
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Elemental mapping of half-sandwich azopyridine osmium arene complexes in cancer cells†
Elizabeth M.
Bolitho
 ab,
Hannah E.
Bridgewater
a,
Russell J.
Needham
a,
James P. C.
Coverdale
a,
Paul D.
Quinn
*b,
Carlos
Sanchez-Cano
*c and
Peter J.
Sadler
*a
ab,
Hannah E.
Bridgewater
a,
Russell J.
Needham
a,
James P. C.
Coverdale
a,
Paul D.
Quinn
*b,
Carlos
Sanchez-Cano
*c and
Peter J.
Sadler
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: P.J.Sadler@warwick.ac.uk
bDiamond Light Source, OX11 0DE, Oxford, OX11 0DE, UK. E-mail: paul.quinn@diamond.ac.uk
cCenter for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), Paseo de Miramon 182, 20014, San Sebastián, Spain. E-mail: csanchez@cicbiogamune.es
First published on 10th June 2021
Abstract
Transition metal complexes are often prodrugs which undergo activation by ligand exchange and redox reactions before they interact with target sites. It is therefore important to understand the roles of both the metal and the ligands in their activation, especially in cells. Here we use a combination of synchrotron nanoprobe X-ray fluorescence (XRF) from Os L3M5 and Br KL3 emissions and inductively coupled plasma-mass spectrometry (ICP-MS) detection of 189Os, 79Br, and 127I, to investigate the time-dependent accumulation and localization of osmium as well as the monodentate ligand and the chelated phenylazopyridine in A2780 human ovarian cancer cells treated with the potent anticancer complexes [Os(η6-p-cymene)(4-R2-phenyl-azopyridine-5-R1)X]PF6, with R2 = NMe2 or OH, R1 = H or Br, and X = Cl or I. The data confirm that the relatively inert iodido complexes are activated rapidly in cancer cells by release of the iodido ligand, probably initiated by attack by the intracellular tripeptide glutathione (γ-L-Glu-l-Cys-Gly) on the azo double bond. The bond between osmium and the azopyridine appears to remain stable in cells for ca. 24 h, although some release of the chelated ligand is observed. Interestingly, the complexes seem to be degraded more rapidly in normal human cells, perhaps providing a possible mechanism for selective cytotoxicity towards cancer cells.
Introduction
Metal complexes capable of undergoing intracellular redox reactions are promising alternative anticancer treatments to Pt(II) agents, which bind to DNA and are currently used in the clinic.1–12 Such drug candidates include inert prodrugs that are activated through metal reduction (i.e. Pt(IV) or Ru(III) complexes), to redox-active complexes that modulate the redox balance in cancer cells producing reactive oxygen species (ROS), or altering the level of key cellular cofactors.Recently, we have reported promising Os(II) arene and Ir(III) cyclopentadienyl half-sandwich anticancer complexes containing azopyridine ligands, which are activated in cancer cells and modulate their metabolism through redox processes. In particular, Os(II) complexes in the family [Os(η6-p-cymene)(4-R2-phenylazopy-5-R1)X]PF6 (where Azpy = p-(phenylazo)pyridine, R2 = NMe2 or OH, R1 = H or Br, and X = Cl or I; Table 1) can be up to 49 times symbol more potent than cisplatin in a panel of over 800-cancer cell lines, including platinum-resistant cancer cells.1,2 Moreover, they exhibit lower in vivo toxicity against zebrafish embryos,13 and act through a different mechanism of action compared to cisplatin.
|
|
|||
|---|---|---|---|
| Complex | R1 | R2 | X |
| 1-PF6 | H | NMe2 | Cl |
| 2-PF6 | H | NMe2 | I |
| 3-PF6 | H | OH | I |
| 4-PF6 | Br | OH | I |
The iodide complexes in particular are inert prodrugs, which are activated intracellularly by hydrolysis of their Os–I bond in presence of GSH, and increase the cellular levels of ROS.13–17 The combination of time-resolved X-ray absorption spectroscopy (XAS) and DFT theoretical approaches has shown that the activation of this family of drug candidates involves a catalytic ligand-mediated mechanism.18 The azo-bond (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) in the chelated azopyridine reacts directly with GSH, promoting the dissociation of the monodentate iodide from osmium. The same reaction can occur when other monodentate ligands such as Cl, H2O, OH− or GS− are coordinated to Os instead of iodide. As such, the nature and chemical properties of the ligands coordinated to the Os are important for the rate of activation of the complexes, as they control the stability of the Os–X bond and overall reactivity of the azo-bond towards GSH. Moreover, this also affects their biological properties.1,2,13,14,19 For example, the Os–Cl bonds are more labile than Os–I bonds, and complexes containing chlorido ligands show slower cellular accumulation19 and are generally significantly less active as anticancer agents than their iodido analogues; 1-PF6 is ca. 12× less potent than 2-PF6 against A2780 ovarian cancer cells; IC50 = 1.8 and 0.15 μM, respectively; Tables 1 and 2.1,2,13,19 Similarly, complexes with phenylazopyridine ligands with R1 = OEt and R2 = H undergo hydrolysis of their Os–X bonds (X = Cl or I) in presence of GSH nearly 3× faster than those with R1 = H and R2 = NMe2 (1-PF6 and 2-PF6, Table 1).15 Complexes with both these types of R1 substituents are activated at a similar rate inside cells, but complexes with R1 = OEt are at least 6× less active than R1 = NMe2 complexes.15 Such differences in activity might be caused by drug deactivation before reaching the intracellular target site, due to an increased lability of the Os–X bond (Os–Cl vs. Os–I) and reactivity of the azopyridine ligand.
N) in the chelated azopyridine reacts directly with GSH, promoting the dissociation of the monodentate iodide from osmium. The same reaction can occur when other monodentate ligands such as Cl, H2O, OH− or GS− are coordinated to Os instead of iodide. As such, the nature and chemical properties of the ligands coordinated to the Os are important for the rate of activation of the complexes, as they control the stability of the Os–X bond and overall reactivity of the azo-bond towards GSH. Moreover, this also affects their biological properties.1,2,13,14,19 For example, the Os–Cl bonds are more labile than Os–I bonds, and complexes containing chlorido ligands show slower cellular accumulation19 and are generally significantly less active as anticancer agents than their iodido analogues; 1-PF6 is ca. 12× less potent than 2-PF6 against A2780 ovarian cancer cells; IC50 = 1.8 and 0.15 μM, respectively; Tables 1 and 2.1,2,13,19 Similarly, complexes with phenylazopyridine ligands with R1 = OEt and R2 = H undergo hydrolysis of their Os–X bonds (X = Cl or I) in presence of GSH nearly 3× faster than those with R1 = H and R2 = NMe2 (1-PF6 and 2-PF6, Table 1).15 Complexes with both these types of R1 substituents are activated at a similar rate inside cells, but complexes with R1 = OEt are at least 6× less active than R1 = NMe2 complexes.15 Such differences in activity might be caused by drug deactivation before reaching the intracellular target site, due to an increased lability of the Os–X bond (Os–Cl vs. Os–I) and reactivity of the azopyridine ligand.
| Complex | R1 | R2 | X | A2780a | A549a | PC3a | MRC-5a | Selectivity factorb |
|---|---|---|---|---|---|---|---|---|
| a Antiproliferative activities (IC50/μM) as determined using the SRB assay 24 h exposure +72 h recovery in drug-free media. b Selectivity factor for A2780 cancer cells as compared to MRC-5 healthy lung fibroblasts. c Literature IC50 concentrations.1,13 | ||||||||
| 1-PF6 | H | NMe2 | Cl | 1.8 ± 0.1c | 36.6 ± 0.8c | 8.8 ± 0.0c | 7.1 ± 0.2 | 3.9 |
| 2-PF6 | H | NMe2 | I | 0.15 ± 0.01c | 1.1 ± 0.2 | 0.62 ± 0.12c | 4.5 ± 0.3 | 30.0 |
| 3-PF6 | H | OH | I | 0.51 ± 0.05 | 0.94 ± 0.1 | 0.91 ± 0.03 | 0.69 ± 0.02 | 1.4 |
| 4-PF6 | Br | OH | I | 0.42 ± 0.03 | 1.1 ± 0.1 | 0.64 ± 0.01 | 0.44 ± 0.03 | 1.0 |
| Cisplatin | — | — | — | 1.20 ± 0.03c | 3.2 ± 0.1c | 21.5 ± 4.1 | 13.5 ± 0.9c | 10.7 |
X-ray Fluorescence (XRF) has allowed studies with subcellular resolution of the localisation of this type of osmium complex in chemically-fixed A2780 human ovarian cancer cells.17,20 This was achieved by mapping Os L3M5 emissions, and showed apparent mitochondrial localisation of the complexes after 24 h treatment.17 Equally, in-cell XAS studies using nanofocused synchrotron radiation have indicated the possible presence of an Os(II)/Os(III) redox cycle in those areas of A2780 cells with high concentration of the complex.21 Such redox reactions may be involved in the generation of the observed ROS.21 Studies are now needed to investigate the importance of the halide and phenylazopyridine ligands coordinated to the Os in the cellular mechanism of action of these complexes. The lability of the Os–X ligand for example, may affect the processes for cellular localisation of these complexes, and their biological activity. Our previous studies suggest that the azopyridine ligand mediates the intracellular activation of the complexes.15,18 Although the redox properties of the phenylazopyridine could also have a role in the ROS production observed in treated cells, it is not known if this chelated ligand remains bound to Os after activation of the complex inside cells. Hence, probing the intracellular stability of these complexes can contribute to our understanding of their detailed cellular mechanism of action.
Recently we reported the tracking of osmium diamine transfer hydrogenation catalysts in cancer cells by monitoring osmium and a bromine substituent on a chelated ligand using XRF and ICP-MS, which provided insight into dissociation of the chelated ligand inside cells.22 Here we have used a similar combination of ICP-MS, for monitoring 189Os, 79Br, 127I, and XRF detecting Os L3M5 and Br KL3 emissions at the I14 nanoprobe beamline (Diamond Light Source) to track and map the time-dependent localisation and ligand dissociation reactions of potent anticancer complexes 1–4 (Table 1) in human ovarian cancer cells and normal human cells. We have monitored osmium, the iodide monodentate ligand and a Br substituent on the chelated phenylazopyridine ligand, in a position that has little effect on activity. This has enabled us to gain new insights into the activation of these complexes in cells, the extent to which the chelated phenylazopyridine ligand remain bound to Os, and the difference in behaviour in normal cells compared to cancer cells.
Experimental
Synthesis of complexes
Osmium azopyridine complexes 1–3 (Table 1) with the general formula [Os(η6-p-cymene)(4-R2-phenyl-azopyridine-5-R1)(X)]PF6, with R2 = NMe2 or OH, R1 = H and X = Cl or I were synthesised from [Os(p-cymene)X2]2 dimer based on a reported procedure.1,23 Brominated complex 4 was synthesised using the following procedures.XRF analysis
General cellular accumulation protocol
10 or 20 mL of a suspension of 4 × 106 cells per mL was seeded in 100 or 145 mm cell culture Petri dishes (respectively) for 24 h (310 K, 5% CO2). Prior to cell treatment, stock solutions of osmium compounds were prepared in 5% v/v DMSO and 95% v/v DMEM, and their Os concentrations determined by ICP-OES (see ESI†). Cells were treated under varying conditions (temperature, concentration, time) as specified below. Then, they were washed with PBS and 0.25% trypsin/EDTA (2 mL) was added to each Petri dish (5 min, 310 K, 5% CO2), which was quenched with a known volume of DMEM to form a single cell suspension. Cells were counted in duplicate (2 × 10 μL) and pelleted by centrifugation (1000 rpm, 5 min, 298 K). The supernatant was removed, pellets re-suspended in PBS (1 mL) for further centrifugation (1200 rpm, 5 min), and the new pellets were analysed by ICP-MS.ICP-MS
Digested cell pellets were analysed on an Agilent ICP-MS 7900 spectrometer instrument in [He] gas mode in either acid (3.6% v/v stabilised nitric acid, using 166Er = 50 ppb internal standard) or alkaline (1% m/v tetramethylammonium hydroxide, using 101Ru = 10 ppb internal standard). Calibrations of osmium (0–1000 ppb) were prepared in stabilised 3.6% v/v stabilised nitric acid (containing 10 mM thiourea and 100 mg L−1 ascorbic acid), and solutions of osmium, bromine and iodine (0–1000 ppb) were prepared in 1% m/v TMAH.(i) Temperature: A2780 (ovarian) cancer cells were treated with 1× IC50 of 3-PF6 (0.51 ± 0.05 μM) or 4-PF6 (0.42 ± 0.03 μM) for 3 or 6 h at (a) 310 K; (b) 277 K, without further recovery period. Cell pellets were digested as previously described.
(ii) Concentration: A2780 (ovarian) cancer cells were treated with 0.25–2× IC50 of 3-PF6 (0.51 ± 0.05 μM) or 4-PF6 (0.42 ± 0.03 μM) for 24 h, without further recovery period. Cell pellets were digested as previously described.
(iii) MRC-5 cells: MRC-5 healthy lung fibroblasts were treated with 1 × IC50 A2780 concentration of 1-PF6 (1.8 ± 0.1 μM), 2-PF6 (0.15 ± 0.01 μM), 3-PF6 (0.51 ± 0.05 μM) or 4-PF6 (0.42 ± 0.03 μM) for 24 h, without further recovery period. Cell pellets were digested as described above.
(i) Time: A2780 cells were treated with 1× IC50 of 4-PF6 (0.42 ± 0.03 μM) for 4, 8 18 or 24 h (without further recovery time), or 24 h followed by a period of 24, 48 or 72 h recovery in drug-free DMEM. Every two Petri dishes were combined, and cells counted from the combined solutions to form larger pellets for reliable bromine detection by ICP-MS. Cell pellets were digested using the alkaline digestion method described above.
(ii) MRC-5 cells: MRC-5 lung fibroblasts were treated with 1× A2780 IC50 concentrations of 4-PF6 (0.42 ± 0.03 μM) for 24 h (without further recovery time). Cell pellets were digested using the alkaline digestion method described above.
Results and discussion
Time-dependent localisation of complexes carrying various halogen ligands in cancer cells
We used nano-focused XRF to probe the effect of different halogen ligands on the time-dependent accumulation and localisation of half-sandwich Os(II) azopyridine complexes in cancer cells. A2780 ovarian cancer cells were treated for various times with equipotent concentrations (7× IC50) of complexes with almost identical structures, but containing Cl (1-PF6; 7× IC50: 12 μM; treated for 4 or 8 h) or I (2-PF6; 7× IC50: 1 μM; treated for 4, 8 or 24 h) as a monodentate coordinated ligand, Table 1. Then, maps based on Os L3M5 emissions were acquired with a 100 × 100 nm2 resolution from cryo-fixed, dehydrated cells (Fig. 1; ESI, Fig. S1–14†). This Os distribution was compared with the localisation of native elements such as P or Zn, which were used to define the cellular limits within the maps (e.g. nucleus and outer membrane), and provide an initial indication of the possible localisation of 1-PF6 and 2-PF6.25–27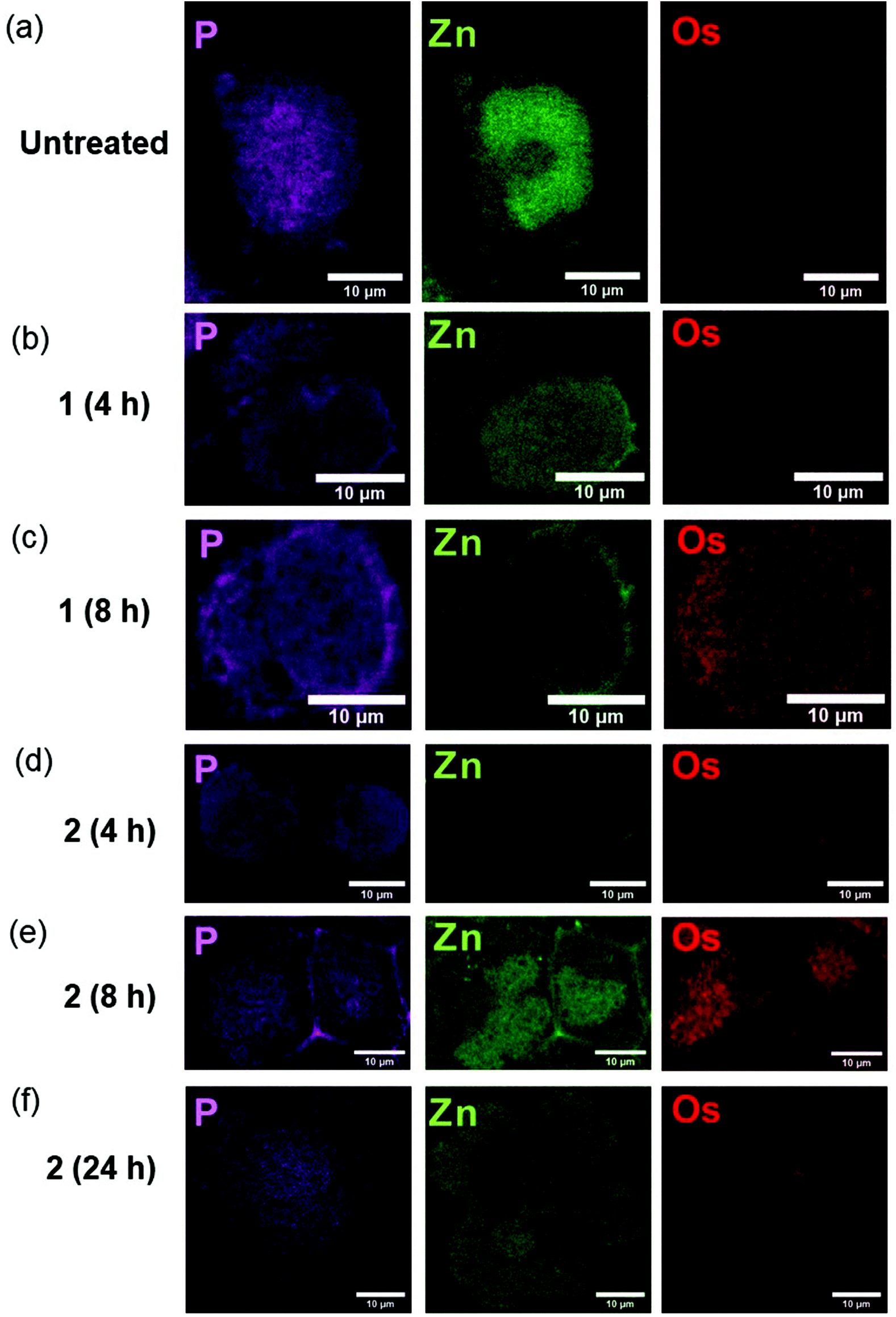 | ||
| Fig. 1 Synchrotron-XRF elemental maps of cryo-fixed and freeze-dried A2780 (human ovarian) cancer cells grown on silicon nitride membranes treated with 7 × IC50 of 1-PF6 (12 μM) or 2-PF6 (1 μM) for 4, 8 or 24 h (no recovery) showing: phosphorus (magenta), zinc (green); and osmium (red) obtained using an incident energy of 12 keV: (a) Untreated (b) 1-PF6 (4 h), (c) 1-PF6 (8 h), (d) 2-PF6 (4 h), (e) 2-PF6 (8 h), (f) 2-PF6 (24 h). Data were fitted in PyMCa,24 and images generated in ImageJ software.33 The scale bar = 10 μm. It is notable that natural Zn is highly concentrated in the cell nucleus, whereas Os from the anticancer complexes is not. | ||
As expected, Os L3M5 emissions were not detected in untreated A2780 cells (ESI, Fig. S1†). Control samples (Fig. 1a; ESI, Fig. S2–4†) also exhibited clear cell outlines with rounded shapes (mean roundness factor, RF: 0.77 ± 0.14; where RF = 1 implies perfect circularity), large cell nuclei, and overall area ranged between 215–439 μm2 (mean area = 303 ± 93 μm2). All of this correlated well with the rounded morphologies and nuclei,28–30 and typical size (<20 μm in diameter; expected area around 314 μm2)31 reported for this cell line. Moreover, observation of the nuclei of the control cells mapped (shown by the concentrated regions of Zn),32 suggested that the wide variation between their areas (ESI, Table S1†) could be explained by their different stages of the cell cycle. This includes cells in the interphase (ESI, Fig. S2†), but also in the anaphase (ESI, Fig. S3†) and telophase/early cytokinesis (where the two daughter cells are separated; ESI, Fig. S4†) of mitosis.
Treatment of A2780 cancer cells with 7× IC501-PF6 or 2-PF6 for 4–8 h did not cause significant alterations to the size (mean area = 327 ± 22, 396 ± 55, 403 ± 275 and 403 ± 275 μm2; ESI, Fig. S5–12, Table S1†) or morphology (mean RF: 0.79 ± 0.04, 0.83 ± 0.04, 0.82 ± 0.05 and 0.84 ± 0.10; ESI, Table S1†) of A2780 cells when compared with untreated controls (t-tests p > 0.05). However, cells treated with iodido complex 2-PF6 for 24 h were significantly larger (mean area = 1156 ± 361 μm2), and presented substantial cellular damage (Fig. 1f; ESI, Fig. S13–14†). Cellular alterations observed included membrane blebbing and the collapse of nuclear integrity, suggesting the initiation of programmed cell death pathways, as previously reported from XRF studies using chemically-fixed A2780 cells.17 Equally, Os maps acquired from cells treated with the complexes confirmed previous ICP-MS studies, and showed that the accumulation of both 1-PF6 and 2-PF6 into A2780 cells was time-dependent and slower for the chlorido complex 1-PF6.19 As such, 2-PF6 was always found at higher levels inside cells than its chlorido analogue 1-PF6 (Fig. 1). Unfortunately, due to synchrotron beamline time-constraints the experimental setup could not be calibrated to an AXO GmbH standard, thus quantities of osmium (pg mm−2) could not be determined. Still, the XRF maps allowed it to be determined that cells treated with 1-PF6 for 4 h contained negligible quantities of Os (Fig. 1b; ESI, Fig. S5–6†), and only small amounts of Os from the same complex were found after 8 h treatment (Fig. 1b; ESI, Fig. S7–8†). A similar trend was observed for 2-PF6, for which osmium accumulation was more pronounced after 8 h compared to 4 h (Fig. 1d and e; ESI, Fig. S9–12†). Yet, there was a decrease in the amounts of intracellular Os after 24 h treatment (Fig. 1f; ESI, Fig. S13–14†), probably caused by the loss of cellular integrity due to the initiation of cell death processes.
XRF maps also indicated cytosolic localisation for both 1-PF6 and 2-PF6 in cells, with no statistical correlation between Os and Zn (as determined by Pearson's R-value, R∼0, and Spearman's Rank Correlation, Rs ∼ 0; ESI, Table S2–3†). The lack of Os reaching the cell nuclei, together with their inability of these complexes to form adducts with nucleobases,16 suggests that chromosomal DNA is not a primary therapeutic target for this type of anticancer compound. Moreover, 2-PF6 was observed to accumulate in small elliptical areas (previously identified as mitochondria)17 after just 8 h (Fig. 1e; ESI, Fig. S12†).17 This finding correlates closely with that observed for glutaraldehyde-fixed A2780 cells after 24 h treatment with iodido complex 2-PF6 and analysed by XRF,17 and supports cell fractionation studies using ICP-MS and proteomic analysis for 2-PF6.2,14 Still, no particular localisation into cellular compartments or organelles could be detected for chlorido complex 1-PF6 under the experimental conditions used. Therefore, the presence of a monodenate Cl bound to Os(II) in these half-sandwich azopyridine complexes both decreases the cellular accumulation and slows down the ability of the complex to reach cellular targets, when compared with their iodido analogues.
Cellular stability of osmium azopyridine complexes in cancer cells
We assessed the intracellular stability of these half-sandwich Os(II) complexes by labelling their phenylazopyridine ligands with bromine-tags and determining the relative Os and Br accumulation and localisation in cancer cells using ICP-MS and nanofocused XRF. A new brominated chelating ligand was synthesised by reacting p-benzoquinone and 5-bromo-2-hydrazinopyridine in the presence of perchloric acid, and used to generate [Os(η6-p-cymene)(4-Br-phenylazopy-5-OH)I]PF64-PF6 a labelled analogue of the active half-sandwich complex [Os(η6-p-cymene)(phenylazopy-5-OH)I]PF63-PF6 (Tables 1 and 2). The Br-tag in the azopyridine ligand of 4-PF6 is located on the pyridine ring, which provides high resistance towards nucleophilic substitution of the C–Br bond by biological thiols.34 This was done to avoid unwanted release of the Br group under cellular conditions, so most of the bromine detected using ICP-MS or XRF on cells treated with 4-PF6 is expected to correspond to the intact ligand or complex, or fragmented chelating ligand (Scheme 1). | ||
| Scheme 1 (a) One step synthesis of Br-AZPY-OH ligand with bromide as a para electron-withdrawing substituent on the pyridine ring and a para hydroxyl substituent on the phenyl ring. (b) Two-step synthesis of complex 4-PF6 from reaction of Br-AZPY-OH ligand with [Os(η6-p-cymene)I2]2. | ||
Bromination of Azpy-OH caused little alteration in the chemical properties of 4-PF6 when compared to non-brominated 3-PF6. Both were stable in phosphate buffer in concentrations of NaCl equivalent to those found in cells (∼25 mM), and showed similar acid dissociation constants (pKa) for their phenolic OH substituents (6.41 ± 0.02 and 6.78 ± 0.02 for 3-PF6 and 4-PF6, respectively; ESI, Fig. S15–16†), which exist as zwitterionic species under physiological conditions (pH = 7.4).
Equally, 3-PF6 and 4-PF6 were readily converted to their chlorido and hydroxido analogues in the presence of 10 mol equiv. of GSH (ESI, Fig. S17–18†). Moreover, they can form thiolato (Os-SG) and sulfenato (Os-SOG) adducts in presence of molar excesses of GSH (as previously observed for 2-PF6; ESI, Fig. S17–18†),15 suggesting that they are activated in cells by the same mechanism. 3-PF6 and 4-PF6 also possess equivalent redox properties linked to the irreversible two-electron reduction of the azo-bond (NN) in their chelating ligands. The half-wave reduction potentials, determined using an Ag/Ag+ reference electrode in acetonitrile with tetrabutylammonium hexafluorophosphate (0.1 M) as a supporting electrolyte (−0.66 and −0.74 V for 3-PF6 and 4-PF6, respectively; ESI Fig. S19†), are comparable with those previously reported for analogous osmium arene complexes (including complex 2-PF6, −0.64 V),35,36 and indicate that reduction of the phenylazopyridine might occur under physiological conditions. However, 4-PF6 was slightly more hydrophobic than 3-PF6 (HPLC capacity factors Kf = 1.51 ± 0.02 and 0.90 ± 0.01, respectively; Fig. 2a), likely owing to the large size, polarisation and increased London dispersion forces of the Br substituent.
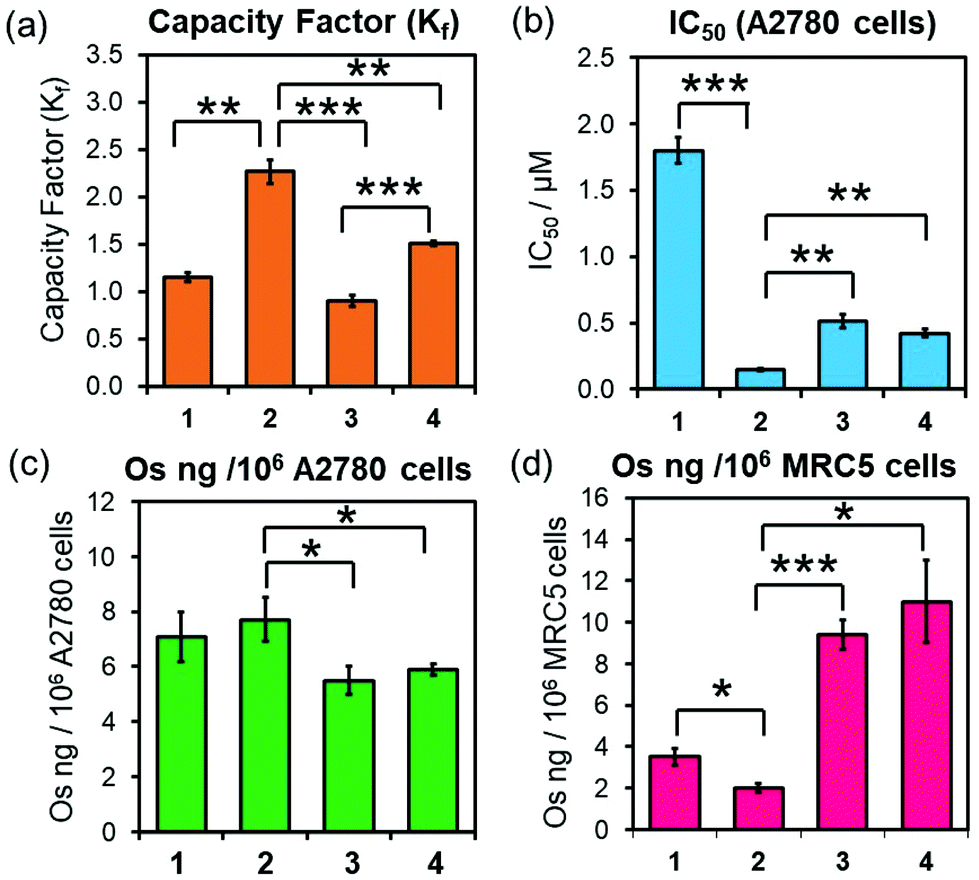 | ||
Fig. 2 (a) HPLC capacity factors (Kf, orange) of 1–4 relative to uracil (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v water:acetonitrile, 50 mM NaCl). (b) IC50/μM (blue) of 1–4 in A2780 (human ovarian) cancer cells upon 24 h exposure, followed by 72 h recovery in complex-free media. (c) Cellular accumulation of 189Os (ng per 106 per cells, green) in A2780 cells treated with 1 × IC50 of 1–4 for 24 h (no recovery). (d) Cellular accumulation of 189Os (ng per 106 per cells, pink) in MRC5 (healthy lung) fibroblasts treated with 1× A2780 IC50 of 1–4 for 24 h (no recovery). Error bars show the standard deviation from the mean. Statistical analysis was performed using Welch's unpaired t-test (assuming unequal variances): * p < 0.05; ** p < 0.01; *** p < 0.001. :1 v/v water:acetonitrile, 50 mM NaCl). (b) IC50/μM (blue) of 1–4 in A2780 (human ovarian) cancer cells upon 24 h exposure, followed by 72 h recovery in complex-free media. (c) Cellular accumulation of 189Os (ng per 106 per cells, green) in A2780 cells treated with 1 × IC50 of 1–4 for 24 h (no recovery). (d) Cellular accumulation of 189Os (ng per 106 per cells, pink) in MRC5 (healthy lung) fibroblasts treated with 1× A2780 IC50 of 1–4 for 24 h (no recovery). Error bars show the standard deviation from the mean. Statistical analysis was performed using Welch's unpaired t-test (assuming unequal variances): * p < 0.05; ** p < 0.01; *** p < 0.001. | ||
Brominated complex 4-PF6 also displayed equivalent biological properties to non-brominated 3-PF6 (Fig. 2, Table 2), with similar in vitro antiproliferative activities (IC50/μM) against A2780 (ovarian) (Fig. 2b, Table 2). Moreover, ICP-MS (189Os) studies showed that both complexes were accumulated equally by A2780 cells when treated with equipotent concentrations of 3-PF6 (0.25–2 × IC50; Fig. 2c; ESI, Fig. S20, Table S4†). Decreasing the incubation temperature (from 310 K to 277 K) reduced significantly the quantity of osmium found inside cells treated with 3-PF6 (1× IC50 concentrations for 3 or 6 h; ESI, Fig. S21†). Suggesting that such cellular accumulation occurs through energy-dependent pathways.
The anticancer properties of 3-PF6 and 4-PF6 correlated strongly with those from other osmium azopyridine complexes of the same family carrying Os–I bonds (Fig. 2, Table 2; ESI Table S4†), which showed similar antiproliferative potencies and cellular accumulations, and were largely taken up by active transport mechanisms such as endocytosis.14,19 Yet, 3-PF6 and 4-PF6 were less selective towards non-cancer cells than 2-PF6. They inhibited the proliferation more (Table 2) and were accumulated in greater quantities by MRC-5 lung fibroblasts (Fig. 2; ESI, Table S4†). This could be due to differences in the interaction between Azpy-OH complexes 3-PF6 and 4-PF6 (zwitterionic at physiological pH) and 2-PF6 (cationic) with the surfaces of membranes from cancer cells (overall negative charge) and healthy cells (neutral).37,38
Complex 4-PF6 is therefore a suitable model to study the in-cell stability and distribution of this type of half-sandwich Os(II) drugs using ICP-MS (189Os, 79Br, 127I) and XRF (Os L3M5, Br KL3). First, the relative cellular accumulation of the different components of the complexes (i.e. Os, chelated azopyridine and halido leaving group) was determined by measuring the quantity of osmium (189Os), bromine (79Br) and iodine (127I) in cancer cells using ICP-MS. A2780 cells were treated with 4-PF6 (1× IC50, 0.42 μM) for 4, 8 and 24 h, or for 24 h followed by 24, 48 or 72 h recovery in complex-free medium (Fig. 3; ESI, Table S5†). Then, cell pellets were collected and digested in tetramethylammonium hydroxide (TMAH). TMAH was used instead of nitric acid to avoid oxidation of bromide to volatile Br2, and to overcome the high ionisation potential of the halogens (Br = 11.8 eV and I = 10.5 eV).39
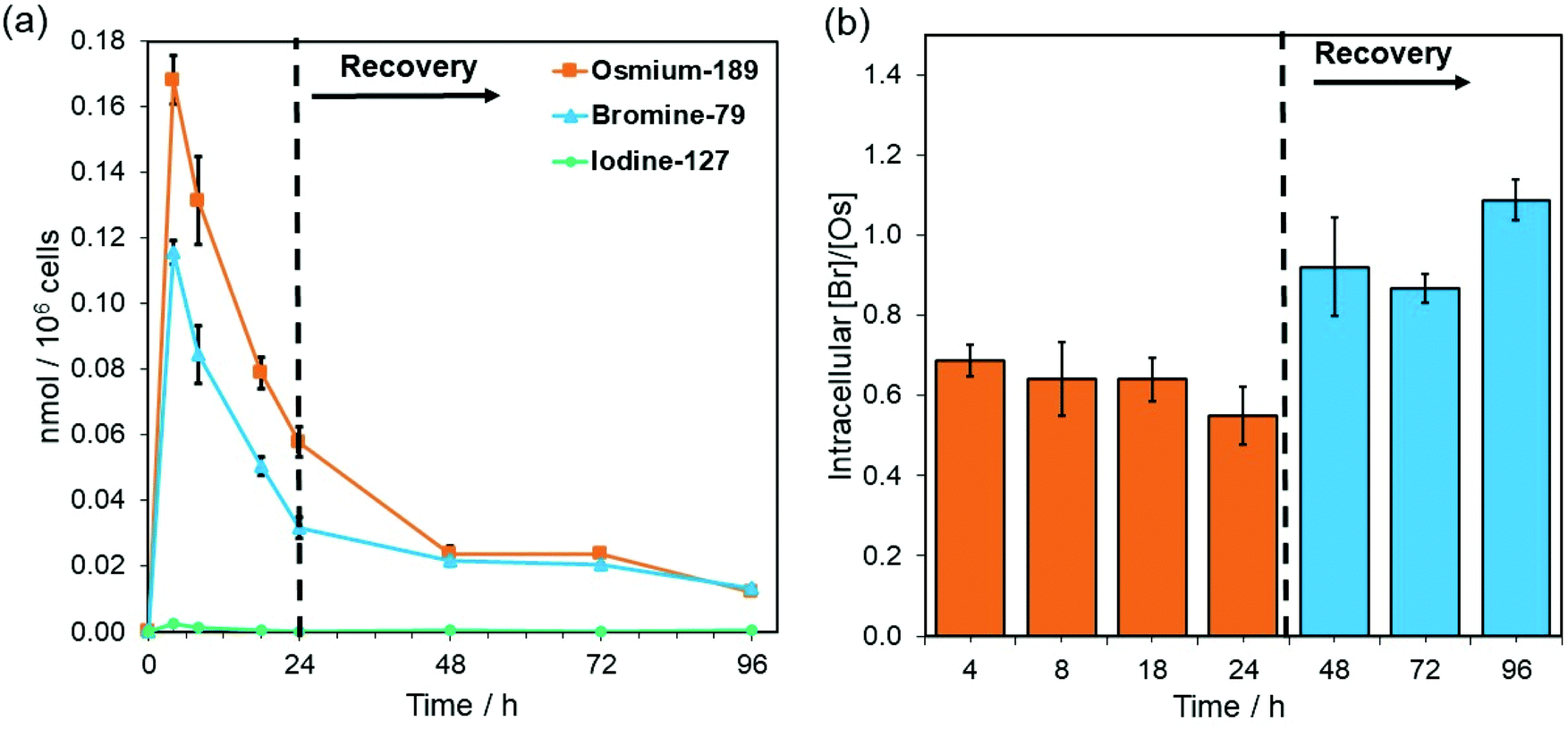 | ||
| Fig. 3 (a) Time-dependent cellular accumulation (nmol/106 cells) of 189Os (orange), 79Br (blue) and 127I (green) in A2780 cells treated with 1 × IC50 of 4-PF6 (0.42 μM) for 4, 8 and 24 h, and additionally 24 h with 24, 48 and 72 h recovery, as determined by ICP-MS. Data were normalised to the number of cells collected in each pellet. Error bars show the standard deviation from the mean. (b) Time-dependent intracellular bromine-to-osmium molar ratios (Br/Os) of A2780 cells treated with 1 × IC50 of 4-PF6 (0.42 μM) for for 4, 8 and 24 h, and additionally 24 h with 24, 48 and 72 h recovery, as determined from the ICP-MS data in (a). | ||
As predicted based on our previous experiments using iodine (131I) radiolabelling, the intracellular activation and cellular efflux of the iodido ligand occurred very fast.15 Hence it was not possible to detect iodine (127I) from 4-PF6 inside A2780 cells even at the shortest exposure time of the experiment (4 h, Fig. 3a). On the contrary, both osmium (189Os) and bromine (79Br) from the azopyridine ligand were detected inside cancer cells treated with 4-PF6. Accumulation of 4-PF6 in A2780 cells followed the same pattern as previously observed for other organo-osmium complexes of the same family (i.e.1-PF6 and 2-PF6),19,40 but it showed faster initial influx and efflux rates. Maximum amounts of intracellular osmium and bromine were found after just 4 h exposure (∼ 32 ± 1 and 9.2 ± 0.3 ng Os or Br/106 cells respectively; Fig. 3a; ESI, Table S5†), and then decreased at a fast rate until the removal of the drug from the medium.
During the exposure of A2780 ovarian cancer cells to 4-PF6 (4–24 h, no recovery), the molar ratio between 79Br/189Os remained relatively constant (ca. 0.65), with less bromine than osmium inside cells (Fig. 3, ESI, Table S5†). This suggests the partial in-cell degradation of the complex. Moreover, the lower amount of intracellular Br might arise from a faster efflux of the brominated azopyridine ligand (or fragments of it) compared to osmium-containing fragments. Yet, efflux of osmium at each timepoint seemed to be always higher than that of bromine (ESI, Table S5†), ruling out that possibility. Alternatively, higher levels of Os than Br might be achieved by rapid internalisation of both intact complex and cationic Os fragments from the degradation of 4-PF6 (occurring inside cells followed by excretion, or in the extracellular medium). Nevertheless, the 79Br/189Os ratio remained constant throughout the incubation with 4-PF6, indicating the presence of significant quantities of azopyridine ligand bound to Os, and probably intact complex inside A2780 cells. The intracellular level of Br and Os approached equal quantities (molar 79Br/189Os became ca. 1) just 24 h after 4-PF6 was removed from the medium, and remained the same even after 72 h recovery (Fig. 3b). As efflux from A2780 of fragments containing Os and Br occurs at different rates, most of the remaining intracellular osmium should be still coordinated to the azopyridine ligand a long time after it was initially internalised by the cancer cells. ICP-MS experiments also showed that MRC-5 healthy lung fibroblast cells accumulated much more Os than Br (molar ratio 79Br/189Os = 0.29) when they were treated for 24 h with the same concentration of 4-PF6 used to treat A2780 cancer cells (1 × IC50 A2780 = 0.42 μM; ESI, Fig. S22†). This could be caused by larger differences between the efflux/influx rates of Os- and Br-carrying fragments compared to those for cancer cells. However, it could also suggest that 4-PF6 displays much lower stability once inside non-cancer cells, caused by the chemical differences in the intracellular environment between cancerous and healthy cells.
The stability of the Os-azopyridine bond found in brominated complex 4-PF6 inside A2780 ovarian cancer cells was further investigated using nanofocused XRF by monitoring the relative distribution of Os (L3M5) and Br (KL3) emissions in a time-dependent manner. Cells were treated with 7× IC50 of 4-PF6 for 4 and 24 h before cryo-fixation and freeze-drying. Acquisition of elemental maps from those cells allowed investigation of whether the complex remains intact once internalised. As expected, XRF emissions from Os and Br were not detected in untreated A2780 cells (Fig. 4a; ESI, Fig. S23–S27†), but were clearly visible even after just 4 h treatment with 4-PF6 (Fig. 4b; ESI, Fig. S28–S33). As previously observed for A2780 cells treated with 1-PF6 and 2-PF6, cells treated with 4-PF6 for 4 h did not show significant alterations in their size and morphology, which were similar to those of the untreated cells (ESI, Table S6†). However, cells treated with the brominated complex for 24 h were significantly damaged, maintaining the roundness of the cells, but with changes in their total area (as seen for 2-PF6, ESI, Table S6†). Moreover, Os and Br co-localised strongly in cells treated with 4-PF6 for both 4 and 24 h exposure times (R = 0.33 ± 0.04 and 0.38 ± 0.08, respectively; ESI Table S7†). This is consistent with the results of ICP-MS studies, reinforcing the suggestion that most of the intracellular osmium is coordinated to the azopyridine ligand. Thus, significant amounts of this type of complex might still remain intact even long periods of time after they have been internalised by cells.
 | ||
| Fig. 4 Synchrotron-XRF elemental maps of cryo-fixed and freeze-dried A2780 (human ovarian) cancer cells grown on silicon nitride membranes treated with 7 × IC50 of brominated complex 4-PF6 (3 μM) for 4, 8 or 24 h (no recovery), showing phosphorus, osmium and bromine as represented using the 16-colour image setting in Image J software.33 Data were obtained using an incident energy of 15 keV: (a) untreated, (b) 4-PF6 (4 h), (c) 4-PF6 (4 h). Data were fitted in PyMCa,24 and images generated in ImageJ software.33 The scale bar = 10 μm. | ||
Conclusions
Metal complexes offer the prospect of novel drugs with new mechanisms of action that can combat resistance to current clinical drugs, especially in the treatment of cancer and microbial infections.10,12,41–46 Improvements in the design of metallodrugs require consideration not only of the role of the metal and its oxidation state, but also that of the ligands, both monodentate and chelating ligands. Investigation of the redox and ligand exchange chemistry of metallodrugs inside cells is a major challenge, but an important step towards optimising structure–activity relationships for both efficacy and unwanted side-effects.Here we have probed reactions of half-sandwich Os(II) anticancer complexes [Os(η6-p-cymene)(R1-PhAzPy-R2)X]PF6 in human ovarian cancer cells by monitoring Os, Br and I by ICP-MS and XRF. These measurements have allowed us to investigate the accumulation and localisation of the complexes in cancer and normal cells, as well as the release of the monodentate iodido ligand and chelated bromo-azopyridine. These results confirm that this family of Os(II) azopyridine complexes are activated rapidly in cancer cells via hydrolysis of the Os–I bond, most likely due to attack by intracellular GSH on the azo-bond. We have also shown for the first time that the osmium-azopyridine fragment generated after activation is remarkably stable in cancer cells over 24 h, although it is still unknown if it is part of a larger fragment also containing the bound arene, ({Os(p-cymene)(azopyridine)}2+). Release of the arene might be expected to facilate oxidation of Os(II) to Os(III).21 These complexes also appear to be degraded more rapidly in normal (non-cancerous) cells, perhaps providing a possible mechanism for selective antiproliferative activity towards cancer cells, which may reduce unwanted patient side effects. This work demonstrates how halogen tags can be used to probe the in-cell stability of metal complexes, providing crucial insights into their mechanisms of action necessary for clinical progression.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC grant no. EP/P030572/1) and Anglo American Platinum for funding. We thank Diamond Light Source and Warwick Collaborative Postgraduate Research Scholarships for a PhD studentship for E. M. B. We thank L. Song for assistance with ICP-MS experiments. C.S.C. thanks Gipuzkoa Foru Aldundia (Gipuzkoa Fellows program; grant number 2019-FELL-000018-01/62/2019) for financial support. This work was performed under the Maria de Maeztu Units of Excellence Programme – Grant No. MDM-2017-0720 Ministry of Science, Innovation and Universities. All synchrotron work was performed at the I14 Beamline (DLS, Oxford) under experiment numbers MG-19838 and SP-20548. We thank S. E. Bakker and I. Hands-Portsman (Advanced Bioimaging RTP) for assistance and training in plunge-freezing, and J. Tod for assistance with freeze-drying. We thank J. Parker and F. Cacho-Nerin for assistance during the experiments at the I14 Beamline.Notes and references
- Y. Fu, A. Habtemariam, A. M. Pizarro, S. H. van Rijt, D. J. Healey, P. A. Cooper, S. D. Shnyder, G. J. Clarkson and P. J. Sadler, Organometallic Osmium Arene Complexes with Potent Cancer Cell Cytotoxicity, J. Med. Chem., 2010, 53, 8192–8196 Search PubMed.
- J. M. Hearn, I. Romero-Canelon, A. F. Munro, Y. Fu, A. M. Pizarro, M. J. Garnett, U. McDermott, N. O. Carragher and P. J. Sadler, Potent organo-osmium compound shifts metabolism in epithelial ovarian cancer cells, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3800–E3805 Search PubMed.
- Y. Adar, M. Stark, E. E. Bram, P. Nowak-Sliwinska, H. van den Bergh, G. Szewczyk, T. Sarna, A. Skladanowski, A. W. Griffioen and Y. G. Assaraf, Cell Death Dis., 2012, 3, e293 Search PubMed.
- A. Bergamo, A. Masi, A. F. A. Peacock, A. Habtemariam, P. J. Sadler and G. Sava, In vivo tumour and metastasis reduction and in vitro effects on invasion assays of the ruthenium RM175 and osmium AFAP51 organometallics in the mammary cancer model, J. Inorg. Biochem., 2010, 104, 79–86 Search PubMed.
- A. Gatti, A. Habtemariam, I. Romero-Canelón, J.-I. Song, B. Heer, G. J. Clarkson, D. Rogolino, P. J. Sadler and M. Carcelli, Half-Sandwich Arene Ruthenium(II) and Osmium(II) Thiosemicarbazone Complexes: Solution Behavior and Antiproliferative Activity, Organometallics, 2018, 37, 891–899 Search PubMed.
- A. F. Peacock, A. Habtemariam, S. A. Moggach, A. Prescimone, S. Parsons and P. J. Sadler, Chloro half-sandwich osmium(II) complexes: influence of chelated N,N-ligands on hydrolysis, guanine binding, and cytotoxicity, Inorg. Chem., 2007, 46, 4049–4059 Search PubMed.
- J. Pracharova, V. Novohradsky, H. Kostrhunova, P. Štarha, Z. Trávníček, J. Kasparkova and V. Brabec, Half-sandwich Os(ii) and Ru(ii) bathophenanthroline complexes: anticancer drug candidates with unusual potency and a cellular activity profile in highly invasive triple-negative breast cancer cells, Dalton Trans., 2018, 47, 12197–12208 Search PubMed.
- J. Kralj, A. Bolje, D. S. Polančec, I. Steiner, T. Gržan, A. Tupek, N. Stojanović, S. Hohloch, D. Urankar, M. Osmak, B. Sarkar, A. Brozovic and J. Košmrlj, Half-Sandwich Ir(III) and Os(II) Complexes of Pyridyl-Mesoionic Carbenes as Potential Anticancer Agents, Organometallics, 2019, 38, 4082–4092 Search PubMed.
- T. Nabiyeva, C. Marschner and B. Blom, Synthesis, structure and anti-cancer activity of osmium complexes bearing π-bound arene substituents and phosphane Co-Ligands: A review, Eur. J. Med. Chem., 2020, 201, 112483 Search PubMed.
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F.-X. Wang, W.-Y. Zhang and Z. Zhang, Metallodrugs are unique: opportunities and challenges of discovery and development, Chem. Sci., 2020, 11, 12888–12917 Search PubMed.
- N. Graf and S. J. Lippard, Redox activation of metal-based prodrugs as a strategy for drug delivery, Adv. Drug Des. Dev., 2012, 64, 993–1004 Search PubMed.
- P. Zhang and P. J. Sadler, Redox-Active Metal Complexes for Anticancer Therapy, Eur. J. Inorg. Chem., 2017, 2017, 1541–1548 Search PubMed.
- J. P. C. Coverdale, H. E. Bridgewater, J. I. Song, N. A. Smith, N. P. E. Barry, I. Bagley, P. J. Sadler and I. Romero-Canelon, In Vivo Selectivity and Localization of Reactive Oxygen Species (ROS) Induction by Osmium Anticancer Complexes That Circumvent Platinum Resistance, J. Med. Chem., 2018, 61, 9246–9255 Search PubMed.
- S. H. van Rijt, I. Romero-Canelon, Y. Fu, S. D. Shnyder and P. J. Sadler, Potent organometallic osmium compounds induce mitochondria-mediated apoptosis and S-phase cell cycle arrest in A549 non-small cell lung cancer cells, Metallomics, 2014, 6, 1014–1022 Search PubMed.
- R. J. Needham, C. Sanchez-Cano, X. Zhang, I. Romero-Canelón, A. Habtemariam, M. S. Cooper, L. Meszaros, G. J. Clarkson, P. J. Blower and P. J. Sadler, Angew. Chem., Int. Ed., 2017, 56, 1017–1020 Search PubMed.
- Y. Fu, M. J. Romero, A. Habtemariam, M. E. Snowden, L. Song, G. J. Clarkson, B. Qamar, A. M. Pizarro, P. R. Unwin and P. J. Sadler, The contrasting chemical reactivity of potent isoelectronic iminopyridine and azopyridine osmium(II) arene anticancer complexes, Chem. Sci., 2012, 3, 2485–2494 Search PubMed.
- C. Sanchez-Cano, I. Romero-Canelón, Y. Yang, I. J. Hands-Portman, S. Bohic, P. Cloetens and P. J. Sadler, Synchrotron X-Ray Fluorescence Nanoprobe Reveals Target Sites for Organo-Osmium Complex in Human Ovarian Cancer Cells, Chem. – Eur. J., 2017, 23, 2512–2516 Search PubMed.
- X. Zhang, F. Ponte, E. Borfecchia, A. Martini, C. Sanchez-Cano, E. Sicilia and P. J. Sadler, Glutathione activation of an organometallic half-sandwich anticancer drug candidate by ligand attack, Chem. Commun., 2019, 55, 14602–14605 Search PubMed.
- A. Ballesta, F. Billy, J. Coverdale, J.-I. Song, C. Sanchez-Cano, I. Romero-Canelón and P. Sadler, Kinetic analysis of the accumulation of a half-sandwich organo-osmium pro-drug in cancer cells, Metallomics, 2019, 11, 1648–1656 Search PubMed.
- C. Sanchez-Cano, I. Romero-Canelon, K. Geraki and P. J. Sadler, Microfocus X-ray fluorescence mapping of tumour penetration by an organo-osmium anticancer complex, J. Inorg. Biochem., 2018, 185, 26–29 Search PubMed.
- C. Sanchez-Cano, D. Gianolio, I. Romero-Canelon, R. Tucoulou and P. J. Sadler, Nanofocused synchrotron X-ray absorption studies of the intracellular redox state of an organometallic complex in cancer cells, Chem. Commun., 2019, 55, 7065–7068 Search PubMed.
- E. M. Bolitho, J. P. C. Coverdale, H. E. Bridgewater, G. J. Clarkson, P. D. Quinn, C. Sanchez-Cano and P. J. Sadler, Tracking Reactions of Asymmetric Organo-Osmium Transfer Hydrogenation Catalysts in Cancer Cells, Angew. Chem., Int. Ed., 2021, 60, 6462–6472 Search PubMed.
- J. Tönnemann, J. Risse, Z. Grote, R. Scopelliti and K. Severin, Efficient and Rapid Synthesis of Chlorido-Bridged Half-Sandwich Complexes of Ruthenium, Rhodium, and Iridium by Microwave Heating, Eur. J. Inorg. Chem., 2013, 2013, 4558–4562 Search PubMed.
- V. A. Solé, E. Papillon, M. Cotte, P. Walter and J. Susini, A multiplatform code for the analysis of energy-dispersive X-ray fluorescence spectra, Spectrochim. Acta, Part B, 2007, 62, 63–68 Search PubMed.
- V. Langlois, in Comprehensive Pediatric Nephrology, eds. D. F. Geary and F. Schaefer, Mosby, Philadelphia, 2008, pp. 39–54 Search PubMed.
- W. Maret, Analyzing free zinc(II) ion concentrations in cell biology with fluorescent chelating molecules, Metallomics, 2015, 7, 202–211 Search PubMed.
- E. Bafaro, Y. Liu, Y. Xu and R. E. Dempski, The emerging role of zinc transporters in cellular homeostasis and cancer, Signal Transduct. Target. Ther., 2017, 2, 17029 Search PubMed.
- P. Tudrej, M. Olbryt, E. Zembala-Nożyńska, K. A. Kujawa, A. J. Cortez, A. Fiszer-Kierzkowska, W. Pigłowski, B. Nikiel, M. Głowala-Kosińska, A. Bartkowska-Chrobok, A. Smagur, W. Fidyk and K. M. Lisowska, Establishment and Characterization of the Novel High-Grade Serous Ovarian Cancer Cell Line OVPA8, Int. J. Mol. Sci., 2018, 19, 2080 Search PubMed.
- A. M. Haslehurst, M. Koti, M. Dharsee, P. Nuin, K. Evans, J. Geraci, T. Childs, J. Chen, J. Li, J. Weberpals, S. Davey, J. Squire, P. C. Park and H. Feilotter, EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer, BMC Cancer, 2012, 12, 91–91 Search PubMed.
- T. Viscarra, K. Buchegger, I. Jofre, I. Riquelme, L. Zanella, M. Abanto, A. C. Parker, S. R. Piccolo, J. C. Roa, C. Ili and P. Brebi, Functional and transcriptomic characterization of carboplatin-resistant A2780 ovarian cancer cell line, Biol. Res., 2019, 52, 13 Search PubMed.
- M. Psurski, A. Łupicka-Słowik, A. Adamczyk-Woźniak, J. Wietrzyk and A. Sporzyński, Discovering simple phenylboronic acid and benzoxaborole derivatives for experimental oncology – phase cycle-specific inducers of apoptosis in A2780 ovarian cancer cells, Invest. New Drugs, 2019, 37, 35–46 Search PubMed.
- R. McRae, B. Lai and C. J. Fahrni, Subcellular redistribution and mitotic inheritance of transition metals in proliferating mouse fibroblast cells, Metallomics, 2013, 5, 52–61 Search PubMed.
- C. T. Rueden, J. Schindelin, M. C. Hiner, B. E. DeZonia, A. E. Walter, E. T. Arena and K. W. Eliceiri, ImageJ2: ImageJ for the next generation of scientific image data, BMC Bioinf., 2017, 18, 529–529 Search PubMed.
- S. Rohrbach, A. J. Smith, J. H. Pang, D. L. Poole, T. Tuttle, S. Chiba and J. A. Murphy, Concerted Nucleophilic Aromatic Substitution Reactions, Angew. Chem., Int. Ed., 2019, 58, 16368–16388 Search PubMed.
- F. Baumann, W. Kaim, G. Denninger, H.-J. Kümmerer and J. Fiedler, Widely Separated Reduction Processes of abpy-Coupled Areneosmium(II) Reaction Centers (abpy=2,2′-Azobispyridine): Stabilization of the Radical Intermediate and of the Os0OsII State, Organometallics, 2005, 24, 1966–1973 Search PubMed.
- Y. Fu, Organometallic osmium arene anticancer complexes, PhD Thesis, University of Warwick, 2011 Search PubMed.
- A. C. Alves, D. Ribeiro, C. Nunes and S. Reis, Biophysics in cancer: The relevance of drug-membrane interaction studies, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 2231–2244 Search PubMed.
- B. Chen, W. Le, Y. Wang, Z. Li, D. Wang, L. Ren, L. Lin, S. Cui, J. J. Hu, Y. Hu, P. Yang, R. C. Ewing, D. Shi and Z. Cui, Targeting Negative Surface Charges of Cancer Cells by Multifunctional Nanoprobes, Theranostics, 2016, 6, 1887–1898 Search PubMed.
- X. Bu, T. Wang and G. Hall, Determination of halogens in organic compounds by high resolution inductively coupled plasma mass spectrometry (HR-ICP-MS), J. Anal. At. Spectrom., 2003, 18, 1443–1451 Search PubMed.
- J. P. C. Coverdale, I. Romero-Canelón, C. Sanchez-Cano, G. J. Clarkson, A. Habtemariam, M. Wills and P. J. Sadler, Asymmetric transfer hydrogenation by synthetic catalysts in cancer cells, Nat. Chem., 2018, 10, 347–354 Search PubMed.
- L. D. Palmer and E. P. Skaar, Transition Metals and Virulence in Bacteria, Annu. Rev. Genet., 2016, 50, 67–91 Search PubMed.
- E. Boros, P. J. Dyson and G. Gasser, Classification of Metal-Based Drugs according to Their Mechanisms of Action, Chem, 2020, 6, 41–60 Search PubMed.
- A. T. Odularu, P. A. Ajibade, J. Z. Mbese and O. O. Oyedeji, Developments in Platinum-Group Metals as Dual Antibacterial and Anticancer Agents, J. Chem., 2019, 2019, 5459461 Search PubMed.
- K. J. Franz and N. Metzler-Nolte, Introduction: Metals in Medicine, Chem. Rev., 2019, 119, 727–729 Search PubMed.
- J. Yang, H. Wang, J. Liu, M. Ding, X. Xie, X. Yang, Y. Peng, S. Zhou, R. Ouyang and Y. Miao, Recent advances in nanosized metal organic frameworks for drug delivery and tumor therapy, RSC Adv., 2021, 11, 3241–3263 Search PubMed.
- A. Frei, J. Zuegg, A. G. Elliott, M. Baker, S. Braese, C. Brown, F. Chen, C. G. Dowson, G. Dujardin, N. Jung, A. P. King, A. M. Mansour, M. Massi, J. Moat, H. A. Mohamed, A. K. Renfrew, P. J. Rutledge, P. J. Sadler, M. H. Todd, C. E. Willans, J. J. Wilson, M. A. Cooper and M. A. T. Blaskovich, Metal complexes as a promising source for new antibiotics, Chem. Sci., 2020, 11, 2627–2639 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1qi00512j |
| This journal is © the Partner Organisations 2021 |