
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced voltammetric anion sensing at halogen and hydrogen bonding ferrocenyl SAMs†
Robert
Hein
 ,
Xiaoxiong
Li
,
Paul D.
Beer
* and
Jason J.
Davis
*
,
Xiaoxiong
Li
,
Paul D.
Beer
* and
Jason J.
Davis
*
Department of Chemistry, University of Oxford, South Parks Road, Oxford OX1 3QZ, UK. E-mail: paul.beer@chem.ox.ac.uk; jason.davis@chem.ox.ac.uk
First published on 15th December 2020
Abstract
Halogen bonding mediated electrochemical anion sensing has very recently been established as a potent platform for the selective and sensitive detection of anions, although the principles that govern binding and subsequent signal transduction remain poorly understood. Herein we address this challenge by providing a comprehensive study of novel redox-active halogen bonding (XB) and hydrogen bonding (HB) ferrocene-isophthalamide-(iodo)triazole receptors in solution and at self-assembled monolayers (SAMs). Under diffusive conditions the sensory performance of the XB sensor was significantly superior. In molecular films the XB and HB binding motifs both display a notably enhanced, but similar, response to specific anions. Importantly, the enhanced response of these films is rationalised by a consideration of the (interfacial) dielectric microenvironment. These effects, and the resolved relationship between anion binding and signal transduction, underpin an improved fundamental understanding of anion sensing at redox-active interfaces which will benefit not just the development of more potent, real-life relevant, sensors but also new tools to study host–guest interactions at interfaces.
Introduction
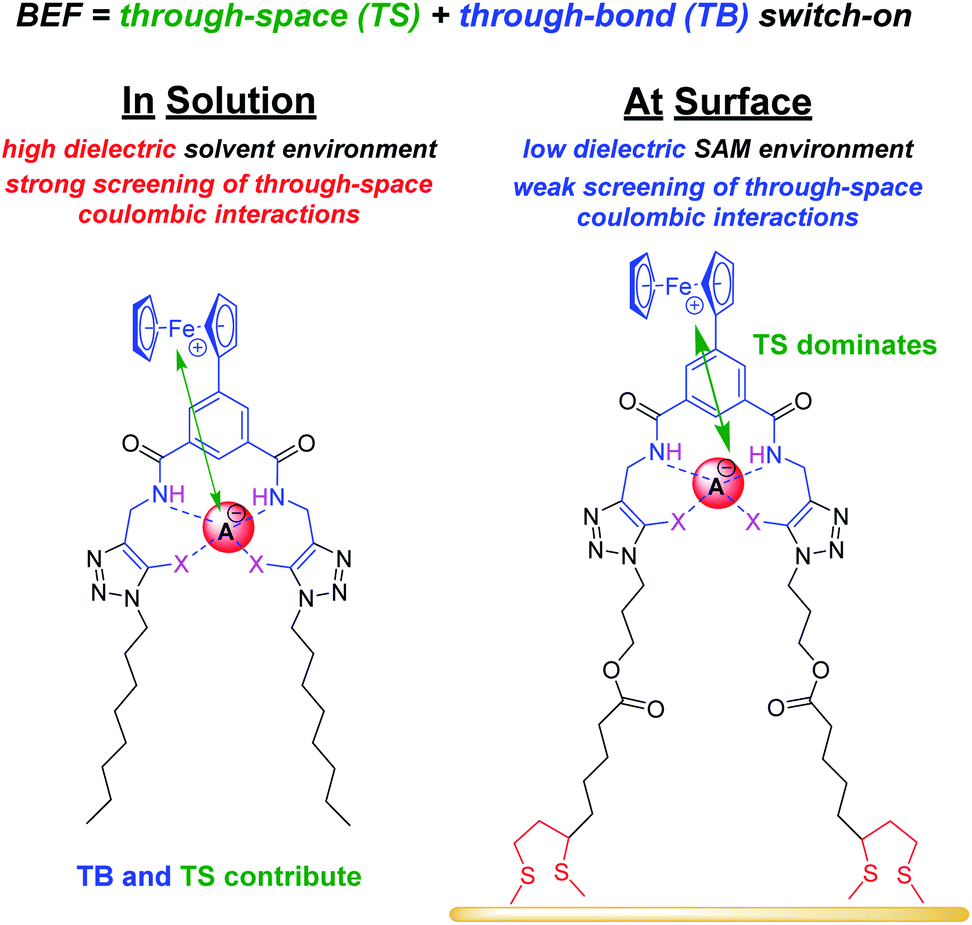
Anions play a crucial role in many environmental and biological settings, necessitating their selective detection. Offering a sensitive, scalable and cheap means of sensing anions, electrochemical methodologies employing synthetic host systems have received considerable attention over the past two decades.1 Most commonly, the electrochemical sensing properties of redox-active anion receptors are exemplified via voltammetric techniques, such as cyclic voltammetry (CV) or square-wave voltammetry (SWV), where, upon anion binding, a cathodic perturbation of the redox-transducer is typically measured. This methodology has been widely applied where, most commonly, a ferrocene (Fc) transducer is appended to hydrogen bonding (HB) receptors.2–4 More recently, halogen bonding (XB) has emerged as a potent non-covalent interaction to drive anion recognition, often displaying enhanced selectivity and binding strength in comparison to HB analogues.5,6 This has also been exploited in electrochemical anion sensors in solution,7–11 and, very recently, at receptive interfaces.12–14The surface-immobilisation of (redox-active) receptors is relevant to the development of real-life relevant sensors,15–18 enabling facile sensor reuse, and sensing both under flow and in (aqueous) solvent media in which many synthetic receptors are not natively soluble.1,19 Our quantitative understanding of anion sensing at redox-active interfaces, does, however, remain underdeveloped.14,18,20–22 Often an enhanced sensory performance (larger signal magnitude) is observed on confining redox-active receptors to interfaces,19,23 but the specific physico-chemical origins of this remain poorly understood. It has been suggested that an enhanced interfacial binding strength, brought about by receptor preorganisation and/or cooperative/chelate effects is the origin of the surface-enhancement effect.14,15,20 A consideration of the relevant binding equilibria and the Nernst equation reveals that these effects cannot be the primary origin of the signal enhancement. Specifically, in its most general form, the voltammetric shift ΔE is determined by eqn (1), where ΔE is not determined by the absolute magnitude of guest binding to either the reduced (KRed) or oxidised receptor (KOx), but rather by their ratio, i.e. the magnitude to which guest binding is affected by a change in redox state (often called the binding enhancement factor (BEF = KOx/KRed)).24,25
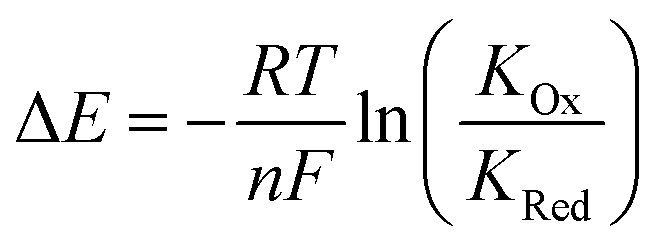 | (1) |
More recently a more refined model has enabled the determination of absolute values of KOx and KRed from fitting of voltammetric binding isotherms.1,7 Herein we report a detailed comparison of novel redox-active XB and HB anion receptors in solution and within self-assembled monolayer (SAM) formats. The resulting insights provide an improved fundamental understanding of anion sensing at redox-active interfaces and specifically highlight the importance of the interfacial binding microenvironment.
Results and discussion
Synthesis
The synthesis of novel amide and (iodo)triazole containing receptors 1.XB/HB and 2.XB/HB (Fig. 1) was carried out as depicted and described in the ESI (Scheme S1†). Briefly, 5-ferrocenylisophthalic acid263 was converted to the bis(iodo)alkyne-appended isophthalamide 4a/b which was subsequently reacted with either octyl azide or disulfide-azide27 in a copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) to yield 1.XB/HB and 2.XB/HB, respectively. All novel compounds were characterised by 1H, 13C NMR and high-resolution mass spectrometry as detailed in the ESI.†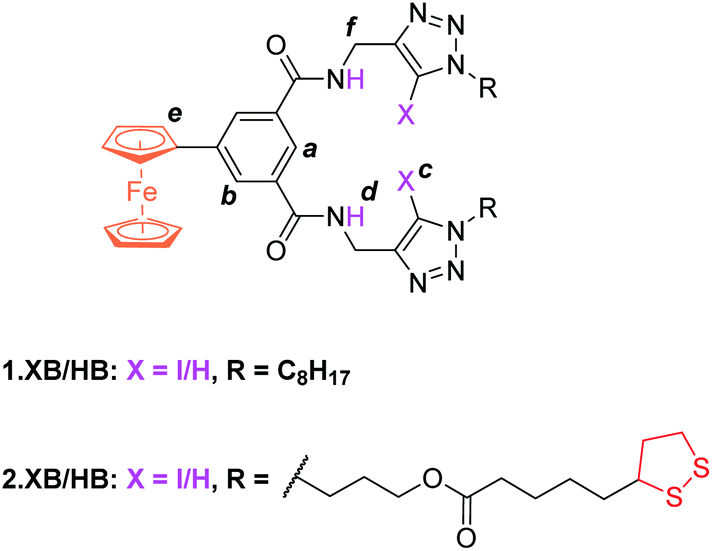 | ||
| Fig. 1 Halogen and hydrogen bonding redox active receptors 1.XB/HB (for solution-phase binding studies) and 2.XB/HB (for SAMs). | ||
Solution-phase binding studies
The solution-phase binding performance of 1.XB/HB was initially investigated by 1H NMR titrations in CD3CN. The addition of TBA salts of various anions induced significant downfield perturbations of the internal aromatic proton Ha, isophthalamide Hd and triazole Hc protons of receptor 1.HB. This is indicative of anion binding within the cavity via multiple, convergent, HB interactions from the isophthalamide as well as the proto-triazole groups. This binding mode is further supported by negligible perturbations of the protons that are further removed (Hb and He) or pointing away from the binding site (Hf). The addition of HSO4-, Cl−, Br−, H2PO4−, and BzO− induced such shift patterns (Fig. S1–S9†) while the presence of ClO4−, ReO4− and NO3− caused minimal shifts indicating a negligible binding consistent with their low basicity. Similar perturbations were observed for the titration of 1.XB with this range of anions, suggesting a comparable binding mode. Quantitative analysis of the 1H NMR binding isotherms (Fig. S10†) determined 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric host–guest binding constants K (M−1), summarised in Table 1. In CD3CN, receptor 1.HB displays a markedly higher affinity for the more basic anions H2PO4− and BzO− in comparison to the halides, HSO4− or NO3−. Of note is a modest preference for the tetrahedral H2PO4− over the trigonal planar BzO−. Surprisingly, the anion binding affinity is significantly diminished for 1.XB in all cases, apart from NO3− (which weakly binds to this receptor but not to 1.HB). This trend is in marked contrast to many previously reported systems in which stronger binding to the XB receptor is normally observed,5,6 and may arise from geometric constraints imposed by the more bulky C–I XB donor groups in 1.XB, sterically impeding convergent binding from all donor groups within the receptor cavity.‡
:1 stoichiometric host–guest model (Fig. S10). N. b. – no binding. — – not carried out. Standard errors from fitting are <10% unless indicated otherwise
:1 stoichiometric host–guest binding constants K (M−1), summarised in Table 1. In CD3CN, receptor 1.HB displays a markedly higher affinity for the more basic anions H2PO4− and BzO− in comparison to the halides, HSO4− or NO3−. Of note is a modest preference for the tetrahedral H2PO4− over the trigonal planar BzO−. Surprisingly, the anion binding affinity is significantly diminished for 1.XB in all cases, apart from NO3− (which weakly binds to this receptor but not to 1.HB). This trend is in marked contrast to many previously reported systems in which stronger binding to the XB receptor is normally observed,5,6 and may arise from geometric constraints imposed by the more bulky C–I XB donor groups in 1.XB, sterically impeding convergent binding from all donor groups within the receptor cavity.‡
:1 stoichiometric host–guest model (Fig. S10). N. b. – no binding. — – not carried out. Standard errors from fitting are <10% unless indicated otherwise
The introduction of 1% D2O to the solvent system greatly reduces the anion binding affinities of both hosts, especially for H2PO4−, HSO4− and BzO−, as a result of the more competitive nature of the solvent medium. In fact, HSO4− and NO3− binding is completely suppressed, while binding of H2PO4− and BzO− is diminished by approximately one order of magnitude for both 1.XB/HB (Table 1). Halide binding is similarly attenuated by the introduction of D2O but less so than observed with oxoanions.
The electrochemical properties of 1.XB/HB were studied in the same solvent systems as the NMR titration experiments (ACN and ACN/H2O 99:1) in the presence of 100 mM TBAClO4 as supporting electrolyte. In all cases a well-defined one-electron redox wave at moderate potentials, corresponding to the Fc/Fc+ couple, was observed (Fig. 2). The CVs at varying scan rates (Fig. S11 and S12†) were consistent with quasi-reversible and diffusion controlled behaviour with minimal adsorption of the receptors onto the working electrodes. The half-wave potential for 1.XB was observed at a slightly more anodic potential of 184 mV in comparison to 1.HB (178 mV; in ACN, vs. Ag|AgNO3), consistent with the more electron withdrawing nature of the iodotriazole groups.9,11
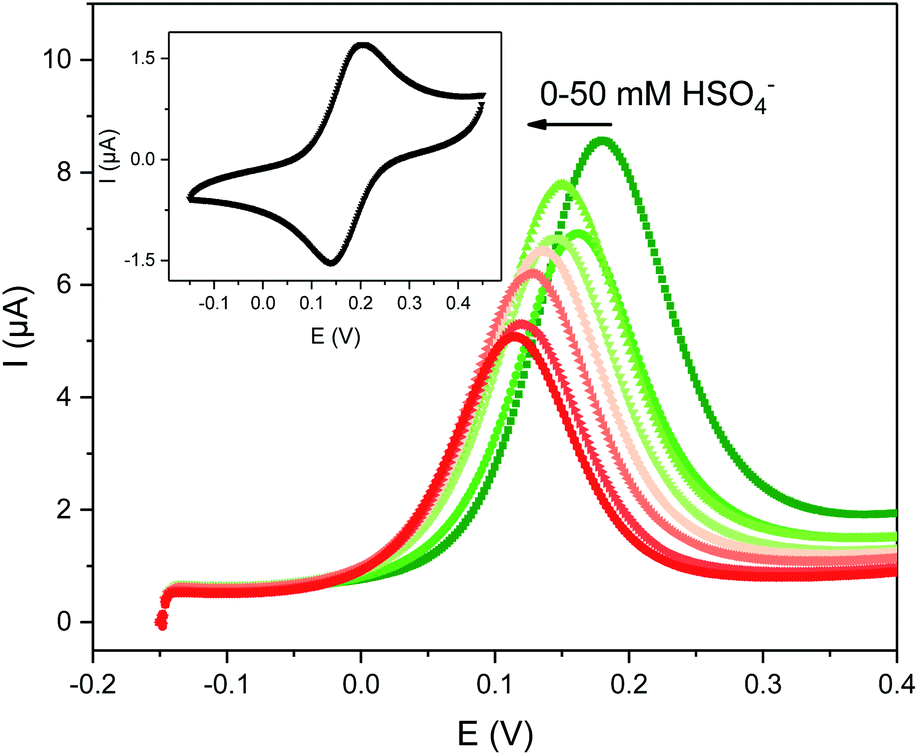 | ||
| Fig. 2 Evolution of the square-wave voltammograms (SWVs) of 0.1 mM 1.XB in ACN, 100 mM TBAClO4 upon titration with TBAHSO4. The inset shows the CV of this receptor at a scan rate of 100 mV s−1. | ||
The solution-phase voltammetric sensing properties of 1.XB/HB in both solvent systems were then investigated by SWV monitoring the changes in the receptor's peak potential upon titration with various anions (Fig. 2 and 3). Significant cathodic perturbations of the Fc/Fc+ couple were observed in ACN upon exposure to HSO4−, Cl−, Br−, H2PO4− and BzO−, while NO3− only induced a response for 1.XB. Importantly, the magnitude of this shift was significantly larger for 1.XB in comparison to 1.HB (Table 2). For example, H2PO4− induces the largest magnitude response for both receptors, but this response is ≈60 mV larger for the XB sensor (−173 mV vs. −109 mV, for 1.XB/HB, respectively).
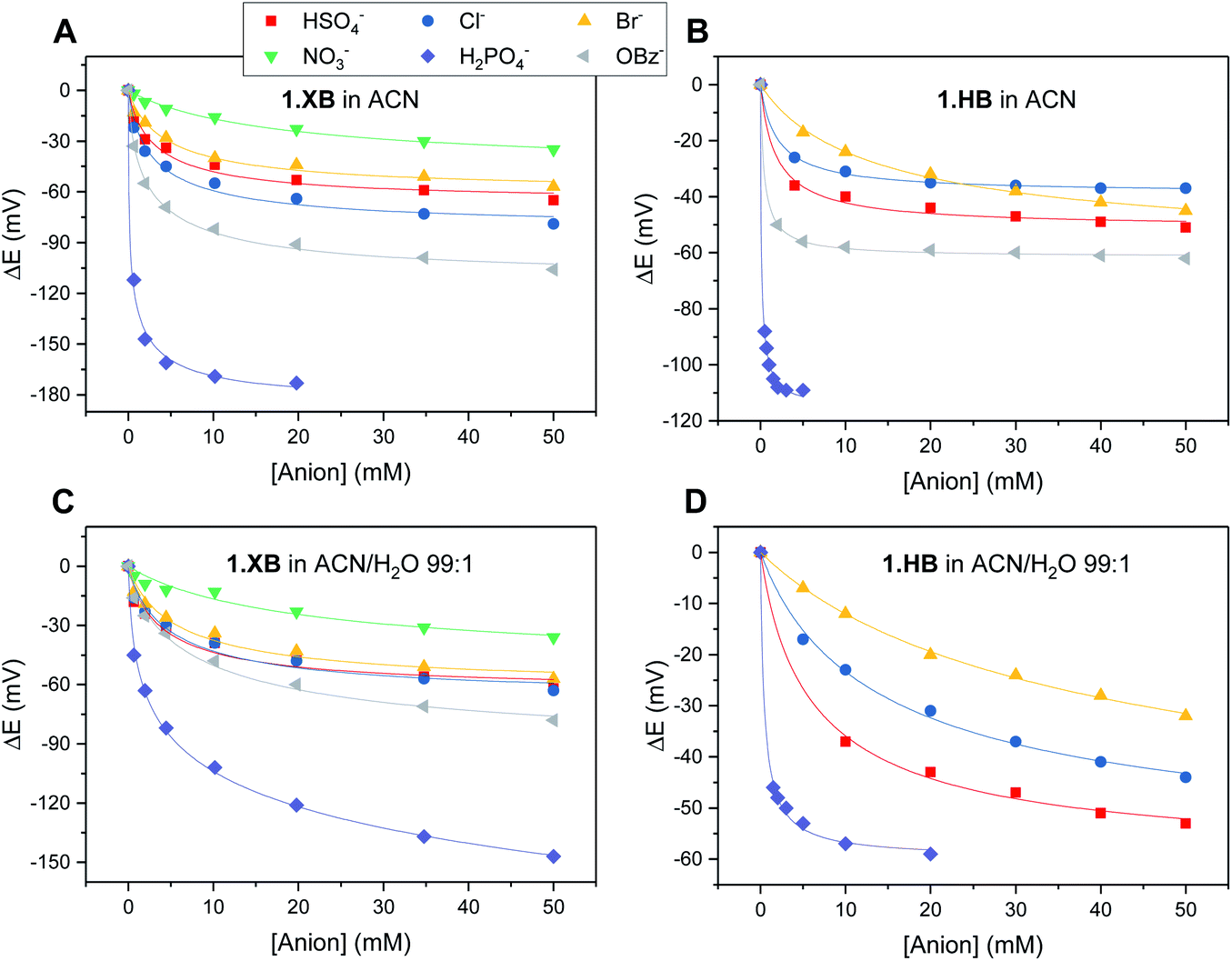 | ||
| Fig. 3 Cathodic voltammetric shifts of 1.XB and 1.HB in ACN (A and B) and in ACN/H2O 99:1 (C and D) upon titration with various anions. [1.XB/HB] = 0.1 mM with 100 mM TBAClO4 supporting electrolyte. The overall ionic strength was kept constant at 100 mM throughout. Solid lines represent fits to a 1:1 host–guest Nernst model (eqn (2)). Note the different y-axis scaling for all graphs. Anions for which no isotherms are shown induce negligible perturbations (i.e. NO3− in (B) and (D) and BzO− in (D)). | ||
:1 in the presence of 50 mM of various anions unless otherwise stated. N.r. – no response. — – not carried out. Estimated error ± 5 mV
| ACN | ACN/H2O 99:1 |
|||||
|---|---|---|---|---|---|---|
| 1.XB | 1.HB | 1.XB | 1.HB | 2.XBSAM | 2.HBSAM | |
| a ΔE at ≈2 mM, response not plateauing. b ΔE at <50 mM, response at plateau. | ||||||
| Cl− | −79 | −37 | −63 | −44 | −118 | −114 |
| Br− | −57 | −45 | −57 | −32 | −34a | −31a |
| HSO4− | −65 | −51 | −62 | −53 | −96 | −106 |
| H2PO4− | −173b | −109b | −147 | −59b | −158b | −175b |
| NO3− | −35 | N.r. | −36 | N.r. | −43 | −45 |
| OBz− | −106 | −62 | −78 | N.r. | — | — |
The overall cathodic anion response trends ΔE shown in Table 2 and Fig. 3 are 1.XB: H2PO4− > BzO− > Cl− > HSO4− ≈ Br− > NO3−; 1.HB: H2PO4− > BzO− > HSO4− > Cl− ≈ Br−, with no response towards NO3−.
In the presence of 1% H2O the response towards all anions was diminished for both receptors (Table 2 and Fig. 3; a direct comparison is also shown in Fig. S13–S18†). Of note here is that 1.XB still responded to all aforementioned anions in this solvent system with the magnitude of cathodic perturbation consistently greater for 1.XB than 1.HB in all cases and across both solvent systems (see also Fig. S19 and S20†). Although in good agreement with previous reports,9,11,28,29 this is, at first glance, perhaps somewhat surprising considering that the anion binding strength to the native receptors, as elucidated by 1H NMR titrations (discussed above), is larger for 1.HB in almost all cases. This importantly illustrates the performance of the sensor (i.e. the magnitude of the ΔE shift) is not simply governed by anion binding strength to the neutral receptor, as discussed in more detail below and in the ESI (Table S1†).
In order to further elucidate the principles that underpin these observations, the voltammetric binding isotherms shown in Fig. 3 were fitted to a 1:1 host–guest stoichiometric Nernst binding model (eqn (2)), which can be considered a special-case of eqn (1) (see ESI†), and is valid under fast-exchange, continuous shift conditions and when [A−] ≫ [H], where [A−] and [H] are the concentrations of the anion and host, respectively.
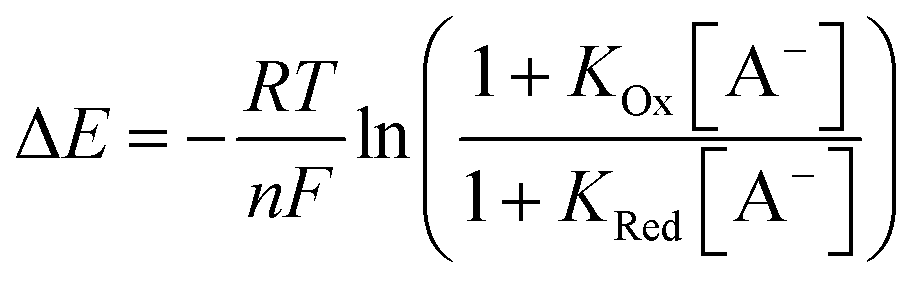 | (2) |
This allows the determination of not only the absolute anion binding constant to the neutral (reduced) receptor (KRed) but also to the oxidised receptor (KOx) and thus also affords the binding enhancement factor (BEF = KOx/KRed). This analysis shows that, in all cases, KOx is, as expected, significantly larger than KRed (Table 3). The anion binding constants of the neutral receptors are, generally, of similar magnitude as those obtained by 1H NMR titrations, as discussed in more detail in the ESI (Section S5, Tables S2 and S3†).
| ACN | ACN/H2O 99:1 |
|||||||
|---|---|---|---|---|---|---|---|---|
| 1.XB | 1.HB | 1.XB | 1.HB | |||||
| K Ox | K Red | K Ox | K Red | K Ox | K Red | K Ox | K Red | |
| a See ESI. | ||||||||
| Cl− | 1600 | 68 | 1030 | 222 | 724 | 54 | 201 | 21 |
| Br− | 683 | 66 | 208 | 20 | 547 | 50 | 70 | 4 |
| HSO4− | 1110 | 85 | 1460 | 201 | 847 | 73 | 503 | 57 |
| H2PO4− | 161000 |
121 | 99200 |
1090 | 5590 | −2a | 7360 | 715 |
| NO3− | 132 | 20 | N. b. | N. b. | 122 | 16 | — | — |
| BzO− | 3930 | 52 | 8200 | 746 | 753 | 20 | N. b. | N. b. |
From this quantitative analysis it can be seen that, as expected, anion binding to both neutral and oxidised forms of the respective receptor is diminished in the more competitive, organic/aqueous solvent system for all anions. Of note are the particularly large binding constants towards H2PO4− in the oxidised state, with KOx of up to 161000 M−1 for 1.XB in ACN (with an associated BEF of 1330, over one order of magnitude larger than that of any of the other anions). This may arise as a result of a unique binding and/or binding enhancement mode towards this oxoanion, potentially due to unique geometric constraints or its charge-density. Further investigations are required to elucidate this high preference for H2PO4−.
Although KOx is not always larger for 1.XB in comparison to 1.HB, the BEF, i.e. the binding switch-on upon oxidation, is consistently larger for 1.XB. This is in excellent agreement with the qualitative voltammetric observations above, confirming that the magnitude of the voltammetric shift (ΔE) is primarily dependent on the BEF and is greater for 1.XB.
The relative redox responses of 1.XB/HB are compared through a BEFXB/BEFHB ratio (Table 4). This XB enhancement can be very substantial. Notably, this not only underlines the uniquely potent nature of XB in voltammetric anion sensors, but also directly reports on a fundamental difference in the nature of the XB/HB interactions. Specifically, the higher sensitivity of XB recognition to the receptors' oxidation state may be indicative of an increased covalent character of the XB-anion binding interaction.30
:1. — – not applicable
| ACN | ACN/H2O 99:1 |
|||||
|---|---|---|---|---|---|---|
| 1.XB | 1.HB | Ratio XB/HB | 1.XB | 1.HB | Ratio XB/HB | |
| a Errors too large for meaningful comparison. | ||||||
| Cl− | 23.5 | 4.64 | 5.07 | 13.4 | 9.57 | 1.40 |
| Br− | 10.4 | 10.4 | 1.00 | 10.9 | 17.5 | 0.63 |
| HSO4− | 13.1 | 7.26 | 1.80 | 11.6 | 8.83 | 1.32 |
| H2PO4− | 1330 | 91.0 | 14.6 | 10.3 | ||
| NO3− | 6.6 | — | — | 7.63 | — | — |
| BzO− | 75.6 | 11.0 | 6.88 | 37.7 | — | — |
SAM formation and characterisation
The receptor immobilisation was achieved by overnight immersion of clean gold electrodes into a solution of 2.XB/HB (0.25 mM in ACN). This afforded well-defined SAMs (2.XB/HBSAM, Fig. 4A) which were characterised by ATR-IR and X-ray photoelectron spectroscopy (XPS), revealing film compositions in excellent agreement with the component atomic ratios (see ESI S6, Fig. S21–S24 and Tables S4, S5†). Water contact angle measurements indicated a moderate hydrophobicity which is somewhat larger for 2.XBSAM, in agreement with the presence of the iodotriazole moiety (Table 5). Electrochemical analysis of 2.XB/HBSAM revealed a single, well-defined redox couple, which showed a linear dependence of the peak currents on the scan rate (Fig. 4B, S25 and S26†) as well as low peak separation of approx. 35 mV as expected for surface-bound redox-centres.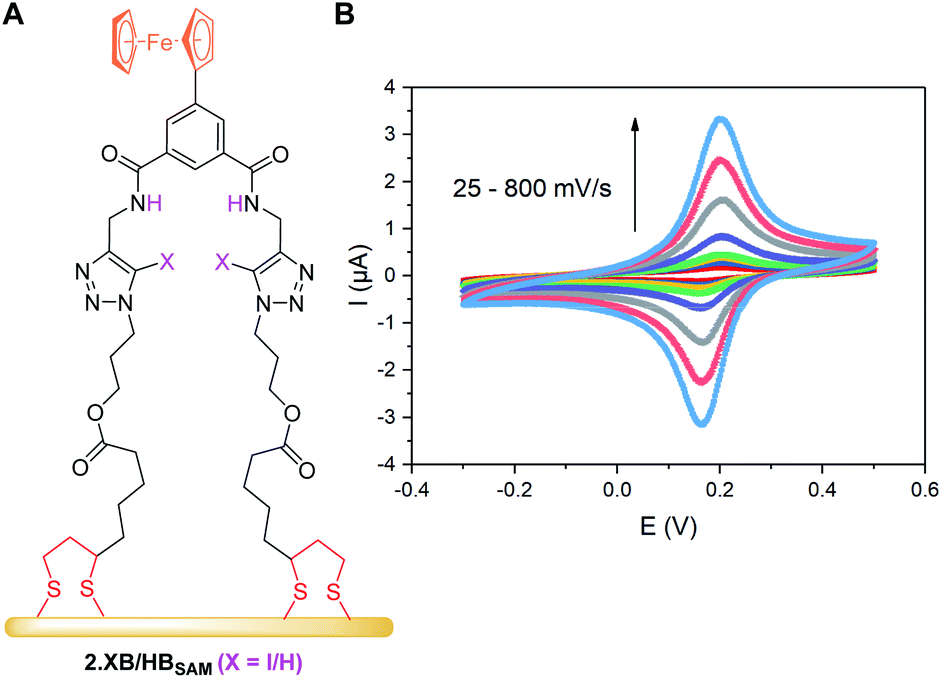 | ||
| Fig. 4 (A) Schematic representation of 2.XB/HBSAM on a gold electrode. (B) CVs at varying scan rate of 2.HBSAM in ACN, 100 mM TBAClO4. The associated anodic and cathodic peak currents as a function of the square-root of the scan rate are shown in Fig. S25.† | ||
:1 containing 100 mM TBAClO4 as electrolyte
| Water contact angle (°)a | Γ (10−10 mol cm−2) | E 1/2 vs. Ag|AgNO3b (mV) | |
|---|---|---|---|
| a Errors represent one standard deviation of 5 repeat measurements. b Obtained from charge integration of the ferrocene peaks. Errors represent one standard deviation of independent experiments on three electrodes. | |||
| 2.XBSAM | 68.4 ± 1.7 | 1.01 ± 0.18 | 193 ± 1 |
| 2.HBSAM | 59.1 ± 1.1 | 1.15 ± 0.22 | 183 ± 2 |
From peak integration, molecular surface coverages Γ of ≈10−10 mol cm−2 were determined for both films (Table 5), corresponding to a molecular footprint of 1.66 nm2, in excellent agreement with the size of the receptors and indicative of densely-packed SAMs in which the receptors adopt an upright conformation as depicted in Fig. 4A.
Upon repeated cycling of these films in ACN or ACN/H2O 99:1 a gradual loss of redox-activity was observed, a well-known problem arising from a non-ideal redox reversibility of the transducer. This was, for the latter solvent system, largely suppressed by the addition of a small amount of acid (100 μM HClO4), as previously reported in purely aqueous electrolytes.31–33 Importantly, this small acid concentration does not significantly affect anion binding but has a very profound effect on redox stability (Fig. S27 and S28†).§
Interfacial anion sensing
As can be seen in Fig. 5, both SAMs respond to all tested anions in this solvent system (ACN/H2O 99:1 + 100 μM H+) with no significant deviations from the expected binding isotherms (for further details see ESI S7†).
 | ||
| Fig. 5 Cathodic voltammetric shifts of 2.XBSAM (filled symbols) and 2.HBSAM (empty symbols) in ACN/H2O 99:1 + 100 μM H+ upon titration with various anions. Solid lines represent fits to a 1:1 host–guest Nernst model (eqn (2)). Note the different x- and y-axis scaling for both graphs. | ||
The overall response trends are similar to those in solution for both XB and HB motifs; the largest response was observed for H2PO4−, with smaller, but significant, cathodic shifts in the presence of HSO4−, Cl− and Br− and a small response to NO3− (Table 6). Interestingly, the difference in response between 2.XB/HBSAM is, in contrast to diffusive conditions, very small in all cases (<17 mV, Table 6), whereby the halides elicit a larger response at 2.XBSAM, while, unexpectedly, 2.HBSAM displays a slightly larger response towards oxoanions.¶ It is noteworthy that in all cases the film responses are significantly larger than the solution phase responses.
:1 (+100 μM H+) as determined by electrochemical titrations. All isotherms were fit to eqn (2) to obtain absolute binding constants (Fig. 5). Mathematical errors from the fitting are generally <20% (see ESI for further details)
| 2.XBSAM | 2.HBSAM | Ratio XB/HB | |||||
|---|---|---|---|---|---|---|---|
| K Ox | K Red | BEF | K Ox | K Red | BEF | ||
| Cl− | 2280 | 3 | 760 | 1960 | 4 | 489 | 1.55 |
| Br− | 1710 | 82 | 20.9 | 1380 | 53 | 26.0 | 0.80 |
| HSO4− | 1590 | 20 | 79.5 | 3920 | 51 | 76.9 | 1.03 |
| H2PO4− | ≈0.9 × 106 | ≈1000 | ≈900 | ≈1.5 × 106 | ≈1000 | ≈1500 | ≈0.6 |
| NO3− | 138 | 11 | 12.5 | 281 | 36 | 7.81 | 1.60 |
Comparison of diffusive and surface-confined sensor response
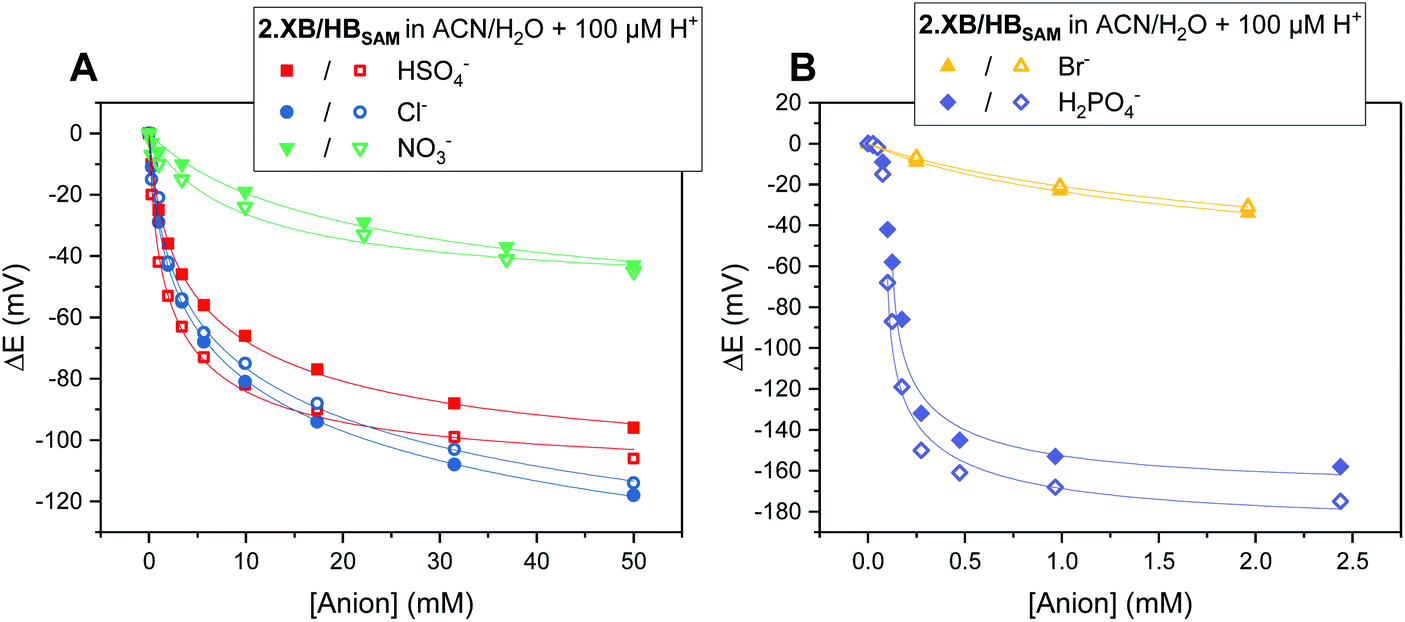
As noted above, in all cases the response of the surface-confined receptors is significantly larger than under diffusive conditions (Table 2; Fig. 3 and 5; for a direct comparison see Fig. S29†), in good agreement with previous studies.15,20,34 Although this has been attributed to surface confined receptor preorganisation and/or cooperative/chelate binding, this does not fully explain the observations herein.14,15,20 Receptor immobilisation within compact films will reduce any entropic penalty associated with anion binding, but other factors, including receptor and anion dehydration, are likely to be important. More importantly, the magnitude of the voltammetric response is determined by the BEF and not the absolute magnitude of the binding (to any one receptor oxidation state).We propose here that the surface BEF is enhanced as a result of diminished charge-screening (dielectric constant) within the hydrophobic SAMs. Pure alkanethiol SAMs possess dielectric constants ε of ≈2–3 (ref. 35) and, although 2.XB/HBSAM are presumably of higher polarity than such alkanethiol SAMs, their dielectric constants are expected to be significantly smaller than that of the solvent (εACN = 37.5).12 Voltammetrically generated Fc+ is thus much less screened in the SAM environment than within the bulk solvent which will translate to significantly enhanced anion binding in the cationic state and hence a more significant binding switch-on (Fig. 6).
 | ||
| Fig. 6 Schematic representation of through-bond (TB, blue) and through-space (TS, green arrows) interactions that drive anion recognition in the cationic state of 1.XB/HB in solution and at 2.XB/HBSAM. In the more polar solvent environment of high dielectric the TS contribution is diminished and TB contributions are significant. In the low dielectric SAM environment, TS contributions are strongly enhanced such that binding in the cationic state is switched-on more strongly, inducing a larger sensor response. | ||
This is directly supported by quantitative analysis (Table 6) where it is evident that the BEF enhancement indeed arises from an increased KOx at the interface (larger in all cases). Note that KRed is similar (or even smaller) in comparison to diffusive conditions (contradicting a model of significantly enhanced film preorganisation; Tables 3, 6 and S6†).
The screening model can also account for the differing performances of the XB/HB sensors when we consider the binding contributions that govern an enhanced anion binding to the oxidised receptor. Specifically, oxidation of Fc to Fc+ is expected to affect anion binding via two main pathways: (1) through-space (TS) electrostatic (i.e. coulombic) interactions between Fc+ and the anion, and (2) through-bond (TB) enhancement of the XB/HB donor strength via an increased electron-withdrawing effect of the electron-deficient Fc+.25,36 Importantly, the relative contributions of these effects will be environmentally dependent. Specifically, the absolute TB contribution is largely constant in both cases (solution/ SAM) as bond polarisation is less likely to be affected by the dielectric environment. In marked contrast, the TS interaction is likely to be much less screened in the low dielectric environment of an organic film such that the coulombic interaction is significantly enhanced (and contributes more towards overall binding). We thus propose that the interfacial binding enhancements are driven largely by coulombic TS effects. These dominate in the films such that the NMR and voltammetric solution phase differences between XB and HB hosts are lost.
A consideration of these screening effects not only provides an in-depth rationalisation for an enhanced interfacial response in voltammetric ion sensors, but can also explain differences in selectivity/response patterns across different XB/HB receptors, such as the similar performance of the SAMs.
The results presented herein highlight the attention that needs to be paid to the dielectric properties of the solvent microenvironment and the interface, an appreciation of which can directly benefit the design of sensors with improved performance (where the dielectric of solution and film microenvironments may be specifically synthetically tuned).
Conclusions
This work provides the first detailed comparison of redox-active XB and HB anion receptors in diffusive and interfacial formats and introduces the use of quantitative Nernst binding isotherm analysis of surface-confined voltammetric anion sensors. From the resolved absolute receptor-anion binding constants (KOx and KRed and their ratio (BEF)), it is apparent that the sensor response is largely dictated by KOx, the importance of through-bond covalent interactions in solution (increasing the XB response), and the extent to which KOx is amplified in a low dielectric environment. Specifically, in solution the XB analogue of novel ferrocene-isophthalamide-triazole receptors 1.XB displayed significantly larger cathodic voltammetric perturbations upon anion binding, attributable to an enhanced binding switch-on upon oxidation of the XB receptor and quantified as an XB enhancement factor (BEFXB/BEFHB). The surface-immobilisation of these receptors via formation of well-defined SAMs 2.XB/HBSAM then enabled anion sensing with a significantly enhanced response in all cases. A detailed analysis of this surface-enhancement afforded unprecedented insights into the transduction principles that govern this amplified response. Specifically, we propose that all observations can be rationalised by through-space transducer – binding site dielectric screening effects. This improved fundamental understanding of anion sensing at redox-active interfaces will benefit the future development of sensitive, real-life relevant sensors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
XL thanks the EPSRC for postdoctoral funding (EPSRC grant number EP/P033490/1). The authors thank Andrew Docker and Sophie Patrick, University of Oxford, for helpful discussions and Dr Philip Holdway, Oxford Materials Characterisation Service, for assistance with XPS measurements.Notes and references
- R. Hein, P. D. Beer and J. J. Davis, Chem. Rev., 2020, 120, 1888–1935 CrossRef CAS.
- N. H. Evans, C. J. Serpell, N. G. White and P. D. Beer, Chem.–Eur. J., 2011, 17, 12347–12354 CrossRef CAS.
- T. Romero, R. A. Orenes, A. Tarraga and P. Molina, Organometallics, 2013, 32, 5740–5753 CrossRef CAS.
- O. Reynes, F. Maillard, J.-C. Moutet, G. Royal, E. Saint-Aman, G. Stanciu, J.-P. Dutasta, I. Gosse and J.-C. Mulatier, J. Organomet. Chem., 2001, 637, 356–363 CrossRef.
- J. Pancholi and P. D. Beer, Coord. Chem. Rev., 2020, 416, 213281 CrossRef CAS.
- A. Brown and P. D. Beer, Chem. Commun., 2016, 52, 8645–8658 RSC.
- R. Oliveira, S. Groni, C. Fave, M. Branca, F. Mavre, D. Lorcy, M. Fourmigue and B. Schollhorn, Phys. Chem. Chem. Phys., 2016, 18, 15867–15873 RSC.
- J. Y. C. Lim and P. D. Beer, Eur. J. Org. Chem., 2019, 2019, 3433–3441 CrossRef CAS.
- J. Y. C. Lim and P. D. Beer, Eur. J. Inorg. Chem., 2017, 2017, 220–224 CrossRef CAS.
- F. Zapata, A. Caballero and P. Molina, Eur. J. Inorg. Chem., 2017, 2017, 237–241 CrossRef CAS.
- J. Y. C. Lim, M. J. Cunningham, J. J. Davis and P. D. Beer, Chem. Commun., 2015, 51, 14640–14643 RSC.
- R. Hein, A. Borissov, M. D. Smith, P. D. Beer and J. J. Davis, Chem. Commun., 2019, 55, 4849–4852 RSC.
- P. R. Bueno, R. Hein, A. Santos and J. J. Davis, Phys. Chem. Chem. Phys., 2020, 22, 3770–3774 RSC.
- H. Hijazi, A. Vacher, S. Groni, D. Lorcy, E. Levillain, C. Fave and B. Schollhorn, Chem. Commun., 2019, 55, 1983–1986 RSC.
- D. P. Cormode, A. J. Evans, J. J. Davis and P. D. Beer, Dalton Trans., 2010, 39, 6532–6541 RSC.
- B. Kaur, C. A. Erdmann, M. Daniëls, W. Dehaen, Z. Rafiński, H. Radecka and J. Radecki, Anal. Chem., 2017, 89, 12756–12763 CrossRef CAS.
- P. Gołębiewski, B. Puciłowski, F. Sommer, S. Kubik, M. Daniels, W. Dehaen, U. Sivasankaran, K. G. Kumar, H. Radecka and J. Radecki, Sens. Actuators, B, 2019, 285, 536–545 CrossRef.
- J. Lehr, T. Lang, O. A. Blackburn, T. A. Barendt, S. Faulkner, J. J. Davis and P. D. Beer, Chem.–Eur. J., 2013, 19, 15898–15906 CrossRef CAS.
- N. H. Evans, H. Rahman, J. J. Davis and P. D. Beer, Anal. Bioanal. Chem., 2012, 402, 1739–1748 CrossRef CAS.
- P. D. Beer, J. J. Davis, D. A. Drillsma-Milgrom and F. Szemes, Chem. Commun., 2002, 1716–1717 RSC.
- C. Adam, L. Faour, V. Bonnin, T. Breton, E. Levillain, M. Sallé, C. Gautier and D. Canevet, Chem. Commun., 2019, 55, 8426–8429 RSC.
- O. Reynes, C. Bucher, J.-C. Moutet, G. Royal and E. Saint-Aman, Chem. Commun., 2004, 428–429 RSC.
- S. Zhang, C. M. Cardona and L. Echegoyen, Chem. Commun., 2006, 4461–4473 RSC.
- S. R. Miller, D. A. Gustowski, Z. H. Chen, G. W. Gokel, L. Echegoyen and A. E. Kaifer, Anal. Chem., 1988, 60, 2021–2024 CrossRef CAS.
- P. D. Beer, P. A. Gale and G. Z. Chen, Coord. Chem. Rev., 1999, 185, 3–36 CrossRef.
- N. H. Evans, H. Rahman, A. V. Leontiev, N. D. Greenham, G. A. Orlowski, Q. Zeng, R. M. Jacobs, C. J. Serpell, N. L. Kilah and J. J. Davis, Chem. Sci., 2012, 3, 1080–1089 RSC.
- P. Fortgang, E. Maisonhaute, C. Amatore, B. Delavaux-Nicot, J. Iehl and J. F. Nierengarten, Angew. Chem., Int. Ed., 2011, 50, 2364–2367 CrossRef CAS.
- J. Y. C. Lim, I. Marques, L. Ferreira, V. Félix and P. D. Beer, Chem. Commun., 2016, 52, 5527–5530 RSC.
- B. R. Mullaney, M. J. Cunningham, J. J. Davis and P. D. Beer, Polyhedron, 2016, 116, 20–25 CrossRef CAS.
- S. W. Robinson, C. L. Mustoe, N. G. White, A. Brown, A. L. Thompson, P. Kennepohl and P. D. Beer, J. Am. Chem. Soc., 2015, 137, 499–507 CrossRef CAS.
- D. D. Popenoe, R. S. Deinhammer and M. D. Porter, Langmuir, 1992, 8, 2521–2530 CrossRef CAS.
- G. Valincius, G. Niaura, B. Kazakevičienė, Z. Talaikytė, M. Kažemėkaitė, E. Butkus and V. Razumas, Langmuir, 2004, 20, 6631–6638 CrossRef CAS.
- L. Zhang, L. A. Godínez, T. Lu, G. W. Gokel and A. E. Kaifer, Angew. Chem., Int. Ed., 1995, 34, 235–237 CrossRef CAS.
- O. Reynes, T. Gulon, J.-C. Moutet, G. Royal and E. Saint-Aman, J. Organomet. Chem., 2002, 656, 116–119 CrossRef CAS.
- M. D. Porter, T. B. Bright, D. L. Allara and C. E. D. Chidsey, J. Am. Chem. Soc., 1987, 109, 3559–3568 CrossRef CAS.
- P. D. Beer, P. A. Gale and G. Z. Chen, J. Chem. Soc., Dalton Trans., 1999, 1897–1910 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of all compounds, surface analysis and detailed comparisons of sensor responses. See DOI: 10.1039/d0sc06210c |
| ‡ It should be noted that both receptors contain a HB isophthalamide core structure with additional XB or HB (iodo)triazole binding sites. Based on the latter we use the XB/HB designations. |
| § A detailed study of the effect of (higher) acid concentrations on the redox stability and sensory properties of these films will be published separately. |
| ¶ The unexpected “inferior” performance of 2.XBSAM relative to 2.HBSAM cannot arise from an increased steric bulk of the iodotriazole groups, as this would only explain a diminished binding strength (in both reduced and oxidised states), but does not justify why binding switch-on (i.e. the BEF) is affected. |
| This journal is © The Royal Society of Chemistry 2021 |