
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing glycan stability via site-selective fluorination: modulating substrate orientation by molecular design†‡
Alexander
Axer
a,
Ravindra P.
Jumde
 b,
Sebastian
Adam
c,
Andreas
Faust
d,
Michael
Schäfers
de,
Manfred
Fobker
f,
Jesko
Koehnke§
*cg,
Anna K. H.
Hirsch
*bg and
Ryan
Gilmour
*a
b,
Sebastian
Adam
c,
Andreas
Faust
d,
Michael
Schäfers
de,
Manfred
Fobker
f,
Jesko
Koehnke§
*cg,
Anna K. H.
Hirsch
*bg and
Ryan
Gilmour
*a
aOrganisch Chemisches Institut, WWU Münster, Corrensstraße 36, 48149 Münster, Germany
bDepartment of Drug Discovery and Optimization, Helmholtz Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Centre for Infection Research (HZI), University Campus E8.1, 66123 Saarbrücken, Germany
cWorkgroup Structural Biology of Biosynthetic Enzymes, Helmholtz Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Institute for Infection Research (HZI), University Campus E8.1, 66123 Saarbrücken, Germany
dEuropean Institute for Molecular Imaging, Münster, Germany
eDepartment of Nuclear Medicine, University Hospital (UKM), Münster, Germany
fCenter for Laboratory Medicine, WWU Münster, Münster, Germany
gDepartment of Pharmacy, Saarland University, 66123 Saarbrücken, Germany
First published on 23rd November 2020
Abstract
Single site OH → F substitution at the termini of maltotetraose leads to significantly improved hydrolytic stability towards α-amylase and α-glucosidase relative to the natural compound. To explore the effect of molecular editing, selectively modified oligosaccharides were prepared via a convergent α-selective strategy. Incubation experiments in purified α-amylase and α-glucosidase, and in human and murine blood serum, provide insight into the influence of fluorine on the hydrolytic stability of these clinically important scaffolds. Enhancements of ca. 1 order of magnitude result from these subtle single point mutations. Modification at the monosaccharide furthest from the probable enzymatic cleavage termini leads to the greatest improvement in stability. In the case of α-amylase, docking studies revealed that retentive C2-fluorination at the reducing end inverts the orientation in which the substrate is bound. A co-crystal structure of human α-amylase revealed maltose units bound at the active-site. In view of the evolving popularity of C(sp3)–F bioisosteres in medicinal chemistry, and the importance of maltodextrins in bacterial imaging, this discovery begins to reconcile the information-rich nature of carbohydrates with their intrinsic hydrolytic vulnerabilities.
Introduction
The abundance of carbohydrates in biology, together with their structural and functional diversity,1 has resulted in a highly specific arsenal of regulatory enzymes.2 This molecular synergy spans the animal, plant and bacteria kingdoms, and manifests itself in processes ranging from subtle post-translational modifications to the high-throughput metabolic processes that are essential for life.3 As workhorses in processing carbohydrate energy sources, the venerable glycoside hydrolases play an essential role in breaking down long-chain glycans into their respective building blocks.4 A pertinent example is the endo-hydrolysis of α-(1 → 4) linked maltodextrins.5 These processes, underpinned by a unique molecular recognition partnership, are essential and repeated iteratively with remarkable precision and fidelity. Regrettably, this juxtaposition of selectivity and intrinsic hydrolytic instability continues to frustrate translational advances to the clinic6 due to the inherent instability of the glycan in vivo: this is particularly relevant in the context of bacterial infections. Specifically, 18F-labelled oligosaccharides have shown great potential due to their selective uptake in prokaryotic (bacterial) cells.7 However, achieving sufficient serum circulation times for maltodextrin PET tracers, to enable satisfactory signal-to-noise ratios, requires the development of metabolically robust scaffolds that are subtly modified so as not to disrupt molecular recognition.8Molecular orientation is a characteristic feature in glycoside hydrolysis with substrates being processed from a specific terminus.9 From the perspective of clinical translation, one conceptual strategy to mitigate this hydrolytic vulnerability is to alter substrate orientation in the enzyme pocket without inhibiting recognition completely (Fig. 1). Fluorine bioisosteres are ideally suited to this purpose,10,11 allowing localised changes to the physicochemical profile of the substrate to be introduced without a dominant steric penalty that would impede recognition completely.12 To expand our interest in modulating small molecule–protein interactions,13 the effect of site-selective fluorination on the hydrolysis of maltotetraoses by α-amylases and α-glucosidases is disclosed (Fig. 2).14
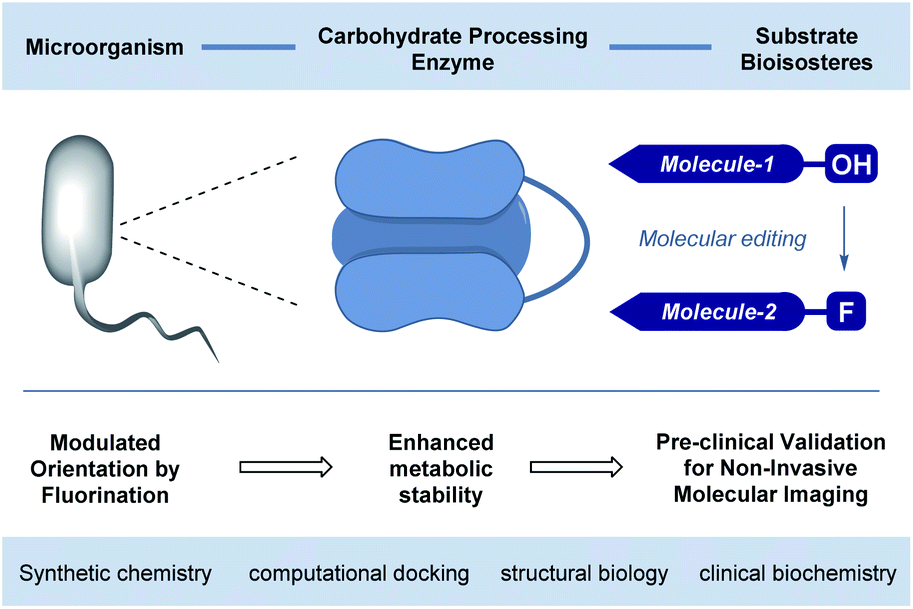 | ||
| Fig. 1 Conceptual framework of this study to validate the preclinical potential of fluorination in rendering tetraoses imaging platforms for bacterial infection. | ||
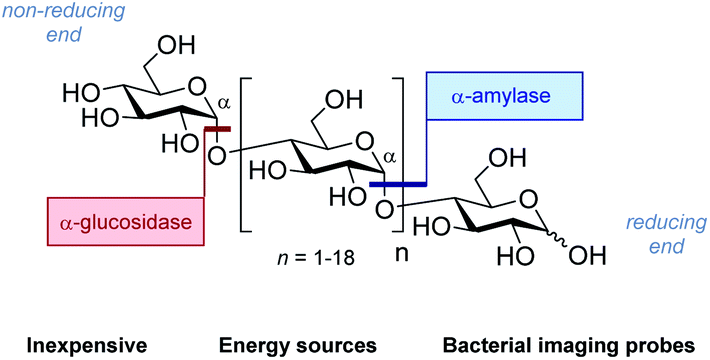 | ||
| Fig. 2 The structure of generic maltodextrins and the location of enzymatic hydrolysis by α-glucosidase and α-amylase. | ||
Consequently, a series of novel tetrasaccharide probes (1–6) were conceived to explore the influence of subtle changes in site-selective fluorine introduction [C(sp3)–OH vs. C(sp3)–F] on hydrolytic stability (Fig. 3).
 | ||
| Fig. 3 The maltotetraose probe set for this study. | ||
Strategic fluorination has a venerable history in the study and inhibition of hydrolases, but, to the best of our knowledge, has never been extended to the maltotetraoses. The maltotetraose core (1) was selected for this analysis since it is the shortest maltodextrin that can be truncated by salivary α-amylase.7b,15 Inspired by Withers' pioneering mono-, di- and tri-saccharide mechanistic enzymology tools,16,17 molecular editing at the C2 position of the terminal monosaccharides was performed. It was envisaged that this would enable an electronic influence to be exerted at the proximal hydrolysis site:18 α-Amylases recognise the reducing end (Fig. 3, blue), and α-glucosidases (Fig. 3, red) process the structure from the non-reducing end. Moreover, C2 fluorination confers additional advantages for carbohydrate drug design, by directing glycosylation selectivity,11 and by providing an NMR active nucleus.19
In the fluorinated structures, both C2 epimers were conceived (gluco- and manno-configured, 3–6) to explore the importance of the stereochemical information encoded at C2. A 2-deoxy species (2) at the non-reducing end was also envisaged as a bioisosteric control species. This would enable the effect of hydrogen bond deletion (C–OH → C–F and C–H) and of partial charge inversion (C–Hδ+ → C–Fδ−) to be assessed.
It is pertinent to note that the rarity of fluorinated natural products has the logical consequence that very few enzymes have evolved to recognise this structural feature.20 Collectively, it was envisaged that site-selectively fluorinated sugars would behave differently compared to their natural counterparts, thereby altering the bound orientation without completely suppressing function.
Results and discussion
Docking studies with human α-amylase
To explore this notion, a computational docking study was performed using the HYDE binding assessment calculation in LeadIT/SeeSAR21 to predict the bound conformations of these fluorinated probes in human α-amylases and α-glucosidases. Based on the sequence alignment and structural superimposition of salivary α-amylase (PDB ID: 1SMD)22 and pancreatic α-amylase (PDB ID: 5U3A),23 it is clear that both structures display a high degree of sequence and structural similarity, especially at the active site. As 5U3A (pancreatic α-amylase) has the highest resolution (0.95 Å), it was used for these docking studies. Initially, the bound conformations of the endogenous substrate 1 and the corresponding deoxy-analogue 2 were investigated in human pancreatic α-amylase (Fig. 4a). Although subtle, this analysis revealed that the deletion of a single hydrogen-bond donor at C2 of the terminal oligosaccharide at the non-reducing end inverts the orientation of the bound molecule in a sub-pocket lined by Asp300, Glu233 and Asp197 residues. Repeating this docking analysis with substrate 5 also revealed a flip in substrate orientation, such that it is repositioned with the fluorinated reducing end situated away from the Asp356 residue (Fig. 4b). To explore the effect of the C2 configuration on molecular orientation, the docked substrate 6 was then compared to 1 in the active site of human pancreatic α-amylase. The consequence of this subtle structural alteration was to re-align the scaffolds as indicated in Fig. 4c. These data demonstrate that the deletion or substitution of the C(sp3)–OH by C(sp3)–F can be harnessed to programme the orientation in which the enzyme binds the substrate. Remarkably, inverting the configuration of this single C(sp3)–F centre causes a flip in the bound orientation that resembles the native substrate 1. Although substrate 6 adopts the same general orientation as 1, a distinct conformation results in which the non-reducing end is positioned away from the Asp-356 residue. On a structural level, the absence of the C2 hydrogen bond donor at the reducing end eliminates the crucial interaction with Glu-233 (Fig. 4, inlayed Fig. c1 and c2). To examine the effects of molecular editing with fluorine at the non-reducing end, substrates 3 and 4 were examined (Fig. 4d and e). Docking studies reveal that substrate 3 (gluco-configured) adopts an orientation that is similar to that of the native substrate 1 in the active site of the pocket. As is evident from Fig. 4d, the non-reducing end is positioned away from Asp-356.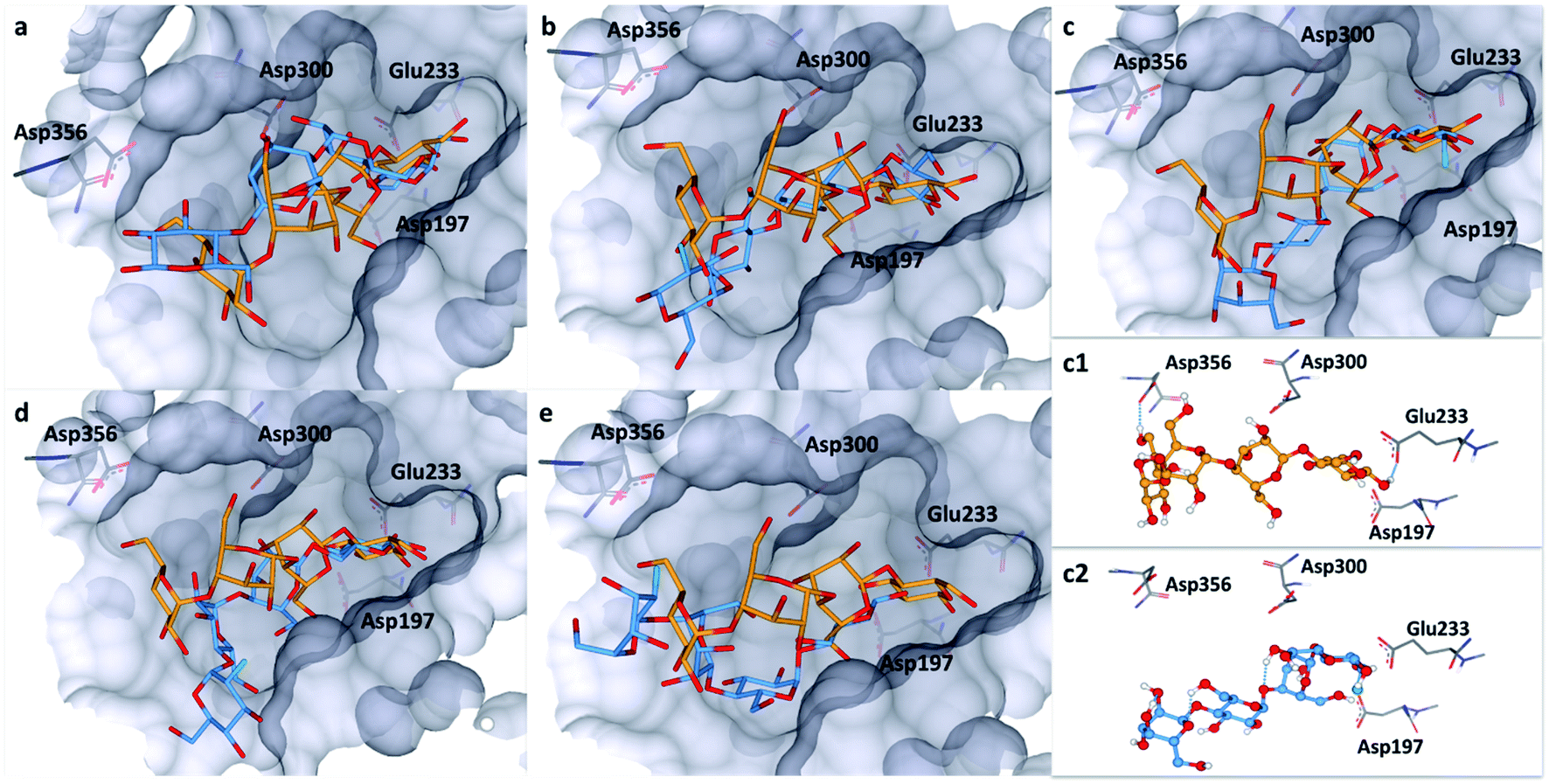 | ||
| Fig. 4 Docking studies: (a) comparison between the native substrate 1 (orange) and deoxy-substrate 2 (blue) in the active site of human pancreatic α-amylase (PDB ID: 5U3A); (b) comparison between substrate 1 (orange) and substrate 5 (blue); (c) comparison of bound substrate 1 (orange) and substrate 6 (blue); (c1) interaction of 1 with residues in the active site; (c2) interaction of 6 with residues in the active site; (d) comparison between substrate 1 (orange) and substrate 3 (blue); (e) comparison between substrate 1 (orange) and substrate 4 (blue). Colour code: protein surface: grey; protein skeleton: C: grey, O: red, N: blue; substrate 1: C: orange, O: red, substrate 2–6: C: blue, O: red, F: green. This figure was generated using SeeSAR 9.1 (BioSolveIT).2 | ||
Finally, an examination of substrate 4 (manno-configured) revealed a bound conformation in which the molecular orientation is similar to the native substrate 1 in the active site. However, the reducing terminus is no longer in close proximity to Asp-300, Glu-233 and Asp-197 (Fig. 4e) and the substrate is generally shifted outwards from the pocket. These docking studies in α-amylase illustrate that molecular editing alters the bound substrate orientation. The absence of these key interactions may thus have a downstream effect on hydrolytic stability.
Docking studies in human α-glucosidase
The bound conformation of substrate 1 and its deoxy-analogue 2 was subsequently investigated in a human intestinal α-glucosidase enzyme (maltase-glucoamylase, PDB ID: 2QMJ).24 Contrary to the bound conformation in α-amylase, the deoxy-substrate 2 was found to be oriented similarly to substrate 1 in the active site (Fig. 5a). Nonetheless, due to the deletion of the OH group relative to 1, a crucial interaction with Asp-542 is precluded (Fig. 5f, sub. 1 and sub. 2). Substrates 3 and 4 are oriented similarly as substrate 1 in the active site (Fig. 5b and f sub. 3; Fig. 5c and f sub. 4). In line with the findings pertaining to substrate 2, the bound conformations of fluorinated probes 3 and 4 (OH → F) also lack the crucial interaction with Asp-542 (Fig. 5f, sub. 1vs. sub. 3 and sub. 4). Oligosaccharides 5 and 6 adopt bound conformations that are similar to that of substrate 1 in the active site (Fig. 5d and e). This docking analysis did not reveal significant differences in the interactions of substrates 1 and 5 or 6 with key active site residues (Fig. 5f, subs 1, 5 and 6).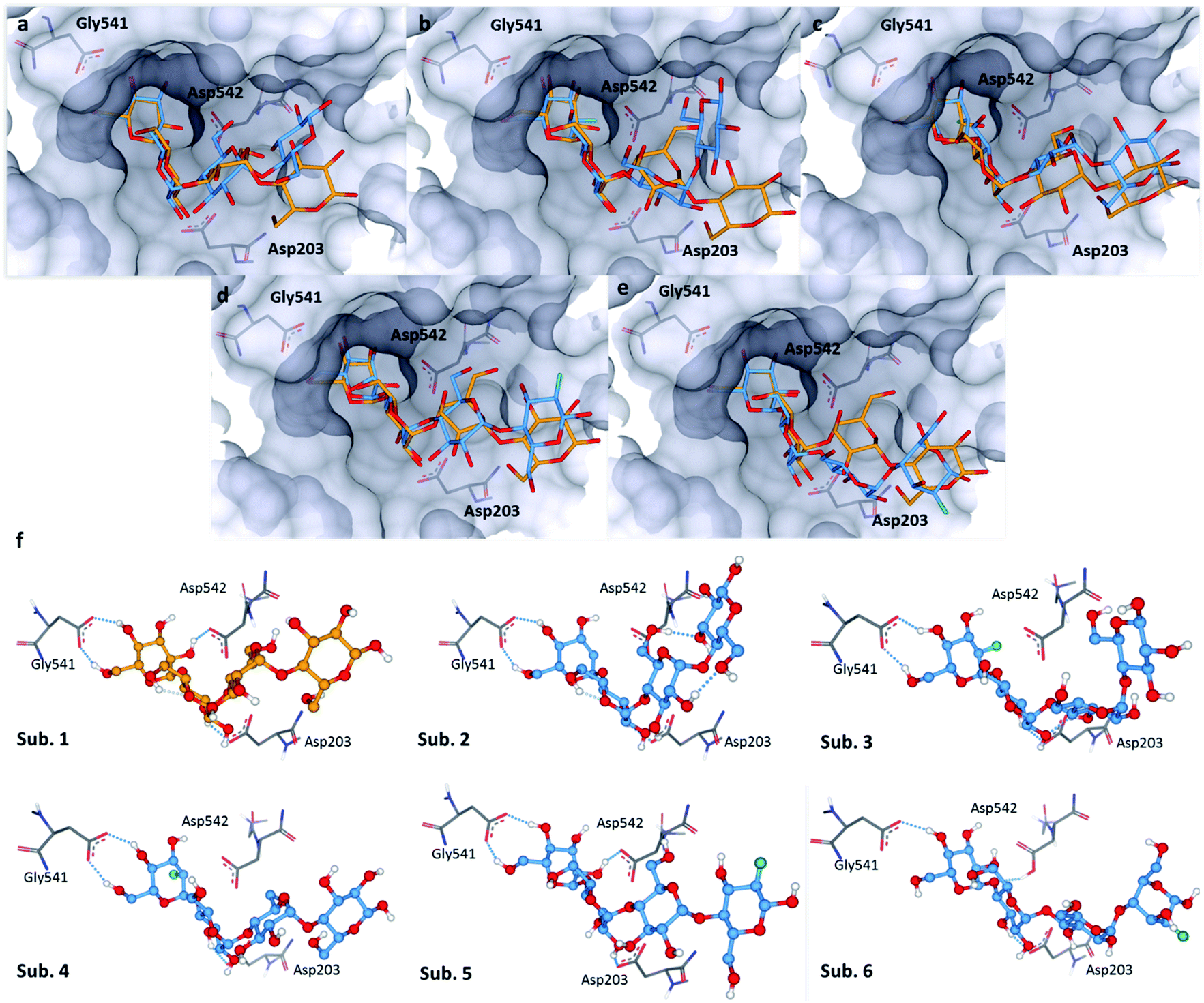 | ||
| Fig. 5 Docking studies: (a) comparison between substrate 1 (orange) and substrate 2 (blue) in the active site of human intestinal α-glucosidase (PDB: 2QMJ); (b) comparison between substrate 1 (orange) and substrate 3 (blue); (c) comparison between substrate 1 (orange) and substrate 4 (blue); (d): comparison between substrate 1 (orange) and substrate 5 (blue); (e) comparison between substrate 1 (orange) and substrate 6 (blue); (f) sub. 1–6: interactions of 1–6 with residues in the active site; colour code: protein surface: grey; protein skeleton: C: grey, O: red, N: blue; substrate 1: C: orange, O: red; substrate 2–6: C: blue, O: red, F: green. This figure was generated using SeeSAR 9.1 (BioSolveIT).21 | ||
Synthesis of the probes 2–6
With a view to exploring the structural ramifications of deletion and fluorination on the stability of these probes towards the target enzymes, a concise synthetic route was conceived (Schemes 1 and 2). The synthesis of the cores 10 and 11 was achieved by treating maltotriose 7 with benzaldehyde dimethyl acetal to generate the benzylidene acetal and subsequent global acetate protection of the trisaccharide (8) (Scheme 1, top). Cleavage of the acetal to unmask positions C4 and C6 allowed for regioselective protection of the primary alcohol affording 10 and 11 thereby completing this synthesis of the glycosyl acceptor.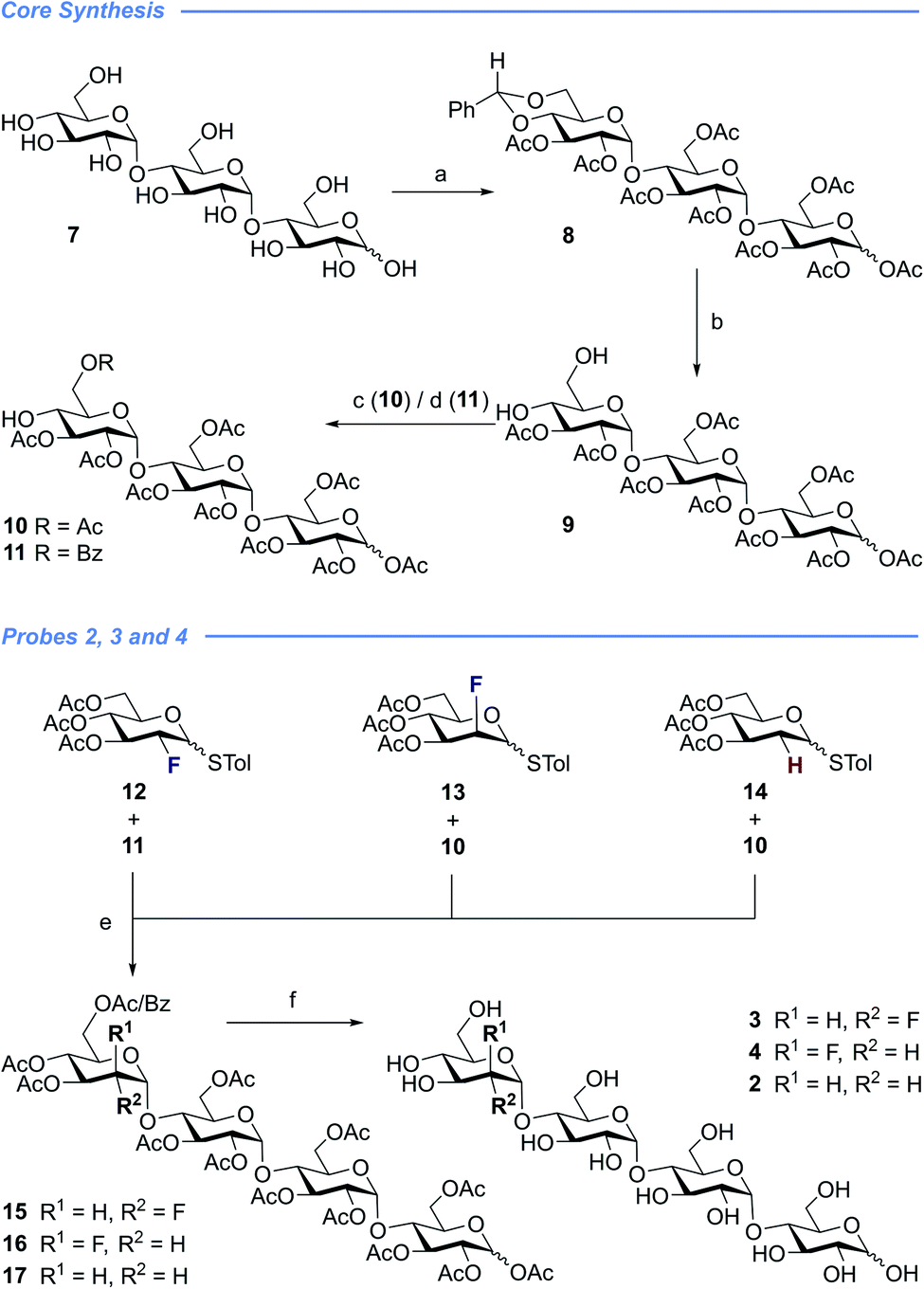 | ||
Scheme 1 Preparation of fluorinated maltotetraose cores 3 and 4, and the control system 2. Conditions: (a) (1) Benzaldehyde dimethyl acetal, p-TsOH, DMF, 4 h, 50 °C, 150 mbar; (2) Ac2O, DMAP, pyridine, 4 h, RT, 69% over two steps. (b) TFA/H2O (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), CH2Cl2, RT, 98%. (c) Acetyl chloride, HOBt, TEA, CH2Cl2, RT, 76%. (d) Benzoyl chloride, pyridine, 3 h, −10 °C, 57%. (e) NIS, TMSOTf, CH2Cl2, 15 min, RT, 15: 33%, 16: 50%, 17: 11%. (f) NaOMe, MeOH, RT, 3: 69%, 4: 86%, 2: 94%. :1), CH2Cl2, RT, 98%. (c) Acetyl chloride, HOBt, TEA, CH2Cl2, RT, 76%. (d) Benzoyl chloride, pyridine, 3 h, −10 °C, 57%. (e) NIS, TMSOTf, CH2Cl2, 15 min, RT, 15: 33%, 16: 50%, 17: 11%. (f) NaOMe, MeOH, RT, 3: 69%, 4: 86%, 2: 94%. | ||
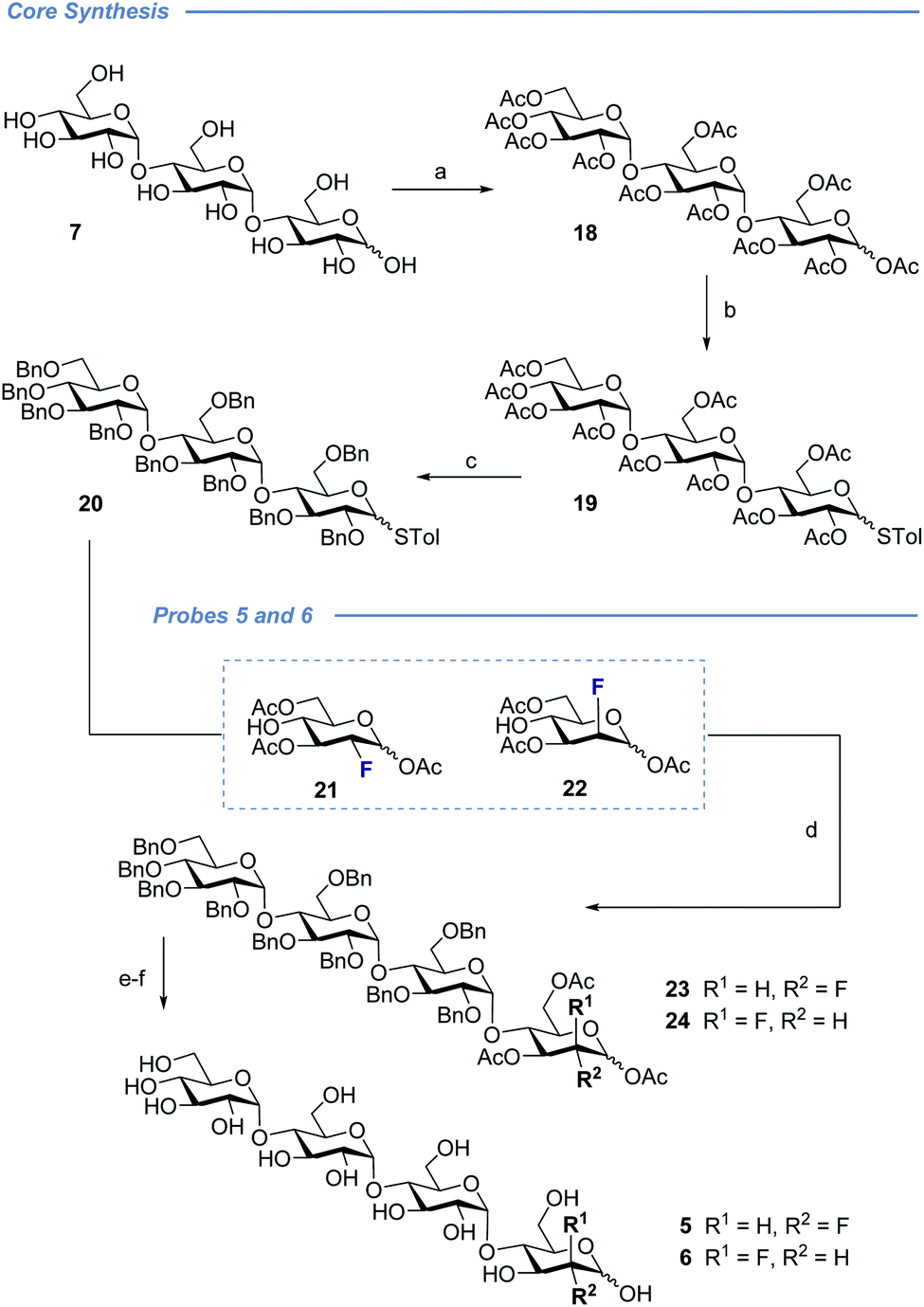 | ||
| Scheme 2 Preparation of fluorinated maltotetraose cores 5 and 6. Conditions: (a) Ac2O, DMAP, pyridine, 4 h, RT, 88%. (b) 4-Methylbenzenethiol, BF3·OEt2, CH2Cl2, 23 h, RT, 78%. (c) (1) NaOMe, MeOH, 2 h, RT, 91%; (2) NaH, BnBr, TBAI, DMF, 20 h, RT, 77%. (d) NIS, BF3·OEt2, CH2Cl2, 15 min, RT, 23: 21%, 24: 25%. (e) Pd/C, H2, MeOH, EtOAc, 24 h, RT; (f) NaOMe, MeOH, 8 h, RT, 5: 88%, 6: 67% over two steps. | ||
Completion of this divergent [3 + 1] strategy was contingent on the preparation of the modified glycosyl donors 12, 13 and 14 (for full preparative details see the ESI‡). These thioglycosides proved to be highly α-selective thereby facilitating the synthesis of the tetraose cores 15, 16 and 17.
Global deprotection proved facile to provide compounds 2 (2-deoxy), 3 (gluco-F) and 4 (manno-F) for biological evaluation. Modification of the reducing end required an adapted glycosyl donor derived from 7. Per-acetylation (18) and thioglycoside formation (19) was followed by global deprotection (Ac)/protection (Bn) sequence (20). Independent α(1 → 4) glycosylation with acceptors 21 and 22 furnished the protected scaffolds 23 and 24, respectively. The desired tetraose derivatives 5 and 6 were obtained by benzyl deprotection and subsequent acetate cleavage.
Enzyme assays
To evaluate the influence of site-selective fluorine introduction on hydrolytic stability, oligosaccharides 1–6 were incubated in human and murine blood serum (please see the ESI‡ for full details). The diversity of glycan-degrading enzymes in serum renders the identification of specific enzymes challenging. Therefore, the stability of maltotetraose conjugates 1–6 was independently examined by incubation with purified α-amylase and α-glucosidase. Both human and murine blood serum contain large concentrations of hydrolytic enzymes which belong to the α-amylase family. It is pertinent to highlight that the concentration in murine serum is considerable.7b This is a consequence of the starch-based diets of small rodents. The stability of the oligosaccharides was established by measuring the liberation of glucose following incubation at 37 °C over one or five hours (purified enzymes or blood serum, Fig. 6 and 7, respectively). At each time point, the glucose level of a 0.6 μL aliquot was measured with a point-of-care glucose meter (ACCU-CHEK®-Inform II; Roche Diagnostic GmbH).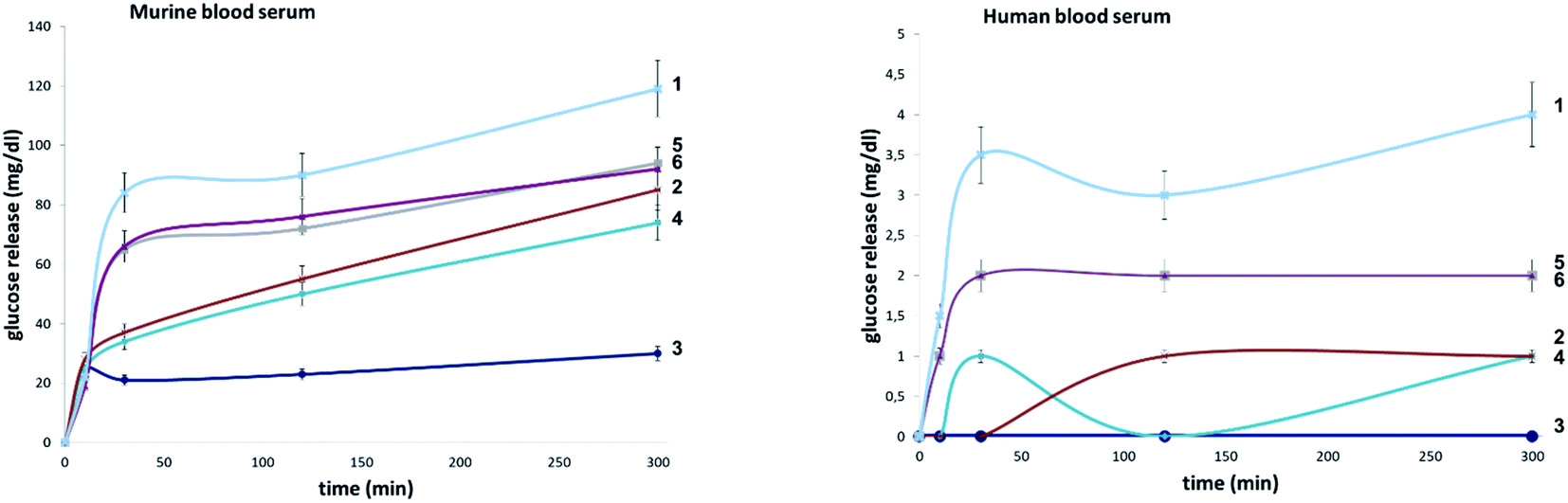 | ||
| Fig. 6 Hydrolytic stability of 1–6 in murine and human blood serum. | ||
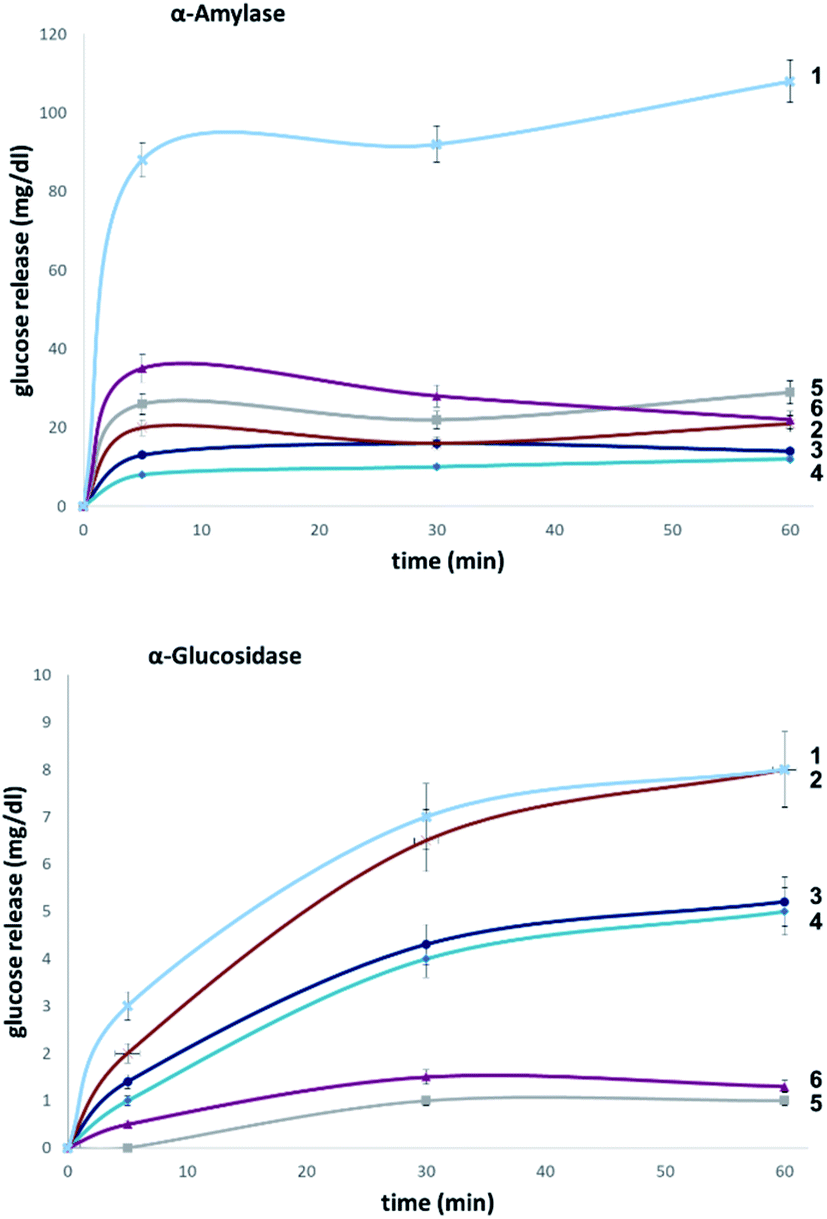 | ||
| Fig. 7 Hydrolytic stability of maltotetraose conjugates in stock solutions of α-amylase (2000 U L−1, top) and α-glucosidase (10 U L−1, bottom). | ||
In both serum test series, wildtype maltotetraose 1 led to the highest release of glucose, followed by the conjugates 5 and 6, which are fluorinated at the reducing end. Modifications at the non-reducing terminus (3 and 4) led to a remarkable stability enhancement, compared to maltotetraose 1. The importance of the C2 configuration also became apparent, with the gluco-configured probe 3 displaying an approximate 5-fold decrease in hydrolysis (murine blood serum) compared to 1, and outcompeting the corresponding epimer 4. The sensitivity of enzyme catalysis towards changes at C2 is further evident from the results of the deletion probe 2, which broadly mirrored the stability of the manno-configured system 4. In comparison to murine serum incubation, the glucose levels resulting from human blood serum incubation are significantly lower reflecting the comparatively lower enzyme concentration of 28–100 units per litre. Gratifyingly, in all of the synthetic maltotetraose derivatives (2–6), enhanced hydrolytic stability was observed with the fluorinated conjugate 3 being entirely stable in human blood serum. The translational implications of this finding are noteworthy, particularly in the field of bacterial imaging.
To explore the stability of the probe molecules in a more controlled environment, the conjugates were incubated in purified α-amylase and α-glucosidase solutions to quantify the effect of a site-selective fluorine installation (Fig. 7). In the case of α-amylase, a stock solution of 2000 units per litre in heat-inactivated blood serum was prepared and the experiments were conducted at 37 °C with gentle agitation.
Natural maltotetraose was rapidly truncated affording a substantial glucose release of 110 mg dL−1. All of the synthetic conjugates exhibited notably improved hydrolytic stability with conjugates 3 (non-reducing gluco-, 14 mg dL−1) and 4 (non-reducing manno-, 12 mg dL−1) being the most robust scaffolds. Given that α-amylase hydrolyses maltodextrin from the reducing end, it is curious that modification at the opposite terminus proved to be most crucial for substrate stability. For completeness, this investigation was conducted with purified acid-α-glucosidase, which metabolises maltodextrins from the non-reducing end. In line with previous observations, the incubation of wildtype maltotetraose resulted in the fastest hydrolysis observed in this study. Interestingly, the 2-deoxy species 2 showed comparable lability to maltotetraose when incubated with α-glucosidase. The two fluorinated conjugates 3 and 4 showed comparable behaviour, with a glucose concentration of 5 mg dL−1 having been measured after an incubation time of 1 hour. Again, fluorination at the reducing end (5 and 6) led to enhanced stability in agreement with the results obtained in the α-amylase assay. Interestingly, tetrasaccharide 5, bearing a 2-deoxy-2-fluoroglycosyl subunit at the reducing end, proved to be most stable, releasing a glucose concentration of 1 mg dL−1. The key kinetic parameters pertaining to the probes analysed in this study were investigated. Values of kcat were determined by dividing vmax values by enzyme concentration using a molecular weight of 53000 (amylase) and 63000 (glucosidase). The catalysed degradation of maltotetraose was initiated by addition of 50–5000 units and 1–100 U enzyme, respectively. Two So concentrations of around 0.1 × Km were used to ensure that substrate hydrolysis was linear with time. The kinetic parameters for the hydrolysis of compounds by α-amylase and α-glucosidase are summarised in Table 1. These were performed by quantitating the glucose formed as a reaction product using glucose dehydrogenase electrochemical detection technique. ΔΔG‡ determined from vmax/Km values indicates transition state destabilisation for a given probe relative to the model substrate 1. ΔΔG‡ for the two analogs with fluorine substituents located at the non-reducing end (3, 4) was in the range 23.1–29.2 kJ mol−1 and for the fluorinated conjugates 5 and 6 between 27.2-52.2 kJ mol−1 for the α-glucosidase and for α-amylase, respectively (Table 1).
| Enzyme/substrate | v max (mM × U−1 × s−1)/Kma (mM) | ΔΔG‡b (kJ mol−1) |
|---|---|---|
|
a
v
max/Km (s−1 × U−1) = vo/EoSo; vo initial reaction rate of hydrolysis, Eo enzyme amount in units, So initial substrate concentration, determination at 37 °C in heat inactivated serum.
b ΔΔG‡ = −RTln[(vmax/Km)derivat/(vmax/Km)maltotetraose] = activation energy increase due to the modified substrate (R gas constant; T absolute temperature) according to the literature;25vmax (the maximum reaction rate) and Km (Michaelis constant; parameter of the enzyme's affinity for the substrate) were determined by fitting initial rates at different substrate concentrations from 0.1 × Km to 4 × Km to the Michaelis–Menten equation.26
|
||
| α-Glucosidase | ||
| 1 | 4 × 10−4 | — |
| 2 | 3.8 × 10−5 | 1.9 |
| 3 | 2.1 × 10−5 | 26.5 |
| 4 | 0.3 × 10−5 | 29.2 |
| 5 | 1.5 × 10−9 | 52.2 |
| 6 | 2.7 × 10−7 | 48.2 |
|
||
| α-Amylase | ||
| 1 | 7.7 × 10−3 | — |
| 2 | 3.1 × 10−4 | 2.8 |
| 3 | 2.6 × 10−4 | 23.4 |
| 4 | 3.8 × 10−4 | 24.6 |
| 5 | 8.9 × 10−4 | 27.2 |
| 6 | 7.6 × 10−4 | 29.7 |
Finally, to glean additional insights into the fate of the tetraose, we attempted to determine the crystal structure of candidate 5 in complex with human α-amylase (see ESI online information for details‡). Co-crystallisation of α-amylase with 5 did not yield electron density for a carbohydrate-like ligand in the proximity of the active-site, so soaking was employed. Data were collected for several crystals and the dataset with clearest electron density for ligands was selected. This crystal allowed data collection to 1.40 Å with excellent statistics (Data collection and refinement statistics can be found in Table 1 in the ESI‡).
To our surprise, we did not find electron density for 5, but were able to identify four maltose molecules and one glucose molecule in the structure. Two maltose molecules are bound at the active-site (Fig. 8), while the other two facilitate crystal contacts between symmetry mates (Fig. 9). Given the very high protein concentration and long incubation time (soaking had to be done for 48 h at 291 K), it is possible that the majority of 5 had degraded. What is intriguing is the presence of maltose units, which may indicate an alternative cleavage site for the fluorinated maltotetraose that results in the very slow degradation observed in biochemical assays. Our data do not hint at the fate of the fluorinated sugar moiety, and we cannot exclude that the observed maltose units are in fact intact 5 with disordered reducing and non-reducing ends. However, if that were the case, the mode of binding of 5 to α-amylase would still be distinct from that of the natural substrate and support the conclusion that fluorination at a single site can have a stabilising effect on maltotetraoses.
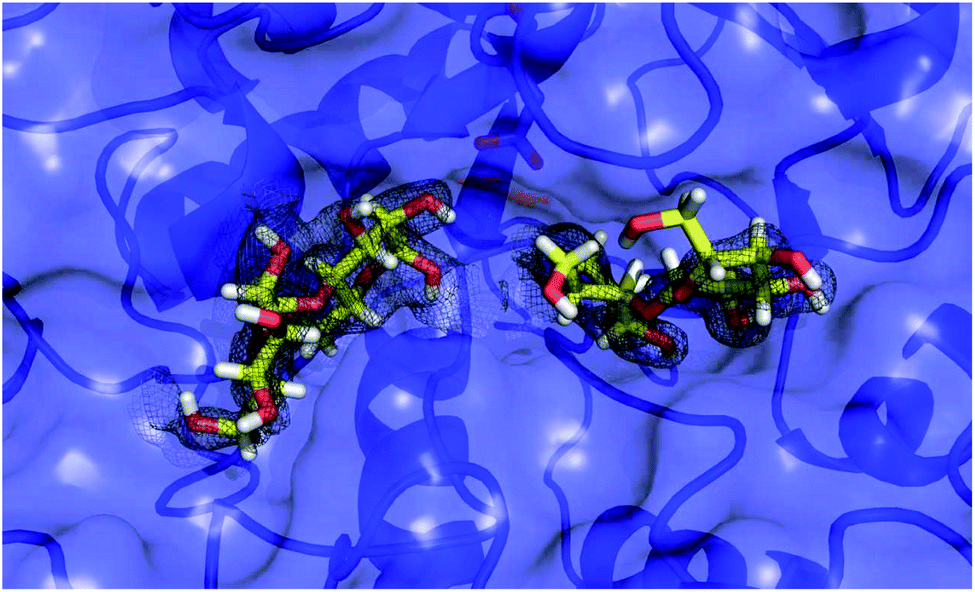 | ||
| Fig. 8 X-ray analysis. Surface representation of α-amylase (blue) with two maltose molecules at the active site (sticks, yellow carbon atoms). A polder map contoured at 3σ is shown as a grey isomesh. Asp300 is shown as sticks (see modelling for comparison). | ||
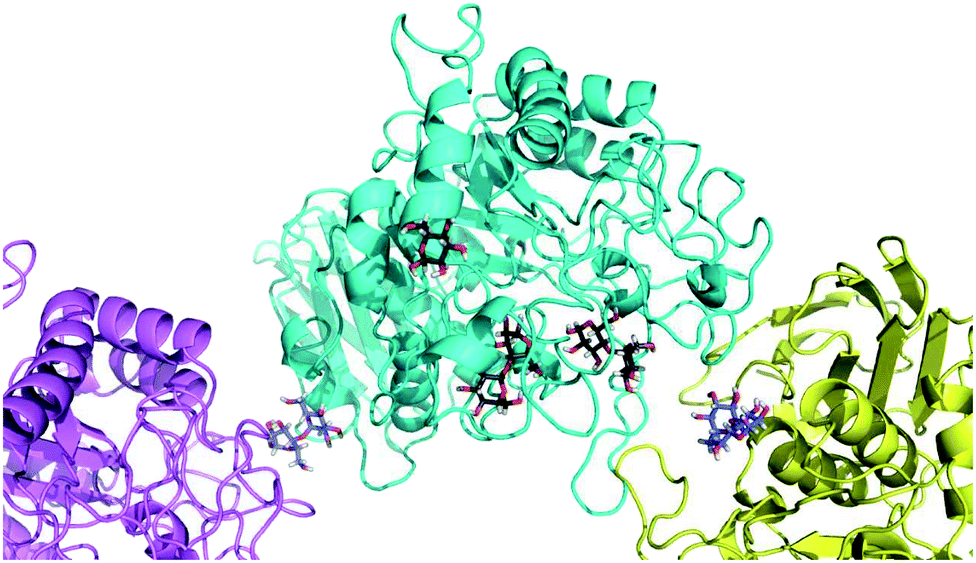 | ||
| Fig. 9 X-ray analysis. Cartoon representation of the co-crystal of α-amylase (cyan) with two symmetry mates (violet and yellow). Carbohydrates are shown as sticks. Maltose units that facilitate crystal contacts are shown with carbon atoms colored in blue, while the other two maltose molecules and the glucose have carbon atoms colored in black. | ||
Conclusions
In conclusion, the effect of molecular editing with fluorine on the bound conformation and resulting hydrolytic stability of maltotetraoses has been assessed. In contrast to natural maltotetraose (1), these conjugates display enhanced metabolic stability towards murine and human blood serum as well as α-amylase and α-glucosidase. The seemingly innocent effect of subtle physicochemical alterations arising from OH → F substitution are sufficient to alter enzyme – small molecule recognition, and this may prove to be expansive in a translational sense.Alterations of the molecule at the opposite end to where enzymatic cleavage occurs actively suppress catalysis (Fig. 10). This phenomenon manifested itself in both assays, whereby fluorination at the “wrong” end of the chain resulted in the greatest hydrolytic stability of the study. Collectively, these data have enabled two probes (3 and 5) to be identified in which stability is enhanced by one order of magnitude relative to the native scaffold. Given the intimate regulatory role that amylases play in metabolic regulation, (e.g., Pompe disease, diabetes and Parkinson's disease),27 their diagnostic significance, and clinical translation to bacterial imaging, it is envisaged that these findings may begin to reconcile the intrinsic hydrolytic instability of complex carbohydrates with their unrealised clinical potential.
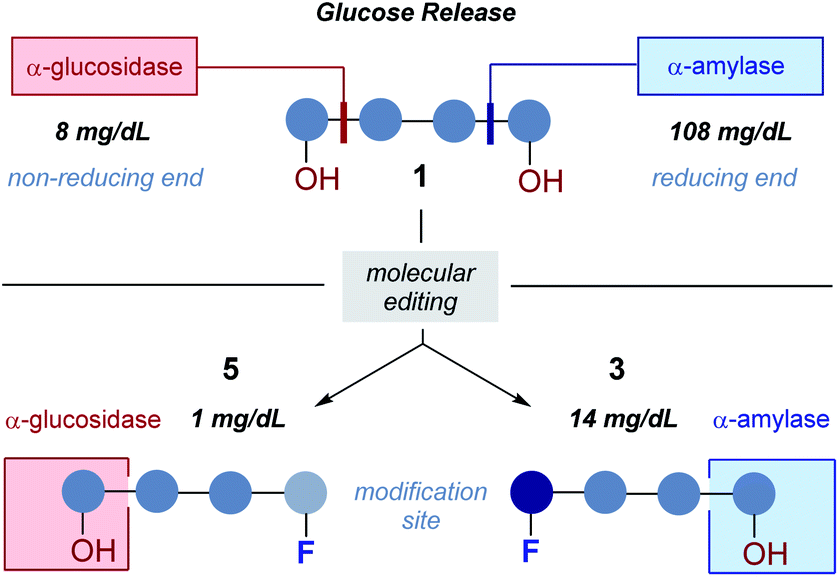 | ||
| Fig. 10 A summary of this study and comparison of 1, 5 and 3. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge generous financial support from the WWU Münster, the European Research Council (ERC Starter Grant - Project number 336376-ChMiFluorS and ERC Consolidator Grant – Project Number 818949-RECON, to RG), the Interdisciplinary Centre for Clinical Research (IZKF), Münster, Germany (Fau2/014/17), the DFG Cluster of Excellence “Cells in Motion - CiM” (FF-2013-10) and a stipend of the CiM-IMPRS graduate programme, Münster, Germany (to A. A.) and the DFG Emmy-Noether programme (KO 4116/3-2 to J. K.). The European Commission is acknowledged for an Intra-European Marie Skłodowska-Curie actions fellowship under Horizon-2020 (796089–NovInDXS, R. P. J.) and the ERC (Starter Grant 757913 to A. K. H. H.). We thank Prof. Dr Andrea Rentmeister (Institute for Biochemistry, WWU Münster) and Ms. Charlotte S. Teschers (Institute for Organic Chemistry, WWU Münster) for helpful suggestions.Notes and references
- (a) D. B. Werz, R. Ranzinger, S. Herget, A. Adibekian, C.-W. Von der Lieth and P. H. Seeberger, ACS Chem. Biol., 2007, 2, 685 CrossRef CAS; (b) A. Adibekian, P. Stallforth, M.-L. Hecht, D. B. Werz, P. Gagneux and P. H. Seeberger, Chem. Sci., 2011, 2, 337 RSC.
- (a) Y. Aoyama, T. Tsuda, E. Hitomi-Ohmura and A. Yoshida, Comp. Biochem. Physiol., 1993, 104, 381 CrossRef CAS; (b) A. Gimeno, P. Valverde, A. Ardá and J. Jiménez-Barbero, Curr. Opin. Struct. Biol., 2020, 62, 22 CrossRef CAS.
- (a) D. H. Dube and C. R. Bertozzi, Nat. Rev., 2005, 4, 477 CAS; (b) P. H. Seeberger, Nat. Chem. Biol., 2009, 5, 368 CrossRef CAS.
- (a) Y. Bourne and B. Henrissat, Curr. Opin. Struct. Biol., 2001, 11, 593 CrossRef CAS; (b) M. L. Sinnott, Chem. Rev., 1990, 90, 1171 CrossRef CAS.
- M. L. Oldham, S. Chen and J. Chen, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18132 CrossRef CAS.
- B. Ernst and J. L. Magnani, Nat. Rev. Drug Discov., 2009, 8, 661 CrossRef CAS.
- (a) W. Wang and N. Murthy, Sci. Transl. Med., 2014, 6, 259fs43, DOI:10.1126/scitranslmed.3010746; (b) For an example from this laboratory see: A. Axer, S. Hermann, G. Kehr, D. Clases, U. Karst, L. Fischer-Riepe, J. Roth, M. Fobker, M. Schäfers, R. Gilmour and A. Faust, ChemMedChem, 2018, 13, 241 CrossRef CAS.
- T. K. Lindhorst, Essentials of Carbohydrate Chemistry and Biochemistry, Wiley-VCH, Weinheim, 2007 Search PubMed.
- V. V. Krasikov, D. V. Karelov and L. M. Firsov, Biochemistry, 2001, 66, 332 Search PubMed.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (c) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (d) L. E. Zimmer, C. Sparr and R. Gilmour, Angew. Chem., Int. Ed., 2011, 50, 11860 CrossRef CAS; (e) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS; (f) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822 CrossRef CAS; (g) M. Aufiero and R. Gilmour, Acc. Chem. Res., 2018, 51, 1701 CrossRef CAS.
- For selected examples for this laboratory, see (a) C. Bucher and R. Gilmour, Angew. Chem., Int. Ed., 2010, 49, 8724 CrossRef CAS; (b) T. Hayashi, A. Axer, G. Kehr, K. Bergander and R. Gilmour, Chem. Sci., 2020, 11, 6527 RSC.
- Y. Yu, T. Tyrikos-Ergas, Y. Zhu, G. Fittolani, V. Bordoni, A. Singhal, R. J. Fair, A. Grafmüller, P. H. Seeberger and M. Delbianco, Angew. Chem., Int. Ed., 2019, 58, 13127 CrossRef CAS.
- P. Bentler, K. Bergander, C. G. Daniliuc, C. Mück-Lichtenfeld, R. P. Jumde, A. K. H. Hirsch and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 10990 CrossRef CAS.
- For studies demonstrating the discrimination of F-acetyl-CoA versus acetyl-CoA (i.e. single F versus H), see: (a) A. W. Weeks and M. C. Y. Chang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19667 CrossRef CAS; (b) A. M. Weeks, N. S. Keddie, R. D. P. Wadoux, D. O'Hagan and M. C. Y. Chang, Biochemistry, 2014, 53, 2053 CrossRef CAS.
- K. Myrbäck and E. Willstädt, Ark. Kemi, 1954, 7, 403 Search PubMed.
- D. L. Zechel and S. G. Withers, Acc. Chem. Res., 2000, 33, 11 CrossRef CAS.
- (a) S. G. Withers, I. P. Street, P. Bird and D. H. Dolphin, J. Am. Chem. Soc., 1987, 109, 7530 CrossRef CAS; (b) M. N. Namchuk, J. D. McCarter, A. Becalski, T. Andrews and S. G. Withers, J. Am. Chem. Soc., 2000, 122, 1270 CrossRef CAS; (c) For examples of di- and tri-saccharides that are fluorinated only at the reducing end, see J. D. McCarter, M. J. Adam, C. Braun, M. Namchuk, D. Tull and S. G. Withers, Carbohydr. Res., 1993, 249, 77 CrossRef CAS.
- Fluorine in Pharmaceutical and Medicinal Chemistry, From Biophysical Aspects to Clinical Applications, ed. V. Gouverneur and K. Müller, Imperial College Press, 2012 Search PubMed.
- S. A. Allman, H. H. Jensen, B. Vijayakrishnan, J. A. Garnet, E. Leon, Y. Liu, D. C. Anthony, N. R. Sibson, T. Feizi, S. Matthews and B. G. Davis, ChemBioChem, 2009, 10, 2522 CrossRef CAS.
- (a) D. O'Hagan and D. Harper, J. Fluorine Chem., 1999, 100, 127–133 CrossRef; (b) D. O'Hagan, C. Schaffrath, S. L. Cobb, J. T. G. Hamilton and C. D. Murphy, Nature, 2002, 416, 279 CrossRef; (c) C. Dong, F. Huang, H. Deng, C. Schaffrath, J. B. Spencer, D. O'Hagan and J. H. Naismith, Nature, 2004, 427, 561 CrossRef CAS; (d) X. Zhu, D. A. Robinson, A. R. McEwan, D. O'Hagan and J. H. Naismith, J. Am. Chem. Soc., 2007, 129, 14597 CrossRef CAS; (e) K. K. Chan and D. O'Hagan, Methods Enzymol., 2012, 516, 219 CAS; (f) H. Aldemir, S. V. Kolhepp, T. Gulder and T. A. M. Gulder, Nat. Prod., 2014, 77, 2331 CrossRef CAS.
- (a) BioSolveIT GmbH, LeadIT, version 2.3.2, Sankt Augustin, http://www.biosolveit.de Search PubMed; (b) BioSolveIT GmbH, SeeSAR, version 9.1, Sankt Augustin, http://www.biosolveit.de Search PubMed.
- N. Ramasubbu, V. Paloth, Y. Luo, G. D. Brayer and M. J. Levine, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1996, 52, 435 CrossRef CAS.
- L. Goldbach, B. J. A. Vermeulen, S. Caner, M. Liu, C. Tysoe, L. van Gijzel, R. Yoshisada, M. Trellet, H. van Ingen, G. D. Brayer, A. M. J. J. Bonvin and S. A. K. Jongkees, ACS Chem. Biol., 2019, 14, 1751 CrossRef CAS.
- L. Sim, R. Quezada-Calvillo, E. E. Sterchi, B. L. Nichols and D. R. Rose, J. Mol. Biol., 2008, 375, 782 CrossRef CAS.
- A. J. Wilkinson, A. R. Fersht, D. M. Blow and G. Winter, Biochemistry, 1983, 22, 3581 CrossRef CAS.
- M. M. Palcic, T. Skrydstrup, K. Bock, N. Le and R. U. Lemieux, Carbohydr. Res., 1993, 250, 87 CrossRef CAS.
- S. P. Cullen, J. Sardi, Y.-H. Ng, Y. Xu, J. J. Sun, P. Tomlinson, I. Kolodziej, P. Kahn and J. Saftig, et al. , Ann. Neurol., 2011, 69, 940 CrossRef.
Footnotes |
| † Dedicated to the memory of Prof. Dr François Diederich (1952–2020). |
| ‡ Electronic supplementary information (ESI) available: The crystal structure reported here has been deposited in the PDB (PDB code 6Z8L). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04297h |
| § Present address: School of Chemistry, University of Glasgow, Joseph Black Building, University Ave, Glasgow G12 8QQ, UK, E-mail: E-mail: Jesko.Koehnke@glasgow.ac.uk. |
| This journal is © The Royal Society of Chemistry 2021 |