
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homolysis/mesolysis of alkoxyamines activated by chemical oxidation and photochemical-triggered radical reactions at room temperature†
Gérard
Audran
*a,
Mitchell T.
Blyth
 b,
Michelle L.
Coote
b,
Georg
Gescheidt
c,
Micael
Hardy
a,
Jeffrey
Havot
a,
Maxence
Holzritter
a,
Samuel
Jacoutot
a,
Jean-Patrick
Joly
*a,
Sylvain R. A.
Marque
*a,
Tataye Moussounda Moussounda
Koumba
a,
Dmytro
Neshchadin
c and
Enzo
Vaiedelich
a
b,
Michelle L.
Coote
b,
Georg
Gescheidt
c,
Micael
Hardy
a,
Jeffrey
Havot
a,
Maxence
Holzritter
a,
Samuel
Jacoutot
a,
Jean-Patrick
Joly
*a,
Sylvain R. A.
Marque
*a,
Tataye Moussounda Moussounda
Koumba
a,
Dmytro
Neshchadin
c and
Enzo
Vaiedelich
a
aAix Marseille Univ, CNRS, ICR, UMR 7273, Case 551, Avenue Escadrille Normandie-Niemen, 13397 Marseille Cedex 20, France. E-mail: sylvain.marque@univ-amu.fr; g.audran@univ-amU.fr; jp.joly@live.fr
bResearch School of Chemistry, Australian National University, Canberra, ACT 2601, Australia
cInstitute of Physical and Theoretical Chemistry, TU Graz, Stremayrgasse 9/Z2, A-8010 Graz, Austria
First published on 14th October 2021
Abstract
Alkoxyamines, which are connected with a phenol moiety by a (substituted) methylene bridge undergo homolytic cleavage upon chemical oxidation or a photo-induced hydrogen transfer. This selectively triggered reaction yields a nitroxide radical. In the presence of an excess of lead dioxide as the oxidant in tert-butylbenzene as solvent, spontaneous, instantaneous and almost quantitative generations of nitroxides from various alkoxyamines are observed at room temperature, which support activation energies for the cleavage lower than 100 kJ mol−1. The rate and the amount of released nitroxide depend on the amount of “catalyst” and the structure of alkoxyamines.
Introduction
In the last decade, the concept of smart alkoxyamines has been developed.1 These are highly stable alkoxyamines with half-lives of several hundred years, which can be selectively transformed into highly labile species with half-lives of seconds or minutes through chemical,2–5 photochemical6–9 or biochemical10,11 events. Such alkoxyamines have interesting properties for potential applications in industry, e.g., safe storage of initiators for radical polymerizations,5,12 or in biology, e.g., drugs for cancer or parasites.10,11,13–18While the focus of these studies has been on triggering alkoxyamine homolysis, to form a nitroxide and carbon-centred radical, recently it has been shown that upon oxidation, through electrochemical19–23 or photoredox24,25 methods, alkoxyamines can spontaneously undergo mesolytic cleavage instead. Depending on the leaving group and other species present, this process can yield nitroxides and carbocations, or alternatively oxoamoniums and carbon-centred radicals. In some cases, oxidation simply generates reactive intermediates capable of undergoing SN2 reactions with nucleophiles. These processes have been developed into successful alkylation methods in small molecule synthesis,21,24 and likely have broader applications for the on-demand generation of carbon-centred radicals and cations. Moreover, it is thought that this mesolytic cleavage pathway may even be operative upon photochemical excitation of certain photoactive alkoxyamines.26
To date, oxidative alkoxyamine cleavage has been achieved using either electrochemical19–23 or photoredox24 methods, but the use of simple chemical oxidation under mild conditions has not yet been reported. Indeed, while PbO2 has been shown to generate phenoxyl radicals from phenol-based anti-oxidants through proton-coupled electron transfer (PCET),27 preliminary results with 3c show only very low conversion into nitroxide 3˙ using PbO2 as oxidant. Here, we report on the preparation phenol-based alkoxyamines 1a–5a,3e (Chart 1) and the benzyl protected homologues 1b–5b,3d (Chart 1). We have investigated their reaction pathways in t-BuPh as the solvent. At room temperature 1a–5a reacted with PbO2 affording almost quantitative conversions to the corresponding nitroxides. Kinetic investigations by EPR, product analysis, NMR/CIDNP, and DFT calculations of 1a–5a are used to unveil the pathways leading both to the decay of alkoxyamines and to the generation of nitroxides.
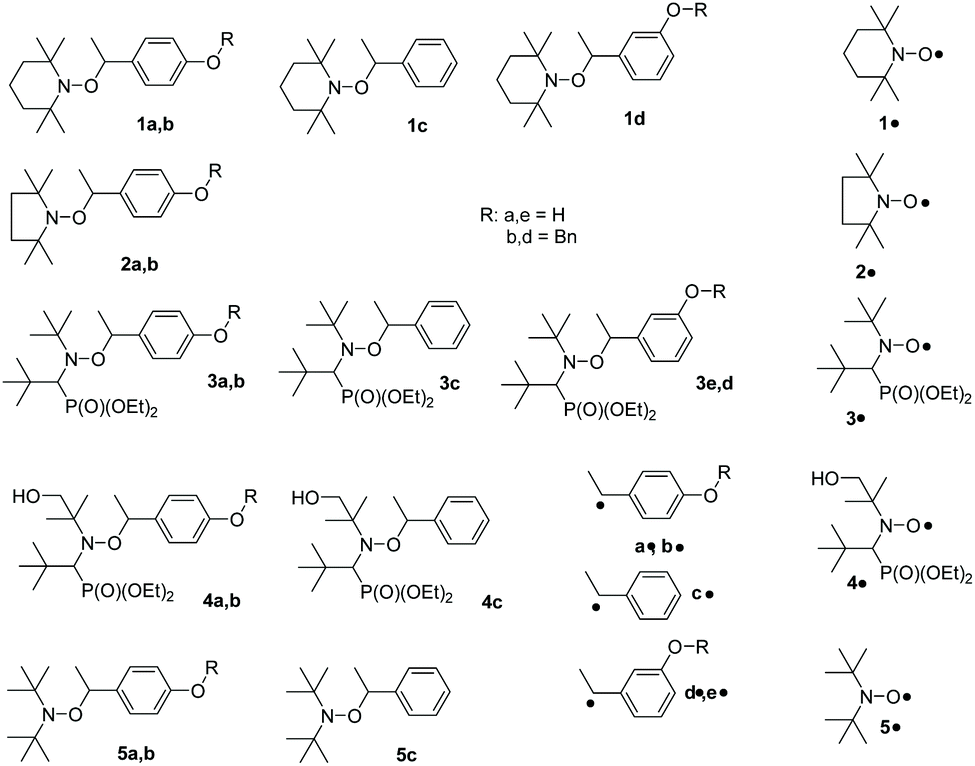 | ||
| Chart 1 Alkoxyamines and nitroxides investigated. | ||
Results
Synthesis
para-Vinyl phenol is protected by benzylation,28‡ to afford 6, which is coupled with nitroxides 1˙–5˙ using the Mn(salen) method to yield 1b–5b.29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) § Then, 1b–5b are deprotected using H2 and the catalyst Pd/C28 to yield 1a–5a (Scheme 1).
§ Then, 1b–5b are deprotected using H2 and the catalyst Pd/C28 to yield 1a–5a (Scheme 1).
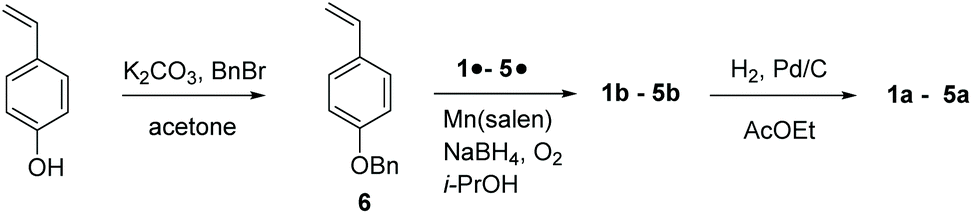 | ||
| Scheme 1 Preparation of 1a–5a. | ||
The same procedures as displayed in Scheme 1 are applied for the preparation of 1d, 3d, and 3e starting from meta-vinylphenol in 25%, 55%, and 74% yields, respectively.
Thermal homolysis
Effects of various substituents attached to the nitroxyl moiety on the homolysis rate constant kd of the C–ON bond in alkoxyamines (Scheme 2) have been thoroughly investigated30–33 and it was shown that steric, polar and stabilization effects are strongly entangled.34,35 Noteworthy, for the thermal C–ON bond homolysis, differences of Ea of ca. 5–7 kJ mol−1 between 1a and 3a, of ca. 5–7 kJ mol−1 between 3a and 4a, of ca. 2–4 kJ mol−1 between 3a and 5a are in the range of differences observed for analogue alkoxyamines. It means that the C–ON bond homolysis of 1a, 3a–5a is ruled by the polarity and bulkiness of the nitroxyl fragment, and stabilization of the released nitroxide as previously reported.34 Therefore, the thermal-triggered homolysis of 1a–5a occurs via the established mechanism34 displayed in Scheme 2. Importantly, at high temperatures, in the presence of O2, the generation of alkyl radicals, peroxyl radicals, alkoxyl radicals and nitroxides do not alter the kinetics. Hence, the mechanism of homolysis via a self-catalytic event in which the phenoxyl moiety would play the role of radical scavenger, analogous to the Denisov cycle,36 is highly unlikely.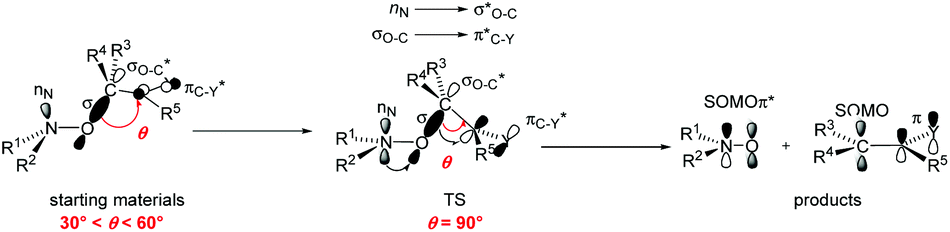 | ||
| Scheme 2 Geometrical parameters and orbital interactions expected in starting materials, TS, and products during the homolysis event. | ||
Effects of para-aryl substituent for 1-37 and 3-based38 alkoxyamines have been thoroughly investigated and linear free energy relationships (LFER) were observed for 1-based alkoxyamines37 with the Hammett constant σp (ref. 39) and for 3-based alkoxyamines38 with localized polar effect for para-substituted phenyl groups σL,4-X-C6H4.40,41 Thus, Ea of 134.3 kJ mol−1¶ and 126.2 kJ mol−1|| are estimated for 1a and 3a, respectively, in nice agreement with the experimental values reported in Table 1. Moreover, very similar values of Ea are expected for 1a and 1b** as well as for 3a and 3b†† agreeing with the values reported in Table 1. The trends in Ea observed for 1a–5a are also observed for 1b–5b. Hence, the C–ON bond homolysis for 1a,b–5a,b occur through TS exhibiting the same features:34 flattening at the N- and C-atoms of the C–ON moiety, rotation around the N–O bond to reach the dihedral angle 〈nitrogen lone pair NOC〉 at 0° ‡‡ and rotation around the C-aryl bond to reach the dihedral angle 〈OCaryl〉 at 90°. These geometric requirements favour the donating interactions nN → σ*O–C of the nitrogen lone pair nN into the C–O antibonding orbital σ*O–C, which in turn favours the donating interaction σO–C → π*aryl of the O–C bonding orbital σO–C into the antibonding orbital π* of the aryl moiety (Scheme 2).
| Alkoxyamines | T (°C) | k d (10−3 s−1)a |
E
ab (kJ mol−1) |
k′d (10−4 s−1)c |
t
1/2d (days) |
|---|---|---|---|---|---|
| a Values determined by EPR and using eqn (2). b Assuming an averaged frequency factor A = 2.4 × 1014 s−1 (see ref. 34) and values of T and k given in second and third columns, respectively. c Values estimated at T = 120 °C assuming an averaged frequency factor A = 2.4 × 1014 s−1 (see ref. 34) and values of Ea given in fourth columns. d Given at 25 °C and using values of kd estimated as described in footnote c. e At T = 139 °C, kd = 3.8 10−3 s−1 (eqn (2)), Ea = 132.7 kJ mol−1 (see footnote b), and at T = 120 °C, kd = 0.9 10−3 s−1 (eqn (2)), Ea = 131.1 kJ mol−1 (see footnote b). f Averaged values of Ea reported in footnote e. g First fraction in chromatography. h Relative configurations were not determined. i Second fraction in chromatography. j See ref. 34. | |||||
| 1a | 128 | 1.82 | 131.4 | 8.2 | 3613 (≈10 years) |
| 2a | —e | —e | 131.9f | 7.0 | 4416 (≈12 years) |
| 3a | 104 | 0.66g,h/1.4h,i | 126.8g,h/124.4h,i | 34.2g,h/70.4h,i | 547g,h/219h,i |
| 4a | 80 | 0.57g,h/0.59h,i | 119.2g,h/119.0h,i | 354.4g,h/365.8h,i | 26g,h/24h,i |
| 5a | 85 | 0.37 | 122.1 | 142.5 | 84 |
| 1b | 138 | 3.80 | 132.2 | 0.65 | 5000 (≈14 years) |
| 2b | 130 | 1.68 | 132.3 | 6.2 | 5183 (≈14 years) |
| 3b | 122 | 4.64g,h/8.94h,i | 126.4g,h/124.3h,i | 38.2g,h/73.8h,i | 475g,h/219h,i |
| 4b | 100 | 2.79g,h/3.37h,i | 121.0g,h/120.4h,i | 202.9g,h/242.7h,i | 55g,h/40h,i |
| 5b | 90 | 0.64 | 122.2 | 140.6 | 88 |
| 1c | — | — | 133.0 | 5.0 | 6882 (≈19 years) |
| 3c | — | — | 124.5 | 68.0 | 218 |
| 4c | — | — | 121.3 | 181.0 | 61 |
| 5c | — | — | 121.8 | 155.0 | 75 |
| 1d | 120 | 0.39 | 133.9 | 3.9 | 9631 (≈26 years) |
| 3d | 100 | 0.59g,h/0.68h,i | 125.4g,h/125.3h,i | 51.5g,h/53.8h,i | 318g,h/301h,i |
| 3e | 100g,h/90h,i | 0.76g,h/0.27h,i | 125.0g,h/124.87h,i | 59.1g,h/62.9h,i | 265g,h/245h,i |
Chemical oxidation
Taking into account the half-life times of 1a–5a at 25 °C (t1/2 = 24 days for 4a is the shortest, Table 1), these alkoxyamines are considered as stable during the 60 seconds of experimental time, i.e., thermal homolysis of 0.1 mM of 4a affords 0.7 nM of nitroxide, below the threshold of detection of EPR at ca. 10 nM. PbO2 is currently used to oxidize phenolic anti-oxidant into their corresponding phenoxyl radical for their detection by EPR.42 Adding PbO2 to 0.1 mM solution of 1a–5a at room temperature does not generate the EPR signal of the corresponding phenoxyl radical but an instantaneous raise of EPR signals corresponding to nitroxides 1˙–5˙. The intensity of the EPR signal of 1˙–5˙ is PbO2-concentration dependent affording bell-shaped curves (Fig. 1). On the left-hand side of the top of bell curve, concentration in PbO2 is too low for complete oxidation§§ whereas on the right-hand side, concentration in PbO2 is too large that it reacts with nitroxides. Nitroxides 4˙ and 5˙ are not displayed in Fig. 1 due to their too high reactivity with PbO2 impeding any significant growth of the EPR signal whatever the concentrations in PbO2. PbO2-oxidation of 3e affords only 17% conversion in 3˙ as expected when less stabilized alkyl species is released from the meta-regioisomer of alkoxyamine.¶¶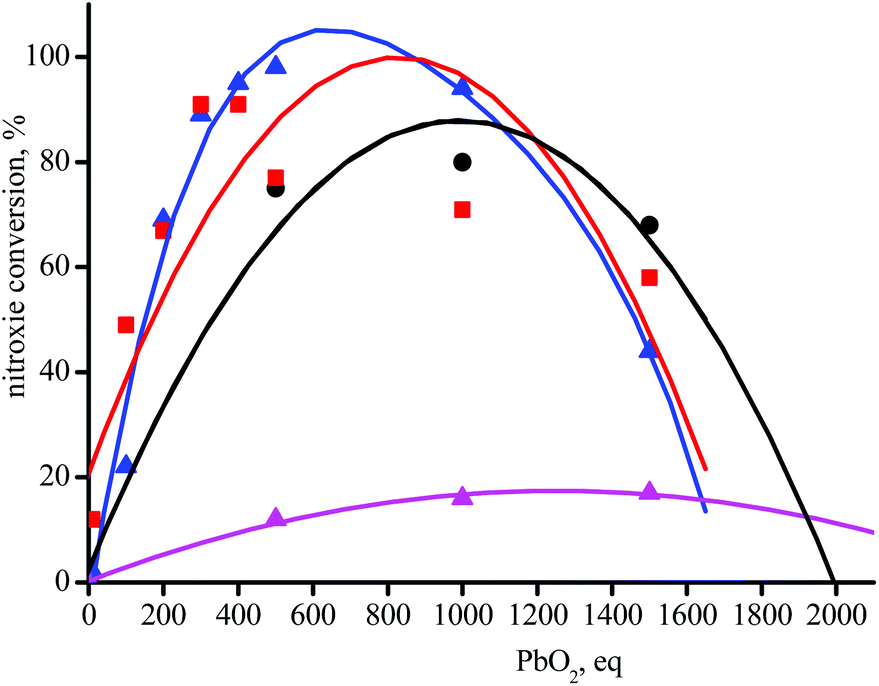 | ||
| Fig. 1 Plots of nitroxide conversion vs. equivalents in PbO2 with t-BuPh as solvent for 1˙ (red squares), 2˙ (black circles), and 3˙ (blue and magenta triangles) from 1a, 2a, 3a and 3e, respectively. Lines (second order polynomes) are for an easy reading of the trend. | ||
Chemical activation via H-abstraction reaction
The use of PbO2 as an oxidant generally also involves the transfer of protons. Since the reduction (of PbO2) is accompanied by proton transfer, it formally corresponds to a H-atom transfer reaction. Accordingly, we have performed H-abstraction reactions induced by the photolysis of di-tertbutyl peroxide. The t-BuO˙ radical (Scheme 3a) is able to primarily abstract the phenolic H-atom to generate a phenoxyl radical (Scheme 3d).43 The latter can undergo fragmentation into a nitroxide and a methyne quinone like-product (Scheme 3e). Indeed, as soon as light is turned on, in a solution containing 3a and di-tertbutyl peroxide EPR signal of 3˙ raised affording around 80% conversion after a very long time (larger than 10 hours) of irradiation and for less than 5% (v/v) of di-tertbutyl peroxide (Table 2).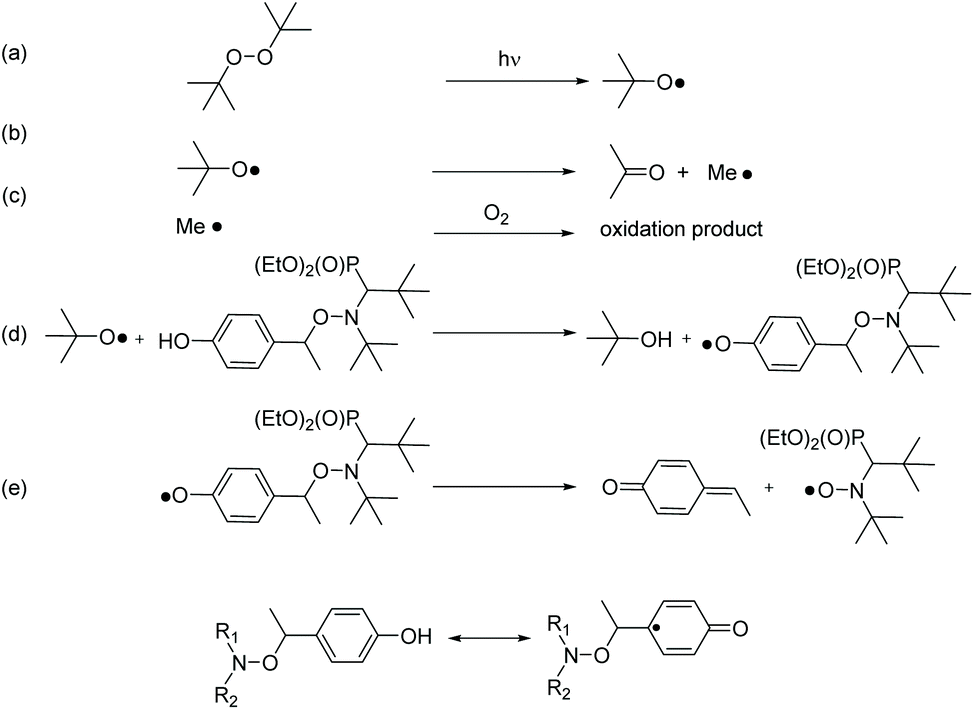 | ||
| Scheme 3 Elementary reactions involved during the generation of 3˙ under light-irradiation of a solution of 3a with (t-BuO)2. | ||
| Alkoxyamines | tBu2O2 (%, v/v) | Equivalentsa | Conversionb (%) |
t
maxc (min) |
|---|---|---|---|---|
| a Density: d = 0.796. b Highest conversion observed. c Time for which the maximum conversion reported in the 4th column is observed. d When experiment was stopped. e When [3a] = 1 mM, no significant differences are observed both in conversion and in kinetics. | ||||
| 1a | 0 | 0 | 24 | 300 |
| 2a | 0 | 0 | 66 | 660 |
| 3a | 0 | 0 | 49d | 720 |
| 3e | 0 | 0 | 43d | 420 |
| 2a | 0.1 | 66 | 63 | 720 |
| 3a | 0.1 | 66 | 68 | 1260 |
| 1a | 0.5 | 330 | 77 | 480 |
| 2a | 0.5 | 330 | 67 | 540 |
| 3a | 0.5 | 330 | 74 | 1020 |
| 3e | 0.5 | 330 | 47 | 300 |
| 2a | 1 | 660 | 62 | 240 |
| 3a | 1 | 660 | 60 | 85 |
| 3a | 5e | 3300 | 49 | 75 |
| 3a | 10 | 6600 | 39 | 85 |
Disappointingly, a large amount of 40% conversion 3a into 3˙ is observed after 12 hours of UV-irradiation in the absence of peroxide. Thus, 3a as well as 1a and 2a are light sensitive. Nevertheless, the kinetics of generation of 3˙ are dramatically faster in the presence of more than 1% of (tBuO)2 than in its absence (Fig. 2), that is, during the first 3 hours of irradiation, kinetics for the generation of 3˙ are the same for 0.1% in (tBuO)2 and without whereas, in this time, amounts of 3˙ reached a plateau and completely decayed for amounts in (tBuO)2 larger than 1% (Fig. 2). Notably, the plateau is reached faster for lower amounts of 3˙, i.e., 30% released in 1000 s in 10% (tBuO)2versus 70% released in 10 hours for 0.5% in (tBuO)2. This striking dependence on time for decomposition of 3a into nitroxide 3˙ is due to several competitive reactions involving 3˙, t-BuO˙, and side-products. Di-tertbutyl peroxide generates tert-butoxyl radicals under UV-irradiation (Scheme 3d). The latter reacts either by H-abstraction on the phenol moiety of 3a to generate a transient phenoxyl radical (Scheme 3b) or by β-fragmentation to afford acetone and methyl radicals (Scheme 3b). These methyl radicals either react with 3˙ by the coupling reaction leading to the formation of stable alkoxyamines, and, hence, to the decay of 3˙ or they react with O2 to afford oxidation products or some other radicals able to react with 3˙. Consequently, several of these reactions depend dramatically on concentration of starting materials (tBu2O2 and 3a), of product (3˙ and possibly acetone and methine quinone like-compound), and also of intermediates such as t-BuO˙ and Me˙ radicals. Therefore, these differences in reactivity, account for the striking differences in conversion and in time to reach maximal conversion (Table 2).
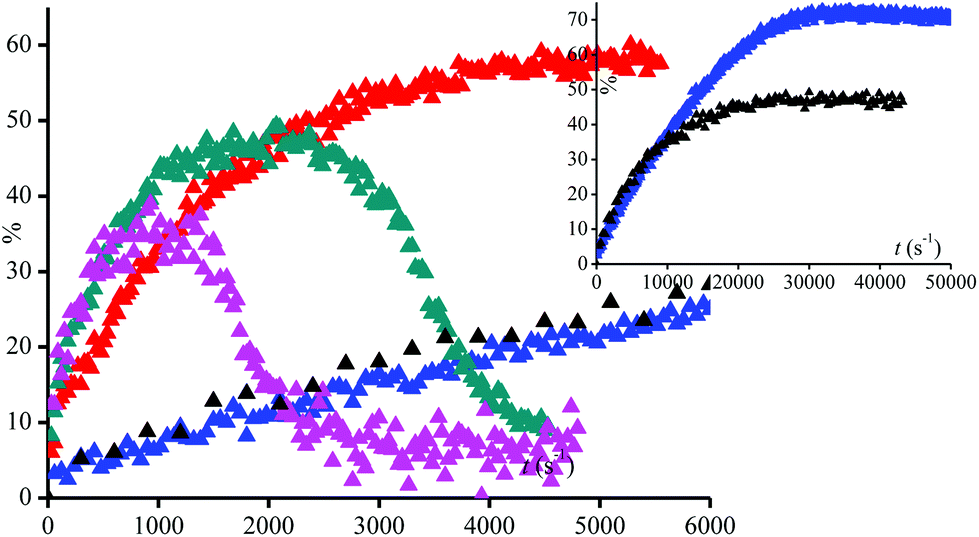 | ||
| Fig. 2 UV-irradiation of 3a (0.1 mM) in t-BuPh at 25 °C with various amounts (% v/v) of (t-BuO)2 (▲) none, (▲) 0.5%, (▲) 1%, (▲) 5%, and (▲) 10%. Inset: Longer kinetics for conditions (▲) no (t-BuO)2, and (▲) 0.5% (t-BuO)2. | ||
HPLC–ESI analysis
For HPLC experiments, chemical oxidation of 3a,e (10−2 M) was performed in a 1:1 (v:v) MeOH/water mixture and afforded the same EPR results as in t-BuPh as solvent when 500 eq. PbO2 were added. Authentic compounds displayed in Chart 2 are either prepared as described in literature (A and B) or purchased (C and D).||||***††† Compounds C–E are expected to be generated in the case of a mesolysis event releasing benzylic type radical and oxonium (route a in Scheme 4).
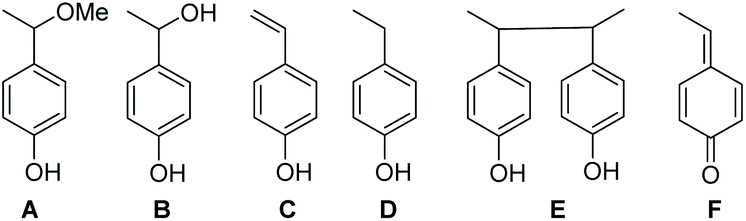 | ||
| Chart 2 Potential product formed from 3a (10−2 M) upon the one-electron oxidation process by PbO2 (500 eq.) in MeOH as solvent, at room temperature. | ||
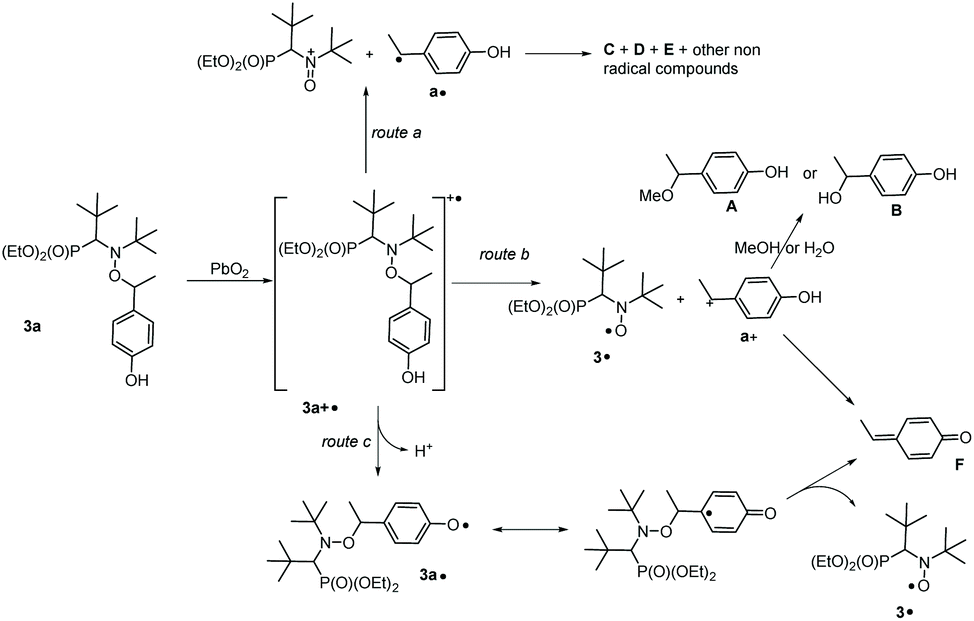 | ||
| Scheme 4 Different routes of decay of 3a+˙ describing HPLC observations reported in Table 3 and ESI.† | ||
HPLC analysis of the decomposition of 3e (Table 3) displays only peaks corresponding to 3e (only 30% conversion) and to some traces of 3˙ (less than 10% in agreement with EPR results) meaning that the pathways of decomposition affording 3˙ are minor events (see section DFT calculations section and Fig. 7).
:v 1:1) solvent mixture
| t | m e/zb | m o/zb |
n
ac |
|
n
oc |
|
|---|---|---|---|---|---|---|
a Retention time in min.
b
m
e/z and mo/z are for the expected and observed masses by ESI-MS, respectively, in MeOH/H2O (1:1 v:v), given at ±0.1 u.
c In 10−8 moles at ±0.01. na, 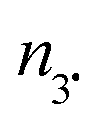 , and no number of moles for alkoxyamines, 3˙, and other molecules, respectively.
d n.a.: not applicable.
e As displayed in Fig. 3, 20% of impurities are detected.
f Mixture of 2 diastereoisomers.
g Only one diastereoisomers was available.
h Amount of A. , and no number of moles for alkoxyamines, 3˙, and other molecules, respectively.
d n.a.: not applicable.
e As displayed in Fig. 3, 20% of impurities are detected.
f Mixture of 2 diastereoisomers.
g Only one diastereoisomers was available.
h Amount of A.
|
||||||
| A | 11.2 | 153.2 | 121.2 | n.a.d | n.a.d | 1.0 |
| C | 12.5 | 121.2 | 121.2 | n.a.d | n.a.d | 1.0 |
| 3˙ | 14.0 | 296.4 | 296.4 | n.a.d | 0.80e | n.a.d |
| 3a | 15.6 | 416.2 | 416.2 | 0.56 | n.a.d | n.a.d |
| 16.2 | 0.44 | |||||
| 3e | 16.5 | 416.2 | 416.2 | 1.0 | n.a.d | n.a.d |
| 3a + PbO2 | 11.1 | n.a.d | 121.2 | n.a.d | n.a.d | 0.51h |
| 14.0 | 296.2 | n.a.d | 0.80 | n.a.d | ||
| 15.6 | 416.2 | 0.05 | n.a.d | n.a.d | ||
| 16.2 | 416.2 | 0.11 | n.a.d | n.a.d | ||
| 3e + PbO2 | 14.0 | n.a.d | 296.2 | n.a.d | 0.07 | n.a.d |
| 16.5 | 416.2 | 0.91 | n.a.d | n.a.d | ||
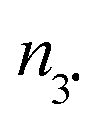
As displayed in Fig. 3, only 3a, 3˙ and A are observed for the decomposition of 3a in the presence of PbO2. No traces of other chemicals displayed in Chart 2 are detected. Mass balances (Table 3) for 3a (84% conversion) and 3˙ (80% conversion) agree with the EPR results. On the other hand, a clear discrepancy is observed in the mass balance for 3˙ (8 nmol against 5.1 nmol for A) meaning that most of the radical cation decomposes into nitroxide and benzylic-type carbocation a+ (route b, Scheme 4) which, is scavenged by MeOH to generate A provided the elimination reaction to afford C does not compete. The discrepancy observed is likely due to the occurrence of route c (Scheme 4) generating a phenoxyl radical 3a˙ which decomposes into nitroxide 3˙ and methine quinone-type compound F or to the loss of H+ from a+ to afford F. The latter cannot be detected under our experimental conditions.
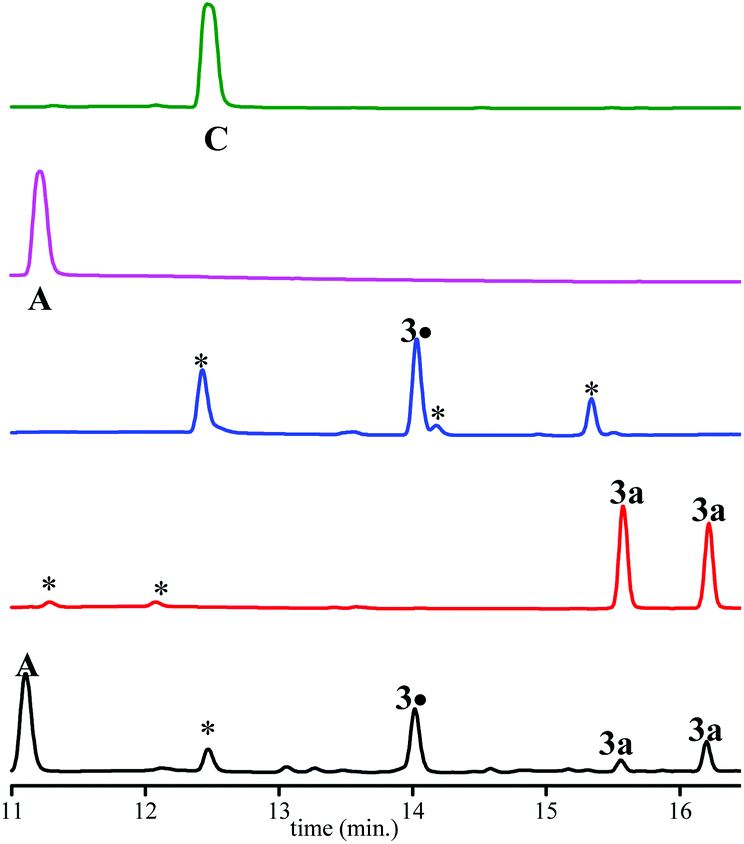 | ||
| Fig. 3 HPLC signal of authentic compounds and of products for the chemical oxidation degradation of 3a. Concentrations are at 10−2 M in MeOH/water (1:1 v:v). From top to bottom, HPLC traces of pure C (magenta), pure A (green); “pure” 3˙ (blue), pure 3a (red), and decomposition (black) of 3a in the presence of PbO2 (500 eq.). | ||
Importantly, only 3a, 3˙, and C are detected by ESI-MS (Table 3). Indeed, for A and B, ESI detects only C, meaning that MeOH- or water-elimination occurs in the course of the ESI experiment. The HPLC mass balance shows that the decomposition of the radical cation of alkoxyamine 3a+˙ does not occur through route a (Scheme 4).
The same experiments have been performed with 3a in THF affording very similar results except that a+ is scavenged by traces of water in THF to yield B (see ESI table and figure†). When experiments are performed with 3e, similar results to those reported in Table 3 are observed (see ESI table†). Thus, all observations for 3a and 3e in MeOH/water mixture apply also to experiments performed in THF as solvent. It was assumed that the same reactivity would be expected for other phenol-based alkoxyamines not investigated by HPLC.
pKa measurements
Due to solubility issues of 3a, THF:D2O (v/v 1/1) mixture was used for 1H NMR (Fig. 4). pKa values of each diastereoisomer of 3a (Fig. 5) are given as 12.3 (±0.02) and 12.8 (±0.05) using eqn (1).44 pKa Values of paravinylphenol are measured as 12.0 when recorded in the same conditions (see ESI†). | (1) |
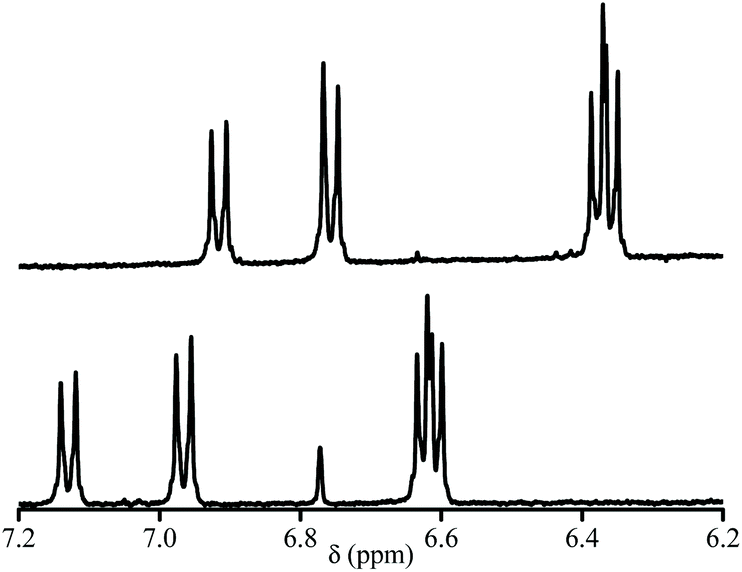 | ||
| Fig. 4 400 MHz 1H NMR signal of the aromatic zone at pH = 13. (Top) and at pH = 1.3 (bottom) of 3a in THF. | ||
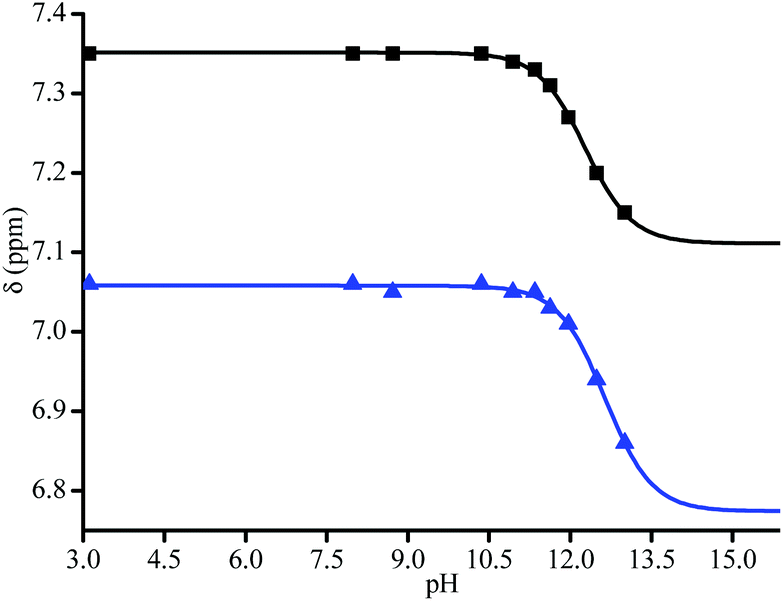 | ||
| Fig. 5 pH curves for diastereoisomers of 3a (▲ and ■) by 2 peaks in the aromatic zone (see Fig. 4). Data were fitted with eqn (1). | ||
CIDNP experiments
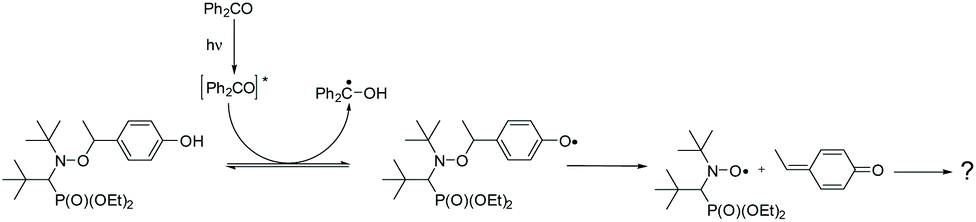
The experiments described above indicate that the formal abstraction of the phenolic H atom induces the formation of the nitroxyl radical. To shed light on the mechanism of the underlying reaction sequence at a μs time scale, we performed photo-induced TR-CIDNP (Time-Resolved Chemically Induced Dynamic Nuclear Polarization) experiments.45,46 As an example, we present the results obtained with 3a (see ESI† for more derivatives). We have used benzophenone (BP) as the photo-activated hydrogen acceptor. It reacts in an analogous way as the t-BuO˙ radical (see above Scheme 3d) and can be conveniently activated at 366 nm (NdYAG laser, used in our CIDNP system). We have utilized this approach to successfully study various H-transfer reactions.47,48In the CIDNP experiment, free radical pairs are generated thermally or photochemically inside the strong magnetic field of the NMR magnet. At the time scale of the CIDNP experiment, the primary radicals undergo reactions, which are spin selective. This leads to a non-Boltzmann population of magnetic energy levels and manifests itself in NMR signals with “unusual” intensities, i.e. enhanced absorption or emission (enhancement factors up to 105) when the corresponding products are formed via radical pairs. The intensities of these polarized signals (of the products) provide information about the isotropic hyperfine coupling constants in the short-lived radical intermediates. To produce primary radical pairs, we used the photo-induced (NdYAG laser, 355 nm, 8 ns) reaction of the triplet-excited state of BP (3*BP) with alkoxyamine in acetonitrile-d3/D2O (Scheme 5). Primarily, 3*BP should abstract the phenolic hydrogen49,50 and the corresponding follow-up products are observed in CIDNP spectrum with the chemical shifts representing the products and the polarized line intensities indicating the intermediate radicals (Scheme 5). As an example, the 1H NMR and CIDNP spectra obtained from 3a together with assignments of selected lines are shown in Fig. 7. The most prominent polarizations in the CIDNP spectrum (7.7 ppm and 8–8.2 ppm) can be attributed to the reversible hydrogen transfer from BPH˙ to 3a˙ re-generating BP and 3a (Scheme 5). This is in concert with an intermediate phenoxyl radical where the predominant spin population is located on the phenyl ring of 3a in agreement with B3LYP/TZVP calculations.51,52 Clearly, an electron transfer producing a basically nitrogen centred radical cation from 3a to 3*BP, would have produced an entirely different polarization pattern than that observed in our experiment (see ESI† for the comparison with additional radicals). The remaining polarizations at 2.55 ppm (doublet) and 7.7 ppm (doublet) can be assigned to a methyne quinone like-product F (Chart 2 and Fig. 6) formed via the homolytic cleavage of the C–O bond in 3a˙. This intermediate is too short lived to be detected in the steady-state NMR taken after the CIDNP experiment. The fragmentation of 3a˙ yielding nitroxyl radical 3˙ is confirmed by the detection of the corresponding EPR spectrum taken from the NMR tube after accomplishing the NMR experiment.
 | ||
| Scheme 5 generation of 3˙ during the irradiation (NdYAG laser, 355 nm, 8 ns) of benzophenone (3 mM) in the presence of 3a (1 mM) at 25 °C in acetonitrile-d3/D2O. | ||
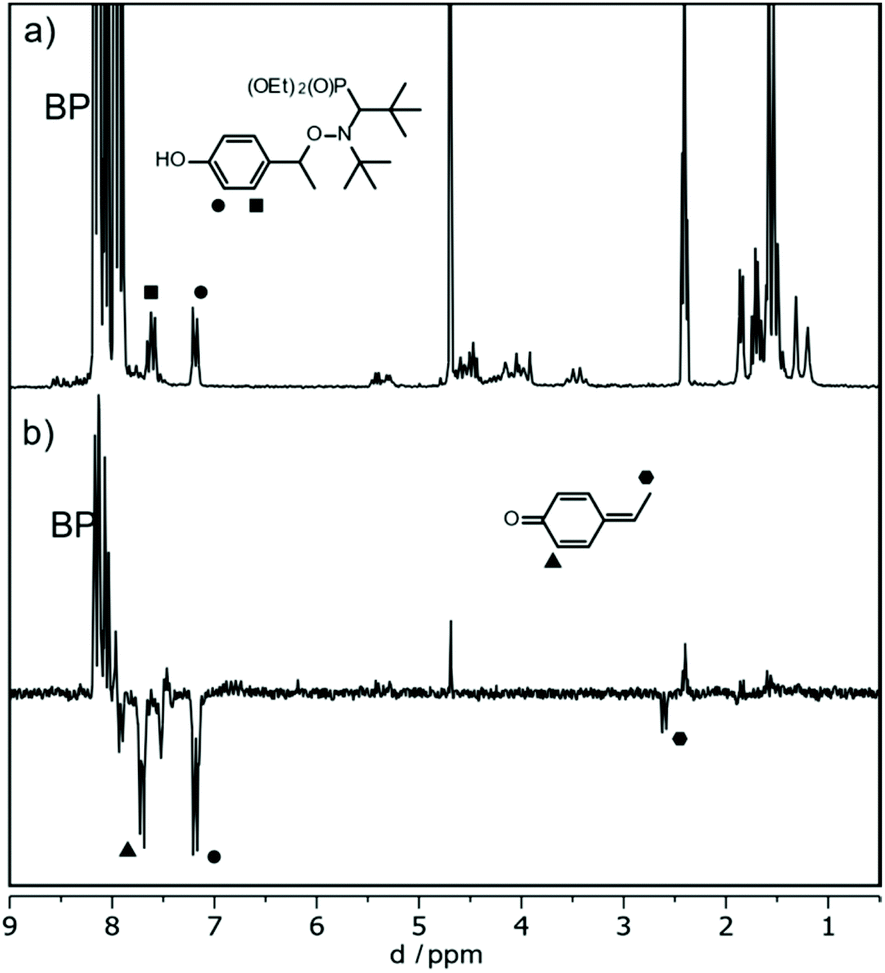 | ||
| Fig. 6 1H NMR (a) and (b) CIDNP spectrum of 3a and BP in Acetonitrile-d3/D2O recorded 2 μs after the laser flash (355 nm, 8 ns). | ||
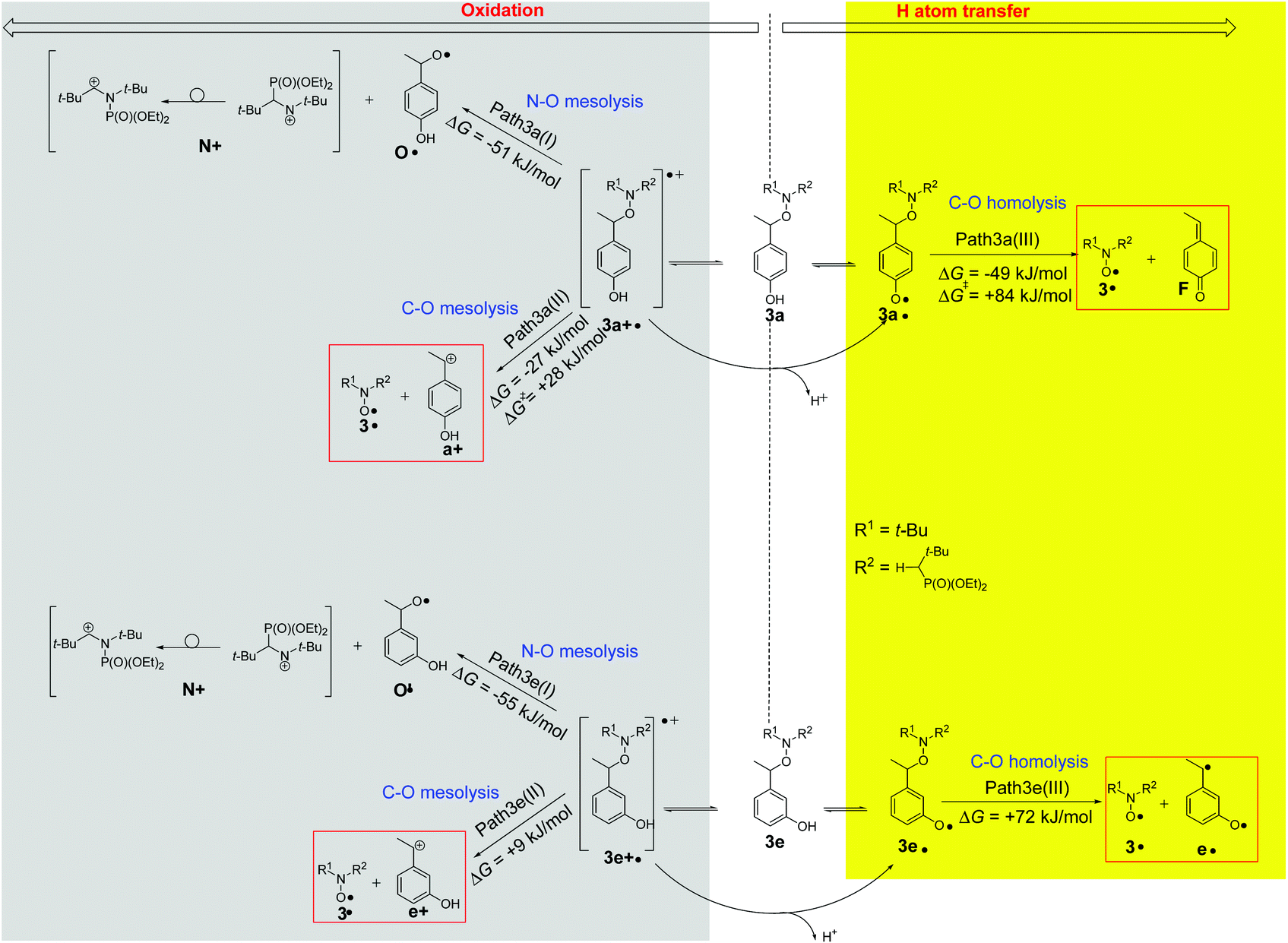 | ||
| Fig. 7 Selected (preferred) pathways of decomposition radicals based on 3a and 3e according to M06-2X/6-31+G(d,p) calculations. Reactions leading to the release of nitroxyl radicals are marked with red boxes. | ||
For derivative 3b, in which the phenolic hydrogen is replaced by the benzyl group, the fragmentation reaction cannot be established by CIDNP (see ESI†); moreover, weak EPR signal is detected. This clearly indicates that the abstraction of the phenolic hydrogen is crucial for the formation of nitroxyl radical 3˙.
DFT calculations
We have performed calculations (M06-2X/6-31+G(d,p) level of theory)53 for radical cations and phenoxyl radicals based on parent 3a and 3e. In Fig. 7, we have displayed the most exergonic (or the lowest endergonic) pathways for the decomposition of 3a and 3e under oxidative conditions (see the ESI† for pathways with highly endergonic ΔG values). For 3a there are three pathways starting from the same radical cation 3a+˙ as intermediate which are preferable. One pathway (Path 3a(I)) involves the cleavage of the N–O bond and the formation of an aminium cation N+ and alkoxyl radical O˙ (ΔG = −51 kJ mol−1). The second radical-cation-based process produces the desired nitroxide together with benzyl cation a+ (Path 3a(II), ΔG = −27 kJ mol−1). The third one, the reaction via neutral phenoxyl radical 3a˙ provides nitroxide 3˙ and 4-ethylidenecyclohexa-2,5-dien-1-one F (Path 3a(III), ΔG = −49 kJ mol−1). The calculations predict two likely reactions for 3e and both are based on radical cation precursors 3e+˙: one being analogous to Path 3a(I) and yielding aminium cation N+ and alkoxyl radical O′˙ (Path 3e(I), ΔG = −55 kJ mol−1); the second resembling Path 3a(II) giving the desired nitroxide 3˙ and benzyl cation e+ (Path 3e(II), ΔG = +9 kJ mol−1). In contrast to 3a, paths 3e(III) is disregarded due to its too high value of ΔG, i.e., 72 kJ mol−1. As delineated in Fig. 7, for both the radical cations 3a+˙ and 3e+˙ issued from the oxidation of 3a and 3e, respectively, there is a connection between oxidation and HAT routes as the radical cation affords phenoxyl radical by loss of proton. Taking into account EPR, CIDNP, and HPLC results, paths 3a(I) and 3e(III) are disregarded.Discussion
Chemical activation via H-abstraction reaction
The generation of 3a˙ through H-abstraction by t-BuO˙ radicals and its β-fragmentation into 3˙ and F, which are entangled with (several) light-initiated reactions, are nicely confirmed by CIDNP experiments by using benzophenone as radical initiator. Moreover, DFT calculations support the occurrence of Path3a(III) which exhibit clear negative value of ΔG with ΔG‡ lower than 90 kJ mol−1 as expected for the occurrence of spontaneous and instantaneous reactions. As the same kinetic trends are observed (Table 2) for 1a, and 2a, it is assumed that the observations for 3a apply to these also.Activation by electron transfer event
Previous studies have examined the behaviour of amoniumyl radical cations generated upon oxidation of alkoxyamines by electrochemical19–23 or photoredox24 methods. Depending on the leaving group and co-reactants present, the oxidation can either be (a) reversible, or lead to mesolytic cleavage of either the (b) CO–N bond to form aminyl radicals, or cleavage of the C–ON to form either (c) an oxoamonium and carbon-centred radical or (d) a nitroxide and carbocation. Alternatively, cleavage can occur via (e) SN2-type reactions with nucleophiles, which may include trace water, the solvent or electrolyte. Importantly, in the case of 1c, only route (d) has been observed experimentally in anhydrous solvents,19,23 and one would expect analogous behaviour for 3c, given that the leaving group is identical.Whatever the amounts of PbO2 added to a solution of 3c in t-BuPh, less than 4% of signal is detected by EPR. When the same experiments are performed with 3e, less than 20% of 3˙ are detected by EPR (Fig. 1) in agreement with HPLC measurements (Table 3), and calculations meaning that path(II) (path3e(II) in Fig. 7) is a minor event in the generation of 3˙ by chemical oxidation process when the released benzylic-cation is not stabilized. This also holds for other alkoxyamines investigated in this article (vide infra).
DFT calculations on the homolysis routes (Fig. 7) show only two relevant processes of fragmentations (other are displayed as ESI†): (i) mesolysis of the C–ON bond in 3a+˙ affording nitroxide 3˙ and benzyl-type carbocation a+ (ΔGb = −27 kJ mol−1, and ΔG‡ = 28 kJ mol−1, Path3a(II) in Fig. 7); and (ii) via proton release from 3a+˙ to generate phenoxyl radical 3a˙‡‡‡ which collapses into nitroxide 3˙ and methine-quinone type compound F (ΔGc = −49 kJ mol−1, route c in Scheme 4 and Path3a(III) in Fig. 7), i.e. Proton-Coupled Electron Transfer (PCET)27,54 followed by a β-fragmentation event. The mesolysis of the C–ON affording oxonium and stabilized styryl-type radical a˙ (see ESI†), is disregarded due to both strongly disfavoured ΔG value (ΔGa = 75 kJ mol−1, see ESI†) and an almost quantitative generation of 3˙.
Indeed, DFT calculated negative ΔG values and ΔG‡ values lower than 100 kJ mol−1 confirm the spontaneous and instantaneous generation of 3˙. Moreover, the lower activation ΔG‡ for path3a(II) (ΔG‡b = 28 kJ mol−1) than for path3a(III) (ΔG‡c = 84 kJ mol−1) in Fig. 7 denotes a favoured path3a(II) over path3a(III) as highlighted by the mass balance in HPLC (Table 3).
The quantitative generation of 1˙ and 2˙ is observed upon addition of PbO2 to a solution of 1a and 2a, meaning that comments done for 3a hold.
Addition of PbO2 to 4a and 5a does not generate significant amounts of the corresponding nitroxides 4˙ and 5˙ due to their low stability in the experimental conditions.¶¶ Nevertheless, it is assumed that the same chemistry as described for 3a holds for 4a and 5a.
Impact on the fragmentation pathway
It has to be mentioned that, whatever the experimental conditions, the phenoxyl radical and radical cation are not detected by EPR meaning a very fast decomposition into the nitroxide. Thus, the generation of nitroxide cannot occur as described in Scheme 2 in which flattening at N- and C-atoms is required. Recent DFT calculations showed that C–ON bond homolysis is controlled by two geometrical requirements (Scheme 2): (i) flattening at N- and C-atoms, and (ii) dihedral angle θ 〈OCaryl〉 close to 90° and dihedral angle θ1 〈nNNOC〉 close to 0°. It was also shown that alkoxyamines exhibiting angle θ close to 90° and θ1 close to 0° are stable at room temperature meaning that the flattening at C- and N-atoms are the key events which must occur during the raise of temperature when homolysis is performed. Thus, two pathways of decay are offered to the radical cation: route (a) for which the radical cation collapses into nitroxide and benzylic-type cation and route (b) which correspond to the release of phenoxyl radical. Interestingly, both routes exhibit strong similarities for stereoelectronic interactions at TS (Scheme 6), i.e., mesomeric forms H of phenoxyl radical G and J of radical cation I display a significant spin population at the position ipso. Such forms H/J favour the rotation around the C–aryl bond to shift θ angle from 60°§§§ to 90° (see TS section in ESI†) aiming to favour hyperconjugative interaction SOMOπ → σ*C–O between the SOMO of π-type and the antibonding orbital σ*C–O of the C–O bond. Such an interaction at TS favours undoubtedly the β-fragmentation event of the O–C bond affording two highly stabilized molecules: a nitroxide and a conjugated keto diene despite aromaticity is lost for the fragmentation of phenoxyl radical, and a nitroxide and a benzylic-type cation for the mesolysis of the radical cation. In such a process involving early TS, i.e., reactant-like TS, neither the unfavoured N-flattening event nor the N–O bond rotation occur leading to a room temperature spontaneously and instantaneously thermodynamically-driven fragmentation.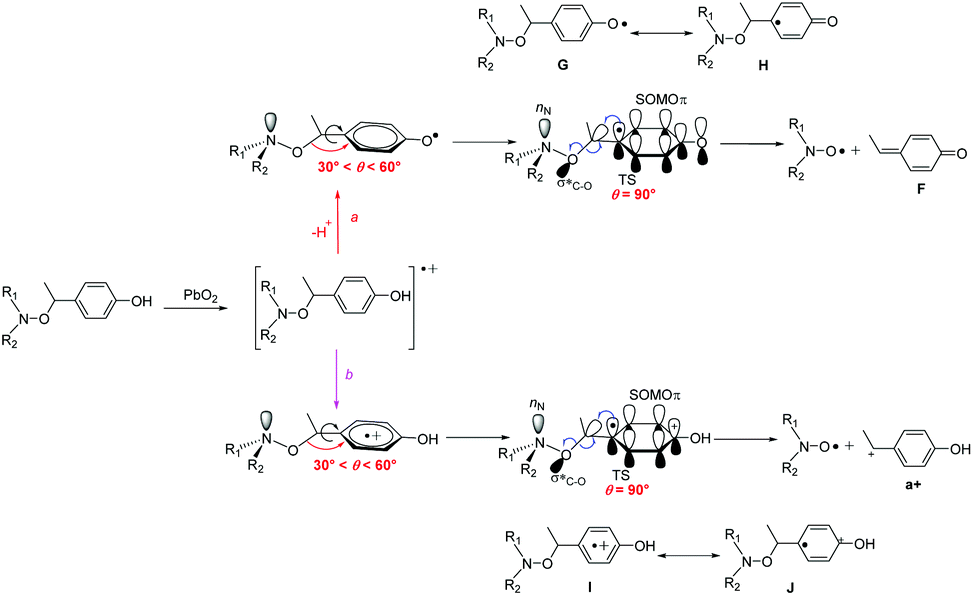 | ||
| Scheme 6 Generation of nitroxide from alkoxyamine through a PCET process coupled with the β-fragmentation of phenoxyl radical (route a) and the mesolysis of radical cation (route b), and geometrical parameters and orbital interaction at their respective TS. | ||
Our experimental observations and the computational results indicate that the very efficient production of the nitroxide (e.g.3˙) is based either on the primary generation of a phenoxyl radical via hydrogen abstraction or PCET (which cannot be distinguished at the time scale of our experiments) or the mesolysis of the corresponding radical cation. The main prerequisite for an efficient reaction, however, is that an alkyl fragment carrying a p-phenolic moiety as exemplified with 1a–5a. Here the release of the nitroxide can be either triggered thermally by PbO2 or by photo-induced reactions. Thus, the caged nitroxides introduced in this work have the potential of being applied in biology and polymer chemistry (e.g., as selectively activable labels). It is likely that the scope of (photo)oxidants can be enhanced.
Experimental
Synthetic procedures
Solvents and reactants for the preparation of alkoxyamines were used as received. Routine reaction monitoring was performed using silica gel 60 F254 TLC plates; spots were visualized upon exposure to UV light and a phosphomolybdic acid solution in EtOH, followed by heating. Purifications were performed on Reveleris® X2 Flash System BUCHI Switzerland. Cartouches flash Reveleris® et GraceResolv™: silica 40 μm. 1H, 13C, and 31P NMR spectra were recorded in CDCl3 on a 300 or 400 MHz spectrometer. Chemical shifts (δ) in ppm were reported using residual nondeuterated solvents as internal references for 1H and 13C NMR spectra, and 85% H3PO4 for 31P-NMR spectra. High-resolution mass spectra (HRMS) were performed on a SYNAPT G2 HDMS (Waters) spectrometer equipped with a pneumatically assisted atmospheric pressure ionization source (API). Positive mode electrospray ionization was used on samples: electrospray voltage (ISV): 2800 V; opening voltage (OR): 20 V; nebulizer gas pressure (nitrogen): 800 L h−1. Low resolution mass spectra were recorded on the ion trap AB SCIEX 3200 QTRAP equipped with an electrospray source. The parent ion [M + H]+ is quoted.:4); 1H NMR (300 MHz, CDCl3) δ 7.49–7.30 (m, 5H), 7.25 (d, J = 8.7 Hz, 2H), 6.93 (d, J = 8.7 Hz, 2H), 5.06 (s, 2H), 4.75 (q, J = 6.7 Hz, 1H), 1.47 (d, J = 6.7 Hz, 3H), 1.45–0.99 (m, 15H), 0.68 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 157.7 (C), 138.3 (C), 137.2 (C), 128.5 (2 × CH), 127.9 (CH), 127.8 (2 × CH), 127.5 (2 × CH), 114.3 (2 × CH), 82.4 (CH), 70.0 (CH2), 59.7 (2 × C), 40.3 (2 × CH2), 34.4 (2 × CH3), 23.2 (CH3), 20.2 (2 × CH3), 17.2 (CH2). HRMS m/z (ESI) Calcd for C24H34NO2+ [M + H]+ 368.2584 Found: 368.2581.
:4); 1H NMR (300 MHz, CDCl3) δ 7.51–7.35 (m, 5H), 7.31 (d, J = 8.7 Hz, 2H), 6.98 (d, J = 8.7 Hz, 2H), 5.09 (s, 2H), 4.67 (q, J = 6.6 Hz, 1H), 1.62–145 (m, 4H), 1.50 (d, J = 6.6 Hz, 3H), 1.31 (s, 3H), 1.21 (s, 3H), 1.08 (s, 3H), 0.73 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 158.0 (C), 137.5 (C), 137.2 (C), 128.5 (2 × CH), 128.2 (2 × CH), 127.8 (CH), 127.4 (2 × CH), 114.2 (2 × CH), 81.6 (CH), 70.0 (CH2), 64.2 (C), 63.2 (C), 36.1 (CH2), 35.4 (CH2), 31.2 (CH3), 31.1 (CH3), 24.3 (CH3), 22.8 (CH3), 22.1 (CH3). HRMS m/z (ESI) Calcd for C23H32NO2+ [M + H]+ 354.2428 Found: 354.2422.
:1); 1H NMR (400 MHz, CDCl3) δ 7.46–7.31 (m, 5H), 7.23 (d, J = 8.6 Hz, 2H), 6.91 (d, J = 8.7 Hz, 2H), 5.05 (s, 2H), 4.96 (q, J = 6.7 Hz, 1H), 4.38–4.31 (m, 1H), 4.23–4.04 (m, 2H), 4.04–3.89 (m, 1H), 3.35 (d, JH–P = 26.2 Hz, 1H), 1.59 (d, J = 6.7 Hz, 3H), 1.35–1.30 (m, 6H), 1.25 (s, 9H), 0.85 (s, 9H). 31P NMR (121 MHz, CDCl3) δ 25.82. 13C NMR (75 MHz, Acetone) δ 152.1 (C), 132.0 (C), 131.3(C), 122.8 (4 × CH), 122.1 (CH), 121.7 (2 × CH), 108.5 (2 × CH), 78.4 (CH), 64.2 (CH2), 64.2 (d, JC–P = 138.9 Hz, CH), 55.8 (d, JC–P = 6.5 Hz, CH2), 55.4 (C), 53.1 (d, JC–P = 8.2 Hz, CH2), 29.8 (d, JC–P = 5.6 Hz, C), 24.5 (d, JC–P = 6.1 Hz, 3 × CH3), 22.8 (3 × CH3), 17.8 (CH3), 11.0 (d, JC–P = 5.7 Hz, CH3), 10.5 (d, JC–P = 6.6 Hz, CH3). HRMS m/z (ESI) Calcd for C28H45NO5P+ [M + H]+ 505.3030 Found: 505.3031. (RS/SR)-3b; pale yellow oil; Rf = 0.67 (EP/EtOAc 4:1); 1H NMR (400 MHz, CDCl3) δ 7.31 (d, J = 8.7 Hz, 2H), 7.35–7.18 (m, 5H), 6.81 (d, J = 8.7 Hz, 2H), 5.13 (q, J = 6.6 Hz, 1H), 4.95 (s, 2H), 4.08–3.84 (m, 1H), 3.85–3.69 (m, 1H), 3.46–3.30 (m, 1H), 3.32 (d, JH–P = 26.0 Hz, 1H), 3.26–3.10 (m, 1H), 1.45 (d, J = 6.6 Hz, 3H), 1.16 (t, J = 7.1 Hz, 3H), 1.13 (s, 9H), 1.11 (s, 9H), 0.81 (t, J = 7.1 Hz, 3H). 31P NMR (121 MHz, CDCl3) δ 24.62. 13C NMR (101 MHz, CDCl3) δ 158.0 (C), 137.1 (C), 135.7 (C), 129.1 (2 × CH), 128.4 (2 × CH), 127.7 (CH), 127.3 (2 × CH), 114.2 (2 × CH), 77.7 (CH), 70.1 (d, JC–P = 139.3 Hz, CH), 69.8 (CH2), 61.6 (d, JC–P = 6.3 Hz, CH2), 61.1 (C), 58.5 (d, JC–P = 7.5 Hz, CH2), 35.3 (d, JC–P = 5.1 Hz, C), 30.6 (d, JC–P = 6.0 Hz, 3 × CH3), 28.1 (3 × CH3), 20.9 (CH3), 16.3 (d, JC–P = 5.9 Hz, CH3), 16.1 (d, JC–P = 6.9 Hz, CH3). HRMS m/z (ESI) Calcd for C28H45NO5P+ [M + H]+ 505.3030 Found: 505.3031
:3); 1H NMR (400 MHz, CDCl3) δ 7.63–7.42 (m, 2H), 7.42–7.36 (m, 2H), 7.34 (d, J = 7.2 Hz, 1H), 7.29 (d, J = 8.6 Hz, 2H), 6.95 (d, J = 8.7 Hz, 2H), 5.06 (s, 2H), 4.94 (q, J = 6.6 Hz, 1H), 4.29 (m, 1H), 4.23–3.92 (m, 3H), 3.67 (d, JH–P = 26.7 Hz, 1H), 3.46 (d, J = 12.0 Hz, 1H), 3.09 (d, J = 12.0 Hz, 1H), 1.56 (d, J = 6.6 Hz, 3H), 1.37 (t, J = 7.1 Hz, 6H), 1.24 (s, 9H), 1.05 (s, 3H), 0.80 (s, 3H). 31P NMR (121 MHz, CDCl3) δ 26.89. 13C NMR (75 MHz, CDCl3) δ 158.0 (C), 137.1 (C), 136.9 (C), 128.5 (2 × CH), 128.0 (2 × CH), 127.8 (CH), 127.5 (2 × CH), 114.4 (2 × CH), 83.85 (CH), 69.8 (CH2), 69.3 (d, JC–P = 139.3 Hz, CH), 67.4 (CH2), 64.7 (C), 61.7 (d, JC–P = 6.3 Hz, CH2), 60.3 (d, JC–P = 6.3 Hz, CH2), 35.6 (d, JC–P = 4.6 Hz, C), 30.1 (d, JC–P = 5.6 Hz, 3 × CH3), 26.6 (CH3), 23.8 (CH3), 23.0 (CH3), 16.5 (d, JC–P = 5.8 Hz, CH3), 16.2 (d, JC–P = 6.6 Hz, CH3). HRMS m/z (ESI) Calcd for C28H45NO6P+ [M + H]+ 522.2979 Found: 522.2977. (RS/SR)-4b; pale yellow oil; Rf = 0.74 (EP/EtOAc 7:3); 1H NMR (400 MHz, CDCl3) δ 7.38–7.16 (m, 5H), 7.26 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 8.7 Hz, 2H), 5.01 (q, J = 6.5 Hz, 1H), 4.94 (s, 2H), 4.48 (s, 1H), 3.90–3.51 (m, 5H), 3.71 (d, JH–P = 27.2 Hz, 1H), 3.44 (d, J = 12.2 Hz, 1H), 1.44 (d, J = 6.6 Hz, 3H), 1.17 (s, 3H), 1.11–1.03 (m, 15H), 1.00 (t, J = 7.0 Hz, 3H). 31P NMR (121 MHz, CDCl3) δ 26.61. 13C NMR (101 MHz, CDCl3) δ 157.7 (C), 137.0 (C), 135.84 (C), 128.4 (2 × CH), 127.7 (CH), 127.7 (2 × CH), 127.3 (2 × CH), 114.3 (2 × CH), 77.6 (CH), 69.8 (CH2), 69.6 (d, JC–P = 136.4 Hz, CH), 67.8 (CH2), 64.5 (C), 61.9 (d, JC–P = 6.5 Hz, CH2), 60.3 (d, JC–P = 7.8 Hz, CH2), 35.2 (d, JC–P = 4.2 Hz, C), 30.5 (d, JC–P = 5.7 Hz, 3 × CH3), 26.5 (d, JC–P = 75.2 Hz, CH3), 24.0 (CH3), 21.7 (CH3), 16.2 (d, JC–P = 5.6 Hz, CH3), 15.9 (d, JC–P = 6.6 Hz, CH3). HRMS m/z (ESI) Calcd for C28H45NO6P+ [M + H]+ 522.2979 Found: 522.2975.
:4); 1H NMR (400 MHz, CDCl3) δ 7.51–7.32 (m, 5H), 7.26 (d, J = 8.6 Hz, 2H), 6.94 (d, J = 8.6 Hz, 2H), 5.07 (s, 2H), 4.81 (q, J = 6.7 Hz, 1H), 1.49 (d, J = 6.7 Hz, 3H), 1.32 (s, 9H), 1.05 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 157.8 (C), 137.7 (C), 137.2 (C), 128.5 (2 × CH), 128.2 (2 × CH), 127.8 (CH), 127.5 (2 × CH), 114.2 (2 × CH), 82.0 (CH), 70.0 (CH2), 61.8 (C), 61.7 (C), 30.6 (6 × CH3), 22.3 (CH3). HRMS m/z (ESI) Calcd for C23H34NO2+ [M + H]+ 356.2584 Found: 356.2579.
:4); 1H NMR (400 MHz, CDCl3) δ 7.37 (m, 2H), 7.28 (m, 3H), 7.14 (m, 1H), 6.89 (m, 1H), 6.83 (m, 1H), 6.77 (m, 1H), 4.99 (s, 2H), 4.67 (q, J = 6.6 Hz, 1H), 1.40 (m, 6H), 1.38 (d, J = 6.7 Hz, 3H), 1.20 (s, 3H), 1.08 (s, 3H), 0.94 (s, 3H), 0.62 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 158.7 (C), 147.6 (C), 137.2 (C), 129.0 (CH), 128.5 (2 × CH), 127.9 (CH), 127.6 (2 × CH), 119.3 (CH), 113.2 (CH), 113.1 (CH), 83.1 (CH), 70.0 (CH2), 59.7 (2 × C), 40.4 (2 × CH2), 34.5 (CH3), 34.1 (CH3), 23.6 (CH3), 20.4 (2 × CH3), 17.2 (CH2). HRMS m/z (ESI) Calcd for C24H34NO2+ [M + H]+ 368.2584 Found: 368.2588.
:1); 1H NMR (400 MHz, CDCl3) δ 7.38 (m, 5H), 7.22 (t, J = 7.8 Hz, 1H), 6.91 (m, 2H), 6.85 (m, 1H), 5.07 (s, 2H), 4.96 (q, J = 6.7 Hz, 1H), 4.35 (m, 1H), 4.10 (m, 2H), 3.98 (m, 1H), 3.34 (d, JH–P = 26.1 Hz, 1H), 1.59 (d, J = 6.8 Hz, 3H), 1.34 (t, J = 7.1 Hz, 3H), 1.31 (t, J = 7.1 Hz, 3H), 1.24 (s, 9H), 0.85 (s, 9H). 31P NMR (121 MHz, CDCl3) δ 25.91. 13C NMR (101 MHz, CDCl3) δ 158.6 (C), 147.3 (C), 137.1 (C), 129.0 (CH), 128.6 (2 × CH), 127.9 (CH), 127.4 (2 × CH), 119.9 (CH), 113.8 (CH), 113.5 (CH), 85.5 (CH), 69.9 (CH2), 69.9 (d, JC–P = 138.5 Hz, CH), 61.6 (d, JC–P = 6.3 Hz, CH2), 61.2 5 (C), 58.8 (d, JC–P = 7.5 Hz, CH2), 35.6 (d, JC–P = 5.8 Hz, C), 30.1 (d, JC–P = 5.8 Hz, 3 × CH3), 28.5 (3 × CH3), 24.1 (CH3), 16.8 (d, JC–P = 5.5 Hz, CH3), 16.3 (d, JC–P = 6.7 Hz, CH3). HRMS m/z (ESI) Calcd for C28H45NO5P+ [M + H]+ 506.3030 Found: 506.3033. (RS/SR)-3d; pale yellow oil; Rf = 0.67 (EP/EtOAc 4:1); 1H NMR (300 MHz, CDCl3) δ 7.38 (m, 5H), 7.19 (t, J = 7.8 Hz, 1H), 7.08 (m, 2H), 6.85 (m, 1H), 5.22 (q, J = 6.5 Hz, 1H), 5.07 (m, 2H), 3.91 (m, 2H), 3.45 (m, 1H), 3.40 (d, JH–P = 26.1 Hz, 1H), 3.29 (m, 1H), 1.54 (d, J = 6.6 Hz, 3H), 1.24 (m, 6H), 1.21 (s, 9H), 1.19 (s, 9H), 0.91 (t, J = 7.1 Hz, 3H). 31P NMR (121 MHz, CDCl3) δ 24.65. 13C NMR (75 MHz, CDCl3) δ 158.5 (C), 145.1 (C), 137.3 (C), 128.9 (CH), 128.5 (2 × CH), 127.8 (2 × CH), 127.4 (CH), 120.8 (CH), 114.1 (CH), 113.9 (CH), 78.2 (CH), 70.1 (CH2), 70.0 (d, JC–P = 139.1 Hz, CH), 61.6 (d, JC–P = 6.4 Hz, CH2), 61.3 (C), 58.7 (d, JC–P = 7.5 Hz, CH2), 35.4 (d, JC–P = 5.1 Hz, C), 30.7 (d, JC–P = 6.0 Hz, 3 × CH3), 28.3 (3 × CH3), 21.3 (CH3), 16.4 (d, JC–P = 5.7 Hz, CH3), 16.2 (d, JC–P = 6.9 Hz, CH3). HRMS m/z (ESI) Calcd for C28H45NO5P+ [M + H]+ 506.3030 Found: 506.3032.
:1); 1H NMR (400 MHz, CDCl3) δ 7.11 (d, J = 7.7 Hz, 2H), 6.71 (d, J = 7.8 Hz, 2H), 4.65 (q, J = 6.7 Hz, 1H), 1.38 (d, J = 6.7 Hz, 3H), 1.56–0.84 (m, 15H), 0.58 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 154.5 (C), 136.8 (C), 127.4 (2 × CH), 114.6 (2 × CH), 81.8 (CH), 59.0 (2 × C), 39.7 (2 × CH2), 33.6 (2 × CH3), 22.5 (CH3), 19.7 (2 × CH3), 16.6 (CH2). HRMS m/z (ESI) Calcd for C17H28NO2+ [M + H]+ 278.2115 Found: 278.2115.
:1); 1H NMR (400 MHz, CDCl3) δ 7.22 (d, J = 7.4 Hz, 2H), 6.78 (d, J = 8.0 Hz, 2H), 4.60 (q, J = 6.7 Hz, 1H), 1.56 (s, 3H), 1 45 (m, 1H), 1.44 (d, J = 6.6 Hz, 3H), 1.25 (s, 3H), 1.15 (s, 3H), 1.02 (s, 3H), 0.67 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 154.4 (C), 137.1 (C), 128.3 (2 × CH), 115.3 (2 × CH), 81.5 (CH), 64.1 (C), 63.0 (C), 35.9 (CH2), 35.2 (CH2), 31.0 (CH3), 30.9 (CH3), 24.1 (CH3), 22.7 (CH3), 21.9 (CH3). HRMS m/z (ESI) Calcd for C16H25NO2+ [M + H]+ 264.1958 Found: 264.1956.
:2); 1H NMR (400 MHz, CDCl3) δ 7.14 (d, J = 7.3 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 4.93 (q, J = 6.6 Hz, 1H), 4.37–4.18 (m, 1H), 4.19–3.89 (m, 3H), 3.37 (d, JH–P = 26.4 Hz, 1H), 1.58 (d, J = 6.6 Hz, 3H), 1.29 (m, 6H), 1.25 (s, 9H), 0.88 (s, 9H). 31P NMR (121 MHz, CDCl3) δ 25.88. 13C NMR (75 MHz, CDCl3) δ 156.3 (C), 135.9 (C), 128.4 (2 × CH), 115.0 (2 × CH), 84.3 (CH), 70.0 (d, JC–P = 139.1 Hz, CH), 61.9 (d, JC–P = 6.3 Hz, CH2), 61.3 (C), 59.3 (d, JC–P = 7.8 Hz, CH2), 35.6 (d, JC–P = 5.5 Hz, C), 30.2 (d, JC–P = 5.9 Hz, 3 × CH3), 28.5 (3 × CH3), 23.5 (CH3), 16.5 (d, JC–P = 5.9 Hz, CH3), 16.2 (d, JC–P = 7.1 Hz, CH3). HRMS m/z (ESI) Calcd for C21H39NO5P+ [M + H]+ 416.2560 Found: 416.2560. (RS/SR)-3a: white solid m.p.: 115 °C (decomp.); Rf = 0.70 (EP/EtOAc 3:2); 1H NMR (400 MHz, CDCl3) δ 7.18 (d, J = 8.5 Hz, 2H), 6.74 (d, J = 8.5 Hz, 2H), 5.17 (q, J = 6.5 Hz, 1H), 4.18–3.70 (m, 3H), 3.50–3.45 (m, 1H), 3.47 (d, JH–P = 26.6 Hz, 1H), 1.52 (d, J = 6.6 Hz, 3H), 1.23 (d, J = 2.4 Hz, 21H), 0.98 (t, J = 7.1 Hz, 3H). 31P NMR (121 MHz, CDCl3) δ 25.05. 13C NMR (75 MHz, CDCl3) δ 156.9 (C), 133.6 (C), 128.7 (2 × CH), 115.2 (2 × CH), 78.5 (CH), 70.5 (d, JC–P = 139.7 Hz, CH), 62.5 (d, JC–P = 6.6 Hz, CH2), 61.2 (C), 59.1 (d, JC–P = 7.5 Hz, CH2), 35.4 (d, JC–P = 5.2 Hz, C), 30.8 (d, JC–P = 6.1 Hz, 3 × CH3), 28.3 (3 × CH3), 21.6 (CH3), 16.4 (d, JC–P = 5.7 Hz, CH3), 16.1 (d, JC–P = 7.1 Hz, CH3). HRMS m/z (ESI) Calcd for C21H39NO5P+ [M + H]+ 416.2560 Found: 416.2558.
:1); 1H NMR (400 MHz, CDCl3) δ 7.12 (d, J = 8.0 Hz, 2H), 6.75 (d, J = 8.2 Hz, 2H), 4.80 (q, J = 6.5 Hz, 1H), 4.37–3.91 (m, 5H), 3.55 (d, JH–P = 26.6 Hz, 1H), 3.39 (d, J = 12.1 Hz, 1H), 3.10 (d, J = 12.1 Hz, 1H), 1.46 (d, J = 6.6 Hz, 2H), 1.27 (m, 6H), 1.15 (s, 9H), 0.97 (s, 3H), 0.78 (s, 3H). 31P NMR (162 MHz, CDCl3) δ 27.06. 13C NMR (101 MHz, CDCl3) δ 156.5 (C), 135.5 (C), 127.9 (2 × CH), 115.2 (2 × CH), 84.5 (CH), 69.6 (d, JC–P = 136.4 Hz, CH), 67.4 (CH2), 64.8 (C), 62.0 (d, JC–P = 6.6 Hz, CH2), 60.8 (d, JC–P = 8.0 Hz, CH2), 35.7 (d, JC–P = 4.5 Hz, C), 30.1 (d, JC–P = 5.6 Hz, 3 × CH3), 26.8 (CH3), 24.1 (CH3), 23.2 (CH3), 16.5 (d, JC–P = 5.8 Hz, CH3), 16.2 (d, JC–P = 6.8 Hz, CH3). (RS/SR)-4a: white solid m.p.: 115 °C (decomp.); Rf = 0.45 (EP/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 9.01 (s, 1H), 7.24 (d, J = 8.1 Hz, 2H), 6.79 (d, J = 8.2 Hz, 2H), 5.11 (q, J = 6.5 Hz, 1H), 4.51 (s, 1H), 4.01–3.64 (m, 6H), 3.79 (d, JH–P = 26.6 Hz, 1H), 1.55 (d, J = 6.5 Hz, 3H), 1.33 (s, 3H), 1.28–1.04 (m, 18H). 31P NMR (162 MHz, CDCl3) δ 26.30. 13C NMR (101 MHz, CDCl3) δ 156.7 (C), 133.6 (C), 127.8 (2 × CH), 115.1 (2 × CH), 78.2 (CH), 70.1 (d, JC–P = 136.7 Hz, CH), 68.2 (CH2), 64.4 (C), 62.5 (d, JC–P = 6.6 Hz, CH2), 60.7 (d, JC–P = 8.0 Hz, CH2), 35.4 (d, JC–P = 4.4 Hz, C), 30.5 (d, JC–P = 5.8 Hz3 × CH3), 26.2 (CH3), 23.9 (CH3), 21.7 (CH3), 16.3 (d, JC–P = 5.8 Hz, CH3), 16.0 (d, JC–P = 6.9 Hz, CH3). HRMS m/z (ESI) Calcd for C21H39NO6P+ [M + H]+ 432.2510 Found: 432.2507 (for a mixture of diastereoisomers).
:1); HRMS m/z (ESI) Calcd for C16H28NO2+ [M + H]+ 266.2115 Found: 266.2114.
:2); 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 7.04 (t, J = 7.7 Hz, 1H), 6.83 (s, 1H), 6.71 (t, J = 7.1 Hz, 2H), 4.85 (q, J = 6.5 Hz, 1H), 4.25 (m, 1H), 4.00 (m, 3H), 3.30 (d, JH–P = 26.1 Hz, 1H), 1.50 (d, J = 6.7 Hz, 3H), 1.23 (m, 6H), 1.17 (s, 9H), 0.83 (s, 9H). 31P NMR (121 MHz, CDCl3) δ 26.14. 13C NMR (101 MHz, CDCl3) δ 156.8 (C), 146.8 (C), 129.0 (CH), 118.3 (CH), 114.2 (CH), 114.2 (CH), 85.5 (CH), 70.0 (d, JC–P = 138.7 Hz, CH), 62.1 (d, JC–P = 6.4 Hz, CH2), 61.4 (C), 59.3 (d, JC–P = 7.5 Hz, CH2), 35.7 (d, JC–P = 5.7 Hz, C), 30.0 (d, JC–P = 5.7 Hz, 3 × CH3), 28.6 (3 × CH3), 24.2 (CH3), 16.7 (d, JC–P = 5.6 Hz, CH3), 16.2 (d, JC–P = 6.9 Hz, CH3). HRMS m/z (ESI) Calcd for C21H39NO5P+ [M + H]+ 416.2560 Found: 416.2562. (RS/SR)-3e: white solid m.p.: 115 °C (decomp.); Rf = 0.70 (EP/EtOAc 3:2); 1H NMR (400 MHz, CDCl3) δ1H NMR (400 MHz, CDCl3) δ 8.73 (s, 1H), 7.10 (t, J = 7.8 Hz, 1H), 7.03 (s, 1H), 6.94 (d, J = 7.6 Hz, 1H), 6.74 (dd, J = 8.0, 1.6 Hz, 2H), 5.17 (q, J = 6.5 Hz, 1H), 3.91 (m, 2H), 3.56 (m, 2H), 3.44 (d, JH–P = 26.3 Hz, 1H), 1.53 (d, J = 6.5 Hz, 3H), 1.22 (m, 21H), 0.97 (t, J = 7.1 Hz, 3H). 31P NMR (121 MHz, CDCl3) δ 24.97 13C NMR (101 MHz, CDCl3) δ 156.9 (C), 144.7 (C), 128.9 (CH), 117.9 (CH), 115.3 (CH), 114.5 (CH), 78.7 (CH), 70.3 (d, J = 139.5 Hz, CH), 62.4 (d, J = 6.5 Hz, CH2), 61.4 (C), 59.1 (d, J = 7.6 Hz, CH2), 35.5 (d, J = 5.1 Hz, C), 30.6 (d, J = 6.0 Hz, 3 × CH3), 28.3 (3 × CH3), 21.66 (CH3), 16.40 (d, J = 5.8 Hz, CH3), 16.08 (d, J = 7.2 Hz, CH3). HRMS m/z (ESI) Calcd for C21H39NO5P+ [M + H]+ 416.2560 Found: 416.2561.
Chemical oxidation as trigger
Addition of several equivalents of lead dioxide PbO2 to a 0.1 mM solution of alkoxyamines 1a–5a, and after a vigorous 20 seconds hand-shaking, provides an EPR signal. Then, using this procedure, effect of 10 equivalents to 1500 equivalents of PbO2 on alkoxyamines 1a–5a was tested (ESI Table 1†). Nitroxides 4˙ and 5˙ are not stable in the presence of PbO2, presumably due to further oxidation to oxoamoniums, impeding the growth of significant EPR signals upon oxidation of the corresponding alkoxyamines (only a few percent of conversion were detected). For low amounts of PbO2, more vigorous hand-shakings do not increase the amount of released nitroxide.CIDNP experiments
1H NMR and CIDNP spectra were recorded on a 200 MHz Bruker AVANCE DPX spectrometer. In situ irradiation of samples inside NMR magnet was carried out by using a frequency-tripled Quantel Brilliant B Nd:YAG laser (355 nm, 8 ns, ca. 70 mJ per pulse) The following pulse sequence was used: presaturation (laser flash)–(RF)–(detection pulse, 2 us)–(free induction decay). Dark spectra (without the laser flash) were always recorded as a reference for a proper suppression of the background NMR transitions.Deuterated solvents were purchased from Sigma-Aldrich and used without additional treatment. All the samples were deaerated by bubbling with N2 for 5 minutes prior to the CIDNP experiment.
DFT calculations
All calculations were performed using the Gaussian 16 electronic structure package55 at the M06-2X/6-31+G(d,p) level of theory,53 and the nature of all stationary points confirmed with frequency calculations at the same level. Gas-phase Gibbs free energies at 298.15 K were calculated using standard textbook formulae based upon the statistical thermodynamics of an ideal gas under the harmonic oscillator and rigid-rotor approximations. Solvent corrections were obtained with the SMD continuum solvent model for methanol,56 and the thermocycle approach was employed to determine Gibbs free energies in solution.57 All energies given are from conformationally-searched and Boltzmann-weighted conformer distributions.HPLC measurements and mass spectral analyses
Alkoxyamines and their homolysed products were separated and monitored by HPLC using an Agilent 1260 apparatus equipped with a UV-Vis absorption and MS detector. Typically, 1 μl of a sample was injected on C18 column (Phenomenex, Luna Omega Polar 1.6 μm, 100 × 2.1 mm) equilibrated with acetonitrile/water mobile phase (15/85 v/v) containing 0.1% formic acid. Compounds were separated by a linear increased of the acetonitrile concentration from 15 to 100% over 8 min. Next, the concentration of acetonitrile was kept at this level for 13 min. All analytes, were eluted at a flow rate of 0.14 ml min−1. The absorption traces were collected at 230 nm. The structural identity of products was confirmed by MS analysis using an Agilent 6120 quadrupole mass spectrometer equipped with electrospray ion source. The compounds were detected in the positive-ion mode.Kinetic measurements
Rate constants kd for the homolysis of the C–ON bond of alkoxyamines 1a,b–5a,b (Scheme 7) are determined using eqn (2) and by monitoring the growth of nitroxide by EPR, using O2 as alkyl radical scavenger as previously reported.58 For all alkoxyamines, a plateau in nitroxides is reached for more than 90% conversion at any temperatures. For the sake of simplicity, activation energies Ea are estimated using the Arrhenius equation in conjunction with the averaged frequency factor A = 2.4 1014 s−1.34 For ease of discussion, values of k′d at 120 °C are obtained from the Arrhenius equation, and half lives (t1/2) are obtained assuming first order kinetics. All data are gathered in Table 1.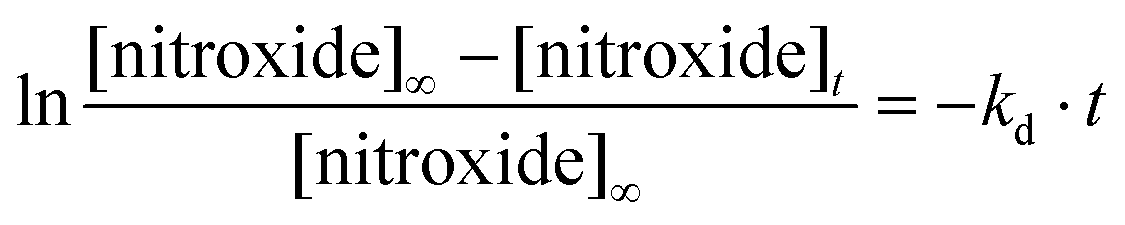 | (2) |
 | ||
| Scheme 7 Thermal homolysis of 1a,b–5a,b in t-BuPh in the presence of O2 as scavenger at various temperatures. | ||
Conclusions
Oxidation of bench stable alkoxyamines yields highly reactive intermediates capable of undergoing SN2 reactions with nucleophiles and/or mesolytic cleavage to form carbon-centred radicals or carbocations for use in chemical synthesis. To date electrochemical and photoredox methods have been used for this purpose; in this contribution we show that chemical oxidation provides a mild and convenient alternative. Moreover, when the nitroxide bears phenolic leaving groups we show that alkoxyamine cleavage can also be triggered by proton coupled electron transfer followed by a β-fragmentation reaction to afford methine quinone like-compounds. Both processes have potential to expand the scope of smart alkoxyamines in chemical synthesis, and as reporter molecules of oxidising environments.Author contributions
All authors contribute equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SRAM, MH and GA are thankful to Aix-Marseille University and CNRS for support. JH and MH are grateful to ANR for support (ANR-17-CE18-0017 and ANR-15-CE18-0012-01). SJ and EV thank Ministry for support. TMMK thank Campus France and government of Gabon for support. MLC gratefully acknowledges generous allocations of supercomputing time on the National Facility of the National Computational Infrastructure and an Australian Research Council Laureate Fellowship (FL170100041).Notes and references
- G. Audran, P. Brémond, J.-M. Franconi, S. R. A. Marque, P. Massot, P. Mellet, E. Parzy and E. Thiaudière, Alkoxyamines: a New Family of Pro-Drugs against Cancer. Concept for Theranostics, Org. Biomol. Chem., 2014, 12, 719 RSC.
- P. Brémond, A. Koïta, S. R. A. Marque, V. Pesce, V. Roubaud and D. Siri, Chemically Trigerred C—ON Bond Homolysis of Alkoxyamines. Quaternization of the Alkyl Fragments, Org. Lett., 2012, 14(1), 358 CrossRef PubMed.
- G. Audran, E. Bagryanskaya, I. Bagryanskaya, P. Brémond, M. Edeleva, S. R. A. Marque, D. Parkhomenko, E. Tretyakov and S. Zhivetyeva, C—ON Bond Homolysis of Alkoxyamines Triggered by Paramagnetic Copper(II) Salts, Inorg. Chem. Front., 2016, 3(11), 1464 RSC.
- G. Gryn'ova, D. L. Marshall, S. J. Blanksby and M. L. Coote, Switching Radical Stability by pH-Induced Orbital Conversion, Nat. Chem., 2013, 5, 474 CrossRef PubMed.
- G. Gryn'ova, L. M. Smith and M. L. Coote, Computational design of pH-switchable control agents for nitroxide mediated polymerization, Phys. Chem. Chem. Phys., 2017, 19, 22678–22683 RSC.
- Y. Guillaneuf, D.-L. Versace, D. Bertin, J. Lalevée, D. Gigmes and J.-P. Fouassier, Importance of the Position of the Chromophore Group on the Dissociation Process of Light Sensitive Alkoxyamine, Macromol. Rapid Commun., 2010, 31, 1909–1913 CrossRef CAS PubMed.
- Y. Guillaneuf, D. Bertin, D. Gigmes, D.-L. Versace, J. Lalevée and J.-P. Fouassier, Toward Nitroxide-Mediated Photopolymerization, Macromolecules, 2010, 43, 2204 CrossRef CAS.
- J. Morris, S. Telitel, K. E. Fairfull-Smith, S. E. Bottle, J. Lalevée, J.-L. Clément, Y. Guillaneuf and D. Gigmes, Novel polymer synthesis methodologies using combinations of thermally- and photochemically-induced nitroxide Mediated Polymerization, Polym. Chem., 2015, 6, 754–763 RSC.
- S. E. Bottle, J.-L. Clement, M. Fleige, E. M. Simpson, Y. Guillaneuf, K. E. Fairfull-Smith, D. Gigmes and J. P. Blinco, Light-Active Azaphenalene Alkoxyamines: Fast and Efficient Mediators of a Photo-Induced Persistent Radical Effect, RSC Adv., 2016, 6, 80328–80333 RSC.
- G. Audran, L. Bosco, P. Brémond, N. Jugniot, S. R. A. Marque, P. Massot, P. Mellet, T. Moussounda Moussounda Koumba, E. Parzy, A. Rivot, E. Thiaudière, P. Voisin, C. Wedl and T. Yamasaki, Controlled Radical Initiation Triggered by Specific Enzymatic Activity, Org. Chem. Front., 2019, 6, 3663 RSC.
- M. Albalat, G. Audran, M. Holzritter, S. R. A. Marque, P. Mellet, N. Vanthuyne and P. Voisin, An Enzymatic Acetal/Hemiacetal Conversion for the Physiological Temperature Activation of the Alkoxyamine C—ON Bond Homolysis, Org. Chem. Front., 2020, 7, 2916 RSC.
- M. Edeleva, D. Morozov, D. Parkhomenko, Y. Polienko, A. Iurchenkova, I. Kirilyuk and E. G. Bagryanskaya, Versatile approach to activation of alkoxyamine homolysis by 1,3-dipolar cycloaddition for efficient and safe nitroxide mediated polymerization, Chem. Commun., 2019, 55, 190 RSC.
- D. Moncelet, P. Voisin, N. Koonjoo, V. Bouchaud, P. Massot, E. Parzy, G. Audran, J.-M. Franconi, E. Thiaudière, S. R. A. Marque, P. Brémond and P. Mellet, Alkoxyamines: Towards a New Family of Theranostic Agents Against Cancer, Mol. Pharmaceutics, 2014, 11, 2412 CrossRef CAS PubMed.
- T. Yamasaki, D. Buric, Ch. Chacon, G. Audran, D. Braguer, S. R. A. Marque, M. Carré and P. Brémond, Chemical modifications of imidazole-containing alkoxyamines increase C–ON bond homolysis rate: effects on cytotoxic properties in glioblastoma cells, Bioorg. Med. Chem., 2019, 27, 1942 CrossRef CAS PubMed.
- A. Yamada, M. Abe, Y. Nishimura, S. Ishizaka, M. Namba, T. Nakashima, K. Shimoji and N. Hattori, Photochemical generation of the 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO radical from, caged nitroxides by near-infrared two-photon irradiation and its cytocidal effect on lung cancer cells, Beilstein J. Org. Chem., 2019, 15, 863 CrossRef CAS PubMed.
- L. Tebben and A. Studer, Nitroxide: Applications in Synthesis and in Polymer Chemistry, Angew. Chem., Int. Ed., 2011, 50, 5034 CrossRef CAS PubMed.
- J. E. Nutting, M. Rafiee and S. S. Stahl, Tertamethylpiperidine N-oxyl (TEMPO)Phtalimide N-Oxyl (PINO), and Related N-Oxyl Species: Electrochemical Properties and Their Use in Electrocatalytic Reactions, Chem. Rev., 2018, 118, 4834 CrossRef CAS PubMed.
- T. Reyser, H. To, C. Egwu, L. Paloque, M. Nguyen, A. Hamouy, J.-L. Stigliani, C. Bijani, J.-M. Augereau, J.-P. Joly, J. Portela, J. Havot, S. R. A. Marque, J. Boissier, A. Robert, F. Benoit-Vical and G. Audran, Alkoxyamines Designed as Potential Drugs Against Plasmodium and Schistosoma Parasites, Molecules, 2020, e3838 CrossRef PubMed.
- B. B. Noble, P. L. Norcott, C. L. Hammill, S. Ciampi and M. L. Coote, Mechanism of Oxidative Alkoxyamine Cleavage: the Surprising Role of the Solvent and Supporting Electrolyte, J. Phys. Chem. C, 2019, 123, 10300–10305 CrossRef CAS.
- C. L. Hammill, B. B. Noble, P. L. Norcott, S. Ciampi and M. L. Coote, Effect of Chemical Structure on the Electrochemical Cleavage of Alkoxyamines, J. Phys. Chem. C, 2019, 123, 5273 CrossRef CAS.
- P. L. Norcott, C. L. Hammill, B. B. Noble, J. C. Robertson, A. Olding, A. C. Bissember and M. L. Coote, TEMPO-Me: an Electrochemically Activated Methylating Agent, J. Am. Chem. Soc., 2019, 141, 15450–15455 CrossRef CAS PubMed.
- F. J. M. Rogers, B. B. Noble and M. L. Coote, Computational optimization of alkoxyamine-based electrochemical methylation, J. Phys. Chem. A, 2020, 124, 6104–6110 CrossRef CAS PubMed.
- L. Zhang, E. Laborda, N. Darwish, B. B. Noble, J. H. Tyrell, S. Pluczyk, A. P. Le Brun, G. G. Wallace, J. Gonzalez, M. L. Coote and S. Ciampi, Electrochemical and Electrostatic Cleavage of Alkoxyamines, J. Am. Chem. Soc., 2018, 140, 766 CrossRef CAS PubMed.
- Q. Zhu, E. C. Gentry and R. R. Knowles, Catalytic ncarbocation Generation Enabled nby the Mesolyticn Cleavage of Alkoxyamine radical Cation, Angew. Chem., Int. Ed., 2016, 55, 9969 CrossRef CAS PubMed.
- E. C. Gentry, L. J. Rono, M. E. Hale, R. Matsuura and R. R. Knowles, Enantioselective Synthesis of Pyrroloindolines via Noncovalent Stabilization of Indole Radical Cations and Applications to the Synthesis of Alkaloid Natural Products, J. Am. Chem. Soc., 2018, 140(9), 3394 CrossRef CAS PubMed.
- N. S. Hill, M. J. Fule, J. Morris, J.-L. Clément, Y. Guillaneuf, D. Gigmes and M. L. Coote, Mesolytic versus homolytic cleavage in photochemical nitroxide mediated polymerization, Macromolecules, 2020, 53, 1567–1572 CrossRef CAS.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Proton-coupled electron transfer, Chem. Rev., 2012, 112, 4016 CrossRef CAS PubMed.
- J. B. Shotwell, E. S. Krygowski, J. Hines, B. Koh, E. W. D. Huntsman, H. W. Choi, J. S. Schneekloth Jr., J. L. Wood and C. M. Crews, Total synthesis of the anti-angiogenic agent Luminacin D, Org. Lett., 2002, 18, 3087 CrossRef PubMed.
- J. Dao, D. Benoît and C. J. Hawker, A Versatile and Efficient Synthesis of Alkoxyamine LFR Initiators via Manganese Based Asymmetric Epoxidation Catalysts, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 2161 CrossRef CAS.
- D. Bertin, D. Gigmes, S. R. A. Marque and P. Tordo, Polar, Steric, and Stabilization Effects in Alkoxyamines C-ON Bond Homolysis: A Multiparameters Analysis, Macromolecules, 2005, 38(7), 2638 CrossRef CAS.
- H. Fischer, A. Kramer, S. R. A. Marque and P. Nesvadba, Steric and Polar Effects of the Cyclic Nitroxyl Fragment on the C-ON Bond Homolysis Rate Constant, Macromolecules, 2005, 38(24), 9974 CrossRef CAS.
- C. Y. Lin, S. R. A. Marque, K. Matyjaszewski and M. L. Coote, Linear-Free energy relationships for modeling structure-reactivity trends in Controlled Radical Polymerization, Macromolecules, 2011, 44(19), 7568–7583 CrossRef CAS.
- J. L. Hodgson, C.-Y. Lin, M. L. Coote, S. R. A. Marque and K. Matyjaszewski, Linear Free Energy Relationship for the Alkyl Affinities of Nitroxides: A Theoretical Study, Macromolecules, 2010, 43(8), 3728 CrossRef CAS.
- S. Marque, The influence of the nitroxide structure on homolysis rate of alkoxyamines: A Taft-Ingold analysis, J. Org. Chem., 2003, 68(20), 7582 CrossRef CAS PubMed.
- E. G. Bagryanskaya and S. R. A. Marque, Kinetic Aspects of Nitroxide-Mediated Polymerization, in Nitroxide Mediated Polymerization: From Fundamentals to Applications in Materials Sciences, RSC Polymer Chemistry Series, n° = 19, ed. D. Gigmes, Royal Society of Chemistry, 2016, ch. 2, pp. 45–113 Search PubMed.
- G. Gryn'ova, K. U. Ingold and M. L. Coote, New insights into the mechanism of amine/nitroxide cycling during the hindered amine light stabilizer inhibited oxidativen degradation of polymers, J. Am. Chem. Soc., 2012, 134, 12979–12988 CrossRef PubMed.
- S. Marque, H. Fischer, E. Baier and A. Studer, Factors influencing the C-O-bond homolysis of alkoxyamines: Effect of H-bonding and polar substituents, J. Org. Chem., 2001, 66(4), 1146 CrossRef CAS PubMed.
- G. Audran, P. Brémond, J.-P. Joly, S. R. A. Marque and T. Yamasaki, C—ON bond homolysis of alkoxyamines. Part 12: Effect of the para-substituent in the 1-phenylethyl fragment, Org. Biomol. Chem., 2016, 14, 3574 RSC.
- C. Hansch, A. Leo and R. W. Taft, A Survey of Hammett substituent constants and resonance and field parameters, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- M. Charton, Adv. Mol. Struct. Res., 1999, 5, 25 CAS.
- D. Bertin, D. Gigmes, S. R. A. Marque, S. Milardo, J. Peri and P. Tordo, Long-range polar effect on the C-ON bond homolysis in SG1 (N-(2-Methyl-2-propyl)-N-(1-diethylphosphono-2,2-dimethylpropyl)-N-oxyl) – alkoxyamines, Collect. Czech. Chem. Commun., 2004, 69(12), 2223 CrossRef CAS.
- G. Audran, S. Dorey, N. Dupuy, F. Gaston and S. R. A. Marque, Degradation of g-Irradiated Polyethylene-etylene Vinyl Alcohol-polyethylene Multilayer Films: An ESR Study, Polym. Degrad. Stab., 2015, 122, 169 CrossRef CAS.
- D. Griller and K. U. Ingold, Electron paramagnetic resonance and the art of physical-organic chemistry, Acc. Chem. Res., 1980, 13(7), 193 CrossRef CAS.
- J. J. Henderson, Concerning the relationship between the strength of acids and their capacity to preserve neutrality, Am. J. Physiol., 1908, 21, 173 CrossRef.
- G. L. Closs and R. J. Miller, Gas-ohase basicities of amides and imidates. Estgim tion of protomeric equilibrium constants by the basicity method in the gas phase, J. Am. Chem. Soc., 1979, 101(6), 1639 CrossRef CAS.
- J.-K. Vollenweider, H. Fischer, J. Hennig and R. Leuschner, Time-resolved CIDNP in laser flash photolysis of aliphatic ketones. A quantitative analysis, Chem. Phys., 1985, 97(2–3), 217 CrossRef CAS.
- D. Neshchadin, F. Palumbo, M. S. Sinicropi, I. Andreu, G. Gescheidt and M. Miranda, Topological control in radical reactions of cholesterol in model dyads, Chem. Sci., 2013, 4(4), 1608 RSC.
- I. Andreu, D. Neshchadin, S. N. Batchelor, M. A. Miranda and G. Gescheidt, Examples for biological reactivity involving free radicals followed by CIDNP, Mol. Phys., 2013, 111(18–19), 2992 CrossRef CAS.
- P. K. Das, M. V. Encinas, S. Steenken and J. C. Scaiano, Reaction of tert-butoxy radicals with phenols. Comparison with the reactions of carbon triplets, J. Am. Chem. Soc., 1981, 103(14), 4162 CrossRef CAS.
- M. V. Encinas and J. C. Scaiano, Reaction of Benzophznone triplets with allylic hydrogens. A laser flash photolysis study, J. Am. Chem. Soc., 1981, 103(21), 6393 CrossRef CAS.
- L. Hermosilla, J. M. G. de la Vega, C. Sieiro and P. Calle, DFT calculations of isotropic hyperfine couplking constants of nitrogen aromatic radicals: The challenge of nitroxide radicals, J. Chem. Theory Comput., 2011, 7(1), 169 CrossRef CAS PubMed.
- H. Kruse, L. Goerigk and S. W. Grimme, Why the standard B3LYP/6-31G* model chemistry should not be used in DFT calculations of molecular thermochemistry: Understanding and correcting the problem, J. Org. Chem., 2012, 77(23), 10824 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functional for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functional and systematic testing of four M06-class functional and 12 other functional, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- J. W. Darcy, B. Koronkiewicz, G. A. Parada and J. M. Mayer, A continuum of proton-coupled electron transfer reactivity, Acc. Chem. Res., 2018, 51, 2391 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, B. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and on continuum model of the solvent defined by the bulk dielectric constant a,d atomic surface tensions, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- J. Ho, A. Klamt and M. L. Coote, Comments on the correct use of continuum solvent models, J. Phys. Chem. A, 2010, 114, 13442 CrossRef CAS PubMed.
- S. Marque, C. Le Mercier, P. Tordo and H. Fischer, Factors influencing the C-O-bond homolysis of trialkylhydroxylamines, Macromolecules, 2000, 33(12), 4403 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: HRMS and NMR spectra of 1a,b–5a,b, 3d and 3e, pKa determination of 4-vinylphenol, HPLC-ESI analysis of 3a and 3e in THF, and DFT calculations. See DOI: 10.1039/d1qo01276b |
| ‡ Disappointingly, all attempts for the preparation of 1a, 2a, and 5a using the procedure described in § failed. |
| § Alkoxyamines 3a and 4a are also prepared by the coupling of nitroxides 3˙ and 4˙ with the commercially available para-vinyl phenol using the procedure Mn(salen) (Scheme 1a). |
| ¶ σp,OH = −0.37, see ref. 39, and equation in Fig. 2 in ref. 37. |
| || σL,OH = 0.24 and σ0R,OH = −049, see ref. 40, and eqn (1) and (5a) in ref. 38. |
| ** It was assumed that σp,OCH2Ph ≈ σp,OEt = −0.32, see ref. 39, and equation in Fig. 2 in ref. 37. |
| †† It was assumed that σL,4-BnOC6H4 ≈ σp,OEt, σL,OEt = 0.28 and σ0R,OEt = −044, see ref. 40, and eqn (1) and (5a) in ref. 38. |
| ‡‡ In general, this angle exhibit a deviation less than ±20° meaning that a very low activation entropic cost is expected. |
| §§ Several vigorous hand-shakings did not increase the EPR signal. |
| ¶¶ Stabilities of 1˙, 3˙–5˙ were investigated by EPR. 1˙ is stable in the presence of PbO2, 3˙ moderately stable, 4˙ and 5˙ decay quickly impeding all chances of detection when generated from their corresponding alkoxyamines. For 3˙ and 4˙, the signal of another radical carrying a phosphorylated moiety is observed but not identified. This species is unstable and decay quickly however, the side species of 3˙ is not obseverved during the oxidation of 3a. |
| |||| Preparation of E and F is tedious, and as no unknown compounds is detected, no more efforts were devoted to their preparations. |
| *** Compounds issued from the degradation of 3e under chemical oxidation conditions were not prepared as the degradation processes are not clearly unveiled. |
| ††† Oxidations of 3a and 3e were performed in THF in the same conditions. In this case, the oxidation of 3a afforded mainly 3˙ and B as main products due to the traces of water in THF. For 3e, results in THF were similar to those in MeOH. See ESI. |
| ‡‡‡ Noteworthily, the addition of PbO2 on molecules exhibiting phenol moieties is well known to generate phenoxyl radical through a Proton-Coupled Electron Transfer (PCET, see ref. 54 and 27) via the generation of radical cation and loss of proton to provide a phenoxyl radical. |
| §§§ For most 3-based alkoxyamines carrying an aryl moiety on the alkyl fragment, the dihedral angle θ is close to 60°. |
| This journal is © the Partner Organisations 2021 |