
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organo-redox-catalysis for the difunctionalization of alkenes and oxidative Ritter reactions by C–H functionalization†
Sensheng
Liu
and
Martin
Klussmann
 *
*
Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany. E-mail: klusi@mpi-muelheim.mpg.de
First published on 3rd April 2021
Abstract
Transition metals are the dominant catalysts for redox-reactions between peroxides and organic substrates. Here, we show that triarylamines can act as organic redox-catalysts, enabling oxidative difunctionalization reactions of alkenes and oxidative Ritter-reactions. Styrene derivatives can be functionalized with alkyl radicals, generated from plain and halogenated hydrocarbons, and with nucleophiles, including nitriles, acetic acid, alcohols and fluoride. An oxidative Ritter reaction can be conducted between allylic C–H bonds as well as fluorene and acetonitrile. Benzoyl peroxide is the oxidant in both reactions. Mechanistic studies suggest that the triarylamines are catalysts and not initiators, mediating the reaction by electron transfer to the peroxide, forming benzoyloxyl radicals, and from C-radical intermediates, forming carbocations.
Introduction
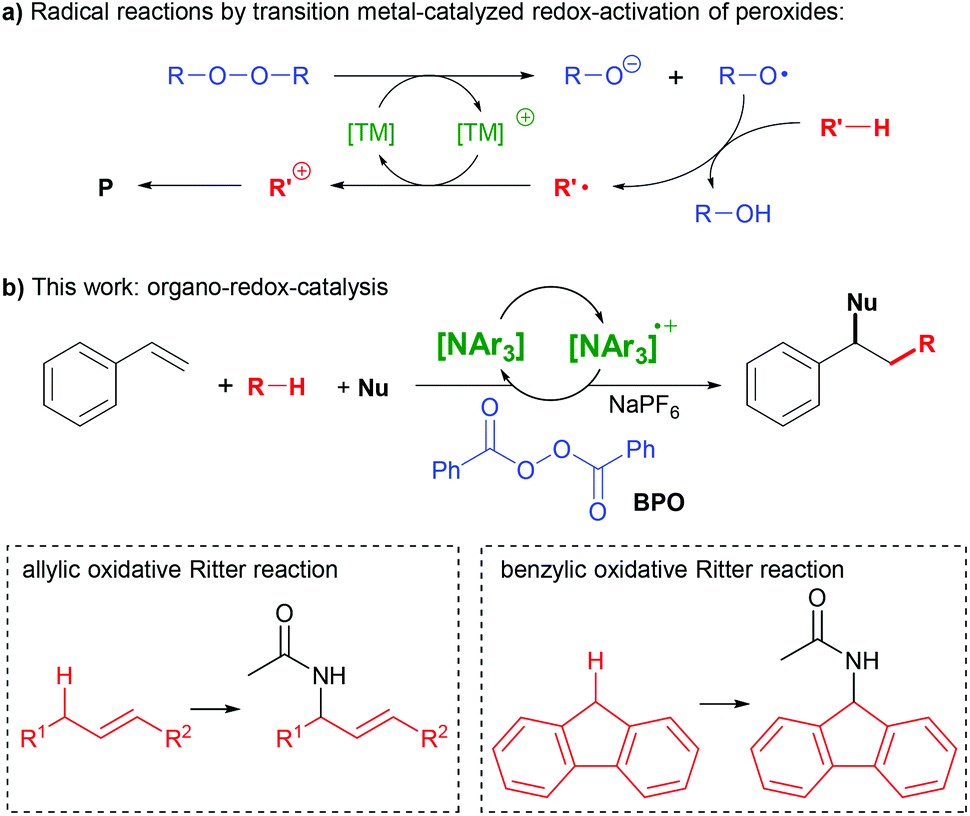
The difunctionalization of alkenes is a powerful method for the construction of C–C and C–X bonds.1–4 A very interesting type amongst those is the successional addition of a radical and a nucleophile.1,3 This method enables functionalizing olefins with a wide variety of reagents in a selective manner, given that radicals and nucleophiles generally react complementarily. However, these reactions are still lacking a truly broad substrate scope, spurring our efforts of further research.A widely used strategy for generating radicals from simple substrates is C–H bond cleavage by hydrogen atom transfer (HAT) to oxyl radicals, which are readily generated from peroxides.5 The subsequent addition of a nucleophile requires an electron transfer (ET) step to convert the radical intermediate into a carbocation. Thus, transition metals are widely used as redox-catalysts in such reactions, as they can mediate peroxide O–O bond cleavage and subsequent ET (Scheme 1a).1,3,6–9
 | ||
| Scheme 1 Activation of peroxides and formation of carbocations by redox-catalysis. | ||
Alternative methods for the consecutive addition of radicals and nucleophiles utilize organic photocatalysts,10,11 hypervalent iodine reagents or iodide as catalyst12–14 and electrochemistry.15,16 We are not aware, however, of the use of an organo-redox catalyst independent of irradiation in such reactions. Here, we report the use of triarylamines as catalysts in the activation of peroxides for synthetic radical reactions (Scheme 1b).
Triarylamines can form stable ammoniumyl radical cation salts by ET, and variation of the aryl-substituents allows for fine-tuning of their properties.17–21 Both the amines and the radical cations are widely applied in electro-optical materials.18–20,22 The radical cations can be used as stoichiometric single-electron oxidants in chemical reactions,23 or in substoichiometric amounts as initiators of radical chain reactions.17,24 Both the amines and the radical cations are also utilized as redox-catalysts in electrochemical25,26 or photochemical reactions,27,28 as well as in aerobic oxidations.29,30 Despite this plethora of applications, we are not aware of amine-based redox catalysis in the activation of peroxides, which would open many opportunities for synthetic applications.
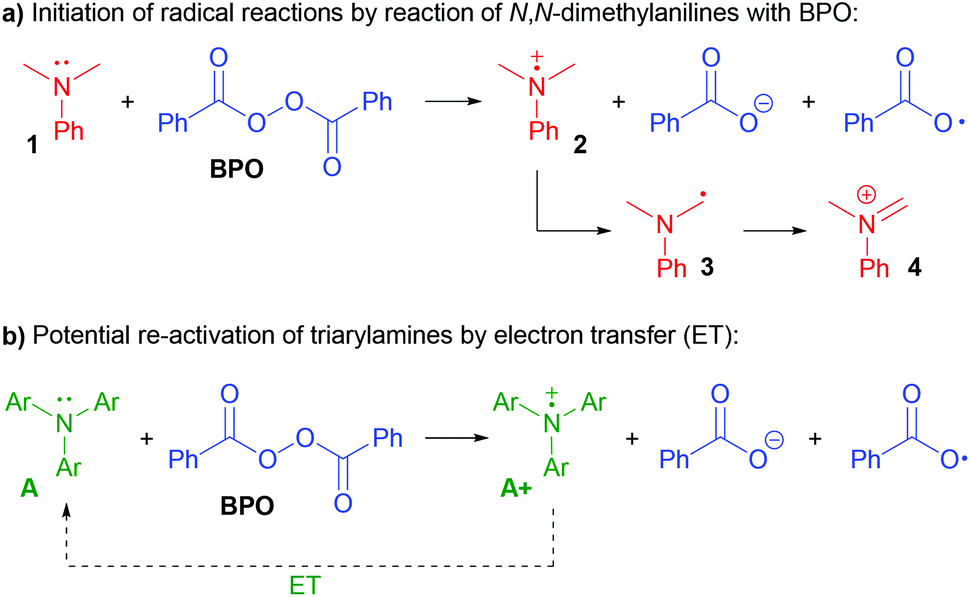
N,N-Dialkylanilines like 1 are well-known to generate radicals from diacylperoxides, especially benzoyl peroxide (BPO, Scheme 2a).31,32 The reaction is irreversible due to the reactivity of the ammoniumyl radical cation 2, which readily forms a C-radical 3, an iminium ion 4 and other products derived thereof.32,33 In contrast, we assumed that triarylamines A could be suitable candidates for catalysis, as the stable radical cation salts A+ could be regenerated by ET (Scheme 2b).
 | ||
| Scheme 2 Activation of BPO with N,N-dimethylaniline and triarylamines. | ||
We had previously utilized BPO in the addition of thioxanthene and similarly facile radical precursors together with nucleophiles to styrenes, which was rationalized as a radical-chain reaction.34 The addition of hexafluorophosphoric acid (HPF6) was found to modulate the redox potential of BPO, and the addition of N,N-dimethylanilines as initiators allowed difunctionalization with acetonitrile with moderate success. We kept working on finding a more efficient method for a broad substrate scope that would also avoid the use of a strong acid.
Results and discussion
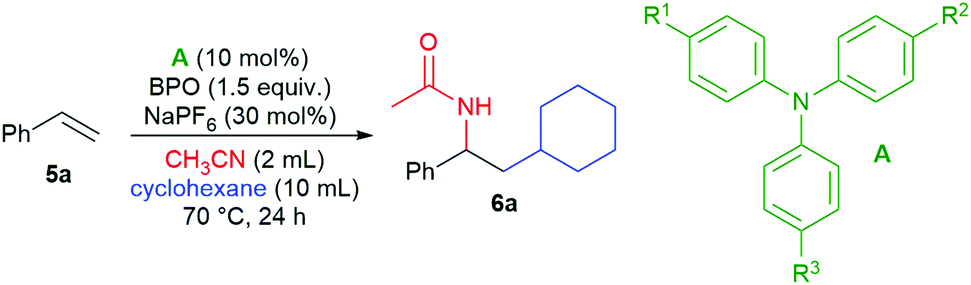
We found that the addition of cyclohexane and acetonitrile to styrene (5a) took place in the presence of catalytic amounts of some triarylamines and NaPF6 as an additive, forming the desired product 6a in good yields (Table 1, entries 1–3). The most effective amine was 4-iodo-N,N-diphenylaniline (A1), closely followed by tris(4-methylphenyl)amine (A2) and tris(4-bromophenyl)amine (A3), other amines were much less efficient. Without triarylamine, product 6a was not formed (entry 4) and the addition of NaPF6 is indispensable (entry 5). A reduction in the product's yield was also seen with other additives and oxidants (see the ESI† for a detailed investigation). The product's structure supported the subsequent addition of a cyclohexyl radical and acetonitrile as a nucleophile in a Ritter reaction.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | R1 | R2 | R3 | Additive | Yieldb (%) |
| a Reaction conditions: 5a (0.5 mmol), A (0.05 mmol, 10 mol%), CH3CN (2 mL), cyclohexane (10 mL), BPO (0.75 mmol, 1.5 equiv.), additive (0.15 mmol, 0.3 equiv.). b Determined by 1H NMR spectroscopic analysis of the crude reaction mixture relative to internal standard 1,3,5-trimethoxybenzene, isolated yield in parentheses. | ||||||
| 1 | A1 | I | H | H | NaPF6 | 91 (86) |
| 2 | A2 | Me | Me | Me | NaPF6 | 58 |
| 3 | A3 | Br | Br | Br | NaPF6 | 86 |
| 4 | — | NaPF6 | 0 | |||
| 5 | A1 | I | H | H | — | 0 |
With these reaction conditions, we investigated the product scope by testing other substrates. Using cyclopentane, cyclohexane, cycloheptane, and cyclooctane as radical precursors with acetonitrile as nucleophile afforded the products 6a–6d in good yields of 80–86% (Scheme 3). Methylcyclohexane gave a mixture of regioisomers from which we could isolate 6e, the major one, in 34% yield. With n-hexane, a mixture of the isomeric products 6f–6h was isolated in 80% overall yield, from which we could isolate the isomer 6f in 4% yield as a pure compound by column chromatography. With 2-methylpentane, the selectivity for the tertiary C–H bond was relatively high, allowing for isolation of the major isomer 6i in 41% yield. With 2,2,4,4-tetramethylpentane, only isomer 6j was isolated, apparently because the methylene group is sterically shielded, resulting in HAT from a primary C–H bond. The haloalkanes dichloromethane, chloroform, dibromomethane and bromoform could also be employed successfully in this reaction, producing the products 6k–6m. When using bromoform, not only 6n was formed by HAT, but also 6m by bromine atom transfer.35,36 When only acetonitrile was used as solvent, 6o was isolated in 86%.
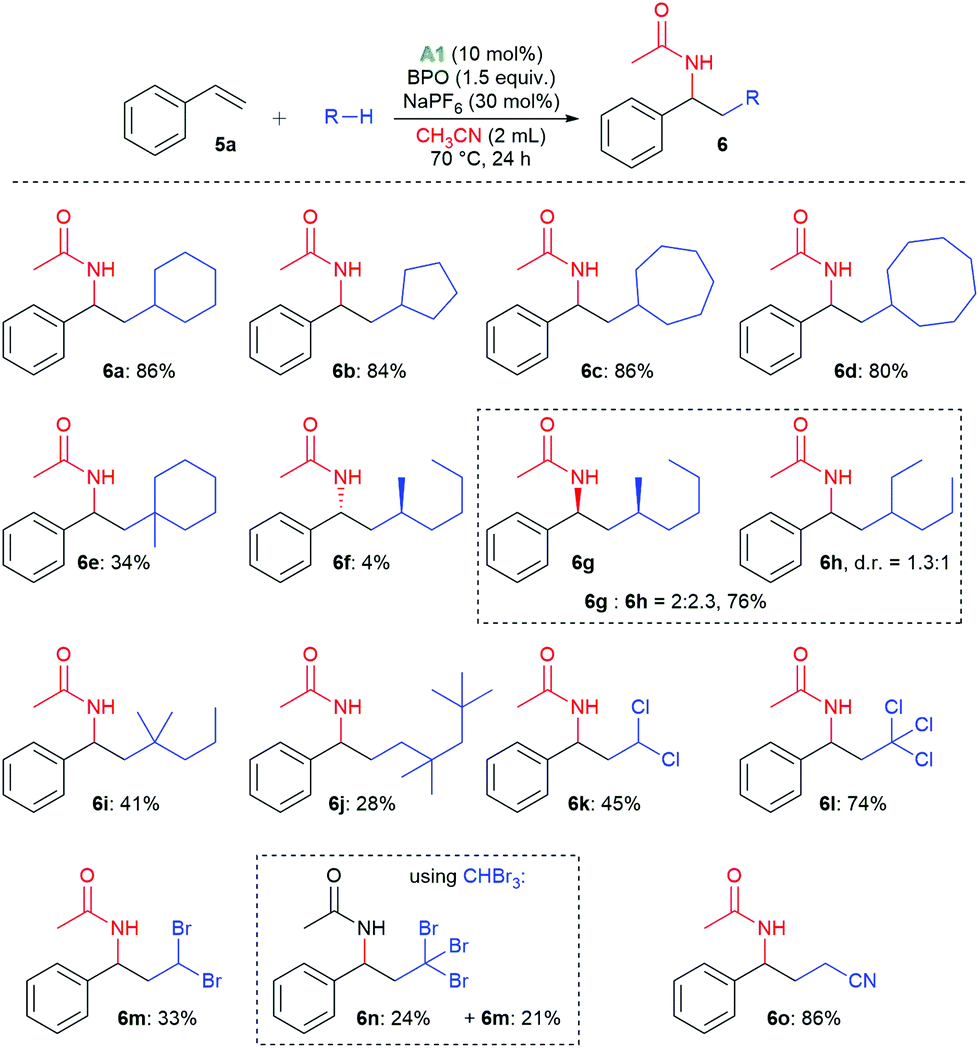 | ||
| Scheme 3 Scope of radicals. | ||
Styrenes with various substituents on the aromatic ring afforded the desired products in generally good yields (Scheme 4). There is no clear electronic substituent effect on the product yields, also substitution in the ortho position was not detrimental (7l–7n). Only in the case of p-methoxystyrene, the desired product was only observed in traces (7b). 2-Methylstyrene and stilbene could also be employed, giving the expected products 7o and 7p in medium yields and as mixtures of diastereomers.
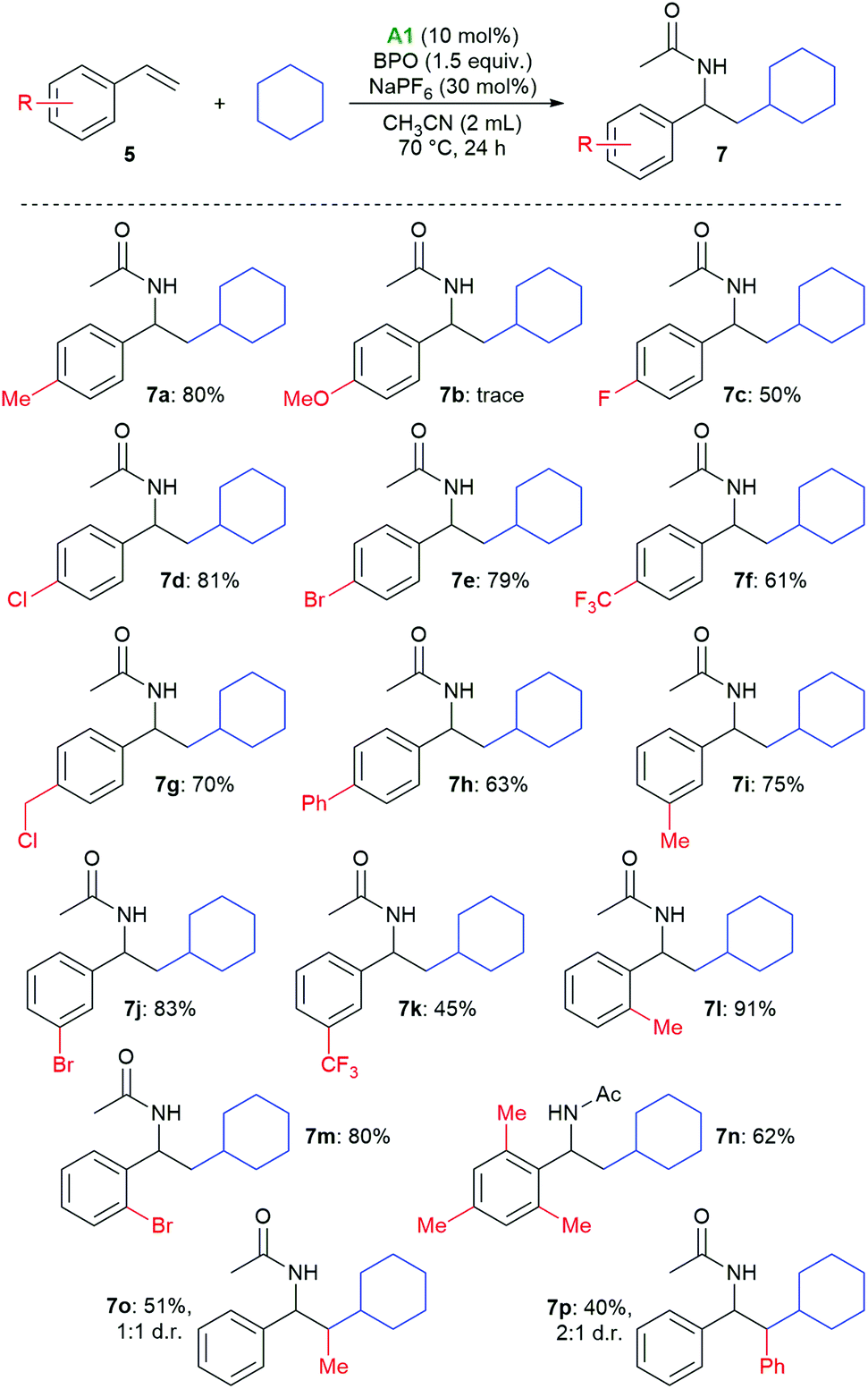 | ||
| Scheme 4 Substrate scope of substituted styrenes. | ||
Next, the scope with respect to nucleophiles was explored. As shown in Scheme 5a, different nitrile solvents and cyclohexane delivered the desired amides 8a–8e in good yields of 48%–76%. Similarly, when acetic acid was used, the corresponding acetate 8f was formed in 63% yield. With 1,1-diphenylethylene, difunctionalization with the α-cyanoalkyl radical from acetonitrile and alcohols was possible. With methanol and ethanol, the products 8g and 8h were isolated in 51% and 41% yields, respectively, however, long-chain alcohols showed a low reactivity. Very similar tertiary alcohols had recently been synthesized by copper-catalysis at higher temperature.37 We also isolated the dimer 9 from unsuccessful tests of other nucleophiles, supporting the occurrence of radical intermediate 15.38,39 In nitromethane as solvent, phenyl groups were incorporated into the products (10a–10c, Scheme 5b). These likely originated from phenyl radicals, formed by decarboxylation of the benzoyloxyl radicals. Acetonitrile and methanol could be used as nucleophiles, and with triethylamine hydrofluoride even fluoride, albeit in low yield (10c).
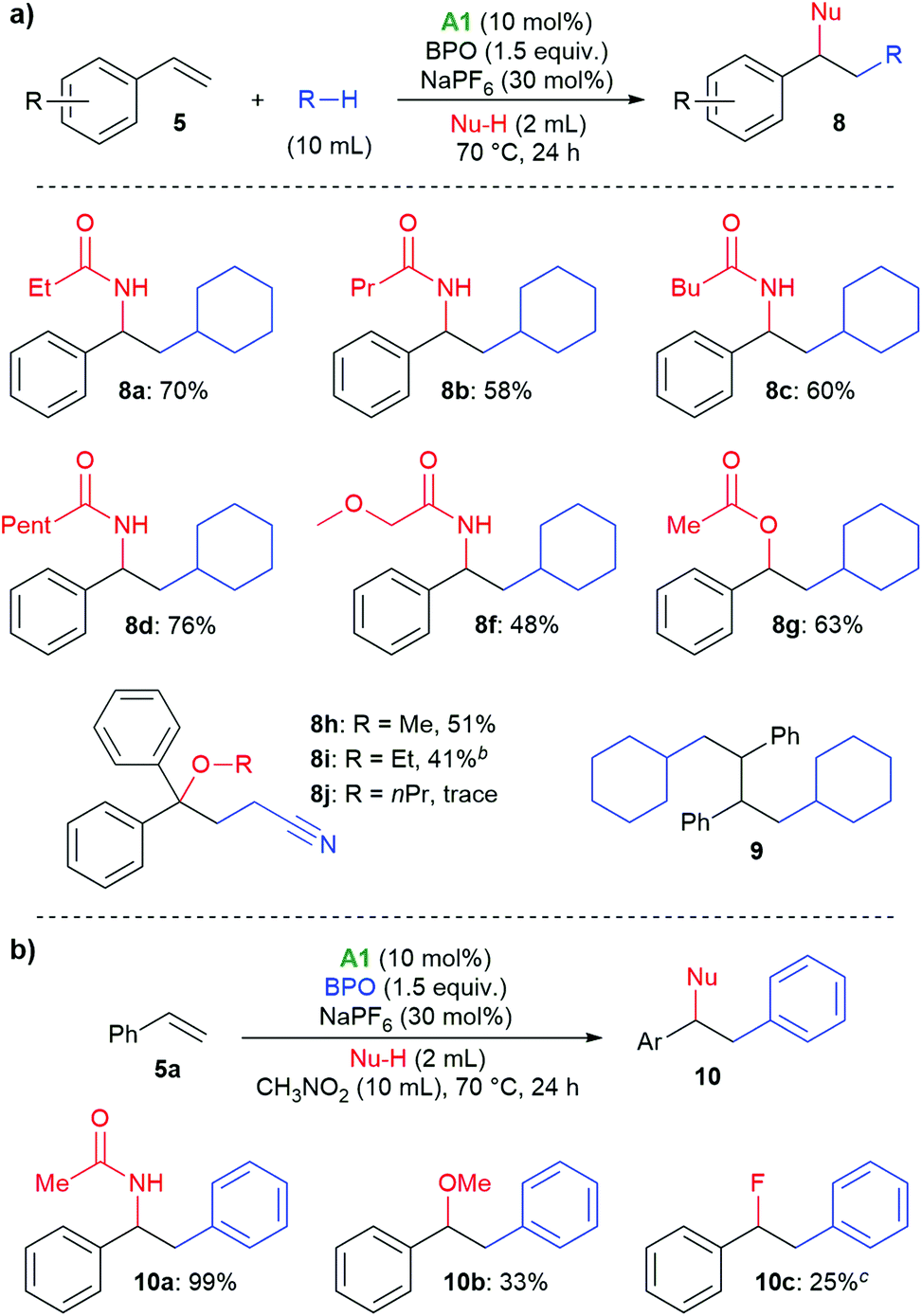 | ||
| Scheme 5 Scope of nucleophiles. | ||
The organo-redox system also proved to catalyze the oxidative Ritter reaction of allylic and benzylic C–H bonds; for these reactions, amine A3 was found to be superior to A1 (see the ESI† for details). As shown in Scheme 6a, cyclohexene and bicyclo[3.2.1]oct-2-ene delivered the amides 11a and 11b in 71% and 72% yield, respectively. When propionitrile was used, the corresponding product 11c was isolated in 33% yield. With (E)-4-octene, 11d and 11e were isolated in 89% yield as a mixture of two isomers with a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.8. When (Z)-4-octene was used, the same products were isolated with a ratio of 1:3. (E)-5-Decene gave the E-isomers 11f and 11g in equimolar amounts. When we used 9H-fluorene, the benzylic methylene group was functionalized, providing the amide 13 in 70% yield (Scheme 6b).
:0.8. When (Z)-4-octene was used, the same products were isolated with a ratio of 1:3. (E)-5-Decene gave the E-isomers 11f and 11g in equimolar amounts. When we used 9H-fluorene, the benzylic methylene group was functionalized, providing the amide 13 in 70% yield (Scheme 6b).
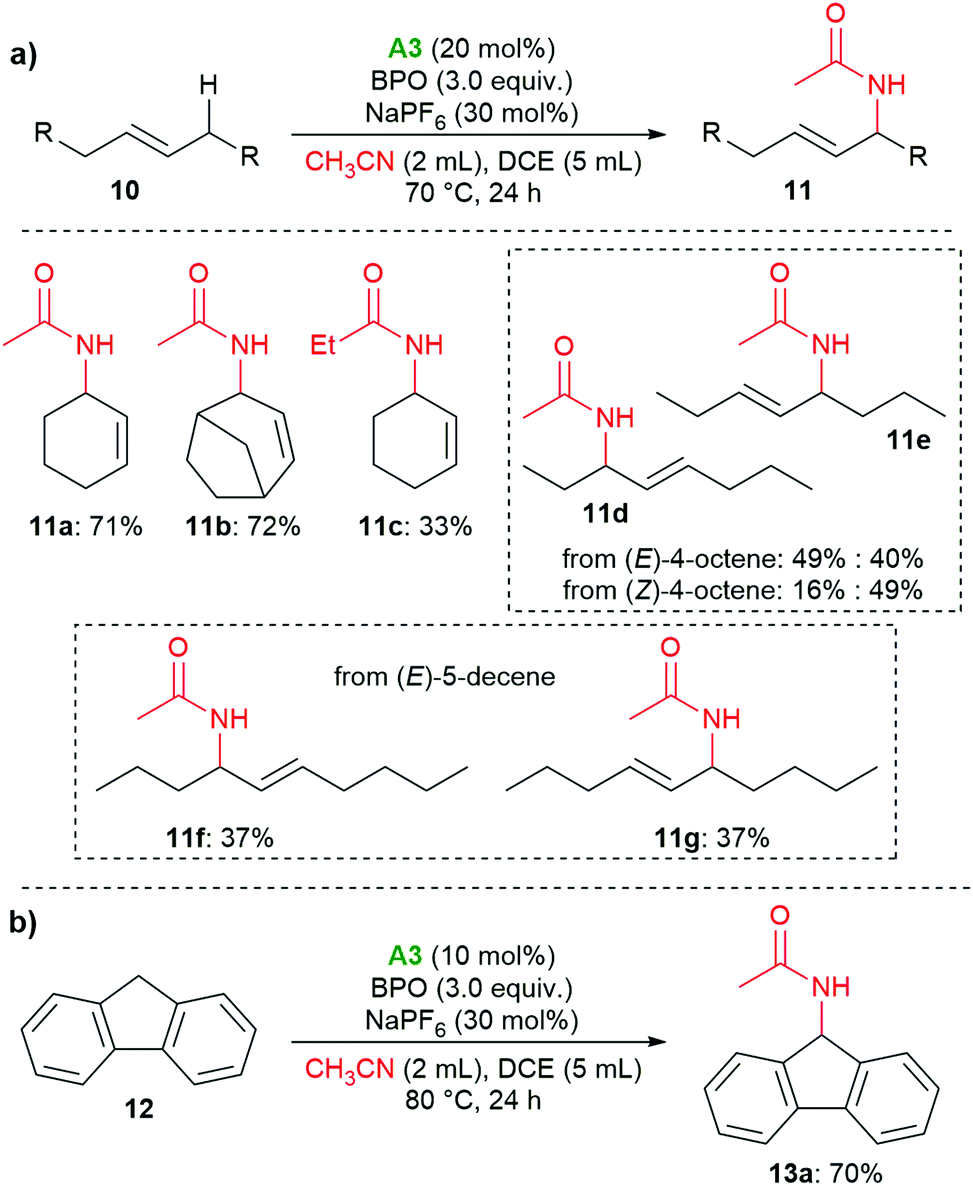 | ||
| Scheme 6 Amination of allylic and benzylic C–H bond. | ||
Are the triarylamines efficient initiators of a radical chain reaction, reacting irreversibly, or are they catalysts, achieving turnover?40 As mentioned above, N,N-dialkylanilines are well-known activators of benzoyl peroxide in radical chain reactions;31 however, they proved to be inefficient for the reactions presented here (Scheme 7a). Also, the reaction did not proceed when employing the well know radical initiator azobisisobutyronitrile (AIBN), suggesting that a radical chain mechanism is not operating. We used two radical cation salts, [(A1+)SbF6−] and [(A3+)SbCl6−], in place of the corresponding triarylamines under otherwise unchanged reaction conditions (Scheme 7b). In both cases, comparable yields of 6a were achieved (89% and 84%, resp.). When A3+ was used, its dark blue colour disappeared and 10% of the reduced form A3 could be isolated, ruling out that the triarylamines are irreversibly consumed and supporting that both the amine and the oxidized radical cation salts are involved in a catalytic reaction.
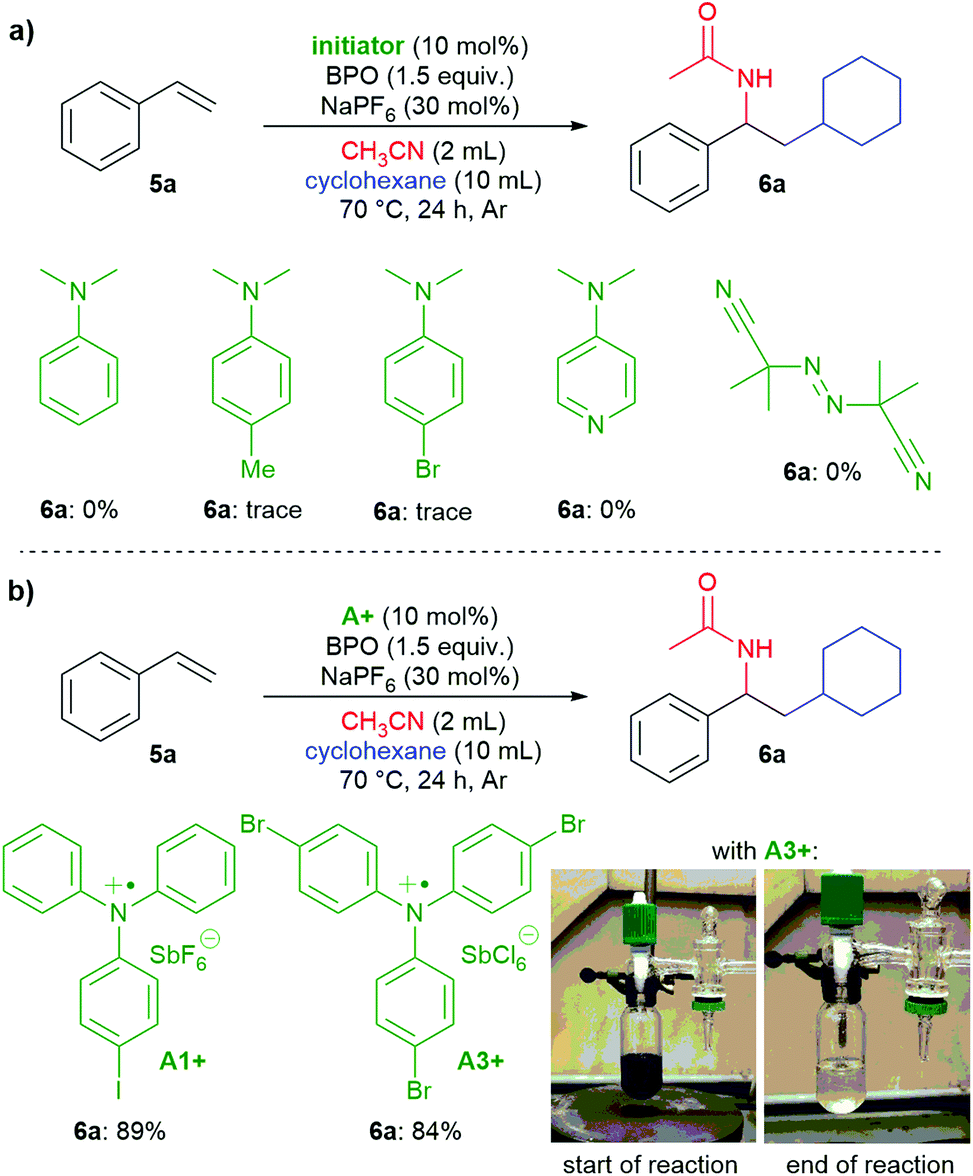 | ||
| Scheme 7 a) Testing N,N-dimethylanilines and AIBN as initiators; (b) triarylamine radical cation salts act as catalysts. | ||
After completion of the reaction, the system remains active. When we added another batch of BPO and styrene to a reaction mixture forming 6a that had gone to completion, a further 85% of these added substrates were converted to 6a after another 30 hours. Also, a third batch of substrates could be converted to product, which further supports that the reaction is catalytic in nature (see the ESI† for details).
We also investigated the system by 1H-NMR at the reaction temperature of 70 °C (Fig. 1). Ca. 40% of BPO on its own had decomposed after 10 h, in line with the reported 10 hours half-life temperature of 73 °C (red bottom line).41 Benzene and benzoic acid were formed in roughly equal amounts, indicating that half of the benzoyloxyl radicals had decarboxylated before they were quenched by HAT. Adding catalytic amounts (10 mol%) of A1 accelerated the decomposition of BPO during the first 2 hours and resulted in forming predominantly benzoic acid (blue upper line). These results indicate that the amine catalyses the decomposition of BPO by an effective ET reaction (see Scheme 7b below and its discussion for details), forming one molecule each of benzoate – observed as benzoic acid by NMR – and benzoyloxyl radical. The latter can decarboxylate to a phenyl radical and both radicals can react by HAT reactions from the medium to form benzoic acid and benzene, respectively. This explains the appearance of significantly more benzoic acid than benzene, compared to the thermal decomposition of BPO.
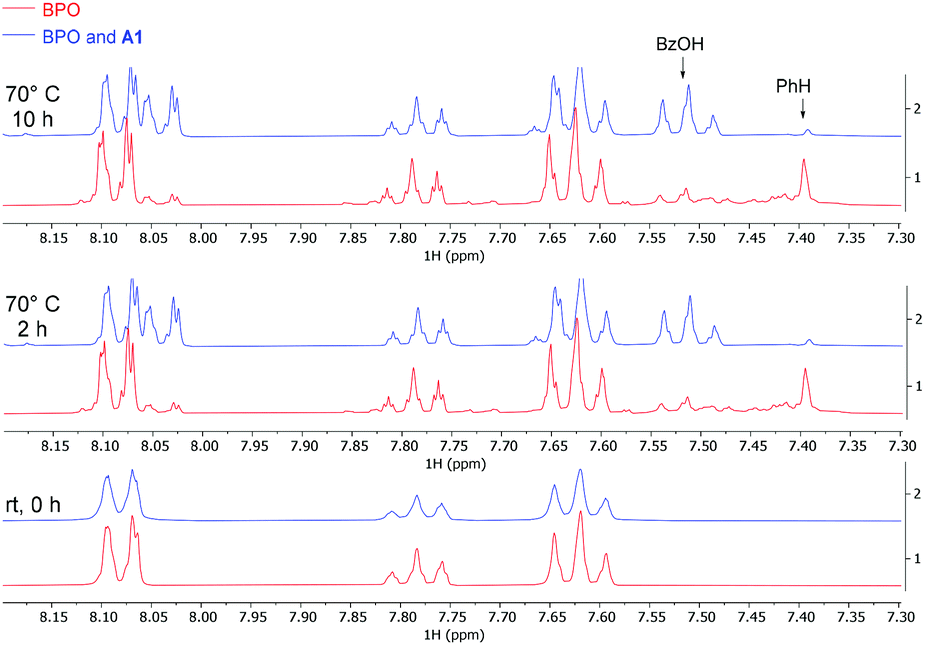 | ||
| Fig. 1 NMR experiments, comparing thermal and A1-induced BPO decomposition (red and blue lines, resp.); conditions: BPO (0.1 mmol), A1 (10 mol%), CD3CN (0.5 mL), 70 °C. | ||
During the difunctionalization reaction, a white precipitate formed, which we found to be sodium benzoate, in line with the suggested cleavage of BPO by ET (see the ESI†). All these results enable us to propose a mechanism for the formation of difunctionalization products like 6a (Scheme 8a) The peroxide bond of BPO is cleaved by an ET reaction from the triarylamine catalyst A, forming the radical cation A+, a benzoate anion and a benzoyloxyl radical. The presence of NaPF6 likely helps stabilizing the radical cation A+ in the form of the salt [A1 + ]PF6−. The benzoyloxyl radical can engage with cyclohexane or other substrates in a HAT reaction, forming a carbon radical 14, which then adds to styrene, forming the benzylic radical 15. Oxidation by the ammoniumyl radical cation A+ regenerates the triarylamine A and forms the carbocation 16, which is attacked by nucleophiles to provide the final product. To some extent, the benzoyloxyl radicals decarboxylate, generating phenyl radicals which can either participate in HAT reactions, too, or add to styrene, as was shown in Scheme 5b above.
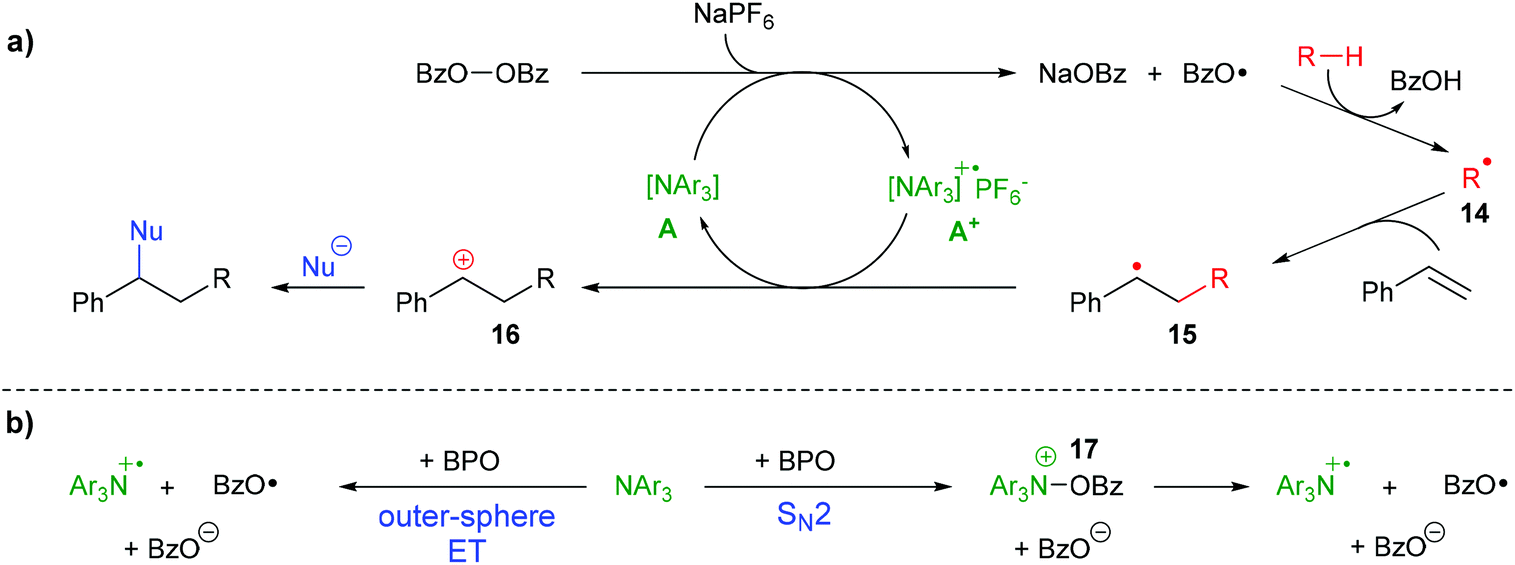 | ||
| Scheme 8 Proposed reaction mechanism: (a) organo-redox-catalysis; (b) effective ET by direct outer-sphere ET or stepwise via SN2 reaction. | ||
Whether the reaction between the triarylamine A and BPO proceeds by an outer-sphere ET, directly forming benzoate and two radicals, or by an SN2-reaction via N-benzoyloxylammonium salt 17, which in a second step decomposes homolytically into the same products (Scheme 8b), is at present unclear. Both pathways have been suggested for reactions between diacylperoxides and amines,32,42–44 but the combination of BPO with the triarylamines used in this study has not been investigated yet.
Additionally, we studied the redox potentials of several triarylamines and the radical cation A1+ as well as BPO by cylic voltammetry (see the ESI†). The reduction potential of A1 was indeed lower than that of BPO, supporting that A1 can reduce BPO by ET. However, BPO has the higher oxidation potential of the two, suggesting that it is the better electron acceptor. However, we consider these results of separate measurements as not fully conclusive for the interpretation of the mechanism, since they are not in agreement with the aforementioned results supporting catalysis by the amine. Furthermore, they do not take potential interactions in the reaction mixture into account. For example, the addition of NaPF6 is crucial for the reaction to occur, which indicates an ionic interaction that might shift the redox potential of the ammoniumyl radical cation.34 A species like 17 could be involved as electron acceptor, or the radical cation A1+ could be transformed into the actual catalyst in situ by attack at its free para-positions.21 Thus, we acknowledge that not all details of the present reactions are understood and we are therefore planning more detailed investigations.
Conclusions
In summary, we have established triarylamines as organo-redox catalysts for oxidative C–H functionalization reactions, with p-iodophenyl diphenylamine as the catalyst of choice in the newly developed method. By using benzoyl peroxide as oxidant, the difunctionalization of styrenes could be accomplished with radicals generated from hydrocarbons by hydrogen atom transfer and with nucleophiles, including nitriles, alcohols, acetic acid and fluoride. Besides, the amination of allylic and benzylic C–H bonds is also achieved under the same reaction conditions. The method does not require irradiation, electrolysis, transition metals nor significantly elevated temperatures. This application of a relatively simple amine might pave the way for further developments of organo-redox-catalysts, which may thus become another established class amongst organocatalysts.45Author contributions
S.L. and M.K. conceived the project, M.K. supervised the project, S.L. executed all experiments, S.L. and M.K. composed the manuscript, S.L. composed the ESI.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. K. thanks the DFG (KL 2221/4-2, Heisenberg scholarship) and S. L. thanks the China Scholarship Council (CSC, doctoral scholarship no. 201808420290). We thank the analytical departments of the Max-Planck-Institut für Kohlenforschung for their support, Dr Jie Ouyang and Dr Qiang Cheng (MPI für Kohlenforschung) for help with cyclic voltammetry and Tobias Greven (Universität zu Köln) for supporting the reaction development. Open Access funding provided by the Max Planck Society.Notes and references
- H. Yao, W. Hu and W. Zhang, Difunctionalization of Alkenes and Alkynes via Intermolecular Radical and Nucleophilic Additions, Molecules, 2021, 26, 105 CrossRef CAS PubMed
.
- J.-S. Zhang, L. Liu, T. Chen and L.-B. Han, Transition-Metal-Catalyzed Three-Component Difunctionalizations of Alkenes, Chem. – Asian J., 2018, 13, 2277–2291 CrossRef CAS PubMed
- X.-W. Lan, N.-X. Wang and Y. Xing, Recent Advances in Radical Difunctionalization of Simple Alkenes, Eur. J. Org. Chem., 2017, 5821–5851 CrossRef CAS
- H. Fischer and L. Radom, Factors Controlling the Addition of Carbon-Centered Radicals to Alkenes—An Experimental and Theoretical Perspective, Angew. Chem., Int. Ed., 2001, 40, 1340–1371 CrossRef CAS PubMed
- M. Salamone and M. Bietti, Tuning Reactivity and Selectivity in Hydrogen Atom Transfer from Aliphatic C–H Bonds to Alkoxyl Radicals: Role of Structural and Medium Effects, Acc. Chem. Res., 2015, 48, 2895–2903 CrossRef CAS PubMed
- A. Bunescu, Q. Wang and J. Zhu, Copper-Catalyzed Cyanomethylation of Allylic Alcohols with Concomitant 1,2-Aryl Migration: Efficient Synthesis of Functionalized Ketones Containing an α-Quaternary Center, Angew. Chem., Int. Ed., 2015, 54, 3132–3135 CrossRef CAS PubMed
- N. Zhu, T. Wang, L. Ge, Y. Li, X. Zhang and H. Bao, γ-Amino Butyric Acid (GABA) Synthesis Enabled by Copper-Catalyzed Carboamination of Alkenes, Org. Lett., 2017, 19, 4718–4721 CrossRef CAS PubMed
- X.-H. Ouyang, Y. Li, R.-J. Song, M. Hu, S. Luo and J.-H. Li, Intermolecular dialkylation of alkenes with two distinct C-H bonds enabled by synergistic photoredox catalysis and iron catalysis, Sci. Adv., 2019, 5, eaav9839 CrossRef CAS PubMed
- M. Lux and M. Klussmann, Additions of Aldehyde-Derived Radicals and Nucleophilic N-Alkylindoles to Styrenes by Photoredox Catalysis, Org. Lett., 2020, 22, 3697–3701 CrossRef CAS PubMed
- T. Koike and M. Akita, New Horizons of Photocatalytic Fluoromethylative Difunctionalization of Alkenes, Chem, 2018, 4, 409–437 CAS
- M.-Y. Cao, X. Ren and Z. Lu, Olefin difunctionalizations via visible light photocatalysis, Tetrahedron Lett., 2015, 56, 3732–3742 CrossRef CAS
- X. Wang and A. Studer, Iodine(III) Reagents in Radical Chemistry, Acc. Chem. Res., 2017, 50, 1712–1724 CrossRef CAS PubMed
- Y. Zheng, Y. He, G. Rong, X. Zhang, Y. Weng, K. Dong, X. Xu and J. Mao, NaI-Mediated Acetamidosulphenylation of Alkenes with Nitriles as the Nucleophiles: A Direct Access to Acetamidosulfides, Org. Lett., 2015, 17, 5444–5447 CrossRef CAS PubMed
- Q. Xue, J. Xie, P. Xu, K. Hu, Y. Cheng and C. Zhu, Metal-Free, n-Bu4NI-Catalyzed Regioselective Difunctionalization of Unactivated Alkenes, ACS Catal., 2013, 3, 1365–1368 CrossRef CAS
- J. C. Siu, N. Fu and S. Lin, Catalyzing Electrosynthesis: A Homogeneous Electrocatalytic Approach to Reaction Discovery, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed
- Y. Yuan, Y. Cao, Y. Lin, Y. Li, Z. Huang and A. Lei, Electrochemical Oxidative Alkoxysulfonylation of Alkenes Using Sulfonyl Hydrazines and Alcohols with Hydrogen Evolution, ACS Catal., 2018, 8, 10871–10875 CrossRef CAS
- X. Jia, Radical Cation Salts: From Single-Electron Oxidation to C–H Activation, Synthesis, 2016, 48, 18–30 CrossRef CAS
- J. Wang, K. Liu, L. Ma and X. Zhan, Triarylamine: Versatile Platform for Organic, Dye-Sensitized, and Perovskite Solar Cells, Chem. Rev., 2016, 116, 14675–14725 CrossRef CAS PubMed
- R. Lartia, C. Allain, G. Bordeau, F. Schmidt, C. Fiorini-Debuisschert, F. Charra and M.-P. Teulade-Fichou, Synthetic Strategies to Derivatizable Triphenylamines Displaying High Two-Photon Absorption, J. Org. Chem., 2008, 73, 1732–1744 CrossRef CAS PubMed
- Y. Shirota and H. Kageyama, Charge Carrier Transporting Molecular Materials and Their Applications in Devices, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS PubMed
- S. Dapperheld, E. Steckhan, K.-H. G. Brinkhaus and T. Esch, Organic Electron Transfer Systems, II Substituted Triarylamine Cation-Radical Redox Systems – Synthesis, Electrochemical and Spectroscopic Properties, Hammet Behavior, and Suitability as Redox Catalysts, Chem. Ber., 1991, 124, 2557–2567 CrossRef CAS
- M. Thelakkat, Star-Shaped, Dendrimeric and Polymeric Triarylamines as Photoconductors and Hole Transport Materials for Electro-Optical Applications, Marcomol. Mater. Eng., 2002, 287, 442–461 CAS
- W. Schmidt and E. Steckhan, Mild Oxidative Removal of the p-Methoxybenzyl Ether Protecting Group by Homogeneous Electron Transfer, Angew. Chem., Int. Ed. Engl., 1978, 17, 673–674 CrossRef
- N. L. Bauld, Cation radical cycloadditions and related sigmatropic reactions, Tetrahedron, 1989, 45, 5307–5363 CrossRef CAS
- C.-Y. Cai and H.-C. Xu, Dehydrogenative reagent-free annulation of alkenes with diols for the synthesis of saturated O-heterocycles, Nat. Commun., 2018, 9, 3551 CrossRef PubMed
- X. Wu, A. P. Davis and A. J. Fry, Electrocatalytic Oxidative Cleavage of Electron-Deficient Substituted Stilbenes in Acetonitrile−Water Employing a New High Oxidation Potential Electrocatalyst. An Electrochemical Equivalent of Ozonolysis, Org. Lett., 2007, 9, 5633–5636 CrossRef CAS PubMed
- L. Wang, R. Li and K. A. I. Zhang, Atom Transfer Radical Polymerization (ATRP) Catalyzed by Visible Light-Absorbed Small Molecule Organic Semiconductors, Macromol. Rapid Commun., 2018, 39, 1800466 CrossRef PubMed
- L. Wang, J. Byun, R. Li, W. Huang and K. A. I. Zhang, Molecular Design of Donor-Acceptor-Type Organic Photocatalysts for Metal-free Aromatic C−C Bond Formations under Visible Light, Adv. Synth. Catal., 2018, 360, 4312–4318 CrossRef CAS
- X. Jia, F. Peng, C. Qing, C. Huo and X. Wang, Catalytic Radical Cation Salt Induced Csp3−H Functionalization of Glycine Derivatives: Synthesis of Substituted Quinolines, Org. Lett., 2012, 14, 4030–4033 CrossRef CAS PubMed
- F. Unglaube, P. Hünemörder, X. Guo, Z. Chen, D. Wang and E. Mejía, Phenazine Radical Cations as Efficient Homogeneous and Heterogeneous Catalysts for the Cross-Dehydrogenative Aza-Henry Reaction, Helv. Chim. Acta, 2020, 103, e2000184 CrossRef CAS
- A. Székely and M. Klussmann, Molecular radical chain initiators for ambient to low temperature applications, Chem. – Asian J., 2019, 14, 105–115 Search PubMed
- K. Kim, N. R. Singstock, K. K. Childress, J. Sinha, A. M. Salazar, S. N. Whitfield, A. M. Holder, J. W. Stansbury and C. B. Musgrave, Rational Design of Efficient Amine Reductant Initiators for Amine–Peroxide Redox Polymerization, J. Am. Chem. Soc., 2019, 141, 6279–6291 CrossRef CAS PubMed
- E. Boess, M. V. Hoof, S. L. Birdsall and M. Klussmann, Investigating the Oxidation Step in the CuCl2-Catalyzed Aerobic Oxidative Coupling Reaction of N-Aryl Tetrahydroisoquinolines, J. Org. Chem., 2020, 85, 1972–1980 CrossRef CAS PubMed
- S. Liu and M. Klussmann, Acid Promoted Radical-Chain Difunctionalization of Styrenes with Stabilized Radicals and (N,O)-Nucleophiles, Chem. Commun., 2020, 56, 1557–1560 RSC
- R. K. Neff, Y.-L. Su, S. Liu, M. Rosado, X. Zhang and M. P. Doyle, Generation of Halomethyl Radicals by Halogen Atom Abstraction and Their Addition Reactions with Alkenes, J. Am. Chem. Soc., 2019, 141, 16643–16650 CrossRef CAS PubMed
- T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Aminoalkyl radicals as halogen-atom transfer agents for activation of alkyl and aryl halides, Science, 2020, 367, 1021–1026 CrossRef CAS PubMed
- C. Chatalova-Sazepin, Q. Wang, G. M. Sammis and J. Zhu, Copper-Catalyzed Intermolecular Carboetherification of Unactivated Alkenes by Alkyl Nitriles and Alcohols, Angew. Chem., Int. Ed., 2015, 54, 5443–5446 CrossRef CAS PubMed
- T. Jun, W. Hiroyasu, M. Masako and K. Nobuaki, Titanocene-Catalyzed Alkylation of Aryl-Substituted Alkenes with Alkyl Halides, Bull. Chem. Soc. Jpn., 2003, 76, 2209–2214 CrossRef
- B. J. Fallon, V. Corcé, M. Amatore, C. Aubert, F. Chemla, F. Ferreira, A. Perez-Luna and M. Petit, A well-defined low-valent cobalt catalyst Co(PMe3)4 with dimethylzinc: a simple catalytic approach for the reductive dimerization of benzyl halides, New J. Chem., 2016, 40, 9912–9916 RSC
- A. Studer and D. P. Curran, Catalysis of Radical Reactions: A Radical Chemistry Perspective, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed
-
C. S. Sheppard, in Encyclopedia of polymer science and engineering, John Wiley & Sons, Inc., 1985, vol. 11, pp. 1–21 Search PubMed
- N. A. Turovskii, I. A. Opeida and O. V. Kushch, Intermediates in Reactions of Diacyl Peroxides with Tertiary Aliphatic Amines, Russ. J. Org. Chem., 2003, 39, 642–645 CrossRef CAS
- S. Srinivas and K. G. Taylor, Amine-induced reactions of diacyl peroxides, J. Org. Chem., 1990, 55, 1779–1786 CrossRef CAS
- D. B. Denney and D. Z. Denney, Studies of the Mechanisms of the Reactions of Benzoyl Peroxide with Secondary Amines and Phenols, J. Am. Chem. Soc., 1960, 82, 1389–1393 CrossRef CAS
- D. W. C. MacMillan, The advent and development of organocatalysis, Nature, 2008, 455, 304–308 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Method development, synthesis, characterization and mechanistic studies. See DOI: 10.1039/d1qo00259g |
| This journal is © the Partner Organisations 2021 |