
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethods of hexagonal boron nitride exfoliation and its functionalization: covalent and non-covalent approaches
Chandkiram Gautam
 *a and
Selvam Chelliah
b
*a and
Selvam Chelliah
b
aAdvanced Glass and Glass Ceramics Research Laboratory, Department of Physics, University of Lucknow, Lucknow, 226007, Uttar Pradesh, India
bDepartment of Pharmaceutical Sciences, Texas Southern University, Houston, USA. E-mail: gautam_ceramic@yahoo.com
First published on 23rd September 2021
Abstract
The exfoliation of two-dimensional (2D) hexagonal boron nitride nanosheets (h-BNNSs) from bulk hexagonal boron nitride (h-BN) materials has received intense interest owing to their fascinating physical, chemical, and biological properties. Numerous exfoliation techniques offer scalable approaches for harvesting single-layer or few-layer h-BNNSs. Their structure is very comparable to graphite, and they have numerous significant applications owing to their superb thermal, electrical, optical, and mechanical performance. Exfoliation from bulk stacked h-BN is the most cost-effective way to obtain large quantities of few layer h-BN. Herein, numerous methods have been discussed to achieve the exfoliation of h-BN, each with advantages and disadvantages. Herein, we describe the existing exfoliation methods used to fabricate single-layer materials. Besides exfoliation methods, various functionalization methods, such as covalent, non-covalent, and Lewis acid–base approaches, including physical and chemical methods, are extensively described for the preparation of several h-BNNS derivatives. Moreover, the unique and potent characteristics of functionalized h-BNNSs, like enhanced solubility in water, improved thermal conductivity, stability, and excellent biocompatibility, lead to certain extensive applications in the areas of biomedical science, electronics, novel polymeric composites, and UV photodetectors, and these are also highlighted.
 Chandkiram Gautam | Dr Chandkiram Gautam is an Assistant Professor (Stage-III) of Physics. He received his BSc in science and MSc in physics (electronics) from C. C. S. University, Meerut in 1995 and 1997, respectively. Dr Gautam obtained an MPhil degree in physics from IIT-Roorkee, India. He received an MTech in materials technology in 2000 and received a PhD in ceramic engineering in 2005 from IIT-BHU, Varanasi, India. He is a recipient of the Raman post-doctoral research award fellowship (2014–2015) from the Department of Nano Engineering and Materials Science, Rice University, Houston Texas, USA. Dr Gautam started his independent career at University of Lucknow and constructed a research program focusing on glass and glass ceramics for biomedical and electronic applications. His main focus is on the synthesis and design of hydroxyapatite and its composites with ZrO2, MgO, Al2O3, and h-BN for dental/bone restoration and sensing applications. |
 Selvam Chelliah | Dr Chelliah Selvam is an Associate Professor of Medicinal Chemistry at the Texas Southern University College of Pharmacy and Health Sciences. He studied pharmacy at Dr MGR Medical University (India) and received his MSc (Pharm) and PhD from National Institute of Pharmaceutical Education and Research, India. Dr Selvam worked as a post-doc researcher at the Universite Paris Descartes (2004–2007), Auckland Cancer Society Research Centre, The University of Auckland (2007–2008), The State University of New York at Binghamton (2008–2011), and South Dakota State University (2011–2012). His research interests focus on the design and synthesis of new chemical entities for the treatment of cancer, computer-aided drug design, and the synthesis of chemically modified RNA/siRNA for gene silencing activity. |
1. Introduction
h-BN is isoelectronic with graphene (it is also referred to as “white graphene”), and it consists of an equal number of boron (B) and nitrogen (N) atoms.1 Moreover, within each layer, B and N atoms are strongly bound together via sp2 covalent bonds. Between the various layers, weak van der Waals forces occur. Moreover, lip–lip ionic bonding interactions can also occur between various neighboring h-BN layers2 that are stronger in comparison to the weak van der Waals forces between layers of graphene, and the extensive band gap of h-BNNSs (∼4–6 eV)3 makes them an insulating material. Hence, the exfoliation of BNNSs from bulk h-BN is much tougher than flaking off graphene from bulk graphite. In contrast to graphene, h-BN shows numerous peculiar characteristics, like high-temperature strength, inherent electrical insulation, anti-corrosion capabilities, etc.4 Thus, these remarkable features have significant possible applications, e.g., in the fabrication of optical and electronic devices.5–9 Additionally, having a wide band gap of ∼5.9 eV, h-BN has also been useful for various technological applications like being used in far-ultraviolet light-emitting devices and high-performance electronic devices.10 There is no doubt that bulk h-BN and its composites have many applications in different areas, such as lubrication,11,12 biomedicine,13,14 electronics, and sensors;15,16 however, the few layer (2D) counterpart has more advanced applications.In the last few decades, graphene was treated as the ultimate material for the design and fabrication of numerous nanodevices. Therefore, it is worthwhile to investigate h-BN nanosheets (BNNSs) in combination with graphene. It was reported that due to the similar lattice parameters of BNNSs and graphene, epitaxial graphene on a BNNS may offer a wide-band-gap graphene material.17 Therefore, h-BN potentially may work as an outstanding gate dielectric material for the construction of graphene transistors, e.g., Dean et al. reported the growth of graphene devices on thin BNNS substrates, revealing mobilities and carrier inhomogeneities that were higher than SiO2 devices.18 Further, Wang et al. investigated the first kind of BN-graphene-BN field-effect transistor (FET) for radiofrequency applications.19
Moreover, in addition to several extensive studies on graphene,20–23 particularly the revolution in the fabrication of electronic (semiconductor) devices,24,25 a variety of research into h-BNNSs, including their exfoliation, functionalizing properties, and applications, has been performed. Numerous techniques,26–31 motivated by those used for the preparation of graphene, have been used to date to synthesize h-BNNSs. Earlier, it was reported that monolayer and few-layer BNNSs could be synthesized from bulk h-BN using various methods like mechanical milling techniques,32 chemical-solution-derived (CSD) techniques,33,34 and chemical vapor deposition (CVD) techniques.35–40 Nevertheless, mechanical milling techniques merely result in inadequate amounts of BNNSs. CSD techniques can harvest large quantities of BNNSs; however, these prepared BNNSs are nanosized with numerous layers. CVD techniques require ultrahigh vacuum systems and a high-temperature atmosphere, and they rely on quite exclusive prototypes, all of which are major difficulties for the preparation of large-scale BNNSs. Therefore, simple and low-cost techniques are now required to harvest large-scale and excellent-quality BNNSs. Current developments in the preparation and uses of h-BNNSs have resulted in enhanced performance when equated with bulk h-BN. For example, h-BNNSs can be utilized as non-wetting coatings,41 2D fillers,42,43 field emitters,44 thin supports for the high-resolution imaging of nanocrystals,45 and ultimate substrates for high-grade graphene electronics.46 Hypothetically, it has been reported that the fabrication of h-BNNSs with triangle defects can result in magnetism and improved half-metallicity,47 and hydrogenated boron nitride nanoribbons (BNNRs) might be used to realize semiconductor-half-metal-metal transition mechanisms and magnetic properties.48 For the preparation of h-BNNSs, mechanical cleavage techniques maybe provide processes with the minimum issues for scaling up. However, sonication-aided exfoliation techniques need suitable solvents, which are very costly, and need extraordinary care during handling. Approaches involving unzipping BNNTs have been accomplished by various researchers. Recently, few-layered BNNSs were generally fabricated via solid-phase mechanical techniques and liquid-phase exfoliation methods.49–53 Moreover, to perform these exfoliation techniques requires high energy and sufficient time. Hence, liquid exfoliation techniques are more effective in comparison to mechanical exfoliation.54–59 Owing to the use of huge quantities of chemicals and lengthy agitation times, and the obtaining of lower yields, these methods are not optimal.60–65 Additionally, several chemical impurities are inexorably present in the final BNNS product; for purification purposes, the product requires further intricate post-treatment. Hence, an ideal approach is urgently required for the fabrication of few-layered h-BN that will provide fast reaction times, nontoxic constituents, scalability, etc.66
Herein, we describe several exfoliation methods, like liquid-phase sonication, mechanical cleavage, controlled gas exfoliation, thermal exfoliation, and surfactant supported exfoliation, for the large-scale production of h-BNNSs, including low-cost, easily manageable, and scalable synthesis techniques for 2D h-BNNSs, which are extremely important. Further, we then describe several successful h-BN functionalization methods and their significant applications. In these methods, H2SO4, KMnO4, H2O2, etc. are utilized to exfoliate few-layer BNNSs from commercially available bulk h-BN powder.
2. Exfoliation
Exfoliation is attained via employing additional exterior force to overcome the attractive van der Waals interactions between material layers. The most frequently used methods are ultrasonication and chemical exfoliation techniques. During ultrasonication, shear forces and cavitation (the growth and collapse of micrometer-sized bubbles) act on the bulk material and induce exfoliation, while in the chemical processing of bulk h-BN compounds there is no need for high-temperature or high-vacuum conditions, with only wet chemical dispensation required. Due to these substantial advantages, this technique could be capable of the fabrication of large-scale BNNSs. The pressure arising due to the disintegration of several functional groups intercalated between the layers overwhelms the van der Waals attractions, assisting exfoliation.Various methods for the preparation of BNNSs may be considered as bottom-up approaches, which produce 2D nanosheets via the association of tiny molecules/used precursors into complex nanostructures. Interestingly, exfoliation methods generate 2D BNNSs from bulk materials and may be designated as top–down approaches. The intrinsic nature of layered materials means that strong in-plane covalent bonds are retained between various essential elements and weak van der Waals interactions occur between the involved layers of atoms. Nevertheless, it is quite possible to directly fabricate 2D nanosheets via refining atomic layers from their parent 3-D crystals, and hence exfoliation can be contrived. Indeed, there are numerous methods for exfoliating layered materials such as graphene and h-BN, either chemically or mechanically, and, therefore, we will deliberate these techniques in turn below.
2.1. Chemical exfoliation
The chemical exfoliation of layered (2D) nanomaterials, for example graphene and transition metal dichalcogenides (TMDs), has been effectively documented, and recently a few of the investigated approaches have gained universal appreciation, e.g., the Hummers' process for the synthesis and exfoliation of reduced graphene oxide (r-GO) from graphite.67 In contrast to techniques produced for the exfoliation of graphite, techniques for exfoliating h-BN to create BNNSs have not been as successful, in spite of the fact that there have been numerous reports on creating different h-BN chemical exfoliation techniques. Li et al. reported a typical synthesis of h-BNNSs using a chemical exfoliation technique with a yield of ∼0.191%.68 In the reported synthesis, the appropriate amounts of NaOH (2.0600 g) and KOH (2.7160 g) were mixed, and then fine h-BN powder (0.2480 g) was incorporated into the mixture for the exfoliation of h-BN. To homogenize the mixture, it was again mixed and transferred into a PTFE stainless-steel autoclave. The autoclave was heated at 180 °C for 2 h and then cooled to room temperature. Thereafter, the autoclave ampule was taken out from the furnace and the synthesized product was frequently washed using a washing process with ethanol and deionized (DI) water until the pH of the product was neutral. Hence, the following reaction take place during the synthesis of exfoliated h-BN:69| BN + 3NaOH → Na3BO3 + NH3↑ | (1) |
The achieved material was then dried in a vacuum oven at 100 °C for a soaking time of 12 h. Finally, a white crystalline material, i.e., exfoliated h-BN, was successfully synthesized. Further, to verify the exfoliation, various characterization techniques such as SEM, AFM, and TEM were used. These characterization techniques revealed the generation of the exfoliated material in the form of typical curved nanosheets and nanoscrolls of BNNSs (Fig. 1).68
 | ||
| Fig. 1 (a and b) Typical curved nanosheets and nanoscrolls of exfoliated BNNSs (circled in black), (c) AFM images of a nanosheet with curved edges (d) h-BN flake with several steps (e) the high-resolution images of the two encircled regions. (f and g) An isolated sheet and the corresponding electron diffraction pattern.68 | ||
Additionally, based on inspiration from a graphene oxide exfoliation method, Du et al. attempted to apply an improved Hummers' technique to the exfoliation of BNNSs.70 They reported that about 65 mg of BNNSs could be achieved from 1 g of h-BN, and this frugal technique produced micro-ordered large-sized BNNSs (2–3 layer thickness), as shown by the SEM results.70 The complete procedure comprises three steps: (i) the mixing of sulfuric acid (H2SO4) into h-BN (powder), which modified the hydrogen ions intercalated into layered h-BN and allowed the layer spacing to be engorged; (ii) the addition of potassium permanganate (KMnO4), with a reaction taking place along with H2SO4 that caused the development of MnO2; and (iii) the removal of O2 and MnO2 to form exfoliated BNNSs. A schematic diagram of the entire process is depicted in Fig. 2(A). Hence, the probable reactions that take place during the exfoliation of h-BN can be described as follows:
| 2H2SO4 + 4KMnO4 → 2K2SO4 + 4MnO2 (nanoparticles) + 3O2↑ + 2H2O | (2) |
 | ||
| Fig. 2 (A) A schematic diagram of the synthesis of BNNSs exfoliated from bulk BN powder using a chemical exfoliation method. (B) An SEM image of the bulk BN powder. (C) An SEM image of exfoliated BNNSs.70 (D) A schematic diagram of a fluorination-supported exfoliation mechanism for achieving F-BNNSs.72 | ||
Additionally, they also recommended that the entire BNNS exfoliation mechanism mainly occurred in two more basic steps, i.e., the first is the effective incorporation of H+ and MnO2 nanoparticles, and the second is the creation of O2, as shown in eqn (3):70
| MnO2 + 2H+ + H2O2 → 2H2O + Mn2+ + O2↑ | (3) |
Moreover, MnO2 nanoparticles play a significant role in protecting the BNNSs from re-stacking and, eventually, the MnO2 nanoparticles are eliminated using H2O2. In this way, BNNSs are exfoliated from bulk BN via the intercalation of MnO2 nanoparticles into the larger interlayers of bulk BN, and this was confirmed using in situ electron microscopy, as depicted in Fig. 2(B) and (C).
Lin et al. have reported the functionalization and exfoliation of h-BN using lipophilic and hydrophilic amine molecules.71 In a typical synthesis, octadecylamine (ODA, CH3(CH2)17NH2) and amine-terminated polyethylene glycol (PEG, O,O′-bis(3-aminopropyl)polyethylene glycol, NH2(CH2)3(OCH2CH2)nO(CH2)3NH2, where n = ∼35) were used. h-BN powder (50 mg) and ODA (500 mg) were taken in a round-bottom flask. These raw materials were mixed and then heated between ∼160 and 180 °C for 96 to 144 h under a nitrogen atmosphere. Further, this mixture was cooled to ambient temperature and approximately 15 mL of tetrahydrofuran (THF) was poured into the mixture. The prepared slurry was sonicated for 10 min and centrifuged; in this way, the supernatant fluid was gathered successfully. From the centrifugation, extraction cycles were repeated several times (5–8) using the residue and, hence, the supernatant fluid was merged as a THF dispersion of the final product, i.e., ODA-functionalized h-BN. Similarly, PEG was used as a functional molecule in a really similar process, except that DI water was used as the removal solvent. Consequently, an aqueous dispersion of PEG-functionalized h-BN (PEG-BN) was achieved as the final product. Herein, ODA/PEG and the h-BN surface played vibrant roles in this tactic for functionalization and exfoliation, which produced a high yield of BNNSs. Later on, Du et al.72 presented a new synthesis mechanism for the exfoliation of BNNSs using NH4F. They successfully exfoliated BNNSs using bulk h-BN and produced fluorinated BNNSs. This synthesis technique was observed to be surface-based and cost-effective. In this process, the entire exfoliation mechanism mainly contained four steps, which are as follows: (i) the fluorination of h-BN; (ii) the buckling of nanosheets on the surface; (iii) the insertion of NH4 ions; and (iv) the exfoliation of the fluorinated nanosheets (Fig. 2(D)).72 Remarkably, it was confirmed that at ambient temperature, the synthesized F-BNNSs showed ferromagnetic characteristics, proving their significant potential for use in the design of spintronic devices.72
Furthermore, Bhimanapati et al. have reported an easy and scalable tactic using a chemical route for the exfoliation and functionalization of h-BN.73 In the beginning, they used 1 g of h-BN powder with a particle size of 1–5 mm, and this was mixed with 6 g of KMnO4 powder in a glass beaker. A mixture of 135 mL of acid was synthesized separately through mixing the two distinct acids H3PO4 and H2SO4 at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8, and this was then transferred into the dry h-BN mixture. The prepared solution was then heated (75 °C) and mixing was performed for 12 h. Afterwards, ∼6 mL of H2O2 and 120 mL of DI water were poured gently into the mixture to inhibit oxidation. The subsequent material was cooled to room temperature and then centrifuged at 6000 rpm for 30 min. Moreover, ∼45 mL of DI water was poured onto this material and it was again centrifuged using the same rpm and time. During centrifugation, non-exfoliated material was eliminated successfully. Further, to achieve a pH > 3 and remove the metal ions, the supernatant solution was washed several times via centrifugation with DI water, ethanol, and HCl, respectively. Finally, the supernatant solution was spin-coated onto Si wafers and then dried to attain the exfoliated and functionalized h-BNNS. Electron microscopy and Raman spectroscopy analysis verified the preparation of exfoliated h-BN (Fig. 3(A–D)).73 Moreover, the broadness and intensity of the Raman peak was reduced significantly when compared with bulk h-BN. Due to a reduction in the layer thickness, a reduction in the peak intensity occurs, which is clearly revealed in Fig. 3(D). Interestingly, a shift in the peak was also observed, which is associated with strain and was produced due to the stretching of the nanosheets. Therefore, a red shift in the peak has been noticed in the Raman spectrum (Fig. 3(D)), which is further potential evidence for the fabrication of mono- and few-layered flakes of exfoliated h-BN.73
:8, and this was then transferred into the dry h-BN mixture. The prepared solution was then heated (75 °C) and mixing was performed for 12 h. Afterwards, ∼6 mL of H2O2 and 120 mL of DI water were poured gently into the mixture to inhibit oxidation. The subsequent material was cooled to room temperature and then centrifuged at 6000 rpm for 30 min. Moreover, ∼45 mL of DI water was poured onto this material and it was again centrifuged using the same rpm and time. During centrifugation, non-exfoliated material was eliminated successfully. Further, to achieve a pH > 3 and remove the metal ions, the supernatant solution was washed several times via centrifugation with DI water, ethanol, and HCl, respectively. Finally, the supernatant solution was spin-coated onto Si wafers and then dried to attain the exfoliated and functionalized h-BNNS. Electron microscopy and Raman spectroscopy analysis verified the preparation of exfoliated h-BN (Fig. 3(A–D)).73 Moreover, the broadness and intensity of the Raman peak was reduced significantly when compared with bulk h-BN. Due to a reduction in the layer thickness, a reduction in the peak intensity occurs, which is clearly revealed in Fig. 3(D). Interestingly, a shift in the peak was also observed, which is associated with strain and was produced due to the stretching of the nanosheets. Therefore, a red shift in the peak has been noticed in the Raman spectrum (Fig. 3(D)), which is further potential evidence for the fabrication of mono- and few-layered flakes of exfoliated h-BN.73
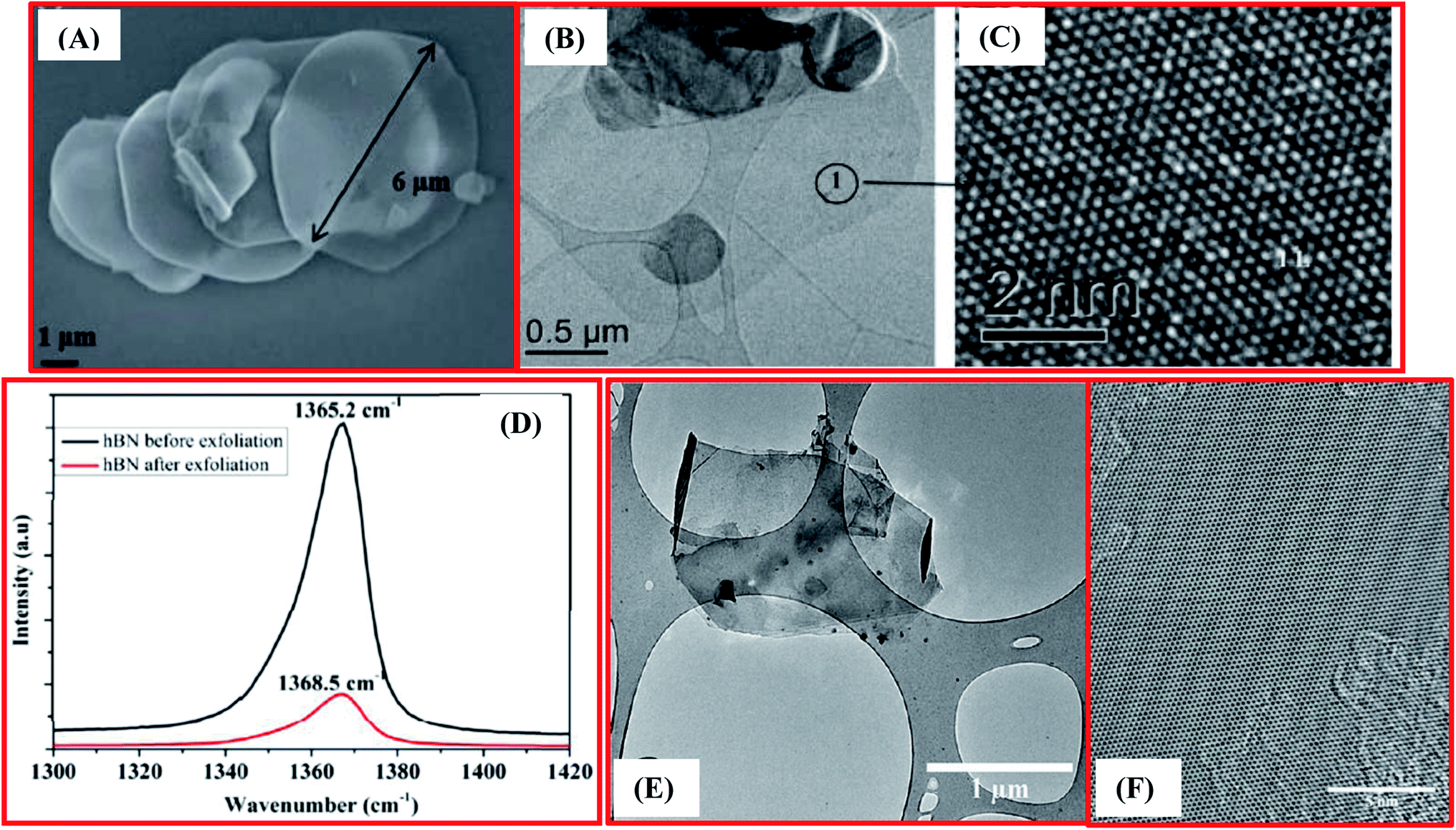 | ||
| Fig. 3 (A) An SEM image displaying exfoliated h-BN with a large flake size of 6 mm (B) TEM images showing monolayer h-BN (C) a high resolutionTEM image showing the atomic structure of h-BN. (D) Raman spectra of bulk h-BN exfoliated h-BN exhibiting a red shift of 3.3 cm−1, demonstrating a substantial reduction in nanosheets thickness.73 (E) TEM image of exfoliated h-BN nanosheets and (F) a HRTEM image of the surface of multilayer h-BN nanosheets.85 | ||
2.2. Exfoliation of h-BN via intercalation approaches
Motivated by the efficacious exfoliation of graphene through graphite intercalation complexes (GICs), various researchers have attempted to exfoliate h-BN using an intercalation approach with h-BN; for example, Doll et al. achieved the intercalation of BN films using potassium metal.74 In the beginning, they grew BN film with a thickness of ∼1–2 μm using a CVD technique. They successfully carried out a reaction with potassium (K), with the fabricated BN film placed into a Pyrex glass container with pure K. Afterwards, the container was evacuated at 10−6 torr, properly sealed, and then transferred into a double-zone electric furnace. BN and K were heat-treated at constant temperatures, i.e., 220 °C and 200 °C, for a soaking time of 24 h. The glass container was taken out from the furnace at room temperature, and the BN film turned a violet color. The change in color indicates that effective optical absorption takes place in the visible region, and this effective optical absorption was absent in pristine BN films. The change in color is due to a reaction between K, O2, and H2O. After the intercalation reaction, K atoms and BN formed a (2 × 2)R 0o in-plane structure, which corresponds with the BN lattice, and an optical transition redshift of 2.7 eV was observed, which was equated with a h-BN band gap of ∼5 eV.74 Further studies revealed the prominent intercalation of K into BN and also the exfoliation of BNNSs. Subsequently, Lin et al. reported a technique for synthesizing monolayer BN quantum dots (QDs) with a lateral size of ∼10 nm via intercalating potassium into h-BN flakes.75 The complete synthesis of monolayer BN QDs mainly involved three steps: (i) the h-BN flakes and potassium (K) were mixed and transferred into a glass tube (Pyrex) and then heat treated from 190–200 °C for 10 h in a vacuum environment until the color of the material entirely changed to gray; (ii) the glass tube was cooled to ambient temperature and taken out from the furnace, and the prepared K-hBN was immediately exposed to atmospheric air and then permitted to react with ethanol/water (EtOH/H2O) using an ultrasonication-supported system; and (iii) residual impurities were eradicated via filtration and centrifugation processes, followed by the use of cation exchange resin.75 Only 2.1 wt% exfoliated BNNS QDs were achieved successfully, and their presence was confirmed using several spectroscopy and electron microscopy characterization methods like Raman, FT-IR, UV-vis, XPS, TEM, and AFM. Moreover, the Raman study revealed a slightly up-shifted E2g phonon mode of h-BN within the wavenumber range of 1366.5–1367 cm−1 compared with raw h-BN flakes under similar measurement conditions, which showed a decrease in the in-plane lattice constant.75,76 In addition to potassium (K) intercalation, lithium (Li) was also used for intercalation and the subsequent exfoliation of h-BN.77 Furthermore, Zeng et al. effectively employed an electrochemical process to intercalate lithium into some 2D nanomaterials, for examples BN, NbSe2, WSe2, and Sb2Se3, to achieve few-layer dense nanosheets.78 Recently, Zhao et al. carried out the electrochemical intercalation of lithium (Li) ions into van der Waals heterostructures sandwiching graphene between h-BN.79 They also revealed that encapsulating graphene in h-BN eradicates parasitic surface reactions while concomitantly producing a new hetero-interface, which allows intercalation between numerous atomically thin layers. Additionally, based on bipolar electrochemistry, Wang et al. demonstrated an accessible and time-effective bipolar electrochemical technique for the exfoliation of a bulk insulator material, i.e., layered h-BN into few-layered h-BNNSs. This technique using a nonconductive substance, h-BN, unlocks the system for various applications in wide-ranging fields, including electronics and biomedical sciences.80Further, Ortiz et al. reported the exfoliation of h-BN in the liquid phase using a green ion-intercalation approach and successfully produced few layered h-BNNSs from pristine h-BN using a two-step process.81 Firstly, the dispersion of pristine h-BN was carried out using an aqueous solution containing gelatin and KCl/ZnCl followed by sonication. During the second step, the elimination of large-size exfoliated h-BNNSs was carried out via a centrifugation process. Further, they found that exfoliation was achieved not only due to the influence of the sonication process but also due to an intercalation process involving K+ and Zn2+ ions. Based on the TEM, XRD, and Raman spectroscopic results, they achieved 2–3-layer h-BNNSs with an appreciable yield of 16.3 ± 0.4%.81 More recently, Kheirabadi et al. theoretically presented the intercalation of BN QDs with Li for lithium-ion battery and spin-dependent photon emission device applications using first-principles DFT concepts. Finally, they revealed that BN works as a protective layer in lithium-ion batteries.82
2.3. Exfoliation of h-BN via liquid-phase sonication methods
Apart from chemical exfoliation techniques, it is worth describing a new kind of technique for the exfoliation of h-BN, i.e., liquid-phase sonication. It was reported for the first time by Han et al. that the exfoliation of BNNSs from highly crystalline transparent h-BN could be effectively achieved using a liquid-phase sonication method.83 In this method, 0.2 mg of crystals of h-BN were mixed with 5 mL of 1,2-dichloroethane solution at a concentration of 1.2 mg/10 mL of poly(m-phenyl-enevinylene-co-2,5-dictoxy-p-phenylenevinylene), and this was then sonicated for exfoliation for up to 1 h. The entire synthesis method was comparable to a method that has been used for the preparation of graphene nanoribbons.84 Afterward, dynamic ultrasonication along with centrifugation was performed by Zhi et al. to exfoliate h-BN using an efficient polar solvent, dimethylformanmide (DMF), and large-scale exfoliated BNNSs were achieved.34 By employing this method, a comparatively high yield of BNNSs can be obtained (∼1 mg).34 In the same way, attempts were made by Warner et al. to carry out the liquid-phase exfoliation of h-BN using the solvent 1,2-dichloroethane in an ultra-sonication bath.85 Usually, this technique is capable of fabricating scalable (microscale) adjacent few-layered BNNSs (Fig. 3(E and F)). Moreover, they studied HRTEM images and compared them with theoretical simulations, verifying the AB stacking of BN bilayers, while AA stacking is excluded.85 Further, Wang et al. reported another method for the liquid-phase exfoliation of h-BN using methane sulfonic acid (MSA).86 In this method, they mixed 0.2 g of h-BN with 100 mL of MSA and kept this in an ultrasonic bath for sonication for up to 8 h; thereafter, the homogeneous mixture was centrifuged at 4000 rpm for 1.5 h, and then the residue was properly washed and dried.86 In this way, with the help of this method, BNNSs with a layer thickness of ∼3 nm and a yield of 0.3 mg can be produced using 21 mL of MSA.86 In order to confirm the preparation of the BNNSs, several microscopic characterization techniques were used, such as SEM and TEM, and the results were compared with pristine h-BN. On the basis of these microscopic results, it was noticed that most of the pristine h-BNs were in the form of thick flakes with lateral sizes from ∼100 nm to 2 μm, as shown in Fig. 4(a). TEM images of the microstructures of large-number and few-layer BNNSs are depicted in Fig. 4(b) and (c). From these TEM images, it is perceived that the thickness of the BNNSs is substantially reduced compared with the h-BN particles. Further, it has also been noticed that the lateral sizes (≥500 nm) of these BNNSs were reduced compared with h-BN, which might be due to the mechanical forces applied on the h-BN sheets (Fig. 4(b and c)). However, the SAED image of few-layered BNNSs exhibits unique six-fold h-BN symmetry, thus signifying the highly polycrystalline nature of the BNNSs (Fig. 4(c)).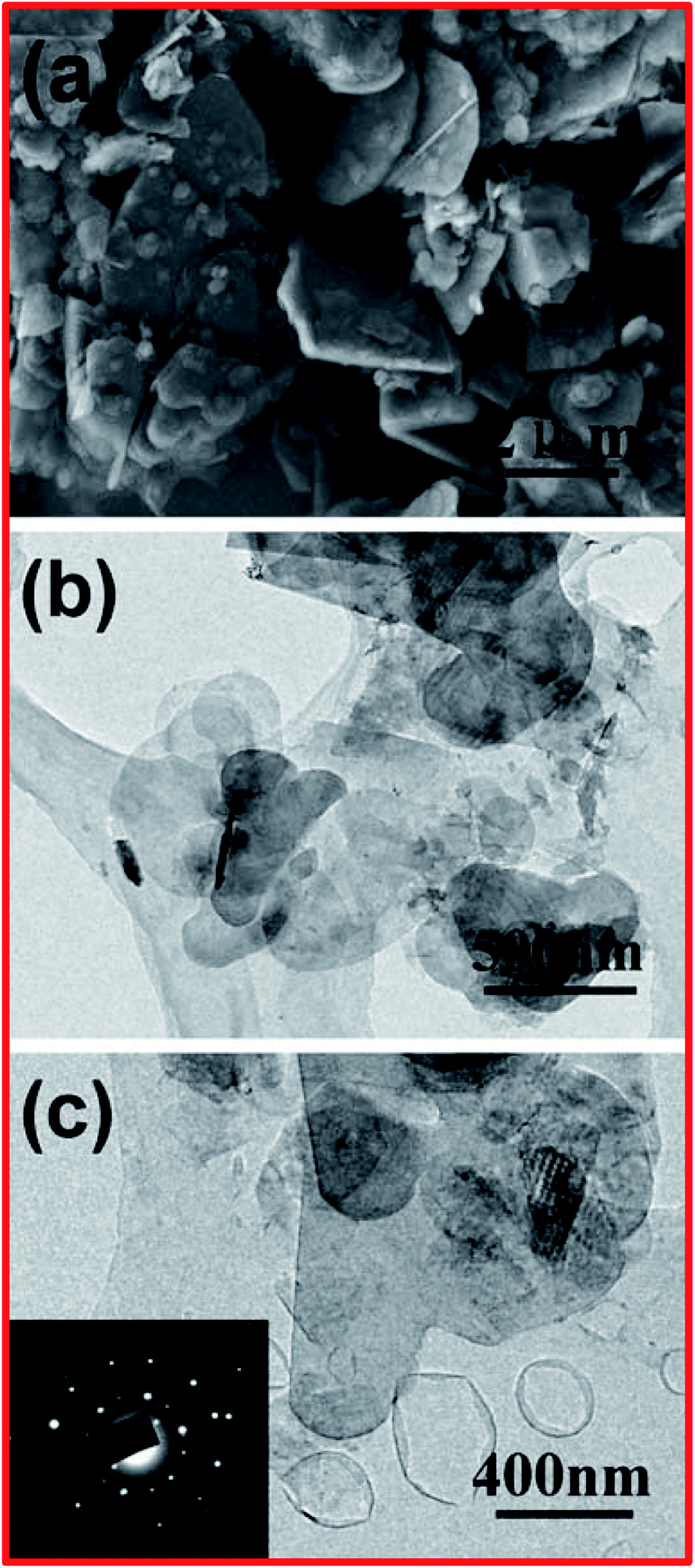 | ||
| Fig. 4 (a) An SEM image of h-BN particles. (b) A TEM image of large-number BNNSs. (c) A TEM image of few-layered BNNSs; the inset shows the selected area electron diffraction (SAED) pattern.86 | ||
Other than carbon-based solvents, it has been revealed that water is also a powerful solvent for the exfoliation of BNNSs during ultra-sonication. Further, Lin et al. revealed that fresh and clean aqueous dispersals of BNNSs could be obtained through sonicating h-BN in DI water, which can be described as a hydrolysis process (sonication-assisted).33 Based on ammonia tests and spectroscopic data, it has been verified that the sonication-assisted method can encourage the formation of exfoliated BNNSs.33
Besides that, Coleman et al. demonstrated the exfoliation of layered materials (2D), for example MoS2, WS2, and BN, via employing an ultrasonic bath.87 They confirmed that bulk transition metal dichalcogenides (TMDs), h-BN, transition metal oxides (TMOs), and other materials (Bi2Te3 and Bi2Se3) might be exfoliated in a common solvent into single-layer/few-layer nanosheets. Moreover, this process exhibited a simple nature, owing to the inertness toward air and water, and it can produce an adequate amount of nanosheets.87 Further, it was reported by Xue et al. that the exfoliation of BNNSs could be achieved from bulk h-BN through the heat treatment of pristine BN in an organic solvent, i.e., isopropanol, at 508 °C for 24 h; ultrasonication was then employed for 20 h along with centrifugation at 14000 rpm.88 The acquired material was used to fabricate fluorinated BNNSs with HBF4, and it was confirmed that the synthesized F-BNNSs offered remarkable electrical conductivity, with the altered nanosheets exhibiting semiconducting properties.88 The exfoliated layered materials, which are known as inorganic analogues of graphene (IAG), e.g., MoS2,89,90 WS2,91 h-BN,92 h-BCN, and graphitic carbon nitride (g-C3N4), are presented in Fig. 5(A).93 Stengl et al. employed a high-intensity ultrasound exfoliation method based on mutual similarities with inorganic graphene, getting a large yield of up to 100% in a few minutes.93 In this method, a highly pressurized ultrasonic reactor was used to produce a cavitation field with extraordinary power, and graphene equivalent materials, like h-BN, WS2, MoS2, and g-C3N4, were synthesized in the suitable solvents of N-methyl-2-pyrrolidone and N,N-dimethylformamide/dimethyl sulfoxide, with sonication carried out for at least 20 min at a reactor pressure of 6 bar.93 Further, to verify the efficiency of exfoliation, several characterization techniques were performed, like XRD, TEM, and AFM, and these validated the efficacy of exfoliation through this synthesis method. Fig. 5(B) reveals the XRD patterns of the synthesized bulk h-BN, h-BCN, and g-C3N4, and the XRD patterns of exfoliated h-BN, h-BCN, and g-C3N4 (inset). The XRD pattern of bulk h-BN revealed a diffraction peak at a 2θ angle of 25.5° and a low intensity peak at 42.7°, which correspond to the (002) and (100) lattice planes, respectively. The h-BCN XRD pattern comprises three different low intensity peaks at 26.4° 42.3°, and 54.8°, which are concomitant with the (002), (100), and (004) lattice planes, respectively. Moreover, g-C3N4 reveals two different peaks, including a high intensity peak at 27.65° which is characteristic of a graphitic material, attributed to the (002) lattice plane, and a low intensity peak at 13.01° (100), giving an interplanar distance of 0.676 nm.94 Once the exfoliated samples were dehydrated, all the characteristic peaks of the raw IAGs, MoS2, WS2, h-BN, h-BCN, and g-C3N4, recurred significantly. The locations of the high intensity peaks corresponding to the (002) plane, with a 2θ value of 14.3° for MoS2 and WS2, 26.0° for h-BN and h-BCN, and 28.0° for g-C3N4, were further used to estimate the particle sizes and interlayer spacings. Therefore, these XRD results verified the exfoliation of IAGs. Moreover, the organic solvents used in this method, like DMF, THF, NMP, and DMSO, played a significant role in improving the effectiveness of exfoliation and the stability of the fabricated BNNSs. Conversely, owing to the toxic and expensive nature of the used solvents, some researchers have tried to substitute them with ecofriendly solvents. Cao et al. investigated the simple, cost-effective, and large-scale production of exfoliated BNNSs in a liquid-phase medium, using a new facile and top-down approach.95 In this approach, they investigated a novel mixture of solvents, i.e., ammonia water solution/isopropyl alcohol, that played a significant role in the large-scale synthesis of stabilized BNNSs in solution. Further, they demonstrated that Lewis acid–base interactions were responsible for the entire mechanism of the exfoliation of h-BN, which is because of the electron-deficient boron atoms.95
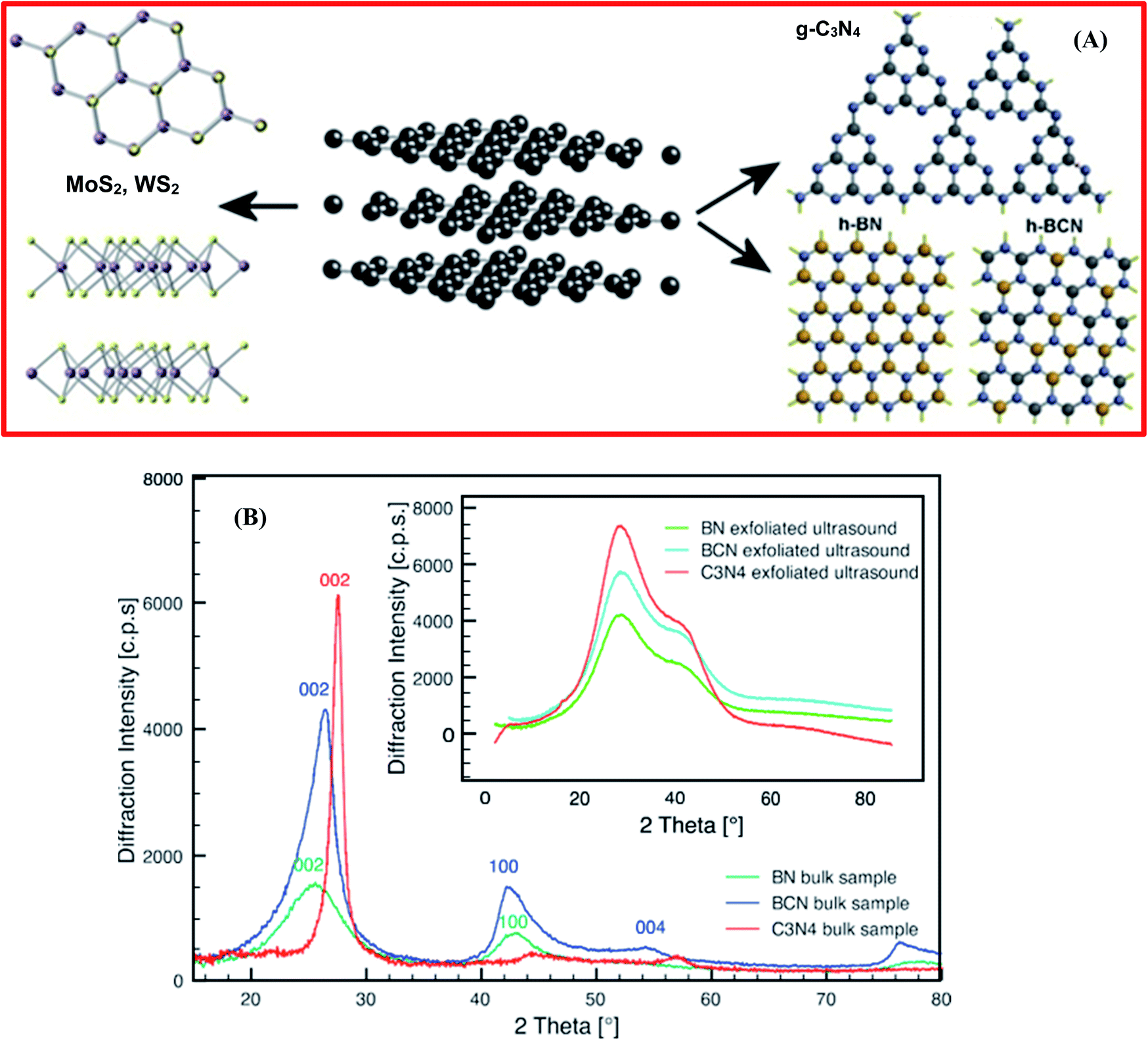 | ||
| Fig. 5 (A) The exfoliated structures of inorganic analogues of graphene – MoS2, WS2, g-C3N4, h-BN, and h-BCN. (B) XRD patterns of bulk synthetic samples of h-BN, h-BCN, and g-C3N4. However, the inset shows the XRD patterns of the ultrasound-exfoliated samples.93 | ||
Zhang et al. employed a highly efficient liquid exfoliation technique to obtain BNNSs using an aqueous solution of monoethanolamine (MEA).96 In order to fabricate the BNNSs, initially, pristine h-BN powder (∼200 mg) and MEA were mixed in a 200 mL beaker, followed by sonication for 4 h at 50 °C using a bath sonicator operated at 40 kHz and 150 W; the resulting solution was then centrifuged at 3500 rpm for a soaking time of 20 min. The supernatant was filtered, washed frequently using ethanol, and then dried under vacuum at 100 °C for 12 h. Thereafter, white BNNS powder was achieved successfully. Moreover, they also reported that a solution of MEA could exfoliate BNNSs more competently than the presently recognized solvents, and a high yield of ∼42% was achieved using an ultrasonication process in 30 wt% MEA aqueous solution.96 Finally, the improved performance of these exfoliated BNNSs in epoxy resin was demonstrated, including improved thermal and mechanical polymer properties. For the synthesis of large-sized BNNSs, Yuan et al. reported a low-temperature thermal-expansion-assisted ultrasonic scalable exfoliation approach.97 Initially, this approach employs the hydrogen-prompted low-temperature thermal reduction of OH-functionalized bulk h-BN (BN–OH) to damage the interlayer interactions between contiguous layers; thereafter, a sonication process is utilized to exfoliate BNNSs from bulk h-BN. Consequently, few-layer and large-sized BNNSs with an adequate yield of 26% were achieved using this approach. Furthermore, the achieved large-area BNNSs could improve the thermal conductivity of thermoplastic polyurethane (TPU) owing to reducing the interfacial thermal resistance.
Besides that, Ye et al. adopted a facile approach for the preparation of BNNSs using a liquid-phase exfoliation technique from bulk h-BN in common organic solvents, including using hyperbranched polyethylene (HBPE) as stabilizer.98 A schematic diagram of the mechanism for the exfoliation of BNNSs from bulk h-BN in chloroform under sonication with HBPE as a stabilizer is displayed in Fig. 6(a). Herein, HBPE enhances h-BN exfoliation in tetrahydrofuran (THF) and chloroform (CHCl3) during the sonication process. Validating the preparation of h-BNNSs, atomic force microscopy and Raman spectroscopy results exhibited the formation of 2D flake-shaped exfoliated BNNSs from the dispersion, and this was compared with pristine h-BN (Fig. 6(b and c)).98 Moreover, regulating the amounts of these solvents can control steady dispersions of fine monolayer/few-layer BNNSs significantly. Further, it was verified that noncovalent CH–π interactions are initiated between HBPE and the BNNS surfaces; irreversible HBPE adsorption averts reaggregation successfully. Hence, the subsequent HBPE-functionalized BNNSs are extremely prone to disintegrate in CHCl3/DMF at a high concentration of ∼10.0 mg mL−1, and they could be utilized as a nanofiller for fluorinated polymeric applications.98 Recently, Zheng et al. reported the preparation of exfoliated h-BN/graphene heterostructures using a facile and scalable liquid-phase exfoliation technique for supercapacitor applications, without the use of any chemical cross-linkers.99 Initially, they dissolved graphite/h-BN powder at a 2:1 molar ratio in a urea/glycerol dispersion. Thereafter, 200 mL of the dispersion of graphite/h-BN was poured into a flat-bottomed 800 mL beaker, in which the graphite/h-BN powder was exfoliated and disseminated via mechanical stirring at 800 rpm for 24 h. The achieved material was poured into a centrifuge tube and centrifuged at 5000 rpm for up to 25 min. The centrifuged graphite/h-BN dispersal was gathered and again disseminated in DMF, followed by filtration and washing with great amounts of DMF and ethanol, and it was then dehydrated using a vacuum atmospheric oven at 60 °C. Further, dispersed solutions of graphene/DMF and h-BN/DMF were mixed with graphene/h-BN mass ratios of 1:2, 1:1, and 2:1. The achieved homogeneous solutions were sonicated for up to 30 min, stirred at RT for 1 day, and then centrifuged at 1000 rpm for half an hour. The residual liquid was taken out and, finally, a solid precipitation was attained as an exfoliated h-BN/graphene material. More recently, Mittal et al. reported an innovative approach for the fabrication of large-scale h-BNNSs using a liquid-phase exfoliation method.100 In this method, they used polar solvents, decreasing the complications that arise when using high-boiling-point solvents.101 The prepared solvent containing a mixture of IPA and DI water at a molar ratio of 3:7 was used for exfoliation, having a surface energy comparable to that of h-BNNSs. This method is a green, basic, and cost-effective way to fabricate large yields of h-BNNSs, and it displayed improved outcomes that were verified using several characterization techniques, viz., XRD, Raman spectroscopy, and SEM. Furthermore, an ion-assisted liquid-phase exfoliation technique was employed to synthesize h-BNNSs by Wang et al.102 They confirmed that the yield of h-BNNSs might be enhanced by up to 12.75% via decreasing the size of the cations in an aqueous solution of LiOH. This is because small-radius cations are easily absorbed on the upper surface of bulk h-BN and then introduced into interlayer spaces, playing a crucial role in the effective synthesis of h-BNNSs in aqueous solutions.102
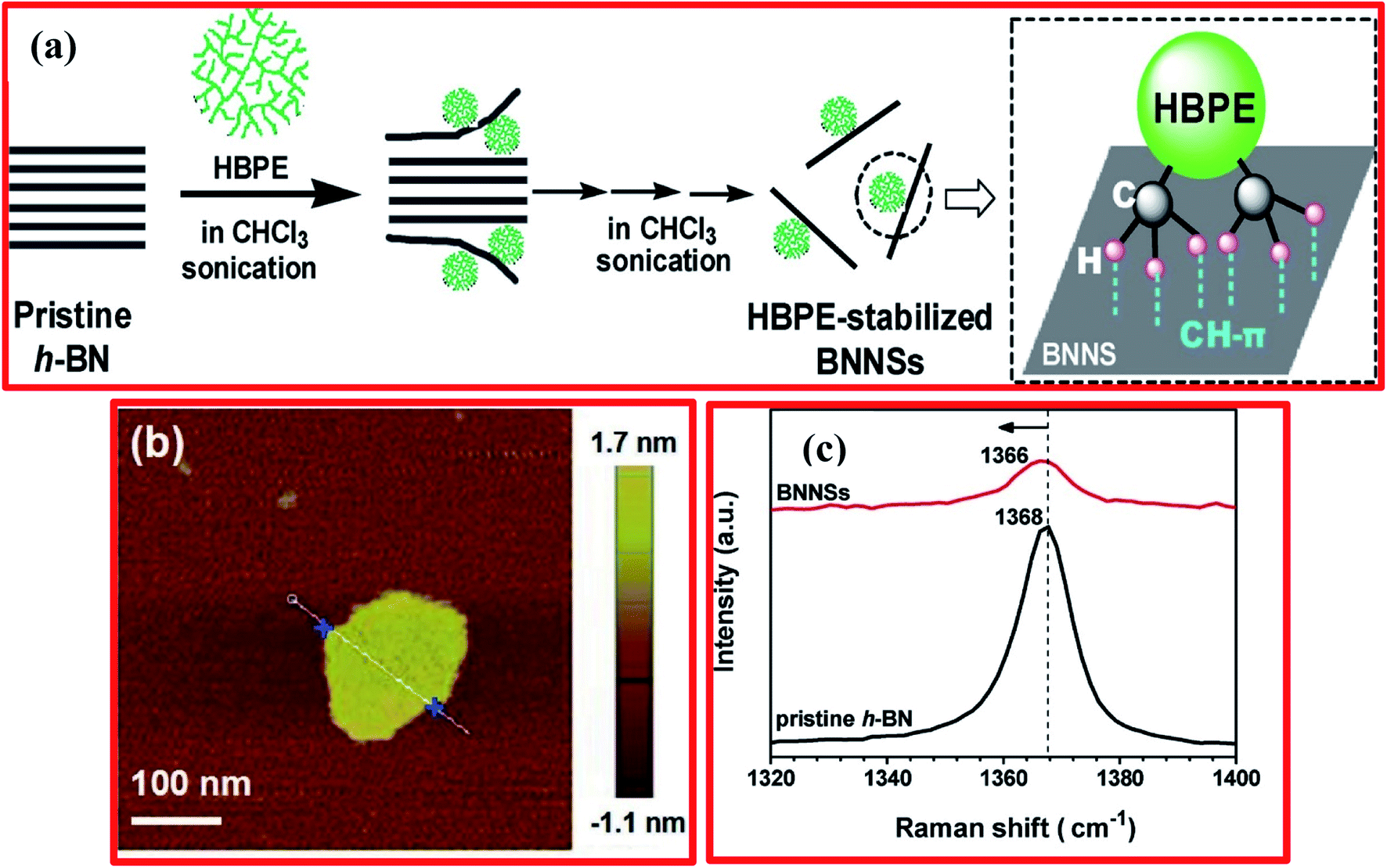 | ||
| Fig. 6 (a) A mechanism for the exfoliation of BNNSs from bulk h-BN in chloroform under sonication with HBPE as the stabilizer. (b) A wide-field AFM image showing the formation of 2D flake-shaped exfoliated BNNSs. (c) The Raman spectra of BNNSs from the dispersion and pristine h-BN.98 | ||
2.4. Mechanical cleavage
Free-standing graphene was prepared for the first time via a mechanical cleavage approach using the Scotch tape method and, similarly, other layered materials like h-BN and MoS2 can be obtained in this way.103–105 In this approach, the pulling energy breaks down the weak van der Waals interfaces between layers of graphene, leaving the sturdy sp2-based in-plane structure, e.g., a shear force can obtain comparable outcomes. Although the liquid-phase exfoliation of few-layered materials (2D) has been extensively employed, mechanical cleavage approaches which exfoliate via generating high-strength shear forces have also gained significant interest. Nevertheless, mechanical cleavage can reduce the creation of good quality h-BNNSs owing to structural defects, and it can also produce extremely low yields.98 Several methods have been studied for exfoliating nanosheets using pristine 3-D bulk materials.106–115 Alem et al. reported an adhesive tape and reactive ion-etching (RIE) technique to fabricate single-layer and multilayered h-BNNSs.116 Initially, they used PT 110 h-BN powder that was mechanically peeled via an adhesive tape method; this was then shifted onto a Si wafer with an oxide layer of 300 nm, and lastly transferred onto a copper grid for recording TEM images. Moreover, exfoliation was carried out using a RIE technique to attain a h-BN monolayer. Further, a layer number map was obtained to validate the monolayer thickness of the synthesized h-BNNSs.116 In order to carry out h-BN exfoliation, Pierret et al. have reported a comparable adhesive tape method followed by a mechanical peeling approach.110 They verified the existence of a strong relationship between defects in h-BNNSs and the recombination intensity of captured excitons. Besides this approach, other approaches were also used such, as shear force and ball-milling techniques, to exfoliate bulk h-BN to h-BNNSs.117In the last few years, scientists have recommended that ball milling (top down) is one of the best approaches for the fabrication of nanomaterials, particularly for the exfoliation of nanomaterials. Li and coworkers described a low-energy ball-milling technique, using a milling agent like benzyl benzoate (C14H12O2) to create moderate shear force to attain the high-yield exfoliation of h-BNNSs.32 It has already been stated that there is a fixed protocol by which BNNSs can be achieved with an in-plane structure.118 Moreover, ball-milling exfoliation successfully decreases the thicknesses of layers and produces some defects in the in-plane structure of BNNSs.32
In the same way, Liu et al. also employed a ball-milling technique for the preparation of few-layered h-BNNSs using a mechanical cleavage approach.119 Ammonia borane (BH6N) was utilized as a milling agent; it was mixed with bulk h-BN at an appropriate ratio and a ball-milling technique was successfully carried out. Hence, by using this method, the formation of 2D h-BNNSs can be scaled up.119 Further, Deepika et al. optimized several milling parameters, i.e., the speed of milling, the ball-to-h-BN ratio, the size of the ball, and the used milling agent, to adjust the grade of the exfoliated h-BNNSs.120 Based on the optimized parameters, it was evaluated that a 13.8% yield of few-layered BNNSs could be achieved successfully using C14H12O2 as the milling agent and milling balls with a diameter of 0.1–0.2 mm. Besides the yield, tribological studies showed that exfoliated BNNSs could reduce the friction coefficient and wear scar diameter of a base oil significantly when used as a dopant.119 Further, the ball-milling process has been modified using NaOH.106 By adopting this technique, the yield was increased to some extent, i.e., to 18% over the aforementioned 13.8%. It was also investigated whether the fabricated OH-BNNSs could be dispersed in distinct solvents, and the exfoliated OH-BNNSs might be used to formulate polymeric-type composites for numerous applications.107 In addition to this technique, Damm et al. have investigated a scalable process for the delamination of h-BN in a non-ionic surfactant solution of TWEEN85 containing ZrO2 as the grinding media via stirred-media milling.112 Based on the AFM results, they noticed that the dimensions and aspect ratio of the h-BNNSs achieved are predominantly correlated to the ZrO2 bead size. As the size of the grinding medium decreased from 0.8 mm to 0.1 mm, the average flake thickness of h-BNNSs was observed to be reduced from 3.5 nm to 1.5 nm, while the value of the aspect ratio increased from 2200 to 5800.112
In order to increase the thermal conductivity of epoxy resin, recently, Jinrui Ma and co-workers demonstrated the exfoliation of few-layered h-BN via surface modification using a wet ball-milling technique.121 In this technique, pristine BN powder was mixed in a urea solution and then ball milled for soaking times from 2–16 h; thereafter, it was centrifuged at 8000 rpm for 30 min, and the same process was repeated two times to attain the urea-free product. Further, to maintain the pH at around 7, DI water was poured into the ball-milled product, followed by centrifuging at 3000 rpm for 30 minutes; then, the supernatant precipitate was collected via a filtration process. The entire synthesis process is depicted in Fig. 7(a). Eventually, they also claimed that the BN gained after 8 h of milling showed an excellent improvement in thermal conductivity, i.e., the conductivity was ∼3 times that of pure epoxy resin.121 For the fabrication of m-BN via a wet ball-milling technique, the mechanism is presented in Fig. 7(b). In this mechanism, the particle size of p-BN was mainly reduced due to large shear forces and due to high impact collisions with the ball bead, resulting in a gradual decrease in the lateral dimensions, including the thickness of BN.
 | ||
| Fig. 7 (a) A flow chart showing the synthesis of small-layer BN and large-layer BN and (b) a schematic illustration of the exfoliation process using a ball-milling technique.121 | ||
After that, An et al. reported the simultaneous production and functionalization of h-BNNs using a solvent-free mechanical exfoliation technique for super-lubricant applications.122 In the beginning, the preparation of the Pebax-BNNSs was done using the above-mentioned technique with bulk h-BN powder and Pebax 1657 (Pebax: a combination of rigid polyamide and soft polyether blocks) as precursors. A schematic diagram of the exfoliation of Pebax-BNNSs and its dispersion is illustrated in Fig. 8(a–d). 13 g of Pebax 1657 powder was taken in an Xplore micro-compounder that was already heated up to 200 °C. Once the Pebax 1657 powder was fully melted, 2 g of h-BN powder was added for mechanical exfoliation with a soaking time of 4 h. Thereafter, the mixer was cooled naturally to room temperature, and the exfoliation procedure was completed. Finally, the mixture of Pebax/h-BN was thawed in water/ethanol (7/3 v/v) solution at 150 °C followed by magnetic stirring for further separation.122
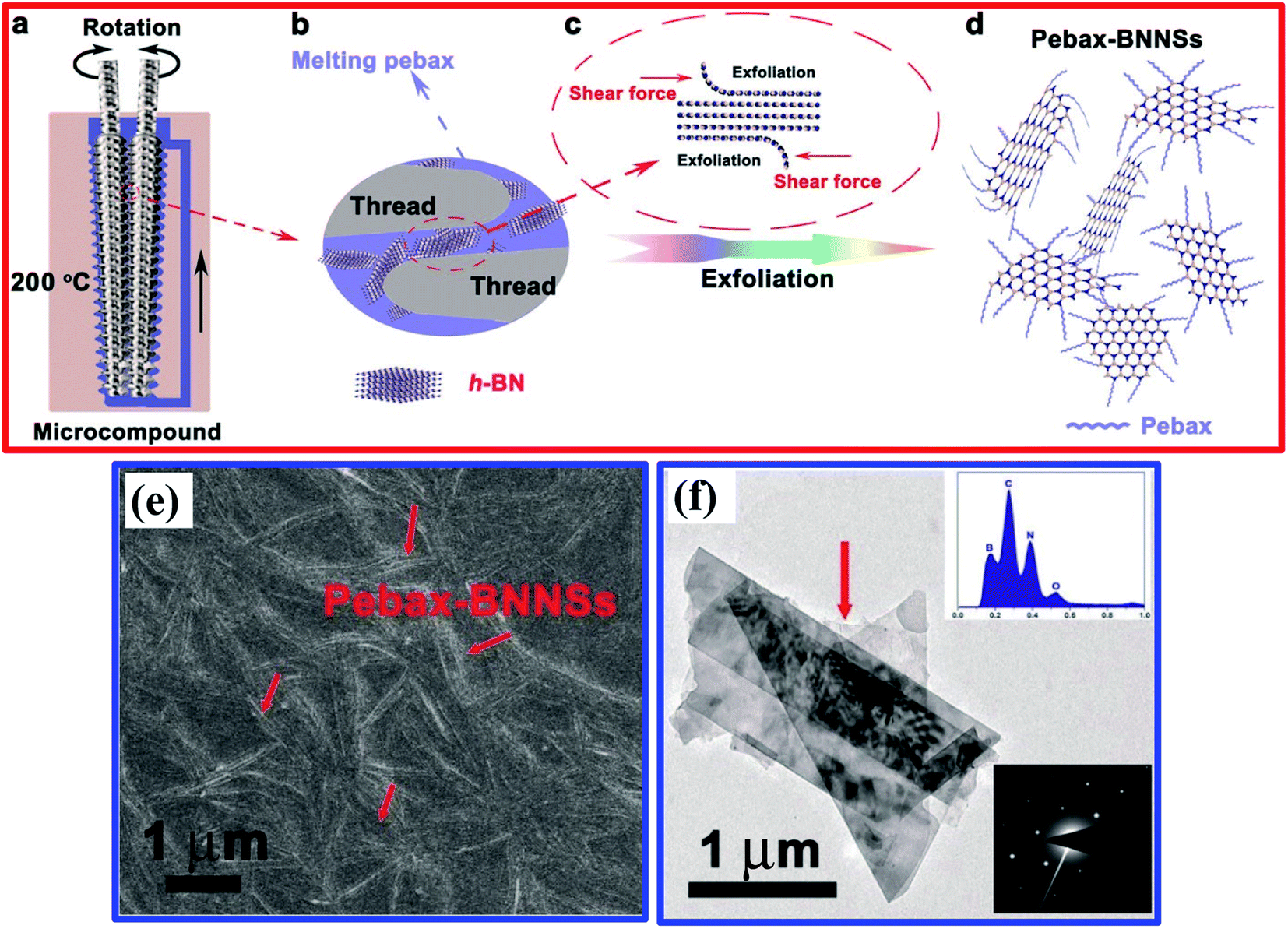 | ||
| Fig. 8 A schematic illustration of the exfoliation and dispersion of Pebax-BNNSs: (a) the chamber for exfoliation and the in situ functionalization of h-BNNSs; (b and c) the exfoliation and in situ functionalization process of h-BNNSs; and (d) the as-obtained Pebax-BNNSs. The Pebax molecules will randomly react with dangling bonds formed during the mechanical exfoliation process. (e) A high-magnification FESEM image of the Pebax-BNNSs randomly distributed on the substrate. (f) A TEM image of the Pebax-BNNSs; the insets show (top) the corresponding EDS spectrum and (bottom) the SAED pattern of the Pebax-BNNSs.122 | ||
The exfoliated h-BNNSs could be clearly seen in high-magnification FESEM images, as shown in Fig. 8(e), in which many large interconnected h-BNNSs are seen to be randomly oriented/distributed throughout the image. A TEM image depicts that the h-BNNSs comprise four layers (Fig. 8(f)). Moreover, a degree of transparency is revealed for the thinner stacked layers of Pebax-BNNSs, which seem to be lighter in color (Fig. 8(f)). The lower inset in Fig. 8(f) shows the SAED pattern, in which typical six-fold h-BN symmetry was present. The EDX pattern of the achieved product (upper inset of Fig. 8(f)) reveals that the elemental constituents are B, N, C, and O. Hence, the large quantities of C and O suggest that Pebax has been incorporated into h-BNNSs through the mechanical exfoliation method.122
More recently, Yang et al. demonstrated the preparation of BN nanoplatelets (h-BNNPs) using an amino-acid-supported ball-milling technique to enhance the thermal conductivity of a Lys@BNNP/PVA hydrogel composite to 0.91 W m−1 K−1, using 11.3 wt% 2-amino-acid-grafted h-BNNPs (Lys@BNNP).123 This was improved by 78% over neat PVA hydrogel. Moreover, to exfoliate h-BN via the L-lysine-supported ball milling (Lys@BNNP) technique, they employed a ball mill (JX-5G) with high energy. Approximately 40 g of h-BN powder (micron-sized), 80 g of L-lysine, and 365 mL of 1.5 M NaOH solution as a mixing medium and for protection during ball milling were transferred into a ball-mill tank with a capacity of 3 L along with 2 kg of ZrO2 ball beads (8 mm:5 mm:3 mm = 1:2:1) at room temperature. The speed of the ball mill was fixed at 350 rpm, and the mixture was minced for a soaking time of 10 h. The exfoliated material was taken out from the centrifuge tube and washed with adequate amounts of DI water and then with ethanol. Further, it was filtered and dried at 60 °C in an oven (vacuum). Thus, white Lys@BNNP powder with a yield of ∼85% was achieved successfully.123
2.5. Thermal exfoliation process
Besides the exfoliation of h-BN via mechanical cleavage, thermal exfoliation techniques are discussed here in brief. Cui et al. described the large-scale thermal exfoliation of h-BN and its functionalization using an easy and scalable thermal oxidation approach.66 Via employing this approach, they noticed that the heating of h-BN in the presence of air produces mass enhancement, as oxygen became embedded into the lattice of h-BN. After heat treatment, stirring the material in DI water for a few minutes resulted in the mixture congealing, hydrolyzing, and then being exfoliated to obtain hydroxylated h-BN (BNO). Further, the BNO sheets form a suspension without any sonication; however, to raise the rate of suspension formation, mild bath sonication was used systematically.66 They demonstrated a systematic study of thermal exfoliation, including the functionalization of single-BNNSs (see Fig. 9(A–C)), and the mechanism of the functionalization of h-BNNSs was also explained in detail. Moreover, it was claimed that this exfoliation method can produce great amounts of material and, owing to its considerably functionalized nature, BNO might act as a filler material, and this could be used for sensing and catalytic applications.66 In addition, Yu and coworkers demonstrated the preparation of h-BN using a thermal exfoliation approach with the effective augmentation of thermal stability and flame retardancy.123 They also reported the smoke suppression properties of epoxy resin nanocomposites fabricated using a sol–gel process. Herein, BNO was fabricated via the thermal treatment of h-BN powder in air. In the beginning, ∼10 g of h-BN powder was transferred to a quartz tube kept inside a tube furnace. The furnace was heated to 1000 °C at a rate of 10 °C min−1, with a soaking time of 2 h. Thereafter, the furnace was cooled to room temperature, and BNO with increased weight (17.9%) was taken from the furnace. Further, 50 g of epoxy resin (EP), 2.5 g of 3-isocyanatopropyl-triethoxysilane (ICTES), 0.1 g of di-n-butyltin dilaurate (DBTDL), and 5 mL of acetone were weighed and mixed in a three-necked flask with the required equipment, for example, a mechanical stirrer, a releasing funnel, a flux condenser, and a N2 inlet. Subsequently, the flask was soaked with nitrogen (N) and the mixture was stirred continuously at a temperature of 50 °C for 6 h, and then a colorless liquid product (MEP) was achieved.124 | ||
| Fig. 9 (A) Unfunctionalized hBN in water, (B) functionalized and exfoliated h-BN in water, and (C) an AFM image of oxidized and exfoliated h-BN nanosheets.66 | ||
Moreover, for the fabrication of BNO/MEP nanocomposites, they used a sol–gel method, as displayed in Fig. 10(a–c). An MEP nanocomposite comprising 1.0 wt% BNO was synthesized using the following methodology. 0.5 g of BNO was disseminated in 40 mL of isopropanol via sonication for 6 h. Afterward, the suspension of BNO was mixed with 41.0 g of MEP and 1.0 mL of ammonia (NH3). Then, the entire mixture was vigorously stirred for 6 h at ambient temperature. When the hydrolysis reaction was completed, the solvents were entirely eliminated via heat treatment at 100 °C. Subsequently, n-dodecyl-β-D-maltoside (DDM) was mixed at an equal epoxide/amino ratio of 1/1, and this mixture was then transferred into a mold (stainless-steel). The blend was cured in two steps: (i) pre-curing at 100 °C in an oven for a soaking time of 2 h; and (ii) post-curing at 150 °C for the same soaking time. Other specimens were fabricated using the same process as mentioned above. The synthesized specimens were denoted as BNO/MEPX, where X represents the BNO wt%.124 SEM studies revealed that BNO was found to be dispersed throughout the EP matrix, and it mainly formed exfoliated intercalated structures, with strong interfacial interactions with the matrix, as presented in Fig. 10(d–g).124
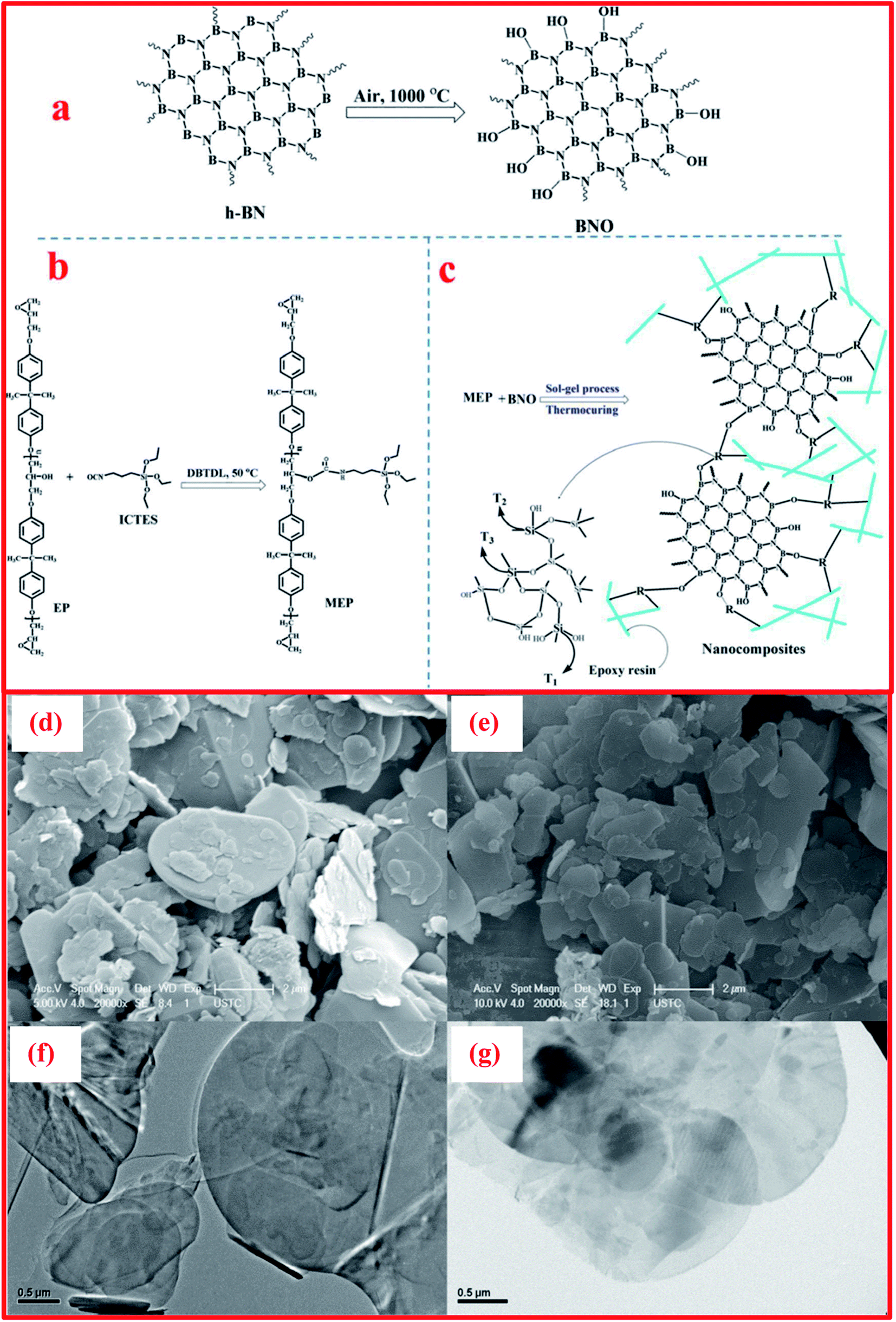 | ||
| Fig. 10 Illustrations of the synthesis processes of (a) BNO, (b) MEP, and (c) BNO/MEP nanocomposites, SEM images of (d) h-BN and (e) BNO, and TEM micrographs of (f) h-BN and (g) BNO.126 | ||
In addition, Ko et al. synthesized h-BN nanosheets using a simple thermal exfoliation method.124 In this typical method, 1 g of h-BN powder was transferred into an alumina crucible, and this was then placed in a thermal furnace at 1000 °C, obtaining using a heating rate of 5 °C min−1, where it was maintained at the same temperature for ∼2.5 h under an air atmosphere before being cooled to room temperature. The material was taken out and washed with adequate water and then collected, followed by filtration and centrifugation for the elimination of bulky materials and remaining large-sized h-BN particles. Finally, the subsequent suspension was kept for conducting other measurements, like structural and morphological investigations, or it was dried to acquire clean BNNSs.125
Recently, Zhi et al. carried out the preparation of a polyaniline (PANI)/thermally exfoliated h-BNO hierarchical composite structure (PANI-BNO) via an in situ deposition technique.126 For the fabrication of PANI-BNO, the preparation of BNO was carried out as already described above.124 Herein, the hybrid PANI-BNO was synthesized via in situ polymerization, using the suitable surfactant of sodium dodecyl sulfonate (SDS). BNO (0.3 g) was dispersed in DI water (500 mL), followed by ultrasonication for 2 h. Afterwards, SDS (0.5 g) and aniline (0.9 g) were poured into the above-mentioned suspension and it was then kept at room temperature for 24 h to complete the reaction. The formed suspension was centrifuged, followed by washing and drying, and then the hybrid material PANI-BNO was successfully obtained. The complete synthesis procedure for PANI−BNO hybrids is demonstrated in Fig. 11(a and b)126 The authors claimed that the fabricated polymer nanocomposite revealed superb dispersion and interfacial h-BN adhesion. Based on this study, they demonstrated that the BNO-PANI hybrid showed flame retardancy behavior, which produced high fire safety performance, including tests on the thermal constancy of polystyrene (PS) and the polar thermoplastic polyurethane (TPU). The overall flame retarding mechanism of the fabricated polymer/PANI-BNO nanocomposites is shown in Fig. 11(c). More recently, Acharya et al. demonstrated the novel preparation of BNNSs from h-BN using a pressure-supported aqueous-phase bi-thermal exfoliation technique.127 For the synthesis of BNNSs, initially, bulk h-BN powder (3.0 g) was added to a pressure cooker (2 L). Further, ∼700 mL of water was poured into the cooker and it was shaken for half an hour and then heated to the attained maximum pressure for 6 h. The dispersion was removed from the pressure cooker at room temperature and subjected to freezing for 10–12 h. Thereafter, any ice was defrosted and the aqueous dispersion comprising the partially exfoliated material was returned back to the cooker for heating under pressure. This consecutive heating and freezing procedure was recognized as one bithermal cycle/stage in this exfoliation technique. Hence, after the recurrence of five such cycles, exfoliated BNNSs were successfully recovered following a filtration process.127
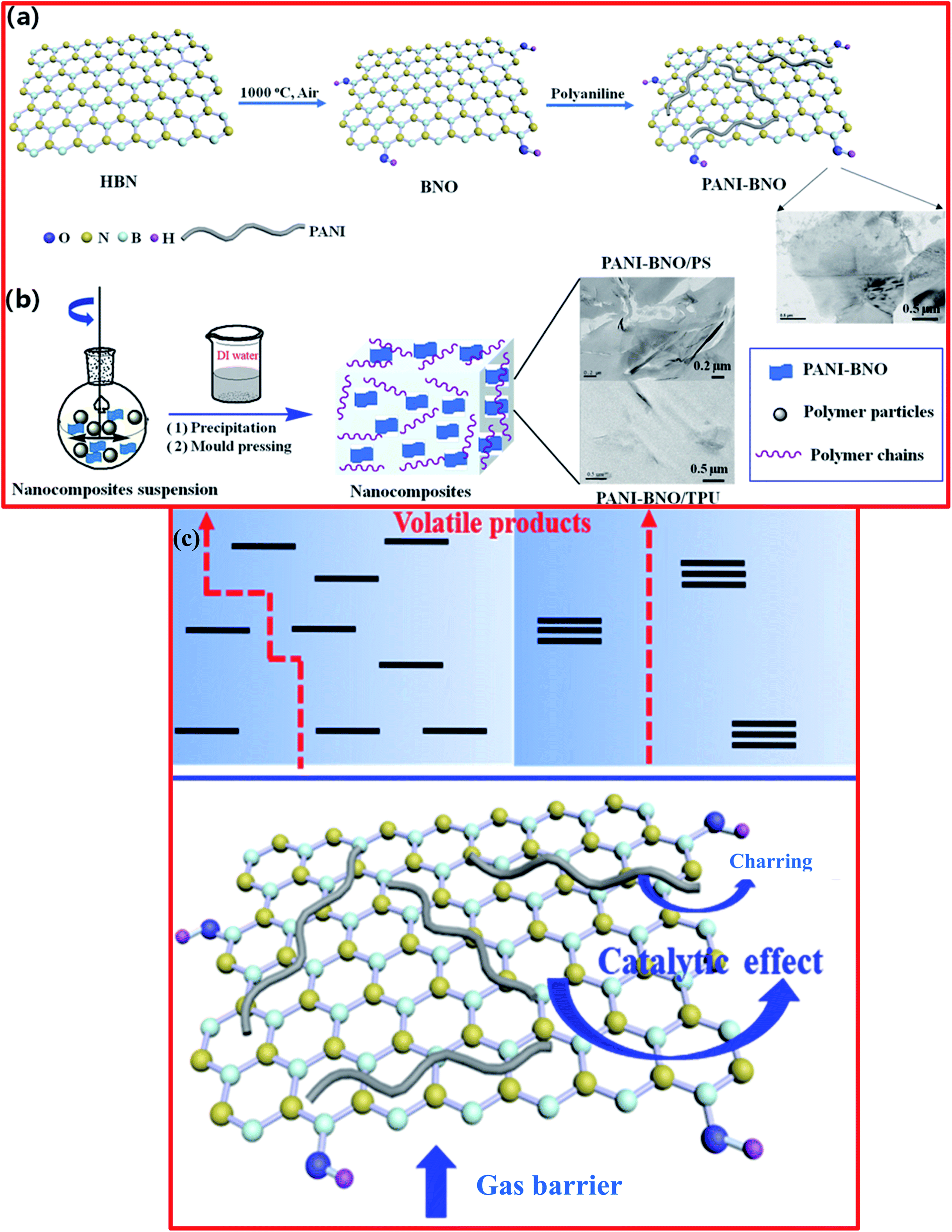 | ||
| Fig. 11 (a) Diagram for the synthesis process of (a) PANI-BNO hybrids and (b) polymer/PANI-BNO nanocomposites, (c) diagram of the flame-retardant mechanism for the nanocomposites of polymer/PANI-BNO.126 | ||
2.6. Controlled gas exfoliation
The controlled gas exfoliation of bulk h-BN into BNNSs is an issue of great interest for researchers. Zhu and coworkers reported the synthesis of few-layer BNNSs using a controlled gas exfoliation technique.128 For a typical synthesis of BNNSs, bulk h-BN powder with a lateral size of 1 mm (1 g) was poured in a quartz boat and kept in a muffle furnace at 800 °C for 5 min under an air atmosphere, it was then quickly transferred in a Dewar bottle including a gasified L-N2 unit. Further, the above-mentioned steps were repeated frequently. The achieved BNNSs after 10 cycles (BNNS-10) consisted mainly of five atomic layers with a large yield of 16–20%. The yield was evaluated using the following procedure. Synthesized BNNS-10 were dispersed in alcohol and then sonicated for half an hour. The dispersion was centrifuged at 800 rpm for 10 min to remove residual bulky particles. Then, the supernatant was obtained and dehydrated in an oven (vacuum) for ∼12 h. Hence, the yield was estimated using the weight ratio relationship between BNNSs and bulk h-BN. A schematic diagram of the entire gas exfoliation process of h-BN activated via thermal expansion is presented in Fig. 12(a).128 SEM images revealed the nanosheet-like morphology of the BNNSs, while a TEM study revealed very thin, clear (transparent), and overlapping layers of BNNS-10 (Fig. 12(b and c)). Moreover, the XRD results exhibited that BNNS-10 showed a highest intensity peak at a 2θ value of 26.70°, corresponding to the (022) plane, while for bulk h-BN, this peak was at 26.82° (Fig. 12(d and e)). Therefore, the enhanced interplanar distance and reduced intensities of the diffraction peaks corresponding to the remaining lattice planes, namely the (100), (101), (102), and (004) planes, confirmed the synthesis of ultrathin h-BN nanosheets.129,130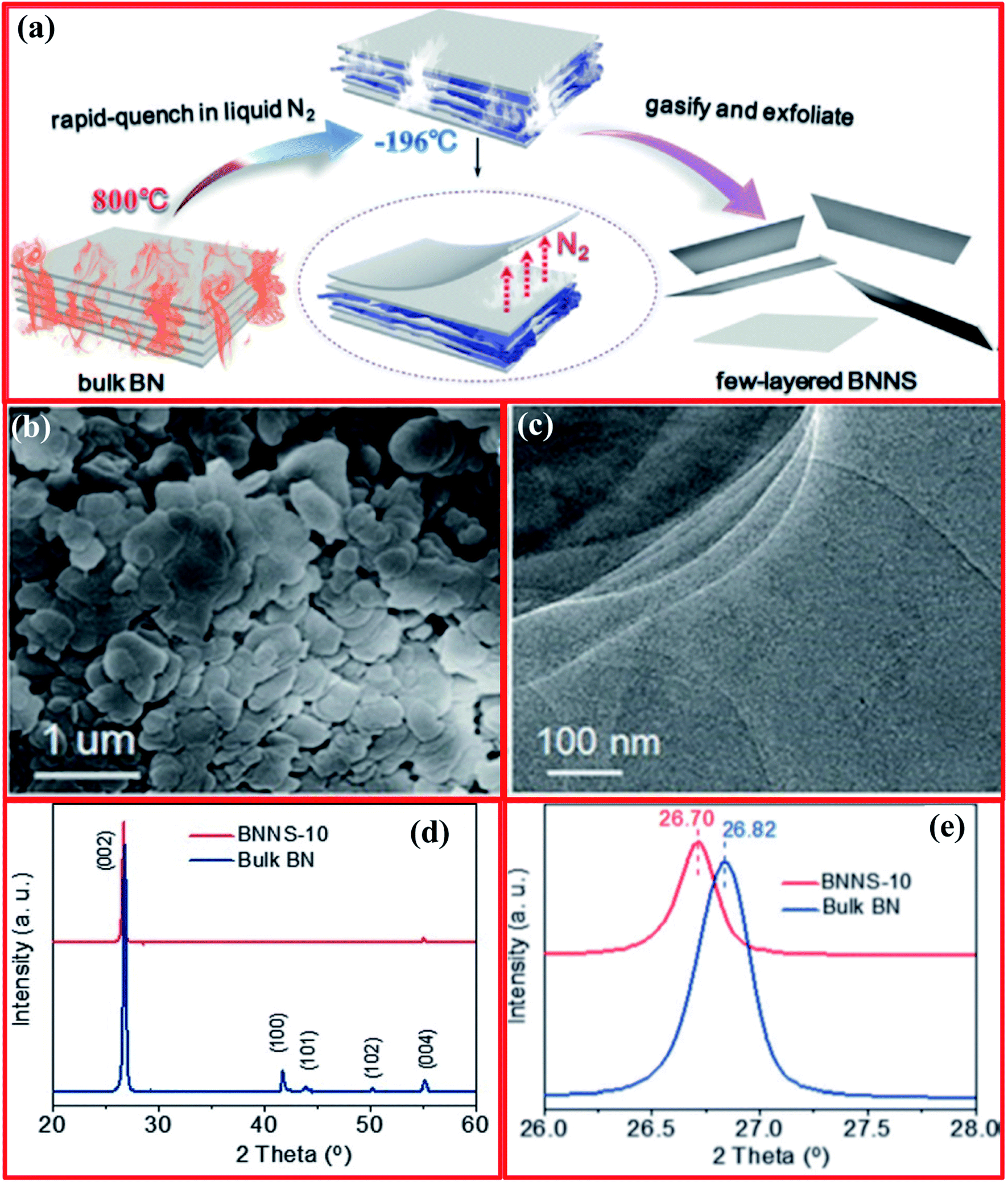 | ||
| Fig. 12 (a) The gas exfoliation of h-BN triggered via thermal expansion, (b) an SEM image of BNNS-10, (c) a TEM image of BNNS-10, and (d and e) structural characterization of bulk h-BN and BNNS-10.128 | ||
2.7. Quadrupole-field-aided exfoliation of h-BN
Earlier, attempts were made by Yang et al. to delaminate graphite into high-quality graphene using an AC approach (two pairs of electrodes and a low-frequency square wave) that allowed high efficiency and large horizontal scalability.131 Using this tactic, the quadrupole-field-assisted exfoliation of h-BN was first demonstrated by Lu et al. in 2018. They used a novel approach to exfoliate bulk h-BN into few-layer h-BNNS via employing a quadrupole field arrangement.132 The entire electrical stimulation arrangement contains three major components: (1) a differential sine wave generator, including a power amplifier of 50 W (Fig. 13(a)), (ii) a quartz-glass-fabricated electrolytic cell consisting of a 4-electrode system (Fig. 13(b–d)), and (iii) a cooling attachment.132 The authors noticed changes in the exfoliation efficiencies and roles of the surfactant as a result of effects arising from frequency and voltage changes. For the exfoliation of h-BN, 40 μL of TAE buffer (50×) and DI water (1960 μL) were poured into the electrolytic cell along with 60 mg of h-BN powder (∼2 μm) and 2 μL of NP-40 (40 wt%). | ||
| Fig. 13 (a) The assembly of the electrical stimulation system. (b) The quadrupole electrolytic cell (U1 and U2 are differential amplification voltages). (c) The rectangular quadrupole electric field (the red lines represent potential lines, and the blue lines represent field intensity lines). (d) The sizes and positions of the orthogonal titanium electrodes. (e) An SEM image of h-BN exfoliated at the optimal frequency of 400 Hz.132 | ||
Further, for h-BN dispersion, the pipette purging method was employed carefully. Then, both electrodes and the cooling system were used, and exfoliation continued for a soaking time of 3 h at 10 °C. During the synthesis process, the temperature was controlled and stabilized using a cooling system. Thus, based on the impedance of the electrolysis system, the amount of h-BN and the electrolyte content were optimized. Therefore, under the optimized conditions, 4–6 nm-thick (10–15 layers) h-BNNSs with a statistical yield of 47.6% were achieved. To attain a high exfoliation efficiency, several parameters, like the ionic medium, frequency, and presence of larger hydrophobic groups in the surfactant, play key roles. For example, at the optimal frequency of 400 Hz, the successful exfoliation of h-BN was performed, as depicted in SEM images (Fig. 13(e)).132
2.8. Laser-plasma-assisted exfoliation of h-BN
The laser plasma deposition (LPD) technique offers the numerous advantages of a low processing temperature, digital control of the specimen thickness, high yields, and scalability. To decrease the synthesis temperature and realize excellent h-BNNS stoichiometry, a pulsed laser deposition (PLD) method was employed for the first time by Glavin et al. in 2014.133 Using this method, they were able to fabricate ultra-thin extremely crystalline h-BN at 700 °C from an amorphous BN target. Moreover, thin films of h-BN were effectively deposited on massively ordered pyrolytic graphite and sapphire substrates using an excimer laser beam with a wavelength of 248 nm (KrF). Further, Velazquez et al. reported single- and few-layer h-BN on crystalline Ag films using a laser beam (Continuum Surelite III-10 YAG) to generate pulses with the following properties: 266 nm, 100 mJ per pulse, 5 ns width.134 They claimed that interactions between few-layer h-BN and the Ag substrate are quite weak, allowing the as-grown h-BN layers to be exfoliated with the help of adhesive tape. Hence, the thin films of h-BN prepared via this technique could be employed for large-scale production without any grain boundaries (deformations), however, the crystallinity is fairly low when equated with CVD and liquid exfoliation techniques.134Thereafter, Zhou et al. reported the synthesis of 2D BNNSs using the LPD technique for deep ultraviolet (DUV) photodetector applications.135,136 The LPD system comprises a CO2 pulse laser and a beam delivery system, including a vacuum chamber, as shown in Fig. 14(a).134 A CO2 laser beam (10.6 μm) was focused via a ZnSe lens (30 cm focal length) on the target at an angle of 45° across its normal surface. A pyrolytic h-BN target (2′′ (diameter) × 0.125′′ (thickness)), having a minimum assay of 99.99% (1.94 g cm−3), was fixed and rotating at 200 rpm through its surface normal.136,137 This fixed laser beam (5 J per pulse at 5 Hz) formed a spot (size: ∼2 mm in diameter) on the target material with a laser energy density of ∼160 J cm−2. Herein, a molybdenum substrate (1.0 cm (diameter) × 0.3 mm (thickness)) was used that was polished using diamond nanoparticles and then cleaned nicely. During the deposition process, the temperature of the substrate was sustained at 400 °C and the distances between targets and the substrate were kept at 4 cm. The entire deposition was carried out within 15 min with ∼4500 laser pulses. To study the surface morphology of the fabricated BNNSs, SEM images reveal an enormous quantity of BNNSs, which covered the entire substrate surface (Fig. 14(b)). A cross-section view of the fabricated BNNSs is revealed in Fig. 14(c), which indicates that the average thickness of the BNNSs is ∼11.5 μm. Therefore, the achieved specimen confirmed the preparation of BNNSs containing an adequate number of randomly orientated single-crystal BNNSs.138 Moreover, the calculated interlayer spacing was noted to be ∼0.33 nm, with a thickness of around 10 nm (Fig. 14(d and e)). The thickness of the BNNSs could be enhanced by using an increased deposition time.
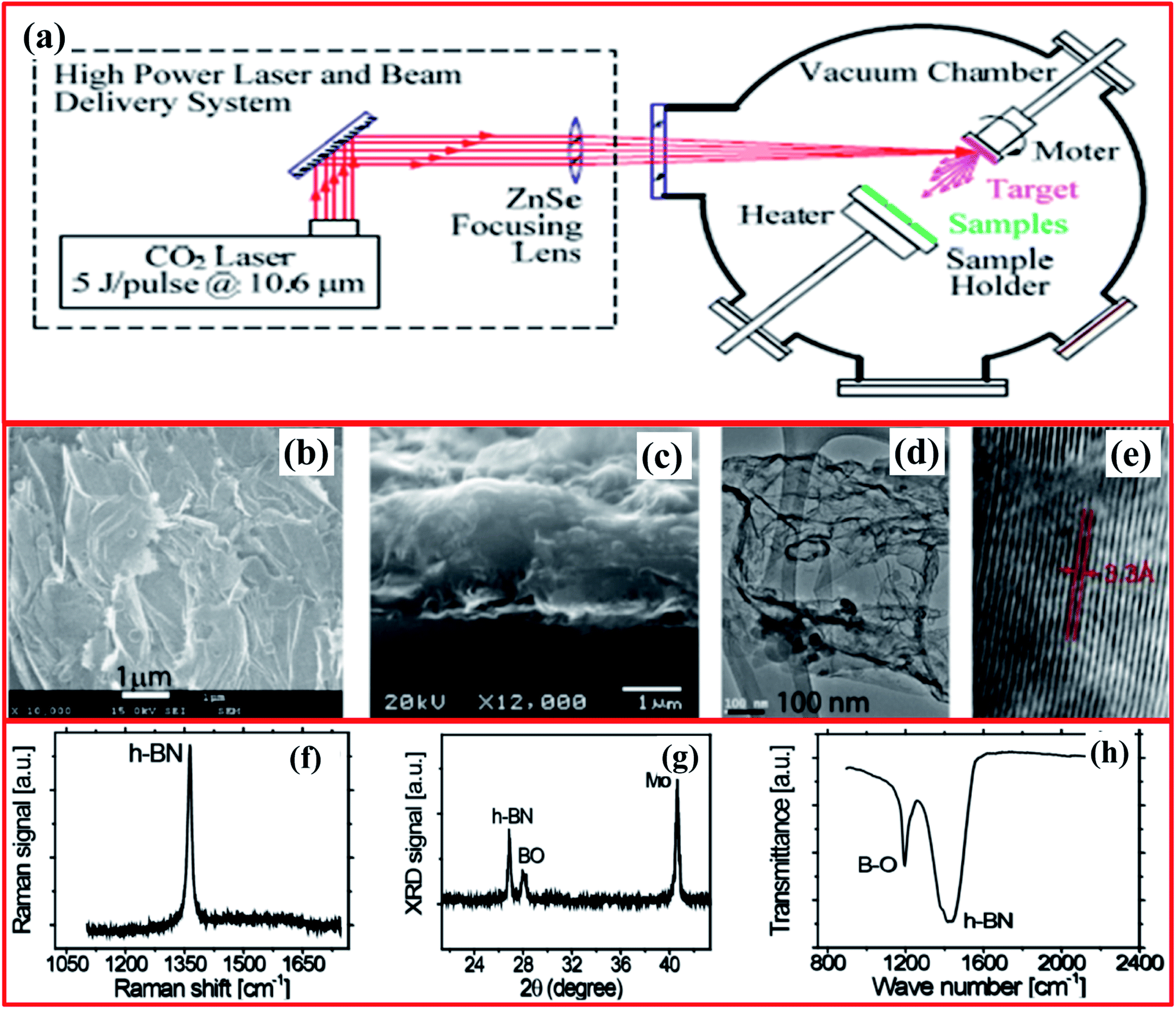 | ||
| Fig. 14 (a) A schematic diagram of the CO2 pulsed laser plasma deposition system. Images of BNNSs deposited on a Mo substrate: (b) a low-magnification top-view SEM image of overlapping BNNSs; (c) a cross-section SEM image of a ∼1.5 μm-thick sample on a Mo substrate; (d) a typical TEM image with visible curved/wrinkled and folded structures; and (e) a high-resolution TEM image of the edge area of a BNNS whose interlayer spacing is ∼0.33 nm. Spectra of BNNSs deposited on a Mo substrate: (f) Raman; (g) XRD; and (h) FTIR.135 | ||
Besides the above study, further structural studies were also performed, and they showed that the Raman active E2g mode was observed at ∼1365 cm−1, which was attributed to in-plane B3N3 vibrations of the h-BNNS structure (Fig. 14(f)). Further, it was verified in Fig. 14(g) that the XRD pattern shows a sharp peak at a 2θ value of 26.9° that is equivalent to an interlayer spacing of about 0.33 nm for the h-BN lattice planes.139 The FTIR spectra showed two distinct absorption peaks at 1200 cm−1 and 1429 cm−1, which were assigned to B2O3-stretching and B–O deformation modes, and the in-plane E1u B–N bonds (stretching/vibration) of sp2-bonded h-BN, respectively (Fig. 14(h)). Later on, Rivera et al. reported the fabrication of high-quality 2D single-crystal BNNSs via a pulsed laser plasma deposition (PLPD) method that was conducted at a low substrate temperature for UV photodetector applications.140 They claimed that the fabricated specimen has a high BNNS content, and the NSs partially and randomly overlap with each other. Recently, Ortiz et al. demonstrated the preparation of carbon-doped 2D h-BNNSs via employing a CO2-PLD technique using SiO2 and Mo substrates.141 Moreover, they also investigated whether the stable hysteresis properties under a low bias voltage at 170 °C make this material a suitable candidate for non-volatile memory device applications.
2.9. Other novel techniques
Due to the need for enormous amounts of atomically thin BNNSs to meet an assortment of research-based and real-world demands, the development of successful new techniques for exfoliating BNNSs is unlikely to cease. Herein, we would like to briefly summarize the plentiful developed techniques below. To prepare few-layered h-BNNs, Yurdakul et al. fabricated 2D BNNSs using high-pressure (207 MPa) microfluidization with a chemical exfoliation method, using large flakes of h-BN with micron-sized-thickness as a precursor. DMF and chloroform (CHCl3) were used as the solvent liquids. Further, based on microscopic characterization techniques, the synthesis of 2D few-layer BNNSs (yield efficiency: ∼45%) was confirmed. Using an EELS study, the calculated thickness of these fabricated 2D BNNSs lies in the range of 8–12 nm and it was suggested that the BNNSs are composed of between 20 and 30 monatomic 2D graphene-comparable h-BN layers.142 Thangasamy et al. reported a simple, accessible, one-pot, rapid, and fascinating supercritical fluid (SCF) supported exfoliation technique.143 Recently, Sun et al. reported a comprehensive approach for the synthesis of 2D nanomaterials via an SCF-assisted exfoliation technique, including outlining the advantages.144 Afterwards, Duong et al. demonstrated the facile production of h-BN nanoparticles (h size > 10 nm) via a cryogenic exfoliation technique.145 To produce small-sized nanoparticles of h-BN, they implemented a well-developed cryogenic exfoliation technique.146 Further, another novel type of exfoliation approach, i.e., the chemically exfoliated synthesis of h-BN doped with Ni, was reported by Ikram et al. For synthesis, BN powder (5 g) was dissolved in 200 mL of DMF to acquire a stock solution. To attain BNNSs, the stock solution was sonicated for up to 12 h and then all floating nanosheets were collected. Furthermore, the fabrication of Ni-doped BNNSs was carried out using a hydrothermal method.147Recently, some researchers have indicated the effectiveness of the ionic-liquid- and surfactant-based exfoliation of 2D layered nanomaterials, like transition metal dichalcogenides, graphene, and h-BN.148,149 Kamath et al. reported an exfoliation and dispersion method for h-BN in an ionic liquid medium via employing molecular dynamics (MD) simulation approaches.150 They recommended the liquid exfoliation of h-BN in an ionic-liquid medium, which can produce BNNSs. For the first time, Khan et al. reported the exfoliation of h-BN into 2D-hBN using sodium cholate as a surfactant in an aqueous medium for the electrochemical reduction of oxygen and capacitance applications.151 They also reported that the fabricated 2D-hBN is a highly active electrochemical material, with a specific capacitance of 1745 F g−1, and it might be useful for the production of supercapacitors.
Based on a literature review, the most common solvent used to exfoliate h-BN into BNNSs is ethyl alcohol (C2H5OH); however, C2H5OH is not a comprehensive solvent for exfoliating h-BN and, hence, there is an essential requirement for a suitable surfactant to be used.152 Kulkarni et al. demonstrated the synthesis of h-BNNSs using a surfactant-supported liquid-phase exfoliation method.152 They used the block copolymer of polyethylene oxide–polypropylene oxide–polyethylene oxide (PEO–PPO–PEO) as a surfactant. A schematic diagram of the preparation of h-BNNSs in a PEO-PPO-PEO surfactant solution is presented in Fig. 15(a). Fine (micron-sized) h-BN powder was mixed with the above-mentioned surfactant (PEO–PPO–PEO block copolymer/ethanol) solution via employing probe sonication, including centrifugation, and only the top 80% of the supernatant was used (Fig. 15(b)). This solution possesses BNNSs, which were studied via UV-vis-NIR spectroscopic techniques (Fig. 15(c)). The recorded UV-vis-NIR spectrum of exfoliated h-BN revealed a sharp peak at 217 nm, which exhibits an increase in the layer separation of the BNNSs owing to exfoliation. The authors also claimed that the estimated absorption coefficient value and yield of the synthesized BNNSs are 1100 mL−1 g−1 and ∼0.45 mg mL−1, respectively. Hence, they could not obtain a decent yield of exfoliated h-BNNSs. Moreover, a TEM study verified the synthesis of BNNSs using a surfactant solution of PEO-PPO-PEO in ethanol (Fig. 15(d)).152 Aside from the surfactant-based exfoliation of h-BN, Deshmukh et al. demonstrated the green synthesis of h-BNNSs via the ultrasound-assisted exfoliation of h-BN using several plant extracts.153 A schematic diagram of the green exfoliation method using plant material extracts is shown in Fig. 16. Moreover, the authors claimed that plant extracts might be utilized for the synthesis of nanoparticles which could play various significant roles as capping, reducing, and stabilizing agents. Likewise, a plant extract may act as a green surfactant via adsorbing onto the h-BN surface and weakening interlayer interactions, gradually exfoliating h-BN into layers of h-BNNSs. They compared the exfoliated h-BNNSs with pristine h-BN and h-BNNSs prepared using isopropanol, and the plant-extract h-BNNSs exhibited greater removal efficiencies toward cationic and anionic dyes. In addition to these studies, the authors successfully enhanced the radical scavenging and mechanical performances in castor-oil-based polyurethane composite materials.153
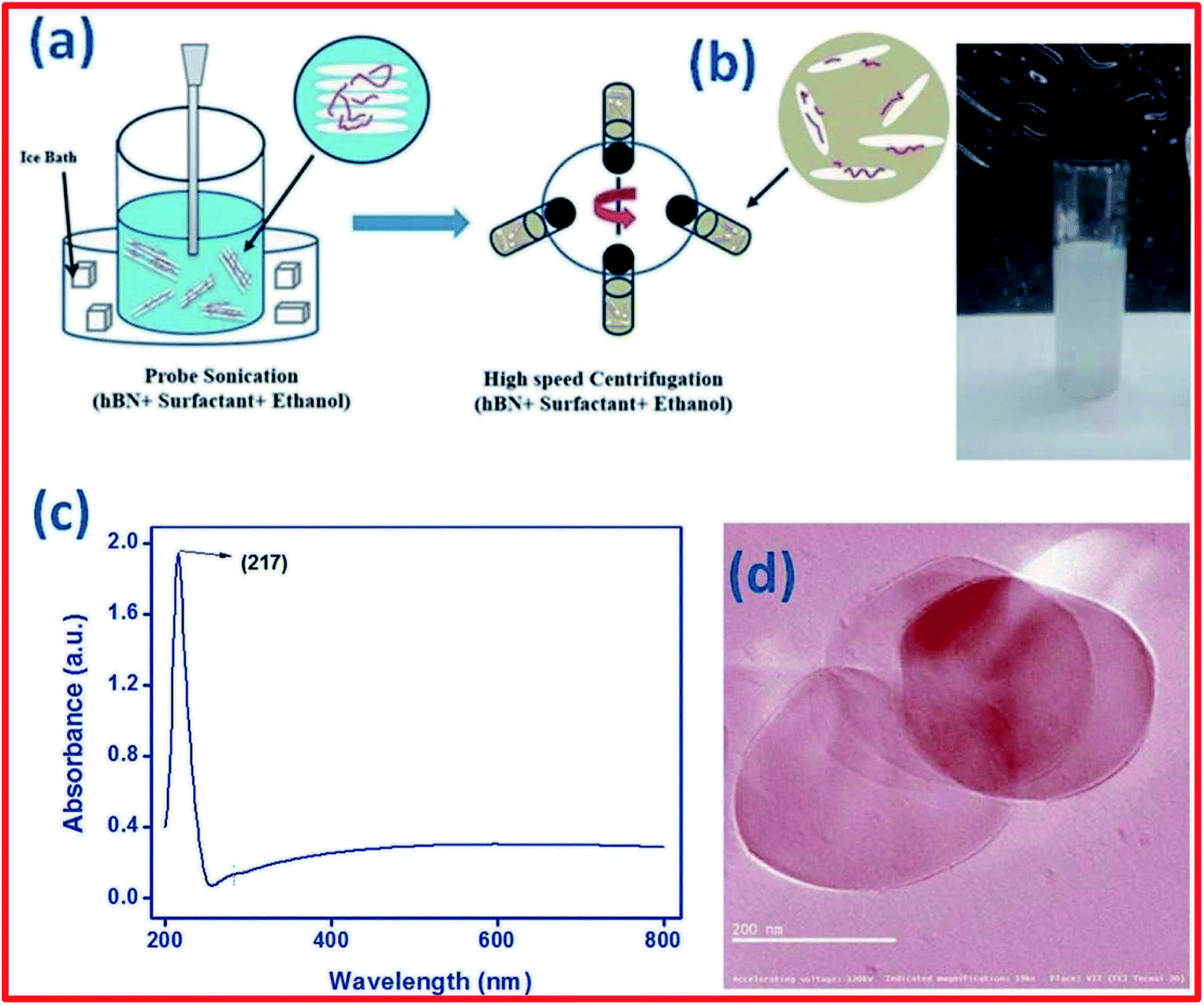 | ||
| Fig. 15 (a) A schematic diagram of the synthesis of h-BN nanosheets in surfactant solution. (b) An image of the centrifuged solution. (c) The UV-vis-NIR spectrum of h-BN nanosheets. (d) A TEM image of exfoliated BNNSs.152 | ||
 | ||
| Fig. 16 A schematic diagram of a green exfoliation synthesis route using plant material extracts.153 | ||
More recently, Zhang et al. reported a novel intermediate-assisted grinding exfoliation (iMAGE) method for the mass production of two-dimensional materials.154 In this method, micron-scale particles have been used as force intermediates to obtain microscopic compressive forces (fi) that can act as minor shear forces, encouraging the highly efficient exfoliation of h-BN, as well as other layered materials. For example, bulk h-BN was exfoliated into high-quality 2D h-BNNSs with adequate flake sizes and a large exfoliation yield ∼67%, including a high production rate of 0.3 g h−1 and low energy expenditure of 3.01 × 106 J g−1. A schematic diagram of this method is shown in Fig. 17(a). Herein, the intermediates change a macroscopic compressive force, Fc, into microscopic forces, fi, on a layer of this material; consequently, 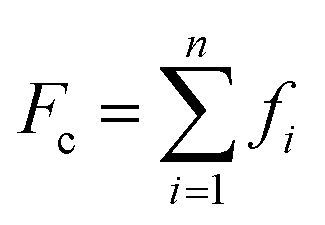 , where n represents the number of microscopic forces in each box surrounded by the dashed lines. Moreover, fi on each layer in the box should be continued in the same direction of pressure, viz.,
, where n represents the number of microscopic forces in each box surrounded by the dashed lines. Moreover, fi on each layer in the box should be continued in the same direction of pressure, viz., 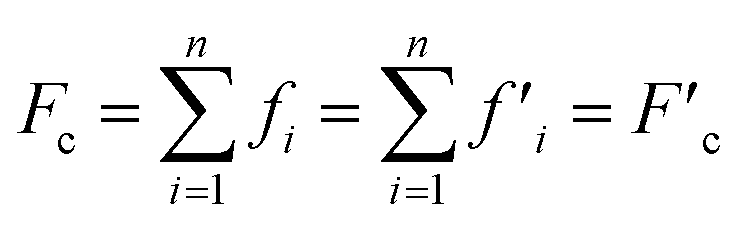 . During grinding, the rotation of the platter (at the bottom) supports slipping between the layers of the material and the force intermediate, and fi is changed into a sliding frictional force ffi, where ffi = μfi, where μ represents the sliding coefficient of friction (COF) between the intermediate and the layers of the material, as depicted in Fig. 17(b). When the shear friction force ffi > bEe, where b and Ee indicate the width and exfoliation energy of the layer material, respectively, the material layers slip and exfoliation take place.154
. During grinding, the rotation of the platter (at the bottom) supports slipping between the layers of the material and the force intermediate, and fi is changed into a sliding frictional force ffi, where ffi = μfi, where μ represents the sliding coefficient of friction (COF) between the intermediate and the layers of the material, as depicted in Fig. 17(b). When the shear friction force ffi > bEe, where b and Ee indicate the width and exfoliation energy of the layer material, respectively, the material layers slip and exfoliation take place.154
 | ||
Fig. 17 The exfoliation mechanism when using the iMAGE method and characterization of as-prepared 2D h-BN. (a) A schematic diagram of the decomposition of the macroscopic compressive forces Fc and  into much smaller microscopic forces fi and into much smaller microscopic forces fi and 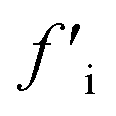 , which were loaded onto the layered material using force intermediates. (b) The exfoliation mechanism of the layered material. fi and , which were loaded onto the layered material using force intermediates. (b) The exfoliation mechanism of the layered material. fi and 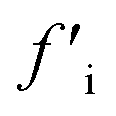 are transformed to the sliding frictional forces ffi and are transformed to the sliding frictional forces ffi and 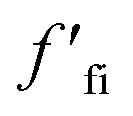 in response to the relative slipping of the intermediates and layered material due to the rotation of the bottom container. When ffi > bEe, the exfoliation of the layered material occurs (here b is the width of the layered material and the unit of bEe is (m)*(J* m−2) = N). (c) An AFM image of 2D h-BN. (d) A HRTEM image of the in-plane structure of exfoliated 2D h-BN and its FFT pattern (inset). (e) The edge structure of exfoliated 2D h-BN, showing that region (ii) separates from region (iii); the inset is an illustration of the sliding and exfoliation process.154 in response to the relative slipping of the intermediates and layered material due to the rotation of the bottom container. When ffi > bEe, the exfoliation of the layered material occurs (here b is the width of the layered material and the unit of bEe is (m)*(J* m−2) = N). (c) An AFM image of 2D h-BN. (d) A HRTEM image of the in-plane structure of exfoliated 2D h-BN and its FFT pattern (inset). (e) The edge structure of exfoliated 2D h-BN, showing that region (ii) separates from region (iii); the inset is an illustration of the sliding and exfoliation process.154 | ||
For preparation purposes, bulk h-BN was crushed with SiC particles; these particles perform as force intermediates, and the used apparatus offers a compressive force of ∼100 N including a rotation speed of 200 rpm. After the completion of the grinding process, the mixture of h-BN + SiC was exfoliated. Further, to isolate exfoliated 2D h-BN, the mixture material was poured into DI water; after 8 h, a green residue comprising SiC and unexfoliated h-BN was clearly observed, and the milky white supernatant was separated. The supernatant includes the colloidal state as exfoliated 2D h-BNNSs in DI water. To confirm the preparation of exfoliated 2D h-BNNSs, particles of silicon carbide (SiC) and bulk h-BN were individually added to DI water, and precipitation of both was completed in 5 min with a clear supernatant. These results suggest the separation of 2D h-BN from the mixture. Based on AFM studies, the fabricated 2D h-BN has an average lateral size of ∼1.2 μm (Fig. 17(c)), including a typical thickness of 4 nm. Besides these results, HRTEM and fast Fourier transform (FFT) studies revealed the excellent quality of the 2D h-BNNSs (Fig. 17(d)) without any defects in the planes or along the edges. The used exfoliation technique produced more highly crystalline 2D h-BNNSs than those synthesized via liquid-phase exfoliation and the technique was observed to be superior to intercalation and ball-milling techniques which result in numerous structural defects and additional functional groups.155–157 During the transformation of bulk h-BN into 2D h-BNNSs, all lattice fringes on the edges show insignificant levels of damage to the layer structure (Fig. 17(e)) when using this exclusive iMAGE process exfoliation technique.154 Later on, Novoselov et al. reported a grinding exfoliation method for the scalable synthesis of 2D materials. Herein, the application of shear forces is an extremely energy-efficient and industrious approach for exfoliation. This method has good advantages, producing enormously high production rates of 0.3 g h−1 with a possible yield of 50%, showing much (ten times) better performance than the ball-milling method.158
In addition, Liu and his co-workers concurrently demonstrated an easy and scalable synthesis method for BNNSs and h-BN quantum dots (BNQDs).159 This synthesis approach involved a high-temperature solvothermal process using bulk h-BN in ethanol for dispersion and aqueous sodium hydroxide (NaOH). The surface morphology and other characteristics of the fabricated BNNSs and BNQDs were tested using several characterization techniques, such as AFM, TEM, XRD, and confocal microscopy, and the results showed that bulk h-BN could be effectively exfoliated into BNNSs and BNQDs. More recently, Zhou and co-workers demonstrated a three-step sorting method to obtain different thicknesses of various 2D nanomaterials, including lateral size sorting. Based on this method, exfoliated h-BN with different thicknesses and lateral sizes is individually sorted into diverse fractions, obtaining 2D h-BN flakes with a thickness of less than 6 nm.160
Therefore, via the use of various fabrication techniques, 2D materials with diverse properties can be obtained. At an industrial level, for the large-scale production of such types of materials, chemical vapor deposition (CVD) techniques are found to be more appropriate; however, numerous exfoliation procedures have also been used for advanced applications, particularly on a research-laboratory scale. Consequently, these materials cannot be obviously equated. Hence, different features of functionalized BN (f-BN) materials, like flake size, the substrate used, and the material thickness, are listed in Table 1 in connection with the synthesis method used. From the listed summary of these 2D materials, it is concluded that the CVD technique offers much thinner and larger sized h-BN layers than those obtained via other exfoliation techniques. Moreover, in the case of other exfoliation methods, generally a bulk material (as a starting material) was used, whereas during the CVD process, an entirely new material is achieved from several molecular precursors. Further, there is no doubt that the properties of the achieved few-layer h-BN are firmly associated with different used substrates. Therefore, based on the above-mentioned results, which are also summarized in Table 2 (the exfoliation synthesis method, the specific operation/medium used, the yield percentage, and observed remarks for the synthesis of BNNSs), we can say that chemical and mechanical exfoliation methods are extremely suitable and repeatable for achieving adequate yields of BNNSs. However, to obtain ultrathin and pure BNNSs, it is still a big challenge to find cost-effective and time- and energy-saving exfoliation methods. Therefore, to overcome these major drawbacks relating to the exfoliation of h-BN, researchers have to investigate novel methods that can produce 2D BNNSs with outstanding performance for the most advanced applications.
| Exfoliation method | Used substrate | Thickness [nm] | Flake size [μm] | Ref. |
|---|---|---|---|---|
| Sonication-centrifugation technique | Dimethylformamide | 3–7 | >Pristine material | 82 |
| Sonication-assisted hydrolysis method | Octadecylamine | 1–2 | 0.3–0.5 | 38 |
| Co-solvent approach | Methanol, ethanol, 1-propanol, 2-propanol, acetone, tert-butanol | 6–10 | — | 62 |
| Two-step method: sonication-assisted intercalation of ions | Zinc chloride and potassium chloride | 2–3 | 0.5 | 81 |
| Chemical-solution-derived method | 1,2-Dichloroethane, poly[(m-phenylenevinylene)-co-(2,5-dioctoxy-p-phenylenevinylene)] | ∼1.2 | Several | 83 |
| High-temperature solvothermal treatment | Ethanol, sodium hydroxide | ∼1 | ∼1.2 | 159 |
| Modified Hummers' method followed by sonication treatment | Potassium permanganate, sulfuric acid, 1-methyl-2-pyrrolidinone | 5 | 0.3–1.2 | 186 |
| Chemical vapor deposition (CVD) and low-pressure chemical vapor deposition (LPCVD) | Cu foil, borazane | 0.42 | 0.05–0.1 | 36 |
| CVD (LPCVD) | Co film, ammonia borane | ∼1 | > 5 | 37 |
| CVD (LPCVD) temperature-dependent | Al2O3, borazane | 40–228 | — | 38 |
| CVD | Fe foil, borazine | 5–15 | 10 × 10 | 39 |
| CVD (AP-CVD) | Pt foil, borazane | 0.32–0.809 | 1–2 | 40 |
| CVD/atmospheric pressure-chemical vapor deposition (AP-CVD) | Ag foil, borazine | 0.7–1.3 | 0.1 | 83 |
| Pulsed laser deposition (PLD) | Single-crystal SrTiO3 (001) and Ag buffer films of 40 nm | 3.33 | — | 134 |
| Pulsed laser deposition (PLD) | Highly ordered pyrolytic graphite (HOPG) and sapphire (0001) | 1.5–2 | — | 133 |
| Laser plasma deposition (LPD) | Molybdenum (1.0 cm dia. × 0.3 mm thick) | 1500 | >10 | 135 |
| Ball-mill technique | — | >5 | 1–10 | 173 |
| Synthesis method | Specific operation/medium | Yield (%) | Remark | Ref. |
|---|---|---|---|---|
| Direct exfoliation (under weak sonication) | Using ionic liquids (highly soluble BNNSs) | ∼50 | Mild, very facile, attractive for the production of BNNSs | 63 |
| Thermal exfoliation | Involves high-temperature treatment | 65 | Water-suspended sheets | 66 |
| Chemical exfoliation | Sodium hydroxide and potassium hydroxide molten salts were used | 0.191 | A one-step, low-cost method | 64 |
| The intercalation of H2 ions and MnO2 nanoparticles plays a key role in exfoliating BNNSs from bulk BN, and the explosion effect of generated O2 gas may accelerate the overall exfoliation of BNNSs | 6.5 | Large-size h-BNNSs | 70 | |
| Sonication method (green approach) | Two-step method: sonication-assisted intercalation of K+ and Zn2+ ions, –OH functions in the h-BNNSs via the intercalation of these ions | 16.3 | Few-layered h-BNNSs | 81 |
| Ultrasound exfoliation | High-intensity | 100 | High-purity h-BNNSs with a minimum content of undesirable functional groups | 93 |
| Modified ball-milling process and low-energy ball-milling method (mechanical peeling) | Mechanical cleavage with the assistance of sodium hydroxide (NaOH) | 18 | Large-sized and highly soluble OH-BNNPs | 106 |
| Lower centrifugal force (benzyl benzoate acts as the milling agent to reduce ball impact and milling contamination) | 67 | High quality h-BNNSs and good efficiency | 109 | |
| Vortex fluidic exfoliation (mechanical cleavage) | A tunable ‘soft energy’ source in the form of shearing. Stewartson/Ekman layers were formed in the rotating tube and the shearing layer was parallel to the rotation axis. | 5 | Top-down fabrication of h-BNNSs | 114 |
| Optimized ball-milling process | Benzyl benzoate as the milling agent and 0.1–0.2 mm-diameter milling balls | 13.8 | h-BNNSs with high crystallinity and chemical purity | 120 |
| Ball-milling technique (high energy) | Amino-acid-assisted method using 2 kg ZrO2 ball beads (8 mm:5 mm:3 mm = 1:2:1) |
∼85 | Boron nitride nanoplatelets (h-BNNPs) | 123 |
| Controlled gas exfoliation | The high-temperature triggered expansion of bulk h-BN and cryogenic l-N2 gasification to exfoliate h-BN | 16–20 | The advantages include using no chemical reagents, the short reaction period, the ease of scaling-up and the low energy consumption. Ultrathin, five atomic layers of BNNSs | 128 |
| Other novel techniques | High-pressure microfluidization | 45 | Thin few-layer BNNSs | 142 |
| Supercritical fluid processing | 10 | A simple, rapid, one-pot exfoliation method which produced surfactant-free BNNSs | 143 | |
| An intermediate-assisted grinding exfoliation (iMAGE) method | SiC particles act as the force intermediate, and the used apparatus offers a compressive force of ∼100 N | 67 | Excellent quality 2D h-BNNSs | 154 |
| High-temperature solvothermal exfoliation | NaOH corrodes the edges of h-BN and this is beneficial for the insertion of Na+ and OH between the h-BN layers. High temperature and pressure weaken the van der Waals forces and further increase the interactions between h-BN and NaOH. | 18.1 | Cheap process which produced BNNSs with excellent colloidal stability | 159 |
| Ball-milling technique | Functionalized with a long alkyl chain amine via Lewis acid–base interactions (defective h-BN) | 40 | Intentionally introduces defects into h-BN, which is more reactive toward Lewis base molecules | 173 |
| Mild ultrasonication exfoliation | Treatment with inorganic reagents such as hydrazine, hydrogen peroxide, a nitric/sulfuric acid mixture, and oleum | ∼70 | Soluble f-BNNSs with few-layer thicknesses | 182 |
| Ultrasonic exfoliation | A low-temperature thermal expansion-assisted method | 26 | High-quality BNNSs | 97 |
| Liquid-phase exfoliation technique | Using a low-molecular weight supramolecular polymer, such as adenine-functionalized polypropylene glycol (A-PPG) | 83.5 | A simple, consistent, and effective method that produces h-BNNSs | 220 |
3. Functionalization of h-BN
Despite the structural similarities between h-BN and graphite, h-BN materials have not been investigated in depth with regards to functionalization and applications. The functionalization of carbon nanomaterials involves numerous chemical reactions, whereas the functionalization of h-BN employing organic chemistry has not been investigated in detail. Reactions involving h-BN and graphite via basal plane sites involve opening congruently conjugated p bonds. Consequently, the generation of new bonds must continually occur with an even number. Once a functional group builds a single bond with B or N, a compensating group must be involved with an unpaired B or N atom to maintain the overall charge. However, other functional groups have the ability to make bonds after a BN p-bond is unlocked. Herein, functionalized h-BN architecture generally contains two groups attached via adjacent B–N units. Nevertheless, experimental and theoretical results based on the chemical modification of connected carbon atoms have demonstrated that once charge and aromaticity are fulfilled, bonded functional groups can be detached.159–167 Therefore, the occurrence of a similar process is also believed to be possible for other layered materials, e.g., h-BN. Subsequently, B–N bonds have exhibited partial similarity within the h-BN structure. In h-BN structures, the B and N atoms reveal moderate positive and negative charge. Hence, this particular characteristic makes the B sites of h-BN more attractive (attackable) to nucleophilic groups, whereas N sites are much reactive toward electrophilic groups. Several functional groups, such as hydroxyl (–OH), amino (–NH2), ether (–OR), amine (–NHR), acyl (–COR), alkyl (–R), and halogen (–X) groups, and heteroatoms (C and O) have been studied in detail for chemical functionalization.168Functionalization is a useful approach by which the properties and applications of a layered material can be modified. It is anticipated that numerous new properties can arise from such functionalized layered materials, such as h-BN, upon using different functionalization approaches, i.e., physical as well as chemical changes. The properties of h-BN could be modified and various unique structural architectures and applications could be obtained via using functionalization approaches. Nevertheless, the high chemical and thermal stability of h-BN hampers its alteration. This creates challenges when it comes to functionalizing h-BN materials using different approaches, making this an interesting research area. In addition, another major challenge is to increase the water dispersibility of h-BN. This could be achieved via generating several functional groups at the surface of exfoliated h-BNNSs. Herein, a detailed discussion of functionalized h-BN structures is provided. Moreover, we include a substantial number of past and currently established functionalization approaches, including the functionalized characteristics and applications.
3.1. Covalent surface functionalization of BNNSs/BNNTs
Several chemical methods comprising weak interactions and covalent bonding approaches have been used to fabricate functionalized boron nitride nanotubes (BNNTs). It has been reported that the covalent functionalization of h-BN using organic molecules will substantially modify its surface chemistry and, therefore, this has the ability to tune the nature of the interactions between h-BN and liquid/matrix constituents. Moreover, it has been reported that the covalent functionalization of BNNSs can be performed based on an oxidation tactic that produces several defect sites, which was carried out via direct functionalization at chemical interfaces.169–172 However, although this tactic is capable of obtaining extremely stable dispersions of BNNSs, the creation of numerous defect sites on BNNSs due to vigorous oxidation results in reduced lateral sizes and the degradation of the inherent performance of BNNSs.173,174 Earlier, He, Holzinger et al. demonstrated the covalent functionalization of graphene and CNTs using nitrene chemistry.175,176 Further, it is well known that h-BN displays outstanding chemical stability in acidic and basic environments;177 numerous approaches have been reported for the covalent functionalization of BNNTs and h-BN using oxygen and nitrogen radical species,177,178 whereas numerous theoretical approaches anticipate the functionalization of h-BN using carbenes.179,180Sainsbury et al. reported the covalent surface functionalization of h-BNNSs through reactive nitrene radicals.181 This functionalization approach was used to covalently assign polymer chains to the surfaces of the h-BNNSs, using the polymer-functionalized h-BNNSs as a filler material within a polymeric matrix. Hexagonal boron nitride was exfoliated in N-methyl pyrrolidone (NMP), as reported earlier.87 The authors used an organo-azide precursor, 4-methoxybenzyloxycarbonyl azide, and a reactive nitrene intermediary produced via a thermolysis process. Herein, nitrene radicals are predicted to target B atoms in the lattice of h-BN that are susceptible to BN bond cleavage. The produced methoxyphenyl carbamate (MPC) has an adequate ability to bind to the h-BN lattice through BN bond formation, as shown in Fig. 18(a). The inset of Fig. 18(b) shows the linear absorption characteristics, which provided an estimated extinction coefficient ε of 3000 mL mg−1 m−1 for pristine and functionalized h-BN. Moreover, the UV/vis absorption patterns of sonicated suspensions showed noticeably improved absorbance of MPC-BNNSs compared with pristine h-BN (Fig. 18(b)). Both curves are very similar in nature and can only be discriminated based on the scattering of h-BNNSs within solution. The differences in the scattering intensity indicate variations in surface chemistry and the functionalized nature of the h-BNNSs.
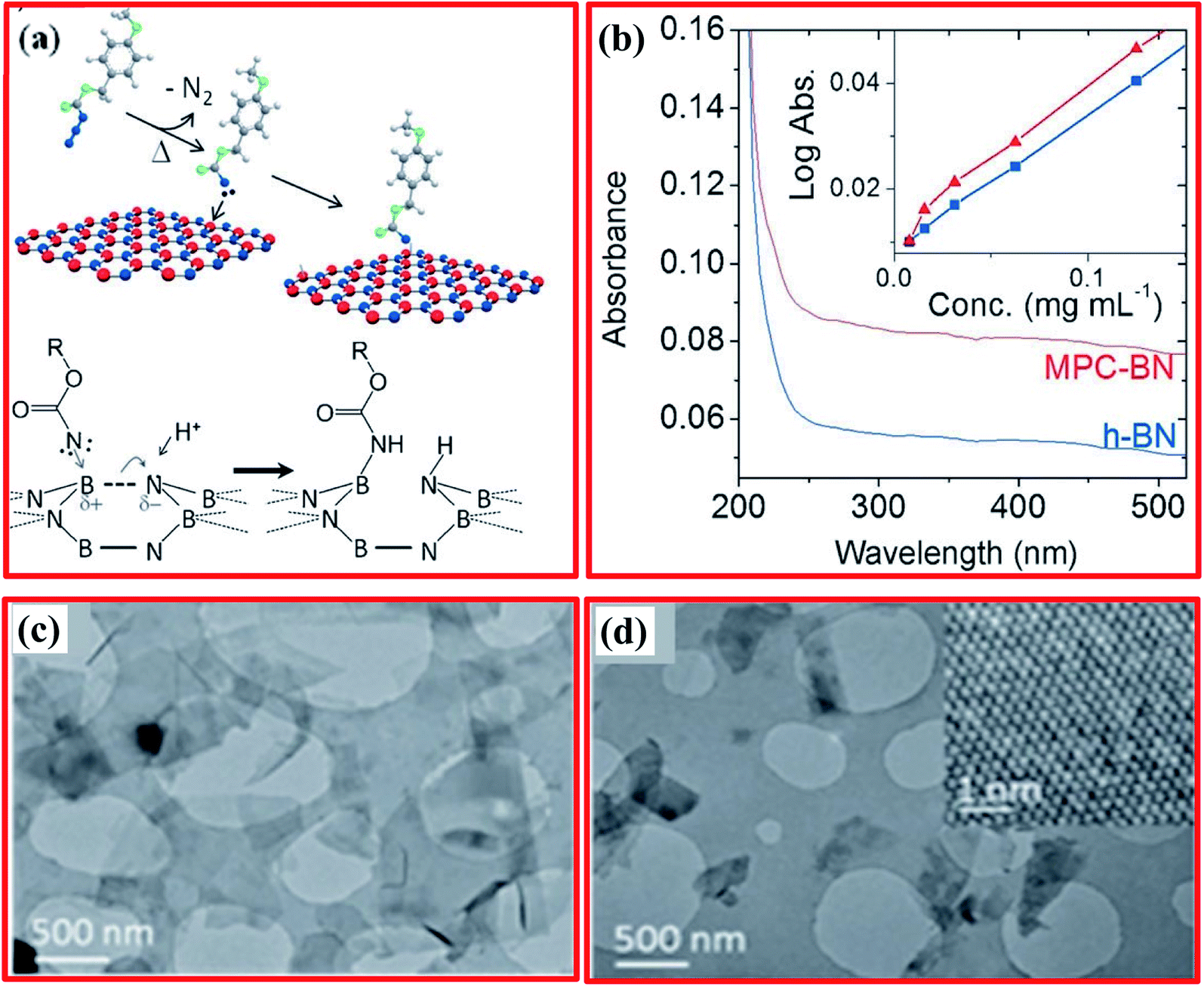 | ||
| Fig. 18 (a) A schematic illustration of the grafting of nitrene species to h-BNNs. (b) Absorbance spectra of h-BN and MPC-BN (1 mm cuvette); inset: Beer–Lambert plots. (c) TEM images of h-BNNs before and (d) after MPC functionalization; inset: a HRTEM image of the MPC-BN hexagonal lattice.87 | ||
Morphological (TEM) studies of h-BN dispersed in N-methyl pyrrolidone (NMP) showed that no obvious damage to h-BNNSs flakes could be observed (comparing before and after functionalization). Nevertheless, after functionalization, MPC-BNNSs accumulate, existing to some extent as thicker and more disordered flakes (Fig. 18(c and d)). Further, the inset of Fig. 18(d) (HR-TEM) exhibits that the MPC-BN crystal structure was not affected during the reaction. In addition to these findings, the authors also reported that MPC-BN could be dispersed in CHCl3, cyclohexyl pyrrolidone, and dimethylformamide solvents at concentrations up to 3 times larger than those obtained using pristine h-BN.87 Nazarov et al. demonstrated a new method for the functionalization and dispersion of bulk h-BN using inorganic reagents, i.e., hydrazine, 30% H2O2, HNO3/H2SO4, and oleum, in response to suitable heat treatment in an autoclave at 100 °C; this produced very high yields (70 wt%) of functionalized BNNSs. To confirm the preparation of functionalized h-BNs, they performed several characterization techniques, such as XRD, FTIR, XPS, SEM, and TEM.182 Further, Jin and coworkers presented the synthesis and surface functionalization of organized hexagonal boron nitride (OhBN) and studied the effects on the structural properties and composite performance upon increasing the concentration of amine groups.183 The entire procedure of OhBN synthesis is presented in Fig. 19(a). Herein, two main steps were involved: during the first step, reactive groups, i.e., isocyanate, were introduced to the surface of h-BN; while during the second step, amine groups were attached along with NCO groups. This process can be explained as follows. Bulk h-BN was dispersed in N,N-dimethylformamide (DMF), followed by ultrasonication under a nitrogen atmosphere to achieve a homogeneous mixture. Thereafter, excess 4,4-methylenebis(phenyl isocyanate) (MDI) was poured into the mixture under continuous stirring with heating at 70 °C for a soaking time of half an hour. Subsequently, the material was immediately vacuum filtered to completely eliminate MDI and attain the raw product; this product was washed with DMF several times (five) and then dried. The subsequent intermediate product material was designated as IhBN. Further, these dried IhBN particles were mixed with DMF to make a slurry; excess diamine diphenyl sulfone (DDS) was gradually poured into this slurry, and it was then heated at 50 °C followed by stirring for 12 h. Finally, the raw product was filtered, washed, and dried, sequentially, and the subsequent product was referred to as OhBN.183 Based on the DSC results, the authors claimed that on the surface of h-BN/OhBN, the amine groups interact with the imide rings of 4,4-bismaleimidodiphenyl methane (BDM) molecules, as depicted in Fig. 19(b); this decreases the amount of BDM homopolymer, and at a lower temperature, a co-reaction occurs rather than homo-polymerization. Therefore, the exothermic peaks of OhBN/BD and h-BN/BD prepolymers seen at low temperatures can be equated to BD prepolymers. Moreover, the OhBN surface contains a large amount of amine groups in comparison to h-BN.183 Denis and Iribarne reported a novel theoretical approach for performing the covalent functionalization of BNNSs via cycloaddition reactions.184 Based on first principles calculations, they validated that the cycloadditions of C6H6, C2H2, and maleic anhydride (C4H2O3) to 2D BN are possible. The cycloaddition products are anticipated to be stable, assuming that the activation energy for the retro reaction is 30.6 kcal mol−1. However, it might be conceivable to detach the functional groups via an annealing process at higher temperature. In addition to these investigations, they also reported that the cycloadditions might reduce the band gap of BN significantly.184 Further, Liu et al. reported an effective and facile approach for the preparation of amino- and silane-functionalized h-BNNSs.185 They revealed that 3-isocyanatopropyltrimethoxysilane (IPTES) can be chemically bonded with amino-functionalized N-BNNSs via a chemical reaction between the isocyanate and amino groups. Moreover, the functionalized h-BNNSs were incorporated into glass via a sol–gel technique. The authors claimed that h-BNNS-doped glass exhibited effective linear optical transmittance within the visible and near-infrared ranges. Moreover, the compatibility and solubility of h-BNNSs in solution and in solid-state matrices were observed to be better after silane functionalization; this research might be used in numerous areas, such as thermal conductivity, lubrication, and optoelectronics.185
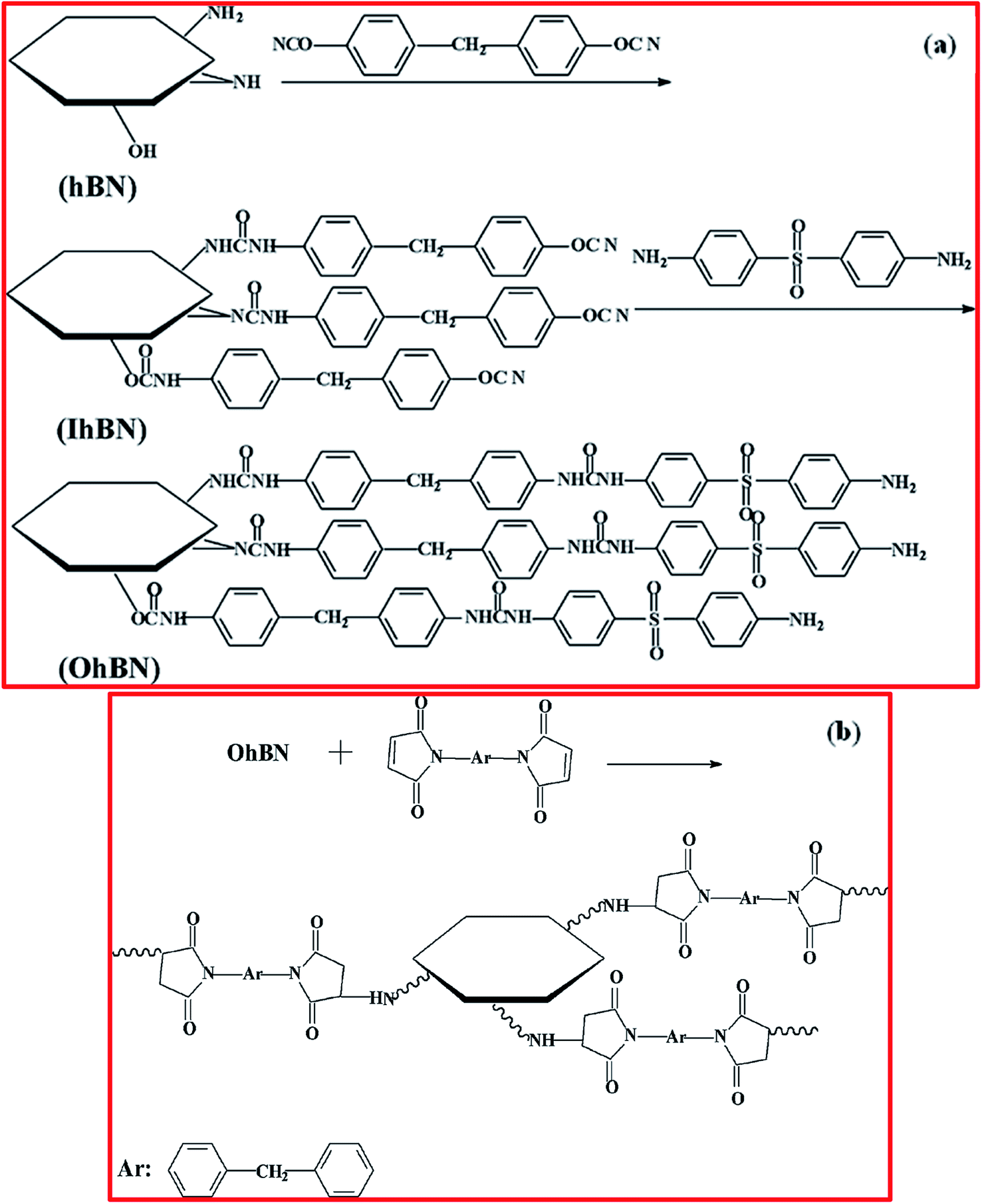 | ||
| Fig. 19 (a) The surface treatment of h-BN and (b) the reactions between h-BN and BDM.183 | ||
Jedrzejczak-Silicka and coworkers demonstrated the exfoliation and functionalization of h-BNNSs using gold nanoparticles (nAu) for biological applications.186 The exfoliation of h-BN was done via a modified Hummers' technique and then exfoliated h-BN was further functionalized with nAu. For the functionalization of h-BN, initially 100 mL of distilled water was poured into 6 mg of h-BN and this was then mixed fully followed by heating at 100 °C in a reflux system. Further, gold(III) chloride trihydrate (∼4 mL) was used at a moderate concentration of 2 mg mL−1, then, after a few minutes, trisodium citrate (∼40 mg) was added and the mixture was heated to 100 °C for a soaking time of 1 h; the mixture was then cooled before purification. Complete purification was achieved through plentiful washing using distilled water with centrifugation at 8000 rpm until the pH reached 7 (10 min).186 Based on several characterization techniques, the authors confirmed the functionalization of h-BN using gold nanoparticles. For example, the TEM results verified that Au nanoparticles were suitably decorated (deposited) on the surface of exfoliated h-BN, as shown in Fig. 20(A–C). Further, the size of these Au nanoparticles was in the range of 10–20 nm, with an average particle size of 12 nm (∼30.5%), as shown in Fig. 20(D). After authenticating the functionalization of h-BN with Au nanoparticles, composite samples exhibited outstanding biological uses; e.g., the effects of functionalized h-BN nanoflakes and fluorescein isothiocyanate (FITC) toward normal and cancerous cells for different time scales, i.e., 24 h, 48 h, and 72 h, respectively, were studied employing confocal microscopy. Fig. 20(E–L) shows the intercellular localization mechanism of h-BN-FITC within cell cytoplasm, which was confirmed based on a green fluorescence signal. Moreover, the authors described that h-BN gathered in the perinuclear section, and its existence was not seen in the nuclei of cells.
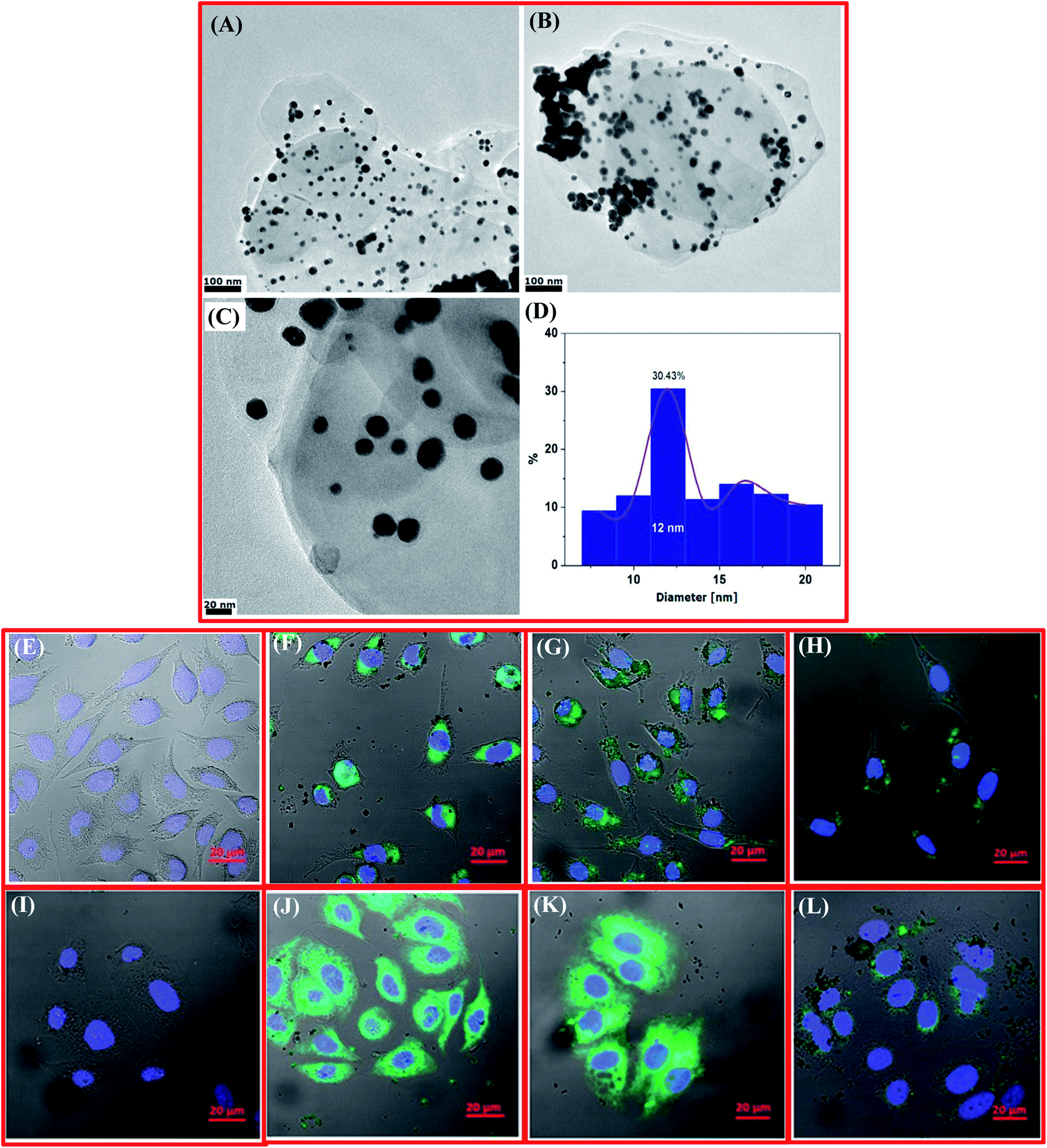 | ||
| Fig. 20 TEM images of the h-BN_AuNP nanocomposite (A–C) and a histogram showing the particle size distribution (D). Confocal laser scanning microscopy images of L929 and MCF-7 cells incubated with h-BN labeled with FITC at a concentration of 50.0 μg mL−1: the L929 control culture (E), and the L929 culture after 24 h (F), 48 h (G), and 72 h (H); the MCF-7 control culture (I), and the MCF-7 culture after 24 h (J), 48 h (K), and 72 h (L).186 | ||
Cytotoxicity testing (CCK-8 and LDH assays) of h-BN_Au particles exhibited that the cellular metabolism was not affected, however they had an impact on the function of lysosomes of normal and cancer cell lines after an exposure time of 24 h. Moreover, lengthier incubation times, i.e., 48 h and 72 h, affected cell viability at 10 μg mL−1. Additionally, the h-BN_Au particles inhibited the cell proliferation of MCF-7 cancer cell more strongly than L929 normal cells after 72 h of incubation time. Therefore, these outcomes certified that Au nanoparticles functionalized with h-BNNSs might be used for anticancer therapy, the design of biosensors, and tissue engineering applications.186
Later, Daneshnia et al. showed the functionalization of 2D h-BN at room temperature and gram-scale using lithium cyclopentadienyl for water-treatment applications.184 They also investigated whether functionalized h-BNNSs had suitable efficacy for removing methylene blue from water rapidly. At an initial concentration of 10–30 mg L−1, the methylene blue removal efficiency was noted to be 100%, while the adsorption capacity achieved was very high (476.3 mg g−1) and it was observed to be larger than that of non-functionalized h-BN.187
Further, Späth et al. performed a detailed experimental and theoretical study of the oxygen functionalization of h-BN using a Ni(111) substrate.188 They found that the interaction between monolayer h-BN on Ni(111) with molecular oxygen from a supersonic molecular beam induced a covalently bonded molecular oxygen species, involving superoxide and peroxide character. Herein, the amount of oxygen functionalization basically depends on the kinetic energy (K. E.). For a supersonic molecular beam with a K. E. of ∼0.7 eV, the oxygen coverage is 0.4 ML. Based on a near-edge X-ray adsorption fine structure (NEXAFS) study, it was reported that a sturdier bond between hexagonal boron nitride and the Ni(111) substrate was formed in the presence of oxygen species (covalently bound). Further, it was also claimed that temperature-programmed XPS techniques exhibited that the bonding between O and B was very stable at ∼306.85 °C during the simultaneous desorption and etching of h-BN.188
Hemmi et al. reported the catalyst-proximity-induced functionalization of h-BN with quaternary ammonium compound derivatives (quats).189 Herein, h-BN monolayers grown on 4-inch Rh(111) thin-film wafers (catalytic metal) can be functionalized with quats, which are widely applied as nonreactive electrolytes. The authors found that treatment with quats enables the electrochemical transfer of 2D nanomaterials, which involves the decay of quat ions leading to covalently bonded quat derivatives on the upper surface of the 2D layer. Moreover, functionalization weakens the interactions between the 2D material and substrate, allowing easy transfer. Further, based on DFT calculations, the mechanism of this functionalization method has been explained and it is observed that the proximity of the catalytic metal substrate can modify the chemical reactivity of the h-BN layers.189
Further, the covalent functionalization of h-BN was reported by Harrison and Alston using an innovative and controlled sonochemical approach.190 In this approach, they used liquid-phase solutions (non-aqueous) along with low amounts of fluorinated alcohol (as a reactant) and h-BN. During the reaction, they conclude that ultrasonic cavitation supports the creation of short-term fluoroalkoxy and hydroxyl radicals. Instantaneously, the cavitation microbubbles collapse in the solution and create a sufficient amount of energy to destabilize the B–N bonds. Hence, the destabilization of B–N bonds results in the enhanced colloidal stabilization of functionalized h-BN. Moreover, FTIR and XPS studies suggested that the fluoroalkyl groups are covalently bonded to the surface and edges of h-BN.190
Recently, Rafiei-Sarmazdeh et al. demonstrated the cost-effective preparation of functionalized BNNSs using a facile green approach. Using this approach, they significantly enhanced the yield of BNNSs (17–20%) along with obtaining an improvement in quality. Herein, hydrogen peroxide (H2O2) was used as an oxidant, and functionalized (liquid exfoliation) h-BN was successfully obtained.191 Besides this green chemical approach, further, Sun et al. reported the covalent functionalization of BNNSs using an easy reductive activation approach.192 In this approach, initially ∼690 mg of sodium and ∼3.84 g of naphthalene (dried) were mixed in THF (120 mL) in a nitrogen (N2) filled glove box, followed by magnetic stirring for up to 24 h to acquire a dark-green solution of sodium/naphthalide. Thereafter, a Schlenk tube containing ∼75 mg of BNNSs (solvent-exfoliated; Na/BN molar ratio of 10) was dried and transferred to a glove box. Then, the Na/naphthalide solution was added to the Schlenk tube along with the BNNSs. The reaction system was sealed properly, and the suspension was transferred into a typical ice bath apparatus and continuously stirred for a soaking time of up to 24 h. Further, liquid alkyl halide was poured dropwise into the tube. Moreover, the mixture was then stirred for up to 48 h at a temperature of 25 °C in a N2 environment. Then, dry O2 was added into this solution for 1 h, followed by stirring for 12 h for the oxidation of remaining groups on the functionalized BNNSs. Thereafter, ethanol was mixed gradually into the prepared solution under stirring and using an adequate water content. After neutralization with HCl (0.1 N), the functionalized BNNSs were extracted into hexane, and then washed using water 3 times. Subsequently, the product was filtered using a Teflon filter with a pore size of 0.1 μm and then washed carefully using hexane, THF, ethanol, and water, respectively. A white material (powder) was obtained after washing again with ethanol and THF, followed by drying (vacuum) for 2 days at a fixed temperature of 70 °C.192
Based on theoretical investigations, Denis et al. reported a comparative analysis of the chemical reactivities of graphene and 2D BN nanosheets.193 The outcomes of this study show that 2D BN could exhibit chemistry very similar to graphene. Experimentally, they observed that the inclusion of carbenes, nitrenes, and amines is also conceivable. Moreover, [2 + 2] cycloadditions are more feasible to occur on 2D BN than on graphene, which means that BN can be more reactive than graphene. Herein, N atoms are inclined to be less reactive in comparison to B atoms. However, this condition is changed once a B atom is functionalized.193 Further, Denis and co-workers described the reduction chemistry of BNNs and graphene via first-principles calculations.194 They observed that the adsorption of alkali atoms intensely enhances the reactivity of BNNs. Indeed, their results revealed that the effect is found to be 3.6 times more than in the case of graphene. The adsorption energies were enhanced to 66.0 kcal mol−1 for 2D BN and to 18.2 kcal mol−1 for graphene during lithium adsorption. Hence, reduced 2D BN is more reactive than rGO when alkalis are used as reducing agents.
3.2. Non-covalent functionalization of h-BN
In contrast to chemical functionalization (covalent) approaches, non-covalent approaches are mainly beneficial due to the occurrence of less structural damage, the large efficacy, and the ease of synthesis. Non-covalent functionalization approaches can retain the intrinsic characteristics of CNTs, e.g., high mechanical strength and thermal conductivity, without interrupting the extended π-conjugation arrangements of CNTs. The chemical functionalization of h-BNNPs (h-BN nanoparticles) permits enhanced solubility, and whether adequate dispersion can be achieved in a polymeric material, aqueous, or organic solution in the absence of substitution with foreign substances is of extreme concern. Besides chemical functionalization, another approach for improving the dispersion of h-BNNPs in aqueous and organic solutions is the non-covalent functionalization of the h-BN surface using various surfactants/polymers. The non-covalent functionalization of h-BN surfaces195–205 involves weak π–π stacking interactions173,196,198,199,205 with an external molecule. Therefore, the h-BN surface offers a π-electronic structure that is implied based on its chemical structure.Hence, in the past few years, attempts have been made by researchers to carry out the non-covalent functionalization of h-BN. Moreover, π–π stacking, which is utilized in 2D materials like graphene, is also useful for the modification of the h-BN structure via non-covalent approaches. Structural imperfections remain unchanged after any kind of substitution within sp2-hybridized structures. In this manner, distinct protein, polymer, and inorganic/organic groups can be attached to the h-BN surface.206 Yu et al. used Lewis acid–base complexations between the various electron-rich amine groups of octadecylamine (ODA) and electron-deficient B atoms to change the surfaces of h-BN nanoplatelets with ODA;207 the resulting material revealed enhanced thermal conductivity (TC) in comparison to pristine h-BN upon reinforcement in epoxy-based composites. Recognizing the π-conjugation performance on the surface of h-BN, conjugated molecules like catechin,208 polyaniline,209 and poly(p-phenylene-ethynylene)210 were employed to attain the non-covalent functionalization of h-BN via strong π–π interactions. Moreover, non-covalently modified h-BN also resulted in superior filler dispersions, including composites with superior TC.208
Gao et al. reported that h-BN could be dispersed easily because of strong π–π interactions between BNNTs and peptides.211 Thereafter, Wu et al. revealed that nanoparticles of h-BN could be modified non-covalently with polydopamine under aqueous (solvent-free) conditions, and the fabricated h-BN/bisphenol E cyanate ester material showed outstanding mechanical and thermal conductivity characteristics.212 Furthermore, the non-covalent functionalization allowed a constant dispersion of BNNSs based on their own inherent properties including several types of diverse interactions, viz., van der Waals forces, Lewis acid–base complexation, π–π interactions, and physical adsorption, which could help in disabling the weak van der Waals interactions between the BNNS layers.213,214
Ma et al. reported the functionalization of BNNSs and their stabilization with organic polymers using a non-covalent approach.215 They used a conjugating polymer, which is mainly based on functionalized polythiophene (PT), as shown in Fig. 21(a). Fig. 21(b and c) shows the structure of polyvinylpyrrolidone (PVP) and a typical model for the π–π-stacking-like interaction between the BNNSs and PT. Herein, PT was used by the researchers because its polymers were reported to act as active components, exhibiting a number of molecular and organic electronics applications for the construction of useful devices.216,217 In addition to the above-mentioned properties of PT, it has also been reported that PT is capable of being used for the non-covalent functionalization of BNNTs.210 Further an extensive study was presented by Emanet et al. on the non-covalent interactions between doxorubicin (Dox) and BNNTs/h-BNNSs, and the possible drug delivery applications.218 For the fabrication of BNNTs, colemanite and Fe2O3 (as a catalyst) were used, as reported earlier.218 Concisely, 2.0 g of colemanite and 160 mg of Fe2O3 were mixed in 2.0 mL of ddH2O; this mixture was then placed in a high-grade alumina (Al2O3) boat. Subsequently, when the water evaporated from this mixture, it was kept inside a furnace (tubular). Hence, BNNTs were prepared under an NH3 atmosphere at 1280 °C for a soaking time of 3 h. In order to obtain pure BNNTs, the mixture was stirred in hydrochloric (HCl) solution (4 M) at 90 °C for 4 h; afterwards, this solution was centrifuged at 14000 rpm for half an hour. For the further purification of BNNTs, a solution of HNO3 (1 M) was added, with stirring at 30 °C for 6 h and centrifugation at 14000 rpm for up to 30 min. In order to eradicate any residual acid content, the obtained BNNTs were washed with ddH2O. The h-BN nanostructure was fabricated using 1.0 g of B2O3 and 3.0 mL of 4.25 M ammonia solution, followed by gradual stirring for ∼5 min; then, the mixture was transferred into an Al2O3 boat. Once the solution of ammonia was vaporized from this mixture, the Al2O3 boat containing the material was kept in an electric furnace (tubular) at 1300 °C for 2 h under an NH3 atmosphere. Moreover, after the furnace was cooled to ambient temperature, the Al2O3 boat was taken out and h-BN was achieved. Thus, the synthesized BNNTs and h-BN were interacted with Dox at a fixed pH value of 7 in PBS. Then, 2.0 mL samples of Dox solution at different concentrations, i.e., 0.5, 1.0, 2.0, 4.0, and 5.0 mM, in PBS were synthesized, and 3.0 mg of BNNTs/h-BN was added to these solutions.218
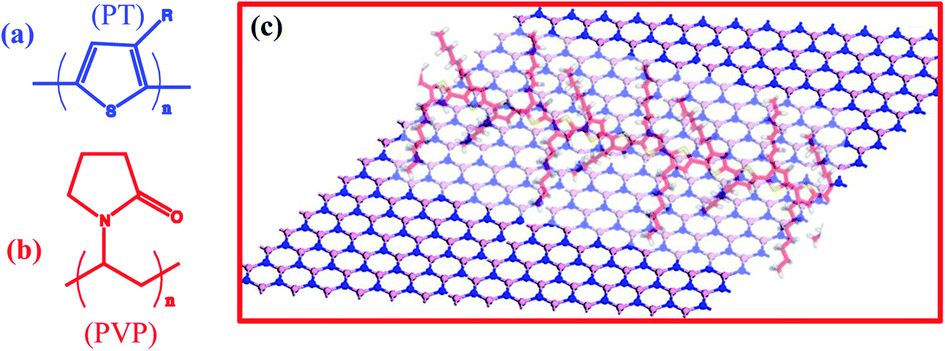 | ||
| Fig. 21 (a) The structure of polythiophene (PT), (b) the structure of polyvinylpyrrolidone (PVP), and (c) a schematic model of the π–π stacking between BNNSs and polythiophene (PT).215 | ||
The structure of Dox and its promising interactions with the B–N sidewalls are illustrated in Fig. 22(A and B). From Fig. 22(A), it is obvious that Dox contains several groups, such as phenyl rings, an amino group, carbonyl groups, ether groups, and numerous OH groups. Moreover, it may be conceivable that the presence of aromatic rings can increase its interactions with hydrophobic BNNTs and h-BN via π–π stacking (Fig. 22(B)), while the amino, carbonyl, ether, and hydroxyl groups can assist its dispersal in aqueous solutions. Probably, such types of interactions could also support an increase in the dispersion of BNNTs/h-BN in aqueous media. In addition to these studies, they also performed the non-covalent conjugation of folate with Dox–BNNTs (F-Dox–BNNTs). First of all, BNNTs were interconnected with Dox, and afterwards folate was decorated onto the surface of Dox–BNNTs to increase the probable matching with folate receptors on the surfaces of cancer cells. Fig. 22(C) reveals the chemical architecture of folate; folate molecules have numerous ionizable groups that might be negatively or positively charged depending on the pH value of the aqueous medium. It is evidently seen in Fig. 22(D) that there are only two possible binding modes between Dox–BNNTs and folate molecules. Folate contains phenyl moieties that can bind to the surfaces of BNNTs (F–BNNTs) via π–π stacking if some regions uncoated with Dox remain. Alternatively, folate molecules can link with Dox molecules and produce electrostatic interaction.218 A systematic comparison of the spectra of free Dox, folate, Dox–BNNTs, F-BNNT, and F-Dox–BNNTs is shown in Fig. 22(E), which reveals a strong absorption band at a wavelength of 230 nm and weak absorption at ∼480 nm for free Dox. Furthermore, several biological (cellular uptake and toxicity) studies were performed for drug delivery applications.218 The outcomes revealed that BNNTs showed three times higher Dox loading capabilities than h-BN. Moreover, the cellular acceptance of folate-Dox–BNNTs was observed to be significantly larger than Dox–BNNTs in the presence of HeLa cells because of the occurrence of folate receptors on these cell surfaces, leading to significantly enhanced levels of cancer cell death.218,219
 | ||
| Fig. 22 The interaction of B–N sidewalls with Dox. (A) The chemical structure of Dox and (B) a depiction of its possible interactions with B–N sidewalls (C) the chemical structure of folate and (D) its possible interactions with Dox–BNNTs. (E) UV/vis spectroscopy analysis data for free Dox, free folate, F-BNNT, and F-Dox–BNNTs. BNNT: boron nitride nanotubes; Dox: doxorubicin.218 | ||
Muhabie et al. reported an extensive study on the functionalization of h-BN mediated using a self-assembled supramolecular polymeric material using a non-covalent approach (a Michael addition reaction).220 Herein, they investigated a simple, consistent, and effective technique for the synthesis of exfoliated h-BNNSs from bulk h-BN via a liquid-phase exfoliation technique using a low-molecular-weight supramolecular polymer, such as adenine-functionalized polypropylene glycol (A-PPG). A-PPG has the ability to undergo self-assembly into long-range ordered lamellae or to form micelle-like nanostructures on the h-BN surface due to the creation of strong and specific interactions between A-PPG and h-BN. Superior h-BNNS exfoliation could be obtained via regulating the amount of A-PPG used.220 It has been explained that adenine moieties possess doublet H-bonding units inside the polymer backbones, as displayed in Fig. 23(a). Moreover, the authors reported that the final yield of the material was 83.5% (Table 2). Owing to its lower glass transition temperature (Tg= −70 °C)221 and excellent solubility within organic solvents, PPG was selected as the amorphous backbone section/segment in A-PPG. The synthesis method of the h-BN/A-PPG composite is shown in Fig. 23(b) and it involves a blending process that results in the exfoliation of h-BN via strong interactions with A-PPG in the solvent of THF. As the inclusion of a large amount of h-BN powder (10–50 wt%) was needed to break down the interplanar van der Waals interactions between layers of h-BN, increasing the time period of sonication was found to be significant, maybe due to an increase in the number and strength of interactions between A-PPG and h-BN. Mechanistically, it was suggested that due to intermolecular adenine–adenine interactions (Fig. 23(c)), the adenine moieties function as required units that guide and drive A-PPG macromers to impulsively accumulate into nanostructures with long-range lamellar order. Nevertheless, when existing in excess, A-PPG spontaneously gathers into micellar structures. Consequently, an excess amount of A-PPG produces spherical micellar structures, and clusters form at the surfaces of the lamellae, as presented in Fig. 23(d).220
 | ||
| Fig. 23 (a) An illustration of the synthesis procedure and self-assembly process for A-PPG macromers [(a) adenine; (b) dimethylformamide and potassium tertbutoxide at 70 °C]. (b) A schematic representation of the direct noncovalent liquid-phase exfoliation of h-BN with the assistance of the A-PPG dispersant. (c) The suggested process for the adsorption of lamellar structures. (d) Spherical micelles of A-PPG on the surface of h-BN.220 | ||
Further, Chen et al. obtained non-covalently functionalized h-BN surfaces using a mussel-inspired approach.222 In order to obtain a reduced interfacial thermal barrier and gain h-BN and composites with enhanced TC, they successfully modified the surface of h-BN via employing dopamine coatings, which reveal self-polymerization behavior. Moreover, when equated with polypropylene (PP) composites that were reinforced with the same amount of pristine BN, TC was observed to be larger for composites reinforced with functionalized BN, which is known as f-BN, and maleic-anhydride-grafted PP owing to enhanced filler dispersion and improved interfacial filler-to-matrix compatibility, which enabled the evolution of additional thermal pathways.222 Chen et al. presented benzoxazine nanocomposites with improved TC based on a non-covalently functionalized h-BN approach.223 Fig. 24(a) depicts the entire modification method for h-BN nanopowder. For the sake of dissolution, initially, 250 mL of DI water and 0.242 g of Tris–HCl buffer solution were decanted into a glass beaker. Thereafter, to maintain the pH at 8.5, dilute NaOH solution was slowly added. Later, ∼1 g of h-BN nanopowder was mixed into this prepared solution followed by ultrasonication dispersion for 1 h. Further, dopamine (0.4 g) was weighed and mixed into the above solution, and this solution was continuously stirred followed by heating at 25 °C for 72 h to attain polymerized dopamine molecules within the h-BNNS surface. After the reaction was completed, the mixture was washed numerous times using centrifugation and DI water until the solution became colorless. Lastly, the dopamine-modified h-BN nanofiller was dehydrated at 80 °C for 24 h in an oven, and the self-polymerization of dopamine took place. During self-polymerization, the nanofiller was found to change, which signifies that the h-BN surface was effectively coated with polydopamine.223
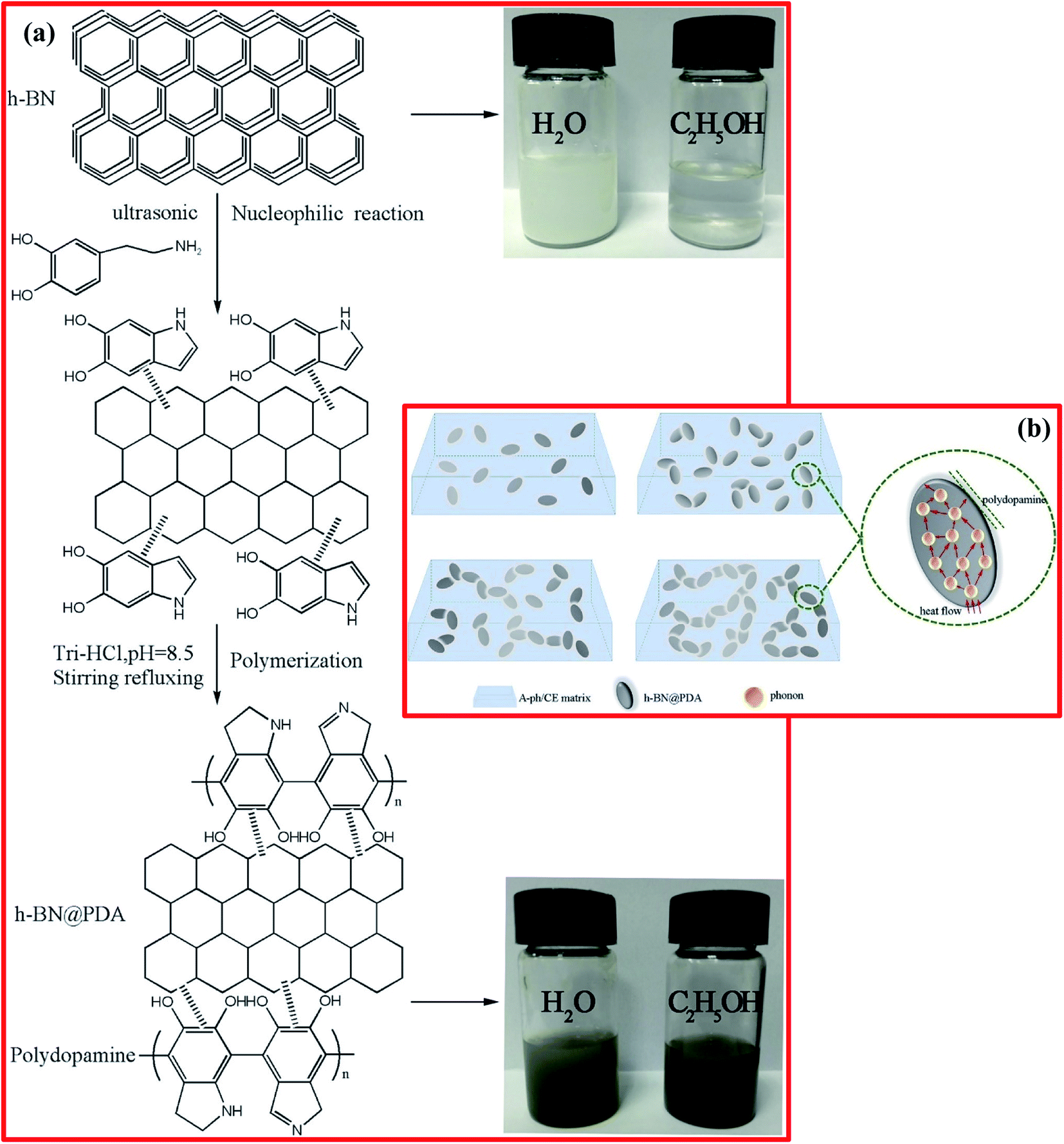 | ||
| Fig. 24 (a) The reaction mechanism of dopamine-modified h-BN nanoparticles and images of dispersions in ethanol and water. (b) A schematic diagram of the heat-conduction mechanism.223 | ||
Further, several polymer nanocomposites with different concentrations (0 wt%, 2 wt%, 5 wt%, 10 wt%, 15 wt%, and 20 wt%) of nanofiller were synthesized using a simple curing casting method. Among all the nanocomposites, the TC of the A-ph/CE/h-BN@PDA composite sample (with 20 wt% h-BN@PDA) was increased to 0.71 W m−1 K−1. As the amount of nanofiller increased, the thermal stability of these nanocomposites increased significantly. During polydopamine coating, there is heat flow through the fabricated nanocomposite. This might be due to a significant reduction in the maximum possibility of collisions and directional deflections of phonon within the nanoparticles; therefore, the reduced phonon scattering and interfacial thermal resistance during heat conduction lead to higher conductivity and the formation of several heat conduction paths,224 as displayed in Fig. 24(b). The dynamic storage modulus increases to 7390 MPa. The dielectric constant, dielectric loss, volume resistivity, and dielectric breakdown strength values for this nanocomposite at 1000 Hz were 5.72, 0.0148, 4.08 × 108 Ω cm, and 61.6 kV mm−1, respectively. Therefore, the nanofiller (h-BN@PDA) reinforced nanocomposite (A-ph/CE/h-BN@PDA) exhibited excellent TC, ideal electrical performance, and outstanding thermal stability, and it might be utilized for various electronic packaging applications. Recently, Kang et al. reported an interesting work on the synthesis of non-covalently functionalized BNNSs with significant stability in water and redispersibility.225 They synthesized BNNTs successfully via an induction thermal plasma method.226 Synthesized BNNT powder (0.1 wt%) was dispersed in water along with a cationic surfactant solution of 0.5 wt% cetyltrimethylammonium 4-vinylbenzoate. The separately formulated and isolated BNNTs were associated with an adsorbed layer, and this mixture was subjected to an ultrasonication process for up to 1 h of soaking time. Afterwards, the in situ polymerization of the used surfactant cetyltrimethylammonium 4-vinylbenzoate (CTVB) was carried out on the surface of the BNNTs via injecting the free radical initiator VA-044 (5 mol%), which led to the stable fixation of CTVB on the surface of the BNNTs. Moreover, for the extraction of these separately isolated BNNTs from primitive p-BNNT solution, the procedure was examined using a variety of different g-forces and centrifugation times, and the centrifugation protocol was fixed (2502 g for 30 min). Approximately 70% of the upper volume of the centrifuged suspension was obtained as p-BNNT. Further, for generating white p-BNNT powder, the synthesized p-BNNT dispersion was freeze-dried at −45 °C for a soaking time of 72 h. On the basis of small-angle neutron scattering (SANS) and AFM results, the authors claimed that p-BNNT was effectively encapsulated in a CTVB surfactant layer possessing a thickness of ∼0.8 nm; BNNTs (diameter: ∼3 nm) were individually isolated successfully.150 The individually isolated BNNTs can be applied in solution-based procedures for industrial applications.
Thereafter, Rice et al. demonstrated the non-covalent functionalization of BNNTs using poly(2,7-carbazole).227 Based on a theoretical approach, i.e., density functional theory (DFT), they suggested that superior interactions occurred between BNNTs and poly(2,7-carbazole) in comparison to fluorene-BNNT interactions. Moreover, homo-poly(2,7-carbazole) and other copolymers with fluorenes were fabricated and utilized to disperse such BNNTs in organic solvents successfully. Thermal and AFM analysis was used to verify the capabilities of such polymers to dissolve adequate amounts (>80 wt%) of modified BNNTs. These highly soluble poly(2,7-carbazole)-BNNT complexes might be harvested for printed electronics, and their transparent composite samples may also be used for simple capacitor applications.227 Another work, by Velayudham et al., demonstrated the non-covalent functionalization of BNNTs with poly(p-phenylene-ethynylene) (PPE) and polythiophene.210 Herein, the functionalized BNNTs were dispersed successfully in different organic solvents, like chloroform (CHCl3), methylene chloride (CH2Cl2), and tetrahydrofuran (CH2)4O, using conjugated PPE and polythiophene ((C4H2S)n) via strong π–π-stacking-like interactions between PPE and BNNTs. Moreover, the functionalization of the BNNTs with PPE improved the planarization of PPE, showing red shifts in the absorbance and emission of composites with reference to free PPEs, while functionalization of the BNNTs with (C4H2S)n interrupts π-conjugation, causing blue shifts in the recorded absorption and emission spectra of composites. Ultimately, the surface morphology (SEM and TEM) of the BNNTs was unaffected during functionalization and, hence, these could be used for the design of high-mechanical-strength fibers.210
Numerous studies have used non-covalent interactions between various organic molecules, polymeric materials, biomolecules, and the surfaces of BNNTs/h-BN.188 The non-covalent functionalization of both h-BN and BNNTs was carried out using alkyl amine, alkyl phosphine, and aromatic groups, involving different molecules and polymers.196,228 In addition to the functionalization of h-BN via covalent and non-covalent approaches, there is another approach that is based on Lewis acid–base interactions. The interactions in the above-mentioned cases are facilitated via electron-rich N and O species networks, electron-deficient B-atoms, and π–π interactions. Hence, B atoms easily act as Lewis acids with respect to electrons (as Lewis bases). This kind of interaction shows an appreciable effect in the presence of extremely reactive radical species; vacant p-orbitals of B atoms can offer an avenue for the formation of bonds and, therefore, covalent functionalization. Upon comparing the use of acids, alkalis, oxidants, and extremely high-temperature conditions for modifying the active surface of h-BN, the use of Lewis acid–base interactions is easier to manage without huge demands for complicated apparatus.124,171,229,230
Using Lewis acid–base interactions, several studies have reported attaching amino functional groups to B atoms in h-BN, which is owing to the occurrence of locally polarized B–N bonds that permit the suitable targeting of B and N atoms71,173,231 Concisely, B atoms have Lewis acid features that are susceptible to attack by Lewis base composites, for example, amines, producing stable Lewis acid–base complexes that are advantageous for the easy dispersion and exfoliation of h-BN. Consequently, the exfoliation of BNNSs and their functionalization can take advantage of solvents with Lewis base performance. Moreover, the existence of several defects in pristine h-BN makes its B atoms too susceptible to attack from Lewis bases during sonication processes. This type of attack can produce more defects during sonication. Lin et al. reported the preparation of h-BN via a ball-milling technique, and it was then functionalized with long-chain alkyl amines through Lewis acid–base interactions between the various amino groups and the B atoms of h-BN, achieving soluble amine-linked nanosheets.173 For BNNT functionalization, a similar mechanism was adopted by several researchers.232–234 Su et al. reported a functionalized-graphene (f-G)/h-BN flexible laminated composite with improved TC as an adhesive material via employing a facile latex approach.235 For the synthesis of the composites, primarily, the authors used a flexible fiber-reinforced laminated composite adhesive which merged with f-G and h-BN layers synthesized via colloid-blending and self-assembly techniques with the aid of secondary forces and hydrophilic variance. Herein, poly(2-ethylhexyl acrylate) (P2EHA) was used as a polymeric matrix that interconnected the layers of f-G with one more layer of h-BN as a cross-linker/adhesive through a self-assembly technique. Moreover, Lewis acid–base (δ+–δ−) interactions and π–π stacking enhanced the compatibility between the filler and polymer matrix. Based on various characterization techniques, like SEM, Raman spectroscopy, and XRD, the authors confirmed the fabrication of these flexible f-G/h-BN laminated composite adhesives. The laminated structure of the f-G/h-BN flexible composite is shown in Fig. 25. The oriented stacking arrangement and laminated structure resulted in outstanding in-plane thermal conductivity of ∼4.20 W m−1 K−1, along with adequate insulative and adhesive properties.235 Recently, Cai and coworkers demonstrated the functionalization of BNNSs via employing Lewis acid–base interactions, in which a possible platform was constructed using a SiO2 coating on the surface of h-BNNSs, which gave a chance for the inclusion of phytic acid (PA).236 The use of the fabricated h-BN nanohybrids can lead to an improvement in the flame retardancy of thermoplastic polyurethane (TPU). A schematic diagram of the SiO2 coating formation and flame-retardant functionalization of h-BN is presented in Fig. 26(a). TEM images of the microstructures of bulk h-BN, h-BN@SiO2, and SiO2 spheres are shown in Fig. 26(b–d), respectively. The TEM image of bulk h-BN revealed a plane edge structure (Fig. 26(b)). Herein, through Lewis acid–base interactions between B atoms of h-BN and amine groups, h-BNNSs were primarily coated with (3-aminopropyl)triethoxysilane (APTES) and, thereafter, a suitable pH value (9.0) supported the hydrolytic condensation of incorporated tetraethyl orthosilicate (TEOS) and APTES. Moreover, the edges of the h-BN nanohybrids were included in a heterogeneous nanostructure with a width of ∼20 nm (Fig. 26(c)). Because of a lack of h-BNNSs, numerous SiO2 spheres were formed via a cross-linking reaction between APTES and TEOS (Fig. 26(d)). The formation of a SiO2 coating might offer an extremely chemically active surface for the advance functionalization of h-BNNSs.236
 | ||
| Fig. 25 The laminated structure of a flexible f-G/h-BN composite.235 | ||
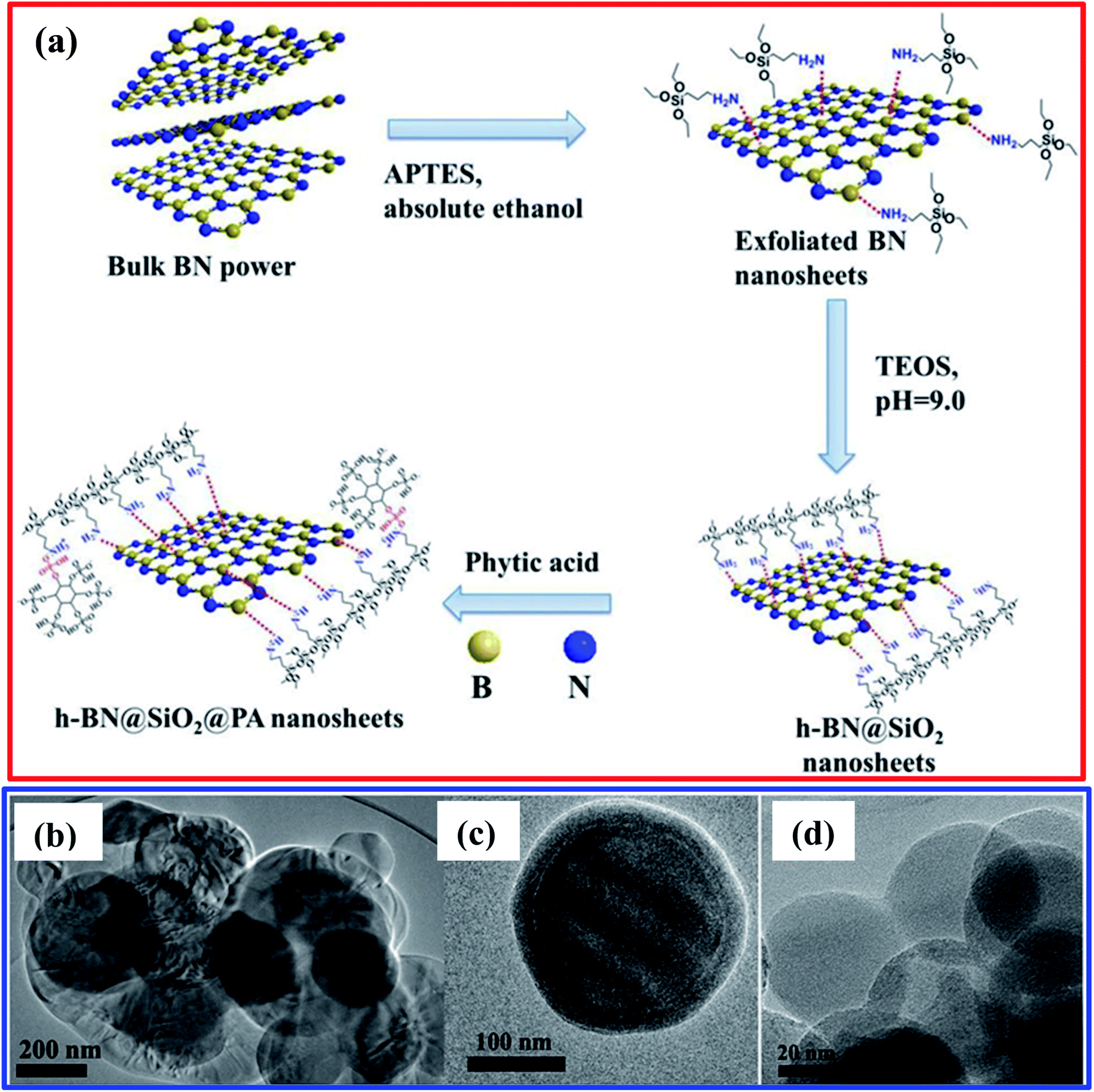 | ||
| Fig. 26 (a) A schematic illustration of the synthesis of functionalized h-BN. (b) TEM images of bulk h-BN, (c) h-BN@SiO2, and (d) SiO2 spheres.236 | ||
More recently, Lee et al. presented an effective process to disperse BNNSs in polar solvents through dual non-covalent interactions by using pyrene-tethered poly(4-vinylpyridine) (P4VP-Py) as a dispersion agent.237 P4VP-Py allows bi-functionalization along with BNNSs via π–π and Lewis acid–base interactions, which are created due to the pyrene and pyridine moieties, causing extremely stable BNNS dispersions in polar solvents. For the preparation of P4VP-Py, it was fabricated using reversible addition–fragmentation chain-transfer polymerization, including a joint reaction with pyrene, as presented in Fig. 27(a). Herein, the P4VP-Py molecules showed concomitant dual Lewis acid–base and π–π interactions with BNNSs owing to the amine group on the pyridine ring in the 4VP monomeric unit and the pyrene moiety at the chain end, hence resulting in outstanding dispersion in diverse alcohol-based solvents at large concentrations. The production of stronger interactions with BN might arise due to optimized geometry achieved using P4VP-Py, which assisted dual Lewis acid–base and π–π interactions, as presented in Fig. 27(b). In addition, the subsequent P4VP-Py-functionalized BNNSs were effectively used in different electrically insulating and thermally conductive polymer composites with enormous filler loading capabilities when mixed with a P4VP-Py matrix. Moreover, a combination of both P4VP-Py-functionalized BNNSs and the pristine P4VP matrix produced improved TC and an improved dielectric constant with higher thermal stability due to a compatible interface between the components.236
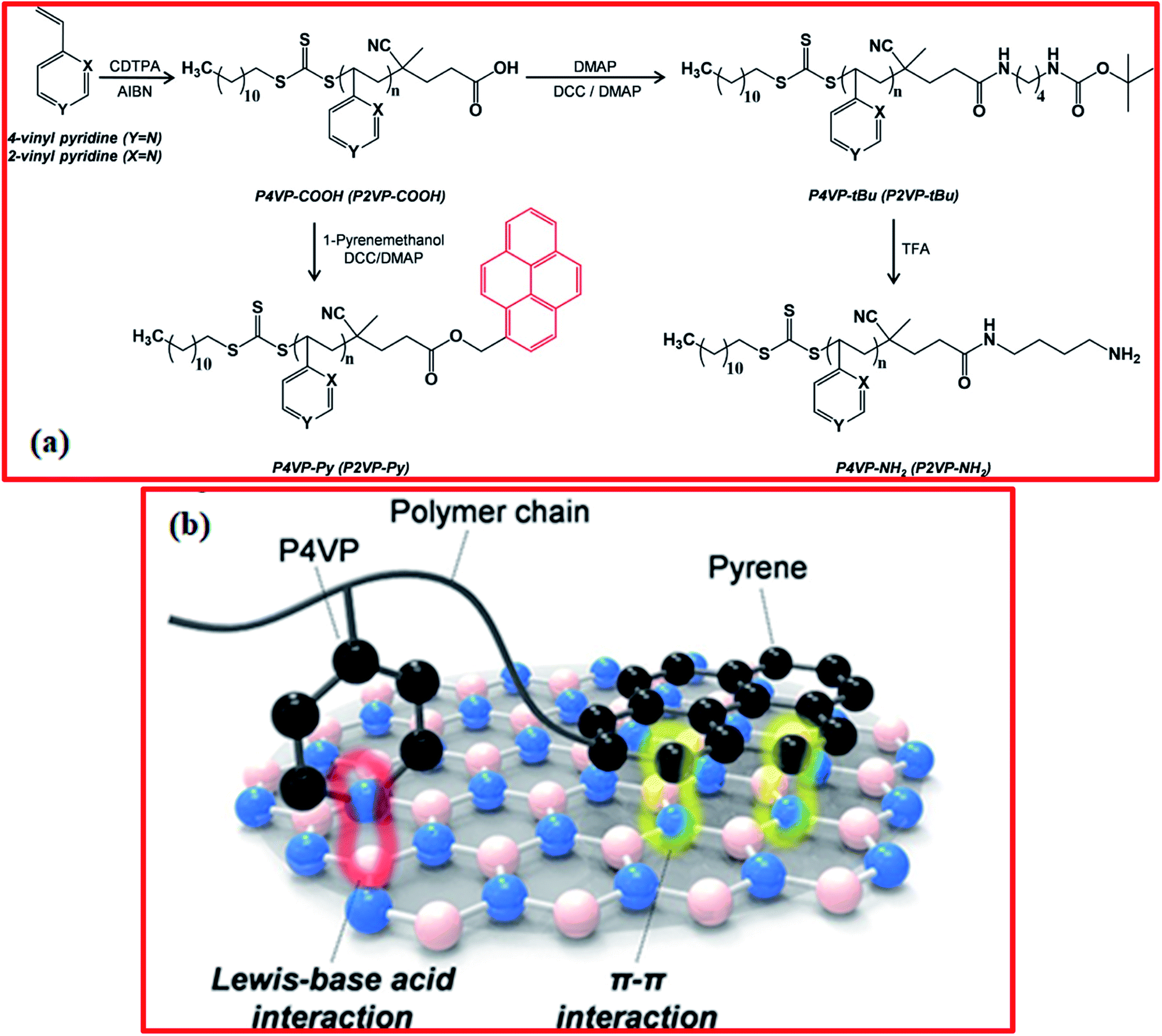 | ||
| Fig. 27 (a) The reaction scheme for PnVP (n = 2 and 4) with different functional groups: carboxylic acid (PnVP-COOH), pyrene (PnVP-Py), tert-butyl (PnVP-TBu), and amine (PnVP-NH2). (b) An illustration of the suggested mechanism for dual Lewis acid–base and π–π interactions using P4VP-Py molecules on BN.237 | ||
4. Applications of 2D h-BN
2D h-BN has numerous uses for different applications, such as in gates and dielectrics for the fabrication of electronic devices, in anticorrosive coatings for iron objects, in layered coatings and paint to reduce friction and wear, and in resistive layers for protection against high temperatures, along with several other technological and advanced 2D material applications.117 The possible applications of 2D h-BN include its use in the fabrication of optoelectronic devices. It is an especially appropriate nanomaterial for use as a photocatalyst, acting as a catalyst in the treatment of waste water.238–240 The global food industry depends strongly on access to reliable clean water supplies.241 Moreover, due to its outstanding electrical insulation properties, excellent thermal stability, chemical inertness, and controllable dimensions, it has been suggested that 2D h-BN is one of the most suitable materials for use in passivation processes and in harsh environments, particularly along with other 2D nanomaterials such as graphene, MoS2, MoSe2, WS2, WSe2, etc.242,243 Moreover, the deterioration of intrinsic properties and the poor characteristics of devices might be efficiently alleviated via encapsulation with h-BN to shield active areas.244Furthermore, the lamellar nanostructures of 2D h-BN/h-BNNSs involve weak van der Waals forces, analogous to graphene and MoS2.32,245 Hence, these types of structure are pretty good lubricant dopant materials, similar to graphene and MoS2. Besides these applications, 2D h-BN reveals excellent biocompatibility and it is also widely used as a food packaging material and as a cosmetic dopant.246–248 This is because h-BN will not harm the living cells and tissues of humans, animals, and plants in case of its environmental release. Lu et al. demonstrated that h-BN-nanoplate-assisted bovine serum albumin (BSA) inspired excellent immune activity in mice and exhibited remarkable biocompatibility and lower reactogenicity. These outcomes showed significant potential for various applications in the area of biomedical tissue engineering, like biomolecular carriers and the design of vaccines.249 In addition, h-BN nanoplates have been loaded covalently with proteins and studied with respect to the immune system of mice as biological nanovectors, and they strongly stimulate an antibody response and are known as adjuvants.250 Adjuvants mainly play a significant role in new kinds of vaccines that are weakly immunogenic. Owing to the large surface area and structural, temperature-based, and chemical stability, including tunability based on additives of different materials and surface functionalization, 2D-hBN also shows many potential applications in the field of nanomedicine, such as in drug delivery, in cell/bone imaging, and as a model for DNA self-assembly, with excellent biocompatibility and adsorption activity.117
Furthermore, the properties of 2D h-BNNSs from a biological perspective have significant potential, showing preferred outcomes upon interaction with cellular materials, e.g., 2D-BNNSs, when used as a therapeutic agent, might be elastic enough to pass through the barrier of the reticuloendothelial system (RES), and they also might be stable enough to oppose primary degradation. Recently, advanced applications of 2D h-BN in the field of electrochemical sensing have received much attention owing to the large surface area, various active edges, and good catalytic characteristics.251 It has been reported in the literature that 2D h-BN can be modified using conductivity-assisting nanomaterials, such as Au, Pt, etc., and the N-pz and B-pz orbitals in 2D h-BN form a network with the dz metal orbitals that can enhance the electrocatalytic properties of the prepared hybrid nanostructures.52,252–254
5. Summary and future advances
In this review article, a systematic summary of past and present developments relating to the preparation of 2D h-BN nanomaterials and their nanocomposites using numerous exfoliation techniques and their functionalization via covalent, non-covalent, and Lewis acid–base (ionic) approaches is provided in detail. Moreover, mechanisms of exfoliation were also described based on the use of external forces to overcome interactions, i.e., attractive van der Waals interactions, between 2D material layers. Even though several significant applications of BNNSs have been studied in the past few years, the preparation and functionalization of BNNSs is quite challenging owing to the low yields, small sample sizes, and characterization issues. The major challenge is executing experiments that reveal the intrinsic dynamics, e.g., temperature-dependent photoluminescence and time-resolved photoluminescence analysis, with spatial resolution.255 To date, many post-synthetic methods for the preparation and functionalization of h-BN have been recognized, but the selectivity and effectiveness of these approaches are not always reliable. Hence, several new and advanced approaches must be developed to meet these challenges.Amongst the numerous established procedures, exfoliation is one of the most widely used techniques to achieve large amounts of BNNSs with high purity. Herein, for the synthesis of BNNSs via exfoliation, all possible exfoliation techniques have been discussed, along with their advantages and disadvantages and a brief summary of relevant applications. A simple exfoliation method is liquid exfoliation, which involves the use of vigorous sonication and suitably designated solvents. Several forces are applicable during this method, such as the use of diverse chemicals, mechanical forces, and other aspects that have been implemented to enhance the effects of exfoliation. In most exfoliation methods involving h-BN, noticeable intercalation could not be significantly detected, which means that most exfoliation methods involving h-BN are based on mechanical force/oxidization. Another major drawback is that centrifugation is always used to separate out the BNNSs and, hence, the separated BNNS material is achieved in smaller amounts than the initially used bulk h-BN powder precursor. Consequently, it has been observed that an intermediate-assisted grinding exfoliation technique produced BNNSs with a very crystalline nature in contrast to samples prepared via liquid phase exfoliation techniques, and these methods were observed to be superior in comparison to intercalation and ball-milling techniques, which were associated with structural defects and additional functional groups. In addition to the exfoliation of h-BN, various functionalization approaches play a significant role in allowing exfoliated BNNSs to be used for versatile technological applications. Hence, covalent functionalization is one of the most fascinating approaches that is more successful, and it can allow the robust modification of the electronic properties of BNNSs. In this review article, we have considered appropriate mechanisms for the functionalization of h-BN. Moreover, this covalent approach permits the assignment of several functional groups to the surface of BNNSs that are very delicate with distinct organic and inorganic groups, thus obtaining suitable 2D nanomaterials for sensing applications. However, non-covalent and covalent functionalization approaches for h-BN are both extremely efficient techniques for controlling numerous aspects, e.g., dispersions in various solutions, the tuning of the transport and biological properties including the electronic band structure, etc. Despite methods for the functionalization of other 2D materials, for example graphene and transition metal dichalcogenides, having already been developed by researchers, the development of functionalization approaches for h-BN is still continuing. Therefore, numerous essential challenges, such as promoting the yields of these 2D materials, developing easy and cost-effective new methods, finding appropriate characterization techniques, and increasing interactions and reactivity between different types of molecules and h-BN layers, remain to be discussed in order to achieve well-connected surface morphologies and chemical activation approaches for these 2D nanomaterials. Moreover, novel functionalities added to these nanomaterials using chemical approaches can pave the way for the significant growth of multifunctional nanomaterials. As discussed in this current review article, BNNSs might be exfoliated using several strategies, such as intercalation, liquid-phase sonication, mechanical cleavage, thermal exfoliation, controlled gas exfoliation, and quadrupole-field-aided exfoliation, and their functionalization can be carried out via covalent, non-covalent, and Lewis acid–base approaches.
Moreover, for the sake of developing new techniques/methods, multiple investigations are urgently needed into intercalating and exfoliating h-BN; this is a big task that is hugely demanded for developing future advanced applications of BNNSs. Therefore, if we become really able to harvest high quantities of high-quality BNNSs via exfoliation, then we can invest in the application of BNNSs with excellent performance in the future.
Conflicts of interest
The authors have no conflicts of interest.Acknowledgements
The authors thankfully acknowledge all publishers of the cited works for their consent to highlight earlier works for academic purposes. C. R. G. is extremely thankful for financial support from the Science and Engineering Research Board, Department of Science and Technology (SERB-DST), New Delhi, India under EEQ (File No. EEQ/2018/000647) a major research project.References
- W. Lei, D. Liu and Y. Chen, Adv. Mater. Interfaces, 2015, 2, 1400529–1400534 CrossRef
.
- D. Golberg, Y. Bando, Y. Huang, T. Terao, M. Mitome, C. C. Tang and C. Y. Zhi, ACS Nano, 2010, 4, 2979–2993 CrossRef CAS PubMed
- A. Zunger, A. Katzir and A. Halperin, Phys. Rev. B: Solid State, 1976, 13, 5560–5573 CrossRef CAS
- C. Zhi, N. Hanagata, Y. Bando and D. Golberg, Chem.–Asian J., 2011, 6, 2530–2535 CrossRef CAS PubMed
- J. G. Kho, K. T. Moon, J. H. Kim and D. P. Kim, J. Am. Ceram. Soc., 2000, 83, 2681–2683 CrossRef CAS
- T. Sugino and T. Tai, Jpn. J. Appl. Phys., 2000, 39, L1101–L1104 CrossRef CAS
- R. Haubner, M. Wilhelm, R. Weissenbacher and B. Lux, Struct. Bonding, 2002, 102, 1–45 CrossRef CAS
- Y. Kubota, K. Watanabe, O. Tsuda and T. Taniguchi, Science, 2007, 317, 932–934 CrossRef CAS PubMed
- K. Watanabe, T. Taniguchi, T. Niiyama, K. Miya and M. Taniguchi, Nat. Photonics, 2009, 3, 591–594 CrossRef CAS
- K. Watanabe, T. Taniguchi and H. Kanda, Nat. Mater., 2004, 3, 404–409 CrossRef CAS PubMed
- Y. Kimura, T. Wakabayashi, K. Okada, T. Wada and H. Nishikawa, Wear, 1999, 232, 199–206 CrossRef CAS
- B. Podgornik, T. Kosec, A. Kocijan and C. Donik, Tribol. Int., 2015, 81, 267–275 CrossRef CAS
- C. R. Gautam, D. Chakravarty, A. Gautam, C. S. Tiwary, C. F. Woellner, V. K. Mishra, N. Ahamad, S. Ozden, S. Jose, S. K. Biradar, R. Vajtai, R. Trivedi, D. S. Galvao and P. M. Ajayan, ACS Omega, 2018, 3, 6013–6021 CrossRef CAS PubMed
- C. R. Gautam, A. Gautam, V. K. Mishra, N. Ahmad, R. Trivedi and S. K. Biradar, Ceram. Int., 2019, 45, 1037–1048 CrossRef CAS
- C. R. Gautam, C. S. Tiwary, L. D. Machado, S. Jose, S. Ozden, S. K. Biradar, D. S. Galvã, R. K. Sonker, B. C. Yadav, R. Vajtai and P. M. Ajayan, RSC Adv., 2016, 6, 87888–87896 RSC
- A. Gautam, C. R. Gautam, U. Kumar and B. C. Yadav, Mater. Res. Express, 2019, 6, 125090–125113 CrossRef CAS
- G. Giovannetti, P. A. Khomyakov, G. Brocks, P. J. Kelly and J. Brink, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 073103–073106 CrossRef
- C. R. Dean, A. F. Young, I. Meric, C. Lee, L. Wang, S. Sorgenfrei, K. Watanabe, T. Taniguchi, P. Kim, K. L. Shepard and J. Hone, Nat. Nanotechnol., 2010, 5, 722–726 CrossRef CAS PubMed
- W. Han, T. Taychatanapat, A. Hsu, K. Watanabe, T. Taniguchi, P. J. Herrero and T. Palacios, IEEE Electron Device Lett., 2011, 32, 1209–1211 Search PubMed
- L. S. Panchakarla, K. S. Subrahmanyam, S. K. Saha, A. Govindaraj, H. R. Krishnamurthy, U. V. Waghmare and C. N. R. Rao, Adv. Mater., 2009, 21, 4726–4730 CAS
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Y. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed
- S. Myung, A. Solanki, C. Kim, J. Park, K. S. Kim and K. B. Lee, Adv. Mater., 2011, 23, 2221–2225 CrossRef CAS PubMed
- R. J. Zou, Z. Y. Zhang, K. B. Xu, L. Jiang, Q. W. Tian, Y. G. Sun, Z. G. Chen and J. Q. Hu, Carbon, 2012, 50, 4965–4972 CrossRef CAS
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS PubMed
- K. Kim, J. Y. Choi, T. Kim, S. H. Cho and H. J. Chung, Nature, 2011, 479, 338–344 CrossRef CAS PubMed
- D. Pacile, J. C. Meyer, C. O. Girit and A. Zettl, Appl. Phys. Lett., 2008, 92, 133107–133109 CrossRef
- C. Zhi, Y. Bando, C. Tang, H. Kuwahara and D. Golberg, Adv. Mater., 2009, 21, 2889–2893 CrossRef CAS
- K. Zhou, N. Mao, H. Wang, Y. Peng and H. Zhang, Angew. Chem., Int. Ed., 2011, 50, 10839–10842 CrossRef CAS PubMed
- L. Song, L. Ci, H. Lu, P. B. Sorokin, C. Jin, J. Ni, A. G. Kvashnin, D. G. Kvashnin, J. Lou, B. I. Yakobson and P. M. Ajayan, Nano Lett., 2010, 10, 3209–3215 CrossRef CAS PubMed
- H. Zeng, C. Zhi, Z. Zhang, X. Wei, X. Wang, W. Guo, Y. Bando and D. Golberg, Nano Lett., 2010, 10, 5049 CrossRef CAS PubMed
- K. J. Erickson, A. L. Gibb, A. Sinitskii, M. Rousseas, N. Alem, J. M. Tour and A. K. Zettl, Nano Lett., 2011, 11, 3221–3226 CrossRef CAS PubMed
- Y. Bai, J. Zhang, Y. Wang, Z. Cao, L. An, B. Zhang, Y. Yu, J. Zhang and C. Wang, ACS Appl. Nano Mater., 2019, 2, 3187–3195 CrossRef CAS
- Y. Lin, T. V. Williams, T. B. Xu, W. Cao, H. E. Elsayed-Ali and J. W. Connell, J. Phys. Chem. C, 2011, 115, 2679–2685 CrossRef CAS
- C. Zhi, Y. Bando, C. Tang, H. Kuwahara and D. Golberg, Adv. Mater., 2009, 21, 2889–2893 CrossRef CAS
- Y. Shi, C. Hamsen, X. Jia, K. K. Kim, A. Reina, M. Hofmann, A. L. Hsu, K. Zhang, H. Li, Z. Y. Juang, M. S. Dresselhaus, L. J. Li and J. Kong, Nano Lett., 2010, 10, 4134–4139 CrossRef CAS PubMed
- K. K. Kim, A. Hsu, X. Jia, S. M. Kim, Y. Shi, M. Hofmann, D. Nezich, J. F. Rodriguez- Nieva, M. Dresselhaus, T. Palacios and J. Kong, Nano Lett., 2012, 12, 161–166 CrossRef PubMed
- C. M. Orofeo, S. Suzuki, H. Kageshima and H. Hibino, Nano Res., 2013, 6, 335–347 CrossRef CAS
- Y. Chen, H. Liang, X. Xia, H. Zhang, J. Shi, Q. Abbas and G. Du, J. Mater. Sci.: Mater. Electron., 2017, 28, 14341–14347 CrossRef CAS
- S. M. Kim, A. Hsu, M. H. Park, S. H. Chae, S. J. Yun, J. S. Lee, D. Cho and W. Fang, Nat. Commun., 2015, 6, 8662 CrossRef CAS PubMed
- T. Gao, X. Song, H. Du, Y. Nie, Y. Chen, Q. Ji, J. Sun, Y. Yang, Y. Zhang and Z. Liu, Nat. Commun., 2015, 6, 6835 CrossRef CAS PubMed
- A. Pakdel, C. Zhi, Y. Bando, T. Nakayama and D. Golberg, ACS Nano, 2011, 5, 6507 CrossRef CAS PubMed
- J. Taha-Tijerina, T. N. Narayanan, G. Gao, M. Rohde, D. A. Tsentalovich, M. Pasquali and P. M. Ajayan, ACS Nano, 2012, 6, 1214–1220 CrossRef CAS PubMed
- W. L. Song, P. Wang, L. Cao, A. Anderson, M. J. Meziani, A. J. Farr and Y. P. Sun, Angew. Chem., Int. Ed., 2012, 124, 6604–6607 CrossRef
- Z. G. Chen and J. Zou, J. Mater. Chem., 2011, 21, 1191–1195 RSC
- Y. A. Wu, A. I. Kirkland, F. Schaffel, K. Porfyrakis, N. P. Young, G. A. D. Briggs and J. H. Warner, Nanotechnology, 2011, 22, 195603–195611 CrossRef PubMed
- C. R. Dean, A. F. Young, I. Meric, C. Lee, L. Wang, S. Sorgenfrei, K. Watanabe, T. Taniguchi, P. Kim, K. L. Shepard and J. Hone, Nat. Nanotechnol., 2010, 5, 722–726 CrossRef CAS PubMed
- A. Du, Y. Chen, Z. Zhu, R. Amal, G. Q. Lu and S. C. Smith, J. Am. Chem. Soc., 2009, 131, 17354–17359 CrossRef CAS PubMed
- W. Chen, Y. Li, G. Yu, C. Z. Li, S. B. Zhang, Z. Zhou and Z. Chen, J. Am. Chem. Soc., 2010, 132, 1699–1705 CrossRef CAS PubMed
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed
- H. Zhang, ACS Nano, 2015, 9, 9451–9469 CrossRef CAS PubMed
- C. Tan, P. Yu, Y. Hu, J. Chen, Y. Huang, Y. Cai, Z. Luo, B. Li, Q. Lu, L. Wang, Z. Liu and H. Zhang, J. Am. Chem. Soc., 2015, 137, 10430–10436 CrossRef CAS PubMed
- A. Pakdel, Y. Bando and D. Golberg, Chem. Soc. Rev., 2014, 43, 934–959 RSC
- X. F. Jiang, Q. Weng, X. B. Wang, X. Li, J. Zhang, D. Golberg and Y. Bando, J. Mater. Sci. Technol., 2015, 31, 589–598 CrossRef CAS
- K. G. Zhou, N. N. Mao, H. X. Wang, Y. Peng and H. L. Zhang, Angew. Chem., 2011, 123, 11031–11034 CrossRef
- G. R. Bhimanapati, D. Kozuch and J. A. Robinson, Nanoscale, 2014, 6, 11671–11675 RSC
- F. Xiao, S. Naficy, G. Casillas, M. H. Khan, T. Katkus, L. Jiang, H. Liu, H. Li and Z. Huang, Adv. Mater., 2015, 27, 7196–7203 CrossRef CAS PubMed
- C. Zhi, Y. Bando, C. Tang, H. Kuwahara and D. Golberg, Adv. Mater., 2009, 21, 2889–2893 CrossRef CAS
- A. Ciesielski, S. Haar, M. El Gemayel, H. Yang, J. Clough, G. Melinte, M. Gobbi, E. Orgiu, M. V. Nardi, G. Ligorio, V. Palermo, N. Koch, O. Ersen, C. Casiraghi and P. Samori, Angew. Chem., Int. Ed., 2014, 53, 10355–10361 CrossRef CAS PubMed
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed
- P. Thangasamy and M. Sathish, CrystEngComm, 2015, 17, 5895–5899 RSC
- R. Bari, D. Parviz, F. Khabaz, C. D. Klaassen, S. D. Metzler, M. J. Hansen, R. Khare and M. J. Green, Phys. Chem. Chem. Phys., 2015, 17, 9383–9393 RSC
- K. L. Marsh, M. Souliman and R. B. Kaner, Chem. Commun., 2015, 51, 187–190 RSC
- T. Morishita, H. Okamoto, Y. Katagiri, M. Matsushita and K. Fukumori, Chem. Commun., 2015, 51, 12068–12071 RSC
- Y. Lin, T. V. Williams and J. W. Connell, J. Phys. Chem. Lett., 2010, 1, 277–283 CrossRef CAS
- Z. Liu, Y. Wang, Z. Wang, Y. Yao, J. Dai, S. Das and L. Hu, Chem. Commun., 2016, 52, 5757–5760 RSC
- Z. Cui, A. J. Oyer, A. J. Glover, H. C. Schniepp and D. H. Adamson, Small, 2014, 10, 2352–2355 CrossRef CAS PubMed
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef
- X. Li, X. Hao, M. Zhao, Y. Wu, J. Yang, Y. Tian and G. Qian, Adv. Mater., 2013, 25, 2200–2204 CrossRef CAS PubMed
- A. V. Lapshin, A. M. Germanskii and S. P. Bogdanov, Glass Phys. Chem., 2004, 30, 202–206 CrossRef CAS
- M. Du, Y. Wu and X. Hao, CrystEngComm, 2013, 15, 1782–1786 RSC
- Y. Lin, T. V. Williams and J. W. Connell, J. Phys. Chem. Lett., 2010, 1, 277–283 CrossRef CAS
- M. Du, X. Li, A. Wang, Y. Wu, X. Hao and M. Zhao, Angew. Chem., Int. Ed., 2014, 53, 3645–3649 CrossRef CAS PubMed
- G. R. Bhimanapati, D. Kozuch and J. A. Robinson, Nanoscale, 2014, 6, 11671–11675 RSC
- G. L. Doll, J. S. Speck, G. Dresselhaus, M. S. Dresselhaus, K. Nakamura and S. I. Tanuma, J. Appl. Phys., 1989, 66, 2554–2558 CrossRef CAS
- L. Lin, Y. Xu, S. Zhang, I. M. Ross, A. C. M. Ong and D. A. Allwood, Small, 2014, 10, 60–65 CrossRef CAS PubMed
- R. Arenal, A. C. Ferrari, S. Reich, L. Wirtz, J. Y. Mevellec, S. Lefrant, A. Rubio and A. Loiseau, Nano Lett., 2006, 6, 1812–1816 CrossRef CAS PubMed
- A. Sumiyoshi, H. Hyodo and K. Kimura, J. Phys. Chem. Solids, 2010, 71, 569–571 CrossRef CAS
- Z. Zeng, T. Sun, J. Zhu, X. Huang, Z. Yin, G. Lu, Z. Fan, Q. Yan, H. H. Hng and H. Zhang, Angew. Chem., Int. Ed., 2012, 51, 9052–9056 CrossRef CAS PubMed
- S. Y. F. Zhao, G. A. Elbaz, D. K. Bediako, C. Yu, D. K. Efetov, Y. Guo, J. Ravichandran, K. A. Min, S. Hong, T. Taniguchi, K. Watanabe, L. E. Brus, X. Roy and P. Kim, Nano Lett., 2018, 18, 460–466 CrossRef CAS PubMed
- Y. Wang, C. C. Mayorga-Martinez, X. Chia, Z. Sofer and M. Pumera, Nanoscale, 2018, 10, 7298–7303 RSC
- G. Ortiz, C. Pochat-Bohatier, J. Cambedouzou, M. Bechelany and P. Miele, Nanomaterials, 2018, 8, 716 CrossRef PubMed
- N. Kheirabadi and A. Shafiekhani, Nanotechnology, 2021, 32, 180001 CrossRef CAS PubMed
- W. Q. Han, L. Wu, Y. Zhu, K. Watanabe and T. Taniguchi, Appl. Phys. Lett., 2008, 93, 223103–223106 CrossRef
- X. Li, X. Wang, L. Zhang, S. Lee and H. Dai, Science, 2008, 319, 1229–1232 CrossRef CAS PubMed
- J. H. Warner, M. H. Rummeli, A. Bachmatiuk and B. Buchner, ACS Nano, 2010, 4, 1299–1304 CrossRef CAS PubMed
- Y. Wang, Z. Shi and J. Yin, J. Mater. Chem., 2011, 21, 11371–11377 RSC
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H. Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS PubMed
- Y. Xue, Q. Liu, G. He, K. Xu, L. Jiang, X. Hu and J. Hu, Nanoscale Res. Lett., 2013, 8, 1 CrossRef PubMed
- G. Eda, H. Yamaguchi, D. Voiry, T. Fujit, M. Chen and M. Chhowalla, Nano Lett., 2011, 11, 5111–5116 CrossRef CAS PubMed
- A. Castellanos-Gomez, M. Poot, G. A. Steele, H. S. J. van der Zant, N. Agrait and G. Rubio- Bollinger, Nanoscale Res. Lett., 2012, 7, 1–4 CrossRef PubMed
- L. Liu, S. B. Kumar, Y. Ouyang and J. Guo, IEEE Trans. Electron Devices, 2011, 58, 3042–3047 CAS
- L. H. Li, M. Petravic, B. C. C. Cowie, T. Xing, R. Peter, Y. Chen, C. Si and W. Duan, Appl. Phys. Lett., 2012, 101, 191604–191608 CrossRef
- V. Stengl, J. Henych, M. Slusna and P. Ecorchard, Nanoscale Res. Lett., 2014, 9, 1–14 CrossRef CAS PubMed
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed
- L. Cao, S. Emami and K. Lafdi, Mater. Express, 2014, 4, 165–171 CrossRef CAS
- B. Zhang, Q. Wu, H. Yu, C. Bulin, H. Sun, R. Li, X. Ge and R. Xing, Nanoscale Res. Lett., 2017, 12, 1–7 CrossRef CAS PubMed
- F. Yuan, W. Jiao, F. Yang, W. Liu, J. Liu, Z. Xu and R. Wang, J. Mater. Chem. C, 2017, 5, 6359–6368 RSC
- H. Ye, T. Lu, C. Xu, B. Han, N. Meng and L. Xu, Macromol. Chem. Phys., 2018, 219, 1700482–1700493 CrossRef
- X. Zheng, G. Wang, F. Huang, H. Liu, C. Gong, S. Wen, Y. Hu, G. Zheng and D. Chen, Front. Chem., 2019, 7, 1–9 CrossRef CAS PubMed
- N. Mittal, G. Kedawat, Kanika, S. Gupta and B. K. Gupta, Chemistry Select, 2020, 5, 12564–12569 CAS
- V. Arunachalam and S. Vasudevan, J. Phys. Chem. C, 2018, 122, 4662–4669 CrossRef CAS
- S. Wang, B. Tao, S. Yu, C. Wei, T. Zhou, X. Chen, C. Han and C. Wang, Mater. Lett., 2020, 269, 127644–127647 CrossRef CAS
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed
- D. Pacile, J. C. Meyer, C. O. Girit and A. Zettl, Appl. Phys. Lett., 2008, 92, 133107–133109 CrossRef
- C. Lee, Q. Li, W. Kalb, X. Z. Liu, H. Berger, R. W. Carpick and J. Hone, Science, 2010, 328, 76–80 CrossRef CAS PubMed
- D. Lee, B. Lee, K. H. Park, H. J. Ryu, S. Jeon and S. H. Hong, Nano Lett., 2015, 15, 1238–1244 CrossRef CAS PubMed
- C. Yu, J. Zhang, W. Tian, X. Fan and Y. Yao, RSC Adv., 2018, 8, 21948–21967 RSC
- L. Liu, Z. Xiong, D. Hu, G. Wu, B. Liu and P. Chen, Chem. Lett., 2013, 42, 1415–1416 CrossRef CAS
- L. H. Li, Y. Chen, G. Behan, H. Zhang, M. Petravic and A. M. Glushenkov, J. Mater. Chem., 2011, 21, 11862–11866 RSC
- A. Pierret, J. Loayza, B. Berini, A. Betz, B. Placais, F. Ducastelle, J. Barjon and A. Loiseau, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 035414–035420 CrossRef
- N. Alem, R. Erni, C. Kisielowski, M. D. Rossell, W. Gannett and A. Zettl, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 155425–155431 CrossRef
- C. Damm, J. Körner and W. Peukert, J. Nanopart. Res., 2013, 15, 1–12 CrossRef
- Y. Tominaga, K. Sato, D. Shimamoto, Y. Imai and Y. Hotta, Ceram. Int., 2015, 41, 10512–10519 CrossRef CAS
- X. Chen, J. F. Dobson and C. L. Raston, Chem. Commun., 2012, 48, 3703–3705 RSC
- Y. Tominaga, K. Sato, D. Shimamoto, Y. Imai and Y. Hotta, Ceram. Int., 2015, 41, 10512–10519 CrossRef CAS
- N. Alem, R. Erni, C. Kisielowski, M. D. Rossell, W. Gannett and A. Zettl, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 155425–155431 CrossRef
- X. Cai, Y. Luo, B. Liu and H. M. Cheng, Chem. Soc. Rev., 2018, 47, 6224–6266 RSC
- K. Zhang, Y. Feng, F. Wang, Z. Yang and J. Wang, J. Mater. Chem. C, 2017, 5, 11992–12022 RSC
- L. Liu, Z. Xiong, D. Hu, G. Wu, B. Liu and P. Chen, Chem. Lett., 2013, 42, 1415–1416 CrossRef CAS
- D. Deepika, L. H. Li, A. M. Glushenkov, S. K. Hait, P. Hodgson and Y. Chen, Sci. Rep., 2014, 4, 1–6 Search PubMed
- J. Ma, N. Luo, Z. Xie, F. Chen and Q. Fu, Mater. Res. Express., 2019, 6, 1050d8–1050d17 CrossRef CAS
- L. An, Y. Yu, C. Bai, Y. Bai, B. Zhang, K. Gao, X. Wang, Z. Lai and J. Zhang, npj 2D Mater. Appl., 2019, 3, 1–9 CrossRef CAS
- N. Yang, H. Ji, X. Jiang, X. Qu, X. Zhang, Y. Zhang and B. Liu, Nanomaterials, 2020, 10(1), 652–1663 Search PubMed
- B. Yu, W. Xing, W. Guo, S. Qiu, X. Wang, S. Lo and Y. Hu, J. Mater. Chem. A, 2016, 4, 7330–7340 RSC
- W. Y. Ko, C. Y. Chen, W. H. Chen and K. J. Lin, J. Chin. Chem. Soc., 2016, 63, 303–307 CrossRef CAS
- Y. R. Zhi, B. Yu, A. C. Y. Yuen, J. Liang, L. Q. Wang, W. Yang, H. D. Lu and G. H. Yeoh, ACS Omega, 2018, 3, 14942–14952 CrossRef CAS PubMed
- L. Acharya, P. Babu, A. Behera, S. P. Pattnaik and K. Parida, Mater. Today, 2021, 35, 239–242 CAS
- W. Zhu, X. Gao, Q. Li, H. Li, Y. Chao, M. Li, S. M. Mahurin, H. Li, H. Zhu and S. Dai, Angew. Chem., 2016, 55, 10766–10770 CrossRef CAS PubMed
- P. Thangasamy and M. Sathish, CrystEngComm, 2015, 17, 5895–5899 RSC
- W. Lei, V. N. Mochalin, D. Liu, S. Qin, Y. Gogotsi and Y. Chen, Nat. Commun., 2015, 6, 1–8 CrossRef PubMed
- S. Yang, A. G. Ricciardulli, S. Liu, R. Dong, M. R. Lohe, A. Becker, M. A. Squillaci, P. Samor, K. Mullen and X. Feng, Angew. Chem., Int. Ed., 2017, 56, 6669–6675 CrossRef CAS PubMed
- H. L. Lu, M. Z. Rong and M. Q. Zhang, Nanotechnology, 2018, 29, 12LT01–12LT13 CrossRef PubMed
- N. R. Glavin, M. L. Jespersen, M. H. Check, J. Hu, A. M. Hilton, T. S. Fisher and A. A. Voevodin, Thin Solid Films, 2014, 572, 245–250 CrossRef CAS
- D. Velazquez, R. Seibert, H. Man, L. Spentzouris and J. Terry, J. Appl. Phys., 2016, 119, 095306–095311 CrossRef
- A. F. Zhou, A. Aldalbahi and P. Feng, Opt. Mater. Express, 2016, 6, 3286–3292 CrossRef CAS
- A. Aldalbahi, A. F. Zhou and P. X. Feng, Sci. Rep., 2015, 5, 16703 CrossRef PubMed
- M. Sajjad, X. Peng, J. Chu, H. Zhang and P. Feng, J. Mater. Res., 2013, 28, 1747–1752 CrossRef CAS
- A. Aldalbahi and P. Feng, IEEE Trans. Electron Devices, 2015, 62, 1885–1890 CAS
- A. Aldalbahi, A. F. Zhou, S. Tan and X. Feng, Rev. Nanosci. Nanotechnol., 2016, 5, 79–92 CrossRef CAS
- M. Rivera, R. Velázquez, A. Aldalbahi, A. F. Zhou and P. Feng, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed
- W. Ortiz, N. J. Ramirez, D. Barrionuevo, M. K. Bhattarai and P. Feng, Nano express, 2021, 2, 010020 CrossRef
- H. Yurdakul, Y. Goncu, O. Durukan, A. Akay, A. T. Seyhan, N. Ay and S. Turan, Ceram. Int., 2012, 38, 2187–2193 CrossRef CAS
- P. Thangasamy and M. Sathish, CrystEngComm, 2015, 17, 5895–5899 RSC
- Z. Sun, Q. Fan, M. Zhang, S. Liu, H. Tao and J. Texter, Adv. Sci., 2019, 6, 1901084–1901117 CrossRef CAS PubMed
- N. M. H. Duong, E. Glushkov, A. Chernev, V. Navikas, J. Comtet, M. A. P. Nguyen, M. Toth, A. Radenovic, T. T. Tran and I. Aharonovich, Nano Lett., 2019, 19, 5417–5422 CrossRef CAS PubMed
- Y. Wang, Y. Liu, J. Zhang, J. Wu, H. Xu, X. Wen, X. Zhang, C. S. Tiwary, W. Yang, R. Vajtai, Y. Zhang, N. Chopra, I. N. Odeh, Y. Wu and P. M. Ajayan, Sci. Adv., 2017, 3, e1701500–e1701506 CrossRef PubMed
- M. Ikram, J. Hassan, M. Imran, J. Haider, A. Ul-Hamid, I. Shahzadi, M. Ikram, A. Raza, U. Qumar and S. Ali, Appl. Nanosci., 2020, 10, 3525–3528 CrossRef CAS
- V. Sahu, S. Grover, B. Tulachan, M. Sharma, G. Srivastava, M. Roy, M. Saxena, N. Sethy, K. Bhargava, D. Philip, H. Kim, G. Singh, S. K. Singh, M. Das and R. K. Sharma, Electrochim. Acta, 2015, 160, 244–253 CrossRef CAS
- M. Matsumoto, Y. Saito, C. Park, T. Fukushima and T. Aida, Nat. Chem., 2015, 7, 730–736 CrossRef CAS PubMed
- G. Kamath and G. A. Baker, RSC Adv., 2013, 3, 8197–8202 RSC
- A. F. Khan, M. P. Down, G. C. Smith and C. W. -8202E. Banks, J. Mater. Chem. A, 2017, 5, 4103–4113 RSC
- H. B. Kulkarni, P. B. Tambe and G. M. Joshi, Compos. Interfac., 2017, 27, 529–550 CrossRef
- A. R. Deshmukh, J. W. Jeong, S. J. Lee, G. U. Park and B. S. Kim, ACS Sustainable Chem. Eng., 2019, 7, 17114–17125 CrossRef CAS
- C. Zhang, J. Tan, Y. Pan, X. Cai, X. Zou, H. M. Cheng and B. Liu, Natl. Sci. Rev., 2020, 7, 324–332 CrossRef CAS
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O'Neill, C. Boland, M. Lotya, O. M. Istrate, P. King, T. Higgins, S. Barwich, P. May, P. Puczkarski, I. Ahmed, M. Moebius, H. Pettersson, E. Long, J. Coelho, S. E. O'Brien, E. K. McGuire, B. M. Sanchez, G. S. Duesberg, N. McEvoy, T. J. Pennycook, C. Downing, A. Crossley, V. Nicolosi and J. N. Coleman, Nat. Mater., 2014, 13, 624–630 CrossRef CAS PubMed
- D. Voiry, A. Mohite and M. Chhowalla, Chem. Soc. Rev., 2015, 44, 2702 RSC
- S. Chen, R. Xu, J. Liu, X. Zou, L. Qiu, F. Kang, B. Liu and H. M. Cheng, Adv. Mater., 2019, 31, 1804810–1804819 CrossRef PubMed
- K. S. Novoselov, Q. Ge and D. V. Andreeva, Natl. Sci. Rev., 2020, 7, 559–560 CrossRef
- Q. Liu, C. Hu and X. Wang, Mater. Lett., 2019, 234, 306–310 CrossRef CAS
- H. Zhou, J. Tan, L. Yang, J. Wang, B. Ding, Y. Pan, X. Yu, M. Liu, C. Yang, L. Qiu, H. M. Cheng and B. Liu, Sci. China Math., 2021, 1–8 Search PubMed
- S. Y. Xie, F. Gao, X. Lu, R. B. Huang, C. R. Wang, X. Zhang, M. L. Liu, S. L. Deng and L. S. Zheng, Science, 2004, 304, 699 CrossRef CAS PubMed
- A. Koshio, M. Inakuma, Z. W. Wang, T. Sugai and H. Shinohara, J. Phys. Chem. B, 2000, 104, 7908–7913 CrossRef CAS
- X. Lu and Z. Chen, Chem. Rev., 2005, 105, 3643–3696 CrossRef CAS PubMed
- Y. Z. Tan, Z. J. Liao, Z. Z. Qian, R. T. Chen, X. Wu, H. Liang, X. Han, F. Zhu, S. J. Zhou, Z. Zheng, X. Lu, S. Y. Xie, R. B. Huang and L. S. Zheng, Nat. Mater., 2008, 7, 790–794 CrossRef CAS PubMed
- Y. Z. Tan, S. Y. Xie, R. B. Huang and L. S. Zheng, Nat. Chem., 2009, 1, 450–460 CrossRef CAS PubMed
- Q. H. Weng, Q. He, T. Liu, H. Y. Huang, J. H. Chen, Z. Y. Gao, S. Y. Xie, X. Lu, R. B. Huang and L. S. Zheng, J. Am. Chem. Soc., 2010, 132, 15093–15095 CrossRef CAS PubMed
- I. V. Kuvychko, A. V. Streletskii, N. B. Shustova, K. Seppelt, T. Drewello, A. A. Popov, S. H. Strauss and O. V. Boltalina, J. Am. Chem. Soc., 2010, 132, 6443 CrossRef CAS PubMed
- Q. Weng, X. Wang, X. Wang, Y. Bandoa and D. Golberg, Chem. Soc. Rev., 2016, 45, 3989–4079 RSC
- J. Ren, L. Stagi and P. Innocenzi, J. Mater. Sci., 2021, 56, 4053–4079 CrossRef CAS
- J. Lee, H. Jung, S. Yu, S. M. Cho, V. K. Tiwari, D. B. Velusamy and C. Park, Chem.–Asian J., 2016, 11, 1921–1928 CrossRef CAS PubMed
- T. Sainsbury, A. Satti, P. May, Z. Wang, I. McGovern, Y. K. Gun’ko and J. Coleman, J. Am. Chem. Soc., 2012, 134, 18758–18771 CrossRef CAS PubMed
- Y. Fan, Z. Yang, W. Hua, D. Liu, T. Tao, M. M. Rahman, W. Lei, S. Huang and Y. Chen, Adv. Energy Mater., 2017, 7, 1602380–1602385 CrossRef
- Y. Lin, T. V. Williams, W. Cao, H. E. Elsayed-Ali and J. W. Connell, J. Phys. Chem. C, 2010, 114, 17434–17439 CrossRef CAS
- O. Şen, M. Emanet and M. Çulha, Front. Bioeng. Biotechnol., 2018, 6, 1–9 CrossRef PubMed
- H. He and C. Gao, Chem. Mater., 2010, 22, 5054–5064 CrossRef CAS
- M. Holzinger, J. Abraha, P. Whelan, R. Graupner, L. Ley, F. Hennrich, M. Kappes and A. Hirsch, J. Am. Chem. Soc., 2003, 125, 8566–8580 CrossRef CAS PubMed
- C. Zhi, Y. Bando, T. Terao, C. Tang, H. Kuwahara and D. Golberg, Chem.–Asian J., 2009, 4, 1536–1540 CrossRef CAS PubMed
- T. Ikuno, T. Sainsbury, D. Okawa, J. M. J. Frechet and A. Zettl, Solid State Commun., 2007, 142, 643–646 CrossRef CAS
- Y. Li, Z. Zhou and J. Zhao, Nanotech, 2008, 19, 015202–015207 CrossRef PubMed
- M. D. Su, J. Phys. Chem. B, 2005, 109, 21647–21657 CrossRef CAS PubMed
- T. Sainsbury, A. Satti, P. May, A. O'Neill, V. Nicolosi, Y. K. Gun’ko and J. N. Coleman, Chem.–Eur. J., 2012, 18, 10808–10812 CrossRef CAS PubMed
- A. S. Nazarov, V. N. Demin, E. D. Grayfer, A. I. Bulavchenko, A. T. Arymbaeva, H. J. Shin, J. Y. Choi and V. E. Fedorov, Chem.–Asian J., 2012, 7, 554–560 CrossRef CAS PubMed
- W. Jin, W. Zhang, Y. Gao, G. Liang, A. Gu and L. Yuan, Appl. Surf. Sci., 2013, 270, 561–571 CrossRef CAS
- P. A. Denis and F. Iribarne, J. Phys. Chem. C, 2018, 122, 18583–18587 CrossRef CAS
- S. Liu, J. Ji, H. Zeng, Z. Xie, X. Song, S. Zhou and P. Chen, 2D Mater, 2018, 5, 035036–035055 CrossRef
- M. Jedrzejczak-Silicka, M. Trukawka, M. Dudziak, K. Piotrowska and E. Mijowska, Nanomaterials, 2018, 8, 605–626 CrossRef PubMed
- S. Daneshnia, M. Adeli and Y. Mansourpanah, Nano: Brief Reports and Reviews 2019, vol. 14, pp. 1950107–1950120 Search PubMed
- F. Späth, H. R. Soni, J. Steinhauer, F. Düll, U. Bauer, P. Bachmann, W. Hieringer, A. Görling, H. P. Steinrück and C. Papp, Chem.–Eur. J., 2019, 25, 8884–8893 Search PubMed
- A. Hemmi, H. Cun, G. Tocci, A. Epprecht, B. Stel, M. Lingenfelder, L. H. de Lima, M. Muntwiler, J. Osterwalder, M. Iannuzzi and T. Greber, Nano Lett., 2019, 19, 5998–6004 CrossRef CAS PubMed
- H. B. Harrison and J. R. Alston, MRS Adv., 2020, 5, 709–716 CrossRef CAS
- Z. Rafiei-Sarmazdeh, S. H. Jafari and S. J. A. Ahmadi, J. Nanostruct., 2020, 10, 64–75 CAS
- C. Sun, J. Zhao, D. Zhang, H. Guo, X. Wang and H. Hu, Nanoscale, 2020, 12, 18379–18389 RSC
- P. A. Denis and F. Iribarne, Comput. Theor. Chem., 2019, 1164, 112538 CrossRef CAS
- P. A. Denis, S. Ullah and F. Iribarne, New J. Chem., 2020, 44(15), 5725–5730 RSC
- S. Y. Xie, W. Wang, K. A. S. Fernando, X. Wang, Y. Lin and Y. P. Sun, Chem. Commun., 2005, 2005, 3670–3672 RSC
- C. Zhi, Y. Bando, C. Tang, R. Xie, T. Sekiguchi and D. Golberg, J. Am. Chem. Soc., 2005, 127, 15996–15997 CrossRef CAS PubMed
- X. Wu, W. An and X. C. Zeng, J. Am. Chem. Soc., 2006, 128, 12001–12006 CrossRef CAS PubMed
- C. Zhi, Y. Bando, C. Tang, S. Honda, K. Sato, H. Kuwahara and D. Golberg, J. Phys. Chem. B, 2006, 110, 1525–1528 CrossRef CAS PubMed
- W. Wang, Y. Bando, C. Zhi, W. Fu, E. Wang and D. Golberg, J. Am. Chem. Soc., 2008, 130, 8144–8145 CrossRef CAS PubMed
- G. Ciofani, V. Raffa, A. Menciassi and A. Cuschieri, Nanoscale Res. Lett., 2009, 4, 113–121 CrossRef CAS PubMed
- J. Yu, Y. Chen and B. M. Cheng, Solid State Commun., 2009, 149, 763–766 CrossRef CAS
- T. L. Li and S. L. C. Hsu, J. Phys. Chem. B, 2010, 114, 6825–6829 CrossRef CAS PubMed
- Z. Gao, C. Zhi, Y. Bando, D. Golberg and T. Serizawa, ACS Appl. Mater. Interfaces, 2011, 3, 627–632 CrossRef CAS PubMed
- C. H. Lee, D. Zhang and Y. K. Yap, J. Phys. Chem. C, 2012, 116, 1798–1804 CrossRef CAS
- H. Yan, Y. Tang, J. Su and X. Yang, Appl. Phys. A: Mater. Sci. Process., 2014, 114, 331–337 CrossRef CAS
- S. Md Sharker, Int. J. Nanomed., 2019, 14, 9983–9993 CrossRef CAS PubMed
- J. Yu, X. Huang, C. Wu, X. Wu, G. Wang and P. Jiang, Polymer, 2012, 53, 471–480 CrossRef CAS
- T. Terao, Y. Bando, M. Mitome, C. Zhi, C. Tang and D. Golberg, J. Phys. Chem. C, 2009, 113, 13605–13609 CrossRef CAS
- C. Zhi, Y. Bando, C. Tang, S. Honda, K. Sato, H. Kuwahara and D. Golberg, Angew. Chem., Int. Ed., 2005, 44, 7929–7932 CrossRef CAS PubMed
- S. Velayudham, C. H. Lee, M. Xie, D. Blair, N. Bauman, Y. K. Yap, S. A. Green and H. Liu, ACS Appl. Mater. Interfaces, 2010, 2, 104–110 CrossRef CAS PubMed
- Z. Gao, C. Zhi, Y. Bando, D. Golberg and T. Serizawa, J. Am. Chem. Soc., 2010, 132, 4976–4977 CrossRef CAS PubMed
- H. Wu and M. R. Kessler, ACS Appl. Mater. Interfaces, 2015, 7, 5915–5926 CrossRef CAS PubMed
- Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856–5857 CrossRef CAS PubMed
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed
- P. Ma and J. T. Spencer, J. Mater. Sci., 2015, 50, 313–323 CrossRef CAS
- D. Fichou, Handbook of Oligo- and Polythiophenes. Wiley-VCH, 1998 Search PubMed
- K. Mllen and G. Wegner, Electronic Materials: the Oligomer Approach. Wiley-VCH, 1998 Search PubMed
- M. Emanet, Ö. Sen and M. Çulha, Nanomedicine, 2017, 12, 797–810 CrossRef CAS PubMed
- S. Kalay, Z. Yilmaz and M. Çulha, Beilstein J. Nanotechnol., 2013, 4, 843–851 CrossRef CAS PubMed
- A. A. Muhabie, C. C. Cheng, J. J. Huang, Z. S. Liao, S. Y. Huang, C. W. Chiu and D. J. Lee, Chem. Mater., 2017, 29, 8513–8520 CrossRef CAS
- E. Piorkowska, Z. Kulinski, A. Galeski and R. Masirek, Polymer, 2006, 47, 7178–7188 CrossRef CAS
- L. Chen, H. F. Xu, S. J. He, Y. H. Du, N. J. Yu, X. Z. Du, J. Lin and S. Nazarenko, PloS One, 2017, 12, e0170523–e0170538 CrossRef PubMed
- L. Chen, K. Li, B. Li, D. Ren, S. Chen, M. Xu and X. Liu, Compos. Sci. Technol., 2019, 182, 107741–107763 CrossRef CAS
- Y. Q. Guo, Z. Y. Lyu, X. T. Yang, Y. J. Lu, K. P. Ruan, Y. L. Wu, J. Kong and J. W. Gu, Composites, Part B, 2019, 164, 732–739 CrossRef CAS
- S. H. Kang, S. W. Jeon, S. Y. Moon, Y. J. Yoon and T. H. Kim, J. Phys. Chem. Lett., 2020, 11, 4511–4516 CrossRef CAS PubMed
- K. S. Kim, C. T. Kingston, A. Hrdina, M. B. Jakubinek, J. Guan, M. Plunkett and B. Simard, ACS Nano, 2014, 8, 6211–6220 CrossRef CAS PubMed
- N. A. Rice, W. J. Bodnaryk, I. Tamblyn, Z. J. Jakubek, J. Lefebvre, G. Lopinski, A. Adronov and C. M. Homenick, J. Polym. Sci., 2020, 58, 1889–1902 CrossRef CAS
- C. Zhi, Y. Bando, C. Tang and D. Golberg, J. Am. Chem. Soc., 2005, 127, 17144–17145 CrossRef CAS PubMed
- T. Morishita and H. Okamoto, ACS Appl. Mater. Interfaces, 2016, 8, 27064–27073 CrossRef CAS PubMed
- L. Fu, G. X. Chen, N. Jiang, J. H. Yu, C. T. Lin and A. M. Yu, J. Mater. Chem. A, 2016, 4, 19107–19115 RSC
- Y. Lin and J. W. Connell, Nanoscale, 2012, 4, 6908–6939 RSC
- D. Golberg, Y. Bando, C. C. Tang and C. Y. Zhi, Adv. Mater., 2007, 19, 2413–2432 CrossRef CAS
- S. Pal, S. R. C. Vivekchand, A. Govindaraj and C. N. R. Rao, J. Mater. Chem., 2007, 17, 450–452 RSC
- A. Maguer, E. Leroy, L. Bresson, E. Doris, A. Loiseau and C. A. Mioskowski, J. Mater. Chem., 2009, 19, 1271–1275 RSC
- Z. Su, H. Wang, X. Ye, K. Tian, W. Huang, C. Xiao and X. Tian, Composites, Part A, 2017, 99, 166–175 CrossRef CAS
- W. Cai, B. Wang, L. Liu, X. Zhou, F. Chu, J. Zhan, Y. Hu, Y. Kan and X. Wang, Composites, Part B, 2019, 178, 107462–107474 CrossRef CAS
- H. Lee, J. Lee, S. H. Joo, S. J. Kang, S. K. Kwak, S. Yu and C. Park, ACS Appl. Nano Mater., 2020, 3, 7633–7642 CrossRef CAS
- A. Rubio, J. L. Corkill and M. L. Cohen, Phys. Rev. B, 1994, 49(7), 5081–5084 CrossRef CAS PubMed
- M. Lu, Photocatalysis and water purification: from fundamentals to recent applications. John Wiley & Sons, 2013 Search PubMed
- G. Centi, P. Ciambelli, S. Perathoner and P. Russo, Catal. Today, 2002, 75(1–4), 3–15 CrossRef CAS
- A. Raza, M. Ikram, M. Aqeel, M. Imran, A. Ul-Hamid, K. N. Riaz and S. Ali, Appl. Nanosci., 2020, 10, 1535–1544 CrossRef CAS
- J. Li, C. Zhang, M. Deng and J. Luo, RSC Adv., 2015, 5, 30861–30868 RSC
- L. H. Li, J. Cervenka, K. Watanabe, T. Taniguchi and Y. Chen, ACS Nano, 2014, 8, 1457–1462 CrossRef CAS PubMed
- A. S. Mayorov, R. V. Gorbachev, S. V. Morozov, L. Britnell, R. Jalil, L. A. Ponomarenko, P. Blake, K. S. Novoselov, K. Watanabe, T. Taniguchi and A. K. Geim, Nano Lett., 2011, 11, 2396–2399 CrossRef CAS PubMed
- D. Fan, J. Feng, J. Liu, T. Gao, Z. Ye, M. Chen and X. Lv, Ceram. Int., 2016, 42(6), 7155–7163 CrossRef CAS
- Y. Bai, J. Zhang, Y. Wang, Z. Cao, L. An, B. Zhang, Y. Yu, J. Zhang and C. Wang, ACS Appl. Nano Mater., 2019, 2, 3187–3195 CrossRef CAS
- A. Merlo, V. R. S. S. Mokkapati, S. Pandit and I. Mijakovic, Biomater. Sci., 2018, 6, 2298–2311 RSC
- G. Ciofani, S. Danti, G. G. Genchi, B. Mazzolai and V. Mattoli, Small, 2013, 9, 1672–1685 CrossRef CAS PubMed
- T. Lu, L. Wang, Y. Jiang, Q. liub and C. Huang, J. Mater. Chem. B, 2016, 4, 6103–6110 RSC
- M. S. Sinyakov, M. Dror, T. Lublin-Tennenbaum, S. Salzberg, S. Margel and R. R. Avtalion, Vaccine, 2006, 24, 6534–6541 CrossRef CAS PubMed
- S. Angizi, M. Khalaj, S. A. A. Alem, A. Pakdel, M. Willander, A. Hatamie and A. Simchi, J. Electrochem. Soc., 2020, 167, 126513 CrossRef CAS
- J. Yin, J. Li, Y. Hang, J. Yu, G. Tai, X. Li, Z. Zhang and W. Guo, Small, 2016, 12, 2942–2968 CrossRef CAS PubMed
- G. Elumalai, H. Noguchi and K. Uosaki, Phys. Chem. Chem. Phys., 2014, 16, 13755–13761 RSC
- K. Uosaki, G. Elumalai, H. C. Dinh, A. Lyalin, T. Taketsugu and H. Noguchi, Sci. Rep., 2016, 6, 32217 CrossRef CAS PubMed
- S. Z. Butler, S. M. Hollen, L. Cao, Y. Cui, J. A. Gupta, H. R. Gutierrez, T. F. Heinz, S. S. Hong, J. Huang, A. F. Ismach, E. Johnston-Halperin, M. Kuno, V. V. Plashnitsa, R. D. Robinson, R. S. Ruoff, S. Salahuddin, J. Shan, L. Shi, M. G. Spencer, M. Terrones, W. Windl and J. E. Goldberger, ACS Nano, 2013, 7(4), 2898–2926 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2021 |