chem. Soc., 2018, 140, 2773 CrossRef CAS PubMed.
- T. Ling, D. Y. Yan, H. Wang, Y. Jiao, Z. Hu, Y. Zheng, L. Zheng, J. Mao, H. Liu, X. W. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2017, 8, 1509 CrossRef PubMed.
- J. R. Petrie, H. Jeen, S. C. Barron, T. L. Meyer and H. N. Lee, J. Am. Chem. Soc., 2016, 138, 7252 CrossRef CAS PubMed.
- J. Hwang, Z. Feng, N. Charles, X. R. Wang, D. Lee, K. A. Stoerzinger, S. Muy, R. R. Rao, D. Lee, R. Jacobs, D. Morgan and Y. Shao-Horn, Mater. Today, 2019, 31, 100 CrossRef CAS.
- Y. Zang, S. Niu, Y. Wu, X. Zheng, J. Cai, J. Ye, Y. Xie, Y. Liu, J. Zhou, J. Zhu, X. Liu, G. Wang and Y. Qian, Nat. Commun., 2019, 10, 1217 CrossRef PubMed.
- J. R. Petrie, V. R. Cooper, J. W. Freeland, T. L. Meyer, Z. Zhang, D. A. Lutterman and H. N. Lee, J. Am. Chem. Soc., 2016, 138, 2488 CrossRef CAS PubMed.
- R. D. L. Smith, B. Sporinova, R. D. Fagan, S. Trudel and C. P. Berlinguette, Chem. Mater., 2014, 26, 1654 CrossRef CAS.
- H. Vrubel, D. Merki and X. Hu, Energy Environ. Sci., 2012, 5, 6136 RSC.
- D. Gonzalez-Flores, I. Sanchez, I. Zaharieva, K. Klingan, J. Heidkamp, P. Chernev, P. W. Menezes, M. Driess, H. Dau and M. L. Montero, Angew. Chem., Int. Ed., 2015, 54, 2472 CrossRef CAS PubMed.
- A. Indra, P. W. Menezes, N. R. Sahraie, A. Bergmann, C. Das, M. Tallarida, D. Schmeisser, P. Strasser and M. Driess, J. Am. Chem. Soc., 2014, 136, 17530 CrossRef CAS PubMed.
- L. Kuai, J. Geng, C. Chen, E. Kan, Y. Liu, Q. Wang and B. Geng, Angew. Chem., Int. Ed., 2014, 53, 7547 CrossRef CAS PubMed.
- T. Gao, Z. Jin, M. Liao, J. Xiao, H. Yuan and D. Xiao, J. Mater. Chem. A, 2015, 3, 17763 RSC.
- Y. Zhang, T. Gao, Z. Jin, X. Chen and D. Xiao, J. Mater. Chem. A, 2016, 4, 15888 RSC.
- Y. R. Hong, S. Mhin, K.-M. Kim, W.-S. Han, H. Choi, G. Ali, K. Y. Chung, H. J. Lee, S.-I. Moon, S. Dutta, S. Sun, Y.-G. Jung, T. Song and H. Han, J. Mater. Chem. A, 2019, 7, 3592 RSC.
- Z. Zhu, H. Yin, C. He, M. Al-Mamun, P. Liu, L. Jiang, Y. Zhao, Y. Wang, H. Yang, Z. Tang, D. Wang, X. Chen and H. Zhao, Adv. Mater., 2018, 30, 1801171 CrossRef PubMed.
- J. Roy, M. Al-Mamun, H. Yin, Y. Dou, L. Zhang, P. Liu, Y. Wang, Y. Zhong and H. Zhao, ChemPlusChem, 2020, 85, 1602 CrossRef CAS PubMed.
- Y. Dou, C. He, L. Zhang, H. Yin, M. Al-Mamun, J. Ma and H. Zhao, Nat. Commun., 2020, 11, 1664 CrossRef CAS PubMed.
- S. Jin, ACS Energy Lett., 2017, 2, 1937 CrossRef CAS.
- L. Stern, L. Feng, F. Song and X. H, Energy Environ. Sci., 2015, 8, 2347 RSC.
- X. Xu, F. Song and X. Hu, Nat. Commun., 2016, 7, 12324 CrossRef CAS PubMed.
- W. Chen, H. Wang, Y. Li, Y. Liu, J. Sun, S. Lee, J. Lee and Y. Cui, ACS Cent. Sci., 2015, 1, 244 CrossRef CAS PubMed.
- L. Gong, D. Zhang, C. Y. Lin, Y. Zhu, Y. Shen, J. Zhang, X. Han, L. Zhang and Z. Xia, Adv. Energy Mater., 2019, 9, 1902625 CrossRef CAS.
- C. Wei, Y. Sun, G. G. Scherer, A. C. Fisher, M. P. Sherburne, J. W. Ager, Z. J. Xu and J. Am, Chem. Soc., 2020, 142, 7765 CrossRef CAS PubMed.
- J. Wan, Z. Zhao, H. Shang, B. Peng, W. Chen, J. Pei, L. Zheng, J. Dong, R. Cao, R. Sarangi, Z. Jiang, D. Zhou, Z. Zhuang, J. Zhang, D. Wang and Y. Li, J. Am. Chem. Soc., 2020, 142, 8431 CrossRef CAS PubMed.
- Z. Jiang, W. Zhou, A. Hong, M. Guo, X. Luo and C. Yuan, ACS Energy Lett., 2019, 4, 2830 CrossRef CAS.
- Y. Pan, X. Xu, Y. Zhong, L. Ge, Y. Chen, J. P. M. Veder, D. Guan, R. O'Hayre, M. Li, G. Wang, H. Wang, W. Zhou and Z. Shao, Nat. Commun., 2020, 11, 2002 CrossRef CAS PubMed.
- J. Li, D. Chu, H. Dong, D. R. Baker and R. Jiang, J. Am. Chem. Soc., 2019, 10, 269 Search PubMed.
- J. T. S. Irvine, D. Neagu, M. C. Verbraeken, C. Chatzichristodoulou, C. Graves and M. B. Mogensen, Nat. Energy, 2016, 1, 15014 CrossRef CAS.
- Y. Luo, L. Tang, U. Khan, Q. Yu, H.-M. Cheng, X. Zou and B. Liu, Nat. Commun., 2020, 11, 2002 CrossRef PubMed.
- B. Zhang, L. Fan, R. B. Ambre, T. Liu, Q. Meng, B. J. J. Timmer and L. Sun, Joule, 2020, 4, 1408 CrossRef CAS.
- D. Li, E. J. Park, W. Zhu, Q. Shi, Y. Zhou, H. Tian, Y. Lin, A. Serov, B. Zulevi, E. D. Baca, C. Fujimoto, H. T. Chung and Y. S. Kim, Nat. Energy, 2020, 5, 378 CrossRef CAS.
- S. J. C. Cleghorn, D. K. Mayfield, D. A. Moore, J. C. Moore, G. Rusch, T. W. Sherman, N. T. Sisofo and U. Beuscher, J. Power Sources, 2006, 158, 446 CrossRef CAS.
- X. Peng, D. Kulkarni, Y. Huang, T. J. Omasta, B. Ng, Y. Zheng, L. Wang, J. M. LaManna, D. S. Hussey, J. R. Varcoe, I. V. Zenyuk and W. E. Mustain, Nat. Commun., 2020, 11, 3561 CrossRef CAS PubMed.
- K. J. May, C. E. Carlton, K. A. Stoerzinger, M. Risch, J. Suntivich, Y.-L. Lee, A. Grimaud and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 3264 CrossRef CAS.
- Y. R. Shen, Nature, 1989, 337, 519 CrossRef CAS.
- S. Haschke, M. Mader, S. Schlicht, A. M. Roberts, A. M. Angeles-Boza, J. A. C. Barth and J. Bachmann, Nat. Commun., 2018, 9, 4565 CrossRef PubMed.
Footnote |
| † Equal contributions. |
| This journal is © The Royal Society of Chemistry 2021 |