
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ real-time monitoring of the mechanism of self-assembly of short peptide supramolecular polymers†
Mari C.
Mañas-Torres
 a,
Cristina
Gila-Vilchez
b,
Juan A.
González-Vera
c,
Francisco
Conejero-Lara
d,
Victor
Blanco
a,
Juan Manuel
Cuerva
a,
Modesto T.
Lopez-Lopez
*be,
Angel
Orte
*c and
Luis
Álvarez de Cienfuegos
*ae
a,
Cristina
Gila-Vilchez
b,
Juan A.
González-Vera
c,
Francisco
Conejero-Lara
d,
Victor
Blanco
a,
Juan Manuel
Cuerva
a,
Modesto T.
Lopez-Lopez
*be,
Angel
Orte
*c and
Luis
Álvarez de Cienfuegos
*ae
aDpto de Química Orgánica, Facultad de Ciencias, Universidad de Granada UGR, 18071-Granada, Spain. E-mail: lac@ugr.es
bDpto de Física Aplicada, Facultad de Ciencias, UGR, Spain. E-mail: modesto@ugr.es
cDpto de FisicoQuímica, Facultad de Farmacia, UGR, 18072, Granada, Spain. E-mail: angelort@ugr.es
dDpto de Química Física, Facultad de Ciencias, UGR, Spain
eInstituto de Investigación Biosanitaria ibs.GRANADA, Spain
First published on 18th May 2021
Abstract
Making use of the combination of multiparametric Fluorescence Lifetime Imaging Microscopy (FLIM) and single-molecule Fluorescence Lifetime Correlation Spectroscopy (FLCS), we have been able to study for the early stages of the fluorenylmethyloxycarbonyl-diphenylalanine (Fmoc-FF) self-assembly process with single-molecule resolution, the kinetics of fiber formation, the packaging of the peptides within the fibers and the capacity of the peptides to reassemble after disruption (self-healing) in the presence of different metallic cations. Other techniques such as FTIR, TEM, DSC and DFT calculations support our findings. The impact that the mechanism of self-assembly has on the physical (rigidity and self-healing) properties of the resulting gels have also been evaluated by rheology. Calcium ions are able to promote the self-assembly of Fmoc-FF faster and more efficiently, forming more rigid hydrogels than do cesium ions. The reasons behind this effect may be explained by the different capacities that these two cations have to coordinate with the peptide, modulate its hydrophobicity and stabilize the water–solute interphase. These findings shed light on the impact that small changes have on the process of self-assembly and can help to understand the influence of the environmental conditions on the in vivo uncontrolled self-assembly of certain proteins.
Introduction
Inspired by nature, researchers have created sophisticated materials through self-assembly.1–4 Their properties are controlled by precise molecular organization from a careful balance of different and usually weak forces. Consequently, pathways involved in the self-assembly process are complex and not well-understood.5–7 On the other hand, such diverse and dynamic non-covalent interactions have given rise to materials showing reversibility, self-healing and the capacity to respond to external stimuli such as temperature, pH, light, solvent composition, concentration, etc.8–14 Nevertheless, the initial formation of the materials is also dynamic, showing a time-dependent evolution in which small initial changes in conditions can have a significant impact on the final result, giving rise to systems that present greater adaptability.15–17 Thus, it is essential to understand the molecular mechanisms by which these molecules self-assemble. Beyond the tremendous complexity of the self-assembly of natural biomolecules,18–24 the study of simpler bioinspired building blocks that are capable of self-assembling under similar conditions can help the understanding of more complex behaviours. The self-assembly of aromatic peptides represents a simpler model to study the interplay between hydrogen bonds and hydrophobic interactions.25–29 Gazit,30 and Yan31 have shown that the mechanism of self-assembly of these peptides goes through consecutive metastable intermediates showing different polymorphic aggregates. Nevertheless, due to their dynamic and complex nature, and the diluted concentrations required to experimentally observe and monitor nucleation and pre-nucleation events, these processes have been difficult to establish. Moreover, traditional techniques used to characterize them, such as nuclear magnetic resonance (NMR), UV-vis spectroscopy, circular dichroism (CD), X-ray diffraction (XRD) and different electronic microscopies, are indirect and/or unable to report events in real-time, which can lead to misinterpretations. A better solution would be to directly visualize the mechanism of growth from nuclei to fibers using fluorescence microscopy. The groups of Meijer,32 De Cola33 and Hamachi,34 among others,35,36 have implemented these techniques for the in situ real-time imaging of the formation of supramolecular fibers and studying the phenomenon of self-sorting and exchange of monomers between fibers, phenomena that are difficult to study using other techniques.Herein, we have monitored and studied in detail the mechanism of the self-assembly of Fmoc-FF in the presence of different metal salts.37–41 Using an environment-sensitive fluorescent reporter for peptide aggregation and the combination of advanced multiparametric fluorescence microscopy and single-molecule fluorescence lifetime correlation spectroscopy (FLCS), we have been able to monitor the mechanism of early self-assembly and the growth of peptide fibers from solutions to gels. Our results are well supported by Transmission Electron Microscopy (TEM), Fourier-Transform Infrared (FT-IR) spectroscopy, DFT calculations, Differential Scanning Calorimetry (DSC) and rheology of the resulting gels. The possibility to in situ monitor the process of self-assembly at the molecular level has allowed us to gain a deeper insight into the mechanism of supramolecular polymerization and understand the influence that small changes can have on the self-assembly process and in the physical properties of the resulting gels.
Fmoc-dipeptides are relevant molecules in materials science due to their great capacity to self-assemble into long fibers, giving rise to hydrogels with applications in fields such as, tissue engineering,42–44 drug delivery,45–47 biomineralization,48,49 protein crystallization,50–53etc. It is interesting to note that the self-assembly of Fmoc-peptides is significantly influenced by the presence of metal ions, which are able to modulate not only their mechanism of growth31 but also the secondary structure of the resulting supramolecular polymers and the properties of the gel.41 Furthermore, the presence of metal ions coordinated with the supramolecular structure gives rise to metallogels with multiple applications.54
Results and discussion
In aqueous solutions, it is known that the formation of supramolecular polymers is driven by the hydrophobic collapse of the monomers.55 Therefore, and taking into account that water–Fmoc-FF interactions are key for the process, we wondered if metal cations could affect such interplay, adjusting to different properties: the hydrophobicity of the cation (kosmotropes or chaotropes) and the binding capabilities.56 Thus, for example, two simple cations such as Ca2+, a kosmotrope, and Cs+, a chaotrope, could be useful for such purposes. It is known that kosmotropes tend to destabilize the water–solute interphase, promoting hydrophobic interactions, while chaotropes have the opposite effect, that is, they stabilize the water–solute interphase, weakening hydrophobic interactions.57,58 For this study, Fmoc-FF presents another remarkable characteristic. Starting from its sodium salt, which is required to make it soluble in water, the promotion of gelation requires only the addition of another salt. Therefore, the resulting properties, from the initial state to the final state, can be easily interpreted since they are modulated by one parameter: the nature of the salts.First, we evaluated the possibility to extract information from Fmoc-FF supramolecular aggregates in the presence of Ca2+ and Cs+ salts using different fluorescence techniques. We employed an environment-sensitive fluorophore that is capable of reporting on the surrounding interactions upon intercalation into proteinaceous fibrils, the 9-azetidinyl-quinolimide derivative AQui [9-(azetidin-1-yl)-5-butyl-4Hbenzo[de][2,6]naphthyridine-4,6(5H)-dione].59 The main advantage of this dye is that it undergoes up to a 10-fold increase in its emission upon binding to the hydrophobic pockets of proteins, also experiencing environment-sensitive changes in both the spectral shift and fluorescence lifetime, τ. These features allow the identification of different interactions by the combination of ratiometric and fluorescence lifetime imaging (FLIM) methodologies (see Fig. 1A and ESI† for experimental details). First, Fmoc-FF (10 mM) aggregates were formed in the presence of dye AQui (25 μM) induced either by Ca2+ or Cs+ (Fig. 1B and C). In both cases, long fibers were detected and a reticulate structure was visible, although it was clearer in the fibers formed with Ca2+. Importantly, the quantitative information in FLIM and I630/I550 ratio images revealed a totally different environment of the dye AQui in the fibers formed with Ca2+ or Cs+. Whereas Fmoc-FF Ca2+ fibers were characterized by long τ values and I630/I550 ratio values of around 1.2 for dye AQui, this dye in Fmoc-FF Cs+ fibers exhibited lower τ values and slightly larger I630/I550 ratio values (Fig. 1D and E). The features of dye AQui indicated a more hydrophilic environment or solvent-exposed pockets for the dye in the fibers of Fmoc-FF Cs+, whereas AQui is more protected in hydrophobic pockets in Fmoc-FF Ca2+ fibers. Interestingly, a second population of aggregates was exclusively found for Fmoc-FF Cs+. A subpopulation of amorphous aggregates was clearly visible (highlighted with arrows in Fig. 1C), characterized by very low τ values and I630/I550 ratio values of >2.5, indicative of a highly solvent-exposed, hydrophilic environment.
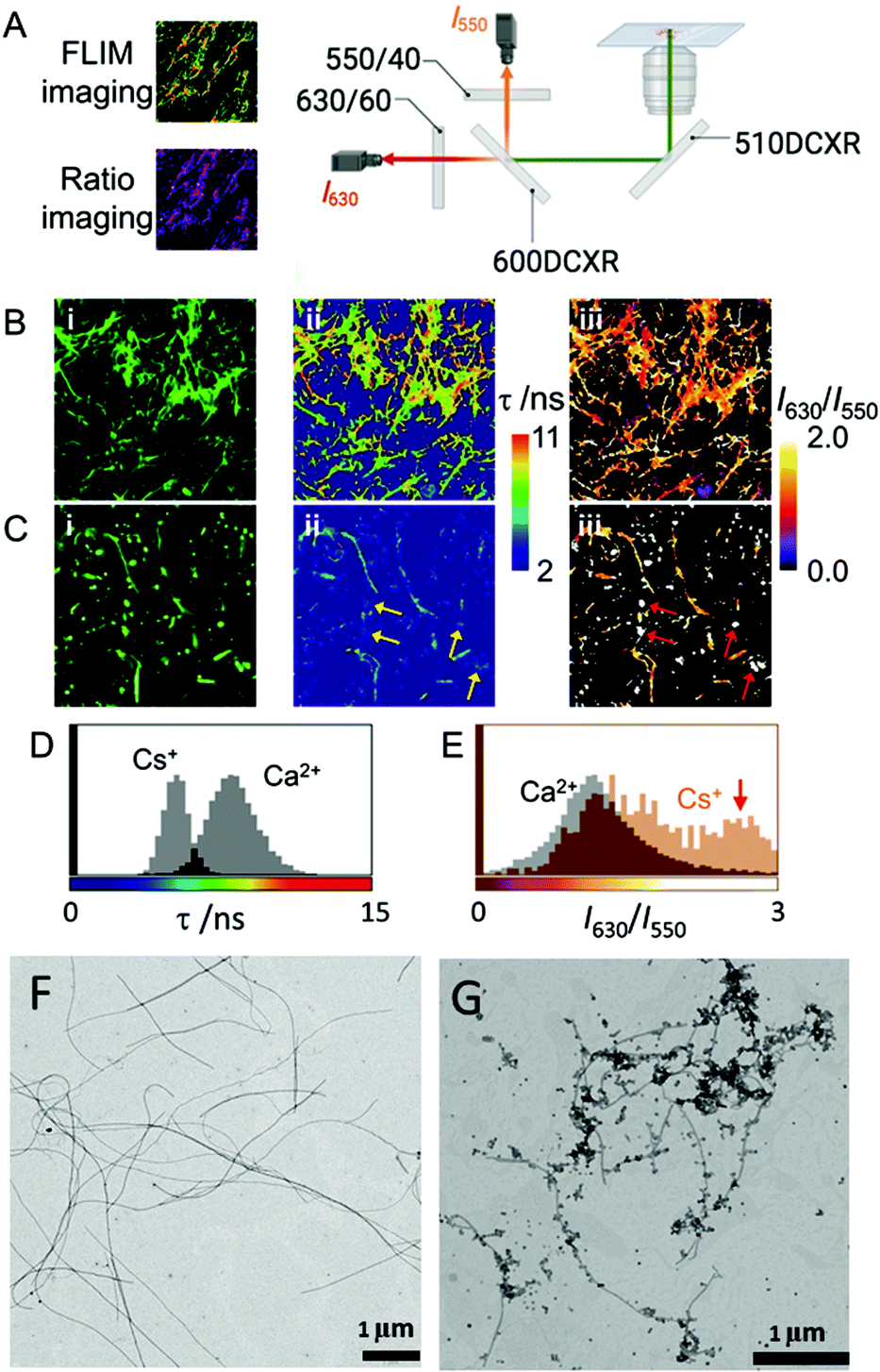 | ||
| Fig. 1 (A) Scheme of the instrumentation employed for multidimensional ratiometric and FLIM imaging. (B and C) Intensity (i), FLIM (ii), and ratiometric (iii) images of dye AQui in Fmoc-FF hydrogels formed with Ca2+ (B) and Cs+ (C). Arrows in (C) indicate the presence of peptide aggregates in which the emission of dye AQui is characterized by a low τ value and high I630/I550 ratio. The size of the images is 10 μm × 10 μm. (D and E) Population histograms of τ values (D) and I630/I550 ratios (E) of AQui in Fmoc-FF hydrogels formed with Ca2+ and Cs+, obtained as the total distribution from at least five different images. (F and G) TEM images of Fmoc-FF Ca2+ salt (F) and Fmoc-FF Cs+ salt (G) at 5 mM. | ||
In fact, the coexistence of nanospheres and fibrils was also found in the TEM images of Fmoc-FF Cs+, whereas they were not detected with Ca2+ ions (Fig. 1F and G). Indeed, nanospheres of homogeneous sizes with diameters of 120–160 nm were found for 2.5 mM Fmoc-FF Cs+ (Fig. S1, ESI†), whereas fibers arose by the coalescence of nanospheres at higher concentrations. Interestingly, Fmoc-FF Na+ fibrillation followed a similar pattern to that with Cs+ ions (Fig. S1, ESI†). On the contrary, TEM images of the peptide solution at 2.5 mM in the presence of Ca2+ showed aggregates of bigger sizes formed by the coalescence of droplets (Fig. S1, ESI†), suggesting the emergence of a liquid–liquid phase separation process at the very early stage (Fig. S2, ESI†),31 but these all became narrow fibrils of 14–20 nm diameter with well-defined edges at higher concentrations (≥5 mM, Fig. S1, ESI†), suggesting that Ca2+ was able to promote peptide self-assembly more efficiently than Na+ or Cs+ (see ESI† for additional HR-TEM and AFM images, Fig. S3 and S4). These results were different with the less hydrophobic Fmoc-AA (Fmoc-dialanine) and Fmoc-GG (Fmoc-diglycine) peptides (Fig. S5, ESI†).
Mechanism of self-assembly
The early stages of self-assembly were investigated using multiparametric imaging and FLCS, a single-molecule technique reporting on the diffusional properties of molecular systems (Fig. 2A and B).60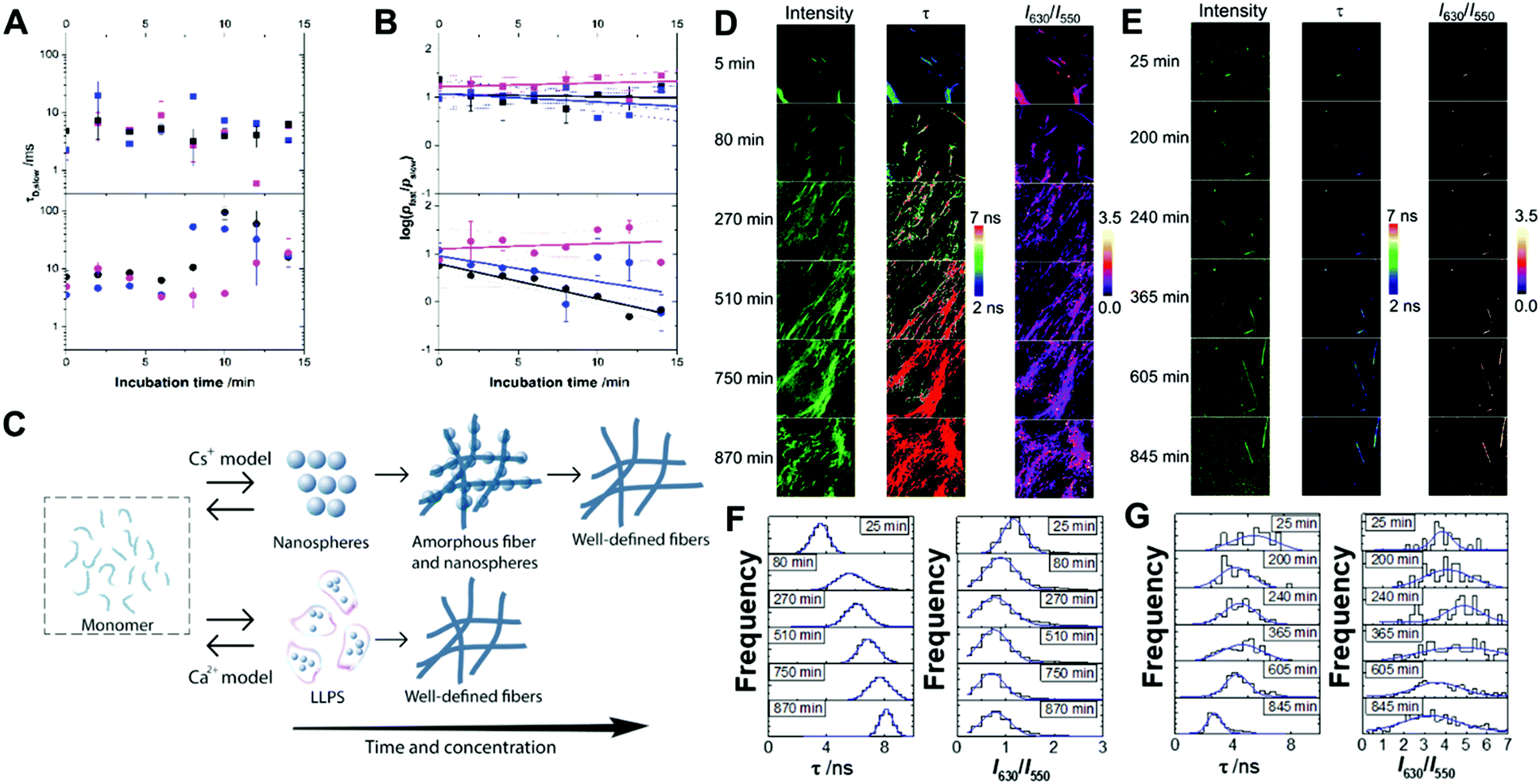 | ||
| Fig. 2 Diffusion time of the slow-diffusing species (τD,slow; A) and plot of the ratio of relative contributions of the fast- and slow-diffusing species on a logarithmic scale (log(pfast/pslow); B) from the fittings of FLCS curves of Fmoc-FF in the presence of Ca2+ (squares) or Cs+ (circles) at an ion concentration of 6.25 mM (magenta), 12.5 mM (blue) or 25 mM (black). Solid lines in panel B represent linear fits to the experimental data. Dashed lines are the upper and lower confidence limits of the fit at 95% confidence. (C) Model of the different pathways of formation of nuclei of Fmoc-FF in the presence of Cs+ or Ca2+ counterions. (D–G) Multiparametric fluorescence microscopy of AQui in incubating Fmoc-FF in the presence of Ca2+ (D and F) or Cs+ (E and G). Panels D and E display the intensity, FLIM (τ) and I630/I550 ratio images as a function of the incubation time. Panels F and G display the extracted distributions of lifetime τ and I630/I550 ratio from the images in C and D. The size of the images is 10 μm × 10 μm. | ||
We studied Fmoc-FF Ca2+ and Fmoc-FF Cs+ at three different concentrations of the dipeptide and counterion, and followed the time evolution of the FLCS correlation curves for the initial 15 minutes of interaction. The analysis of the correlation curves demonstrated the presence of two different diffusing states, a fast-diffusing form with a diffusion time, τD,fast, of 55 ± 3 μs, and a slow-diffusing state with different τD,slow values depending on the concentration and incubation time. The τD,fast values correspond to the diffusional properties of small organic molecules, such as individual fluorophores; hence, we assigned this form to the Fmoc-FF monomer. Then, the slow-diffusing species can be attributed to specific growing nuclei and aggregates. Hence, we compared the different behaviour of Fmoc-FF Ca2+ and Fmoc-FF Cs+ salts, in terms of τD,slow values, which can be related to the average size of the aggregates, and the relative population of the slow and fast components.
The FLCS curves of incubated Fmoc-FF Ca2+ showed a constant contribution of large, high-order aggregates of around 7 ± 3% and an average τD,slow value of (7 ± 5) ms at the three concentrations tested (Fig. 2A and B). This behaviour is consistent with a continuous equilibrium between monomer and aggregates in a micellar-like fashion, similar to what happens for other aggregating small peptides.22,37,39,40 Strikingly, this situation remained constant over a long period of time (>30 min), indicating a stable nucleus size in equilibrium with the monomer serving as a reservoir for subsequent fibril growth (Fig. 2C). By contrast, the FLCS curves of incubated Fmoc-FF Cs+ pictured a clearly different scenario. The τD,slow values exhibited time and concentration dependence, with values increasing up to several tens of ms (Fig. 2A), reaching higher values at 10 mM Fmoc-FF and 25 mM Cs+. This indicates that large species, responsible for τD,slow, underwent a growth within the 15 min time frame. Interestingly, at the lowest concentration used (2.5 mM Fmoc-FF), the relative contribution of the slow-diffusing species was around 6 ± 3% and did not vary with the incubation time; this is similar to the situation found for the supramolecular polymer forming in the presence of Ca2+. Nevertheless, at 5 and 10 mM Fmoc-FF, the relative contribution of the large species showed a remarkable increase with time as depicted in the ratio of the two relative populations (Fig. 2B). We obtained the apparent kinetics of pfast → pslow equilibrium shift from the slopes of plots in Fig. 2B. The apparent rates were (12 ± 2) × 10−4 s−1 and (9 ± 5) × 10−4 s−1 for the 10 mM and 5 mM concentrations, respectively; around 30% faster at the higher concentration of Fmoc-FF. These findings support a different mechanism for the self-assembly of Fmoc-FF in the presence of Cs+ ions: there is a critical nucleus size in equilibrium with monomers; and as the incorporation of large aggregates into even larger amorphous fibers (which would not diffuse in a time regime accessible by FLCS) proceeds, a shift in the monomer ⇆ nucleus aggregate equilibrium toward the formation of more aggregates allows fibrillization (Fig. 2C).
Kinetics of self-assembly
We also followed the self-assembly kinetics of Fmoc-FF Ca2+ and Fmoc-FF Cs+ solutions (10 mM Fmoc-FF peptide, 25 mM Ca2+ or Cs+ and 25 μM of AQui) by fluorescence spectrometry and multiparametric microscopy through intensity, FLIM and ratiometric images of AQui during the formation of the supramolecular polymers. The AQui dye exhibited a more pronounced emission enhancement and a further blue-shift in Fmoc-FF Ca2+ fibers than it did in Fmoc-FF Cs+ (Fig. S6, ESI†), confirming that fibrils formed with Ca2+ counterions were more hydrophobic than those formed in the presence of Cs+, as has already been discussed for the mature fibrils. Fibril formation with Ca2+ ions showed faster kinetics, with an apparent rate constant of (1.8 ± 0.1) × 10−3 s−1, whereas the growth rate was (2.1 ± 0.5) × 10−4 s−1 for Cs+-promoted fibrils, one order of magnitude slower than those formed with Ca2+ (Fig. S6, ESI†). In addition, real-time FLIM and ratiometric images of AQui during the formation of Fmoc-FF Ca2+ and Fmoc-FF Cs+ fibers (Fig. 2D and E) demonstrated that the dye was in a hydrophilic environment in the early stages, characterized by low intensity emission and low fluorescence lifetime values, but as the formation of mature fibers occurred, AQui found more hydrophobic pockets to interact, hence resulting in enhanced fluorescence emission and longer τ values. This was also noteworthy in the I630/I550 ratio images, which exhibited a shift towards higher ratio values as fibrillar growth proceeded. These experiments also evidenced differences in the type of fibrils formed in the presence of Ca2+ or Cs+. Quantitatively, the population histograms demonstrated how fibrils formed with Ca2+ evolved to more hydrophobic pockets with time (characterized by higher τ values and lower I630/I550 ratios, Fig. 2F). By contrast, fibrils formed in the presence of Cs+ ions always showed low τ values and high I630/I550 ratios (Fig. 2G), supporting that the reporter AQui dye interacts with more hydrophilic sites.Our results so far clearly demonstrated distinct interactions, structure and kinetics in Fmoc-FF hydrogel fibrils formed with Ca2+ or Cs+ ions. To shed more light onto these differences, we employed a combination of experimental and computational techniques. FTIR also supported the significant structural differences between the gels formed in the presence of Cs+ and Ca2+ (Fig. 3). The spectra showed two separate bands at 1638 cm−1 and 1690 cm−1, attributable to peptidic and carbamate groups, respectively.61,62 In the presence of Cs+ the gel formation resulted in an initial sharp drop in absorbance for both bands and a slow recovery for the peptide band, although the band positions did not change significantly. By contrast, the formation of gel in the presence of Ca2+ clearly showed a simultaneous and opposite shift for both bands to 1635 and 1692 cm−1, respectively. This suggests the formation and strengthening of intermolecular H-bonds between the peptides in the gel that are not present in the Fmoc-FF Cs+ gel.
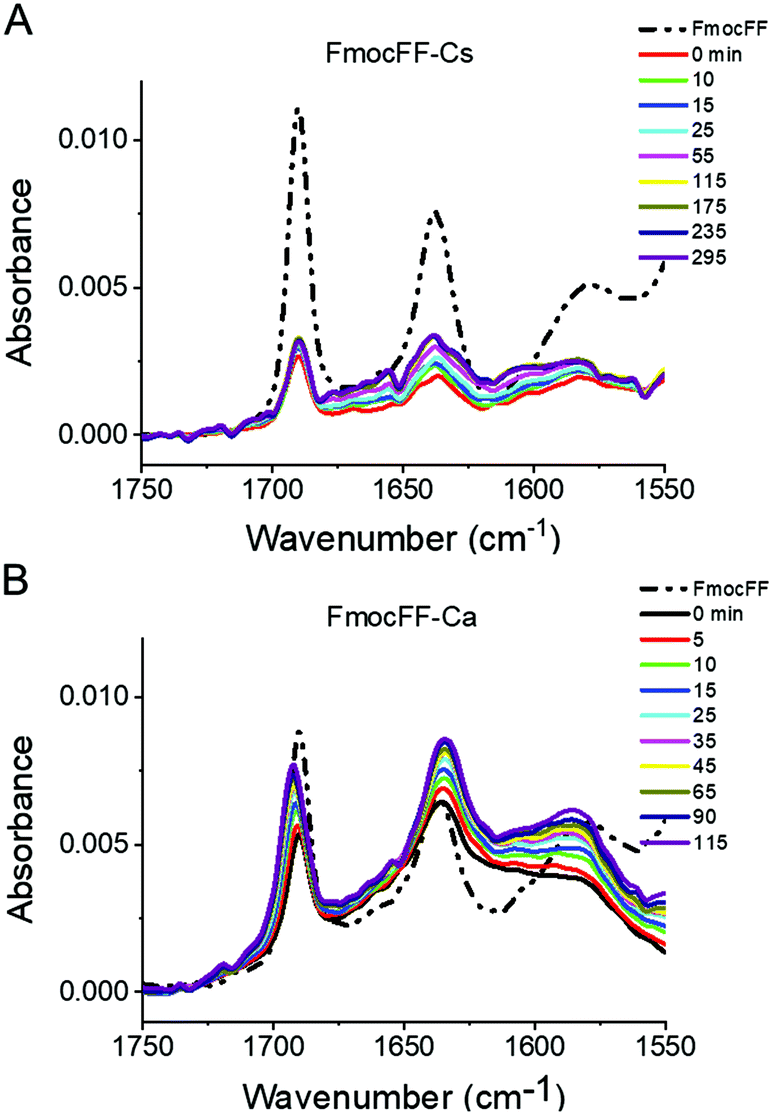 | ||
| Fig. 3 Time-dependent FTIR spectra of Fmoc-FF Cs+ (A) and Fmoc-FF Ca2+ (B) salt hydrogels (10 mM peptide conc.). | ||
The different fibrillation mechanism was also confirmed in DSC measurements investigating the energy exchanges involved in the formation of Fmoc-FF hydrogels. Soluble Fmoc-FF samples at different concentrations in the presence of different salts were heated within the DSC calorimeter at 2 °C min−1 to induce their aggregation. At the 2.5 mM Fmoc-FF concentration, in the presence of Cs+ or Ca2+, a single endothermic peak was observed around 41–44 °C, which could be attributed to the disaggregation of nanospheres and droplets observed by TEM. In the presence of Na+ these aggregates showed a similar peak but at a lower temperature, suggesting that the nanospheres were less stable under these conditions although they became stabilized at a higher Fmoc-FF concentration (Fig. 4). At 5 mM and 10 mM concentration, Fmoc-FF-Cs+ showed more complex DSC thermograms with several maxima and minima at higher temperatures (Fig. 4), suggesting the formation of different, more stable, aggregates during the heating.
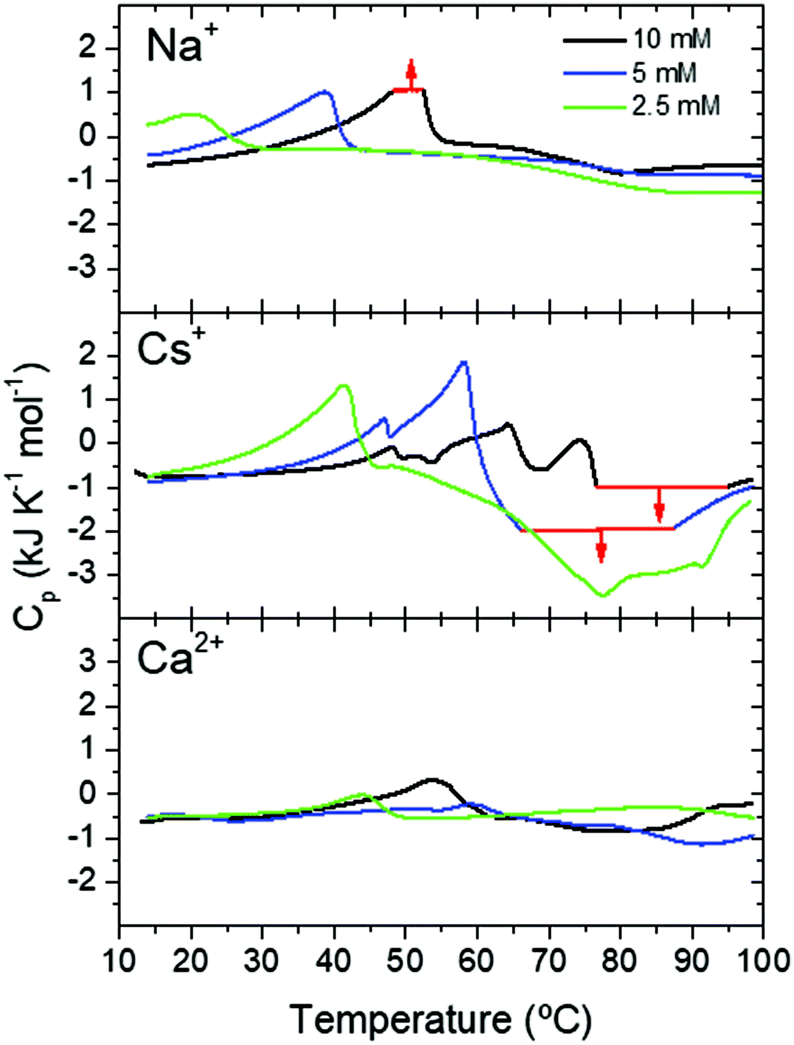 | ||
| Fig. 4 DSC scans of Fmoc-FF at different concentrations in the presence of different cations. Thermogram regions in red indicate signal saturation of the calorimeter. | ||
These aggregate conversions were time-dependent, irreversible processes, as indicated by experiments at different scan rates and with consecutive re-heatings (Fig. S7, ESI†). Interestingly, the last endothermic peak was followed by a strong exothermic effect at high temperature, attributable to extensive gel formation. A second consecutive full heating showed no peaks of any kind, indicating the high thermal stability of the gel formed (Fig. S7, ESI†). In the case of Fmoc-FF-Ca2+ the DSC profiles were less complex and the endothermic and exothermic effects were much less intense (Fig. 4), despite the formation of a highly rigid and stable hydrogel. This indicates that the gel formation of Fmoc-FF Ca2+ passes through a less complex pathway than in the case of Fmoc-FF Cs+. These results suggest that in the presence of Cs+ several increasingly stable intermediate species are formed, favored by an increase in Fmoc-FF and Cs+ concentrations. Since these intermediates form by consecutive endothermic processes, they accumulate enthalpy, which is released by their subsequent exothermic conversion into the hydrogel. In the presence of Ca2+, however, the precursor states of gelification appear to accumulate less enthalpy and, accordingly, gel formation is clearly less exothermic. All these results are in agreement with the mechanistic model in Fig. 2C.
To shed light on the peptide–metal coordination interactions that may be involved in the different types of fibril formation, the exchange of Na+ in Fmoc-FF Na+ by the other cations was investigated using DFT calculations at the ωB97XD/def2SVP level of theory, using the LanL2DZ basis set and pseudopotentials for the metal cations. Two possible conformations were considered for the Fmoc-FF backbone, an extended β-sheet-like conformation, and a more folded conformation (see the ESI†). We studied the interaction of deprotonated Fmoc-FF with Na+, showing that the β-sheet-like structure with the cation interacting with the terminal carboxylate was more stable than the folded one. We then investigated the exchange of Na+ by Cs+ or Ca2+. The corresponding calculations showed that the replacement of Na+ by a Ca2+ ion was favoured (ΔG = −3.56 kcal mol−1), indicating that Ca2+ binds more strongly to the Fmoc-FF residue through hydrogen bonding-ready sites due to its kosmotrope character. Moreover, the extended structure, which should favour the supramolecular assembly, remains more stable. On the contrary, the interaction with Cs+ was less favoured and would also point to a more folded conformation, which is less suitable for the establishment of supramolecular interactions.
Mechanical properties
Applications of hydrogels as new materials require a thorough characterization of their mechanical properties by rheological measurements under shear.40,63 Given that Fmoc-FF hydrogels formed with either Ca2+ or Cs+ ions presented different self-assembly mechanisms, it is important to correlate them with the corresponding rheological properties.38,64,65 The approximate independence of the viscoelastic moduli with shear strain amplitude at low values of this quantity, exhibited in amplitude sweeps (Fig. 5A and D), defines the linear viscoelastic region (LVR). This region is more clearly observed for Ca2+ samples and for the most concentrated Cs+ sample, whereas it can only be approximately inferred for gels with lower concentrations of Fmoc-FF Cs+ samples (2.5 mM and 5 mM), indicating weaker interactions in these two samples. For gel-like samples, the storage modulus (G′) must be appreciably larger than the loss modulus (G′′) within the LVR. Nevertheless, for the two lowest concentrations in the case of Fmoc-FF Cs+, G′ was barely larger than G′′, which can be taken as additional proof of the extreme weakness of the resulting gels, or even their liquid-like (instead of gel-like) character. For Fmoc-FF Cs+, at a concentration of 10 mM, G′ was markedly higher than G′′ (about a factor of 2), although by much less than one order of magnitude, which is typical of weak gels. On the contrary, for Ca2+ samples (Fig. 5A), G′ was larger than G′′ by almost an order of magnitude, which is normally taken as the interface between weak and strong gels.66 This can be further supported by frequency sweeps (Fig. 5C and F), which for the Cs+ samples exhibited a growing trend with frequency, especially for the two lowest concentrations, which is more typical of polymeric solutions,66 corroborating the mostly liquid-like nature of the Fmoc-FF Cs+ samples at low concentrations. On the contrary, frequency sweeps of Ca2+ samples showed values of G′ and G′′ approximately independent of frequency, with G′ > G′′, as expected for gels. For Fmoc-FF Cs+ (10 mM), an intermediate behaviour between liquid-like and gel-like character was demonstrated. Finally, the strength of the samples can be quantified by the average values of the storage modulus corresponding to the LVR (Fig. 5B and E). As observed, Ca2+ samples exhibited values of the storage modulus about 500 times larger than for Cs+ samples, demonstrating the much larger strength of the former. For both salts, the strength of the samples largely increased with peptide content (remember that the peptide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :salt ratio was 1:2.5 in all samples). This enhancement with peptide (and salt) content can be explained as follows. Fmoc-FF Ca2+ samples demonstrated gel-like behaviour and thus, according to the classical theory for gels, the elastic modulus of the gel network is given by67,68
:salt ratio was 1:2.5 in all samples). This enhancement with peptide (and salt) content can be explained as follows. Fmoc-FF Ca2+ samples demonstrated gel-like behaviour and thus, according to the classical theory for gels, the elastic modulus of the gel network is given by67,68| G ≈ νkT |
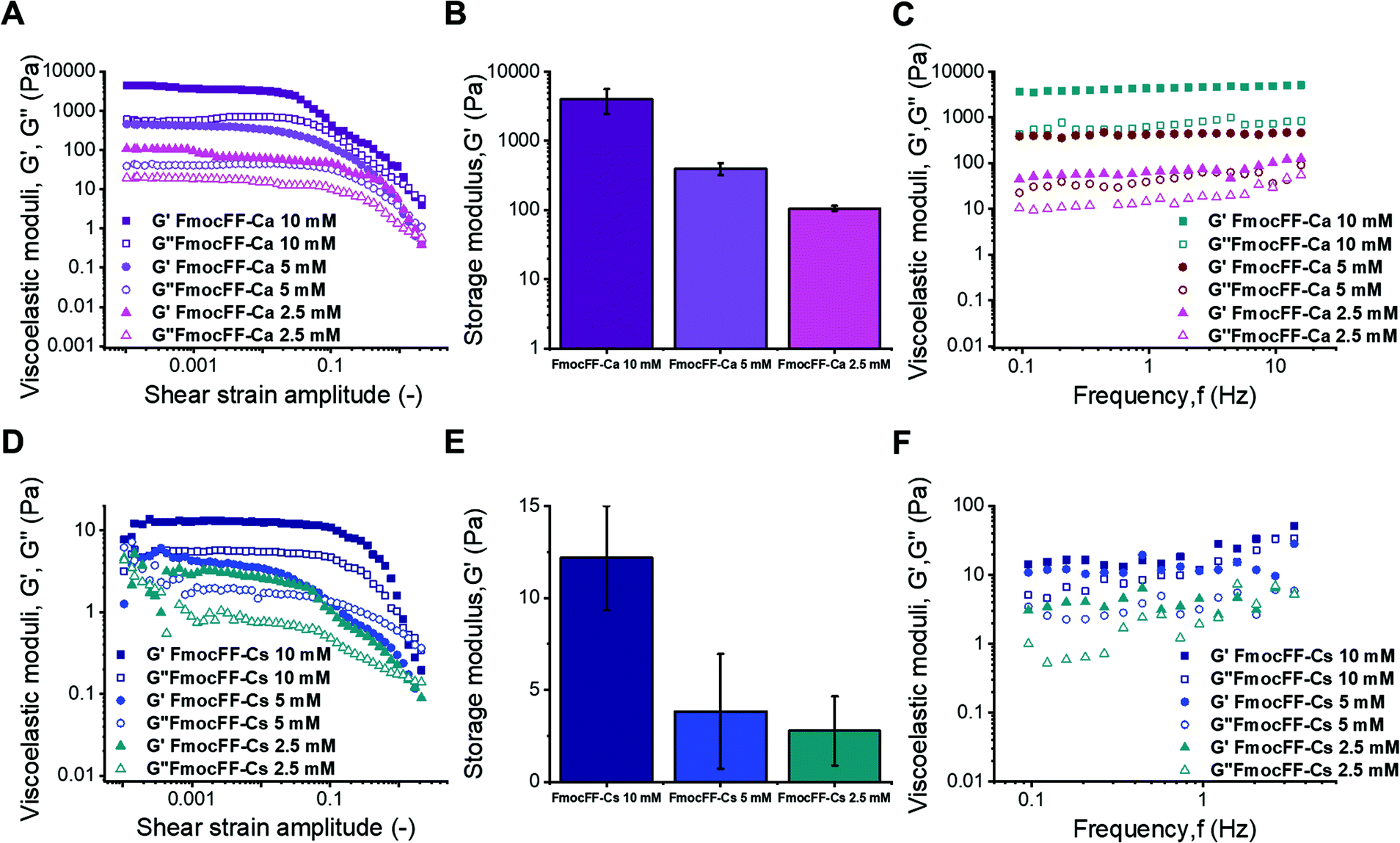 | ||
| Fig. 5 Mechanical characterization of the samples under oscillatory shear. (A) and (C) show the amplitude and frequency sweeps for Ca2+ samples, and (B) the mean and standard deviation of the storage modulus corresponding to the linear viscoelastic region. (D)–(F) show the same results but for Cs+ samples. | ||
Self-healing studies
The ability of the hydrogels to recover the reticulate structure after disrupting the fibers was also probed using fluorescence microscopy. We treated Fmoc-FF (10 mM conc.) gels, formed in the presence of Ca2+ or Cs+ counterions, in an ultrasound bath for 10 min, in ice to avoid excessive heating of the samples, which may irreversibly damage the gels. The hydrogel samples were then imaged for several minutes using multidimensional FLIM microscopy, adding dye AQui as a reporter (Fig. 6). In both cases, the reticular structure of the gels appeared broken, and only short fibers were visible. Moreover, in both types of gels, elongation of the fibers was clearly detected within a time frame of 20–30 min. This time frame is much shorter than the overall fibrillation process, and indicates that pre-formed fibril fragments are capable of acting as secondary nucleation centers. The features of AQui in the hydrogels exhibited the same trends as in mature gels; this is, a more hydrophobic environment in Fmoc-FF Ca2+ hydrogels compared with gels formed with Cs+, and the presence of a large population of amorphous, solvent-exposed aggregates in the later gels, in which dye AQui showed low τ values and high I630/I550 ratios (Fig. 6B). Importantly, these amorphous aggregates seemed to have an important role in the self-healing process, as they tend to connect fibers with each other.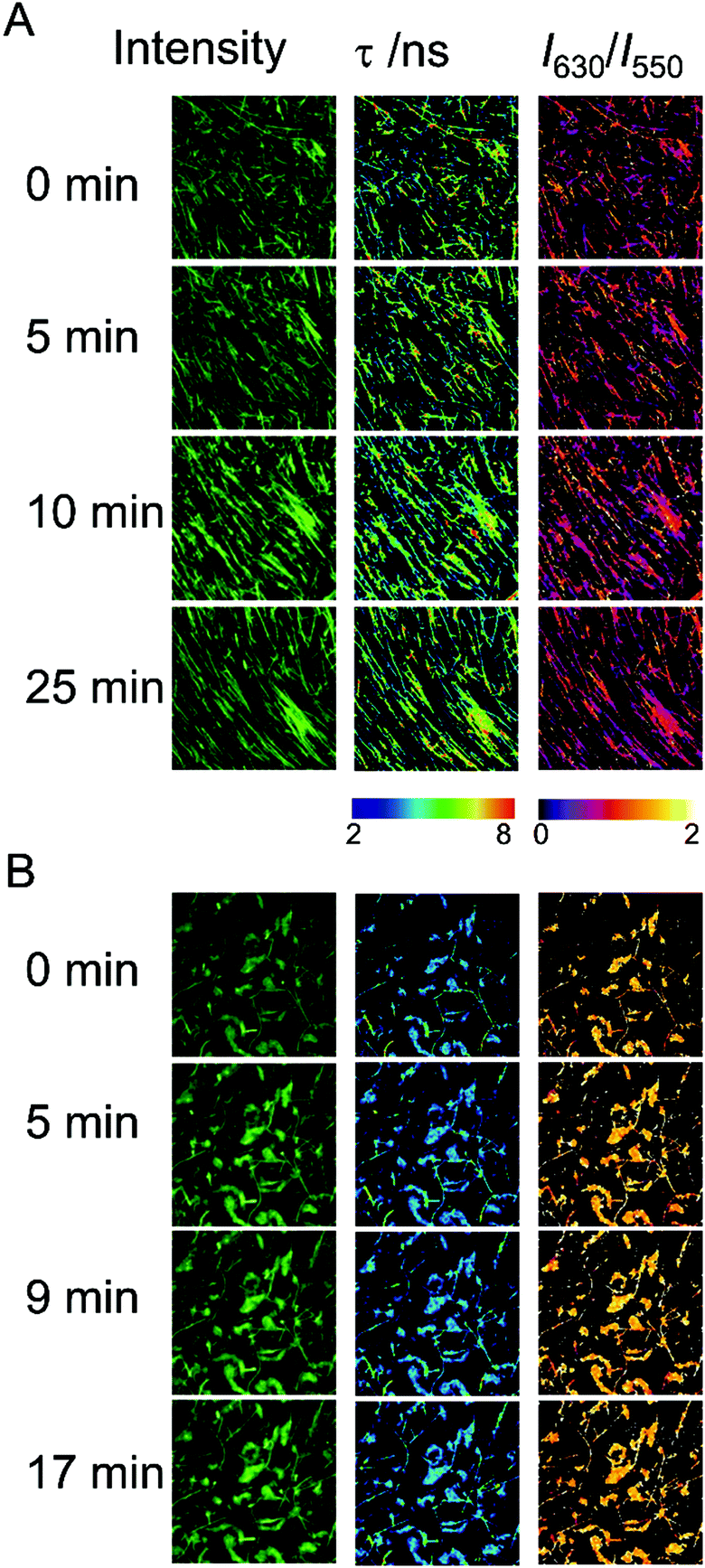 | ||
| Fig. 6 Intensity, FLIM, and ratiometric images of the dye AQui in self-healing Fmoc-FF hydrogels formed with Ca2+ (A) and Cs+ (B) after ultrasound treatment, as a function of time. The size of the images is 10 μm × 10 μm. | ||
We also checked the self-recovery process by following the time evolution of the emission spectra of AQui of the gels placed in a cuvette. The dye AQui exhibited a 76% recovery in the emission intensity after 100 min of self-healing of the Fmoc-FF Ca2+ gel (Fig. S8A, ESI†), whereas the emission recovery was only 25% in the Fmoc-FF Cs+ gel (Fig. S8B, ESI†). The fact that fibrillary fragments may act as secondary seeds for continuing fibrillary growth was also evidenced by following the process for 16 h of incubation, after sonication. After 16 h, AQui exhibited a 5.5-fold increase and a 1.8-fold increase in the emission with respect to the pre-formed gels in the Fmoc-FF Ca2+ and Fmoc-FF Cs+ gels, respectively. This indicates that the gels continued forming and growing, after sonication, even to a larger extent than the pre-formed gel, as expected for heterogeneous nucleation in which the seeds promote the fibril growth more efficiently under kinetic control. In any case, this recovery and subsequent growth were always more noticeable when Ca2+ ions were present in the medium.
Self-healing was also investigated by rheological methods. For this aim, we formed the most concentrated Ca2+ and Cs+ samples directly in the plate–plate geometry of the rheometer, and after the formation time (24 hours), we subjected the samples to intense shearing for 120 s at a shear rate of 20 s−1 to provoke the breakage of the samples. We monitored the corresponding shear stress during this time (Fig. S9A and C, ESI†). As observed, a strong decrease in shear stress with time during the first 10–20 s was obtained, evidencing the intense and extensive destruction of the gel network. After 20–30 s the shear stress tended towards constant values. After 120 s, we stopped the shearing and immediately subjected the samples to oscillatory shear strain of a very low amplitude (0.1%) and 1 Hz of frequency in order to monitor the self-healing of the destroyed samples (Fig. S9B and D, ESI†). For both Ca2+ and Cs+ samples, a rapid increase of G′ and G′′ was demonstrated, evidencing the rapid self-healing of the gels. What is more, in the case of Ca2+, G′ was greater than G′′ from the very beginning, which demonstrated that the gel-like character was recovered as soon as the shearing was stopped. Interestingly, as can be concluded by comparison of Fig. S9D with Fig. 5D, for the Cs+ sample, at the end of the self-healing test G′ reached larger values, and a more intense gel-like character, than before breakage, showing that, in this case, the heterogeneous nucleation had a stronger effect of promoting self-assembly and minimizing the formation of the metastable intermediates observed with this cation. This was not observed for the Ca2+ sample during the time of study (compare Fig. S9B, ESI† with Fig. 5A).
Conclusions
Thanks to the use of advanced fluorescence techniques, we have been able to monitor the mechanism of fibril growth and self-healing of Fmoc-FF in the presence of different salts. Our results have shown that the nature of the metallic counterion has a strong impact on the mechanism of self-assembly and, as a result of this, the physical properties of the resulting gels change completely. Ca2+ ions are able to promote a liquid–liquid separation phase that promotes peptide self-assembly more quickly and more efficiently, giving rise to more stable hydrophobic fibrils at lower peptide concentrations. On the other hand, Cs+ ions modulate the self-assembly process very differently from Ca2+. In this case, the mechanism of fibril growth goes through well-identified different metastable intermediates. This different mechanism of formation is also reflected in the physical properties of the resulting gels. Thus, gels formed in the presence of Cs+ are 500 times weaker than those formed by Ca2+. This difference can be rationalized by the different features of the counterions regarding their coordination abilities and their capacity to stabilize the water–solute interphase. As a large, soft cation, Cs+ tends to stabilize the water–solute interphase, weakening hydrophobic interactions and, therefore, favoring the formation of metastable intermediates. By contrast, kosmotrope Ca2+ ions destabilize the water–solute interphase, promoting hydrophobic interactions. This means that the peptide–peptide interaction must occur through pure hydrophobic interactions, resulting in more hydrophobic fibrils.These results show the influence of the different metal ions on the mechanism of nucleation and growth of amphiphilic peptides. In addition, the impact of the mechanism of growth on the physical properties of the resulting gels is reported. These findings provide meaningful insights into the self-assembly process of amphiphilic peptides, and thus allow a better control of the resulting self-assembled materials, expanding the scope of their applications.
Author contributions
M. C. M.-T.: conceptualization, formal analysis, investigation, validation, visualization. C. G.-V.: formal analysis, investigation, validation. J. A. G.-V.: conceptualization, formal analysis, investigation, supervision. F. C.-L.: formal analysis, methodology, writing – original draft. V. B.: investigation, formal analysis, writing – original draft. J. M. C.: conceptualization, formal analysis, writing – original draft. M. T. L.-L.: conceptualization, methodology, formal analysis, writing – original draft. A. O.: formal analysis, funding acquisition, methodology, supervision, writing – original draft, writing – review. L. A. C.: conceptualization, funding acquisition, methodology, project administration, supervision, writing – original draft, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by projects CTQ2017-86568-R and FIS2017-85954-R (Ministerio de Economía, Industria y Competitividad, MINECO, and Agencia Estatal de Investigación, AEI, Spain, cofunded by Fondo Europeo de Desarrollo Regional, FEDER, European Union), and by FEDER/Junta de Andalucía-Consejería de Transformación Económica, Industria, Conocimiento y Universidades (Spain) project P18-FR-3533. We want to thank “Unidad de Excelencia Química aplicada a Biomedicina y Medioambiente” of the University of Granada. Thanks go to the CIC personnel of the University of Granada for technical assistance. We thank the Centro de Servicios de Informática y Redes de Comunicaciones (CSIRC), Universidad de Granada, for providing the computing time. Authors acknowledge to Dr Rosario Herranz and Dr Francisco Fueyo-Gonzalez for the fluorophore 9-azetidinyl-5-butyl-quinolimide (AQui) used in this study, which was synthesized at Instituto de Química Médica-CSIC (IQM-CSIC) with the support of the Ministerio de Ciencia e Innovación/Agencia Estatal de Investigación grant FU2015-67284-R.References
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Supramolecular polymers, Chem. Rev., 2001, 101, 4071–4097 Search PubMed.
- J. R. Nitschke, Systems chemistry: molecular networks come of age, Nature, 2009, 462, 736–738 Search PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Functional supramolecular polymers, Science, 2012, 335, 813–817 Search PubMed.
- E. Mattia and S. Otto, Supramolecular systems chemistry, Nat. Nanotechnol., 2015, 10, 111–119 Search PubMed.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Pathway complexity in supramolecular polymerization, Nature, 2012, 481, 492–496 Search PubMed.
- P. A. Korevaar, T. F. A. De Greef and E. W. Meijer, Pathway complexity in π-conjugated materials, Chem. Mater., 2014, 26, 576–586 Search PubMed.
- T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Control over differentiation of a metastable supramolecular assembly in one and two dimensions, Nat. Chem., 2017, 9, 493–499 Search PubMed.
- S. Zhang, Fabrication of novel biomaterials through molecular self-assembly, Nat. Biotechnol., 2003, 21, 1171–1178 Search PubMed.
- S. Mann, Self-assembly and transformation of hybrid nano-objects and nanostructures under equilibrium and non-equilibrium conditions, Nat. Mater., 2009, 8, 781–792 Search PubMed.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Optically healable supramolecular polymers, Nature, 2011, 472, 334–337 Search PubMed.
- Y. Liu, Z. Wang and X. Zhang, Characterization of supramolecular polymers, Chem. Soc. Rev., 2012, 41, 5922–5932 Search PubMed.
- A. Döring, W. Birnbaum and D. Kuckling, Responsive hydrogels – structurally and dimensionally optimized smart frameworks for applications in catalysis, micro-system technology and material science, Chem. Soc. Rev., 2013, 42, 7391–7420 Search PubMed.
- C. D. Jones and J. W. Steed, Gels with sense: Supramolecular materials that respond to heat, light and sound, Chem. Soc. Rev., 2016, 45, 6546–6596 Search PubMed.
- A. Wang, W. Shi, J. Huang and Y. Yan, Adaptive soft molecular self-assemblies, Soft Matter, 2016, 12, 337–357 Search PubMed.
- X. Huang, S. R. Raghavan, P. Terech and R. G. Weiss, Distinct kinetic pathways generate organogel networks with contrasting fractality and thixotropic properties, J. Am. Chem. Soc., 2006, 128, 15341–15352 Search PubMed.
- T. D. Do, W. M. Kincannon and M. T. Bowers, Phenylalanine oligomers and fibrils: The mechanism of assembly and the importance of tetramers and counterions, J. Am. Chem. Soc., 2015, 137, 10080–10083 Search PubMed.
- G. Fichman, T. Guterman, J. Damron, L. Adler-Abramovich, J. Schmidt, E. Kesselman, L. J. W. Shimon, A. Ramamoorthy, Y. Talmon and E. Gazit, Supramolecular Chemistry: Spontaneous structural transition and crystal formation in minimal supramolecular polymer model, Sci. Adv., 2016, 2, 1–10 Search PubMed.
- P. R. Ten Wolde and D. Frenkel, Enhancement of protein crystal nucleation by critical density fluctuations, Science, 1997, 277, 1975–1978 Search PubMed.
- T. P. J. Knowles, C. A. Waudby, G. L. Devlin, S. I. A. Cohen, A. Aguzzi, M. Vendruscolo, E. M. Terentjev, M. E. Welland and C. M. Dobson, An analytical solution to the kinetics of breakable filament assembly, Science, 2009, 326, 1533–1537 Search PubMed.
- J. Adamcik, J. M. Jung, J. Flakowski, P. De Los Rios, G. Dietler and R. Mezzenga, Understanding amyloid aggregation by statistical analysis of atomic force microscopy images, Nat. Nanotechnol., 2010, 5, 423–428 Search PubMed.
- S. I. A. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758–9763 Search PubMed.
- B. Morel, M. P. Carrasco, S. Jurado, C. Marco and F. Conejero-Lara, Dynamic micellar oligomers of amyloid beta peptides play a crucial role in their aggregation mechanisms, Phys. Chem. Chem. Phys., 2018, 20, 20597–20614 Search PubMed.
- J. C. Sang, J. E. Lee, A. J. Dear, S. De, G. Meisl, A. M. Thackray, R. Bujdoso, T. P. J. Knowles and D. Klenerman, Direct observation of prion protein oligomer formation reveals an aggregation mechanism with multiple conformationally distinct species, Chem. Sci., 2019, 10, 4588–4597 Search PubMed.
- T. C. T. Michaels, A. Šarić, S. Curk, K. Bernfur, P. Arosio, G. Meisl, A. J. Dear, S. I. A. Cohen, C. M. Dobson, M. Vendruscolo, S. Linse and T. P. J. Knowles, Dynamics of oligomer populations formed during the aggregation of Alzheimer's Aβ42 peptide, Nat. Chem., 2020, 12, 445–451 Search PubMed.
- G. Wei, Z. Su, N. P. Reynolds, P. Arosio, I. W. Hamley, E. Gazit and R. Mezzenga, Self-assembling peptide and protein amyloids: From structure to tailored function in nanotechnology, Chem. Soc. Rev., 2017, 46, 4661–4708 Search PubMed.
- E. R. Draper and D. J. Adams, Low-Molecular-Weight Gels: The State of the Art, Chem, 2017, 3, 390–410 Search PubMed.
- C. Yuan, W. Ji, R. Xing, J. Li, E. Gazit and X. Yan, Hierarchically oriented organization in supramolecular peptide crystals, Nat. Rev. Chem., 2019, 3, 567–588 Search PubMed.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Biomimetic peptide self-assembly for functional materials, Nat. Rev. Chem., 2020, 4, 615–634 Search PubMed.
- A. Lampel, Biology-Inspired Supramolecular Peptide Systems, Chem, 2020, 6, 1222–1236 Search PubMed.
- A. Levin, T. O. Mason, L. Adler-Abramovich, A. K. Buell, G. Meisl, C. Galvagnion, Y. Bram, S. A. Stratford, C. M. Dobson, T. P. J. Knowles and E. Gazit, Ostwalds rule of stages governs structural transitions and morphology of dipeptide supramolecular polymers, Nat. Commun., 2014, 5, 5219 Search PubMed.
- C. Yuan, A. Levin, W. Chen, R. Xing, Q. Zou, T. W. Herling, P. K. Challa, T. P. J. Knowles and X. Yan, Nucleation and Growth of Amino Acid and Peptide Supramolecular Polymers through Liquid–Liquid Phase Separation, Angew. Chem., Int. Ed., 2019, 58, 18116–18123 Search PubMed.
- L. Albertazzi, D. Van Der Zwaag, C. M. A. Leenders, R. Fitzner, R. W. Van Der Hofstad and E. W. Meijer, Probing exchange pathways in one-dimensional aggregates with super-resolution microscopy, Science, 2014, 344, 491–495 Search PubMed.
- A. Aliprandi, M. Mauro and L. De Cola, Controlling and imaging biomimetic self-assembly, Nat. Chem., 2016, 8, 10–15 Search PubMed.
- S. Onogi, H. Shigemitsu, T. Yoshii, T. Tanida, M. Ikeda, R. Kubota and I. Hamachi, In situ real-time imaging of self-sorted supramolecular nanofibres, Nat. Chem., 2016, 8, 743–752 Search PubMed.
- Y. Liang, D. G. Lynn and K. M. Berland, Direct observation of nucleation and growth in amyloid self-assembly, J. Am. Chem. Soc., 2010, 132, 6306–6308 Search PubMed.
- Z. Huang, T. Jiang, J. Wang, X. Ma and H. Tian, Real-Time Visual Monitoring of Kinetically Controlled Self-Assembly, Angew. Chem., Int. Ed., 2021, 60, 2855–2860 Search PubMed.
- L. Chen, G. Pont, K. Morris, G. Lotze, A. Squires, L. C. Serpell and D. J. Adams, Salt-induced hydrogelation of functionalised-dipeptides at high pH, Chem. Commun., 2011, 47, 12071–12073 Search PubMed.
- L. Chen, T. O. McDonald and D. J. Adams, Salt-induced hydrogels from functionalised-dipeptides, RSC Adv., 2013, 3, 8714–8720 Search PubMed.
- A. Z. Cardoso, L. L. E. Mears, B. N. Cattoz, P. C. Griffiths, R. Schweins and D. J. Adams, Linking micellar structures to hydrogelation for salt-triggered dipeptide gelators, Soft Matter, 2016, 12, 3612–3621 Search PubMed.
- K. McAulay, P. A. Ucha, H. Wang, A. M. Fuentes-Caparrós, L. Thomson, O. Maklad, N. Khunti, N. Cowieson, M. Wallace, H. Cui, R. J. Poole, A. Seddon and D. J. Adams, Controlling the properties of the micellar and gel phase by varying the counterion in functionalised-dipeptide systems, Chem. Commun., 2020, 56, 4094–4097 Search PubMed.
- W. Ji, C. Yuan, S. Zilberzwige-Tal, R. Xing, P. Chakraborty, K. Tao, S. Gilead, X. Yan and E. Gazit, Metal-Ion Modulated Structural Transformation of Amyloid-Like Dipeptide Supramolecular Self-Assembly, ACS Nano, 2019, 13, 7300–7309 Search PubMed.
- J. J. Panda, R. Dua, A. Mishra, B. Mittra and V. S. Chauhan, 3D cell growth and proliferation on a RGD functionalized nanofibrillar hydrogel based on a conformationally restricted residue containing dipeptide, ACS Appl. Mater. Interfaces, 2010, 2, 2839–2848 Search PubMed.
- E. J. Berns, Z. Álvarez, J. E. Goldberger, J. Boekhoven, J. A. Kessler, H. G. Kuhn and S. I. Stupp, A tenascin-C mimetic peptide amphiphile nanofiber gel promotes neurite outgrowth and cell migration of neurosphere-derived cells, Acta Biomater., 2016, 37, 50–58 Search PubMed.
- J. Liu, Z. Sun, Y. Yuan, X. Tian, X. Liu, G. Duan, Y. Yang, L. Yuan, H. C. Lin and X. Li, Peptide Glycosylation Generates Supramolecular Assemblies from Glycopeptides as Biomimetic Scaffolds for Cell Adhesion and Proliferation, ACS Appl. Mater. Interfaces, 2016, 8, 6917–6924 Search PubMed.
- A. Friggeri, B. L. Feringa and J. Van Esch, Entrapment and release of quinoline derivatives using a hydrogel of a low molecular weight gelator, J. Controlled Release, 2004, 97, 241–248 Search PubMed.
- J. J. Panda, A. Mishra, A. Basu and V. S. Chauhan, Stimuli responsive self-assembled hydrogel of a low Molecular weight free dipeptide with potential for tunable drug delivery, Biomacromolecules, 2008, 9, 2244–2250 Search PubMed.
- R. Huang, W. Qi, L. Feng, R. Su and Z. He, Self-assembling peptide-polysaccharide hybrid hydrogel as a potential carrier for drug delivery, Soft Matter, 2011, 7, 6222–6230 Search PubMed.
- C. J. Newcomb, R. Bitton, Y. S. Velichko, M. L. Snead and S. I. Stupp, The role of nanoscale architecture in supramolecular templating of biomimetic hydroxyapatite mineralization, Small, 2012, 8, 2195–2202 Search PubMed.
- S. Kocabey, H. Ceylan, A. B. Tekinay and M. O. Guler, Glycosaminoglycan mimetic peptide nanofibers promote mineralization by osteogenic cells, Acta Biomater., 2013, 9, 9075–9085 Search PubMed.
- M. Conejero-Muriel, J. A. Gavira, E. Pineda-Molina, A. Belsom, M. Bradley, M. Moral, J. D. García López Durán, A. Luque González, J. J. Díaz-Mochón, R. Contreras-Montoya, Á. Martínez-Peragón, J. M. Cuerva and L. Álvarez De Cienfuegos, Influence of the chirality of short peptide supramolecular hydrogels in protein crystallogenesis, Chem. Commun., 2015, 51, 3862–3865 Search PubMed.
- R. Contreras-Montoya, G. Escolano, S. Roy, M. T. Lopez-Lopez, J. M. Delgado-López, J. M. Cuerva, J. J. Díaz-Mochón, N. Ashkenasy, J. A. Gavira and L. Álvarez de Cienfuegos, Catalytic and Electron Conducting Carbon Nanotube–Reinforced Lysozyme Crystals, Adv. Funct. Mater., 2019, 29, 1807351 Search PubMed.
- R. Contreras-Montoya, A. Castellví, G. Escolano-Casado, J. Juanhuix, M. Conejero-Muriel, M. T. Lopez-Lopez, J. M. Cuerva, L. Álvarez De Cienfuegos and J. A. Gavira, Enhanced Stability against Radiation Damage of Lysozyme Crystals Grown in Fmoc-CF Hydrogels, Cryst. Growth Des., 2019, 19, 4229–4233 Search PubMed.
- R. Contreras-Montoya, M. Arredondo-Amador, G. Escolano-Casado, M. C. Mañas-Torres, M. González, M. Conejero-Muriel, V. Bhatia, J. J. Díaz-Mochón, O. Martínez-Augustin, F. Sánchez de Medina, M. T. Lopez-Lopez, F. Conejero-Lara, J. A. Gavira and L. Álvarez De Cienfuegos, Insulin Crystals Grown in Short-Peptide Supramolecular Hydrogels Show Enhanced Thermal Stability and Slower Release Profile, ACS Appl. Mater. Interfaces, 2021, 13, 11672–11682 Search PubMed.
- T. Shao, N. Falcone and H. B. Kraatz, Supramolecular Peptide Gels: Influencing Properties by Metal Ion Coordination and Their Wide-Ranging Applications, ACS Omega, 2020, 5, 1312–1317 Search PubMed.
- M. F. J. Mabesoone, A. R. A. Palmans and E. W. Meijer, Solute–Solvent Interactions in Modern Physical Organic Chemistry: Supramolecular Polymers as a Muse, J. Am. Chem. Soc., 2020, 142, 19781–19798 Search PubMed.
- S. Roy, N. Javid, P. W. J. M. Frederix, D. A. Lamprou, A. J. Urquhart, N. T. Hunt, P. J. Halling and R. V. Ulijn, Dramatic specific-ion effect in supramolecular hydrogels, Chem. – Eur. J., 2012, 18, 11723–11731 Search PubMed.
- S. Moelbert, B. Normand and P. De Los Rios, Kosmotropes and chaotropes: Modelling preferential exclusion, binding and aggregate stability, Biophys. Chem., 2004, 112, 45–57 Search PubMed.
- P. Lo Nostro and B. W. Ninham, Hofmeister phenomena: An update on ion specificity in biology, Chem. Rev., 2012, 112, 2286–2322 Search PubMed.
- F. Fueyo-González, J. A. González-Vera, I. Alkorta, L. Infantes, M. L. Jimeno, P. Aranda, D. Acuña-Castroviejo, A. Ruiz-Arias, A. Orte and R. Herranz, Environment-Sensitive Probes for Illuminating Amyloid Aggregation in Vitro and in Zebrafish, ACS Sens., 2020, 5, 2792–2799 Search PubMed.
- S. Rüttinger, P. Kapusta, M. Patting, M. Wahl and R. Macdonald, On the resolution capabilities and limits of fluorescence lifetime correlation spectroscopy (FLCS) measurements, J. Fluoresc., 2010, 20, 105–114 Search PubMed.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Fmoc-Diphenylalanine Self-ASsembly Mechanism Induces Apparent pK a Shifts, Langmuir, 2009, 25, 9447–9453 Search PubMed.
- S. Fleming, P. W. J. M. Frederix, I. Ramos Sasselli, N. T. Hunt, R. V. Ulijn and T. Tuttle, Assessing the utility of infrared spectroscopy as a structural diagnostic tool for β-sheets in self-assembling aromatic peptide amphiphiles, Langmuir, 2013, 29, 9510–9515 Search PubMed.
- R. Contreras-Montoya, A. B. Bonhome-Espinosa, A. Orte, D. Miguel, J. M. Delgado-López, J. D. G. Duran, J. M. Cuerva, M. T. Lopez-Lopez and L. Álvarez de Cienfuegos, Iron nanoparticles-based supramolecular hydrogels to originate anisotropic hybrid materials with enhanced mechanical strength, Mater. Chem. Front., 2018, 2, 686–699 Search PubMed.
- J. Raeburn, G. Pont, L. Chen, Y. Cesbron, R. Lévy and D. J. Adams, Fmoc-diphenylalanine hydrogels: Understanding the variability in reported mechanical properties, Soft Matter, 2012, 8, 1168–1174 Search PubMed.
- E. V. Alakpa, V. Jayawarna, A. Lampel, K. V. Burgess, C. C. West, S. C. J. Bakker, S. Roy, N. Javid, S. Fleming, D. A. Lamprou, J. Yang, A. Miller, A. J. Urquhart, P. W. J. M. Frederix, N. T. Hunt, B. Péault, R. V. Ulijn and M. J. Dalby, Tunable Supramolecular Hydrogels for Selection of Lineage-Guiding Metabolites in Stem Cell Cultures, Chem, 2016, 1, 298–319 Search PubMed.
- A. Borzacchiello, F. Della Sala and L. A. Ambrosio, Rheometry of Polymeric Biomaterials, Woodhead Publishing, 2017, pp. 233–253 Search PubMed.
- M. Rubistein and R. H. Colby, Polymer Physics, Oxford University, New York, 2003 Search PubMed.
- R. G. Larson, The structure and rheology of complex fluids, Oxford University Press, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental part and supplementary figures. See DOI: 10.1039/d1qm00477h |
| This journal is © the Partner Organisations 2021 |