
Photoactive rose bengal-based latex via RAFT emulsion polymerization-induced self-assembly†
Charlène
Boussiron
,
Mickaël
Le Bechec
 ,
Julia
Sabalot
,
Sylvie
Lacombe
and
Maud
Save
*
,
Julia
Sabalot
,
Sylvie
Lacombe
and
Maud
Save
*
CNRS, University Pau & Pays Adour, E2S UPPA, Institut des Sciences Analytiques et de Physico-Chimie pour l'Environnement et les Matériaux, IPREM, UMR5254, 64000, PAU, France. E-mail: maud.save@univ-pau.fr
First published on 13th October 2020
Abstract
Copolymerization of acrylic acid (AA) with a photosensitizer-based comonomer by reversible addition fragmentation transfer (RAFT) polymerization produces a reactive stabilizer for emulsion polymerization-induced self-assembly (PISA). This polymerization in aqueous dispersed medium is a surfactant-free and solvent-free environmentally friendly process for the efficient design of dye-supported polymer colloids dispersed in water. Rose Bengal (RB) was selected as a photosensitizer for the synthesis of two types of comonomers (MRB), vinyl benzyl rose bengal (VBRB) and ethyl acrylate rose bengal (EARB). The effects of the structure of MRB and solvent for RAFT copolymerization with AA were investigated. While VBRB induced strong retardation in 1-4-dioxane, to a greater extent compared to EARB, such retardation was overcome in polar dimethyl sulfoxide. The high level of chain end fidelity of PAA-based macromolecular chain transfer agent, as highlighted by UV-visible spectroscopy and proton NMR spectroscopy, allowed for efficient chain extension. Self-assembled amphiphilic block copolymers synthesized by RAFT-mediated emulsion polymerization were stable, monodisperse core–shell particles of 90–100 nm diameter. The shell is a hydrophilic poly(ammonium acrylate-co-MRB) statistical copolymer and the core is a film-forming poly(alkyl acrylate) of poly(n-butyl acrylate), poly(ethyl acrylate) or poly(n-butyl acrylate-co-ethyl acrylate). A second synthetic route is proposed to label the particle core with a photosensitizer. Interfacial singlet oxygen production was monitored by the degradation of furfuryl alcohol quencher under visible light irradiation. The average quantum yield of supported rose bengal (ϕΔ = 0.64 ± 0.10) in a similar range to that of free rose bengal in water (ϕΔ = 0.76 ± 0.10) proves the photoactivity of photosensitizer-grafted waterborne latexes.
Introduction
Photosensitized reactions are defined as the absorption of radiation by an organic molecular species, called a photosensitizer, which induces the modification of another molecular species, either by electron transfer (type I mechanism) or by energy transfer (type II mechanism), with regeneration of the photosensitizer to its ground state without chemical alteration. Singlet oxygen (1O2) is a powerful and selective oxidant, mainly produced by energy transfer from the triplet state of the photoactivated sensitizer to the ground state of oxygen.1,2 Singlet oxygen is a versatile synthetic reagent used for fine chemical synthesis to produce, for instance, pharmaceutical ingredients like artemisinin.3,4 Photooxidation is also of interest in wastewater treatment to promote the degradation of insecticides and pesticides.5,6 Under specific irradiation of a photosensitizer, the produced singlet oxygen has the ability to damage living tissue by oxidation of the cell wall, thus acting as an oxidizing agent in photodynamic processes, including blood sterilization, photodynamic therapy (PDT),7 and photodynamic inactivation (PDI) (i.e. light activated antimicrobial agent).8–12 In the present work, Rose Bengal (RB) was selected as a non-toxic, commercially available photosensitizer.13 It exhibits an absorption band in the visible range (540–560 nm), a high singlet oxygen quantum yield (ϕΔ ∼ 0.6–0.75) in polar solvents and a sodium carboxylate function as a platform for chemical reaction (Scheme S1†). Immobilization of photosensitizers on solid substrates improves their handling, recyclability and stability and facilitates purification steps to remove the photocatalyst from reactants in fine chemistry.14 Among the substrates for organic photosensitizers, polymers have attracted increasing interest because of their versatility, in terms of mechanical properties and processability. Especially, polymer colloids can confer additional properties to the photoactive materials, such as higher interfacial area, swelling ability, high stability in water or alcoholic medium, ON/OFF activity or film-forming properties.15–18 Waterborne latexes produced by emulsion polymerization are a class of polymer colloids directly synthesized in aqueous dispersed media. Emulsion polymerization19 is an inherently safe and environmentally friendly process that is widely implemented at an industrial level to produce stable polymer colloids dispersed in a continuous water phase. Waterborne latexes are used for a myriad of applications in paints and coatings, adhesives and sealants, paper, cement and concrete additives, floor polish, textiles, synthetic rubbers, and reinforced plastics, but also in cosmetics, biomaterials and other high-tech areas. By producing one material, waterborne latex grafted with a photosensitizer could be handled as a photoactive colloid or cast to produce films. Designing the RB monomer should allow the synthesis of photosensitizer-based waterborne latex by conventional emulsion polymerization, but one has to address the solubility of the powder form of chromophore-based monomer either in water or in the monomer phase. Polymerization-induced self-assembly (PISA)20–24 is a versatile method to control the molar mass and location of the RB monomer units. For instance, PISA was implemented to control the design of fluorescently labelled particles either by post-modification with the fluorescent tag,25,26 or by copolymerization of the dye monomer with the core-forming monomer.27 Moreover, the synthesis of waterborne latex by PISA has other advantages like suppression of labile molecular surfactant, direct synthesis of core–shell particles, control of macromolecular features of the core-forming polymer (molar mass, dispersity, branching…) or control of particle morphology (sphere, fiber, vesicle).20–24 The anchored hydrophilic shell of amphiphilic block copolymer particles synthesized by PISA also offer the opportunity to reinforce mechanical properties or water barrier properties of the final polymer films.28,29Herein, poly(acrylic acid)-b-poly(alkyl acrylate) will be the polymeric system of choice in the present work to synthesize photosensitizer-based core–shell particles by emulsion PISA. Since the pioneering work on PISA involving oligo(acrylic acid) as the macromolecular chain transfer agent (macroCTA) and polymerization of n-butyl acrylate (nBA) by emulsion polymerization,30 reversible addition–fragmentation chain transfer (RAFT) polymerization has been the main method to mediate emulsion PISA of alkyl acrylates by poly(acrylic acid) (PAA) macro(CTA).31–39 Recently, poly(2-(N-acryloyloxy)ethyl pyrrolidone) (PNAEP) was involved as a non-ionic stabilizer block in the synthesis of poly(n-butyl acrylate) latex by RAFT emulsion polymerization.40 Nitroxide-mediated polymerization also produced polyacrylic diblock copolymer latex from PAA macroalkoxyamine.41,42
A versatile way to covalently anchor rose bengal photosensitizer is to synthesize a polymerizable monomer (MRB) that can be copolymerized with either acrylic acid (AA) to functionalize the particle shell or with alkyl acrylate for core functionalization (Scheme 1). For this purpose, two types of MRB were synthesized in the present work, vinyl benzyl rose bengal (VBRB, Scheme S1†) and ethyl acrylate rose bengal (EARB, Scheme S2†). Herein, the alkyl acrylate forming the particle core will range from n-butyl acrylate (nBA), ethyl acrylate (EA) or nBA/EA mixture to tune the glass transition temperature from −15 °C to −45 °C.43 This study will investigate whether the chemical nature of the polymer core or the location of the photosensitizer (shell versus core) might affect interfacial 1O2 production. In addition to being used as a colloidal substrate in the present work, the design of poly(alkyl acrylate) particles offers a versatile strategy to use the supported photosensitizer as a polymer film by simple casting of film-forming waterborne latex.18 It has been previously reported that the mechanical properties of polyacrylics vary according to the pendant chain length,44 and could be reinforced by the presence of H-bonding poly(acrylic acid) percolated nanophase.29 Poly(acrylic acid) has been widely used in emulsion polymerization. Herein, we will take advantage of the hydrophilic character of neutralized PAA to transport the covalently bonded hydrophobic rose bengal photosensitizer moiety into the aqueous continuous phase of emulsion polymerization (route 1 in Scheme 1). The main route of this work relies indeed on the introduction of the rose bengal photosensitizer in the outer shell via the design of P(AA-co-MRB) macromolecular chain transfer agent (macroCTA) (route 1 in Scheme 1), but the copolymerization of MRB with the hydrophobic core monomer will be also studied to a lesser extent (route 2 in Scheme 1). Prior to implementing emulsion PISA, it was relevant to investigate the control of AA RAFT polymerization and its copolymerization with MRB comonomers in three different solvents (1,4-dioxane, DMF, DMSO) to produce living macromolecular chain transfer agent. Since the early work of Chiefari et al.,45 RAFT polymerization has been the most widely implemented reversible deactivation radical polymerization (RDRP) technique for AA polymerization carried out in solvents such as 1,4-dioxane,46–48 ethanol,47 2-propanol,47 DMF49 or water.50,51 DMSO is an interesting polar solvent for MRB solubilisation, as an alternative to more hazardous solvents, such as 1,4-dioxane or DMF. To the best of our knowledge, RAFT polymerization of AA in DMSO has never been reported so far. Measurement of the quantum yield of singlet oxygen is a means to accurately compare the photoactivity of each synthesized latex to produce singlet oxygen at the interface with water.
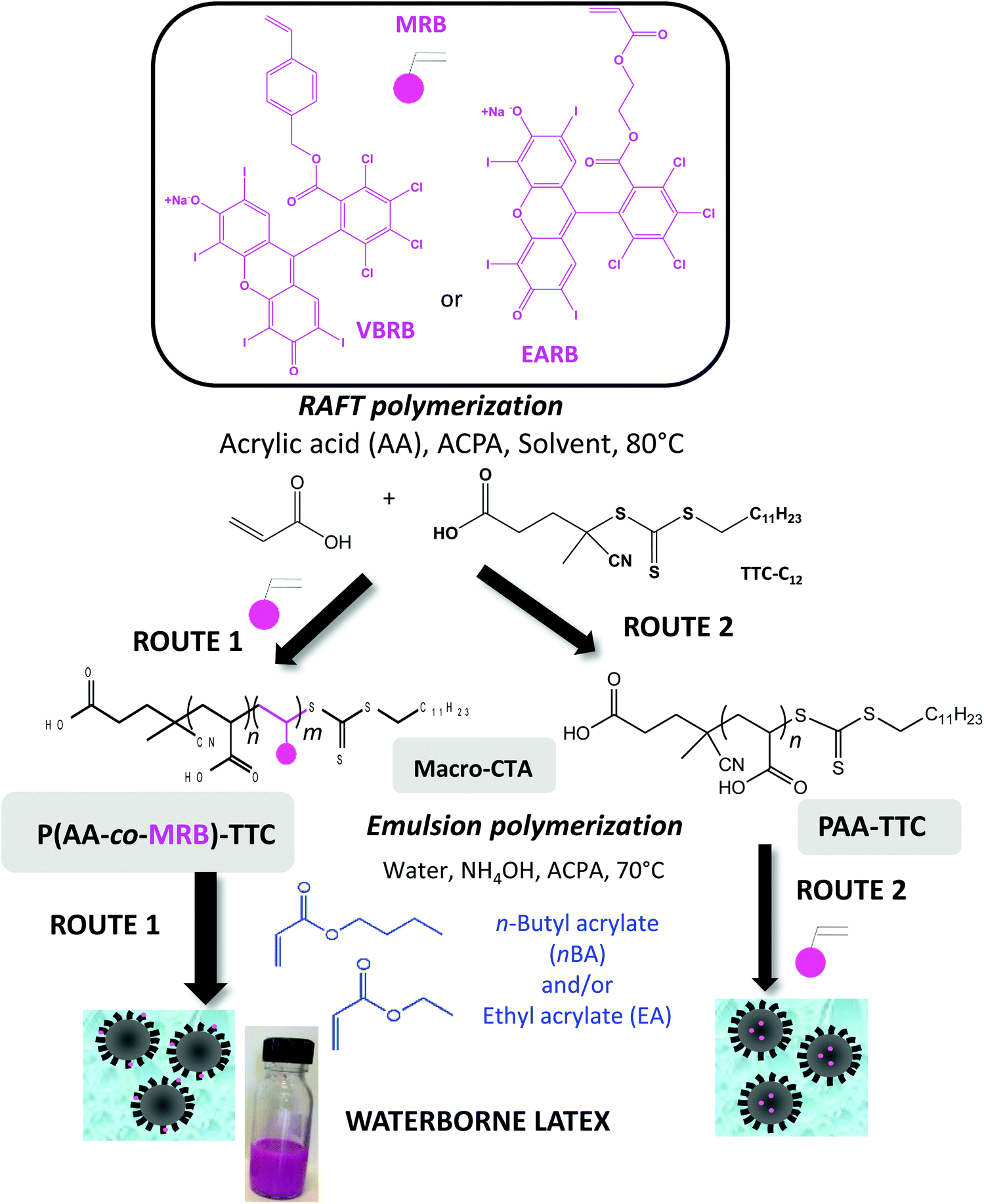 | ||
| Scheme 1 Syntheses of P(AA-co-MRB)-TTC and PAA-TTC by RAFT polymerization used as reactive stabilizers for the synthesis of photosensitizer-based waterborne latex by emulsion PISA. Route 1 for RB in the shell and route 2 for RB in the core. | ||
Experimental section
Materials
Acrylic acid (AA) (Sigma-Aldrich, 99%), ethyl acrylate (EA) (Sigma-Aldrich, 99%) and n-butyl acrylate (nBA) (Sigma-Aldrich, >99%) were mixed with inhibitor removers (Sigma-Aldrich) for 30 min prior to being filtered and used for polymerization. Acryloyl chloride (Sigma-Aldrich, 97%), 2-bromoethanol (Sigma-Aldrich, 95%), triethylamine (TEA) (Sigma-Aldrich, 99.5%), 4-vinylbenzyl chloride (VBC) (Sigma-Aldrich, 90%), Rose Bengal (RB) (Sigma-Aldrich, 95%), 1,3,5-trioxane (Sigma-Aldrich, ≥99%), 4,4′-azobis(4-cyanopentanoic acid) (ACPA) (Fluka, ≥98%), 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (TTC) (Sigma-Aldrich, 97%), (trimethylsilyl)diazomethane solution (Sigma-Aldrich, 2.0 M in diethyl ether), hydrochloric acid (HCl) (Sigma-Aldrich, ACS reagent, 37%), ammonium hydroxide (NH4OH) (Acros Organic, 28–30% in water), anhydrous magnesium sulfate (MgSO4) (Sigma-Aldrich, ≥99.5%), sodium chloride (NaCl) (Sigma-Aldrich, ≥99%), furfuryl alcohol (Sigma-Aldrich, 98%), 1,4-dioxane (Sigma-Aldrich, 99.8%), dichloromethane (DCM) (VWR), diethyl ether (VWR), dimethylformamide (DMF) (Sigma-Aldrich), dimethyl sulfoxide (DMSO) (VWR), tetrahydrofuran (THF) (VWR), toluene (Sigma-Aldrich), and deuterated chloroform (CDCl3) (Sigma-Aldrich) were used as received. Deionized (DI) water was used for all emulsion polymerizations. Syntheses of vinyl benzyl rose bengal (VBRB) and ethyl acrylate rose bengal (EARB) are described in the ESI (Schemes S1 and S2†).Synthesis of macromolecular chain transfer agent (PAA-TTC, P(AA-co-VBRB)-TTC, P(AA-co-EARB)-TTC)
In a typical experiment (PAA-3), AA (2.355 g, 3.27 × 10−2 mol), TTC (188 mg, 4.67 × 10−4 mol), ACPA (13 mg, 4.67 × 10−5 mol), 1,3,5-trioxane (160 mg, 1.78 × 10−3 mol) and 1,4-dioxane (9.725 g, 9.442 mL) were introduced into a 50 mL round-bottomed flask and stirred magnetically until complete dissolution (Scheme S3†). The mixture was degassed by nitrogen bubbling over 20 min. A sample was withdrawn under nitrogen at time t = 0. The round-bottomed flask was placed into an oil bath previously heated to 80 °C. Samples were withdrawn every 15 min until 1 h and then every 30 min until the reaction was stopped (3 h) by cooling down in an ice-water bath and by the introduction of oxygen into the mixture. The polymer was precipitated in diethyl ether and dried. A part of each sample was reacted with trimethylsilyldiazomethane to methylate the acid group of PAA, and thus, allow size exclusion chromatography (SEC) analyses. The procedure used to synthesize P(AA-co-MRB)-TTC macromolecular RAFT agents was similar, except that VBRB or EARB, as MRB monomers, was added to the initial mixture.Monomer conversion (x) was calculated from proton nuclear magnetic resonance (1H NMR) spectra using 1,3,5-trioxane as an internal standard (eqn (1)).
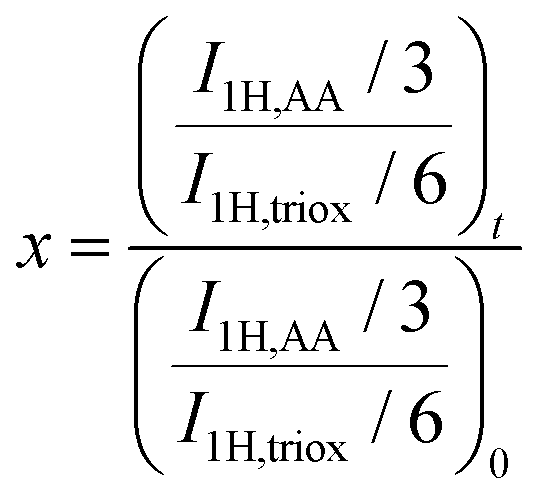 | (1) |
Synthesis of poly(alkyl acrylate)-based latexes by emulsion PISA
Emulsion polymerization of nBA is described in detail, but the procedures also apply to the emulsion polymerization of EA and the emulsion copolymerization of nBA and EA. Emulsion polymerizations were performed by targeting 23 wt% of solid content at 100% monomer conversion.Syntheses of PAA-b-PnBA and P(AA-co-MRB)-b-PnBA latexes
In a typical experiment (LBA-12 in Table S7†), PAA-TTC macroCTA (546 mg, 1.6 × 10−4 mol) was dissolved in deionized water (42.5 g). Then, the pH of the solution was adjusted to 5.4 by adding 1.4 g of NH4OH 28–30% in water. The equivalent of 8.5 mg (3 × 10−5 mol) of ACPA initiator was added via 1.1 g of a stock solution (38.6 mg of ACPA in 5.0 g of a stock solution of 1 mL NH4OH 28–30% and 62.2 g deionized water), to target a molar ratio between macroCTA and initiator of 5.2. Finally, nBA (13.8 g, 0.11 mol) was introduced and a biphasic system was formed. The emulsion was simultaneously stirred and purged with argon for 45 min in an ice bath to prevent monomer evaporation. The flask was introduced into an oil bath preheated to 70 °C and the reaction was stirred using a collapsible blade stirrer at 300 rpm. After 4 h, the flask was dipped into an ice bath and opened to air. For the latexes synthesized from a macroCTA synthesized in DMSO or in DMF, the residual solvent contained in PAA was removed by dialysis of the latex for 2 days.The monomer conversion for emulsion polymerization was determined by gravimetry. Around 1 mL of latex was withdrawn and dried overnight at room temperature in an aluminium cap. Conversions were calculated based on eqn (2) with τPnBA,exp as the experimental solids content calculated from eqn (3) and τPnBA,theo as the theoretical solids content at 100% monomer conversion (eqn (4)). Note that this calculation holds true for PAA-b-PEA latexes and RB-containing latexes.
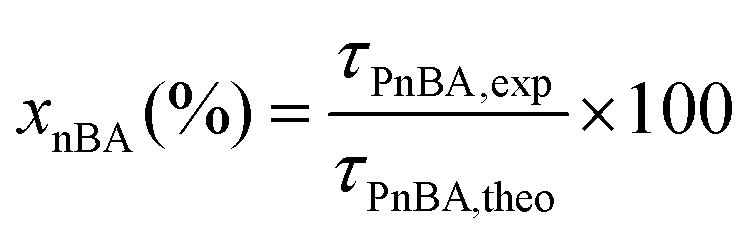 | (2) |
| τPBA,exp = Dry extract − τsalts | (3) |
| τPnBA,theo = mBA,0/mlatex,0 | (4) |
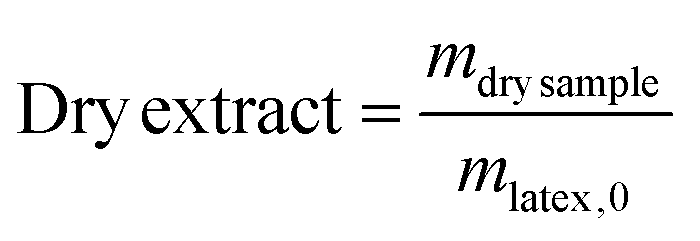 | (5) |
 | (6) |
Syntheses of PAA-b-P(nBA-co-MRB) latexes
In a typical experiment (LBA-VBRB3 in Table 3), PAA-TTC macroCTA (PAA-8, 546 mg, 1.6 × 10−4 mol) was dissolved in deionized water (42.5 g). Then, the pH of the solution was adjusted to 5.4 by adding 1.6 g of NH4OH 28–30% in water. The equivalent of 8.7 mg (3.1 × 10−5 mol) of ACPA initiator was added via 0.9 g of a stock solution (57.9 mg of ACPA in 5.9 g of a stock solution of 0.95 g NH4OH 28–30% and 25 g deionized water), to target a molar ratio between macroCTA and initiator of 5.1. Finally, a mixture of VBRB (7 mg) and nBA (13.8 g, 0.11 mol) was introduced and a biphasic system was formed. The next steps are the same as those for PAA-b-PnBA synthesis.Methylation of PAA-b-poly(alkyl acrylate) block copolymer
Prior to performing size exclusion chromatography (SEC) analyses in THF, polymers were methylated to derivatize poly(acrylic acid) into poly(methyl acrylate). For that purpose, (trimethylsilyl)diazomethane (TMS) was used as an efficient reactant. In a typical methylation, 100 mg of latex (23 mg of polymer) and 16 mL of THF were mixed. One drop of 1 M hydrochloric acid (HCl) solution was added to ensure that all the acid units were protonated. The solution of TMS was added dropwise to the mixture until the yellow coloration persisted and there were no more bubbles (approximately 10 drops). The mixture was stirred for 3 hours at room temperature then dried under the fume hood. The procedure was repeated one more time and the dried polymer was dissolved in dichloromethane to be extracted with DI water three times in order to remove the residual salts. After evaporation of dichloromethane and drying, copolymers were dissolved in THF to be analysed by SEC.Characterization methods
NMR spectra were recorded using a Bruker 400 MHz spectrometer at 25 °C. The polymers were analysed by size exclusion chromatography (SEC) operating in THF, at 30 °C (flow rate: 1 mL min−1). The SEC apparatus is equipped with a Viscotek-Malvern VE 1122 automatic injector, Styragel columns (HR 5E and 4E, 7.8 × 300 mm, noted as set no. 1) working in series, a Wyatt Heleos II Multi Angle Laser Light Scattering detector (MALLS, 18 angles, λ0 = 664.4 nm), a viscometer (Wyatt Viscostar II), a Viscotek-Malvern VE 3210 UV-visible detector and a Viscotek-Malvern VE 3580 refractive index (RI) detector. A second set of SEC Shodex columns were used (KF 803L, KF 804L, KF 806L, 8 × 300 mm, noted as set no. 2). Toluene was used as a flow marker. Polymer samples were prepared at concentrations from 3 to 5 g L−1. The number-average molar mass (Mn) and molar mass distribution (Đ) were obtained either from a calibration based on polystyrene (PS) standards or from MALLS (Astra software). See the last part of the ESI† for details on (i) the calculation of number-average molar mass (Mn) of PAA-based macroCTA calculated from the experimental molar mass of the methylated samples, (ii) the error between the molar mass of polystyrene standards and poly(alkyl acrylate) based on Mark–Houwink parameters, (iii) refractive index increments (dn/dc) of polymers for MALLS analysis, and (iv) equation for theoretical Mn. UV-visible absorption spectra were recorded at 25 °C using a PerkinElmer Lambda 850 spectrophotometer. The hydrodynamic diameter (Dh) was measured by dynamic light scattering (DLS) on a Nano-ZS (ZEN3600 Malvern) zetasizer, at a concentration of particles of 0.05 g L−1. A He–Ne laser (4.0 mW power) was used, operating at a wavelength of 633 nm at an angle of 173°. The average number of latex particles per litre of latex (Np) was calculated from the latex average hydrodynamic diameter (eqn (7)). The polydispersity index (PDI) was measured with the cumulant model for monomodal distributions.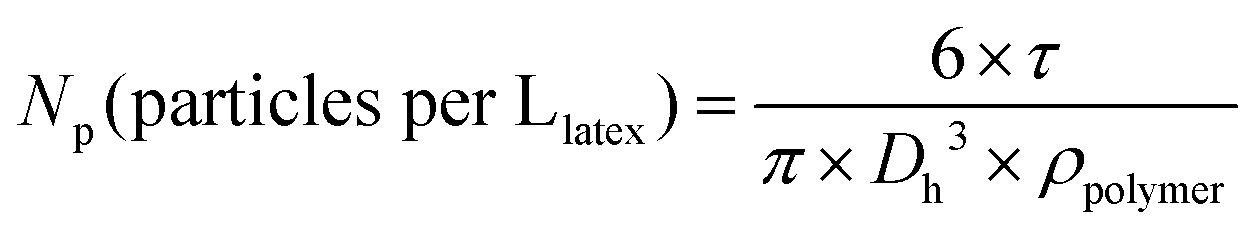 | (7) |
Differential scanning calorimetry (DSC) was carried out using the Q100 apparatus from TA Instruments. Analysis was conducted at a heating rate of 10 °C min−1, under nitrogen gas flow, from −100 °C to 150 °C. The measurements method included a first heating step from −100 to 150 °C at 10 °C min−1, a cooling step from 150 °C to −100 °C at 10 °C min−1, and a second heating step from −100 °C to 150 °C at 10 °C min−1. Glass transition temperatures (Tg) of the copolymer were measured during the second heating step.
The setup for monitoring of singlet oxygen production is detailed in the ESI.†
Results and discussion
Syntheses of reactive stabilizers by RAFT polymerization: PAA-TTC and P(AA-co-MRB)-TTC macroCTA
The high reactivity of acryclic acid in radical polymerization induces the occurrence of irreversible transfer reactions to solvent, even for RDRP processes.47,52 Such side reactions can be minimized for RAFT polymerization of AA by using 1,4-dioxane instead of protic alcoholic solvents.47 In the present work, more polar solvents, such as DMF or DMSO, were useful to solubilize rose bengal-based comonomer, as MRBs are less soluble in 1,4-dioxane and insoluble in water. Consequently, RAFT homopolymerization of AA mediated by TTC was first carried out in three different solvents (1,4-dioxane, DMF and DMSO, Table S1†) to compare the level of control prior to copolymerization of AA with rose bengal-based comonomer (Table 1 and Table S3†). Lai et al. have successfully controlled the polymerization of AA in DMF by RAFT polymerization mediated by a trithiocarbonate chain transfer agent,49 but, to the best of our knowledge, RAFT polymerization of AA in DMSO has never been reported so far. While DMF is a carcinogenic solvent, DMSO is a non-hazardous solvent widely used in industry. Herein, AA was successfully polymerized at 80 °C by RAFT polymerization mediated by 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (trithiocarbonate chain transfer agent, TTC) (Scheme S3 in the ESI†) using 4,4′-azobis(4-cyanopentanoic acid) (ACPA) initiator, leading to high conversions (88–93%) in the three different solvents (1,4-dioxane, DMSO or DMF) (Table S1†). Kinetics of AA RAFT polymerization in 1,4-dioxane and in DMF perfectly superimposed with a linear increase of logarithmic conversion versus time up to 80% conversion, indicating a constant concentration of propagating radicals (Fig. S2† and Fig. 1). The good reproducibility of AA kinetics is reported in Fig. S1.† At higher conversions, the loss of linearity suggests a slight occurrence of irreversible termination reactions or transfer to a polymer, producing less reactive radicals. Interestingly, DMSO induced a 2.7-fold increase of AA polymerization rate, while maintaining a constant concentration of radicals up to 80% conversion (Fig. S2† and Fig. 1). Such discrepancies between the polymerization rates could be due to intrinsic characteristics of different solvents. The polarity of DMSO, expressed either by the normalized polarity (ENT) values53 or by the dipolarity/polarizability parameters,54 is higher than those for 1,4-dioxane and DMF (Table S2†). Previous studies reported an effect of the solvent polarity on the radical polymerization process. For instance, Beuermann et al.55 showed an increase of the propagation rate coefficient of methyl methacrylate with the solvent dipolarity/polarizability of ionic liquids or polar organic solvents like DMSO. Horn et al.56 also evidenced the relationship between the dipolarity/polarizability of the solvent and the activation rate constant in atom transfer radical polymerization (ATRP). In the present work, the increase of AA polymerization rate with the solvent polarity may arise from a better solvation of the polar polymeric radicals, and thus a higher reactivity. The increase of polymerization rate in DMSO is particularly relevant for the implementation of RAFT copolymerization of AA with rose bengal-based comonomer (MRB) (Fig. 1, Table 1 and Table S3†). Indeed, 0.2–0.5 mol% of VBRB induces a strong retardation of AA RAFT polymerization in 1,4-dioxane (to a lesser extent for EARB), which is fully overcome in DMSO and observed at an intermediate level (40% slope decrease) in DMF (Fig. 1).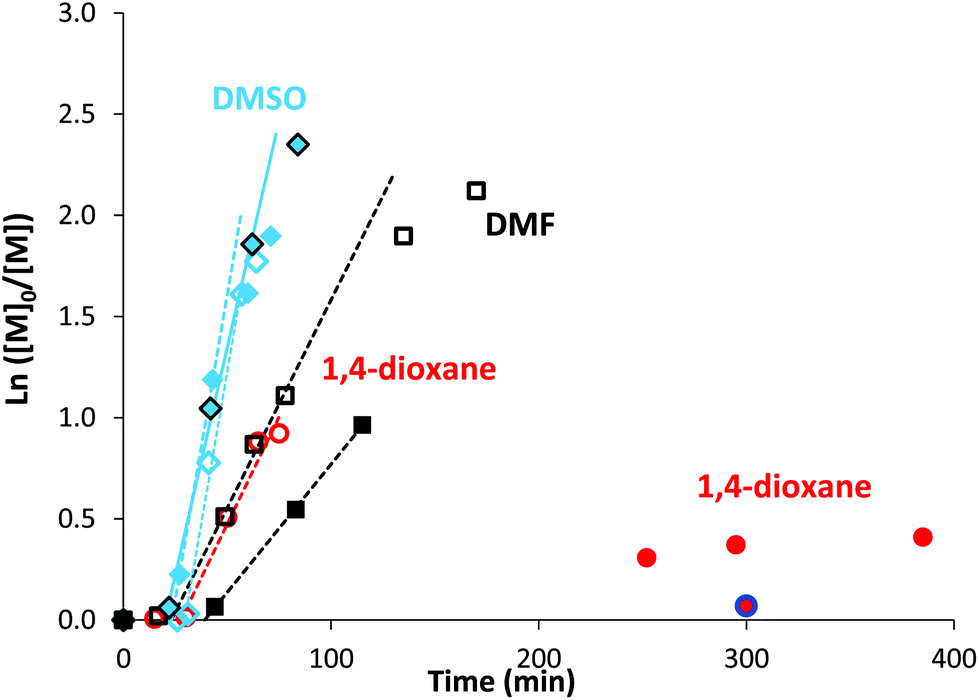 | ||
Fig. 1 Comparison of semilogarithmic conversion of AA versus time for the RAFT polymerization carried out at T = 80 °C in different solvents: diamonds for DMSO, squares for DMF and circles for 1,4-dioxane. Empty signals for 0 mol% of MRB ( PAA-8, □ PAA-10, PAA-8, □ PAA-10,  PAA-7), and plain symbols for copolymerization of AA with 0.25 mol% of EARB ( PAA-7), and plain symbols for copolymerization of AA with 0.25 mol% of EARB ( PAA-EARB8), 0.25 mol% of VBRB ( PAA-EARB8), 0.25 mol% of VBRB ( PAA-VBRB6, in dark), 0.25 mol% of VBRB (■ PAA-VBRB7), 0.21 mol% of EARB ( PAA-VBRB6, in dark), 0.25 mol% of VBRB (■ PAA-VBRB7), 0.21 mol% of EARB ( PAA-EARB6), and 0.50 mol% of VBRB ( PAA-EARB6), and 0.50 mol% of VBRB ( PAA-VBRB3). See Table 1, Tables S1 and S3† for polymer descriptions. PAA-VBRB3). See Table 1, Tables S1 and S3† for polymer descriptions. | ||
| Expt | Solvent | MRB |
f
MRB,0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (mol%) b (mol%) |
[ACPA]0 (10−3 mol L−1) | [TTC]0 (10−2 mol L−1) |
|
|
|
Convc (%) | M n,th (g mol−1) | M n,PS (g mol−1) | Đ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Synthesis carried out in the dark. b Initial molar fraction of MRB versus AA, fMRB,0 = nMRB,0/(nMRB,0 + nAA,0). c Conversion determined by 1H NMR spectroscopy in DMSO-d6 (eqn (1)). d Theoretical Mn of P(AA-co-MRB)-TTC calculated from eqn (S16).† e M n of P(AA-co-MRB)-TTC recalculated from Mn of P(MA-co-MRB)-TTC determined by SEC with polystyrene calibration in THF (eqn (S6)†). f Dispersity (Đ = Mw/Mn). g Analysed with set no. 1 of SEC columns. h Analysed with set no. 2 of SEC columns (see the Experimental section). See Table S3† for additional experiments. | ||||||||||||
| PAA-EARB1 | 1,4-Dioxane | EARB | 0.10 | 4.4 | 4.0 | 0.6 | 9.0 | 70 | 92 | 5060 | 4320g | 1.4 |
| PAA-EARB2 | 1,4-Dioxane | EARB | 0.10 | 4.3 | 4.0 | 0.7 | 9.3 | 70 | 70 | 3930 | 4720h | 1.1 |
| PAA-EARB3 | 1,4-Dioxane | EARB | 0.10 | 2.7 | 2.4 | 1.2 | 8.9 | 120 | 85 | 7790 | 5510h | 1.4 |
| PAA-VBRB1 | 1,4-Dioxane | VBRB | 0.10 | 4.2 | 3.9 | 0.7 | 9.3 | 72 | 87 | 4900 | 4020g | 1.9 |
| PAA-EARB4a | DMSO | EARB | 0.36 | 4.3 | 4.0 | 2.4 | 9.3 | 70 | 83 | 4560 | 4430h | 1.2 |
| PAA-VBRB2a | DMSO | VBRB | 0.25 | 4.2 | 4.0 | 1.7 | 9.5 | 70 | 90 | 4900 | 5210h | 1.4 |
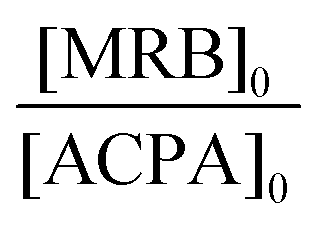
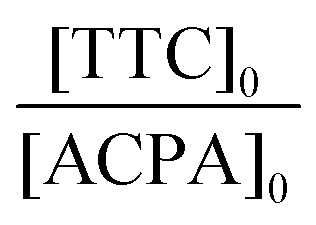
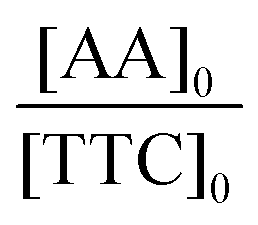
It should be noticed that minor retardation was observed for polymerization carried out in 1,4-dioxane in the presence of 0.5 mol% of free RB compared to inhibition of polymerization for the copolymerization of 0.5 mol% of VBRB (Fig. S6†). Also, RAFT copolymerization of AA and MRB carried out in the dark exhibited a comparable polymerization rate to that of copolymerization under ambient light in both DMSO (Fig. 1) and 1,4-dioxane (Fig. S6†). Increasing the molar fraction of MRB comonomer induced a decrease of the polymerization rate (Fig. 1, Fig. S6 and S7†). In 1,4-dioxane, the retardation of AA copolymerization is particularly obvious for a molar ratio of MRB comonomer over ACPA initiator above unity (Table 1 and Table S3†). The hypothesis of light-activated side reactions inducing retardation of the RAFT AA/MRB copolymerization is inconsistent with the literature. Indeed, while xanthene dyes (including RB) are able to act as an initiating system, producing radicals from the trithiocarbonate chain transfer agent by photoinduced electron/energy transfer (PET) under specific LED-driven light irradiation, xanthene dyes are inactive in radical production under simple ambient visible light.57 As no specific irradiation was used for the RAFT AA/MRB copolymerization reported in the present study, it is unlikely that a side PET-RAFT polymerization process occurs. Thus, the retardation of polymerization is due to the chemical structure of MRB, rather than MRB-induced side photochemical processes. This is confirmed by a more pronounced polymerization retardation induced by vinyl benzyl rose bengal compared to that of ethyl acrylate rose bengal in 1,4-dioxane (Fig. 1 and Fig. S6†). A fraction of 0.1 mol% (∼1.5 wt%) of EARB or VBRB induced a decrease of the apparent rate constant (i.e. slope of ln([M]0/[M]) = f(t)) of 25% or 45%, respectively, compared to AA RAFT polymerization (Fig. S6†). A quasi-inhibition of polymerization is observed for 6 hours with 0.5 mol% of VBRB (6.9 wt%) and polymerization kinetics in the presence of 0.1 mol% of VBRB is typical of slow initiation (PAA-VBRB3 in Fig. S6†). This inhibition is not ascribed to a VBRB solubility issue, as the limiting concentration for the solubility of VBRB measured in 1,4-dioxane and DMSO (see Table S12†) is far beyond the VBRB concentration in AA RAFT copolymerization (0.3 × 10−2 mol L−1 < [VBRB] < 1.7 × 10−2 mol L−1). It has been previously reported that protons at the benzylic position are rather labile and prone to radical abstraction or other transfer reactions.58–61 While these previous studies showed an impact on macromolecular features, like a broader molar mass distribution, they did not show such inhibition of polymerization kinetics. The RB pendant group probably has an additional effect on the radical produced after the transfer reaction. The higher polymerization rate of AA/MRB RAFT copolymerization in polar DMSO might kinetically counterbalance such side reactions, as no retardation is observed by adding VBRB or EARB (Fig. 1). Increasing the fraction of both rose bengal-based comonomer did not affect the length of the inhibition period of RAFT polymerization, but induced an earlier deviation from linearity characteristics of a decrease of radical concentration (Fig. S7†). As an intermediate conclusion, high monomer conversions are reached in DMSO for the successful synthesis of P(AA-co-MRB)-TTC statistical copolymer by RAFT polymerization mediated by TTC.
The linear increase of experimental Mnversus conversion, close to that of the theoretical Mn (Fig. S3†), supports a good level of control of the RAFT homopolymerization of AA mediated by TTC in DMSO, 1,4-dioxane and DMF. The slight deviation of Mnversus linearity observed above 75% conversion (Fig. S3†) can be ascribed to transfer reactions to the solvent, as reported by Loiseau et al.47 and/or inaccuracy in the SEC values due to long-chain branching (LCB). Indeed, radical polymerization of AA is prone to transfer reactions to polymers, leading to branching.62 In the case of long-chain branching (LCB), the variation of the hydrodynamic volume of chains may affect the determination of Mn. An increase of Mnversus conversion is still observed for RAFT copolymerization of AA with MRB in DMSO (Fig. 2).
 | ||
Fig. 2 Evolution of the number-average molar mass and the dispersity versus monomer conversion for the RAFT (co)polymerization of AA in DMSO with 0 mol% of MRB ( PAA-8), 0.36 mol% of EARB ( PAA-8), 0.36 mol% of EARB ( PAA-EARB4) or 0.25 mol% of VBRB ( PAA-EARB4) or 0.25 mol% of VBRB ( PAA-VBRB6). Dotted line: theoretical number-average molar mass. Mn of PAA-TTC recalculated from Mn of PMA-TTC (eqn (S7)†), as determined by SEC/PS calibration in THF. PAA-VBRB6). Dotted line: theoretical number-average molar mass. Mn of PAA-TTC recalculated from Mn of PMA-TTC (eqn (S7)†), as determined by SEC/PS calibration in THF. | ||
The series of PAA-TTC and P(AA-co-MRB)-TTC synthesized in the three different solvents exhibited monomodal SEC chromatograms with dispersity values below 1.4 at final conversion (Table 1, Fig. S4, S8, Tables S1 and S3†). The shift of the narrow SEC chromatograms of PAA-TTC towards lower elution volumes throughout polymerization confirmed the control of polymerization in different solvents (Fig. S5†).
Characterization of chain end fidelity of PAA-TTC and P(AA-co-MRB)-TTC macroCTA
Among the criteria for controlled radical polymerization, most important for the synthesis of latex particles by PISA is to assess the retention of the reactive trithiocarbonate end groups of both PAA-TTC and P(AA-co-MRB)-TTC polymers further used as reactive stabilizers in emulsion polymerization (Scheme 1). For this purpose, the end group fidelity of the macromolecular chain transfer agents was first analyzed by UV-visible spectroscopy in order to calculate the average number of moles of TTC end groups per number of moles of PAA chains (NTTC/chain, Fig. S11 and S12†). The TTC function exhibits a maximum absorption band, assigned to a π–π* transition, at 307 nm in ethanol (Fig. S9†). The average values of NTTC/chain for PAA-TTC are 1.0 ± 0.2 (standard deviation based on 8 samples), 1.2 ± 0.1 (based on 2 samples) and 0.8 (based on 1 sample) for polymerization carried out, respectively, in 1,4-dioxane, DMSO and DMF (Table S4†). The good level of chain end fidelity is also maintained for copolymerization of AA with either EARB or VBRB, as the average values of NTTC/chain are 1.0 ± 0.4 (standard deviation based on 8 samples), 1.3 ± 0.2 (based on 5 samples) and 1.2 ± 0.2 (based on 2 samples) for polymerization carried out in, respectively, 1,4-dioxane, DMSO and DMF (Fig. S11 and Table S4†). The TTC chain end of the macroCTA was also evidenced by 1H NMR spectroscopy with the presence of additional signals corresponding to the carbon chain of the Z stabilizing fragment of the initial TTC chain transfer agent (Fig. S10†). A similar trend for the degree of functionalization was observed (Fig. S12†). So, considering the standard deviation (±0.2 or ±0.4) for each technique and the relative error between NTTC/chain calculated by UV-visible spectroscopy and 1H NMR spectroscopy (see Table S4 and Fig. S11†), it can be concluded that the solvent used for RAFT polymerization does not compromise chain end retention. Both PAA-TTC and P(AA-co-MRB)-TTC could be considered as reactive macromolecular chain transfer agent for emulsion PISA, as the average values of NTTC/chain close to 1 depict a high degree of the trithiocarbonate chain end functionalization (see Fig. S11 and Table S4†). Monitoring the individual conversion of MRB and AA by 1H NMR spectroscopy to assess the relative reactivity of both monomers is not possible at such low levels of molar concentration (<0.5 mol%, <7 wt% based on AA) to prove effective copolymerization. The overlay of the UV-visible (570 nm) and refractometer traces of SEC chromatograms observed for P(AA-co-EARB0.21%)-TTC copolymer indicates the presence of EARB monomer in the copolymer (Fig. S13†). The covalent bond between the RB moiety and the polymer was also confirmed by thin-layer chromatography (TLC) for P(AA-co-EARB0.21%)-TTC and P(AA-co-EARB0.4%)-TTC copolymers. While the pink spot, characteristic of free EARB dye, moved up the TLC plate with the eluent (Fig. S14 left, spot (1)†), the main fractions of P(AA-co-EARB0.21%)-TTC and P(AA-co-EARB0.4%)-TTC copolymers (PAA-EARB6 and PAA-EARB7) were retained by the stationary phase, confirming the successful copolymerization of AA with EARB. For the sake of comparison, when a mixture of PAA-TTC and EARB was cast onto the TLC plate and subjected to eluent migration, EARB moved up the plate, indicating that the polymer did not retain the RB-based co-monomer (spot (3) in Fig. S14 left and spot (4) in Fig. S14† right). Due to the low molar ratio of MRB, no quantifiable characteristic signal of MRB was observed by 1H NMR spectroscopic analysis of precipitated P(AA-co-MRB). On the other hand, such a composition can be evaluated by UV-visible spectroscopy, since the MRB moiety in the copolymer exhibits a distinct absorbance band at λmax = 568–569 nm in ethanol (see Fig. S9†). Detailed calculations on theoretical and experimental MRB loading are provided in the ESI.† The results are gathered in Table S5† to show an experimental MRB weight loading in the precipitated copolymer ranging from 5 to 33 μmol gpolymer−1. The composition of copolymer is indeed tuned by the initial molar fraction of MRB, as a linear increase of experimental MRB loading versus a theoretical one is observed with a slope of 0.9 (Fig. S15†).Emulsion PISA for the synthesis of waterborne latex by route 1 (Scheme 1)
In the first part of this study, we highlighted, by UV-visible and 1H NMR spectroscopy, that the chain end fidelity of PAA-based polymers was maintained at a good level, regardless of the solvent used for RAFT polymerization (1,4-dioxane, DMSO, DMF), but also in the presence of the photoactive MRB comonomer. In the present work, the main route chosen to synthesize the photoactive latexes was based on emulsion PISA mediated by P(AA-co-MRB)-TTC macroCTA in its neutralized form (route 1 in Scheme 1). In order to confirm the efficacy of poly(ammonium acrylate) (PA−NH4+) to act as a reactive stabilizer for in situ synthesis of amphiphilic block copolymers, regardless of the solvent used for PAA-TTC synthesis (1,4-dioxane, DMSO, DMF), a series of RB-free PnBA, P(nBA-co-EA) and PEA latexes were first synthesized by emulsion PISA (Tables S6 and S7†). Note that the synthesis of PAA-b-PnBA block copolymer latex has already been reported by Chenal et al.48,63 from PAA synthesized by RAFT in 1,4-dioxane, but neither EA homopolymerization nor nBA/EA copolymerization by emulsion PISA has been reported so far. In addition to interest in tuning the mechanical properties of polyacrylics via the pendant chain length,44 EA monomer is also interesting for comparison with nBA in emulsion PISA to investigate the impact of both the limit of monomer solubility in water and the polymerization rate constant on emulsion kinetics. While the saturation concentration in water intuitively increases upon decreasing the number of carbon atoms in the ester side chain of n-alkyl acrylates,19 the propagation rate coefficient (kp) decreases less significantly for shorter lengths of the alkyl side chains (Fig. S16†). It can be concluded from Fig. 3 that the kinetics of emulsion PISA mediated by neutralized PAA-TTC or P(AA-co-EARB0.1%)-TTC is mainly affected by the monomer saturation concentration in water, as the length of the inhibition period is three times shorter for EA compared to that for BA.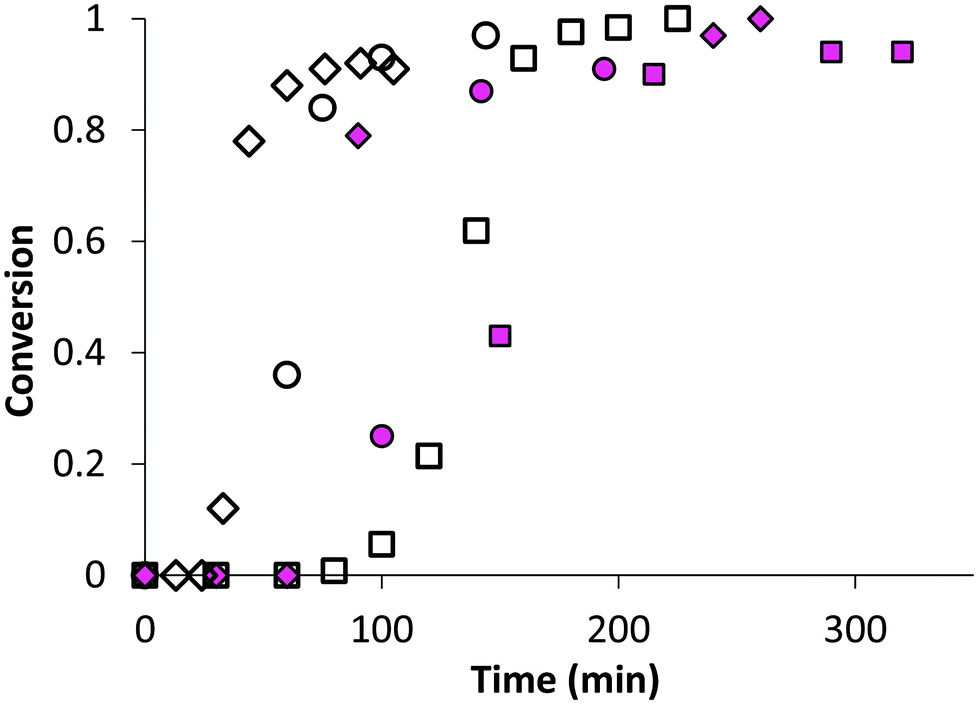 | ||
| Fig. 3 Monomer conversion versus time for emulsion PISA: comparison of monomer (◇) EA (LEA-14), (◯) nBA70%/EA30% (LBAEA-13), (□) nBA (LBA-8) from either PAA-TTC (empty symbols) or P(AA-co-EARB0.1%-TTC) (pink plain symbols) (LEA-EARB1, LBAEA-EARB1, LBA-EARB1). See Table 2 and Table S6.† | ||
This inhibition period is characteristic of the nucleation period of emulsion polymerization. It corresponds to the creation of the initial particles (i.e. nuclei), so for the PISA process it is the time for polymerization to reach a sufficient length of the hydrophobic second block to induce block copolymer self-assembly. The apparent rate of propagation, estimated from the slope of the linear part after the nucleation period, is also four times higher for EA polymerization. The EA/nBA copolymerization exhibits an intermediate trend. Analogously to AA/EARB copolymerization carried out in the less polar 1,4-dioxane solvent, emulsion polymerization of n-alkyl acrylates faced slight retardation in the presence of 0.1 mol% EARB in the initial macroCTA (Fig. 3). It should be mentioned that all waterborne latexes of the present work are synthesized with final monomer conversion above 90% within 7 hours of polymerization (see Tables 2 and 3, Table S7, Fig. S17 and S18†).
| Macro-CTA | Latex expt | Polymer | ω (wt%) | [M]0 (mol LH20−1) | [MacroCTA]0 [10−3 mol LH20−1] |
|
Conv.b (%) |
M
n,thc (kg mol−1) |
M
n,PSd (kg mol−1) (Đ) |
M
n,MALLSe (kg mol−1) (Đ) |
D
hf (nm) (PDI) |
N
pg (1017 Llatex−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Final P(AA-co-MRB)-TTC weight fraction = m0(P(AA-co-MRB)-TTC)/[m0(P(AA-co-MRB)-TTC) + m0(nBA or EA) × conversion]. b Conversion calculated by gravimetry (eqn (2)). c Theoretical Mn of block copolymers calculated from eqn (S15) and (S16).† d Experimental Mn of block copolymers recalculated from Mn of methylated block copolymers determined by SEC in THF with polystyrene calibration (eqn (S7)†). Values obtained from a refractometer detector using PS calibration. e Experimental Mn of block copolymers recalculated from Mn of methylated block copolymers determined by SEC in THF by MALLS (eqn (S7)†).Values obtained from a MALLS detector. f Hydrodynamic diameter of particles measured by DLS. g Average number of particles determined from Dh (eqn (7)). h Analysed with set no. 1 of SEC columns. i Analysed with set no. 2 of SEC columns (see the Experimental section). | ||||||||||||
| PAA-EARB2 | LBA-EARB1 | P(AA-co-EARB0.1%)-b-PnBA | 3.4 | 2.5 | 2.3 | 3.1 | 94 | 139 | 136i (5.3) | 426i (2.1) | 76 (0.08) | 9.2 |
| PAA-EARB3 | LBA-EARB2 | P(AA-co-EARB0.1%)-b-PnBA | 6.2 | 2.5 | 3.6 | 5.0 | 93 | 89 | 100i (7.2) | 373i (2.3) | 80 (0.05) | 7.9 |
| PAA-EARB4 | LBA-EARB3 | P(AA-co-EARB0.36%)-b-PnBA | 5.7 | 2.6 | 4.2 | 5.6 | 93 | 78 | 202i (5.7) | 203i (1.8) | 83 (0.06) | 7.0 |
| PAA-VBRB1 | LBA-VBRB1 | P(AA-co-VBRB0.1%)-b-PnBA | 4.3 | 2.6 | 3.6 | 5.0 | 98 | 93 | 135 (3.9) | 343h (1.6) | 96h (0.04) | 4.8 |
| PAA-VBRB2 | LBA-VBRB2 | P(AA-co-VBRB0.25%)-b-PnBA | 5.6 | 2.6 | 3.7 | 4.8 | 98 | 93 | 88i (3.2) | 237i (1.5) | 140 (0.17) | 1.5 |
| PAA-EARB2 | LBAEA-EARB1 | P(AA-co-EARB0.1%)-b-P(nBA70%/EA30%) | 3.8 | 2.7 | 1.8 | 3.2 | 91 | 125 | 141i (4.3) | 322i (2.0) | 74 (0.06) | 9.3 |
| PAA-EARB1 | LEA-EARB1 | P(AA-co-EARB0.1%)-b-PEA | 6.2 | 3.0 | 4.7 | 5.3 | 100 | 69 | 136i (3.2) | 243i (1.8) | 95 (0.04) | 4.2 |
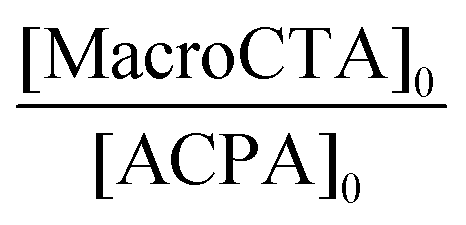
| Macro-CTA | Latex expt | Polymera | ω (wt%) | [M]0 (mol LH20−1) | [MacroCTA]0 [10−3 mol LH20−1] |
|
Conv.c (%) |
M
n,thd (kg mol−1) |
M
n,PSe,i (kg mol−1) (Đ) |
M
n,MALLSf,i (kg mol−1) (Đ) |
D
hg (nm) (PDI) |
N
ph (1017 Llatex−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a MRB molar fraction of 0.1 mol% noted in PAA-b-P(nBA-co-MRB0.1%) corresponds to the molar fraction of MRB compared to PAA units in order to target similar ranges of final RB loading compared to latexes synthesized by route 1 (see Table 2 and experimental RB loadings in Table S8†); [MRB]nBA ∼ 4.2 × 10−4 mol L−1. b Final PAA-TTC weight fraction = m0(PAA-TTC)/[m0(PAA-TTC) + m0(nBA-co-MRB) × conversion]. c Conversion calculated by gravimetry (eqn (2)). d Theoretical Mn of block copolymers calculated from eqn (S15) and (S16).† e Experimental Mn of block copolymers recalculated from Mn of methylated block copolymers determined by SEC in THF (eqn (S7)†). Values obtained from a refractometer detector using PS calibration. f Experimental Mn of block copolymers recalculated from Mn of methylated block copolymers determined by SEC in THF (eqn (S7)†). Values obtained from a MALLS detector. g Hydrodynamic diameter of particles measured by DLS. h Average number of particles determined from Dh (eqn (7)). i Analysed with set no. 2 of SEC columns. | ||||||||||||
| PAA-8 | LBA-VBRB3 | PAA-b-P(nBA-co-VBRB0.1%) | 4.6 | 2.5 | 4.3 | 5.9 | 94 | 79 | 101 (2.9) | 152 (2.1) | 90 (0.06) | 7.3 |
| PAA-9 | LBA-EARB4 | PAA-b-P(nBA-co-EARB0.1%) | 2.7 | 2.4 | 2.1 | 3.2 | 100 | 154 | 210 (3.3) | 531 (1.7) | 88 (0.07) | 6.4 |
| PAA-9 | LBA-EARB5 | PAA-b-P(nBA-co-EARB0.1%) | 4.7 | 2.6 | 3.7 | 5.1 | 92 | 86 | 125 (2.6) | 235 (1.5) | 69 (0.08) | 12.0 |
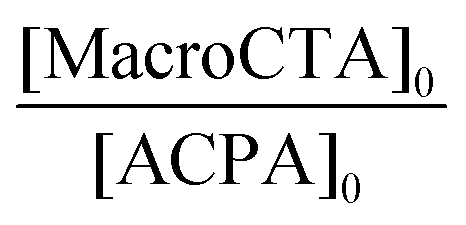
The overlay of emulsion PISA kinetics, regardless of the solvent used for the synthesis of the PAA-based stabilizers (Fig. S17 and S18†), is in accordance with the high level of chain end functionalization of the initial macroCTA. Moreover, the obvious shift of SEC chromatogram after emulsion polymerization confirms the chain end fidelity of macroCTA, allowing the successful synthesis of block copolymers (Fig. 4, Fig. S19 and S20†). Chain extension is observed from PAA-TTC synthesized in either in DMSO (Fig. 4a) or 1,4-dioxane (Fig. S20†). Note that chain extension is also effective from the photoactive P(AA-co-EARB0.36%)-TTC in the absence of residual macroCTA (Fig. 4b).
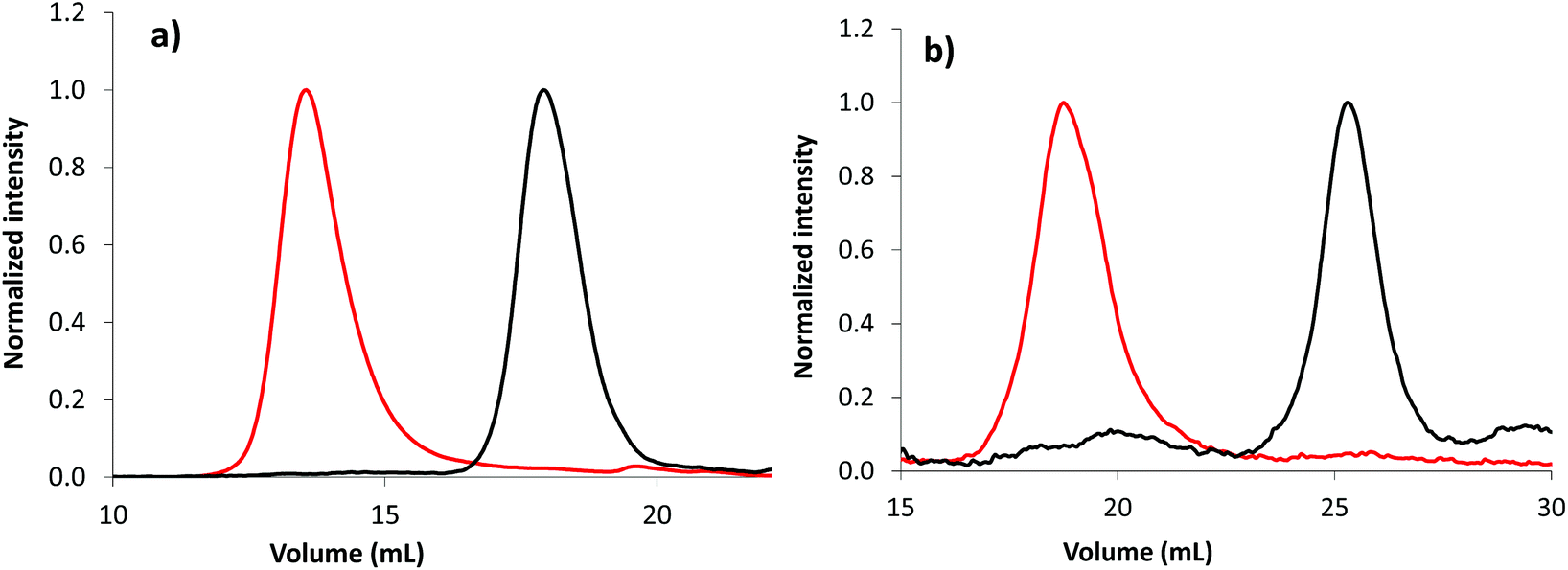 | ||
| Fig. 4 Normalized SEC chromatograms of (a) PAA-b-PnBA block copolymer (LBA-11 in Table S7,† set no. 1 of SEC columns) and (b) P(AA-co-EARB0.36%)-b-PnBA block copolymer (LBA-EARB3 in Table 2, set no. 2 of SEC columns) synthesized by emulsion PISA. The black traces correspond to the first PAA-based macroCTA and the red traces to the diblock copolymer. Light scattering traces at θ = 90° are depicted; see the corresponding RI traces in Fig. S19.† | ||
As depicted in Table 2 and Table S7,† the number-average molar masses of the diblock copolymer measured by SEC from a PS calibration (named Mn,PS) were in the range of the theoretical molar masses, whereas Mn,MALLS were systematically higher. Moreover, the poly(alkyl acrylate) diblock copolymer exhibited broad molar mass distributions (2.7 < Đ < 7.0). The significant differences in molar masses provided by conventional calibration and MALLS, as well as high dispersity, are likely to be due to chain transfer to polymers, producing statistically branched polyacrylates64,65 and/or irreversible chain-chain termination reactions favoured at high monomer conversions.48 Indeed, poly(alkyl acrylate) is prone to intramolecular and intermolecular chain transfer reactions, leading to short-chain branching (SCB) and long-chain branching (LCB), respectively.64,65 Branching induces polymer chain compaction compared to a linear polymer of the same molar mass. LCB leads to inaccuracy of molar masses determined by SEC, as linear and branched polymer chains with different molar masses can co-elute at the same hydrodynamic volume, inducing local dispersity.66,67
Syntheses of PAA-b-poly(alkyl acrylate) and P(AA-co-MRB)-b-poly(alkyl acrylate) block copolymers by RAFT-mediated emulsion PISA produced latex particles with hydrodynamic diameter (Dh) ranging from 75 to 105 nm and narrow particle size distributions (polydispersity values < 0.1) (Fig. 5, Table 2 and Table S7†).
 | ||
Fig. 5 Particle size distribution of waterborne latexes in water. Green dotted lines for PAA-b-PnBA particles (see Table S7†) and plain lines correspond to RB-based latex:  P(AA-co-VBRB0.1%)-b-PnBA (LBA-EARB2), P(AA-co-VBRB0.1%)-b-PnBA (LBA-EARB2),  P(AA-co-EARB0.36%)-b-PnBA (LBA-EARB3) and P(AA-co-EARB0.36%)-b-PnBA (LBA-EARB3) and  P(AA-co-VBRB0.25%)-b-PnBA (LBA-VBRB2) (see Table 2). Inset shows higher Dh values. DLS measurements performed at [polymer] = 0.05 g L−1. P(AA-co-VBRB0.25%)-b-PnBA (LBA-VBRB2) (see Table 2). Inset shows higher Dh values. DLS measurements performed at [polymer] = 0.05 g L−1. | ||
The final number of particles (Np) is between 3.7 × 1017 and 1.0 × 1018 particles per litre of latex (Table 2 and Table S7†). Latexes are free from coagulum and aggregates, except for the P(AA-co-VBRB0.25%)-b-PnBA diblock copolymer particles, which contain 0.25 mol% of the more hydrophobic VBRB comonomer in the outer shell (Fig. 5). Indeed, very few aggregates are observed for P(AA-co-VBRB0.25%)-b-PnBA latex together with a higher polydispersity of 0.17 and an average Dh of 140 nm, as measured for the main population, excluding aggregates (non-linear least squares method). It reveals a slight influence of the increasing fraction of more hydrophobic VBRB (see Fig. S21†), but still produces a stable latex. Note that both series of RB-free and RB-based waterborne latexes are stable over 12 months of ageing (Fig. S22†).
Emulsion PISA for the synthesis of waterborne latex by route 2 (Scheme 1)
The main strategy of the present work, as described above, involved the copolymerization of MRB with AA in order to take advantage of PISA to directly produce core–shell particles (route 1 in Scheme 1). The location of the RB photosensitizer in the outer hydrophilic shell is expected to improve the interfacial energy transfer between oxygen and photosensitizer via a better vicinity. The diffusion length of 1O2 is a compromise between its lifetime in the surrounding medium and its diffusion rate. At the solid/liquid interface, it has been reported that the singlet oxygen diffusion length is limited to several tens of nanometers,9,68 but, as diameters of the waterborne latexes synthesized in the present work are below 100 nm, we attempted to design a second type of latex with the core functionalized by RB in order to investigate the effect of RB location on the quantum yield of singlet oxygen production. For this purpose, some latexes were synthesized by emulsion copolymerization of MRB and nBA mediated by P(A−NH4+)-TTC macroCTA (route 2 in Scheme 1). The PnBA-based latexes were synthesized by emulsion PISA from PAA-TTC synthesized in DMSO (Table 3).It should also be mentioned that route 1 facilitated the transportation of the hydrophobic MRB comonomer in the water phase via the hydrophilic poly(ammonium acrylate-co-MRB) stabilizer. For route 2, MRB was solubilized in n-butyl acrylate before the addition of water to circumvent the non-solubility of MRB powder monomer in water. Note that the MRB molar fraction of 0.1 mol% corresponds to the molar fraction of MRB compared to PAA units in order to target a similar range of final RB loadings compared to latexes synthesized by route 1 (see experimental RB loadings in Table S8†). This corresponds to the MRB concentration in nBA of [MRB]nBA = 4.2 × 10−4 mol L−1 (see Table S12† for the average range of solubility of VBRB in monomers). Stable monodisperse latex particles were recovered with hydrodynamic diameters and particle numbers in a range comparable to those of P(AA-co-MRB)-b-PnBA latexes (Tables 2 and 3). Blocking efficiency is not compromised by the copolymerization of MRB with nBA, as shown in Fig. S23.† It can be however noticed that, conversely to route 1, for which the effective MRB copolymerization was proven by characterizing the low molar mass of macroCTA (Fig. S13 and S14†), it is too challenging to prove the effective copolymerization of MRB with nBA in emulsion polymerization (route 2) due to the extremely low molar fraction of MRB in the final block copolymer (<0.006 mol%, <0.05 wt% based on final polymer) and poor MRB water solubility, which hinders fully effective dialysis. Moreover, emulsion copolymerization of nBA with VBRB (route 2 in Scheme 1) is associated with a long inhibition period of 6 hours and retardation of emulsion polymerization compared to the synthesis of P(AA-co-VBRB0.1%)-b-PnBA latex by route 1 (Fig S24†). As for AA/MRB copolymerization in 1,4-dioxane, emulsion copolymerization of EARB induces less retardation compared to that of VBRB (Fig S24†). As an intermediate conclusion, route 1 is a more relevant strategy to tune the synthesis of rose bengal-based latex. A further study, beyond the scope of the present manuscript, will investigate the detrimental impact of MRB located in the core on the properties of the films formed from the RB-based waterborne latexes.
Spectroscopic and photochemical properties of RB-based waterborne latex particles
The absorbance spectra of the series of RB-based waterborne latexes exhibit the characteristic absorbances of both the conjugated rose bengal moiety (450–600 nm) and the trithiocarbonate end group (280–340 nm) (Fig. S25†). The maximum absorbance of the MRB moiety at 560–570 nm was used to calculate the experimental loadings of MRB in the latex (0.32–1.80 μmol g−1, Table S8†), which were comparable to the theoretical loadings. The maximum absorbance of MRB located either in the shell or in the core is associated with a bathochromic shift (Fig. S25 and Table S8†) relative to that of free RB in water (549 nm). Also, note that mixing RB-free latex with RB (λmax, water = 548 nm) or P(AA-co-EARB0.1%)-TTC macroCTA (λmax, water = 564 nm) did not induce any shift of the absorption maxima of these species (Fig. S25 and Table S8†). It should be taken into account that the bathochromic shift of the maximum absorbance and emission wavelengths was observed in the early VBRB synthesis step, as the maximum of VBRB in ethanol (λabs, max, VBRB = 559 nm, λem, max, VBRB = 573 nm) red-shifted relative to that of free RB in ethanol (λabs, max; RB = 548 nm, λem, max, RB = 569 nm).16 The bathochromic shift of the MRB moiety in the waterborne latex is slightly higher for MRB located in the apolar PnBA core, in comparison with MRB surrounded by the P(A−NH4+) shell (Table S8†). The MRB group is characterized by two absorbance bands (noted S1 at ∼520 nm and S2 at ∼548–560 nm in Fig. S25†). The values of S2/S1 intensity ratio between the two vibrational components of the MRB main band are given in Table S8.† It was first checked that mixing free RB or P(AA-co-EARB0.1%)-TTC macroCTA with PAA-b-PnBA latex provided an S2/S1 ratio of 3.0, similar to that of free RB in water. The lower S2/S1 values for RB-grafted latex (S2/S1 = 1.9–2.6, Table S8†) reflect an effect of the latex surroundings, but with no particular trend for MRB located in the particle core or particle shell. More interesting is the very good level of photoactivity of all the waterborne latexes covalently bonded with the RB moiety. Indeed, the quantum yield of singlet oxygen production (ΦΔ), determined by an indirect method based on the photooxidation reaction of furfuryl alcohol (FFA) in water (see the ESI†), of most of the RB-based latexes ranges between 0.57 ± 0.06 and 0.74 ± 0.07 (Fig. 6 and Table S8†). The low quantum yield of P(AA-co-EARB0.1%)-b-P(nBA-co-EA) LBAEA-EARB1 latex (ΦΔ = 0.41) is an exception that is not explained. These values are close to the quantum yield of singlet oxygen production by free RB (ΦΔ = 0.76), which reveals a particularly high photoactivity of the supported photosensitizer. The ΦΔ value measured for the mixture of free RB and PAA-b-PnBA waterborne latex (ΦΔ = 0.74), which is very close to the one of free RB (ΦΔ = 0.76), validates the methodology based on correction of the contribution from the light scattering effect on the absorbance spectra (see details in the ESI and in Table S8†). The quantum yield of singlet oxygen, corresponding to the ratio of the number of moles of produced singlet oxygen per mole of photons absorbed, provides an absolute value of photosensitizer efficiency, regardless of its concentration. Fig. 6 shows that the efficiency of the supported RB is similar for all ranges of MRB molar fraction (from 0.1 to 0.36 mol% based on AA units) and for both EARB and VBRB comonomers. | ||
Fig. 6 Singlet oxygen production quantum yield versus experimental RB loading for latexes  P(AA-co-EARB)-b-PnBA (LBA-EARB1, LBA-EARB2, LBA-EARB3), P(AA-co-EARB)-b-PnBA (LBA-EARB1, LBA-EARB2, LBA-EARB3),  PAA-b-P(nBA-co-EARB0.1%) (LBA-EARB5), PAA-b-P(nBA-co-EARB0.1%) (LBA-EARB5),  P(AA-co-VBRB)-b-PnBA (LBA-VBRB1, LBA-VBRB2), P(AA-co-VBRB)-b-PnBA (LBA-VBRB1, LBA-VBRB2),  PAA-b-P(nBA-co-VBRB0.1%) (LBA-VBRB3), PAA-b-P(nBA-co-VBRB0.1%) (LBA-VBRB3),  P(AA-co-EARB0.1%)-b-PEA (LEA-EARB1), P(AA-co-EARB0.1%)-b-PEA (LEA-EARB1),  P(AA-co-EARB0.1%)-b-P(nBA70%-co-EA30%) (LBAEA-EARB1), ■ mixture of PAA-b-PnBA (LBA-8) and RB, P(AA-co-EARB0.1%)-b-P(nBA70%-co-EA30%) (LBAEA-EARB1), ■ mixture of PAA-b-PnBA (LBA-8) and RB,  mixture of PAA-b-PnBA (LBA-8) and P(AA-co-EARB0.1%)-TTC (PAA-EARB1). The dotted line represents the reference ϕΔ value of RB. Error bars on experimental RB loading are determined from several measurements; error bars on ϕΔ are estimated at 10%. mixture of PAA-b-PnBA (LBA-8) and P(AA-co-EARB0.1%)-TTC (PAA-EARB1). The dotted line represents the reference ϕΔ value of RB. Error bars on experimental RB loading are determined from several measurements; error bars on ϕΔ are estimated at 10%. | ||
The environmentally friendly emulsion PISA process produced photoactive core–shell particles with supported RB photosensitizer that was highly efficient at the water/colloid interface. The P(AA-co-MRB)-b-poly(alkyl acrylate) block copolymers based on nBA, EA/nBA or EA exhibit glass transition temperatures of the poly(alkyl acrylate) block ranging between −45 °C and −12 °C (Table S9†). These film-forming block copolymer polyacrylic latexes pave the way for the preparation of photoactive polymer films/coating with tuneable mechanical properties by simple casting of waterborne latexes. The covalent bonding of rose bengal with the polymer colloids via straightforward copolymerization will confer strong anchorage of the organic photocatalyst to the polymer film, hence limiting any leaching, multi-step syntheses or the use of halogenated solvents, as previously reported for the grafting of a photosensitizer onto polymer surfaces.69–72 Film formation and characterization is beyond the scope of the present manuscript and will be the object of another subsequent study.
Conclusion
The RAFT polymerization of AA mediated by a trithiocarbonate chain transfer agent proved to be controlled at similar levels in 1,4-dioxane, DMF and DMSO. The more polar DMSO solvent overcomes the retardation of AA polymerization observed in the presence of the MRB photosensitizer-based comonomer, especially for vinyl benzyl rose bengal compared to ethyl acrylate RB. A high degree of chain end fidelity was observed for both PAA-TTC and P(AA-co-MRB)-TTC. These well-defined hydrophilic macromolecular chain transfer agents were further involved in emulsion PISA to control the synthesis of stable and monodisperse block copolymer particles of diameters ranging between 85 and 100 nm. The incorporation of 0.1 to 0.36 mol% of EARB or VBRB comonomer, with respect to AA units, either in the hydrophilic shell or in the hydrophobic core of nanoparticles, slightly affected the polymerization kinetics. Complete monomer conversions were reached within a few hours for all latexes. The colloidal features of the series of latexes were not impacted by the MRB, except for the highest content of the more hydrophobic VBRB comonomer in the shell, which produced latexes with slightly larger sizes and higher dispersity. Emulsion PISA is a very efficient methodology to produce core–shell particles with the rose bengal photosensitizer straightforwardly anchored on the outer shell through the MRB-based reactive macromolecular stabilizer. The synthesized photoactive colloids are efficient in producing singlet oxygen at the water interface under visible light irradiation (λLED = 515 nm), as the average quantum yield of singlet oxygen production (ϕΔ = 0.64 ± 0.10) almost reaches the level of that of free rose bengal (ϕΔ = 0.76 ± 0.10).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Agence Nationale de la Recherche (ANR) for funding the FUNPOLYSURF project and DLS equipment (ANR-15-CE08-0005-01), and CNRS and UPPA for financial support. The IPREM laboratory received funding from the Equipex Xyloforest program (ANR-10-EQPX-16 XYLOFOREST) for NMR probe equipment. This manuscript is in honour of the 50th anniversary of the French Polymer Group (Groupe Français des Polymères – GFP).References
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233, 351–371 CrossRef
.
- M. L. Marin, L. Santos-Juanes, A. Arques, A. M. Amat and M. A. Miranda, Chem. Rev., 2012, 112, 1710–1750 CrossRef CAS
- A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS
- S. Triemer, K. Gilmore, G. T. Vu, P. H. Seeberger and A. Seidel-Morgenstern, Angew. Chem., Int. Ed., 2018, 57, 5525–5528 CrossRef CAS
- E. Díez-Mato, F. C. Cortezón-Tamarit, S. Bogialli, D. García-Fresnadillo and M. D. Marazuela, Appl. Catal., B, 2014, 160–161, 445–455 CrossRef
- F. Manjon, L. Villen, D. Garcia-Fresnadillo and G. Orellana, Environ. Sci. Technol., 2008, 42, 301–307 CrossRef CAS
- Z. Zhou, J. Song, L. Nie and X. Chen, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC
- K. Page, M. Wilson and I. P. Parkin, J. Mater. Chem., 2009, 19, 3819–3831 RSC
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC
- K. Zerdin, M. A. Horsham, R. Durham, P. Wormell and A. D. Scully, React. Funct. Polym., 2009, 69, 821–827 CrossRef CAS
- A. Beltrán, M. Mikhailov, M. N. Sokolov, V. Pérez-Laguna, A. Rezusta, M. J. Revillo and F. Galindo, J. Mater. Chem. B, 2016, 4, 5975–5979 RSC
- P. Sautrot-Ba, A. Contreras, S. A. Andaloussi, T. Coradin, C. Hélary, N. Razza, M. Sangermano, P.-E. Mazeran, J.-P. Malval and D.-L. Versace, J. Mater. Chem. B, 2017, 5, 7572–7582 RSC
- L. Moczek and M. Nowakowska, Biomacromolecules, 2007, 8, 433–438 CrossRef CAS
- S. Lacombe and T. Pigot, Catal. Sci. Technol., 2016, 6, 1571–1592 RSC
- Y. Shiraishi, Y. Kimata, H. Koizumi and T. Hirai, Langmuir, 2008, 24, 9832–9836 CrossRef CAS
- L. Petrizza, M. Le Bechec, E. Decompte, H. El Hadri, S. Lacombe and M. Save, Polym. Chem., 2019, 10, 3170–3179 RSC
- C. T. J. Ferguson, N. Huber, K. Landfester and K. A. I. Zhang, Angew. Chem., Int. Ed., 2019, 58, 10567–10571 CrossRef CAS
- C. Boussiron, M. Le Bechec, L. Petrizza, J. Sabalot, S. Lacombe and M. Save, Macromol. Rapid Commun., 2019, 40, 1800329 CrossRef
-
A. Van Herk, in Chemistry and Technology of Emulsion Polymerization, Blackwell Publishing Ltd, 2005 Search PubMed
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., 2020, 59, 8368–8392 CrossRef
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS
- P. B. Zetterlund, S. C. Thickett, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 CrossRef CAS
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS
- J. Rosselgong, A. Blanazs, P. Chambon, M. Williams, M. Semsarilar, J. Madsen, G. Battaglia and S. P. Armes, ACS Macro Lett., 2012, 1, 1041–1045 CrossRef CAS
- P. Gurnani, A. B. Cook, R. A. E. Richardson and S. Perrier, Polym. Chem., 2019, 10, 1452–1459 RSC
- S. Y. Khor, M. N. Vu, E. H. Pilkington, A. P. R. Johnston, M. R. Whittaker, J. F. Quinn, N. P. Truong and T. P. Davis, Small, 2018, 14, 13 CrossRef
- J. Lesage de la Haye, I. Martin-Fabiani, M. Schulz, J. L. Keddie, F. D'Agosto and M. Lansalot, Macromolecules, 2017, 50, 9315–9328 CrossRef CAS
- M. Chenal, C. Vechambre, J. M. Chenal, L. Chazeau, V. Humblot, L. Bouteiller, C. Creton and J. Rieger, Polymer, 2017, 109, 187–196 CrossRef CAS
- C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R. G. Gilbert, A. K. Serelis and C. H. Such, Macromolecules, 2002, 35, 9243–9245 CrossRef CAS
- N. S. Serkhacheva, N. I. Prokopov, E. V. Chernikova, E. Y. Kozhunova, I. O. Lebedeva and O. V. Borisov, Polym. Int., 2019, 68, 1303–1314 CrossRef CAS
- J. Zhou, R. He and J. Ma, Polymers, 2016, 8, 207 CrossRef
- Z. Qiao, T. Qiu, W. Liu, L. Guo and X. Li, Polym. Chem., 2016, 7, 3993–3997 RSC
- C. St Thomas, R. Guerrero-Santos and F. D'Agosto, Polym. Chem., 2015, 6, 5405–5413 RSC
- E. V. Chernikova, A. V. Plutalova, K. O. Mineeva, I. R. Nasimova, E. Y. Kozhunova, A. V. Bol'shakova, A. V. Tolkachev, N. S. Serkhacheva, S. D. Zaitsev, N. I. Prokopov and A. B. Zezin, Polym. Sci., Ser. B, 2015, 57, 547–559 CrossRef CAS
- X. Zhang, F. Boisson, O. Colombani, C. Chassenieux and B. Charleux, Macromolecules, 2013, 47, 51–60 CrossRef
- M. Chenal, J. Rieger, C. Vechambre, J. M. Chenal, L. Chazeau, C. Creton and L. Bouteiller, Macromol. Rapid Commun., 2013, 34, 1524–1529 CrossRef CAS
- D. E. Ganeva, E. Sprong, H. de Bruyn, G. G. Warr, C. H. Such and B. S. Hawkett, Macromolecules, 2007, 40, 6181–6189 CrossRef CAS
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS
- O. J. Deane, O. M. Musa, A. Fernyhough and S. P. Armes, Macromolecules, 2020, 53, 1422–1434 CrossRef CAS
- G. Delaittre, J. Nicolas, C. Lefay, M. Save and B. Charleux, Chem. Commun., 2005, 5, 614–616 RSC
- G. Delaittre, J. Nicolas, C. Lefay, M. Save and B. Charleux, Soft Matter, 2006, 2, 223–231 RSC
- M. Gaborieau, R. Graf, S. Kahle, T. Pakula and H. W. Spiess, Macromolecules, 2007, 40, 6249–6256 CrossRef CAS
- G. D. Airey and M. H. Mohammed, Rheol. Acta, 2008, 47, 751–763 CrossRef CAS
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS
- C. Ladavière, N. Dörr and J. P. Claverie, Macromolecules, 2001, 34, 5370–5372 CrossRef
- J. Loiseau, N. Doërr, J. M. Suau, J. B. Egraz, M. F. Llauro, C. Ladavière and J. Claverie, Macromolecules, 2003, 36, 3066–3077 CrossRef CAS
- M. Chenal, L. Bouteiller and J. Rieger, Polym. Chem., 2013, 4, 752–762 RSC
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS
- J. Ji, L. Jia, L. Yan and P. R. Bangal, J. Macromol. Sci., Part A: Pure Appl.Chem., 2010, 47, 445–451 CrossRef CAS
- I. Chaduc, W. Zhang, J. Rieger, M. Lansalot, F. D'Agosto and B. Charleux, Macromol. Rapid Commun., 2011, 32, 1270–1276 CrossRef CAS
- L. Couvreur, C. Lefay, J. Belleney, B. Charleux, O. Guerret and S. Magnet, Macromolecules, 2003, 36, 8260–8267 CrossRef CAS
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS
- M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877–2887 CrossRef CAS
- A. Jeličić, N. García, H.-G. Löhmannsröben and S. Beuermann, Macromolecules, 2009, 42, 8801–8808 CrossRef
- M. Horn and K. Matyjaszewski, Macromolecules, 2013, 46, 3350–3357 CrossRef CAS
- C. Wu, N. Corrigan, C. H. Lim, K. Jung, J. Zhu, G. Miyake, J. Xu and C. Boyer, Macromolecules, 2019, 52, 236–248 CrossRef CAS
- E. L. Madruga, J. San Roman, M. A. Lavia and M. C. Fernandez-Monreal, Eur. Polym. J., 1986, 22, 357–360 CrossRef CAS
- U. Wendler, J. Bohrisch, W. Jaeger, G. Rother and H. Dautzenberg, Macromol. Rapid Commun., 1998, 19, 185–192 CrossRef CAS
- P. Lacroix-Desmazes, T. Delair, C. Pichot and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3845–3854 CrossRef CAS
- P. J. M. Stals, T. N. T. Phan, D. Gigmes, T. F. E. Paffen, E. W. Meijer and A. R. A. Palmans, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 780–791 CrossRef CAS
- A. R. Maniego, A. T. Sutton, Y. Guillaneuf, C. Lefay, M. Destarac, C. M. Fellows, P. Castignolles and M. Gaborieau, Polym. Chem., 2019, 10, 2469–2476 RSC
- M. Chenal, J. Rieger, C. Vechambre, J. M. Chenal, L. Chazeau, C. Creton and L. Bouteiller, Macromol. Rapid Commun., 2013, 34, 1524–1529 CrossRef CAS
- N. M. Ahmad, D. Britton, F. Heatley and P. A. Lovell, Macromol. Symp., 1999, 143, 231–241 CrossRef CAS
- N. Ballard and J. M. Asua, Prog. Polym. Sci., 2018, 79, 40–60 CrossRef CAS
- M. Gaborieau, J. Nicolas, M. Save, B. Charleux, J. P. Vairon, R. G. Gilbert and P. Castignolles, J. Chromatogr., A, 2008, 1190, 215–223 CrossRef CAS
- M. Gaborieau and P. Castignolles, Anal. Bioanal. Chem., 2011, 399, 1413–1423 CrossRef CAS
- C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685–1758 CrossRef CAS
- C. Piccirillo, S. Perni, J. Gil-Thomas, P. Prokopovich, M. Wilson, J. Pratten and I. P. Parkin, J. Mater. Chem., 2009, 19, 6167–6171 RSC
- H. Hettegger, M. Gorfer, S. Sortino, A. Fraix, D. Bandian, C. Rohrer, W. Harreither, A. Potthast and T. Rosenau, Cellulose, 2015, 22, 3291–3304 CrossRef CAS
- W. J. Peveler, S. Noimark, H. Al-Azawi, G. B. Hwang, C. R. Crick, E. Allan, J. B. Edel, A. P. Ivanov, A. J. MacRobert and I. P. Parkin, ACS Appl. Mater. Interfaces, 2018, 10, 98–104 CrossRef CAS
- L. Pessoni, S. Lacombe, L. Billon, R. Brown and M. Save, Langmuir, 2013, 29, 10264–10271 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01128b |
| This journal is © The Royal Society of Chemistry 2021 |