
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadical cations and dications of bis[1]benzothieno[1,4]thiazine isomers†
Simone T.
Hauer
a,
Arno P. W.
Schneeweis
a,
Sven D.
Waniek
b,
Lukas P.
Sorge
b,
Katja
Heinze
 b and
Thomas J. J.
Müller
*a
b and
Thomas J. J.
Müller
*a
aHeinrich-Heine-Universität Düsseldorf, Institut für Organische Chemie und Makromolekulare Chemie, Universitätsstraße 1, D-40225 Düsseldorf, Germany. E-mail: ThomasJJ.Mueller@hhu.de
bJohannes Gutenberg University Mainz, Department of Chemistry, Duesbergweg 10-14, D-55128 Mainz, Germany
First published on 22nd July 2021
Abstract
Bis[1]benzothieno[1,4]thiazines (BBTT) are particularly electron-rich S,N-heteropentacenes and their radical cations and dications can be relevant intermediates in charge transport materials. All three regioisomers of N-p-fluorophenyl-BBTT (syn–syn, syn–anti, and anti–anti) were studied. A reliable preparation of radical cations and dications using antimony pentachloride as an oxidant gives deeply colored salts. The electronic structure of the radical cations was assessed by EPR spectroscopy, whereas dicationic structures were characterized by NMR spectroscopy. In addition, a deeper insight into the electronic structure was experimentally and computationally obtained by UV/Vis spectroscopy as well as UV/Vis spectroelectrochemistry and DFT and TDDFT calculations. BBTTs can be considered as highly polarizable donor π-systems with significant charge transfer contributions in neutral, cationic and dicationic state.
Introduction
Molecular electronics based upon organic molecules has become increasingly important over the past decade and the quest for tailor-made functional small molecules is an ongoing task for chemistry.1,2 Since the efficiency of electronic devices correlates with electronic properties of the constituting molecules, a rational design of novel molecule based semiconductors commences with high lying HOMO levels, which in turn correspond to low oxidation potentials.3–8Electron-rich heterocycles, such as phenothiazine, are prominent structural motifs used in electroactive materials for batteries,9–11 OLEDs,12–14 and DSSCs.15–19 They possess low oxidation potentials with fully chemically reversible one-electron oxidations, low band gaps, and luminescent properties.20–22 In addition to their convenient accessibility, their intrinsic properties are easily fine tunable by synthetic functionalization.22,23 Furthermore, the remarkable stability of their radical cations is crucial for providing efficient charge transfer as hole conductors.24,25 Recently, we successfully proposed the two-fold thieno expansion of phenothiazine to bis[1]benzothieno[1,4]thiazines (BBTT) that can be considered to be novel electron-enriched phenothiazine congeners (Scheme 1).26
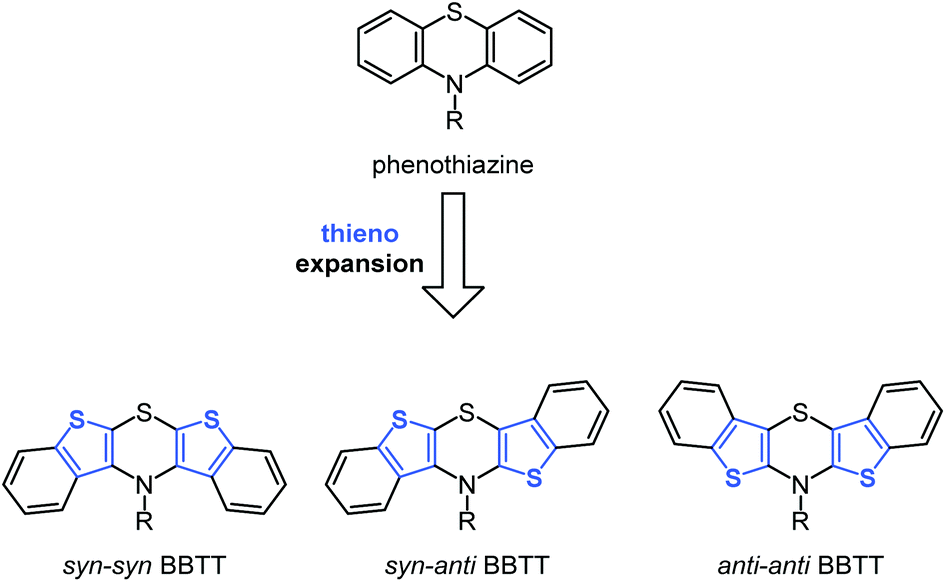 | ||
| Scheme 1 Regioisomeric BBTTs resulting by formal thieno expansion of phenothiazine. | ||
The resulting compounds likewise possess discretely adjustable properties that depend on the mode of anellation, as for dithienothiazines,27 as well as on the substitution pattern. Different steric hindrance unequivocally causes the absence of measurable luminescence in syn–syn BBTTs (bis[1]benzo-thieno[2,3-b:3′,2′-e][1,4]thiazines), whereas inverse anellation of benzo[b]thiophene units leads to luminescence for syn–anti BBTTs (bis[1]benzothieno[2,3-b:2′,3′-e][1,4] thiazines) peaking for anti–anti BBTTs (bis[1]benzo-thieno[3,2-b:2′,3′-e][1,4]thiazines) with fluorescence quantum yields drastically increased to 45%.26–30 Furthermore, anti–anti BBTTs represent the first known fused thiazines with antiaromatic character in the crystalline solid state. Likewise, being identified as Weitz-type-redox systems,31 BBTTs are outscoring their progenitors, i.e. phenothiazines, by significantly lower oxidation potentials with reversible two step oxidations, forming radical cations and dications.26,28 Herein, we report the first synthesis of BBTTs’ radical cations and dications by chemical and electrochemical oxidation as well as the detailed investigation of structural and electronic properties.
Results and discussion
Synthesis and ground state structure
N-p-Fluorophenyl-BBTTs were prepared according to our previously published procedure28 by twofold Buchwald–Hartwig amination32–34 (Scheme 2). | ||
Scheme 2 Selective synthesis of regioisomeric N-p-fluorophenyl-BBTTs 5, 6, and 7 prepared by cyclizing Buchwald–Hartwig amination (for the synthesis of compounds 5 and 6 DCPF (1,1′-bis(![[d with combining low line]](https://www.rsc.org/images/entities/char_0064_0332.gif) i i![[c with combining low line]](https://www.rsc.org/images/entities/char_0063_0332.gif) yclohexyl yclohexyl![[p with combining low line]](https://www.rsc.org/images/entities/char_0070_0332.gif) hosphano) hosphano)![[f with combining low line]](https://www.rsc.org/images/entities/char_0066_0332.gif) errocene) was employed as a ligand, and for compound 7 DPPF (1,1′-bis(ihenylhosphano)errocene) was used as a ligand).28 errocene) was employed as a ligand, and for compound 7 DPPF (1,1′-bis(ihenylhosphano)errocene) was used as a ligand).28 | ||
Starting from respective dibrominated dibenzo[b]thienyl sulfides 1, 2, or 328,35–37 and p-fluoroaniline (4), all regioisomeric BBTTs 5–7 were reliably synthesized. The original syntheses were upscaled by a factor of ten and even upon reducing catalyst loading the yields for compounds 5 (49%), 6 (45%), and 7 (67%) were maintained. Fluorinated compounds were chosen due to their synthetic accessibility of the neutral BBTT, as well as the opportunity to monitor conversion and easily identify the products by 19F NMR spectroscopy.
As supported previously by CV measurements, the BBTTs’ radical cations (5: E00/1+ = 0.21 V, 6: E00/1+ = 0.05 V, 7: E00/1+ = 0.01 V; against ferrocene E00/1+ = 0.00 V) and dications (5: E01+/2+ = 0.68 V, 6: E01+/2+ = 0.72 V, 7: E01+/2+ = 0.75 V; against ferrocene E00/1+ = 0.00 V) can be readily generated in solution and they are stable over short periods.26,28 In addition, the radical cations reveal high semiquinone formation constants KSEM (5: 9.2 × 107, 6: 2.3 × 1011, 7: 3.5 × 1012)28 that claim remarkably stable radical cations against disproportionation into reduced BBTT and dication.28,31 Consequently, we attempted the preparation of the oxidized species of all three BBTTs. Analogously to dithieno[1,4]thiazine (DTT)27,30,38–41 chemical oxidation was carried out with antimony pentachloride as oxidant (Scheme 3).41 By choice of solvent, temperature and equivalents of antimony pentachloride the oxidation stage can be reliably controlled.
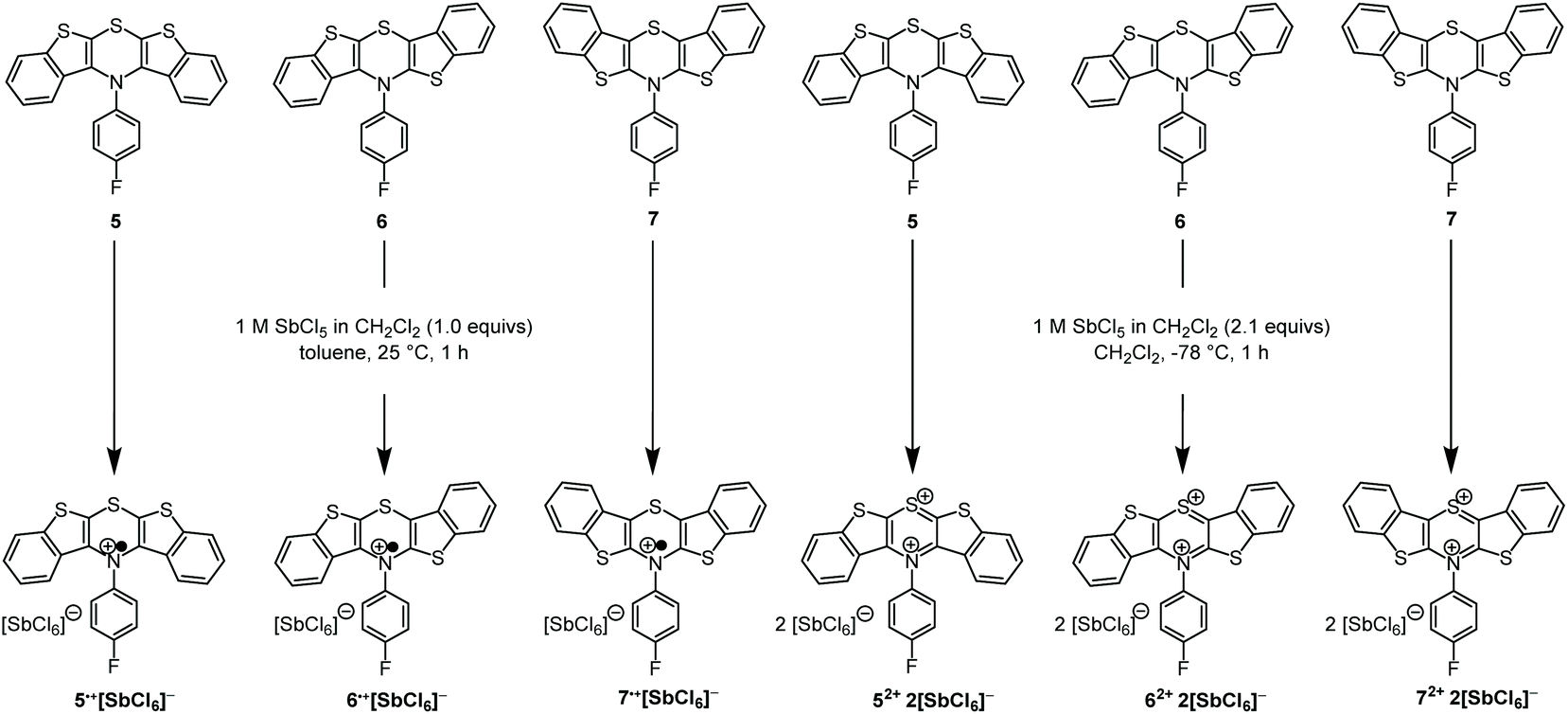 | ||
| Scheme 3 Controlled preparation of oxidized BBTT-compounds 5˙+, 6˙+, 7˙+, and 52+, 62+, 72+ as hexachloroantimonates using antimony pentachloride as an oxidant. | ||
BBTTs 5–7 are soluble in toluene and dichloromethane, whereas the corresponding hexachloroantimonate salts of the radical cations 5˙+, 6˙+, and 7˙+ precipitated from toluene and thereby removed from the reaction equilibrium. Additionally, over-oxidation was prevented by a stoichiometric deficiency of oxidant. If dichloromethane was used as a solvent, the hexachloroantimonate salts of the radical cations remained soluble, forming dication salts upon increasing the amount of antimony pentachloride. The desired oxidation products precipitated from the respective reaction mixture.
For assessing the electronic structure, the molecules’ geometries were calculated on the DFT level of theory using the uB3LYP functional42,43 and the 6-311G** basis set44,45 with SCRF (IEFPCM, CH2Cl2)46–48 as implemented in the Gaussian 09 program package.49 Indeed the BBTT units are entirely planarized with a perpendicular orientation of the N-p-fluorophenyl substituents for all three radical cations 5˙+, 6˙+, and 7˙+. Depending on the anellation mode, different spin density distributions were found, however, with highest spin density located on the nitrogen atoms (Mulliken atomic spin density at N-atom: 5˙+: 0.284, 6˙+: 0.248, 7˙+: 0.228). The highest delocalization of spin density is anticipated for the anti–anti derivative 7˙+, whereas the lowest delocalization is predicted for the syn–syn derivative 5˙+ (Fig. 1).
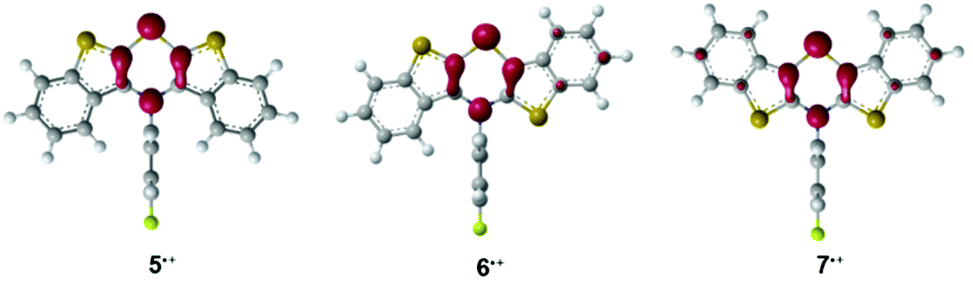 | ||
| Fig. 1 DFT calculated spin density distribution plots of radical cations 5˙+, 6˙+, and 7˙+ (uB3LYP/6-311++G**, IEFPCM CH2Cl2, isosurface value = 0.06 a.u.). | ||
The radical nature of the obtained salts 5˙+, 6˙+, and 7˙+ was proven by X-band EPR spectroscopy and by comparison with the simulated spectra (Fig. 2).
 | ||
| Fig. 2 Simulated and experimental EPR spectra of hexachloroantimonate salts of 5˙+, 6˙+, and 7˙+ (recorded in THF, T = 298 K, g-factor referenced to external Mn2+ in ZnS). | ||
Spectra with hyperfine coupling patterns of three equidistant lines reflect the coupling of the unpaired electron with the nitrogen nucleus in each case. Furthermore, the range of the g-factors (5˙+: 2.0106, 6˙+: 2.0050, 7˙+: 2.0078) indicates the predominant localization of the radicals on the nitrogen atoms.41,50 The Mulliken atomic spin density on the nitrogen atoms decreases from syn–syn over syn–anti to anti–anti derivative, which is in accordance with the descending order of hyperfine coupling constants. Hence, an increasing spin delocalization across the BBTT units for anti-anellated benzo[b]thiophene wings can be deduced. The stronger delocalization matches with an increased stability of the corresponding radical cation 7˙+, which is consistent with the experimentally determined semiquinone formation constants KSEM, which also give largest values for anti–anti radical cation 7˙+.
The delocalization of spin density of the radical cations is unequivocally supported by the calculated Wiberg bond orders (Fig. 3).51
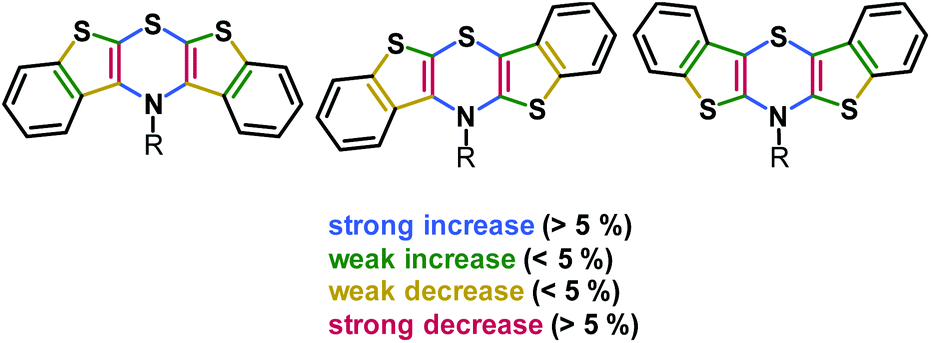 | ||
| Fig. 3 Relative change of Wiberg bond orders Δ(BOX˙+-BOX) from neutral precursors 5, 6, and 7 to radical cations 5˙+, 6˙+, and 7˙+ in the BBTT core (R = p-fluorophenyl; uB3LYP/6-311G**) (A detailed list of bond orders is given in the ESI†). | ||
The bond orders of the systems should gradually converge upon oxidation due to increasing orbital overlap by planarization resulting in an increased resonance stabilization. All three regioisomers show the most significant changes in bond orders in the 1,4-thiazine core, where CN and CS bond orders increase while CC bond orders decrease. This effect is most pronounced for the syn–syn regioisomer 5/5˙+ while the anti–anti isomer 7/7˙+ is most marginally affected. However, anti-fused benzo[b]-thiophene wings result in higher bond order changes on the fused thiophene in turn. This also confirms the previously observed increased spin delocalization upon introduction of anti-anellated wings. Nevertheless, even with anti-anellation, the spin density on the benzo ring is not high enough for causing observable coupling with the protons’ nuclear spins. Although the calculated spin density distributions (Fig. 1) suggest this conclusion, no hyperfine coupling patterns of the protons are experimentally found.
For hexachloroantimonate salts of dications 52+, 62+, and 72+, DFT calculations claim formation of singlet dications being thermodynamically favored over triplet dications by energy differences of −92 kJ mol−1 (52+), −89 kJ mol−1 (62+), and −90 kJ mol−1 (72+). Consequently, EPR-silent but NMR-active target compounds should be present. NMR spectra of the bis(hexachloroantimonate) salts of 52+, 62+, and 72+ were recorded and compared to the spectra of the native compounds 5, 6, and 7, thus, confirming the formation of diamagnetic dications, i.e. in their singlet ground states (Fig. 4).
 | ||
| Fig. 4 Sections of the 1H NMR spectra of the neutral compounds 5, 6, and 7 (blue) and of the bis(hexachloro-antimonate) salts of 52+, 62+, and 72+ (black) (300 MHz, acetone-d6, T = 298 K). | ||
As expected, the majority of the multiplets shift towards lower field, underlining the electron-deficient dicationic nature of the compounds. Due to structural differences of the regioisomers, parts of the signals appear shielded in high field. DFT-calculated geometry optimizations show planar BBTT units with orthogonally twisted N-p-fluorophenyl substituents, in analogy to the radical cations. syn-Anellated wings place benzo protons into the anisotropy cone of the N-aryl substituent's diamagnetic ring current. Thereby, their signals are shifted to high field.
Photophysical properties and spectroelectrochemistry
For further insight into electronic transitions of oxidized species, i.e. radical cations and dications, spectroelectrochemical measurements were performed in an OTTLE (Optically Transparent Thin Layer Electrochemical) cell, monitoring the in situ formation of radical cations and dications from the neutral precursors 5, 6, and 7 (Table 1). Hence, spectroelectrochemical UV/Vis absorption spectra were recorded by steadily increasing the applied potential in steps of 0.1 V.| Compound | Absorption maxima λmax [nm] | Isosbestic points [nm] | ||
|---|---|---|---|---|
| syn–syn | 5 | 230, 265, 300, 309 | ||
| 5˙+ | 226, 234, 258, 284, 295 (sh), 311, 328, 382, 427, 494, 680 | 5→5˙+ | 243, 276, 342, 357 | |
| 52+ | 227, 233, 272, 346, 494, 526, 672 | 5˙+→52+ | 266, 279, 320, 372, 401 | |
| syn–anti | 6 | 236, 263, 311 | ||
| 6˙+ | 228, 262, 285, 315(sh), 328, 360 (sh), 377, 398, 566 (sh), 617 (sh), 694 | 6→6˙+ | 271, 317, 351 | |
| 62+ | 224, 236 (sh), 253, 324, 349 (sh), 395, 595, 709 (sh) | 6˙+→62+ | 249, 303, 368, 418, 649, 763 | |
| anti–anti | 7 | 236, 268 (sh), 317, 424 | ||
| 7˙+ | 229, 259, 276, 293, 308, 318, 352, 369, 450 (sh), 490, 502, 534, 626 (sh), 717 | 7→7˙+ | 255, 264, 275, 326, 361, 374, 459 | |
| 72+ | 223, 255, 271, 337, 561, 657 | 7˙+→72+ | 316, 444, 542, 697 |
Initially, UV/Vis spectra of all three parent regioisomers 5, 6, and 7 were recorded showing multiple absorptions bands in the UV region with the longest wavelength maxima at 309 nm (5) and 311 nm (6). For compound 7 the longest wavelength maximum is found in the visible at 424 nm (Fig. 5).
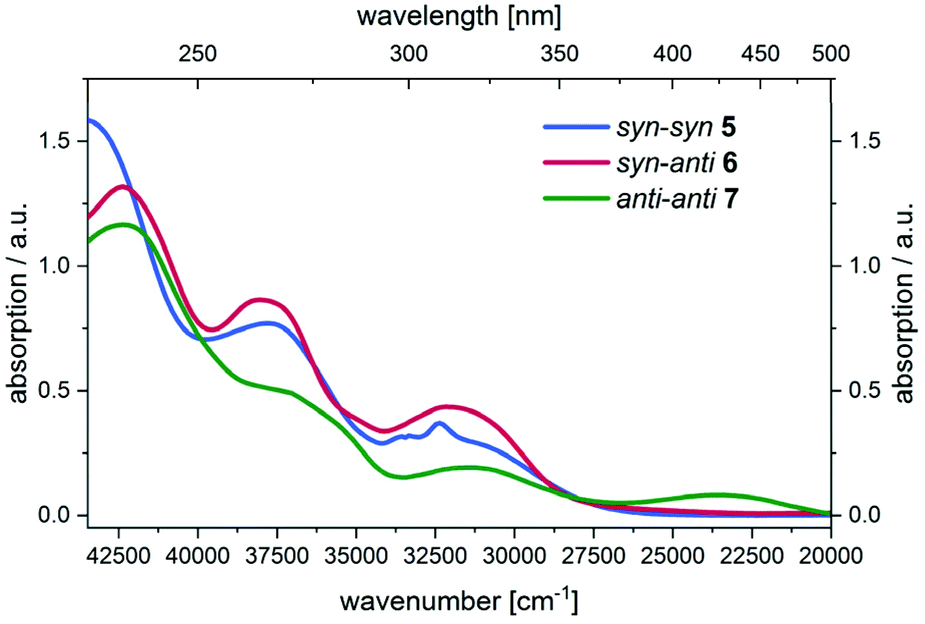 | ||
| Fig. 5 UV/Vis spectra of compounds 5, 6, and 7 in the spectroelectrochemical cell at zero potential (CH2Cl2, T = 293 K). | ||
All oxidation processes proceed without formation of intermediates, as supported by the occurrence of isosbestic points. Moreover, the oxidations from the neutral BBTTs to the radical cations reflect the largest changes in the UV/Vis spectra and consequently in the electronic structure (vide infra). This originates from major geometry changes from butterfly structures to the planar alignments of the BBTT units (cf. phenothiazines and DTT).38,52,53 Consequently, oxidized compounds yielded by chemical as well as electrochemical oxidation are deeply colored (Fig. 6).
 | ||
| Fig. 6 From left to right: photographs of the solutions of parent compounds 5, 6, and 7 in dichloromethane (c = 5.5 × 10−4 M, left), hexachloroantimonate salts of 5˙+, 6˙+, and 7˙+ in dichloromethane (c = 5.5 × 10−4 M, center) and of 52+, 62+, and 72+ in nitromethane (c = 5.5 × 10−4 M, right). | ||
By gradual increase of the potential the formation of radical cations is accomplished, revealing significantly bathochromically shifted absorption bands (Fig. 7).
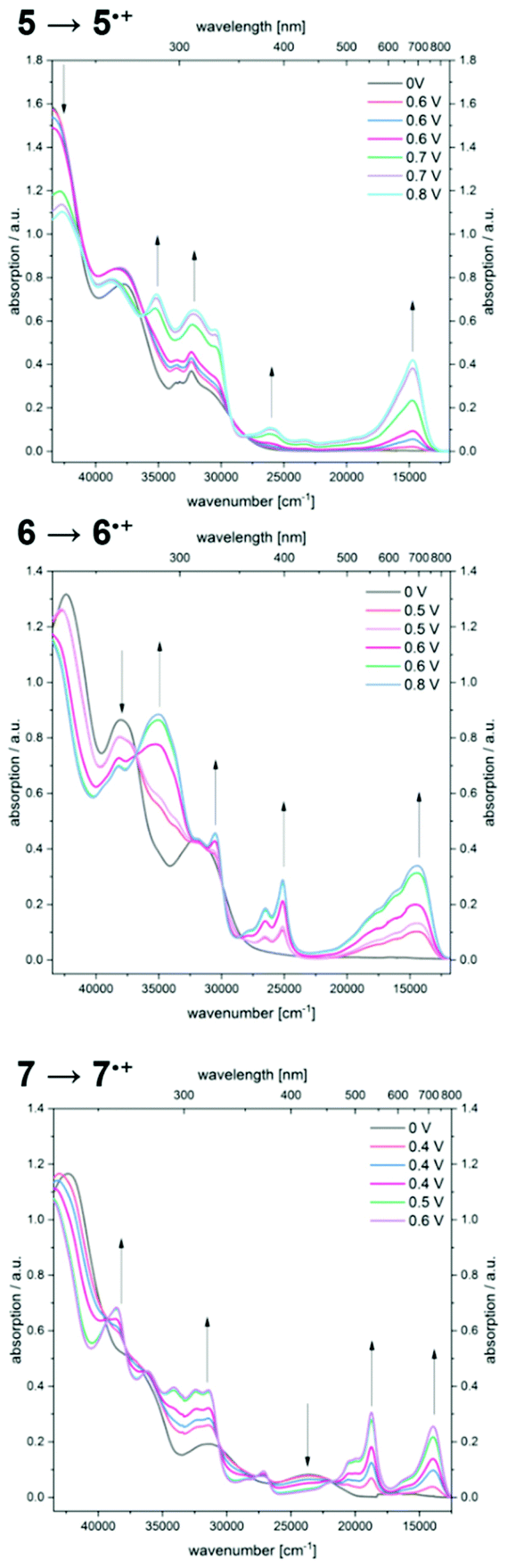 | ||
| Fig. 7 UV/Vis spectroelectrochemical measurements of compounds 5, 6, and 7 forming the corresponding radical cations 5˙+, 6˙+, and 7˙+ (CH2Cl2, T = 293 K; electrolyte 0.1 M [nBu4N][PF6]; Pt working electrode, Pt counter electrode and Ag/AgCl pseudo reference electrode). | ||
Therefore, compound 5˙+ shows an intense maximum at 680 nm. A strongly broadened absorption band ranging from 450 to 850 nm with shoulders at 566 and 617 nm in conjunction with a maximum at 694 nm is found for radical cation 6˙+. The two prominent absorption maxima for oxidation product 7˙+ appear at 534 and 717 nm.
Further increase of the potential allows monitoring of the formation of dications (Fig. 8). For dication 52+, the longest wavelength absorption maximum at 680 nm shifts slightly hypsochromically, resulting in a band at 672 nm while simultaneously a second band of lower intensity appears at 526 nm. In addition, the most intense band appears at 346 nm. A comparable absorption band is found at 324 nm for dication 62+. The shoulders observed for radical cation 6˙+ merge into a new maximum at 595 nm, whereas the maximum at 694 nm decreases to a shoulder at 709 nm. Upon oxidation of radical cation 7˙+ to dication 72+, the two intense bathochromically shifted maxima of the radical cation result in the formation of two new maxima at 561 and 657 nm for the dication. Likewise, an intense band appears in the UV at 337 nm.
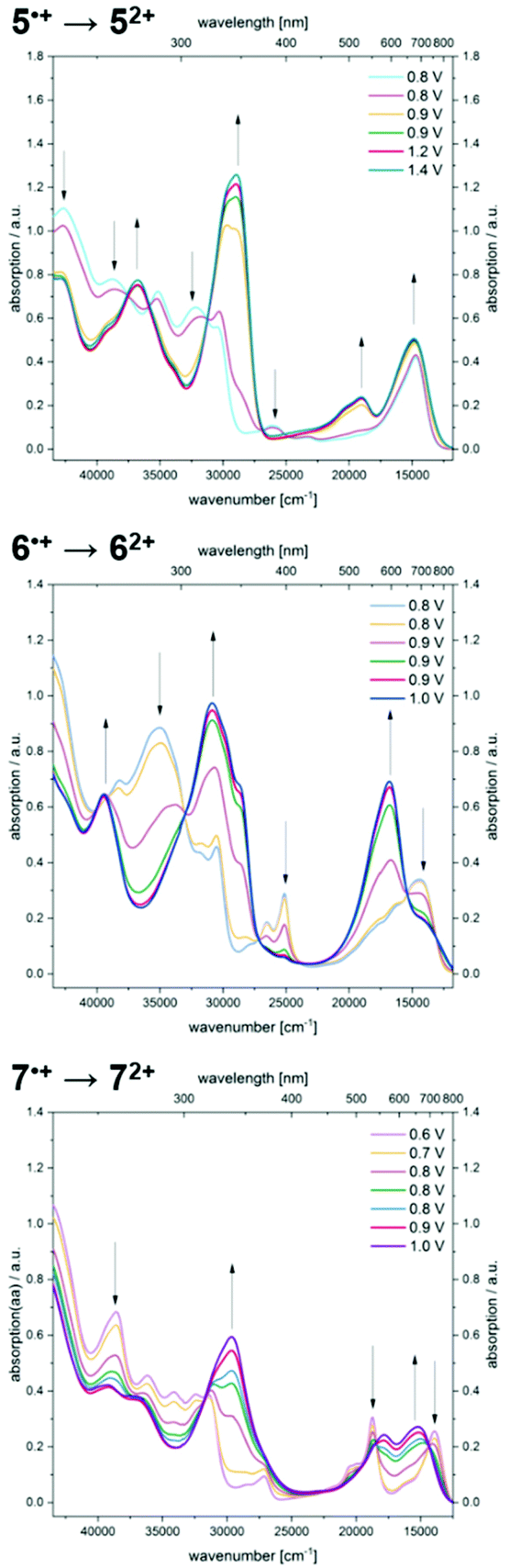 | ||
| Fig. 8 UV/Vis spectroelectrochemical measurements of radical cations 5˙+, 6˙+, and 7˙+ forming the corresponding dications 52+, 62+, and 72+ (CH2Cl2, T = 293 K; electrolyte 0.1 M [nBu4N][PF6]; Pt working electrode, Pt counter electrode and Ag/AgCl pseudo reference electrode). | ||
Finally, the spectra from spectroelectrochemical measurements were compared with the spectra of the synthesized hexachloroantimonate radical cation salts (Fig. 9).
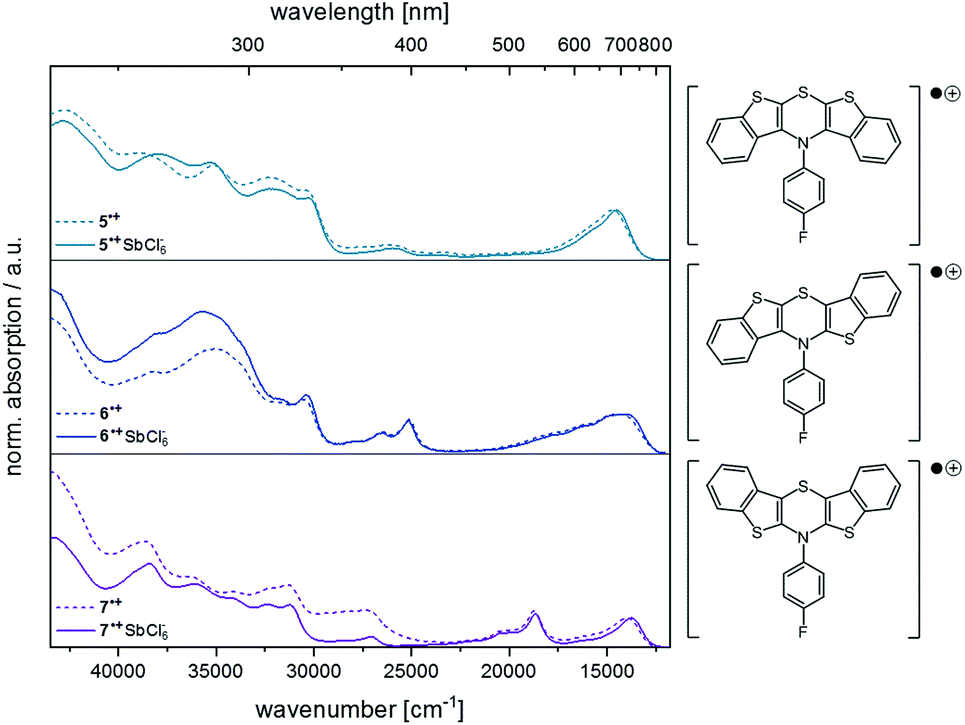 | ||
| Fig. 9 Comparison of the UV/Vis spectra from measurements of the radical cation salts (solid lines) and from spectroelectrochemical measurements (dotted lines) (CH2Cl2, T = 293 K, for further details, see ESI†). | ||
The superposition shows that the salts unambiguously correspond to the spectroelectrochemically generated specimen and a good overall fit is obtained. Differences in the intensity of the absorption bands in the UV region of the spectra (230–380 nm) can be accounted to counter ion effects.25 Likewise, the longest wavelength absorption maxima for the hexachloroantimonate salts of the radical cations are only slightly bathochromically shifted (5˙+ SbCl6−: Δ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = +171 cm−1, 6˙+ SbCl6−: Δ = +305 cm−1, 7˙+ SbCl6−: Δ = +173 cm−1). As known for Weitz-type redox systems, the radical ions are remarkably stable due to their high semiquinone formation constants KSEM (vide supra).31 However, the dications could not be successfully measured due lacking stability under prolonged measuring conditions.
= +171 cm−1, 6˙+ SbCl6−: Δ = +305 cm−1, 7˙+ SbCl6−: Δ = +173 cm−1). As known for Weitz-type redox systems, the radical ions are remarkably stable due to their high semiquinone formation constants KSEM (vide supra).31 However, the dications could not be successfully measured due lacking stability under prolonged measuring conditions.
Electronic structure of the radical cations and dications
For a deeper insight into the electronic structure of the absorption behavior of neutral, radical cation and dication BBTTs TDDFT calculations54,55 ((u)B3LYP43,44,56/6-311++G**, IEFPCM CH2Cl2) were performed.First, the optical transitions of the experimentally observed absorption bands of the parent compounds 5, 6, and 7 were assigned (Table 2, Fig. 10).
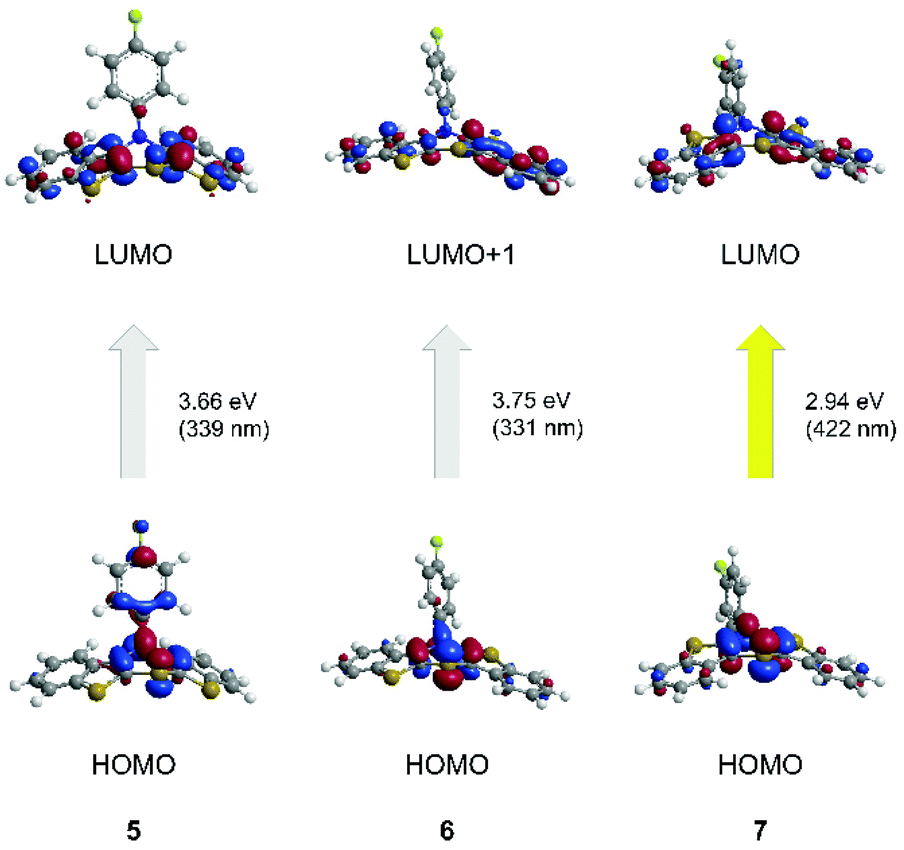 | ||
| Fig. 10 TDDFT calculated frontier molecular orbital (FMO) transitions of the longest wavelength absorptions of the neutral compounds 5, 6, and 7 (B3LYP/6-311++G**, IEFPCM CH2Cl2, isosurface value 0.05 a.u.). | ||
| Compound | λ max,abs | λ max,calcd (oscillatory strength) | Most dominant contributions |
|---|---|---|---|
| [nm] | [nm] | ||
| a Recorded in CH2Cl2, c = 10−5M, T = 293 K. | |||
| syn–syn 5 | 309 | 339 (0.075) | HOMO → LUMO (94%) |
| syn–anti6 | 311 | 331 (0.143) | HOMO → LUMO+1 (68%) |
| anti–anti 7 | 424 | 422 (0.144) | HOMO → LUMO (95%) |
The longest wavelength absorption bands are reasonably well reproduced by transitions at 339 nm (5) and 422 nm (7). They predominantly consist of HOMO → LUMO transitions representing a charge transfer (CT) from the thiazine core to the benzo[b]thiophene wings. The calculated longest wavelength absorption band of 6 appears at 331 nm and can be predominantly assigned to a HOMO → LUMO+1 transition. Again, this transition is accompanied by a CT from the central thiazine to the benzo[b]thiophene units, with a dominant shift of coefficient density due to anti-anellation.
Likewise, for assigning the observed UV/Vis absorption of the radical cations, TDDFT calculations with the uB3LYP/6-311++G** functional were performed to prevent spin pairing and to cause separate orbitals occupation (Table 3, Fig. 11).
 | ||
| Fig. 11 TDDFT calculated molecular orbital transitions of most bathochromic absorptions of radical cations 5˙+, 6˙+, and 7˙+ (uB3LYP/6-311++G**, IEFPCM CH2Cl2, isosurface value 0.05 a.u.) (For further molecular orbital transitions, see SI). | ||
| Compound | λ max,abs | λ max,calcd (oscillatory strength) | Most dominant contributions |
|---|---|---|---|
| [nm] | [nm] | ||
| a Recorded in CH2Cl2, c = 10−5 M, T = 293 K. | |||
| syn–syn 5˙+ | 680 | 665 (0.191) | HOMO−1 → SOMO (100%) |
| syn–anti6˙+ | 694 | 714 (0.086) | HOMO−1 → SOMO (97%) |
| 617 | 617 (0.156) | HOMO−2 → SOMO (95%) | |
| 566 | 577 (0.022) | HOMO−3 → SOMO (94%) | |
| anti–anti 7˙+ | 717 | 701 (0.154) | HOMO−1 → SOMO (98%) |
| 534 | 538 (0.111) | HOMO−3 → SOMO (100%) | |
This leads to a singly occupied molecular orbital (SOMO). The longest wavelength absorption bands are almost accurately reproduced by HOMO−1 → SOMO transitions at wavelengths of 665 nm (5˙+), 714 nm (6˙+) and 701 nm (7˙+). For cation 7˙+, the measured second absorption band at 534 nm is best represented by a HOMO−3 → SOMO transition at 538 nm, while the shoulders of structure 6˙+ are best reproduced by calculated bands at 617 nm (HOMO−2 → SOMO) and 577 nm (HOMO−3 → SOMO). All these transitions are predominantly characterized by CT contributions from the anellated benzo[b]thiophene wings to the central thiazine core, while coefficient density on the N-aryl substituents is absent.
Finally, TDDFT calculations (uB3LYP/6-311++G**) were also carried out to assign the absorption bands of the dications 52+, 62+, and 72+ (Table 4, Fig. 12).
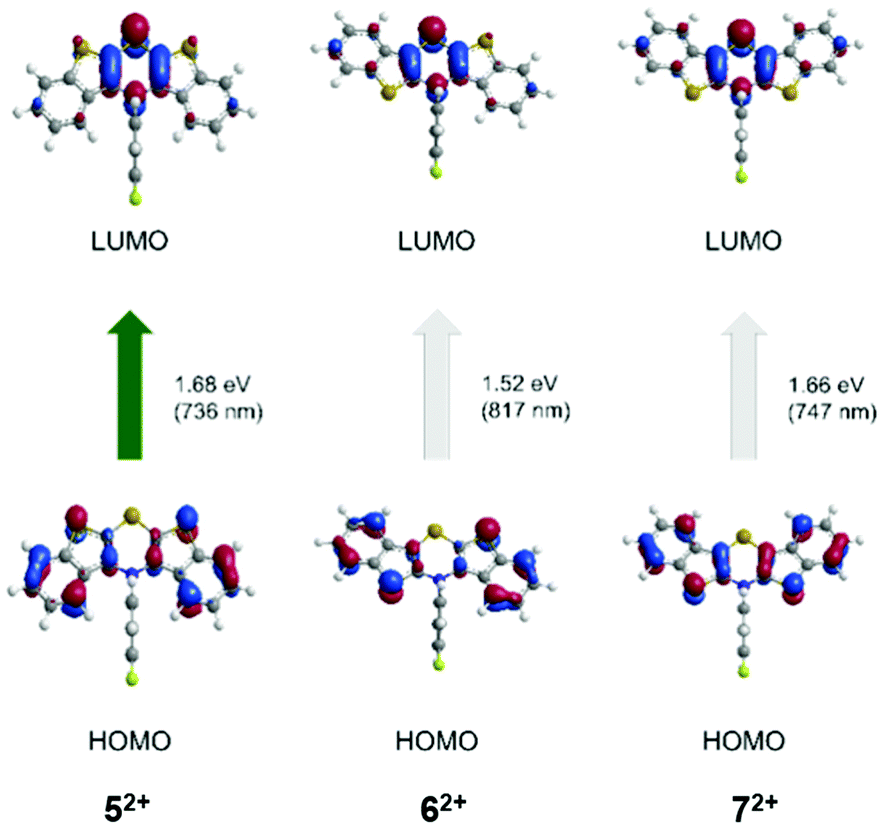 | ||
| Fig. 12 TDDFT calculated molecular orbital transitions of most bathochromic absorptions of the dications 52+, 62+, and 72+ (uB3LYP/6-311++G**, IEFPCM CH2Cl2, isosurface value 0.05 a.u.) (For further molecular orbital transitions, see ESI†). | ||
| Compound | λ max,abs | λ max,calcd (oscillatory strength) | Most dominant contributions |
|---|---|---|---|
| [nm] | [nm] | ||
| a Recorded in CH2Cl2, c = 10−5 M, T = 293 K. | |||
| syn–syn 52+ | 672 | 736 (0.226) | HOMO → LUMO (92%) |
| 526 | 517 (0.078) | HOMO−4 → LUMO (91%) | |
| 346 | 325 (0.578) | HOMO−1 → LUMO+1 (91%) | |
| syn–anti62+ | 650–850 (br) | 817 (0.063) | HOMO → LUMO (78%) |
| 595 | 617 (0.345) | HOMO−1 → LUMO (64%) | |
| 324 | 311 (0.704) | HOMO−1 → LUMO+1 (100%) | |
| anti–anti 72+ | 657 | 747 (0.163) | HOMO → LUMO (83%) |
| 561 | 579 (0.363) | HOMO−2 → LUMO (84%) | |
| 337 | 317 (0.490) | HOMO−1 → LUMO+1 (100%) | |
The longest wavelength absorption bands are best represented by calculated HOMO → LUMO transitions at wavelengths of 736 (52+), 709 (62+), and 747 nm (72+) that can be interpreted as CT transitions from the outer benzo[b]thiophene part to the central thiazine unit. The following experimental bands in the visible are assigned to transitions arising from HOMO−4 → LUMO at 517 nm (52+), HOMO−1 → LUMO at 617 nm (62+), and HOMO−2 → LUMO at 579 nm (72+). These transitions are characterized by CT contributions from the N-aryl moiety to the thiazine core for structure 52+, whereas for structure 62+ by a shift of coefficient density from the benzo[b]thiophene parts to the thiazine core. For structure 72+, the transition can be accounted as a combination of both preceding coefficient density shifts.
The preceding calculations reveal that BBTT are strongly polarizable systems with large CT contributions. Accordingly, neutral BBTT are potent donors due to their electron richness, whereas the oxidized compounds are accompanied by strong acceptor properties.
Aromaticity of the dications
Furthermore, the dications’ thiazine cores should inhere aromatic character due to their Weitz-type nature. Proving this, starting with the previously mentioned optimized planar geometries, NICS (0) and NICS (1) (above (+1) and under (−1) the plane) calculations were performed. Using the GIAO protocol with B3LYP functional and 6-311+G** basis set44,45 calculations were carried out for all five rings of the BBTT core, giving NICS values indicating diatropic ring current stating aromaticity (Table 5).|
|
|||||
|---|---|---|---|---|---|
| Ring no. | NICS (0/+1/−1) | Ring current | Aromaticity | ||
| syn–syn52+ | syn–anti62+ | anti–anti72+ | |||
| 1a | −8.16/−9.83/−9.83 | −8.26/−10.23/−10.14 | −6.02/−8.20/−8.20 | Diatropic | Aromatic |
| 1b | −8.16/−9.83/−9.83 | −5.99/−8.33/−8.35 | −6.02/−8.20/−8.20 | Diatropic | Aromatic |
| 2a | −7.29/−6.17/−6.17 | −7.38/−6.46/−6.35 | −6.50/−5.96/−5.96 | Diatropic | Aromatic |
| 2b | −7.29/−6.17/−6.17 | −6.39/−5.66/−5.72 | −6.50/−5.96/−5.96 | Diatropic | Aromatic |
| 3 | −6.94/−7.22/−7.22 | −7.19/−7.43/−7.54 | −6.64/−7.12/−7.12 | Diatropic | Aromatic |
Experimental
1H and 19F NMR spectra of compounds 5, 6, 7, 52+, 62+, and 72+, EPR spectra of 5˙+, 6˙+, and 7˙+, spectroelectrochemistry of compounds 5, 6, and 7, and TDDFT calculations of optimized structures 5, 6, 7, 5˙+, 6˙+, 7˙+, 52+, 62+, and 72+ are compiled in the ESI.†General procedure – synthesis of bis[1]benzothieno[1,4]thiazines (BBTTs)
In a dry Schlenk tube with a magnetic stir bar under Ar atmosphere were placed dibrominated sulfane 1, 2, or 3 (2.3 g, 5.0 mmol), Pd(dba)2 (0.14 g, 5.0 mol%) and ligand (10 mol%). By glovebox technique sodium tert-butoxide (1.4 g, 15 mmol) was added into the reaction tube. Subsequently, p-fluoroaniline (0.47 mL, 5.0 mmol) und dry toluene (30 mL) were added by syringe before stirring the reaction mixture at 110 °C (oil bath) for 65 h. After cooling to room temp the reaction was terminated by addition of saturated aqueous sodium sulfite solution (400 mL). The aqueous phase was extracted with dichloromethane (3 × 300 mL), the combined organic phases were dried with anhydrous magnesium sulfate and the solvents were removed under vacuum. After adsorption to Celite® the residue was purified by chromatography on silica gel. After recrystallization from n-hexane the products 5, 6, or 7 were isolated as crystals. Deviations from this general procedure are individually highlighted.N-(4-Fluorophenyl)bis[1]benzothieno[2,3-b:3′,2′-e][1,4]thiazine (syn–syn5)26
According to the general procedure with bis(3-bromobenzo[b]thiophene-2-yl)sulfane (1) and 1,1′-bis(dicyclo-hexylphosphano)ferrocene (0.29 g, 0.50 mmol) as a ligand and after chromatography (n-hexane) and recrystallization from n-hexane (250 mL) compound 5 (0.92 g, 49%) was obtained as colorless needles, Mp 215 °C (DSC measurement). 1H NMR (300 MHz, acetone-d6) δ 6.92–7.04 (m, 2 H), 7.42–7.55 (m, 5 H), 7.89–7.99 (m, 5 H); 19F NMR (282 MHz, acetone-d6) δ 53.42; 1H NMR (600 MHz, THF-d8) δ 6.85–6.93 (m, 4 H), 7.35–7.45 (m, 4 H), 7.80–7.88 (m, 4 H); 13C{1H} NMR (151 MHz, THF-d8) δ 116.4 (CH, d, 2JCF = 22.7 Hz), 119.1 (CH, d, 3JCF = 7.7 Hz), 122.0 (CH), 124.0 (CH), 126.0 (CH), 126.0 (CH), 132.7 (Cquat), 136.1 (Cquat), 139.6 (Cquat), 141.0 (Cquat), 144.6 (Cquat, d, 4JCF = 2.4 Hz), 159.2 (Cquat, d, 1JCF = 239.4 Hz); 19F NMR (565 MHz, THF-d8) δ −124.4; HRMS(ESI) m/z calcd for [C22H12FNS3]+: 405.0110; Found: 405.0112; Anal. calcd for C22H12FNS3 (405.5): C 65.16, H 2.98, N 3.45, S 23.72; Found: C 65.38, H 2.98, N 3.44, S 23.49.N-(4-Fluorophenyl)bis[1]benzothieno[2,3-b:2′,3′-e][1,4]-thiazine (syn–anti6)26
According to the general procedure with 2-bromo-3-((3-bromobenzo[b]thiophen-2-yl)thio)benzo[b]thiophene (2) and 1,1′-bis(dicyclohexyl-phosphano)ferrocene (0.29 g, 0.50 mmol) as a ligand and after chromatography (n-hexane) and recrystallization from n-hexane (550 mL) compound 6 (0.92 g, 2.3 mmol, 45%) was obtained as yellow crystals, Mp 201 °C. 1H NMR (300 MHz, acetone-d6) δ 6.92–6.98 (m, 1 H), 7.17–7.24 (m, 1 H), 7.24–7.40 (m, 4 H), 7.44–7.48 (m, 2 H), 7.63–7.71 (m, 2 H), 7.80–7.85 (m, 1 H), 7.85–7.90 (m, 1 H); 19F NMR (282 MHz, acetone-d6) δ 61.08; 1H NMR (600 MHz, THF-d8) δ 6.93–6.96 (m, 1 H), 7.10–7.20 (m, 3 H), 7.20–7.24 (m, 1 H), 7.25–7.30 (m, 1 H), 7.35–7.43 (m, 2 H), 7.56–7.61 (m, 2 H), 7.69–7.77 (m, 2 H); 13C{1H} NMR (151 MHz, THF-d8) δ 109.0 (Cquat), 117.3 (CH, d, 2JCF = 22.8 Hz), 120.6 (CH), 121.1 (Cquat), 121.8 (CH), 123.3 (CH), 123.6 (CH), 125.2 (CH), 125.3 (CH), 125.5 (CH), 126.1 (CH), 128.1 (CH, d, 3JCF = 8.4 Hz), 133.8 (Cquat), 135.1 (Cquat), 136.0 (Cquat), 136.3 (Cquat), 140.0 (Cquat), 143.3 (Cquat, d, 4JCF = 2.7 Hz), 145.3 (Cquat), 161.9 (Cquat, d, 1JCF = 245.2 Hz); 19F NMR (565 MHz, THF-d8) δ −116.7; HRMS (ESI) m/z calcd for [C22H12FNS3]+: 405.0110; Found: 405.0111; Anal. calcd for C22H12FNS3 (405.5): C 65.16, H 2.98, N 3.45, S 23.72; Found: C 65.42, H 2.89, N 3.47, S 23.96.N-(4-Fluorophenyl)bis[1]benzothieno[3,2-b:2′,3′-e][1,4]thiazine (anti–anti7)26
According to the general procedure with bis(2-bromobenzo[b]thiophen-3-yl)sulfane (3) and 1,1′-bis(diphenyl-hexylphosphano)ferrocene (0.29 mg, 0.50 mmol) as a ligand and after chromatography (n-hexane/dichloromethane/triethylamine 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3) and recrystallization from n-hexane (450 mL) compound 7 (1.35 g, 67%) was obtained as orange crystals, Mp 177 °C (DSC measurement); 1H NMR (300 MHz, acetone-d6) δ 7.26–7.33 (m, 2 H), 7.35–7.39 (m, 2 H), 7.43–7.51 (m, 4 H), 7.72–7.77 (m, 2 H), 7.79–7.88 (m, 2 H); 19F NMR (282 MHz, acetone-d6) δ 65.73; 1H NMR (600 MHz, THF-d8) δ 7.18–7.22 (m, 2 H), 7.31–7.38 (m, 6 H), 7.58–7.62 (m, 2 H), 7.70–7.74 (m, 2 H); 13C{1H} NMR (151 MHz, THF-d8) δ 100.3 (Cquat), 118.3 (d, 2JCF = 23.3 Hz, CH), 120.2 (CH), 122.9 (CH), 124.2 (CH), 126.2 (CH), 132.2 (d, 3JCF = 9.1 Hz, CH), 134.6 (Cquat), 137.3 (Cquat), 140.0 (d, 4JCF = 3.2 Hz, Cquat), 142.3 (Cquat), 164.1 (d, 1JCF = 249.0 Hz, Cquat); 19F NMR (565 MHz, THF-d8) δ −111.7; HRMS (ESI) m/z calcd for [C22H12FNS3]+: 405.0110; Found: 405.0110; Anal. calcd for C22H12FNS3 (405.5): C 65.16, H 2.98, N 3.45, S 23.72; Found: C 65.31, H 2.97, N 3.43, S 23.53.
:1:3) and recrystallization from n-hexane (450 mL) compound 7 (1.35 g, 67%) was obtained as orange crystals, Mp 177 °C (DSC measurement); 1H NMR (300 MHz, acetone-d6) δ 7.26–7.33 (m, 2 H), 7.35–7.39 (m, 2 H), 7.43–7.51 (m, 4 H), 7.72–7.77 (m, 2 H), 7.79–7.88 (m, 2 H); 19F NMR (282 MHz, acetone-d6) δ 65.73; 1H NMR (600 MHz, THF-d8) δ 7.18–7.22 (m, 2 H), 7.31–7.38 (m, 6 H), 7.58–7.62 (m, 2 H), 7.70–7.74 (m, 2 H); 13C{1H} NMR (151 MHz, THF-d8) δ 100.3 (Cquat), 118.3 (d, 2JCF = 23.3 Hz, CH), 120.2 (CH), 122.9 (CH), 124.2 (CH), 126.2 (CH), 132.2 (d, 3JCF = 9.1 Hz, CH), 134.6 (Cquat), 137.3 (Cquat), 140.0 (d, 4JCF = 3.2 Hz, Cquat), 142.3 (Cquat), 164.1 (d, 1JCF = 249.0 Hz, Cquat); 19F NMR (565 MHz, THF-d8) δ −111.7; HRMS (ESI) m/z calcd for [C22H12FNS3]+: 405.0110; Found: 405.0110; Anal. calcd for C22H12FNS3 (405.5): C 65.16, H 2.98, N 3.45, S 23.72; Found: C 65.31, H 2.97, N 3.43, S 23.53.
General procedure – synthesis of bis[1]benzothieno[1,4]thiazine radical cations (BBTT˙+)
In a dry Schlenk tube with a magnetic stir bar the N-4-fluorophenyl-BBTT 5, 6, or 7 (0.12 g, 0.30 mmol) was dissolved in dry toluene (60 mL). Under vigorous stirring, a 1 M solution of antimony pentachloride in dichloromethane (0.30 mL, 0.30 mmol) was added dropwise to the solution at room temp. After 45 min the precipitate was filtered off and washed with dry toluene (5 × 10 mL) and dry n-pentane (2 × 10 mL). Deviations from this general procedure are individually highlighted.12-(4-Fluorophenyl)benzo[4,5]thieno[2,3-b]benzo[4,5]-thieno-[3,2-e][1,4]thiazinyl-12-ium hexachloroantimonate (syn–syn5˙+SbCl6−)
According to the general procedure with N-(4-fluorophenyl)-bis[1]benzothieno[2,3-b:3′,2′-e][1,4]thiazine (5) compound 5˙+SbCl6− (0.12 g, 80%) was obtained as a dark turquoise solid. EPR (THF): g = 2.0106 (A(14N) = 6.6 G); HRMS (ESI) m/z calcd for [C22H12FNS3]+: 405.0110; Found: 405.0106, ESI-MS m/z for [SbCl6]−: 334.7 [121Sb35Cl437Cl2/123Sb35Cl537Cl]−; IR: [cm−1] = 3109 (w), 3067 (w), 1585 (w), 1548 (w), 1506 (m), 1443 (m), 1423 (w), 1358 (m), 1315 (s), 1304 (s), 1265 (m), 1233 (m), 1152 (w), 1134 (w), 1111 (w), 1063 (w), 1043 (w), 1018 (m), 856 (w), 772 (s), 754 (s), 719 (s), 653 (w), 619 (w); Anal. calcd for C22H12Cl6NS3Sb (739.98): C 35.71, H 1.63, N 1.89, S 13.00; Found: C 35.99, H 1.58, N 1.88, S 13.29.
12-(4-Fluorophenyl)benzo[4,5]thieno[2,3-b]benzo[4,5]thieno[2,3-e][1,4]thiazinyl-12-ium hexachloroantimonate (syn–anti6˙+SbCl6−)
According to the general procedure with N-(4-fluorophenyl)bis[1]benzothieno[2,3-b:2′,3′-e][1,4]thiazine (6) compound 6˙+ SbCl6− (0.10 g, 71%) was obtained as an ink blue solid. EPR (THF): g = 2.0050 (A(14N) = 6.4 G); HRMS (ESI) m/z calcd for [C22H12FNS3]+: 405.0110; Found: 405.0114; ESI-MS m/z for [SbCl6]−: 334.7 [121Sb35Cl437Cl2/123Sb35Cl537Cl]−; IR: [cm−1] = 2972 (w), 2901 (w), 1601 (w), 1584 (w), 1520 (w), 1506 (m), 1445 (m), 1427 (w), 1368 (w), 1335 (s), 1315 (s), 1291 (w), 1256 (m), 1240 (m), 1236 (w), 1221 (w), 1157 (m), 1111 (w), 1059 (w), 1029 (w), 980 (w), 843 (m), 756 (s), 727 (m), 717 (m), 632 (w), 610 (m), 602 (s); Anal. calcd for C22H12Cl6NS3Sb (739.98): C 35.71, H 1.63, N 1.89, S 13.00; Found: C 35.76, H 1.67, N 1.81, S 12.79.
6-(4-Fluorophenyl)benzo[4,5]thieno[3,2-b]benzo[4,5]thieno[2,3-e][1,4]thiazinyl-6-ium hexachloroantimonate (anti–anti7˙+SbCl6−)
According to the general procedure with N-(4-fluorophenyl)-bis[1]benzothieno[3,2-b:2′,3′-e][1,4]thiazine (7) and after additional washing of the collected precipitate with toluene/ethyl acetate (25 mL (10:1)) in an ultrasound bath, filtration and washing with toluene/ethyl acetate (20 mL (10:1)), 2 mL (1:1)) compound 7˙+ SbCl6− (0.14 g, 97%) was obtained as a dark violet solid. EPR (THF): g = 2.0078 (A(14N) = 5.5 G); HRMS (ESI) m/z calcd for [C22H12FNS3]+: 405.0110; Found: 405.0115; ESI-MS m/z for [SbCl6]−: 334.7 [121Sb35Cl437Cl2/123Sb35Cl537Cl]−; IR: [cm−1] = 3065 (w), 2990 (w), 2974 (w), 2899 (w), 1738 (m), 1585 (w), 1525 (w), 1501 (m), 1481 (m), 1433 (m), 1340 (w), 1329 (s), 1304 (m), 1250 (m), 1233 (s), 1186 (w), 1167 (w), 1153 (m), 1132 (w), 1089 (w), 1065 (w), 1045 (w), 1016 (w), 849 (m), 755 (m), 750 (s), 718 (m), 620 (w); Anal. calcd for C22H12Cl6NS3Sb (739.98): C 35.71, H 1.63, N 1.89, S 13.00; Found: C 37.07, H 1.60, N 1.82, S 12.98.
General procedure – of bis[1]benzothieno[1,4]thiazine dications (BBTT2+)
In a dry Schlenk tube with a magnetic stir bar was dissolved N-4-fluorophenyl-BBTT 5, 6, or 7 (0.12 g, 0.30 mmol) in dry dichloromethane (9 mL). Under vigorous stirring a 1 m solution of antimony pentachloride in dichloromethane (0.60 mL, 0.60 mmol) was added dropwise to the solution at −78 °C (dry ice/acetone). After 45 min the mixture was allowed to warm to room temp. The precipitate was filtered off under Ar atmosphere and washed with dry dichloromethane (5 × 10 mL). Deviations from this general procedure are individually highlighted.12-(4-Fluorophenyl)benzo[4,5]thieno[2,3-b]benzo[4,5]thieno[3,2-e][1,4]thiazine-6,12-diium bishexachloroantimonate (syn–syn52+ (SbCl6-)2)
According to the general procedure with N-(4-fluorophenyl)-bis[1]benzothieno[2,3-b:3′,2′-e][1,4]thiazine (5) and after additional washing with dry toluene (2 × 10 mL) and dry diethyl ether (2 × 10 mL) compound 52+ (SbCl6−)2 (0.10 g, 48%) was obtained as a dark blue solid. 1H NMR (300 MHz, acetone-d6) δ 6.32–6.34 (m, 1 H), 7.27–7.30 (m, 1 H), 7.59–7.62 (m, 1 H), 7.68–7.73 (m, 1 H), 8.07–8.13 (m, 1 H), 8.15–8.20 (m, 1 H); HRMS (ESI) calcd for [C22H12FNS3]+: 405.0110; Found: 405.0101; ESI-MS m/z for [SbCl6]−: 330.8 [121Sb35Cl6]−, 332.8 [121Sb35Cl537Cl]−, 334.8 [121Sb35Cl437Cl2]−, 336.8 [121Sb35Cl337Cl3]−, 338.8 [121Sb35Cl237Cl4], 340.8 [121Sb35Cl37Cl5], 342.8 [121Sb37Cl6]; IR: [cm−1] = 3082 (w), 1578 (m), 1565 (w), 1501 (m), 1481 (w), 1451 (w), 1445 (w), 1431 (w), 1428 (w), 1416 (w), 1315 (w), 1285 (m), 1256 (w), 1212 (w), 1196 (s), 1184 (s), 1179 (s), 1155 (s), 1144 (m), 1125 (m), 1103 (w), 1076 (w), 1042 (s), 1036 (s), 1013 (w), 993 (w), 951 (w), 945 (w), 849 (w), 822 (w), 775 (s), 762 (s), 721 (s), 711 (m); Anal. calcd for C22H12Cl12FNS3Sb2 (1074.4): C 24.59, H 1.13, N 1.30, S 8.95; found: C 24.82, H 1.12, N 1.34, S 8.82.
12-(4-Fluorophenyl)benzo[4,5]thieno[2,3-b]benzo[4,5]thieno[2,3-e][1,4]thiazine-6,12-diium bishexachloroantimonate (syn–anti62+ (SbCl6-)2)
According to the general procedure with N-(4-fluorophenyl)-bis[1]benzothieno[2,3-b:2′,3′-e][1,4]thiazine (6) and after additional washing with dry tetrahydrofuran (2 × 2 mL) and dry pentane (2 × 10 mL) compound 62+ (SbCl6−)2 (0.13 g, 62%) was obtained as a blue solid. 1H NMR (300 MHz, acetone-d6) δ 6.42–6.49 (m, 1 H), 7.26–7.37 (m, 1 H), 7.49–7.56 (m, 1 H), 7.59–7.72 (m, 4 H), 7.98–8.04 (m, 1 H), 8.05–8.11 (m, 2 H), 8.11–8.16 (m, 1 H), 8.18–8.23 (m, 1 H); HRMS (ESI) calcd for [C22H12FNS3]+: 405.0110; Found: 405.0115; ESI-MS m/z for [SbCl6]−: 330.8 [121Sb35Cl6]−, 332.8 [121Sb35Cl537Cl]−, 334.8 [121Sb35Cl437Cl2]−, 336.8 [121Sb35Cl337Cl3]−, 338.8 [121Sb35Cl237Cl4]−; IR: [cm−1] = 3107 (w), 3052 (w), 1580 (m), 1573 (m), 1554 (w), 1508 (w), 1499 (m), 1485 (w), 1464 (m), 1442 (w), 1404 (w), 1346 (m), 1337 (m), 1320 (w), 1312 (w), 1275 (s), 1254 (m), 1190 (s), 1162 (m), 1150 (m), 1125 (m), 1076 (w), 1043 (m), 1012 (w), 988 (w), 845 (w), 754 (s), 756 (s), 729 (w), 716 (m), 621 (w), 604 (w); Anal. calcd for C22H12Cl12FNS3Sb2 (1074.4): C 24.59, H 1.13, N 1.30, S 8.95; Found: C 24.88, H 1.28, N 1.27, S 9.06.
6-(4-Fluorophenyl)benzo[4,5]thieno[3,2-b]benzo[4,5]thieno[2,3-e][1,4]thiazine-6,12-diium bishexachloroantimonate (anti–anti72+ (SbCl6-)2)
According to the general procedure with N-(4-fluorophenyl)bis[1]benzothieno[3,2-b:2′,3′-e][1,4]thiazine (7) after additional washing with dry pentane (2 × 10 mL) compound 72+ (SbCl6−)2 (0.18 g, 82%) was obtained as a dark violet solid. 1H NMR (300 MHz, acetone-d6) δ 7.52–7.58 (m, 2 H), 7.66–7.72 (m, 4 H), 8.00–8.05 (m, 2 H), 8.05–8.13 (m, 2 H), 8.18–8.22 (m, 2 H); HRMS (ESI) calcd for [C22H12FNS3]+: 405.0105; found: 405.0113; HRMS (ESI) calcd for [SbCl6]−: 330.7175; Found: 330.7131; IR: [cm−1] = 3095 (w), 1578 (m), 1497 (m), 1454 (w), 1437 (w), 1420 (w), 1406 (w), 1325 (m), 1297 (w), 1269 (m), 1246 (w), 1211 (s), 1167 (w), 1155 (m), 1130 (m), 1092 (w), 1063 (w), 1024 (w), 993 (w), 943 (w), 866 (w), 845 (m), 756 (s), 712 (m), 670 (w), 764 (w), 635 (w), 627 (w), 620 (w); Anal. calcd for C22H12Cl12FNS3Sb2 (1074.4): C 24.59, H 1.13, N 1.30, S 8.95; Found: C 28.38, H 1.25, N 1.54, S 10.47.
Conclusions
The electronic properties of oxidized species of all three anellative bis[1]benzothieno[1,4]thiazine (BBTT) isomers were extensively characterized by experimental and computational methods. All three regioisomeric N-p-fluorophenyl-BBTTs (syn–syn, syn–anti and anti–anti) were successfully transformed into the corresponding radical cation and dication salts by using antimony pentachloride as an oxidant on a preparative scale. By EPR spectroscopy the radical character was unambiguously proven and the predominant localization of the unpaired spin on the central 1,4-thiazine moiety was supported by the resulting hyperfine coupling with the nitrogen nucleus. Moreover, experimentally determined hyperfine coupling constants and computationally determined spin density distributions and Wiberg bond orders underline that a stronger delocalization arises from anti-alignment of the benzo[b]-thiophene units. The diamagnetic singlet character as well as geometric structure of the dications was assigned by NMR spectroscopy, especially supported by high-field shifted signals in diamagnetic anisotropy cones of syn-aligned wings due to planarization of the BBTT core.Spectroelectrochemical measurements by in situ generation of the oxidized species allowed characterization of the electronic absorption bands. Finally, the experimentally observed absorption bands could be reproduced TDDFT calculations and assigned to the underlying molecular orbital transitions. The calculated electronic structure of the radical cations and dications underlines for all transitions a high polarizability of BBTT upon photonic excitation, as revealed by pronounced charge transfer from the benzo[b]thiophene moieties or the N-aryl unit to the central 1,4-thiazine core, stating a strong acceptor character of oxidized BBTT. Further studies on the synthesis of functionalized BBTTs focusing on enhancing their neutral antiaromatic character as well as addressing their electroactive and electro-optical applications are currently underway.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors cordially thank the Fonds der Chemischen Industrie. The “Centre for Information and Media Technology” (ZIM) at the University of Düsseldorf (Germany) provided computational support and the infrastructure.Notes and references
- P. Li, C. Jia and X. Guo, Molecule-Based Transistors: From Macroscale to Single Molecule, Chem. Rec., 2020, 20, 1–17 CrossRef
.
- D. Xiang, X. Wang, C. Jia, T. Lee and X. Guo, Molecular-Scale Electronics: From Concept to Function, Chem. Rev., 2016, 116, 4318–4440 CrossRef CAS PubMed
- Y. Shirota and H. Kageyama, Charge Carrier Transporting Molecular Materials and Their Applications in Devices, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS PubMed
-
Functional Organic Materials – Synthesis, Strategies, and Applications, ed. T. J. J. Müller and U. H. F. Bunz, Wiley-VCH, Weinheim, 2007, vol. 1 Search PubMed
- G. P. Neupane, W. Ma, T. Yildirim, Y. Tang, L. Zhang and Y. Lu, 2D organic semiconductors, the future of green nanotechnology, Nano Mater. Sci., 2019, 1, 246–259 CrossRef
- G. Gryn'ova, K.-H. Lin and C. Corminboeuf, Read between the Molecules: Computational Insights into Organic Semiconductors, J. Am. Chem. Soc., 2018, 140, 16370–16386 CrossRef PubMed
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, The role of chemical design in the performance of organic semiconductors, Nat. Rev. Chem., 2020, 4, 66–77 CrossRef CAS
- G. Schweicher, G. Garbay, R. Jouclas, F. Vibert, F. Devaux and Y. H. Geerts, Molecular Semiconductors for Logic Operations: Dead-End or Bright Future?, Adv. Mater., 2020, 32, 1905909 CrossRef CAS PubMed
- Y. Zhang, P. Gao, X. Guo, H. Chen, R. Zhang, Y. Du, B. Wang and H. Yang, Hypercrosslinked phenothiazine-based polymers as high redox potential organic cathode materials for lithium-ion batteries, RSC Adv., 2020, 10, 16732–16736 RSC
- C. Zhang, Z. Niu, S. Peng, Y. Ding, L. Zhang, X. Guo, Y. Zhao and G. Yu, Phenothiazine-Based Organic Catholyte for High-Capacity and Long-Life Aqueous Redox Flow Batteries, Adv. Mater., 2019, 31, 1901052 CrossRef PubMed
- K. A. Narayana, M. D. Casselman, C. F. Elliott, S. Ergun, S. R. Parkin, C. Risko and S. A. Odom, N-Substituted Phenothiazine Derivatives: How the Stability of the Neutral and Radical Cation Forms Affects Overcharge Performance in Lithium-Ion Batteries, ChemPhysChem, 2015, 16, 1179–1189 CrossRef CAS PubMed
- F. Khan, E. Urbonas, D. Volyniuk, J. V. Grazulevicius, S. M. Mobin and R. Misra, White hyperelectrofluorescence from solution-processable OLEDs based on phenothiazine substituted tetraphenylethylene derivatives, J. Mater. Chem. C, 2020, 8, 13375–13388 RSC
- Y. Park, B. Kim, C. Lee, A. Hyun, S. Jang, J.-H. Lee, Y.-S. Gal, T. H. Kim, K.-S. Kim and J. Park, Highly Efficient New Hole Injection Materials for OLEDs Based on Dimeric Phenothiazine and Phenoxazine Derivatives, J. Phys. Chem. C, 2011, 115, 4843–4850 CrossRef CAS
- S. Xiang, Z. Huang, S. Sun, X. Lv, L. Fan, S. Ye, H. Chen, R. Guo and L. Wang, Highly efficient non-doped OLEDs using aggregation-induced delayed fluorescence materials based on 10-phenyl-10H-phenothiazine 5,5-dioxide derivatives, J. Mater. Chem. C, 2018, 6, 11436–11443 RSC
- L. Mao, Y. Wu, J. Jiang, X. Guo, P. Heng, L. Wang and J. Zhang, Rational Design of Phenothiazine-Based Organic Dyes for Dye-Sensitized Solar Cells: The Influence of π-Spacers and Intermolecular Aggregation on Their Photovoltaic Performances, J. Phys. Chem. C, 2020, 124, 9233–9242 CrossRef CAS
- Z.-S. Huang, H. Meier and D. Cao, Phenothiazine-based dyes for efficient dye-sensitized solar cells, J. Mater. Chem. C, 2016, 4, 2404–2426 RSC
- B. Moll, T. Tichelkamp, S. Wegner, B. Francis, T. J. J. Müller and C. Janiak, Near-infrared (NIR) surface-enhanced Raman spectroscopy (SERS) study of novel functional phenothiazines for potential use in dye sensitized solar cells (DSSC), RSC Adv., 2019, 9, 37365–37375 RSC
- T. Meyer and T. J. J. Müller, Consecutive Three-Component Synthesis of Donor-Substituted Merocyanines by a One-Pot Suzuki–Knoevenagel Condensation Sequence, Org. Mater., 2020, 2, 064–070 CrossRef CAS
- Y.-L. Weng, Y.-C. Li, C.-P. Chen and Y. J. Chang, Effect of intermolecular interaction with phenothiazine core on inverted organic photovoltaics by using different acceptor moiety, Dyes Pigm., 2017, 146, 374–385 CrossRef CAS
- M. Hauck, M. Stolte, J. Schönhaber, H.-G. Kuball and T. J. J. Müller, Synthesis, Electronic, and Electro-Optical Properties of Emissive Solvatochromic Phenothiazinyl Merocyanine Dyes, Chem. – Eur. J., 2011, 17, 9984–9998 CrossRef PubMed
-
R. A. Aitken and K. M. Aitken, 1,4-Thiazines and their Benzo Derivatives, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, pp. 607–675 Search PubMed
- C. F. Elliott, K. E. Fraser, S. A. Odom and C. Risko, Steric Manipulation as a Mechanism for Tuning the Reduction and Oxidation Potentials of Phenothiazines, J. Phys. Chem. A, 2021, 125, 272–278 CrossRef CAS PubMed
- L. Mayer, L. May and T. J. J. Müller, The interplay of conformations and electronic properties in N-aryl phenothiazines, Org. Chem. Front., 2020, 7, 1206–1217 RSC
- U. Tokiko, I. Masanori and K. Kozo, Crystal Structure and Related Properties of Phenothiazine Cation Radical-Hexachloroantimonate. Monoclinic(I) Form, Bull. Chem. Soc. Jpn., 1983, 56, 577–582 CrossRef
- I. Yôichi, The Cation Radical Salts of Phenothiazine and Related Compounds, Bull. Chem. Soc. Jpn., 1971, 44, 663–667 CrossRef
- A. P. W. Schneeweis, S. T. Hauer, D. A. Lopez, B. von Dressler, G. J. Reiss and T. J. J. Müller, Game of Isomers: Bifurcation in the Catalytic Formation of Bis[1]benzothieno[1,4]thiazines with Conformation-Dependent Electronic Properties, J. Org. Chem., 2019, 84, 5582–5595 CrossRef CAS PubMed
- L. May and T. J. J. Müller, Electron-Rich Phenothiazine Congeners and Beyond: Synthesis and Electronic Properties of Isomeric Dithieno[1,4]thiazines, Chem. – Eur. J., 2020, 26, 12111–12118 CrossRef CAS PubMed
- A. P. W. Schneeweis, S. T. Hauer, G. J. Reiss and T. J. J. Müller, Bis[1]benzothieno[1,4]thiazines: Planarity, Enhanced Redox Activity and Luminescence by Thieno-Expansion of Phenothiazine, Chem. – Eur. J., 2019, 25, 3582–3590 CrossRef CAS PubMed
- H. R. V. Berens, K. Mohammad, G. J. Reiss and T. J. J. Müller, 3,9-Disubstituted Bis[1]benzothieno[3,2- b;2′,3′- e][1,4]thiazines with Low Oxidation Potentials and Enhanced Emission, J. Org. Chem., 2021, 86, 8000–8014 CrossRef CAS PubMed
- L. May and T. J. J. Müller, Dithieno[1,4]thiazines and Bis[1]benzothieno[1,4]thiazines—Organometallic Synthesis and Functionalization of Electron Density Enriched Congeners of Phenothiazine, Molecules, 2020, 25, 2180 CrossRef CAS PubMed
- K. Deuchert and S. Hünig, Multistage Organic Redox Systems—A General Structural Principle, Angew. Chem., Int. Ed., 1978, 17, 875–886 CrossRef
- J. F. Hartwig, Palladium-Catalyzed Amination of Aryl Halides: Mechanism and Rational Catalyst Design, Synlett, 1997, 329–340 CrossRef CAS
- A. S. Guram, R. A. Rennels and S. L. Buchwald, A Simple Catalytic Method for the Conversion of Aryl Bromides to Arylamines, Angew. Chem., Int. Ed., 1995, 34, 1348–1350 CrossRef CAS
- R. Dorel, C. P. Grugel and A. M. Haydl, The Buchwald–Hartwig Amination After 25 Years, Angew. Chem., Int. Ed., 2019, 58, 17118–17129 CrossRef CAS PubMed
- Y. Ren and T. Baumgartner, Ladder-Type π-Conjugated 4-Hetero-1,4-dihydrophosphinines: A Structure–Property Study, Chem. – Asian J., 2010, 5, 1918–1929 CrossRef CAS PubMed
- T. Yamamoto, S. Ogawa and R. Sato, Selective synthesis, structure and oxidation properties of isomeric 1,4-dithiins fused to two benzo[b]thiophenes, Tetrahedron Lett., 2004, 45, 7943–7946 CrossRef CAS
- R. P. Dickinson and B. Iddon, Synthesis and reactions of 3-benzo[b]thienyl-lithium, J. Chem. Soc. C, 1968, 2733–2737 RSC
- C. Dostert, C. Wanstrath, W. Frank and T. J. J. Müller, 4H-Dithieno[2,3-b:3′,2′-e][1,4]thiazines – synthesis and electronic properties of a novel class of electron rich redox systems, Chem. Commun., 2012, 48, 7271–7273 RSC
- C. Dostert, D. Czajkowski and T. J. J. Müller, 2,6-Difunctionalization of N-Substituted Dithienothiazines via Dilithiation, Synlett, 2014, 371–374 CAS
- J. Nau, A. P. W. Schneeweis and T. J. J. Müller, Dithienothiazine dimers, trimers and polymers – novel electron-rich donors with red-shifted luminescence, Mater. Chem. Front., 2020, 4, 621–630 RSC
- A. P. W. Schneeweis, A. Neidlinger, G. J. Reiss, W. Frank, K. Heinze and T. J. J. Müller, Radical cation and dication of a 4H-dithieno[2,3-b:3′,2′-e][1,4]-thiazine, Org. Chem. Front., 2017, 4, 839–846 RSC
- A. D. Becke, A new mixing of Hartree–Fock and local density–functional theories, J. Phys. Chem., 1993, 98, 1372–1377 CrossRef CAS
- A. D. Becke, Density–functional thermochemistry. III. The role of exact exchange, J. Phys. Chem., 1993, 98, 5648–5652 CrossRef CAS
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self–consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Phys. Chem., 1980, 72, 650–654 CrossRef CAS
- A. D. McLean and G. S. Chandler, Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18, J. Phys. Chem., 1980, 72, 5639–5648 CrossRef CAS
- J. L. Pascual-ahuir, E. Silla and I. Tuñon, GEPOL: An improved description of molecular surfaces. III. A new algorithm for the computation of a solvent-excluding surface, J. Comput. Chem., 1994, 15, 1127–1138 CrossRef CAS
- S. Miertuš, E. Scrocco and J. Tomasi, Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects, Chem. Phys., 1981, 55, 117–129 CrossRef
- S. Miertuš and J. Tomasi, Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes, Chem. Phys., 1982, 65, 239–245 CrossRef
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed
- F. L. Rupérez, J. C. Conesa and J. Soria, Electron spin resonance study of the influence of the nitrogen substituent on the conformation and spin density distribution of phenothiazine derivatives, J. Chem. Soc., Perkin Trans. 2, 1986, 391–395 RSC
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
- J. McDowell, The crystal and molecular structure of phenothiazine, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 5–10 CrossRef
- J. D. Bell, J. F. Blount, O. V. Briscoe and H. C. Freeman, The crystal structure of phenothiazine, Chem. Commun., 1968, 24, 1656–1657 RSC
- R. Bauernschmitt and R. Ahlrichs, Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules, J. Phys. Chem., 1998, 109, 8218–8224 CrossRef CAS
- J.-L. Calais, Density-functional theory of atoms and molecules, Int. J. Quantum Chem., 1993, 47, 101–101 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1qo00867f |
| This journal is © the Partner Organisations 2021 |