
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling selectivity in N-heterocycle directed borylation of indoles†
S. A.
Iqbal
,
K.
Yuan
,
J.
Cid
 ,
J.
Pahl
and
M. J.
Ingleson
*
,
J.
Pahl
and
M. J.
Ingleson
*
EaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: michael.ingleson@ed.ac.uk
First published on 11th March 2021
Abstract
Electrophilic borylation of indoles with BX3 (X = Cl or Br) using directing groups installed at N1 can proceed at the C2 or the C7 position. The six membered heterocycle directing groups utilised herein, pyridines and pyrimidine, result in indole C2 borylation being the dominant outcome (in the absence of a C2-substituent). In contrast, C7 borylation was achieved using five membered heterocycle directing groups, such as thiazole and benzoxazole. Calculations on the borylation of indole substituted with a five (thiazole) and a six (pyrimidine) membered heterocycle directing group indicated that borylation proceeds via borenium cations with arenium cation formation having the highest barrier in both cases. The C7 borylated isomer was calculated to be the thermodynamically favoured product with both five and six membered heterocycle directing groups, but for pyrimidine directed indole borylation the C2 product was calculated to be the kinetic product. This is in contrast to thiazole directed indole borylation with BCl3 where the C7 borylated isomer is the kinetic product too. Thus, heterocycle ring size is a useful way to control C2 vs. C7 selectivity in N-heterocycle directed indole C–H borylation.
Introduction
Derivatives of the heterocycle indole are core motifs in a variety of bioactive compounds such as Hippadine, Chloropeptin I and Chuangxinmycin,1 while functionalised indoles also have been utilised in organic materials applications.2 Therefore, the functionalisation of indole derivatives in a selective manner is important. An efficient route to generate selectively functionalised heteroarenes, including indoles, is directed C–H borylation,3 with the borylated products extremely useful in synthesis.4 In the past decade directed electrophilic C–H borylation (e.g.Fig. 1A) has been widely used including to form boron containing organic materials,3c,5 borylated intermediates for synthesis,3c and boron containing bioactive compounds.3c,6 Electrophilic borylation of indoles using directing groups installed on N1 can lead to C2 or C7 borylation (Fig. 1B). High C7 selectivity was reported only recently, by some of us,7 and concomitantly Shi, Houk and co-workers.8 Both groups used the N-pivaloyl directing group to achieve electrophilic C–H borylation of indoles with BBr3, with this directing group providing good selectivity for the C7 position. In this case, regioselectivity stems from unfavourable interactions between the pivaloyl tBu group and the C7-H,9 this orients the Lewis basic carbonyl group closer to C7 than C2.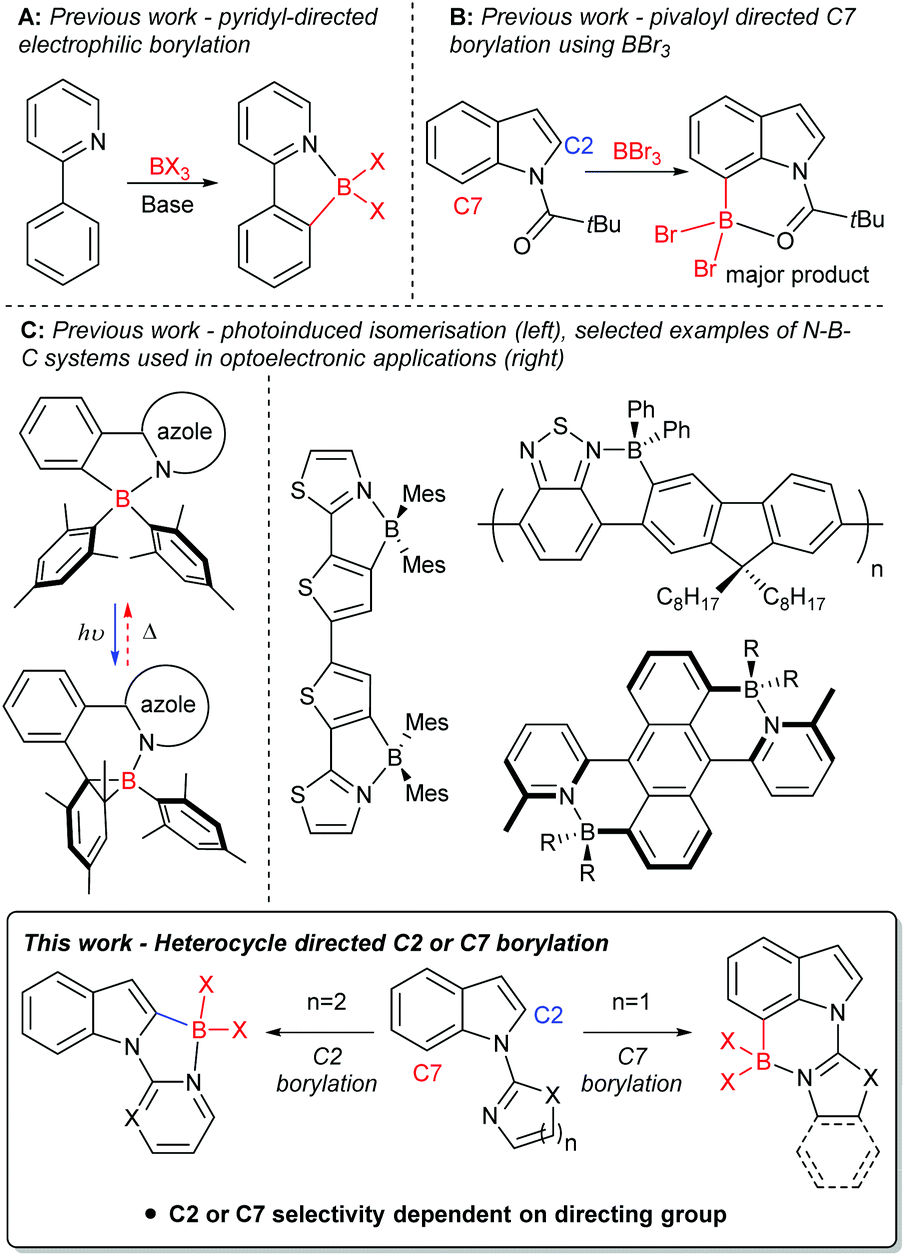 | ||
| Fig. 1 Directed electrophilic C–H borylation of 2-phenylpyridine (top, left) and N-pivaloyl indole at C7 (top, right). Photo/thermal induced isomerisation of 4-coordinate N–B–C compounds (middle, left). Selected examples of 4-coordinate N–B–C compounds used in optoelectronic applications (middle, right). This work: heterocycle directed electrophilic C–H borylation at C2 or C7 (bottom). | ||
We were interested in probing the electrophilic C–H borylation of indoles using heterocycle based directing groups installed on N1. Directing groups are necessary to overcome the C3 selectivity otherwise observed using BX3 derived electrophiles.7b Selectivity (C2 versus C7) in heterocycle directed borylation could vary depending on heterocycle ring size and heterocycle substituents. The targeted borylated products are potentially of interest for use in synthesis and in their own right as fused four coordinate boron containing materials. Four-coordinate organoboron containing compounds are of current interest, in part due to the incorporation of LB → BR3 (LB = Lewis base) units into conjugated materials being an effective method to significantly lower the LUMO energy.5,10 The incorporation of LB → B units into a π-conjugated system has been utilised to produce a range of materials (Fig. 1C) with interesting properties, such as: small HOMO–LUMO gaps; high photoluminescence quantum yields;5,10 photo/thermal isomerisation11 to name a few. Herein we demonstrate that Lewis basic N-heterocycles installed on indole-N1 are viable directing groups for the selective electrophilic C–H borylation of indoles at either the C2 or C7 position (Fig. 1, bottom). Rational selection of the heterocyclic directing group (by using a five membered or a six membered heterocycle) enables the borylation reaction to be switched between C2 and C7.
Results and discussion
Indole C–H borylation with different directing groups
Since the key goal of this study was to understand selectivity in directed borylation, it was important to determine C2 vs. C7 selectivity at the primary product stage (e.g. the BX2 boracycle containing compound, X = Cl or Br). This was necessary as subsequent functionalisation at boron, e.g. installation of pinacol, has been observed to lead to isomerisation and/or protodeboronation,7 with the latter more prevalent for the C2 isomer than the C7.12 During this work it was found in multiple borylation reactions that solid precipitated. This solid could be an N → BX3 Lewis adduct, borylated products or other species such as protonated starting material (with the proton generated as the by-product from SEAr). Therefore to determine the selectivity in the C–H borylation step reaction mixtures were dried in-vacuo and a portion of the resultant solid was dissolved for analysis by NMR spectroscopy. The discussion of borylation selectivity throughout is based on analysis of these fully homogeneous solutions.N-Pyrimidine indole, 1, was combined with BCl3 under a range of conditions (see ESI†). In all cases, the major borylated indole product observed by NMR spectroscopy was from C2 borylation (δ11B = 2.7 ppm); a minor product was observed at ca. 10–15% conversion, which was tentatively assigned as the C7 borylation product (δ11B = 5.9 ppm). This minor product was not protonated 1 as the minor product resonances persisted in the presence of the hindered base 2,6-ditertbutyl-4-methylpyridine (DTP). Furthermore, it was not the Lewis adduct, 1-BCl3 (inset Scheme 1), which was observed in reaction mixtures analysed at short times (ca. 5 min) and had a δ11B = 7.2 ppm. The C2-borylated product, 2-Cl, displayed a diagnostic singlet in the 1H NMR spectrum at 6.88 ppm (for the C3-H) while the δ11B of 2.7 ppm is consistent with a four coordinate boron centre. Highest conversions to 2-Cl at room temperature were obtained in the presence of DTP. Notably, heating the reaction of 1/BCl3 in a sealed tube (to prevent loss of HCl) did not lead to any increase in the species assigned as the C7 borylated product suggesting that C2-B to C7-B isomerisation is not occurring under these conditions.
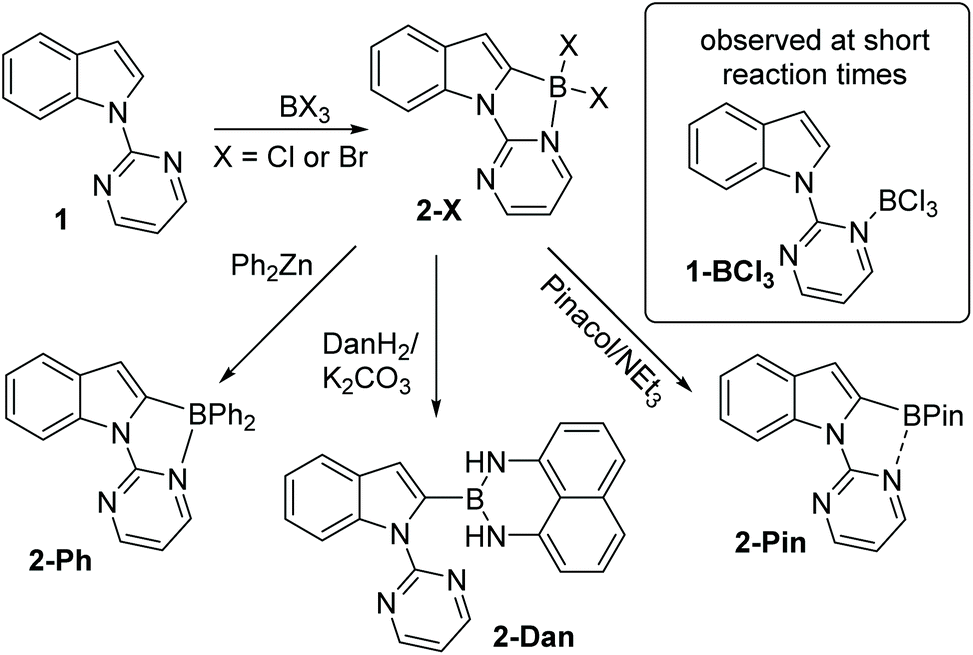 | ||
| Scheme 1 Pyrimidine directed borylation and subsequent functionalisation at boron. | ||
Addition of pinacol/NEt3 solutions to the crude reaction mixtures containing 2-Cl also produced one major new borylated indole with NMR spectroscopy consistent with 2-Pin (Scheme 1). The 11B NMR spectrum of 2-Pin showed a peak at 26 ppm, this resonance is shifted upfield relative to heteroaryl-BPin species (∼31 ppm) presumably due to some N–B interaction with pyrimidine. Isolation of 2-Pin proved challenging due to sensitivity towards protodeboronation, although the corresponding BPh2 compound, 2-Ph, was isolable. Borylation of 1 using BBr3 led to exclusive (by NMR spectroscopy) formation of 2-Br with no C7-borylation observed in this case. Compound 2-Br was converted to 2-Ph by addition of ZnPh2 and into the 1,8-diaminonaphthalene derivative, 2-Dan, which proved more robust to isolation than the pinacol congener.
The pyridyl analogue, 3 (Scheme 2), also was readily borylated with BCl3 and analysis of this reaction revealed formation of the C2 borylation product, 4-Cl, as the only observable borylated product by 1H NMR spectroscopy (singlet for C3-H observed at 6.91 ppm is consistent with C2 borylation). 11B NMR spectroscopy again confirmed the presence of a four coordinate boron centre (δ11B = 3.0 ppm). Compounds 2-X and 4-Cl were all bench stable indicating a strong N → B dative bond. For 4-Cl, slow evaporation of solvent led to crystals suitable for X-ray diffraction analysis which confirmed the formulation as the C2 borylated product (vide infra for further discussion). Furthermore, addition of ZnPh2 to 4-Cl led to formation of 4-Ph. It should be noted that no minor products derived from C7 borylation were isolated during the purification of compounds 2 and 4 by column chromatography.
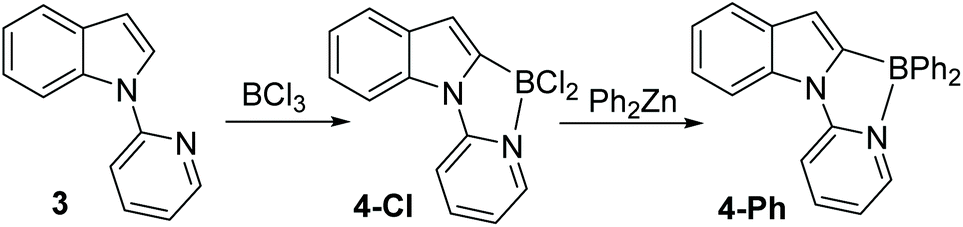 | ||
| Scheme 2 Borylation of indole functionalised with a pyridine directing group. | ||
Due to the absence of any definitively characterised six membered boracycles from pyrimidine/pyridine directed C7-H indole borylation, confirmation of the accessibility of six membered boracycles using these borylation conditions was sought in a related system. Thus N-pyrimidine-carbazole, 5, a substrate where no five membered boracycle is accessible, was synthesised. Borylation of 5 with BCl3 proceeded readily to form 6-Cl. Addition of pinacol/NEt3 to 6-Cl yielded 6-Pin in 67% isolated yield (Scheme 3). Compound 6-Pin proved more robust to protic species than 2-Pin, consistent with the previously reported relatively high sensitivity of C2/C3 borylated indoles to protodeboronation.12 The δ11B for 6-Pin (9.9 ppm) suggested a stronger interaction between pyrimidine and the BPin unit than that present in 2-Pin (δ11B = 26 ppm), which may also contribute to the enhanced stability of 6-Pin towards protic species.
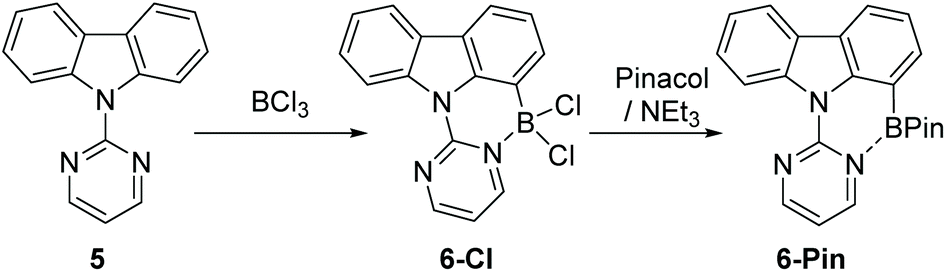 | ||
| Scheme 3 Pyrimidine directed carbazole borylation. | ||
Following the successful formation of the six membered boracycle in 6, an N-heterocycle-indole derivative, 7, that only permits C7-borylation, due to blocking of the C2 position, was synthesised (Scheme 4). Compound 7 was combined with 1.5 equiv. of BCl3 at room temperature. This led to rapid borylation, with a new resonance at δ11B = 6.0 ppm, assigned as the C7 borylated product 8-Cl. Protection at boron by addition of pinacol/NEt3 enabled isolation of 8-Pin in 80% yield. The observed δ11B for 8-BPin at 11.1 ppm indicated a significant N–B interaction, with this chemical shift more comparable to 6-Pin (containing a 6 membered boracycle) than 2-Pin (containing a five membered boracycle). The formation of 8-Cl confirmed the feasibility of N-heterocycle-directed indole C7 borylation when functionalisation at the C2 site is blocked. Furthermore, the δ11B of 8-Cl is very close to that of the minor product observed during the borylation of 1, supporting the assignment of this minor product as derived from C7-borylation.
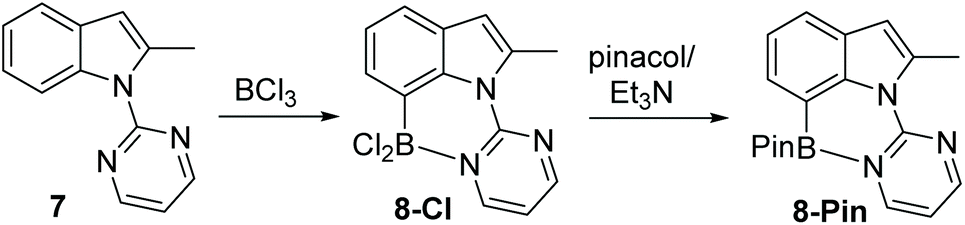 | ||
| Scheme 4 Pyrimidine directed borylation of 2-methyl indole. | ||
N-Heterocycle substituted indoles were targeted that would undergo selective C7 borylation even when the C2 position is not blocked. Thus, a 3-methyl pyridyl directing group was installed on indole to give compound 9 (Scheme 5). It was hypothesised that due to unfavourable interactions between the C7-H and the pyridyl-methyl group the pyridyl would rotate to position the methyl group closer to C2-H than C7-H. This would orientate the Lewis basic N more towards C7 than C2. However, compound 9 underwent borylation with BCl3 to form one major new borylated indole species consistent with C2 borylation, 10-Cl, along with protonated 9 as the by-product. Repeating borylation in the presence of the hindered base DTP led to formation of 10-Cl as the only new indole containing product, with no C7-borylated product, 11-Cl, observed. While the δ11B of 10-Cl was observed at 2.5 ppm, comparable to that for 2-Cl for example, 10-Cl proved much more sensitive to protodeboronation than 2-X and 4-Cl. We attribute this to the C7-H/Me interaction destabilising the N–B dative bond in 10-Cl leading to more facile decomposition via protodeboronation. Wang and co-workers have previously noted that weak dative bonds in related Ar3B ← N containing compounds leads to much less stable (with respect to protodeboronation) compounds than those with stronger dative bonds.13 The failure to observe any 11-Cl indicates the methyl groups is not large enough to force 9 to adopt a geometry where the pyridyl N is proximal to C7 (Scheme 4). This is consistent with the requirement for bulkier groups, such as tBu in pivaloyl, in previously reported C7 selective indole electrophilic borylation reactions.
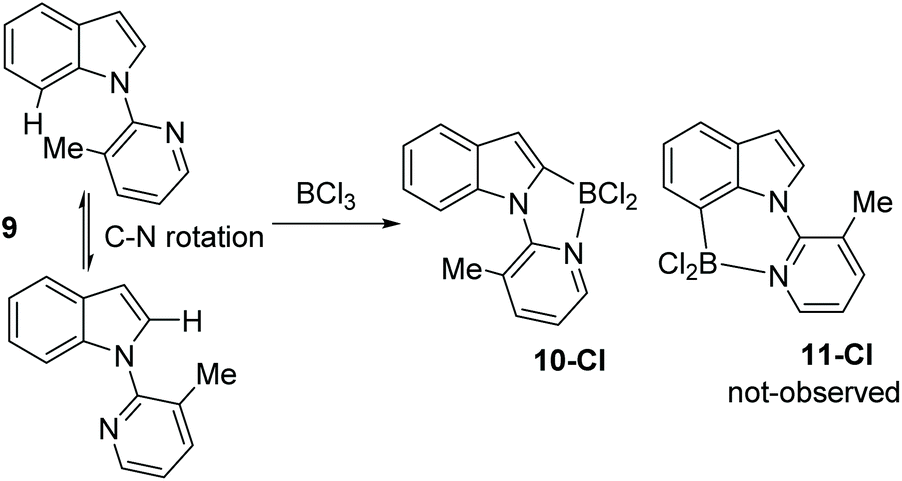 | ||
| Scheme 5 3-Methyl pyridine directed C–H borylation of indole at C2. | ||
A different approach to realise selective C7 borylation was targeted, using five membered heterocyclic directing groups. It was hypothesised that replacing six-membered N-heterocycle directing groups with five membered analogues, as in compound 12 (Scheme 6), would disfavour C2 functionalisation. The greater strain expected in the product from C2 borylation, 13-Cl, due to the presence of the three fused five membered rings, and the greater distortion expected during the C2 borylation process, would lead to higher energy barriers for C2 borylation relative to C7 borylation and a less stable borylated product (for the C2 isomer 13-Cl relative to the C7 isomer 14-Cl).
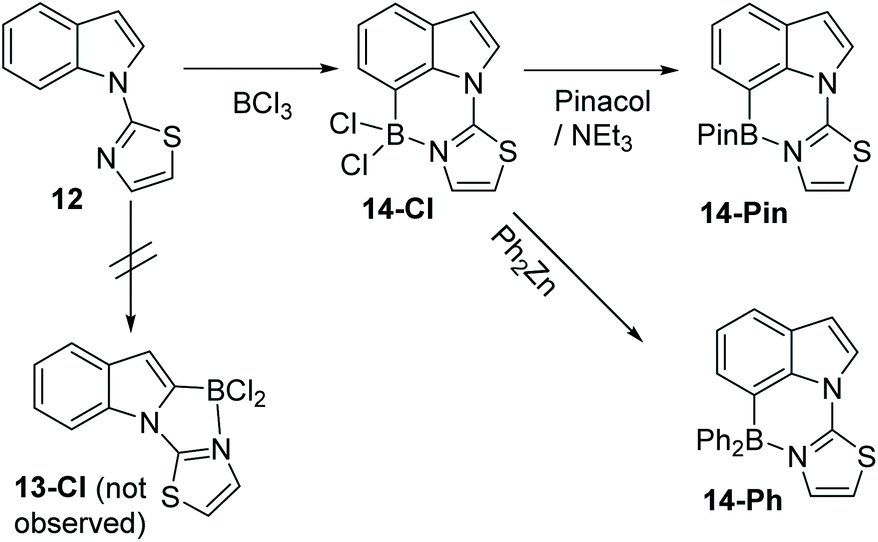 | ||
| Scheme 6 Thiazole directed borylation selectively at indole C7. | ||
Directed electrophilic borylation of 12 proved selective for the C7 position with no C2 borylation observed by 1H NMR spectroscopy. C7 borylated product 14-Cl has an 11B resonance at 6.2 ppm consistent with a four coordinate boron centre. Pinacol installation on to boron in 14-Cl could be performed and a modest yield (55%) of the C7-BPin product, 14-Pin, was isolated (δ11B 13.8 ppm). ZnPh2 was utilised to install phenyl groups on boron in 14-Cl which enabled isolation of the C7-BPh2 product 14-Ph (Scheme 6). To the best of our knowledge, this is the first example of five membered heterocycle directing groups being used to enable selective C7-H functionalisation of indoles.14
C7 selective indole borylation using BCl3 or BBr3 was also observed with benzoxazole (15) and benzothiazole (16) as the directing groups – thus it appears a general outcome with five membered heterocycle directing groups. Compound 15 produced 17-X (X = Cl or Br) as the only observed product by NMR spectroscopy on addition of BX3 (Scheme 7). Slow cooling of a DCM solution of 17-Cl yielded crystals suitable for X-ray diffraction analysis which further confirmed the formulation as the C7 borylated product (vide infra for discussion of metrics). The reaction of 17-Cl with ZnPh2 yielded the C7-BPh2 product 17-Ph. The borylation of the benzothiazole derivative, 16, also was achieved on addition of BX3. However, the extremely low solubility of 18-X in chlorinated organic solvents meant that only 18-Ph could be characterised, so the selectivity in the borylation step for this compound cannot be readily determined. Nevertheless, borylation is likely also to be C7-selective by analogy to 12 and 15.
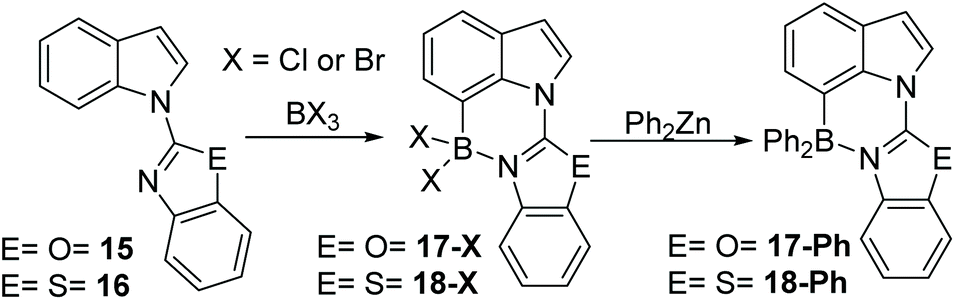 | ||
| Scheme 7 Benzoxazole and benzothiazole directed borylation at C7. | ||
In all the BPh2 products isolated in this work, there was a significant N–B interaction as indicated by the 11B NMR spectra (δ11B in the region 0–2 ppm), which showed noticeable upfield shifts relative to Ar3B species. As expected, N → BPh2 coordination modulates the electronic properties of the substituted indoles, with the first reduction potential for the BPh2 functionalised compounds being less negative by ca. 0.5 V than the non-borylated precursors, as observed previously for related compounds.11 Similar changes in the first reduction process were observed on installation of BPh2 at C2 or C7.
Solid state structures
Direct comparison of the solid state structures of a C2 and a C7 borylated product, 4-Cl and 17-Cl, is informative (Fig. 2). Both 4-Cl and 17-Cl feature effectively planar fused polycyclic cores, with small angles between the planes of the five-membered ring in indole and the heterocyclic directing group (for 4-Cl = 3.50°, for 17-Cl = 7.44°). The boron atom in both are 4-coordinate, however the N2–B1 bond distance in 4-Cl is 1.589(2) Å, slightly longer than the analogous bond distance in 17-Cl (1.561(6) Å), possibly due to the greater strain in five-membered boracycle. In contrast, the C–B distances are effectively identical. Increased strain in 4-Cl also is indicated by 17-Cl having a C7–B1–N2 angle of 107.7(4)° (close to the ideal for a tetrahedral boron centre), while 4-Cl has a contracted comparable angle (N2–B1–C2 angle of 97.7(1)°) imposed by the five membered boracycle constructed of three shorter bonds (three CN bonds) and two longer bonds (the CB and NB bonds). In the extended structure of 4-Cl face-to-face π stacking is seen with a short distance of 3.379 Å between stacked adjacent molecules, whereas no close face to face stacking is observed in the extended solid state structure for 17-Cl.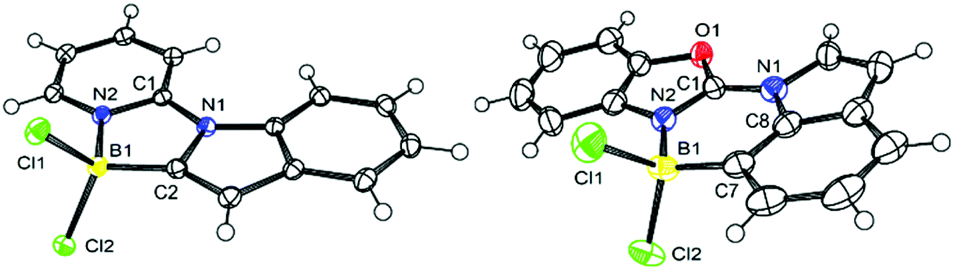 | ||
| Fig. 2 Solid state structures of left, 4-Cl, and right 17-Cl, ellipsoids at 50% probability. Selected distances (Å) and angles (°) for 4-Cl: B1–N2 = 1.589(2); C2–B1 = 1.587(2); B–Cl1 = 1.856(1), B1–Cl2 = 1.848(2); N1–C1 = 1.375(2) N1–C1–N2 = 109.7(1); N1–C2–B1 = 107.3(1); N2–B1–C2 = 97.7(1); C1–N2–B1 = 111.8(1). For 17-Cl: N2–B1 = 1.561(6); C7–B1 = 1.595(7); B1–Cl1 = 1.875(6); B1–Cl2 = 1.861(6); N1–C1 = 1.334(5); N1–C1–N2 124.0(4); C1–N2–B1 = 125.2(4); C7–B1–N2 = 107.7(4); C1–N1–C8 = 118.7(4). | ||
DFT calculations
DFT calculations at the M06-2X/6-311+G(d,p)//(PCM(CH2Cl2)) (PCM = polarisable continuum model) level were performed to gain further insight into the origin of the C2 vs. C7 borylation selectivity with five and six membered N-heterocycle directing groups. The calculations indicated the mechanism of borylation is very similar irrespective of the heterocyclic directing group employed, proceeding via borenium cation intermediates, with arenium cation formation being rate limiting. It should be noted that the metrics for the five and six membered boracycles in the solid state structures of 4-Cl and 17-Cl are closely comparable to the metrics of the boracycles in the calculated C2-BCl2 and C7-BCl2 structures derived from pyrimidine and thiazole directed indole borylation.Addition of BCl3 to 12 forms complex INT1 (Fig. 3a), with this step calculated to be exergonic by 12.8 kcal mol−1. Borenium cation (INT2) formation through halide transfer to a second molecule of BCl3, forming [BCl4]−, is then endergonic (by 4.3 kcal mol−1 relative to 12 + two equiv. BCl3), consistent with no borenium intermediates being observed by NMR spectroscopy. The formation of the arenium cation at C7 then proceeds through a 6-membered transition state, TS‡1A, with an overall energy barrier of 23.9 kcal mol−1 from INT1. TS‡1A leads to arenium cation, INT3A, at which point there is a small energy barrier for deprotonation with [BCl4]− as base. The formation of the C7 borylated product, 14-Cl, and HCl is energetically downhill by −15.8 kcal mol−1 relative to 12 and BCl3. The mechanism has many similarities to that calculated for pivaloyl directed indole borylation,7,8 and an earlier study by Uchiyama and co-workers on imidazole directed electrophilic borylation.15
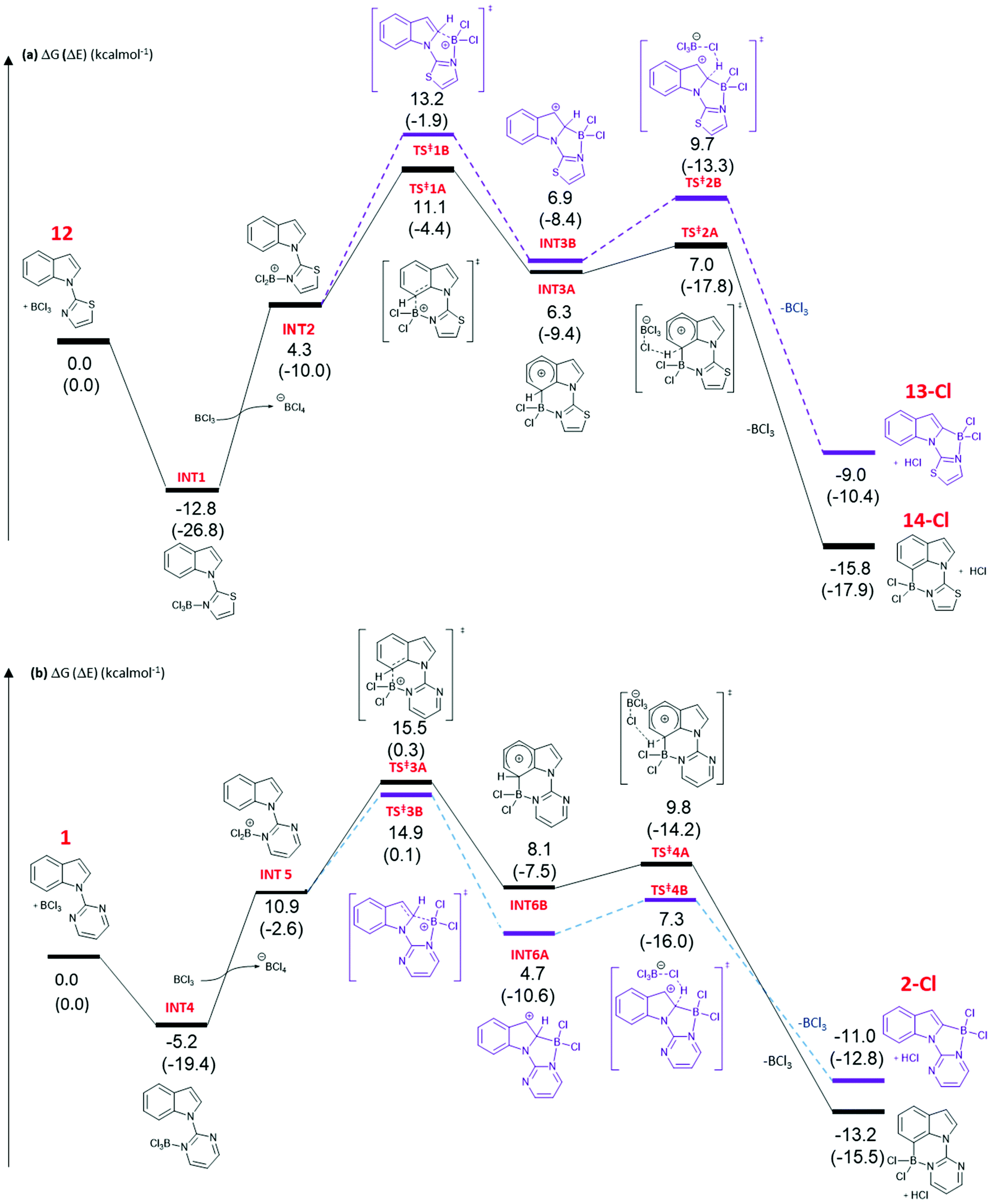 | ||
| Fig. 3 DFT calculated energy profiles for addition of BCl3 to 12 (a) and 1 (b) at M06-2X/6-311+G(d,p)//PCM (CH2Cl2). Route for C2 borylation: purple. C7: black. Values are for ΔG in kcal mol−1, with ΔE provided in parentheses. | ||
The observed C7 selectivity in the borylation of 12 is reproduced by the calculations, with the formation of the C2 borylated isomer having higher barriers throughout (e.g. energy of TS‡1B > TS‡1A). Comparing the calculated structures for TS‡1A and TS‡1B with 12 (or INT2) shows that during borylation there is a larger distortion of the thiazole-indole unit in TS‡1B relative to 12 (or to INT2) than in TS‡1A. This is demonstrated by the relative change in the N–C–N and C–N–B bond angles (INT2 to TS‡1BΔ = 7.38° and 9.27°, respectively), with a smaller distortion in TS‡1A relative to INT2 (for the equivalent angles Δ = 2.2° and 0.95°). The different degrees of distortion leads to an energy difference of 3 kcal mol−1 between the two geometries of the thiazole-indole unit in the respective transition states (Fig. 4). This presumably is a significant contributor to the observed δΔE between TS‡1A and TS‡1B (2.5 kcal mol−1).16 This confirms that increasing distortion energy during C2 borylation by using five membered heterocycle directing groups is an effective route to enable highly selective C7 functionalisation. It is also noteworthy that the C7 borylated isomer, 14-Cl, is the kinetic and thermodynamic product starting from 12, with a significant energy difference (6.8 kcal mol−1) between 13-Cl and 14-Cl. This is presumably due to the greater strain present in the C2 borylated product (as exemplified by N–C–N = 112.97° in the calculated structure of 13-Clvs. 121.59° in that of 14-Cl).
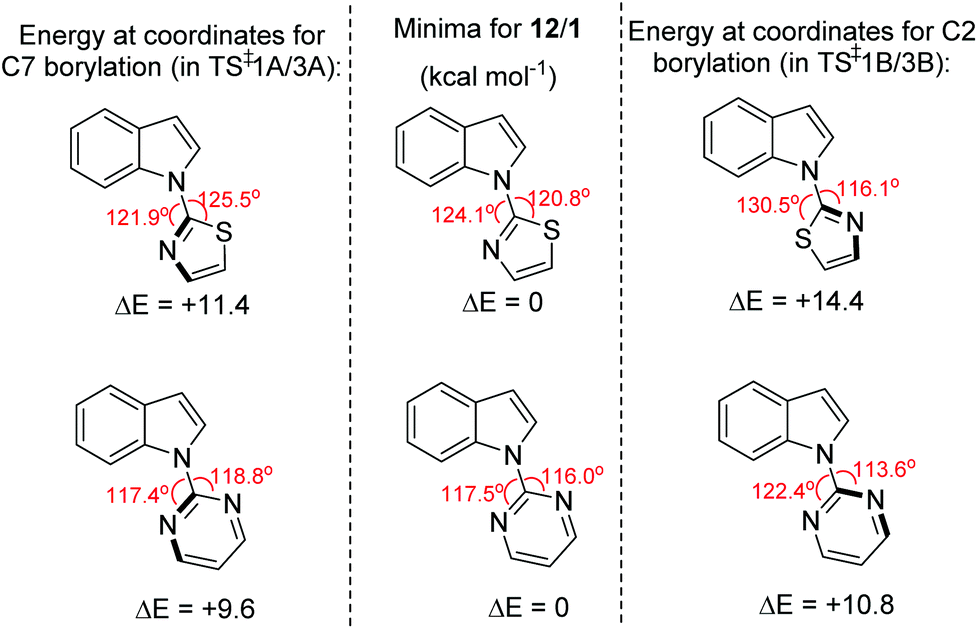 | ||
| Fig. 4 Change in energy on distorting the N-heterocycle-indole 12/1 from the optimised geometry (centre) to the geometry observed in the key transition state for C2 (right) and C7 (left) borylation. | ||
The borylation of 1 (Fig. 3b) follows a similar pathway to that of 12, although borenium cation formation is more endergonic starting from 1, possibly due to the lower basicity of pyrimidine relative to thiazole (pKa values of the conjugate acids are 1.3 and 2.5, respectively). While the relative energies of the two product isomers show the C7 product to be 2.2 kcal mol−1 lower in energy than the C2 isomer (2-Cl), the reaction is proceeding under kinetic control (as observed in many other N-directed borylation reactions using BX3)3c to give the C2 borylated product as the major product. However, the key transition state for C2 borylation is only slightly lower in energy relative to that for C7 borylation (compare TS‡3A and 3B), consistent with the observed formation of minor amounts of a product derived from 1 assigned as the C7-borylated product. These observations are consistent with the documented kinetic preference for functionalisation of indoles at C2 in preference to C7 in the absence of other factors (e.g. ring strain as in 12 or steric bulk as in the pivaloyl functionalised indoles). The smaller difference in ΔE between the key transition states for determining C2 vs. C7 selectivity (e.g. ΔE between TS‡1A/B is > than ΔE between TS‡3A/B) is due, at least in part, to a smaller difference in the distortion energy of the N-heterocycle-indole units in the two borylation transition states starting from 1 (Fig. 4 bottom) relative to that starting from 12 (Fig. 4 top). Thus, the relatively high distortion energy (14.4 kcal mol−1) of the thiazole-indole moiety in the key transition state for C2 borylation is a significant factor in the borylation of 12 being selective for C7.
Finally, consistent with pivaloyl directed indole borylation, the C7 borylated product is more stable than the C2 analogue in both the cases calculated herein. This is attributed to the 6-membered boracycle derived from C7 borylation having less strain, as indicated by calculated C–B–N angles being close to 109°, whereas borylation at C2 to form a five-membered ring gives calculated C–B–N angles of ca. 97°, significantly more acute than the optimal angle for a 4-coordinate boron compound. This is consistent with relative angles observed in the solid state structures of 4-Cl and 17-Cl. It is noteworthy that the energy difference between 13-Cl and 14-Cl is considerably greater than that between the C2 and C7 pyrimidine isomers, again highlighting the greater impact on relative energies of the two isomers imposed by five membered directing groups.
Conclusions
Modification of the heterocycle directing group installed at N1 on indoles can afford selective electrophilic borylation at either the C2 or the C7 position. Six membered heterocyclic directing groups lead to preferential C2 borylation using BX3, with borylation calculated to proceed via a borenium cation mediated mechanism. Calculations indicate with a six membered heterocycle directing group that C2 borylation is the kinetically favoured pathway, but C7 borylation leads to the thermodynamic product. In contrast, five membered heterocycle directing groups lead to selective C7 borylation, with the smaller internal angles in five membered rings resulting in higher barriers to borylation at C2 relative to C7. Thus with five membered heterocycle directing groups, C7 indole borylation with BX3 is the kinetically and the thermodynamically favoured outcome. These results indicate that five membered heterocyclic directing groups have been overlooked as a means to target selective C7 functionalisation in directed indole functionalisation. To date this area has been dominated by six-membered heterocycle based directing groups and thus C2 functionalisation.17Experimental
General
All reactions were performed under an inert atmosphere using standard Schlenk techniques unless otherwise stated. All chemicals were purchased from commercial sources and used without further purification unless stated otherwise. BCl3 and BBr3 solutions were transferred to Schlenks fitted with J. Youngs valves prior to use. Dry solvents were obtained from an Inert PureSolv MD5 SPS machine or dried over CaH2 and stored over 3 Å molecular sieves. Bruker 300, Bruker 400 and Bruker 500 MHz NMR spectrometers were used to obtain 13C{1H}, 1H and 11B NMR spectra. CDCl3 or CD2Cl2 was used as the solvent in all cases and the residual CHCl3 or CH2Cl2 resonance was used as reference for 13C{1H} and 1H NMR spectra. 11B NMR spectra were referenced to external BF3-Et2O. NMR Spectroscopy was undertaken at room temperature (∼20 °C), spin–spin J coupling constants are reported in hertz (Hz) and the chemical shifts δ are reported in ppm. C–B bonded and C–(N)313C resonances were not detected in the 13C{1H} NMR spectra presumably due to their being very broad resonances due to quadrupolar effects. Column chromatography was performed on 40–63 μm silica gel manually or using a CombiFlash NextGen 300+ Autocolumn system. Mass spectrometry was performed by the mass spectrometry services at either the University of Manchester or the University of Edinburgh using electrospray or APCI ionisation modes. Cyclic voltammetry measurements were conducted under an N2 atmosphere using a CH-Instrument 1110C Electrochemical/Analyser potentiostat. THF (1 mM) was used as the solvent in all cases and tetrabutylammonium hexafluorophosphate (0.1 M) was used as the electrolyte. A glassy carbon working electrode was used and platinum wire as the counter and reference electrodes. All potentials were calibrated against the ferrocene/ferrocenium (Fc/Fc+) redox couple.Synthesis of 2-Br
To an ampule fitted with a J-Youngs tap was added compound 1 (0.039 g, 0.2 mmol) which was dissolved in DCM (0.7 mL). BBr3 (0.44 mL, 1 M in DCM) was added, the ampule was sealed and the mixture was stirred at room temperature for 0.5 hours. The solvent/volatiles were removed under vacuum and the solid dried. The product was dissolved in DCM, passed through a filter and the volatiles were removed to give a solid which was washed with pentane and dried to give the pure product, 2-Br (0.054 g, 74%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.97 (dd, J = 4.7, 2.2 Hz, 1H), 8.82 (dd, J = 6.1, 2.3 Hz, 1H), 8.19–8.10 (m, 1H), 7.60 (dt, J = 7.4, 1.0 Hz, 1H), 7.40–7.26 (m, 3H), 6.93 (d, J = 0.8 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3) δ 165.9, 151.8, 135.7, 132.7, 125.1, 124.8, 122.4, 114.8, 113.9, 113.3. 11B NMR (160 MHz, CDCl3) δ −6.82. [Acc. Mass] calculated [M + H]+: 363.92508, observed [M + H]+: 363.92830.Synthesis of 2-Ph
To an ampule fitted with a J-Youngs tap was added compound 1 (0.030 g, 0.15 mmol) which was dissolved in o-DCB (0.5 mL). BCl3 1 M in hexanes (0.33 mL, 0.33 mmol) was added, the tube was sealed and the mixture was heated to 80 °C for 16 hours. The solvents and excess BCl3 were removed under vacuum and ZnPh2 (0.077 g, 0.35 mmol) was added followed by toluene (0.5 mL). The reaction mixture was stirred for 2 days at room temperature. The product was purified by column chromatography on silica gel (EtOAc/petroleum ether) to give 2-Ph (0.011 g, 22%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.90 (dd, J = 4.8, 2.3 Hz, 1H), 8.54 (dd, J = 5.8, 2.3 Hz, 1H), 8.31–8.22 (m, 1H), 7.62–7.52 (m, 1H), 7.42–7.31 (m, 4H), 7.29–7.26 (m, 2H), 7.25–7.16 (m, 6H), 7.11 (dd, J = 5.8, 4.8 Hz, 1H), 6.67 (d, J = 0.9 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.3, 154.6, 152.4, 136.7, 133.0, 127.8, 126.5, 123.9, 122.6, 120.8, 113.8, 113.6, 108.9. 11B NMR (128 MHz, CDCl3) δ 0.36. [Acc. Mass] calculated [M + H]+: 360.1667, observed [M + H]+: 360.1655.Synthesis of 2-Dan
To an ampule fitted with a J-Youngs tap was added compound 1 (0.028 g, 0.14 mmol) which was dissolved in DCM (0.35 mL). BBr3 (0.33 mL, 1 M in DCM) was added and the ampule was sealed and stirred at room temperature for 0.75 hours. The solvent/volatiles were removed under vacuum and the product dried. A mixture of 1,8-diaminonapthalene (0.025 g, 0.14 mmol) in DCM (1 mL) and K2CO3 (0.020 g, 0.7 mmol) in H2O (0.7 mL) was prepared and stirred vigorously for 1 hour and then added to the reaction ampule containing the borylated indole at 0 °C, after stirring at 0 °C for 10 minutes the ampule was warmed to room temperature and stirred for a further 2 hours. The reaction mixture was poured into a conical flask and dried over MgSO4, the crude product was purified by column chromatography on silica-gel (EtOAc/petroleum ether) to give the pure product, 2-Dan (0.016 g, 31%) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 8.77 (dd, J = 8.2, 0.9 Hz, 1H), 8.68 (d, J = 4.8 Hz, 2H), 7.66 (dt, J = 7.7, 1.1 Hz, 1H), 7.45–7.34 (m, 1H), 7.34–7.24 (m, 1H), 7.19–7.11 (m, 2H), 7.07 (dd, J = 8.3, 1.0 Hz, 2H), 7.04 (t, J = 4.7 Hz, 1H), 6.95 (s, 1H), 6.32 (dd, J = 7.1, 1.0 Hz, 2H), 5.80 (br s, 2H, N–![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ). 13C{1H} NMR (126 MHz, CDCl3) δ 158.0, 141.7, 137.5, 136.6, 131.6, 127.7, 124.3, 122.5, 120.9, 119.6, 117.5, 116.4, 115.8, 114.7, 105.8.11B NMR (128 MHz, CDCl3) δ 27.64. [Acc. Mass] calculated [M + H]+: 362.15715, observed [M + H]+: 362.15720.
). 13C{1H} NMR (126 MHz, CDCl3) δ 158.0, 141.7, 137.5, 136.6, 131.6, 127.7, 124.3, 122.5, 120.9, 119.6, 117.5, 116.4, 115.8, 114.7, 105.8.11B NMR (128 MHz, CDCl3) δ 27.64. [Acc. Mass] calculated [M + H]+: 362.15715, observed [M + H]+: 362.15720.
Synthesis of 4-Cl
To an ampule fitted with a J-Youngs tap was added compound 3 (0.019 g, 0.10 mmol) which was dissolved in DCM (0.35 mL). BCl3 (0.33 mL, 1 M in DCM) was added to and the ampule was sealed and stirred at room temperature for 3 hours. The solvent/volatiles were removed under vacuum until dryness to give the pure product, 4-Cl (0.024 g, 87%) as a yellow solid. Crystals were grown by slow evaporation of a DCM/pentane solution of the product. 1H NMR (400 MHz, CDCl3) δ 8.53 (d, J = 5.8 Hz, 1H), 8.22–8.06 (m, 1H), 7.72–7.59 (m, 3H), 7.36–7.18 (m, 3H), 6.91 (s, 1H).13C{1H} NMR (126 MHz, CDCl3) δ 148.8, 145.3, 142.3, 136.0, 132.7, 124.3, 123.7, 122.8, 118.4, 111.4, 110.7, 109.8. 11B NMR (128 MHz, CDCl3) δ 3.03. [Acc. Mass] calculated [M + Na]+: 297.01281, observed [M + Na]+: 297.01270.Synthesis of 4-Ph
To an ampule fitted with a J-Youngs tap was added compound 4-Cl (0.056 g, 0.2 mmol) and ZnPh2 (0.100 g, 0.46 mmol) followed by toluene (1.5 mL). The ampule was sealed and the reaction was stirred at room temperature for 16 hours. The crude product was purified by column chromatography on silica-gel (EtOAc: petroleum ether) to give the pure product, 4-Ph (0.011 g, 15%) as a brown solid. 1H NMR (400 MHz, CDCl3) δ 8.34–8.30 (m, 1H), 8.10–8.00 (m, 1H), 7.80 (dt, J = 8.6, 1.1 Hz, 1H), 7.77–7.66 (m, 1H), 7.66–7.53 (m, 1H), 7.35 (dd, J = 8.1, 1.5 Hz, 4H), 7.29–7.15 (m, 8H), 7.17–7.08 (m, 1H), 6.66 (d, J = 0.9 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3) δ 150.3, 143.9, 142.8, 136.9, 133.1, 132.8, 127.7, 126.2, 122.8, 121.9, 121.3, 117.3, 111.2, 109.6, 107.2. 11B NMR (128 MHz, CDCl3) δ 0.02. [Acc. Mass] calculated [M + H]+: 359.17141, observed [M + H]+: 359.17100.Synthesis of 6-Pin
BCl3 (0.11 mL, 1 M in DCM) was added to a solution of compound 5 (0.024 g, 0.1 mmol) in DCM (1 mL) and the reaction mixture was stirred at room temperature for 1 hour. Pinacol (0.018 g, 0.15 mmol) and NEt3 (0.21 mL, 1.5 mmol) were added and the mixture was stirred for 1 hour. Volatiles were removed under vacuum and the crude product was purified by column chromatography on silica-gel (EtOAc: hexane) to yield the pure product, 6-Pin (0.025 g, 67%) as a white solid. 1H NMR (400 MHz, CD2Cl2) δ 9.10 (d, J = 5.2 Hz, 2H), 8.83 (d, J = 8.3 Hz, 1H), 8.05 (d, J = 7.7 Hz, 1H), 7.95 (d, J = 7.6 Hz, 1H), 7.75 (d, J = 7.2 Hz, 1H), 7.57–7.41 (m, 3H), 7.28 (t, J = 5.2 Hz, 1H), 1.31 (s, 12H). 13C{1H} NMR (101 MHz, CD2Cl2) δ 156.8, 154.0, 141.5, 138.9, 129.9, 128.6, 127.2, 125.3, 124.6, 124.0, 120.8, 119.0, 119.0, 114.8, 81.4, 27.1. 11B NMR (128 MHz, CD2Cl2) δ 9.90. [Acc. Mass] calculated [M + H]+: 372.1878, observed [M + H]+: 372.1869.Synthesis of 8-Pin
To an ampule fitted with a J-Youngs tap was added compound 7 (0.042 g, 0.2 mmol) which was dissolved in DCM (1.6 mL). BCl3 (0.3 mL, 1 M in DCM) was added and the ampule was sealed and stirred at room temperature for 0.25 hours. The volatiles were removed and the crude dried under vacuum. NEt3 (0.42 mL, 3 mmol) was added followed by pinacol (0.029 g, 0.24 mmol) and DCM (2.5 mL), the ampule was sealed and stirred for 1 h. The crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether) to give the pure product (0.054 g, 80%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 9.00 (d, J = 5.1 Hz, 2H), 7.55 (dd, J = 7.2, 1.3 Hz, 1H), 7.41–7.35 (m, 1H), 7.32–7.27 (m, 1H), 7.19 (t, J = 5.1 Hz, 1H), 6.48–6.45 (m, 1H), 2.84 (d, J = 1.2 Hz, 3H), 1.31 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 156.2, 154.1, 139.9, 138.0, 127.3, 126.5, 124.6, 118.9, 114.3, 111.5, 81.1, 26.8, 17.9. 11B NMR (128 MHz, CDCl3) δ 11.19. [Acc. Mass] calculated [M]+ 335.17996, observed [M]+: 335.18117.Synthesis of 10-Cl
To an ampule fitted with a J-Youngs tap was added compound 9 (0.020 g, 0.1 mmol) and 2,6-di-tert-butyl-4-methylpyridine (0.21 g, 0.01 mmol) which were dissolved in DCM (0.35 mL) followed by addition of BCl3 (3.2 eq., 1 M in DCM, 0.33 mL). The mixture was stirred at room temperature for 3 hours after which the solvent/volatiles were removed under inert conditions by vacuum and the product dried. Crude NMR spectra in CDCl3 showed the major product as 10-Cl (88% yield determined by 1H NMR spectrosocpy. 55 mg crude solid obtained, 1 mg dissolved fully in CD2Cl2 and 1 μL mesitylene internal standard added enabling yield determination). 1H NMR (500 MHz, CD2Cl2) δ 8.43 (d, J = 5.7 Hz, 1H), 8.05–7.90 (m, 2H), 7.68–7.53 (m, 1H), 7.34–7.21 (m, 3H), 6.91 (s, 1H), 2.98 (s, 3H).11B NMR (128 MHz, CDCl3) δ 2.45. [Acc. Mass] calculated [M]+: 288.03869, observed [M]+: 288.03925. 13C{1H} NMR spectrum was not obtained due to poor solubility of 9-Cl.Synthesis of 14-Ph
To an ampule fitted with a J-Youngs tap was added compound 12 (0.029 g, 0.15 mmol) which was dissolved in DCM (0.4 mL). BCl3 (0.33 mL, 1 M in DCM) was added, the ampule was sealed and the mixture was stirred at 60 °C for 3 hours. The solvent and excess BCl3 were removed under vacuum and ZnPh2 (0.077 g, 0.35 mmol) was added followed by DCM (1.5 mL). The reaction mixture was stirred for 1 hour at room temperature. The solids were filtered off and the crude product was purified by column chromatography on silica gel (EtOAc/petroleum ether) to give the product, 14-Ph (0.035 g, 66%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.37 (d, J = 4.0 Hz, 1H), 7.35 (dd, J = 7.3, 1.5 Hz, 1H), 7.29 (m, 6H), 7.24–7.18 (m, 5H), 7.18–7.11 (m, 2H), 6.82 (dd, J = 7.5, 3.8 Hz, 2H).13C{1H} NMR (101 MHz, CDCl3) δ 159.2, 137.2, 136.2, 133.7, 129.39, 127.4, 125.9, 125.7, 125.4, 121.6, 117.9, 112.7, 108.8. 11B NMR (128 MHz, CDCl3) δ 0.22. [Acc. Mass] calculated [M + H]+: 365.1278, observed [M + H]+: 365.1269.Synthesis of 14-Pin
To an ampule fitted with a J-Youngs tap was added compound 12 (0.037 g, 0.18 mmol). DCM (0.4 mL) was added followed by BCl3 (0.22 mL, 1 M in DCM). The ampule was sealed and the reaction mixture was stirred for 3 hours at 60 °C after which the solvent/volatiles were removed under vacuum. NEt3 (0.38 mL, 2.7 mmol) was added followed by pinacol (0.021 g, 0.18 mmol) and the reaction mixture was stirred vigorously overnight at room temperature. The crude product was purified on silica-gel (EtOAc/petroleum ether) to give 14-Pin (0.032 g, 55%) as an orange oil. 1H NMR (500 MHz, CDCl3) δ 7.83 (d, J = 4.0 Hz, 1H), 7.70 (d, J = 7.2 Hz, 1H), 7.54 (d, J = 7.8 Hz, 1H), 7.33 (t, J = 7.5 Hz, 1H), 7.29–7.18 (m, 1H), 6.96 (d, J = 4.0 Hz, 1H), 6.77 (d, J = 3.6 Hz, 1H), 1.37 (s, 12H). 13C{1H} NMR (126 MHz, CDCl3) δ 161.1, 138.8, 136.5, 129.8, 127.1, 124.6, 123.8, 120.8, 111.4, 110.5, 81.3, 27.1. 11B NMR (160 MHz, CDCl3) δ 13.75. [Acc. Mass] calculated [M + H]+: 327.1333, observed [M + H]+: 327.1324.Synthesis of 17-Ph
To an NMR tube fitted with a J-Youngs tap was added compound 15 (0.024 g, 0.1 mmol). DCM (0.35 mL) was added followed by BCl3 1 M in DCM (0.22 mL, 0.22 mmol). The tube was sealed and heated to 60 °C for 2.75 hours after which it was cooled and excess BCl3 and DCM were removed under vacuum. ZnPh2 (0.050 g, 0.23 mmol) was added followed by DCM (0.5 mL) and the reaction mixture was left overnight at room temperature. The crude product was purified by column chromatography on silica gel (EtOAc/Hexanes) to give 17-Ph (0.026 g, 64%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.59–7.53 (m, 2H), 7.42–7.38 (m, 5H), 7.34–7.27 (m, 3H), 7.25–7.18 (m, 6H), 7.17–7.11 (m, 2H), 6.89 (d, J = 3.6 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3) δ 152.2, 146.9, 136.6, 134.0, 134.0, 129.7, 127.3, 126.5, 126.5, 126.1, 125.6, 125.0, 119.7, 118.3, 118.3, 114.1, 110.9. 11B NMR (160 MHz, CDCl3) δ 0.08. [Acc. Mass] calculated [M + H]+: 399.16632, observed [M + H]+: 399.16540.Synthesis of 17-Cl
To an ampule fitted with a J-Youngs tap was added compound 15 (0.024 g, 0.1 mmol). DCM (0.35 mL) was added followed by BCl3 (0.22 mL, 1 M in DCM). The ampule was sealed and the reaction mixture was heated to 60 °C for 2.75 hours, after which it was cooled and the solvent/volatiles removed under vacuum and the solid dried to give 17-Cl (0.030 g, 73%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.38 (m, J = 8.2, 1.3, 0.6 Hz, 1H), 7.92 (d, J = 7.2 Hz, 1H), 7.65 (dt, J = 8.3, 0.9 Hz, 1H), 7.62–7.58 (m, 3H), 7.56–7.46 (m, 2H), 6.97 (d, J = 3.7 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3) δ 149.9, 146.8, 134.3, 131.2, 129.9, 127.6, 127.5, 126.8, 126.4, 121.6, 119.9, 118.7, 115.4, 111.3. 11B NMR (128 MHz, CDCl3) δ 3.92. [Acc. Mass] calculated [M]+: 314.01795, observed [M]+: 314.01728.Synthesis of 18-Ph
To an NMR tube fitted with a J-Youngs tap was added compound 16 (0.0250 g, 0.1 mmol DCM (0.35 mL) was added followed by BBr3 (0.22 mL, 1 M in DCM), the tube was sealed, and the reaction mixed for 2 hours at room temperature followed by 1 hour at 60 °C. The solvent/volatiles were removed under vacuum and the crude material dried. ZnPh2 (20 mg, 0.1 mmol) was added followed by DCM (1 mL), the tube was sealed and mixed overnight at room temperature. The product was purified on silica-gel (EtOAc/petroleum ether) to give the pure product, 18-Ph (0.012 g, 29%) as a grey solid. 1H NMR (400 MHz, CDCl3) δ 7.73–7.68 (m, 1H), 7.60 (dt, J = 8.4, 0.7 Hz, 1H), 7.44 (dd, J = 8.1, 1.5 Hz, 4H), 7.33–7.25 (m, 3H), 7.23–7.14 (m, 7H), 7.14–7.08 (m, 2H), 6.85 (d, J = 3.7 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3) δ 159.6, 144.7, 135.4, 133.8, 129.4, 127.5, 127.3, 126.5, 125.5, 125.5, 125.4, 125.2, 122.4, 122.2, 121.7, 117.8, 114.0. 11B NMR (128 MHz, CDCl3) δ 0.88. [Acc. Mass] calculated [M + H]+: 415.14348, observed [M + H]+: 415.14370.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no 769599). We acknowledge SIRCAMS at University of Edinburgh for performing mass spectrometry and Dr G. Nichol for collecting single crystal X-ray diffraction data.References
- (a) T. A. Shah, P. B. De, S. Pradhan and T. Punniyamurthy, Chem. Commun., 2019, 55, 572 RSC; (b) J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618 CrossRef CAS; (c) J. Kalepu, P. Gandeepan, L. Ackermann and L. T. Pilarksi, Chem. Sci., 2018, 9, 4203 RSC.
- For select recent examples: (a) M. Liu, M. Shi and H. Meng, New J. Chem., 2020, 44, 2961–2965 RSC; (b) Y. Qin, G. Li, T. Qi and H. Huang, Mater. Chem. Front., 2020, 4, 1554–1568 RSC.
- (a) For select work on metal catalysed directed indole C–H borylation see: D. W. Robbins, T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 4068 CrossRef CAS. For reviews on directed C–H borylation see: (b) A. Ros, R. Fernandez and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229 RSC; (c) S. A. Iqbal, J. Pahl, K. Yuan and M. J. Ingleson, Chem. Soc. Rev., 2020, 49, 4564–4591 RSC.
- Boronic Acids: Preparation and Applications, ed. D. Hall, Wiley-VCH, Weinheim, 2011 Search PubMed.
- (a) F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed; (b) D. Li, H. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416–8433 RSC; (c) A. Wakamiya and S. Yamaguchi, Bull. Chem. Soc. Jpn., 2015, 88, 1357–1377 CrossRef CAS; (d) A. Escande and M. J. Ingleson, Chem. Commun., 2015, 51, 6257–6274 RSC; (e) L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC; (f) Z. X. Giustra and S. Y. Liu, J. Am. Chem. Soc., 2018, 140, 1184–1194 CrossRef CAS PubMed; (g) E. von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS; (h) S. K. Mellerup and S. Wang, Chem. Soc. Rev., 2019, 48, 3537–3549 RSC; (i) X. Y. Wang, X. Yao and K. Müllen, Sci. China: Chem., 2019, 62, 1099–1144 CrossRef CAS.
- F. Yang, M. Zhu, J. Zhang and H. Zhou, MedChemComm, 2018, 9, 201–211 RSC.
- (a) S. A. Iqbal, J. Cid, R. J. Procter, M. Uzelac, K. Yuan and M. J. Ingleson, Angew. Chem., Int. Ed., 2019, 58, 15381–15385 CrossRef CAS PubMed; (b) In the absence of directing groups C3 borylation dominates see: V. Bagutski, A. Del Grosso, J. Ayuso Carrillo, I. A. Cade, M. D. Helm, J. R. Lawson, P. J. Singleton, S. A. Solomon, T. Marcelli and M. J. Ingleson, J. Am. Oil Chem. Soc., 2013, 135, 474 CrossRef CAS PubMed.
- (a) J. Lv, X. Chen, X.-S. Xue, B. Zhao, Y. Liang, M. Wang, L. Jin, Y. Yuan, Y. Han, Y. Zhao, Y. Lu, J. Zhao, W.-Y. Sun, K. N. Houk and Z. Shi, Nature, 2019, 575, 336–340 CrossRef CAS PubMed; (b) J. Lv, B. Zhao, Y. Yuan, Y. Han and Z. Shi, Nat. Commun., 2020, 11, 1316 CrossRef CAS PubMed.
- T. Fukuda, R. Maeda and M. Iwao, Tetrahedron, 1999, 55, 9151 CrossRef CAS.
- For review articles covering four coordinate boron containing organic materials see: (a) S. K. Mellerup and S. Wang, Trends Chem., 2019, 1, 77–89 CrossRef CAS; (b) Main Group Strategies towards Functional Hybrid Materials, ed. T. Baumgartner and F. Jäkle, WILEY-VCH, 2018 Search PubMed; (c) A. Lorbach, A. Huebner and M. Wagner, Dalton Trans., 2012, 41, 6048–6063 RSC; (d) A. Escande and M. J. Ingleson, Chem. Commun., 2015, 51, 6257–6274 RSC; (e) Y. Rao and S. Wang, Inorg. Chem., 2011, 50, 12263–12274 CrossRef CAS PubMed.
- For select references on this area see (for fuller coverage see review articles in ref. 10): (a) A. Wakamiya, T. Taniguchi and S. Yamaguchi, Angew. Chem., Int. Ed., 2006, 45, 3170–3173 CrossRef CAS PubMed; (b) T. Matsumoto, K. Tanaka and Y. Chujo, Dalton Trans., 2015, 44, 8697–8707 RSC; (c) H. Amarne, C. Baik, S. K. Murphy and S. Wang, Chem. – Eur. J., 2010, 16, 4750–4761 CrossRef CAS; (d) Z. M. Heiden, M. Schedler and D. W. Stephan, Inorg. Chem., 2011, 50, 1470–1479 CrossRef CAS PubMed; (e) A.-C. Smith, D. S. Ranade, S. Thorat, A. Maity, P. P. Kulkarni, R. G. Gonnade, P. Munshi and N. T. Patil, Chem. Commun., 2015, 51, 16115 RSC; (f) C. Dou, Z. Ding, Z. Zhang, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2015, 54, 3648–3652 CrossRef CAS PubMed; (g) A. Job, A. Wakamiya, G. Kehr, G. Erker and S. Yamaguchi, Org. Lett., 2010, 12, 5470–5473 CrossRef CAS; (h) K. Liu, R. A. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2017, 139, 18170–18173 CrossRef CAS; (i) D. L. Crossley, I. A. Cade, E. R. Clark, A. Escande, M. J. Humphries, S. M. King, I. Vitorica-Yrezabal, M. J. Ingleson and M. L. Turner, Chem. Sci., 2015, 6, 5144–5151 RSC; (j) D. L. Crossley, L. Urbano, R. Neumann, S. Bourke, J. Jones, L. A. Dailey, M. Green, M. J. Humphries, S. M. King, M. L. Turner and M. J. Ingleson, ACS Appl. Mater. Interfaces, 2017, 9, 28243–28249 CrossRef CAS PubMed; (k) Y. Rao, H. Amarne, L. D. Chen, M. L. Brown, N. J. Moses and S. Wang, J. Am. Chem. Soc., 2013, 135, 3407 CrossRef CAS PubMed.
- (a) E. Tyrrell and P. Brookes, Synthesis, 2003, 469 CrossRef CAS; (b) P. A. Cox, A. G. Leach, A. D. Campbell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2016, 138, 9145 CrossRef CAS PubMed.
- Y.-L. Rao, T. Kusamoto, R. Sakamoto, H. Nishihara and S. Wang, Organometallics, 2014, 33, 1787 CrossRef CAS.
- C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC.
- D.-Y. Wang, H. I. Minami, C. Wang and M. Uchiyama, Chem. Lett., 2015, 44, 1380 CrossRef CAS.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070 CrossRef CAS.
- R. A. Jagtap and B. Punji, Asian J. Org. Chem., 2020, 9, 326 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental and computational details, copies of NMR spectra for isolated compounds and key in situ NMR spectra. CCDC 2051058 and 2051059. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ob00018g |
| This journal is © The Royal Society of Chemistry 2021 |