
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModelling drug adsorption in metal–organic frameworks: the role of solvent†
Abhishek T. Sose‡
 a,
Hannah D. Cornell‡b,
Bradley J. Gibbonsb,
Ashley A. Burrisb,
Amanda J. Morris*b and
Sanket A. Deshmukh*a
a,
Hannah D. Cornell‡b,
Bradley J. Gibbonsb,
Ashley A. Burrisb,
Amanda J. Morris*b and
Sanket A. Deshmukh*a
aDepartment of Chemical Engineering, Virginia Tech, Blacksburg, VA 24060, USA. E-mail: sanketad@vt.edu
bDepartment of Chemistry, Virginia Tech, Blacksburg, VA,. 24060, USA. E-mail: ajmorris@vt.edu
First published on 10th May 2021
Abstract
Solvent plays a key role in biological functions, catalysis, and drug delivery. Metal–organic frameworks (MOFs) due to their tunable functionalities, porosities and surface areas have been recently used as drug delivery vehicles. To investigate the effect of solvent on drug adsorption in MOFs, we have performed integrated computational and experimental studies in selected biocompatible MOFs, specifically, UiO-AZB, HKUST-1 (or CuBTC) and NH2-MIL-53(Al). The adsorption of three drugs, namely, 5-fluorouracil (5-FU), ibuprofen (IBU), and hydroxyurea (HU) were performed in the presence and absence of the ethanol. Our computational predictions, at 1 atmospheric pressure, showed a reasonable agreement with experimental studies performed in the presence of ethanol. We find that in the presence of ethanol the drug molecules were adsorbed at the interface of solvent and MOFs. Moreover, the computationally calculated adsorption isotherms suggested that the drug adsorption was driven by electrostatic interactions at lower pressures (<10−4 Pa). Our computational predictions in the absence of ethanol were higher compared to those in the presence of ethanol. The MOF–adsorbate interaction (UHA) energy decreased with decrease in the size of a drug molecule in all three MOFs at all simulated pressures. At high pressure the interaction energy increases with increase in the MOFs pore size as the number of molecules adsorbed increases. Thus, our research shows the important role played by solvent in drug adsorption and suggests that it is critical to consider solvent while performing computational studies.
Introduction
Solvent has shown its prime importance in biological functions, polymer science, catalysis, and biomedical applications.1–7 For example, in drug delivery vehicle (DDV) development, the interactions between the drug and relevant body fluids (solvent) play a critical role in driving drug release from the carrier.8 Similarly, the drug loading in a DDV is determined by the interactions between solvent and drugs, between solvent and the DDV, and between drug and DDV.9–11 At present, our atomic-level understanding of drug loading in DDVs, such as polymers and porous materials, is very limited. Specifically, the precise role of the interactions between solvent and the drugs, and between solvent and DDV is unknown.12–14 Without knowledge of these interactions, it is difficult to understand if the drug adsorption occurs at the interface between solvent and the DDV or if the drug molecules reside primarily in the pore volume of the DDV. An understanding of the interplay of drug, solvent, and carrier interactions is, therefore, important for the design of new DDVs with higher uptake and efficient drug release.Recently, metal–organic frameworks (MOFs) have been used in the field of catalysis, gas storage, and drug delivery due to their adjustable functionalities, tunable porosities, and high surface area.13,15–21 In these applications, it has been reported that solvent plays an important role in determining the stability and function (e.g. adsorption capacity) of a MOF.22,23 A number of experimental and computational studies also show that MOFs have higher drug storage capacity than other porous materials due to high surface area and the formation of non-bonded interactions with drugs.17 Grand canonical Monte Carlo (GCMC) simulations have been used to study drug adsorption both in the presence and absence of solvent at the molecular-level.13,15,24–28 For example, adsorption of the anticancer drugs 5-FU and HU in ZIFs was studied by Gomar et al.,25 while Erucar et al. performed a computational study on methotrexate and 5-FU uptake in MOF-74 in the absence of solvent.15 Bernini et al. employed GCMC simulations to study the adsorption of IBU in a series of bio-compatible MOFs.24 In the computational studies performed in the absence of solvent, drug adsorption is often overestimated compared to experiment, which uses solvent to dissolve and adsorb drugs in MOFs. The differences are attributed to experimental limitations, such as the non-accessibility of the cavities and poor activation of the MOF samples.13 However, Proenza et al. performed GCMC simulations to study adsorption of 5-FU and caffeine (CAF) in ZIF-8 analogs, and identified that the lack of consideration for solvent and surface effects in adsorption study contributes majorly towards the overestimation of drug adsorption.29
Result and discussion
Motivated by elucidating the importance of solvent on drug adsorption in MOFs at the molecular-level, we performed a combined computational and experimental study of drug adsorption in MOFs in the presence of explicit solvent. Specifically, we employ GCMC simulations to study adsorption of 5-FU, IBU, and HU in three MOFs of interest, namely, HKUST-1, NH2-MIL-53 and UiO-AZB in the presence of ethanol. HKUST-1 and NH2-MIL-53 were selected because of their established biocompatibility and wide-use throughout the literature.30,31 UiO-AZB is a light-responsive framework developed in our lab that shows promise as a versatile drug delivery vehicle.32 Tables S1–S3 and Fig. S1 in Section S1† discuss details on drug and solvent molecules used in our study. Table 1 summarizes the textural properties of these MOFs and a detailed computational and experimental methodology used to calculate the properties is listed in Section S2 of the ESI.† The molecular-level structures of all three MOFs and force-field parameters are shown in Fig. S2 and Tables S5–S7 in the ESI.† As can be seen from Table 1, the calculated surface area for all three MOFs is higher than experimental measurements. This is expected, as simulations are performed on perfect crystalline frameworks and do not account for defects.33 The differences in pore volume can be accounted for with scaling factors that reconcile experimental and simulated adsorption.33,34 In addition, Epley et al. highlights differences in the experimental and theoretical surface area observations in UiO-AZB, which can be due to the introduction of macroporosity within the samples.32 The pore size distribution (PSD) suggests that the HKUST-1, NH2-MIL-53, and UiO-AZB are microporous (pore size upto 20 Å).35 More details on PSD calculations can be found in the ESI, Section S2.†| Calculated void volume (cm3 g−1) | Calculated pore-cavity size (Å) | Calculated surface area (m2 g−1) | Experimental surface area (m2 g−1) | Literature reported surface area (m2 g−1) | Literature reported void volume (cm3 g−1) | |
|---|---|---|---|---|---|---|
| HKUST-1 | 0.9066 | 5.1, 10.6, 12.2 | 2365 | 1830 | 1507 (ref. 36), 1843 (ref. 37), 1600 (ref. 38), 1740 (ref. 39) | 0.75 (ref. 40), 0.79 (ref. 41) |
| NH2-MIL-53(Al) | 0.6272 | 6.36 | 1395 | 725 | 675 (ref. 42), 712 (ref. 43), 947 (ref. 44) | 0.83 (ref. 45) |
| UiO-AZB | 1.4057 | 10.8, 11.9, 13.7 | 3692 | 1959 | 903–2687 (ref. 32) | — |
To probe the effect of solvent, we performed GCMC simulations in the absence and presence of ethanol. Table 2 lists the maximum drug uptake calculated with GCMC simulations and experimentally measured at 300 K and 1 atm. To study drug adsorption in the presence of ethanol, initially, GCMC simulations were performed to evaluate the number of ethanol molecules adsorbed in the MOFs at 1 bar and 300 K for 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps. The adsorption of ethanol in HKUST-1, NH2-MIL-53 and UiOAZB was found out to be 509.1 (mg g−1), 296.67 (mg g−1) and 920.45 (mg g−1), respectively. This corresponds to 107 mol uc−1, 5.75 mol uc−1 and 183 mol uc−1 in HKUST-1, NH2-MIL-53 and UiOAZB, respectively. The number of molecules of ethanol obtained from these simulations were used as a starting number of molecules in initial configuration for drug adsorption studies in the presence of ethanol. During these two components (both drug and ethanol) simulations, equal probabilities of both molecules for insertion, deletion, translation, and rotation were used. These simulations were also performed at 300 K for 1500000 steps. This process here is referred to as the simulations in presence of ethanol. More details on computational methods are discussed in Section S3 of the ESI.† In the absence of ethanol in GCMC simulations the uptake of 5-FU, IBU and HU in Cu–BTC was 886.86 mg g−1, 1118.73 mg g−1 and 943.22 mg g−1, respectively. In presence of ethanol, the maximum simulated uptake of 5-FU, IBU and HU in HKUST-1 decreased to 174.7 mg g−1, 86.05 mg g−1, and 564.21 mg g−1, respectively, suggesting that solvent occupies MOF pores during drug loading. The results are in qualitative agreement with our drug loading experiments, performed in the presence of ethanol that showed the maximum adsorption of 150 ± 10 mg g−1, 130 ± 20 mg g−1 and 300 ± 70 mg g−1 for 5-FU, IBU and HU in HKUST-1, respectively. For NH2-MIL-53 the maximum simulated uptake of 5-FU, IBU and HU in the absence of ethanol was 502.77 mg g−1, 466.59 mg g−1 and 510.92 mg g−1, respectively. However, in the presence of ethanol the uptake was 36.43 mg g−1, 23.32 mg g−1 and 221.61 mg g−1, respectively. In the experiments, uptake of 5-FU, IBU and HU in NH2-MIL-53 was 190 ± 90 mg g−1, 100 ± 10 mg g−1 and 440 ± 150 mg g−1, which was significantly higher to that of calculated adsorption in our GCMC simulations. We attribute the higher adsorption to the breathing behavior of NH2-MIL-53, which can be controlled and instigated by adsorbate loading.46–50 Thus, due to its breathable nature, in our experiments, the pores in NH2-MIL-53 might open in the presence of ethanol and drug molecules, resulting in higher adsorption of drug molecules compared to our simulations, which do not account for breathability.46
000 steps. The adsorption of ethanol in HKUST-1, NH2-MIL-53 and UiOAZB was found out to be 509.1 (mg g−1), 296.67 (mg g−1) and 920.45 (mg g−1), respectively. This corresponds to 107 mol uc−1, 5.75 mol uc−1 and 183 mol uc−1 in HKUST-1, NH2-MIL-53 and UiOAZB, respectively. The number of molecules of ethanol obtained from these simulations were used as a starting number of molecules in initial configuration for drug adsorption studies in the presence of ethanol. During these two components (both drug and ethanol) simulations, equal probabilities of both molecules for insertion, deletion, translation, and rotation were used. These simulations were also performed at 300 K for 1500000 steps. This process here is referred to as the simulations in presence of ethanol. More details on computational methods are discussed in Section S3 of the ESI.† In the absence of ethanol in GCMC simulations the uptake of 5-FU, IBU and HU in Cu–BTC was 886.86 mg g−1, 1118.73 mg g−1 and 943.22 mg g−1, respectively. In presence of ethanol, the maximum simulated uptake of 5-FU, IBU and HU in HKUST-1 decreased to 174.7 mg g−1, 86.05 mg g−1, and 564.21 mg g−1, respectively, suggesting that solvent occupies MOF pores during drug loading. The results are in qualitative agreement with our drug loading experiments, performed in the presence of ethanol that showed the maximum adsorption of 150 ± 10 mg g−1, 130 ± 20 mg g−1 and 300 ± 70 mg g−1 for 5-FU, IBU and HU in HKUST-1, respectively. For NH2-MIL-53 the maximum simulated uptake of 5-FU, IBU and HU in the absence of ethanol was 502.77 mg g−1, 466.59 mg g−1 and 510.92 mg g−1, respectively. However, in the presence of ethanol the uptake was 36.43 mg g−1, 23.32 mg g−1 and 221.61 mg g−1, respectively. In the experiments, uptake of 5-FU, IBU and HU in NH2-MIL-53 was 190 ± 90 mg g−1, 100 ± 10 mg g−1 and 440 ± 150 mg g−1, which was significantly higher to that of calculated adsorption in our GCMC simulations. We attribute the higher adsorption to the breathing behavior of NH2-MIL-53, which can be controlled and instigated by adsorbate loading.46–50 Thus, due to its breathable nature, in our experiments, the pores in NH2-MIL-53 might open in the presence of ethanol and drug molecules, resulting in higher adsorption of drug molecules compared to our simulations, which do not account for breathability.46
| Maximum simulated uptake in the presence of ethanol (mg g−1) | Maximum simulated uptake in the absence of ethanol (mg g−1) | Maximum experimental uptake (mg g−1) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 5-FU | IBU | HU | 5-FU | IBU | HU | 5-FU | IBU | HU | |
| HKUST-1 | 174.7 | 86.1 | 564.2 | 886.9 | 1118.7 | 943.2 | 150 ± 10 | 130 ± 20 | 300 ± 70 |
| NH2-MIL-53(Al) | 36.4 | 23.3 | 221.6 | 502.8 | 466.6 | 510.9 | 190 ± 90 | 100 ± 10 | 440 ± 150 |
| UiO-AZB | 397.8 | 238.6 | 1071.9 | 1534.3 | 1660.3 | 1620.3 | 120 ± 20 | 180 ± 40 | 290 ± 20 |
In the case of UiO-AZB, in GCMC simulations performed in the absence of ethanol, the uptake of 5-FU, IBU and HU was 1534.32 mg g−1, 1660.26 mg g−1 and 1620.30 mg g−1, respectively. On the other hand, in the presence of ethanol adsorption decreased to 397.78 mg g−1, 238.65 mg g−1, and 1071.89 mg g−1, respectively. In our experiments, the uptake of 5-FU, IBU and HU in UiO-AZB was 120 ± 20 mg g−1, 180 ± 40 mg g−1, and 290 ± 20 mg g−1, respectively. Experimentally, to access the larger interior pore (ca. 13.7 Å) of UiO-AZB, drug molecules must first diffuse through the smaller exterior pores (ca. 11.9 Å and 10.8 Å). However, diffusion of larger IBU molecules compared to smaller ethanol can be hindered as IBU may block the entrance for the larger interior pore.32 In addition, in presence of ethanol, displacing ethanol in these pores becomes intricate in the experiments.29 In GCMC simulations, however, all the pores are accessible to drug molecules, which may result in quantitatively higher adsorption than experiments. To further investigate the origin of the notably higher adsorption of 5-FU, IBU, and HU in UiO-AZB, in our GCMC simulations, we calculated the number of drug molecules adsorbed in different pores. We found that 13.7 Å sized (interior) pore adsorbed 7.21 molecules of IBU, while the exterior pores had 4.8 IBU molecules. Adsorption in the exterior pores accounts for the 109.15 (mg g−1) adsorption and is ∼40% lower compared to our experimental adsorption of IBU in UiO-AZB. This higher adsorption in experiments can be attributed to the stronger interaction of the carboxylic group with the defect sites on the zirconium nodes of the UiO-AZB in our experiments.51,52 Similarly, the adsorption of 5-FU and HU in the exterior pores was 8.59 and 72.86 molecules accounting for 121.92 mg g−1 and 605.41 mg g−1. Thus, the adsorption of 5-FU and HU is within 2% and 110% to that of experimentally measured adsorption, respectively. This further suggests that the both 5-FU and HU might have been only adsorbed in the exterior pores of UiO-AZB in our experiments.
In general, maximum uptake capacity for all three drugs follows the trend HU > 5-FU > IBU, which can be attributed to their size. The smaller sized molecules (e.g. HU) are adsorbed more readily compared to the larger sized molecules (e.g. IBU). To further investigate the drug adsorption sites in the MOFs, we visually inspected the simulation trajectories. The snapshots shown in Fig. 1 suggest that in the presence of ethanol, the preferential site for drug adsorption is the interface between ethanol and MOF, while ethanol is adsorbed at the core of the MOF pores. In the absence of solvent, drug molecules can also be adsorbed at the core of MOF pores. We found that the smallest pore of HKUST-1 (ca. 5 Å) is occupied by only smaller molecules like ethanol, HU and 5-FU. In general, drugs are strongly confined in the one-dimensional pore of NH2-MIL-53 as shown in Fig. 1(c) and (d).
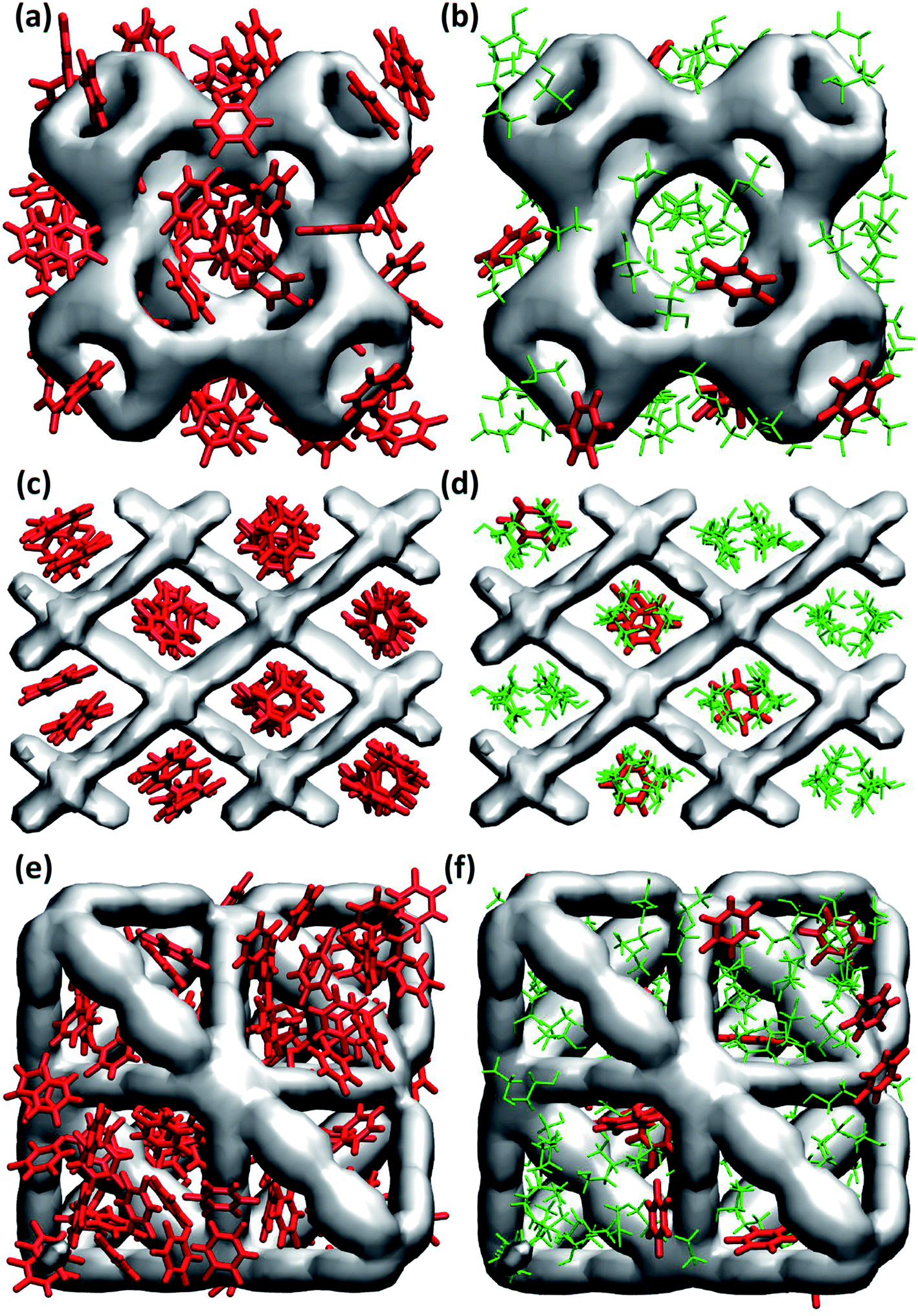 | ||
| Fig. 1 Snapshots of adsorbed 5-FU molecules in (a) and (b) HKUST-1, (c) and (d) NH2-MIL-53, (e) and (f) UiO-AZB without ethanol (left) and with ethanol (right). (5-FU is represented in red and ethanol in green). | ||
To quantify the atomic-level structure and local arrangement of specific atoms of drugs and solvent in MOFs, we performed radial distribution function (RDF) analysis.53,54 Fig. 2(a)–(c) show the RDF of metal atoms in HKUST-1, NH2-MIL-53 and UiO-AZB, respectively, with selected atoms of 5-FU (oxygen, nitrogen and fluorine), IBU (all atoms of carboxylic group) and HU (nitrogen and oxygens). See ESI Fig. S1† for the definition/names of drug atoms. The first peak for Cu–O1, Cu–N2, and Cu–F1 pairs appears at 2.89 Å, 3.59 Å, and 3.54 Å, respectively (Fig. 2(a)(i)). The distances suggest a stronger structural correlation of Cu with O1 atoms compared to with N2 and F1 atoms of 5-FU. Indeed, Cu metal atoms are expected to bind stronger to more polar oxygen sites (i.e., the O1 of 5-FU).55 Similarly, peaks for IBU and HU suggest a stronger correlation between Cu–OH_ib and Cu–O2 pairs, respectively (Fig. 2(a)(ii) and (iii)), suggesting stronger binding of hydroxyl groups with the copper metal sites.
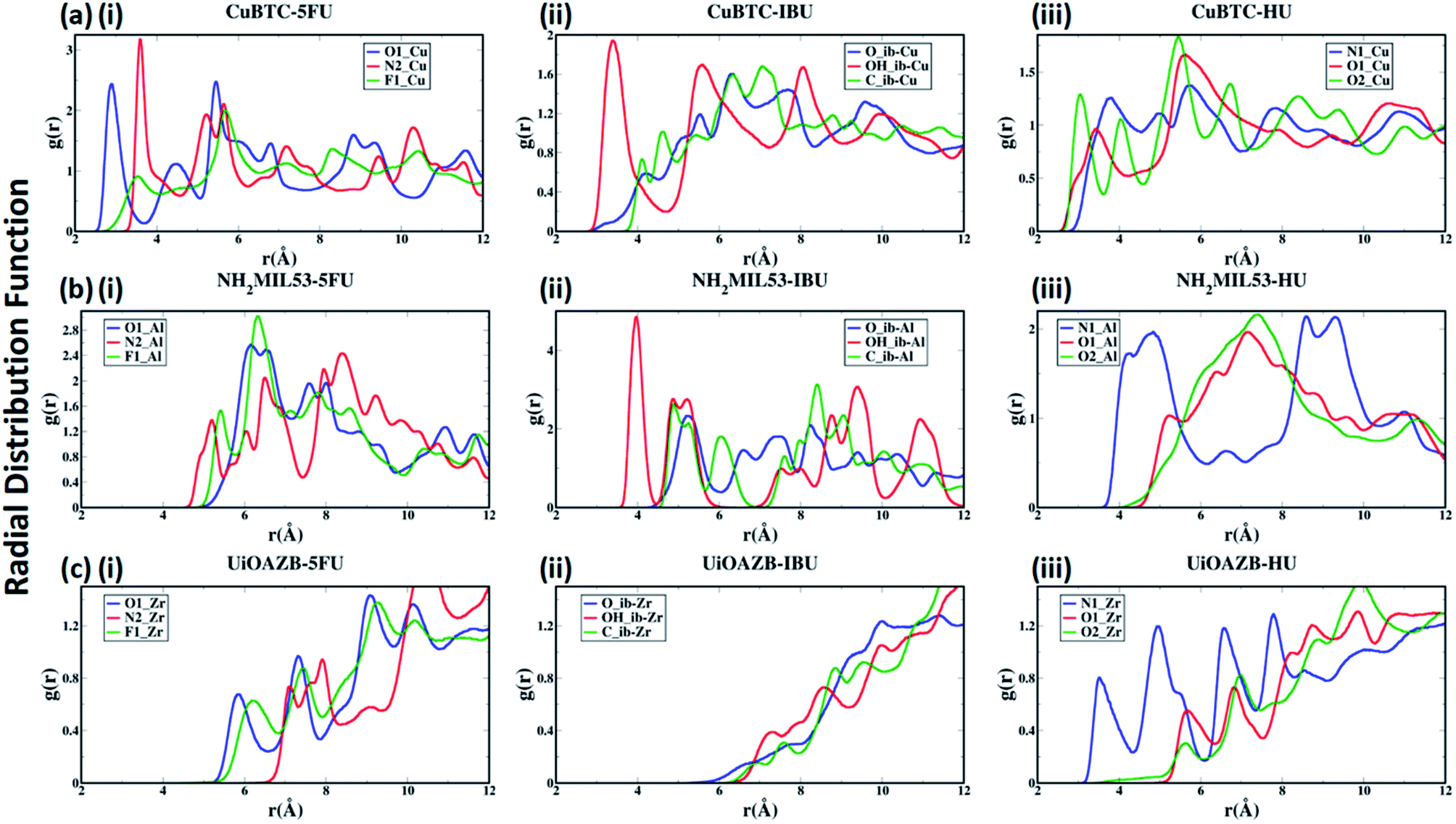 | ||
| Fig. 2 Radial distribution functional (RDF) analysis of selected atoms of drugs (i) 5-FU, (ii) IBU and (iii) HU with unsaturated metal sites of (a) HKUST-1, (b) NH2-MIL-53 and (c) UiO-AZB. | ||
In the case of NH2-MIL-53, a strong structural correlation between Al and –NH sites (N2 atom of 5-FU) is apparent compared to the correlations between Al and F1 and between Al and O1 atoms of 5-FU (Fig. 2(b)(i)). A stronger affinity of –COOH group of IBU and –NH2 group of HU with Al of NH2-MIL-53 can be observed from their RDFs. Similarly, for UiO-AZB the Zr has stronger structural correlation with O1 compared to the F1 and N2 of 5-FU (Fig. 2(c)(i)). The carboxylic group of IBU didn't show any structural correlation with the Zr metal sites, which could be because Zr is not exposed in UiO-AZB.56,57 However, the Zr metal site shows strongly ordered structure with –NH2 groups of HU (Fig. 2(c)(iii)), thus exhibiting strong affinity towards it. The 1st peak distances of drug atoms with MOF follows the order HKUST-1 > NH2-MIL-53 > UiO-AZB, which can be accredited to the decreasing order of exposure of unsaturated metal atoms of MOFs.32,58,59 Fig. S22 and S23 of ESI† describes the RDF of selected atoms of drugs with oxygen and nitrogen present in the MOF structures. Here, we observe that atom-pairs that have the potential to form hydrogen bonds have stronger structural correlations as compared to RDFs with unsaturated metal sites in the MOF.24 For example, the oxygen atoms of drugs and nitrogen in the –NH2 functional groups in MOF can serve as hydrogen bond donors and acceptors, respectively, resulting in strong structural correlations.60,61
Further, the RDFs calculated for drugs in absence of ethanol show larger intensities of second and third peaks as in Fig. S24 of ESI,† which suggests that the most of the drugs are adsorbed in an empty pore volume. More details on structural correlations can be found out in Section S4 of ESI.†
The adsorption isotherms of drugs, the knowledge of preferred drug adsorption sites and the effect of solvent on drug adsorption at a range of different pressures still remains unexplored. Both temperature and pressure is known to play an important role in both drug solubility in a solvent as well as in its loading and drug release.62,63 The study of dependence of drug uptake on pressure is important as it may allow us to tune the amount of drug adsorbed in MOFs by simply tuning the surrounding pressure.62 As we have used three MOFs and three drugs with different stability and solubility, respectively, we have used a wide range of pressure to study drug adsorption. Specifically, we calculated adsorption isotherms in the external pressure range of 10−12 to 106 pascal at 27 °C in presence and absence of ethanol using GCMC simulations. These results are shown in Fig. 3. At atmospheric pressure, drug adsorption, in the presence of ethanol, follows the following trend: UiO-AZB > HKUST-1 > NH2-MIL-53, which is consistent with the PSD results, surface area, and surface chemistry of MOFs. In general, at high pressure (>102 Pa), drug adsorption in all frameworks is higher in the absence of ethanol. As we increase the external pressure in simulations in the presence of ethanol, adsorption of drug molecules decreases while ethanol increases. Interestingly, at low pressures (<10−2 Pa), adsorption of drug molecules is similar in the presence and absence of ethanol. The phenomenon is attributed to stronger drug–MOF electrostatic interactions compared to drug–solvent interactions at low pressures.24 IBU in HKUST-1 was adsorbed at lower pressure compared to both 5-FU and HU, which can be attributed to the stronger electrostatic interactions between IBU and HKUST-1. All three selected drugs were adsorbed in NH2-MIL-53 at reasonably lower pressures, which can be attributed to its smaller pore size and strong electrostatic interactions through the –NH2 functional group. Due to stronger electrostatic interactions between 5-FU and IBU with UiO-AZB, they are both adsorbed at lower pressures compared to HU. In general, NH2-MIL-53 starts adsorbing all three drug molecules at lower pressure compared to UiO-AZB and HKUST-1. This could be because in addition to the electrostatic interactions, NH2-MIL-53's smaller pore size can also play a significant role in enhancing drug adsorption.64
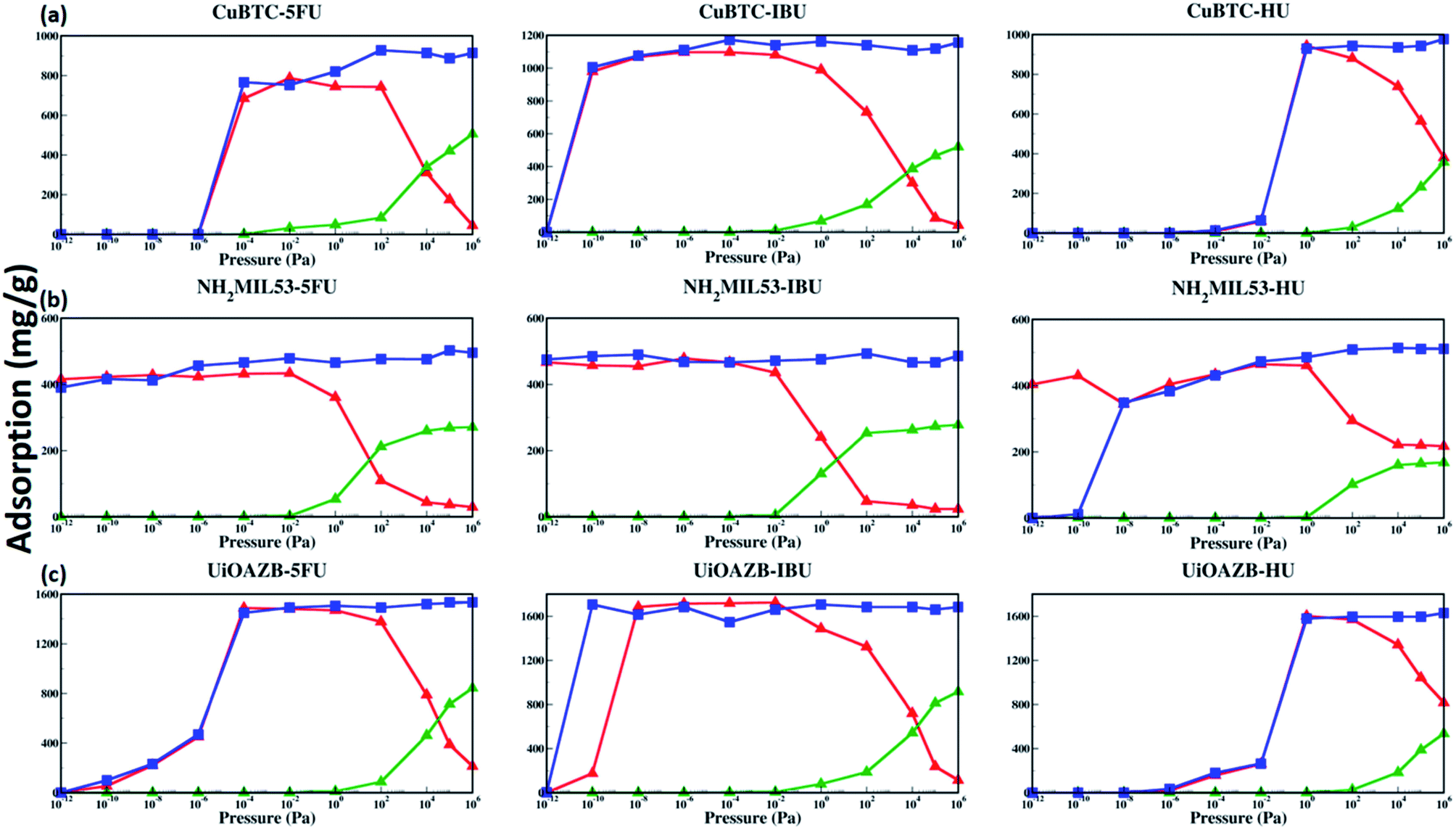 | ||
| Fig. 3 Adsorption isotherm of the drugs in the presence (red triangles) and absence (blue squares) of ethanol in (a) HKUST-1, (b) NH2-MIL-53 and (c) UiO-AZB. Adsorption of ethanol is also shown (green triangles). | ||
The host–adsorbate energy (UHA) can be a major contributing factor in determining the adsorption of adsorbate drug and solvent molecules in MOFs.29 The total potential energy of drugs in a MOF (i.e., total sum of host–adsorbate and adsorbate–adsorbate energy) regulates the drug entrapment and release mechanism.24 Therefore, by using the production run of the last 500000 GCMC cycles, we calculated UHA for all the systems at all simulated pressures as presented in Fig. 4. In general, UHA decreases with increase in the number of adsorbate molecules in a MOF. In the case of all MOFs, the UHA decreases with increasing pressure up to a certain value, and then it remains almost constant before and after the ethanol molecules are adsorbed in the MOFs. However, in all MOFs, at higher pressures adsorption of ethanol molecules increases compared to drug molecules, resulting in higher contribution of ethanol–MOF interactions in UHA. At these high pressures the host–adsorbate interaction energy for all three MOFs follows the trend HU + ethanol < 5-FU + ethanol < IBU + ethanol. This could be because, at high pressure, adsorption is dependent on the size of adsorbate molecules, and as HU is the smallest among all three drugs, more HU molecules are adsorbed.65–68 As expected, UHA was zero when no drug was adsorbed in HKUST-1 and UiO-AZB. However, for NH2-MIL-53 a non-zero UHA was observed at the lowest calculated pressures, which was attributed to the non-zero drug adsorption. Specifically, for HU with NH2-MIL-53, the lower pressure range (<10−8 Pa) shows larger peaks despite similar drug adsorption, which can be attributed to preferential electrostatic interactions within drugs and smaller pore size MOF.24 Moreover, despite the decrease in the adsorption of drugs at higher pressures (>104 Pa), the energy fluctuates around a certain constant value, indicating that ethanol–MOF energy contribution in UHA increases at higher pressure. For all three drugs, both at low and high pressures, the general trend of interaction energy is as follows: NH2-MIL-53 < UiO-AZB < HKUST-1. Indeed, at low pressure the number of drug molecules adsorbed in NH2-MIL-53 is higher compared to the other two MOFs. This can be attributed to stronger interactions between NH2-MIL-53, and drug and ethanol molecules due to its smaller pores.24 However, at high pressure the number of drug molecules adsorbed in MOFs is as follows: UiO-AZB > HKUST-1 > NH2-MIL-53, suggesting that adsorption increases with increase in MOFs pore size.
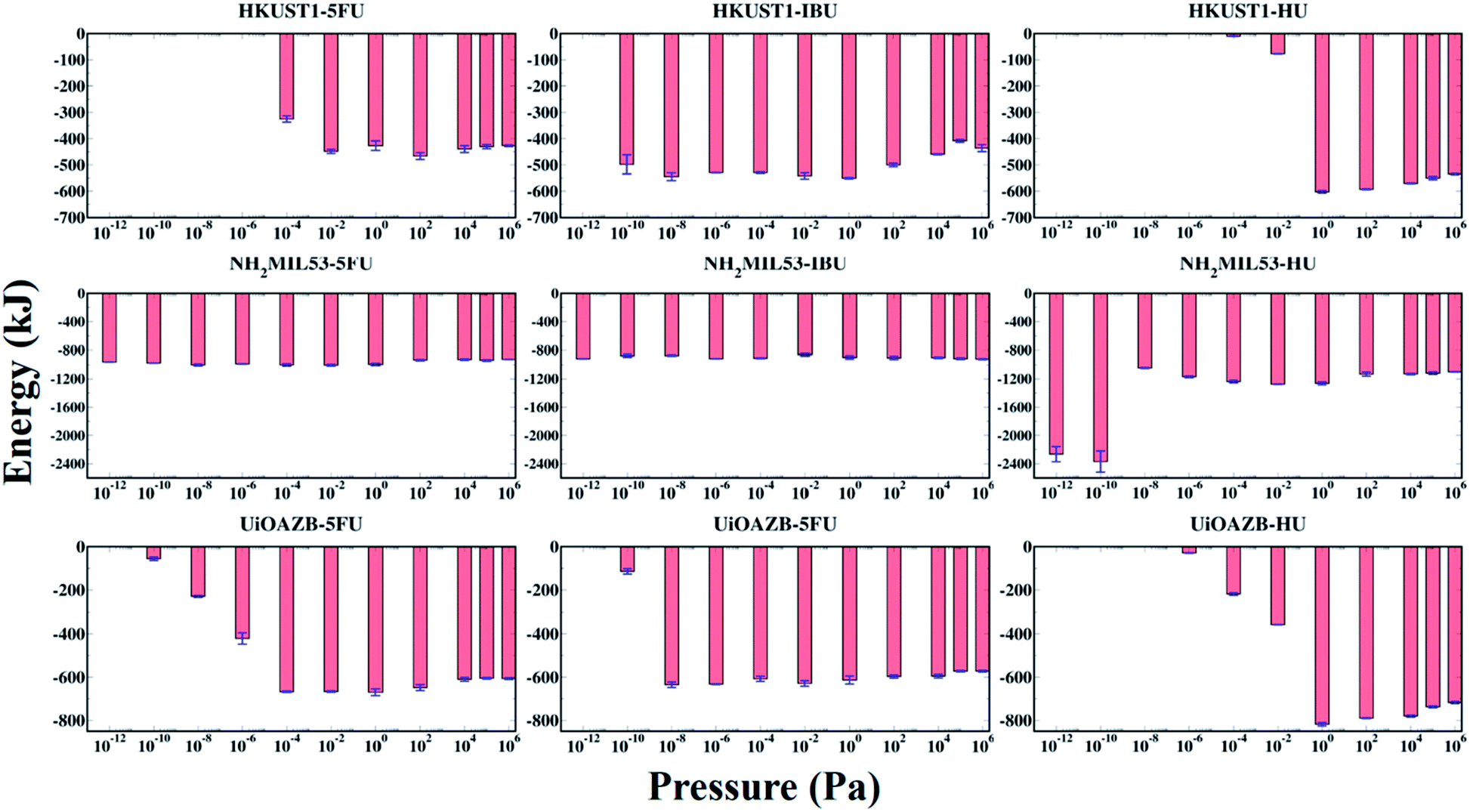 | ||
| Fig. 4 Host (MOF)–adsorbate (drug + ethanol) interaction energy (UHA) of all systems understudy. | ||
Conclusions
A combined computational and experimental study aimed to unravel the effect of solvent on drug adsorption in MOFs was carried out. We performed GCMC simulations to study adsorption of 5-FU, IBU and HU in UiO-AZB, HKUST-1 and NH2-MIL-53 MOFs in the presence and absence of ethanol at a range of pressures. Drug adsorption in the absence of ethanol was significantly overestimated when compared to experimental results. In the presence of ethanol, simulated adsorption showed better agreement with our experimentally measured drug adsorption at 300 K and 105 Pa. In general, at high pressures, the maximum uptake capacity for all MOFs, in the presence and absence of ethanol, was proportional to the size of the drug, with HU showing consistently higher adsorption than 5-FU and IBU. We also found that in the presence of ethanol at 105 Pa, drug molecules are adsorbed at the interface of MOF and ethanol through stronger structural correlation with the MOF metal atoms. In the absence of ethanol, however, drugs can be adsorbed inside the MOF pores. Interestingly, at lower pressures (<10−4 Pa) drug adsorption in the presence and absence of solvent was driven by electrostatic interactions between drugs and MOFs. As a result, solvent plays a critical role in the drug adsorption at atmospheric pressure while it does not have any impact at lower pressures. The host–adsorbate interaction (UHA) energy was affected by the drug size, MOF pore diameter, and simulated pressure. At low pressure, with decrease in pore size and increase in drug size UHA decreases. On the other hand, at high pressure, UHA decreases up to a certain value and then stabilizes, which also decreases with the pore size while the adsorption of drug molecules increases. Our study implies that solvent is very critical in adsorption of small molecules in porous materials and the impact of solvent on small molecules should be considered when designing new materials investigation, both experimentally and computationally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge Advanced Research Computing (ARC), Virginia Tech for providing the computational resources. This research used resources of the National Energy Research Scientific Computing Center (NERSC), a U.S. Department of Energy Office of Science User Facility located at Lawrence Berkeley National Laboratory, operated under Contract No. DE-AC02-05CH11231. This work was made possible by the use of Virginia Tech's Materials Characterization Facility, which is supported by the Institute for Critical Technology and Applied Science, the Macromolecules Innovation Institute, and the Office of the Vice President for Research and Innovation. The authors are also thankful to Hazel Thorpe Carman and George Gay Carman Trust for financial support.References
- A. Ben-Naim, Biopolymers, 1990, 29, 567–596 CrossRef CAS PubMed
.
- F. Gräter, P. Heider, R. Zangi and B. J. Berne, J. Am. Chem. Soc., 2008, 130, 11578–11579 CrossRef PubMed
- P. A. Bartlett, N. Yusuff, A. C. Rico and M. K. Lindvall, J. Am. Chem. Soc., 2002, 124, 3853–3857 CrossRef CAS PubMed
- C.-P. Li and M. Du, Chem. Commun., 2011, 47, 5958–5972 RSC
- X. Yang and A. E. Clark, Inorg. Chem., 2014, 53, 8930–8940 CrossRef CAS PubMed
- N. Perunov and J. L. England, Protein Sci., 2014, 23, 387–399 CrossRef CAS PubMed
- M. M. Teeter, Annu. Rev. Biophys. Biophys. Chem., 1991, 20, 577–600 CrossRef CAS PubMed
- B. U. Riebesehl, in The Practice of Medicinal Chemistry, ed. C. G. Wermuth, D. Aldous, P. Raboisson and D. Rognan, Academic Press, San Diego, 4th edn, 2015, pp. 699–722 Search PubMed
- Y. Liu, Y. Zhao and X. Chen, Theranostics, 2019, 9, 3122–3133 CrossRef CAS PubMed
- M.-X. Wu and Y.-W. Yang, Adv. Mater., 2017, 29, 1606134 CrossRef PubMed
- Y. Sun, L. Zheng, Y. Yang, X. Qian, T. Fu, X. Li, Z. Yang, H. Yan, C. Cui and W. Tan, Nano-Micro Lett., 2020, 12, 103 CrossRef CAS PubMed
- J. G. Vitillo and G. Ricchiardi, J. Phys. Chem. C, 2017, 121, 22762–22772 CrossRef CAS
- I. Erucar and S. Keskin, Ind. Eng. Chem. Res., 2016, 55, 1929–1939 CrossRef CAS
- D. Cunha, M. Ben Yahia, S. Hall, S. R. Miller, H. Chevreau, E. Elkaïm, G. Maurin, P. Horcajada and C. Serre, Chem. Mater., 2013, 25, 2767–2776 CrossRef CAS
- I. Erucar and S. Keskin, J. Mater. Chem. B, 2017, 5, 7342–7351 RSC
- J.-Q. Liu, X.-F. Li, C.-Y. Gu, J. C. S. da Silva, A. L. Barros, S. Alves Jr, B.-H. Li, F. Ren, S. R. Batten and T. A. Soares, Dalton Trans., 2015, 44, 19370–19382 RSC
- P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, J. Am. Chem. Soc., 2008, 130, 6774–6780 CrossRef CAS PubMed
- A. U. Czaja, N. Trukhan and U. Müller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC
- M. Ranocchiari and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2011, 13, 6388–6396 RSC
- B. Wang, L.-H. Xie, X. Wang, X.-M. Liu, J. Li and J.-R. Li, Green Energy Environ., 2018, 3, 191–228 CrossRef
- H. Li, K. Wang, Y. Sun, C. T. Lollar, J. Li and H.-C. Zhou, Mater. Today, 2018, 21, 108–121 CrossRef CAS
- R. Seetharaj, P. V. Vandana, P. Arya and S. Mathew, Arab. J. Chem., 2019, 12, 295–315 CrossRef CAS
- B. Zhang, J. Zhang, C. Liu, X. Sang, L. Peng, X. Ma, T. Wu, B. Han and G. Yang, RSC Adv., 2015, 5, 37691–37696 RSC
- M. C. Bernini, D. Fairen-Jimenez, M. Pasinetti, A. J. Ramirez-Pastor and R. Q. Snurr, J. Mater. Chem. B, 2014, 2, 766–774 RSC
- M. Gomar and S. Yeganegi, Microporous Mesoporous Mater., 2017, 252, 167–172 CrossRef CAS
- F. Li, B. Li, C. Wang, Y. Zeng, J. Liu, C.-Y. Gu, P. Lu and L. Mei, RSC Adv., 2016, 6, 47959–47965 RSC
- L. Bei, L. Yuanhui, L. Zhi and C. Guangjin, Acta Chim. Sin., 2014, 72, 942–948 CrossRef
- R. Babarao and J. Jiang, J. Phys. Chem. C, 2009, 113, 18287–18291 CrossRef CAS
- Y. G. Proenza and R. L. Longo, J. Chem. Inf. Model., 2020, 60, 644–652 CrossRef CAS PubMed
- L. R. Parent, C. Huy Pham, J. P. Patterson, M. S. Denny, S. M. Cohen, N. C. Gianneschi and F. Paesani, J. Am. Chem. Soc., 2017, 139, 13973–13976 CrossRef CAS PubMed
- Y. Li, X. Li, Q. Guan, C. Zhang, T. Xu, Y. Dong, X. Bai and W. Zhang, Int. J. Nanomed., 2017, 12, 1465–1474 CrossRef CAS PubMed
- C. C. Epley, M. D. Love and A. J. Morris, Inorg. Chem., 2017, 56, 13777–13784 CrossRef CAS PubMed
- M. Jorge, M. Fischer, J. R. B. Gomes, C. Siquet, J. C. Santos and A. E. Rodrigues, Ind. Eng. Chem. Res., 2014, 53, 15475–15487 CrossRef CAS
- M. Fischer, J. R. B. Gomes, M. Fröba and M. Jorge, Langmuir, 2012, 28, 8537–8549 CrossRef CAS PubMed
- J. Rouquerol, D. Avnir, C. W. Fairbridge, D. H. Everett, J. M. Haynes, N. Pernicone, J. D. F. Ramsay, K. S. W. Sing and K. K. Unger, J. Macromol. Sci., Part A: Pure Appl.Chem., 2009, 66, 1739–1758 Search PubMed
- N. Al-Janabi, P. Hill, L. Torrente-Murciano, A. Garforth, P. Gorgojo, F. Siperstein and X. Fan, Chem. Eng. J., 2015, 281, 669–677 CrossRef CAS
- S. Yang, L. Peng, D. T. Sun, M. Asgari, E. Oveisi, O. Trukhina, S. Bulut, A. Jamali and W. L. Queen, Chem. Sci., 2019, 10, 4542–4549 RSC
- K.-J. Kim, Y. J. Li, P. B. Kreider, C.-H. Chang, N. Wannenmacher, P. K. Thallapally and H.-G. Ahn, Chem. Commun., 2013, 49, 11518–11520 RSC
- H. K. Kim, W. S. Yun, M.-B. Kim, J. Y. Kim, Y.-S. Bae, J. Lee and N. C. Jeong, J. Am. Chem. Soc., 2015, 137, 10009–10015 CrossRef CAS PubMed
- J. L. C. Rowsell and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 1304–1315 CrossRef CAS PubMed
- M. Schlesinger, S. Schulze, M. Hietschold and M. Mehring, Microporous Mesoporous Mater., 2010, 132, 121–127 CrossRef CAS
- J. Gascon, U. Aktay, M. D. Hernandez-Alonso, G. P. M. van Klink and F. Kapteijn, J. Catal., 2009, 261, 75–87 CrossRef CAS
- S. Rostamnia and M. Jafari, Appl. Organomet. Chem., 2017, 31, e3584 CrossRef
- L. Bolinois, T. Kundu, X. Wang, Y. Wang, Z. Hu, K. Koh and D. Zhao, Chem. Commun., 2017, 53, 8118–8121 RSC
- X. Cheng, A. Zhang, K. Hou, M. Liu, Y. Wang, C. Song, G. Zhang and X. Guo, Dalton Trans., 2013, 42, 13698 RSC
- O. Weser and V. Veryazov, Front. Chem., 2017, 5, 111 CrossRef PubMed
- A. Vimont, A. Travert, P. Bazin, J.-C. Lavalley, M. Daturi, C. Serre, G. Férey, S. Bourrelly and P. L. Llewellyn, Chem. Commun., 2007, 3291 RSC
- L. Hamon, H. Leclerc, A. Ghoufi, L. Oliviero, A. Travert, J.-C. Lavalley, T. Devic, C. Serre, G. Férey, G. De Weireld, A. Vimont and G. Maurin, J. Phys. Chem. C, 2011, 115, 2047–2056 CrossRef CAS
- D. Bousquet, F.-X. Coudert, A. G. J. Fossati, A. V. Neimark, A. H. Fuchs and A. Boutin, J. Chem. Phys., 2013, 138, 174706 CrossRef PubMed
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC
- J. J. Wardzala, J. P. Ruffley, I. Goodenough, A. M. Schmidt, P. B. Shukla, X. Wei, A. Bagusetty, M. De Souza, P. Das, D. J. Thompson, C. J. Karwacki, C. E. Wilmer, E. Borguet, N. L. Rosi and J. K. Johnson, J. Phys. Chem. C, 2020, 124, 28469–28478 CrossRef CAS
- S. Wang, G. Zhou, Y. Sun and L. Huang, AIChE J., 2021, 67(3) DOI:10.1002/aic.17035
- S. A. Deshmukh, S. K. R. S. Sankaranarayanan, K. Suthar and D. C. Mancini, J. Phys. Chem. B, 2012, 116, 2651–2663 CrossRef CAS PubMed
- S. A. Deshmukh, Z. Li, G. Kamath, K. J. Suthar, S. K. R. S. Sankaranarayanan and D. C. Mancini, Polymer, 2013, 54, 210–222 CrossRef CAS
- C. H. Hendon and A. Walsh, Chem. Sci., 2015, 6, 3674–3683 RSC
- G. Gonzalez, A. Sagarzazu and T. Zoltan, J. Drug Delivery, 2013, 2013, 803585 Search PubMed
- R. Bueno-Perez, A. Martin-Calvo, P. Gómez-Álvarez, J. J. Gutiérrez-Sevillano, P. J. Merkling, T. J. H. Vlugt, T. S. van Erp, D. Dubbeldam and S. Calero, Chem. Commun., 2014, 50, 10849–10852 RSC
- S. S. Chui, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed
- E. Stavitski, E. A. Pidko, S. Couck, T. Remy, E. J. M. Hensen, B. M. Weckhuysen, J. Denayer, J. Gascon and F. Kapteijn, Langmuir, 2011, 27, 3970–3976 CrossRef CAS PubMed
- K. A. Connors, Chem. Rev., 1997, 97, 1325–1358 CrossRef CAS PubMed
- J. Zhong, Y. Zhao, L. Li, H. Li, J. S. Francisco and X. C. Zeng, J. Am. Chem. Soc., 2015, 137, 12070–12078 CrossRef CAS PubMed
- K. Jiang, L. Zhang, Q. Hu, D. Zhao, T. Xia, W. Lin, Y. Yang, Y. Cui, Y. Yang and G. Qian, J. Mater. Chem. B, 2016, 4, 6398–6401 RSC
- N. Kaur, P. Tiwari, K. S. Kapoor, A. K. Saini, V. Sharma and S. M. Mobin, CrystEngComm, 2020, 22, 7513–7527 RSC
- J. R. Karra and K. S. Walton, Langmuir, 2008, 24, 8620–8626 CrossRef CAS PubMed
- S. Li, Y. G. Chung and R. Q. Snurr, Langmuir, 2016, 32, 10368–10376 CrossRef CAS PubMed
- H. Daglar and S. Keskin, J. Phys. Chem. C, 2018, 122, 17347–17357 CrossRef CAS PubMed
- S. Keskin, in Metal-Organic Frameworks, InTech, 2016 Search PubMed
- S. Gautam and D. Cole, Nanomaterials, 2020 DOI:10.3390/nano10112274
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01746b |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |