
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-concentrated aqueous electrolytes with organic solvent additives for stable zinc batteries†‡
Yang
Dong§
a,
Licheng
Miao§
bc,
Guoqiang
Ma
a,
Shengli
Di
a,
Yuanyuan
Wang
a,
Liubin
Wang
ab,
Jianzhong
Xu
a and
Ning
Zhang
 *ab
*ab
aCollege of Chemistry & Environmental Science, Key Laboratory of Analytical Science and Technology of Hebei Province, Hebei University, Baoding, 071002, China. E-mail: ningzhang@hbu.edu.cn
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), College of Chemistry, Nankai University, Tianjin, 300071, China
cCollege of Physics and Optoelectronic Engineering, Shenzhen University, Shenzhen, 518060, China
First published on 15th March 2021
Abstract
Rechargeable aqueous zinc batteries (RAZBs) are promising for large-scale energy storage because of their superiority in addressing cost and safety concerns. However, their practical realization is hampered by issues including dendrite growth, poor reversibility and low coulombic efficiency (CE) of Zn anodes due to parasitic reactions. Here, we report a non-concentrated aqueous electrolyte composed of 2 m zinc trifluoromethanesulfonate (Zn(OTf)2) and the organic dimethyl carbonate (DMC) additive to stabilize the Zn electrochemistry. Unlike the case in conventional aqueous electrolytes featuring typical Zn[H2O]62+ solvation, a solvation sheath of Zn2+ with the co-participation of the DMC solvent and OTf− anion is found in the formulated H2O + DMC electrolyte, which contributes to the formation of a robust ZnF2 and ZnCO3-rich interphase on Zn. The resultant Zn anode exhibits a high average CE of Zn plating/stripping (99.8% at an areal capacity of 2.5 mA h cm−2) and dendrite-free cycling over 1000 cycles. Furthermore, the H2O + DMC electrolytes sustain stable operation of RAZBs pairing Zn anodes with diverse cathode materials such as vanadium pentoxide, manganese dioxide, and zinc hexacyanoferrate. Rational electrolyte design with organic solvent additives would promote building better aqueous batteries.
Introduction
Safe, reliable, and cost-effective electrochemical energy storage technologies are demanded for the efficient utilization of renewable energy sources such as solar and wind.1,2 Among various options, rechargeable aqueous Zn batteries (RAZBs) hold great promise because of the advantages of metallic Zn anodes including abundant resources, environmental benignancy, low cost, and high theoretical specific/volumetric capacity (820 mA h g−1 and 5855 mA h cm−3).3–5 Moreover, compared with flammable organic electrolytes widely adopted in lithium-ion batteries (LIBs), aqueous electrolytes intrinsically provide improved safety, and their higher ionic conductivities favor high rate capability.6,7 These features promote the recent renaissance of RAZBs with extensive research on a variety of cathode materials, including manganese oxides,8–10 Prussian blue analogues,11,12 vanadium oxides,13–16 and organic compounds.17–20Nonetheless, state-of-the-art RAZBs are plagued by the issues associated with metallic Zn anodes, such as low plating/stripping efficiency, dendrite growth, and unstable Zn–electrolyte interface along with water-induced side reactions (e.g., H2 evolution and surface passivation).4,21,22 To address these challenges, an efficient strategy is to construct hierarchical structures23–26 or modification layers27–30 on Zn anodes. Besides, electrolyte modulation is considered to be a facile approach to stabilize metal anodes by regulating the interface chemistry.31–35 Recently, highly concentrated electrolytes (e.g., 1 m Zn(TFSI)2 + 20 m LiTFSI (m: mol kg−1)36) and deep eutectic electrolytes (e.g., ∼4.2 m Zn(TFSI)2 in nonaqueous acetamide37) were proposed to form an anion-derived solid electrolyte interphase (SEI) layer on the Zn anode. This SEI allows rapid Zn2+ diffusion while blocking solvents and electrons, and thus suppressing water-induced side reactions.36,37 However, the higher cost, reduced ionic conductivity, and increased viscosity of these salt-concentrated electrolytes raise much concern towards their practical use. In a conventional aqueous electrolyte (concentration generally below 2 mol L−1), the typical solvation structure of Zn[H2O]62+ without anion coordination makes it infeasible to form a protective SEI from the reductive decomposition of anions before Zn deposition.35,36 Instead, competitive water decomposition with H2 evolution occurred (H2O → H2 + OH−), which increases the local pH and induces the generation of Zn hydroxides or zincates to passivate the Zn anode.35,38 Therefore, it is desirable to stabilize the Zn electrochemistry in non-concentrated aqueous electrolytes.
Herein, an aqueous electrolyte composed of 2 m Zn(CF3SO3)2 (Zn(OTf)2) in water with the dimethyl carbonate (DMC) additive is developed and demonstrated to form a stable SEI layer on the Zn anode. Based on molecular dynamics (MD) simulations, density functional theory (DFT) computations and spectroscopic investigations (i.e., FTIR, 17O NMR, and Raman), a new solvation shell of Zn2+ with the co-participation of the DMC solvent and OTf− anion is found in the formulated H2O + DMC electrolyte. The co-solvation process enables the formation of an organic–inorganic hybrid SEI with rich ZnF2 and ZnCO3, which is proposed to derive from the reductive decomposition of Zn2+–DMC and Zn2+–OTf− coordinations before Zn deposition. This hybrid SEI effectively suppresses water-induced parasitic reactions on the Zn anode, and thus enables high-efficiency Zn plating/stripping and extended dendrite-free cycling. In addition, the Zn(OTf)2 in the H2O + DMC electrolyte is versatilely compatible with different cathode materials such as transition metal oxides and hexacyanoferrate.
Results and discussion
Electrolyte formulation and characterization
Dimethyl carbonate (DMC) as a primary co-solvent has been widely used in LIBs, and its favorable reduction results in the formation of the SEI on the anode surface, which can stabilize the electrode/electrolyte interface and support cation transfer.39 Inspired by this co-solvent design, we have constructed aqueous electrolytes with the DMC additive for RAZBs by mixing water and DMC in varied molar ratios and then adding Zn(OTf)2 salt into the mixed solvent with a constant concentration of 2 m (m: mol kg−1, Fig. S1, ESI‡). The molar ratios between water and DMC solvents are tentatively selected at 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 6:1, 4:1, and 2:1, and the corresponding electrolytes are denoted as W8D1, W6D1, W4D1, and W2D1, respectively. A 2 m Zn(OTf)2 aqueous electrolyte without DMC (denoted as 2 m W) was also prepared for comparison. The compositions of these electrolytes are summarized in Table S1 (ESI‡). As a non-polar solvent, DMC is immiscible with polar water (Fig. 1a) and it is not readily soluble with Zn(OTf)2 salt (Fig. S2, ESI‡). Interestingly, the miscibility between DMC and water can be regulated (Fig. 1b) by adding Zn(OTf)2 salt, which acts as a linker interplaying with both DMC and H2O (discussed later). Molecular dynamics (MD) simulations reveal the homogeneous solution state (Fig. 1c) of 2 m Zn(OTf)2 in the H2O + DMC electrolyte and the apparent phase separation (Fig. 1d) of the H2O + DMC mixture without salt. Ignition tests intuitively identify the non-flammable nature of W4D1 and W8D1 electrolytes (Fig. 1e and S3, ESI‡), whereas, further increasing the DMC content in the electrolyte (e.g., W2D1) leads to electrolyte combustion (Fig. 1f). Although the ionic conductivity of Zn(OTf)2 in H2O + DMC electrolytes gradually decreases with increasing DMC ratio, a considerable conductivity of 25.4 mS cm−1 is obtained in W4D1 (Fig. S4, ESI‡). This value is much higher than those of typical non-aqueous electrolytes (1–10 mS cm−1).1,39
:1, 6:1, 4:1, and 2:1, and the corresponding electrolytes are denoted as W8D1, W6D1, W4D1, and W2D1, respectively. A 2 m Zn(OTf)2 aqueous electrolyte without DMC (denoted as 2 m W) was also prepared for comparison. The compositions of these electrolytes are summarized in Table S1 (ESI‡). As a non-polar solvent, DMC is immiscible with polar water (Fig. 1a) and it is not readily soluble with Zn(OTf)2 salt (Fig. S2, ESI‡). Interestingly, the miscibility between DMC and water can be regulated (Fig. 1b) by adding Zn(OTf)2 salt, which acts as a linker interplaying with both DMC and H2O (discussed later). Molecular dynamics (MD) simulations reveal the homogeneous solution state (Fig. 1c) of 2 m Zn(OTf)2 in the H2O + DMC electrolyte and the apparent phase separation (Fig. 1d) of the H2O + DMC mixture without salt. Ignition tests intuitively identify the non-flammable nature of W4D1 and W8D1 electrolytes (Fig. 1e and S3, ESI‡), whereas, further increasing the DMC content in the electrolyte (e.g., W2D1) leads to electrolyte combustion (Fig. 1f). Although the ionic conductivity of Zn(OTf)2 in H2O + DMC electrolytes gradually decreases with increasing DMC ratio, a considerable conductivity of 25.4 mS cm−1 is obtained in W4D1 (Fig. S4, ESI‡). This value is much higher than those of typical non-aqueous electrolytes (1–10 mS cm−1).1,39
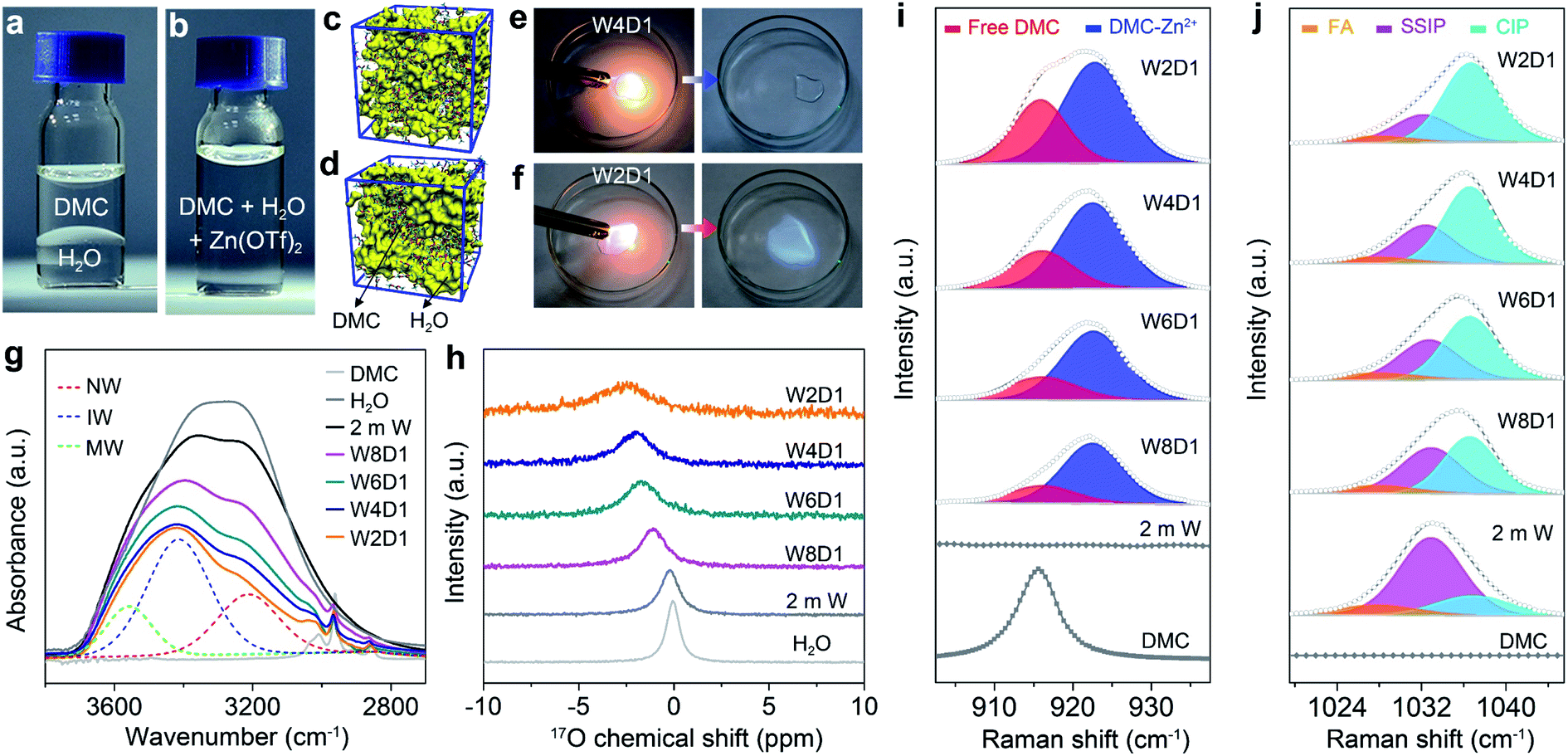 | ||
| Fig. 1 Electrolyte characterization. Digital photos of (a) the immiscibility between H2O and DMC solvents, and (b) the homogeneous 2 m Zn(OTf)2 in a H2O + DMC electrolyte (W4D1). 3D snapshots obtained from MD simulations showing (c) the miscibility of the W4D1 electrolyte and (d) the phase separation of H2O and DMC solvents (H2O domain in yellow and DMC as the wireframe). Ignition tests of (e) W4D1 and (f) W2D1. (g) FTIR spectra of 2 m W, W8D1, W6D1, W4D1 and W2D1 electrolytes, and typical fitted curves (dashed line) of W2D1. (h) Chemical shifts of 17O nuclei of H2O in H2O + DMC electrolytes. Raman spectra between (i) 903 and 937 cm−1 (CH3–O stretching vibration of DMC) and (j) 1019 and 1045 cm−1 (SO3 stretching mode) of different H2O + DMC electrolytes. | ||
The solvation structure of H2O + DMC electrolytes and the interaction between Zn(OTf)2 and the solvent were investigated by different spectroscopic methods. Fig. 1g presents the Fourier transform infrared (FTIR) spectra of H2O + DMC electrolytes along with the neat DMC and H2O solvents. The broad O–H stretching band can be ascribed to three states of water molecules using the Gaussian function:40 (1) “network water (NW)” (∼3205 cm−1) associated with water molecules with a strong H-bond coordination (coordination number close to four); (2) “intermediate water (IW)” (∼3410 cm−1) corresponding to water molecules with distorted H-bonds and somewhat connected to other water molecules though unable to build a fully connected network; (3) “multimer water (MW)” (∼3560 cm−1) ascribed to water molecules standing as free monomers or dimers or trimers and being poorly connected to their environment. Accordingly, the addition of DMC significantly disrupts the strong H-bonding network as confirmed by the significant decrease of the “network water” fraction from 52.9% in pristine 2 m W to 27.5% in W2D1 (Fig. S5, ESI‡). Nuclear magnetic resonance (NMR) spectra demonstrate that the 17O chemical shift in H2O (0 ppm) experiences a downshift (high filed) accompanied by peak broadening upon the addition of DMC (Fig. 1h). This evolution reveals that the increase of electron density on oxygen (water) caused by the breaking of the H-bond network results in the shielding effect.41,42 The 17O chemical shift of DMC solvent is provided in Fig. S6 (ESI‡).
Raman spectra of the H2O + DMC series also unveil the evolution of the solvation configuration. As shown in Fig. 1i, pure DMC shows a broad peak at ∼916 cm−1, which is associated with the CH3–O stretching mode of the free DMC.43,44 With the addition of DMC into aqueous electrolytes, a new band located at ∼923 cm−1 appears owing to the coordination of DMC with Zn2+. Note that Lewis acidic Zn2+ can interact with the lone-pair of electrons on O from DMC (Lewis base), making the DMC–Zn2+ coordination feasible. Moreover, the calculated binding energy of the DMC–Zn2+ complex is lower than that of the H2O–Zn2+ counterpart (Fig. S7, ESI‡), indicating that DMC can favourably coordinate with Zn2+. The fitting results demonstrate that the contents of DMC bound to Zn2+ in the electrolyte samples (Table S1, ESI‡) gradually increase with the addition of DMC (from W8D1 to W2D1, Table S3, ESI‡). Further increasing the DMC ratio (from W4D1 to W2D1) leads to the increase of the free DMC fraction, which could explain the flammable nature of the W2D1 electrolyte (Fig. 1e). In the Raman region of the OTf− anion (Fig. 1j), three broad peaks at ∼1028, 1032, and 1037 cm−1 represent the free anion (FA), solvent-separated ion pairs (SSIP, Zn2+–(H2O)x(DMC)y–OTf−) and contact ion pairs (CIP, Zn2+–OTf−), respectively.45 Similar to previous reports,36,37 the majority of OTf− anions do not directly connect with Zn2+ in the non-concentrated 2 m W electrolyte. Upon adding DMC into aqueous electrolytes, the portion of the Zn2+–OTf− coordination significantly strengthens in W4D1 as compared to that in 2 m W (Table S4, ESI‡), suggesting the involvement of the OTf− anion in the Zn2+ solvation sheath. Additionally, the SSIP signal experiences a slight redshift with the addition of DMC (from W8D1 to W2D1), indicating that the reduced coulombic interaction may be due to the barriers of DMC.
MD simulations were performed to study the Zn2+-solvation sheath in 2 m W and H2O + DMC (W4D1) electrolytes. The particular number of ions and molecules in the simulation box is summarized in Table S2 (ESI‡). In the non-concentrated aqueous electrolyte (2 m W), Zn2+ is considered to coordinate with six H2O molecules (Zn[H2O]62+) without considerable contribution from OTf− anions. In W4D1, the DMC solvent and OTf− anion occur in the Zn2+-solvation structure (schematically shown in Fig. 2a), due to the strong interaction among Zn2+, OTf− and DMC, and the decreased ratio of water. This evolution of the Zn2+ structure agrees with the spectroscopic results. Radial distribution functions (RDFs) revealing the distributions of nearest-neighbor molecules19 were further employed to elucidate the solvation information of W4D1 (Fig. 2b). RDFs show that the primary solvation shell of Zn2+ (central ion) is around 0.4 nm in distance. Two peaks of the Zn-O (DMC) pair and Zn-O (OTf−) pair are identified at 0.33 and 0.37 nm, respectively, supporting the participation of DMC and OTf− in the Zn2+-solvation structure. In W4D1, the first Zn2+-solvation sheath is occupied primarily by 3.9 H2O molecules, 0.7 DMC solvent, and 1.4 OTf− anions on average (i.e., Zn2+[H2O]3.9[DMC]0.7[OTf−]1.4). The RDFs in W8D1 are provided in Fig. S8 (ESI‡). Furthermore, the fraction of free water is ∼82% in 2 m W and decreases to ∼55% in the W4D1 electrolyte (Fig. 2c). This decrease suggests significantly reduced water activity, which favors the suppression of parasitical Zn–water reactions. In addition, the predicted ionic conductivities of the electrolytes are in good agreement with the experimental values (Fig. S4, ESI‡), demonstrating the validity of MD simulations.
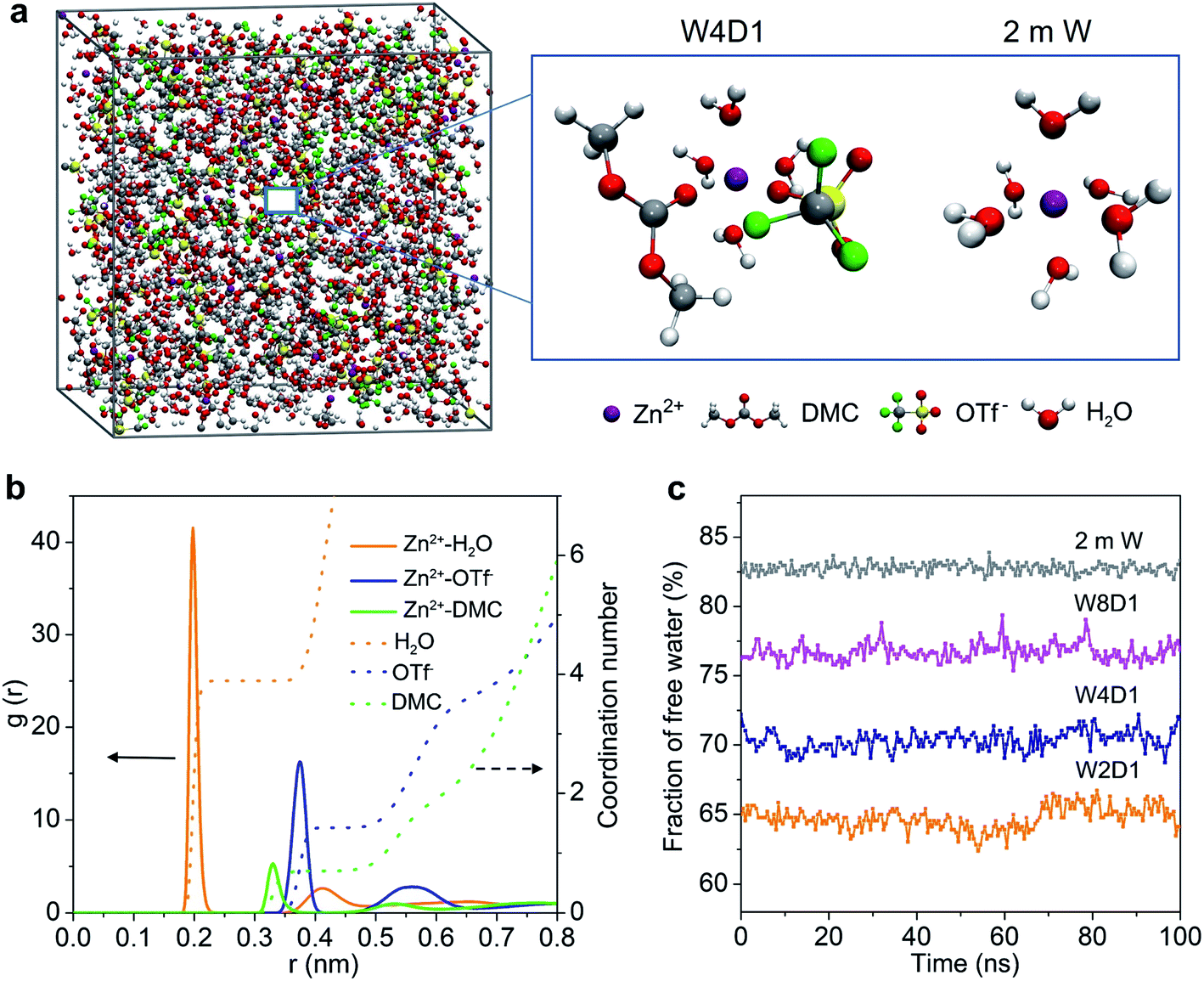 | ||
| Fig. 2 MD studies of the Zn2+ solvation structure. (a) Snapshot of the MD simulation cell for the W4D1 electrolyte, and the representative Zn2+-solvation structures in W4D1 and 2 m W electrolytes. (b) RDFs of Zn-O (H2O), Zn-O (DMC) and Zn-O (OTf−) pairs, and the coordination number in the W4D1 electrolyte. (c) Fraction of free water (not solvating to Zn2+) in the H2O + DMC electrolytes with a 100 ns simulation time. | ||
According to the combination of experimental (FTIR, 17O-NMR, and Raman spectra) and modeling investigations, we propose that the significant interaction among Zn2+, OTf−, and DMC provides a unique Zn2+-solvation structure in the W4D1 electrolyte with the co-participation of the DMC solvent and OTf− anion to form Zn2+[H2O]3.9[DMC]0.7[OTf−]1.4. This solvation structure differs from that of the typical solvation shell (i.e., Zn[H2O]62+) in a non-concentrated aqueous electrolyte without DMC solvent. The co-contribution of anion OTf− and DMC further enables a new interface chemistry induced by the reductive decomposition of OTf− and DMC and stabilizes Zn electrochemistry, as discussed below.
Reversibility and stability of the Zn anode in H2O + DMC electrolytes
The Zn plating/stripping properties in H2O + DMC electrolytes were evaluated using Zn/Ti asymmetric cells and Zn/Zn symmetric cells. Fig. 3a shows the CE of Zn plating/stripping in Zn/Ti cells recorded at a current density of 1.0 mA cm−2, a deposition capacity of 1.0 mA h cm−2 and a cut-off voltage of 0.5 V (vs. Zn2+/Zn). The CE value is defined as the ratio of the stripping capacity to the plating capacity. Clearly, the reversibility and stability of the Zn anode improve with increasing DMC content. The Zn/Ti cell in the W4D1 electrolyte displays an initial CE of 84.4%, and then it increases to 99.6% after 30 cycles. Impressively, an average CE of 99.8% is maintained after 200 cycles. In sharp contrast, the cell in the 2 m W electrolyte shows rapid capacity decay and CE below 58% after 100 cycles, which could be attributed to the side reactions between H2O and the Zn anode. Fig. 3b displays the typical Zn plating/stripping profiles. Stable voltage curves are characterized in W4D1, where the separation of charge and discharge potentials decreases initially and stabilizes at ∼110 mV after 50 cycles. A lower CE with an unstable stripping process is observed in the 2 m W counterpart. Although the W2D1 electrolyte also affords stable cycling of Zn, the overpotential (∼172 mV at the 50th cycle, Fig. S9, ESI‡) is much higher than that in W4D1, due to the lower ionic conductivity. Typical charge/discharge profiles of Zn/Ti cells in W8D1 and W6D1 electrolytes (Fig. S10 and S11, ESI‡) suggest better performances than 2 m W but inferior stability to W4D1.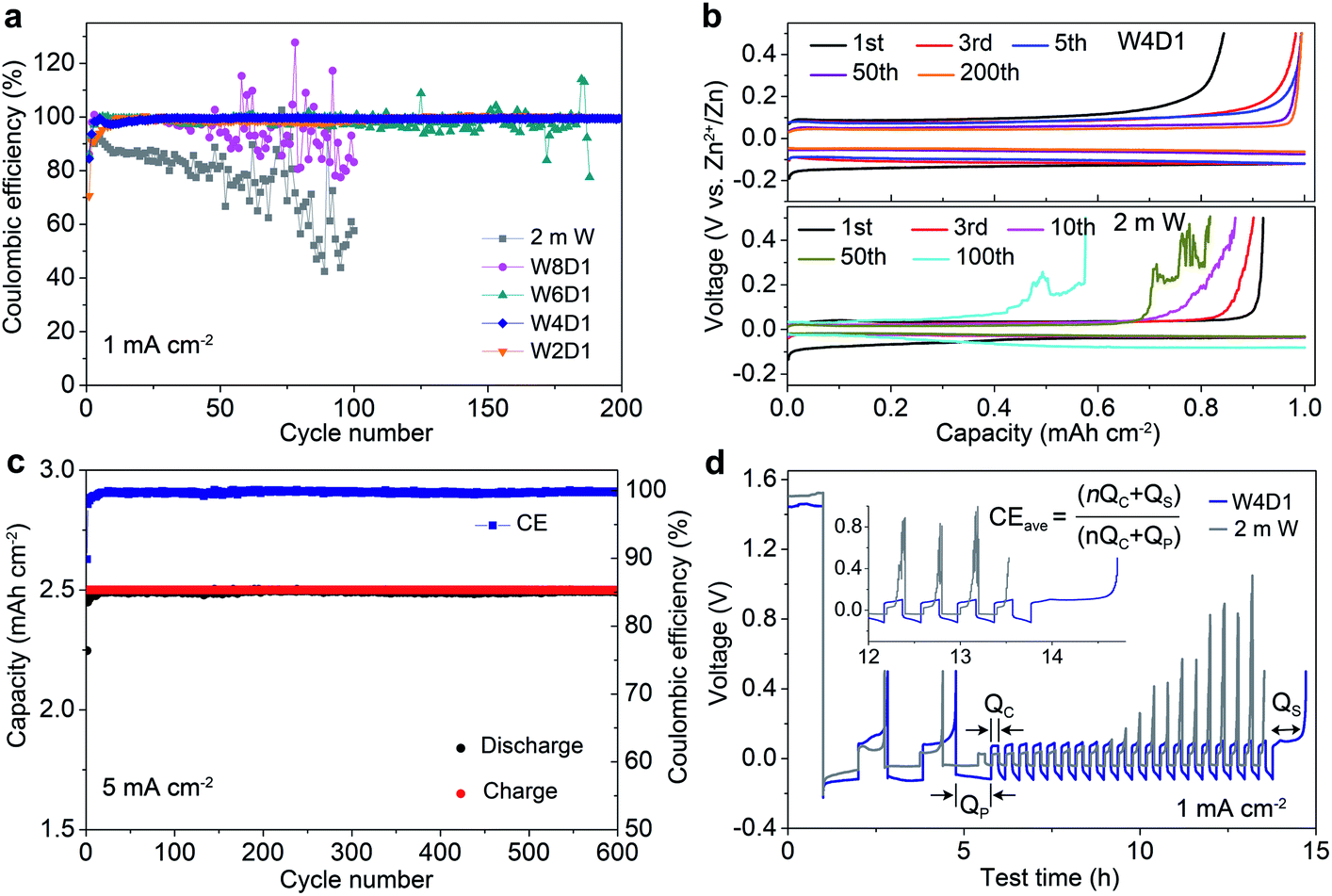 | ||
| Fig. 3 Zn2+ plating/stripping behaviors in H2O + DMC electrolytes. (a) Zn plating/stripping CE values of Zn/Ti cells in different electrolytes. (b) Typical voltage profiles of Zn/Ti cells in W4D1 and 2 m W at 1 mA cm−2. (c) Cycling stability and Zn plating/stripping CE of the Zn/Ti cell in W4D1 at 5 mA cm−2. (d) Evolution of voltage profiles of Zn/Ti cells in W4D1 and 2 m W electrolytes under a ‘reservoir half-cell’ method. | ||
At a high current of 5.0 mA cm−2 with an areal capacity of 2.5 mA h cm−2, the Zn/Ti cell in W4D1 sustains durability over 600 cycles with a high average CE of 99.8% (Fig. 3c). Moreover, stable Zn plating/stripping plateaus without fluctuations are detected (Fig. S12, ESI‡), suggesting effective suppression of the side reactions on Zn (e.g., H2 evolution and Zn passivation). Such good reversibility significantly stands out among those of the reported Zn anodes (Table S5, ESI‡). Fig. 3d shows the voltage profiles of Zn/Ti cells to further evaluate the reversibility of Zn plating/stripping in W4D1 and 2 m W electrolytes, according to the ‘reservoir half-cell’ protocol.21 The Zn/Ti cell firstly undergoes two cycles of charge/discharge to mitigate the substrate effects (e.g., lattice mismatch and interphase effects etc.) at 1 mA cm−2 with an areal capacity of 1 mA h cm−2 and an upper cut-off voltage of 0.5 V. Subsequently, 1 mA h cm−2 of Zn (Qp) is plated on Ti as a Zn reservoir, which is cycled at a fixed capacity of 0.2 mA h cm−2 (Qc). After n (e.g., 20) cycles, this cell is fully charged to 0.5 V to strip all removable Zn (Qs). The average CE can be calculated by the equation CE = (nQc + Qs)/(nQc + Qp). The W4D1 electrolyte supports significantly more stable plating/stripping and higher average CE than 2 m W (99.2% vs. 82.5%).
Furthermore, Zn/Zn symmetric cells were assembled to investigate Zn plating/stripping behaviors. The thickness of Zn foil is 50 μm (areal capacity: ∼29.2 mA h cm−2). Fig. 4a shows the voltage profiles at 1 mA cm−2 with a discharge/charge time of 0.5 h. The cell with W4D1 exhibits a high cycling stability over 1000 h without any evident voltage hysteresis or overpotential increase, in contrast to those in W8D1 and 2 m W with short lifespans and apparent voltage fluctuations. Enlarged discharge/charge curves are provided in Fig. S13 (ESI‡). The gradually reduced polarization in W4D1 during the initial 50 cycles is ascribed to the electrode activation process, as confirmed by the decrease of charge transfer resistance from electrochemical impedance spectroscopy (EIS, Fig. S14, ESI‡). At 5 mA cm−2 with an areal capacity of 2.5 mA h cm−2 (corresponding to ∼8.5% depth of discharge (DOD)), the durability and reversibility of the Zn anode can be sustained up to 800 h in W4D1 (Fig. 4b and Fig. S15, ESI‡). Fig. 4c shows the rate capability of Zn/Zn cells with W4D1 and W2D1 electrolytes. The W4D1 cell delivers a stable and flat voltage plateau with a smaller increase of overpotential relative to the W2D1 cell (Fig. S16, ESI‡). Remarkably, as the current increases to 20 mA cm−2, the cell sustains a high areal capacity of 10 mA h cm−2 with a considerable DOD of 34.2% and low overpotential of ∼270 mV. The electrochemical performance of the Zn/Zn cell in this H2O + DMC system is competitive in comparison with those of recently reported Zn-based symmetric cells (Table S6, ESI‡).
 | ||
| Fig. 4 Electrochemical test and characterization of Zn anodes in Zn/Zn cells. (a) Comparison of voltage profiles in W4D1, W8D1 and 2 m W electrolytes at 1 mA cm−2. (b) Cycling performance in the W4D1 electrolyte at 5 mA cm−2. (c) Rate capability of Zn/Zn cells with W4D1 and W2D1. (d, e) SEM images (scale bar, 10 μm) and (f) XRD patterns of Zn anodes after 300 cycles. Insets of (d, e) show the optical images of cycled Zn anodes. | ||
Scanning electron microscopy (SEM) imaging demonstrates that the Zn electrode in the W4D1 electrolyte maintains a dense and smooth surface after 300 cycles at 1C (Fig. 4d). Impressively, a dendrite-free and uniform Zn surface is also characterized after 1050 cycles, ensuring the high stability of the Zn anode (Fig. S17, ESI‡). In sharp contrast, the cycled Zn electrode in the 2 m W electrolyte shows an uneven porous structure with cracks and dendrites (Fig. 4e). Moreover, there is no discernible by-product in Zn after 300 cycles in W4D1, as evidenced by the X-ray diffraction (XRD) analysis (Fig. 4f), while, strong XRD signals related to the zinc triflate hydroxide hydrate (ZnxOTfy(OH)2x–y·nH2O, ZOTH) are detected in 2 m W. The formation of ZOTH might follow a similar process to that of zinc sulfate hydroxide hydrate (Zn4SO4(OH)6·5H2O), which has been often observed in aqueous ZnSO4 electrolytes.30,38,46 Specifically, the H2 evolution reaction leads to an increase of OH− concentration in the vicinity of the electrode, triggering the precipitation reaction (xZn2+ + yOTf− + (2x–y)OH− + nH2O = ZnxOTfy(OH)2x–y·nH2O). Furthermore, the hydrophilicity of different electrolytes with respect to the Zn electrode was probed by contact angle measurements at ambient temperature (Fig. S18, ESI‡). The initial contact angle of the W4D1 electrolyte on Zn foil is 58.7°, which drops to 36.2° after 1 min. Much higher contact angles are observed in 2 m W (initial 96.8° and 80.8° after 1 min). In addition, the contact angle of W4D1 on the cycled Zn is further decreased to 24.1° (Fig. S19, ESI‡). The enhanced hydrophilicity would reduce the interfacial free energy between Zn and the electrolyte, favoring the homogeneous Zn2+ plating/stripping process.30,38
Formation and composition of the SEI layer on the Zn anode
To analyze the influence of the Zn2+-solvation structure on the formation mechanism and chemical composition of the SEI layer, DFT calculations were performed by adopting a cation–solvent/anion complex model. The energy levels of the lowest unoccupied molecular orbital (LUMO) of the free DMC solvent and OTf− anion are 0.77 and 2.42 eV, respectively. When Zn2+ is coordinated with DMC and OTf−, their LUMO energy levels are significantly decreased to −2.75 and −2.79 eV, respectively (Fig. 5a). The cation–solvent or cation–anion complexes with a lower LUMO level indicate their easier electron reception from the anode than the free solvent/anion.43,47,48 The predicted reduction potentials of the free DMC solvent (−2.39 V) and free OTf− anion (−2.46 V) are much lower than that of the Zn2+/Zn redox couple (Fig. 5b). Thus, the reductive decomposition of free OTf− (or free DMC) can hardly occur before Zn deposition, resulting in the absence of a protective SEI layer. With cation–solvent/anion coordination, the Zn2+–DMC and Zn2+–OTf− complexes become reductively unstable below 0.47 and 0.14 V (vs. Zn2+/Zn), where Zn2+ reduction will dominate, as further supported by the Mulliken charge analysis (Fig. S20, ESI‡). It is well known that the electron could preferably transfer from the cation to the solvent (or anion) in the cation–solvent (or cation–anion) unit and induce the solvent/anion decomposition, where the SEI could be formed at the electrode interface.37,39,48 Herein, the introduction of the DMC solvent into the aqueous Zn(OTf)2 electrolyte leads to a significant change of the Zn2+ solvation environment from a typical Zn[H2O]62+ (in 2 m W) to Zn2+[H2O]3.9[DMC]0.7[OTf−]1.4 (in W4D1). Accordingly, both coordinated DMC and OTf− participate in the formation of the SEI on the Zn anode in W4D1. | ||
| Fig. 5 SEI characterization on the Zn anode. (a) LUMO energy levels with corresponding LUMO isosurfaces and (b) the predicted reduction potential with geometric structures of the free OTf− anion, free DMC solvent, and Zn2+–OTf− and Zn2+–DMC coordinations from DFT calculations. (c) LSV profiles of W4D1 and 2 m W electrolytes at 5 mV s−1. (d) XPS spectral regions for C 1s, F 1s, S 2p and Zn 2p with corresponding Ar+ sputtering times on the Zn electrode in W4D1 after 10 cycles. Schematic illustration of the surface chemistry on the Zn electrode in (e) W4D1 and (f) 2 m W electrolytes. | ||
To support the above mechanism, LSV measurements were conducted at 5 mV s−1 using a three-electrode configuration employing Ti foil as the working electrode, and Zn foil as the reference and counter electrodes (Fig. 5c and S21, ESI‡). The W4D1 electrolyte exhibits two discernible reduction peaks at ∼0.3 and 0 V (vs. Zn2+/Zn) prior to Zn2+ plating (around −0.14 V), corresponding to the stepwise reduction of the Zn2+–DMC coordination and Zn2+–OTf− coordination, respectively. The difference of the reduction potential between the modeling and experimental data results from the overpotential in the practical cell. Besides, the decomposition of DMC could push back the reduction potential of OTf−. The preferential decomposition of DMC and OTf− before Zn deposition in W4D1 would contribute to the in situ formation of an interphase on the Zn anode. In contrast, a much weaker current response with a broad peak at ∼0.15 V in the 2 m W electrolyte indicates inefficient anion reduction.
The surface composition of the SEI formed on Zn after 10 cycles was analyzed by X-ray photoelectron spectroscopy (XPS, Fig. 5d). Compared with the C 1s spectrum in the 2 m W electrolyte (Fig. S22a, ESI‡), the signals related to organic (ROCO2)2Zn (289.3 eV), inorganic ZnCO3 (290.2 eV), and ethereal (C–O) species (285.8 eV) intensify strongly in W4D1, mainly arising from the decomposition of the Zn2+–DMC complex. The reduction product of the DMC solvent is similar to that of DMC in a Li-based electrolyte.39 The strong signal of C–C/C–H at the top layer mainly comes from the residual DMC and the adventitious carbon adsorbed on the substrate surface. After 25 s sputtering, the inorganic ZnCO3 content increases largely from 4.25% to 39.7%. In the F 1s spectra, besides the –CF3 species (688.8 eV) from either the incomplete reduction of OTf− or the trace salt on the Zn surface, the distinct peak of inorganic ZnF2 at 684.7 eV (ref. 3 and 37) is clearly observed in W4D1, which results from the decomposition of Zn2+–OTf− aggregates. When the top layer of the interphase is removed under Ar+ sputtering, ZnF2 becomes the dominant species. In sharp contrast, no apparent ZnF2 signal can be detected on the cycled Zn anode in 2 m W (Fig. S22b, ESI‡). For the S 2p spectra, the signals of sulfide (e.g., ZnS)35,37 apparently appear as the Ar+ sputtering time increases, further confirming the decomposition of the Zn2+–OTf− coordination and the involvement of decomposed species in the SEI formation. Additionally, the intensifying of the Zn peak coincides with the evolution of C and F signals, suggesting the enrichment of ZnCO3 and ZnF2 components in the inner SEI layer.
Consequently, the unique Zn2+ solvation structure in the W4D1 electrolyte enables the decomposition of Zn2+–DMC and Zn2+–OTf− complexes and is involved in the formation of the SEI layer containing components such as ZnCO3, ZnF2, (ROCO2)2Zn, sulfide, organic compounds and/or their derivatives (Fig. 5e). The in situ generated interphase on the Zn anode in W4D1 suppresses H2 evolution and Zn passivation and favors uniform Zn2+ electrodeposition, thus improving the reversibility and stability of the Zn electrode. In contrast, the 2 m W electrolyte cannot provide stable interface chemistry at the Zn electrode, leading to continuous parasitic reactions. After prolonged cycling, H2 evolution increases the local pH on Zn, thus inducing the by-product generation of zinc triflate hydroxide hydrate (Fig. 5f and S23, ESI‡).
Electrochemical performance of Zn-ion full batteries
We then evaluated the application of this H2O + DMC electrolyte in Zn-ion full batteries composed of a Zn foil anode and different cathode materials (such as V2O5, zinc hexacyanoferrate (ZnHCF), and layered manganese dioxide (δ-MnO2)). Phase and morphology characterization studies of as-prepared V2O5, ZnHCF, and δ-MnO2 are given in Fig. S24–S26 (ESI‡), respectively. Fig. 6a compares the cycling performances of Zn/V2O5 batteries in W4D1, W8D1, and 2 m W electrolytes at 0.2 A g−1. In W4D1, the V2O5 cathode delivers an initial discharge capacity of 210.5 mA h g−1, which increases to 519.1 mA h g−1 with ∼100% CE after 50 cycles. Impressively, the Zn/W4D1/V2O5 system displays a reversible capacity of 512.9 mA h g−1 after 200 cycles (over 1000 h) with a high capacity retention of 98.8%. For comparison, the W8D1 electrolyte with a smaller amount of DMC moderately improves the stability of a Zn/V2O5 battery with 410 mA h g−1 capacity after 100 cycles. As expected, the 2 m W cell displays a rapid capacity decay due to the severe dissolution of active materials. Fig. 6b shows the typical discharge/charge curves of a Zn/V2O5 battery in the W4D1 electrolyte between 0.3 and 1.6 V. The gradual overpotential decrease and capacity increase upon cycling suggest an activation process of V2O5 and an increase of active mass utilization.15,49 An average discharge voltage plateau of 0.75 V is obtained at the 50th cycle, corresponding to an energy density of 415.2 W h kgcathode−1 and surpassing that of Zn/V2O5 batteries in 2 m W and W8D1 electrolytes (Fig. S27 and S28, ESI‡).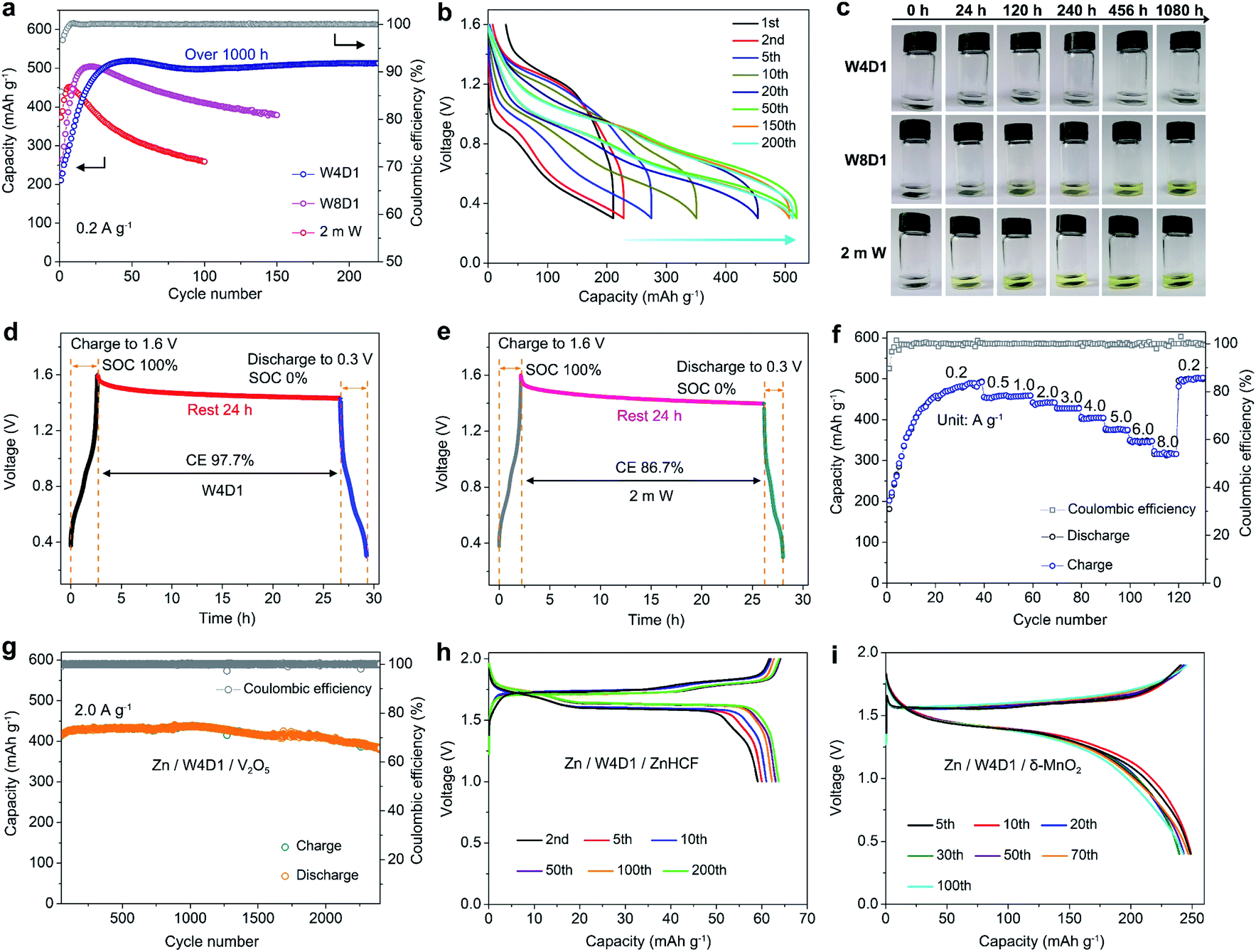 | ||
| Fig. 6 Electrochemical performance of Zn-ion full batteries. (a) Cycling stability of Zn/V2O5 batteries in W4D1, W8D1, and 2 m W electrolytes at 0.2 A g−1. (b) Selected charge/discharge profiles in W4D1. (c) Optical images of V2O5 electrodes immersed in W4D1, W8D1, and 2 m W solutions for different periods. Storage performance comparison of Zn/V2O5 batteries in (d) W4D1 and (e) 2 m W evaluated by resting for 24 h at 100% state of charge (SOC) at 0.2 A g−1, followed by full discharge. (f) Rate capability. (g) Long-term cycling performance of the Zn/W4D1/V2O5 battery at 2.0 A g−1. Typical discharge/charge profiles of the (h) Zn/W4D1/ZnHCF battery at 50 mA g−1 and (i) Zn/W4D1/δ-MnO2 battery at 100 mA g−1. | ||
To identify the solubility of electrode materials in electrolytes, the static soaking experiment was conducted by loading V2O5 electrodes in W4D1, W8D1, and 2 m W for different periods. As shown in Fig. 6c, the W4D1 electrolyte remains colorless and transparent after 1080 h of electrode immersion, whereas W8D1 and 2 m W turn light yellow after 120 h and 24 h, respectively. Additionally, parasitic reactions in Zn/V2O5 cells were assessed by monitoring the open circuit voltage drop of a cell at the fully charged state and at the discharged state after 24 h of storage. A high CE of 97.7% is achieved in W4D1 (Fig. 6d), much higher than that in 2 m W (86.7%, Fig. 6e). These comparative results indicate significant suppression of V2O5 dissolution in the hybrid electrolyte with an organic solvent.
The rate capability of Zn/W4D1/V2O5 was recorded by progressively increasing the current densities from 0.2 to 8.0 A g−1 (Fig. 6f and S29, ESI‡). The first 40 cycles were tested at 0.2 A g−1 to activate the cell. Reversible capacities of 457, 440, 427, 404, 375, and 343 mA h g−1 are delivered at rates of 1.0, 2.0, 3.0, 4.0, 5.0, and 6.0 A g−1, respectively. A high capacity of 319 mA h g−1 is achieved at 8.0 A g−1. The average capacity recovers to 500 mA h g−1 as the rate resumes to 0.2 A g−1. Besides, Fig. 6g shows the long-term cycling performance of the Zn/W4D1/V2O5 cell at 2.0 A g−1. The initial 50 cycles were tested at 0.5 A g−1 (Fig. S30a, ESI‡). A remarkable capacity of 381.9 mA h g−1 is achieved after 2400 cycles. The differential capacity (dQ/dV) curves exhibit two pairs of well overlapped broad peaks (Fig. S30b, c, ESI‡), further indicating the excellent reversibility. Note that the Zn anode (50 μm thickness) in the Zn/W4D1/V2O5 cell is much more excessive, which compromises the whole energy density of the full cell. Thus, we attempted to estimate the battery performance on a more practical basis with a high-mass-loading V2O5 cathode (∼11 mg cm−2) and a thin Zn foil anode (20 μm thickness, ∼14.28 mg cm−2). The assembled cell achieves a reversible capacity of 175 mA h g−1 with an energy density of 131 W h kg−1 based on the total mass of the anode and the cathode, and the DOD of the Zn anode reaches ∼37.7% (Fig. S31, ESI‡).
In addition to V2O5, full batteries pairing a Zn anode with ZnHCF and δ-MnO2 cathodes were assembled to demonstrate the universality of W4D1 for RAZB applications. The ZnHCF-based cell exhibits a high discharge voltage of 1.62 V and a stable capacity of 64 mA h g−1 with a 99% CE after 200 cycles at 50 mA g−1 (Fig. 6h and S32a, ESI‡). The δ-MnO2 cell delivers a reversible capacity of 243 mA h g−1 with an average discharge voltage of 1.35 V after 100 cycles at 100 mA g−1 (Fig. 6i and S32b, ESI‡). At a rate of 500 mA g−1, the capacity retention reaches 89.5% after 500 cycles (Fig. S33, ESI‡). These results indicate the favorable compatibility between the W4D1 electrolyte and ZnHCF or δ-MnO2 cathode materials.
Conclusions
In summary, we report a non-concentrated aqueous electrolyte with the organic DMC additive to stabilize Zn electrochemistry. A Zn2+-solvation structure with the co-contribution of the DMC solvent and OTf− anion is found in the hybrid H2O + DMC electrolyte, as verified by structural and spectroscopic studies and MD simulations. This unique Zn2+-solvation-sheath allows the formation of a ZnF2–ZnCO3-rich SEI layer on the Zn electrode induced by the decomposition of Zn2+–DMC and Zn2+–OTf− coordinations. As a result, parasitic reactions on the Zn anode are effectively suppressed, enabling reversible plating/stripping of Zn with 99.8% CE at 2.5 mA h cm−2 over 600 cycles and dendrite-free cycling of the Zn electrode over 1000 cycles. Furthermore, the formulated electrolyte (W4D1) consisting of 2 mol kg−1 Zn(OTf)2 in 4H2O + 1DMC (molar ratio) features non-flammability and is applicable to support the stable operation of full batteries that couple a Zn anode with diverse cathode materials (e.g., V2O5, MnO2 and ZnHCF). This work provides an alternative avenue to improve the performance of rechargeable Zn-based batteries. The strategy of addition of organic solvents can also inspire the design of efficient electrolytes for other aqueous battery chemistries.Author contributions
N. Z. proposed the concept and supervised the work; Y. D. and N. Z. carried out the experimental design, characterization and electrochemical measurements; Y. D., G. M. and S. D. prepared the cathode materials; L. M. performed the theoretical simulations; N. Z. wrote the manuscript; Y. W., L. W. and J. X. helped to discuss and analyze the data; all authors discussed and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22075067 and 21805066), Natural Science Foundation of Hebei Province (B202001001 and B2019201160), Top Young Talents Project of Hebei Education Department (BJ2019052), Advanced Talents Incubation Program of Hebei University (521000981138), and Chunhui Program of the Ministry of Education of China.References
- N. Zhang, X. Chen, M. Yu, Z. Niu, F. Cheng and J. Chen, Chem. Soc. Rev., 2020, 49, 4203–4219 RSC
.
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS
- L. Ma, T. P. Pollard, Y. Zhang, M. A. Schroeder, M. S. Ding, A. V. Cresce, R. Sun, D. R. Baker, B. A. Helms, E. J. Maginn, C. Wang, O. Borodin and K. Xu, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202017020
- Q. Yang, Q. Li, Z. Liu, D. Wang, Y. Guo, X. Li, Y. Tang, H. Li, B. Dong and C. Zhi, Adv. Mater., 2020, 32, 2001854 CrossRef CAS PubMed
- J. Shin, J. Lee, Y. Park and J. W. Choi, Chem. Sci., 2020, 11, 2028–2044 RSC
- D. Chao, W. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S. Qiao, Sci. Adv., 2020, 6, eaba4098 CrossRef CAS PubMed
- A. Konarov, N. Voronina, J. H. Jo, Z. Bakenov, Y.-K. Sun and S.-T. Myung, ACS Energy Lett., 2018, 3, 2620–2640 CrossRef CAS
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS
- D. Chao, W. Zhou, C. Ye, Q. Zhang, Y. Chen, L. Gu, K. Davey and S.-Z. Qiao, Angew. Chem., Int. Ed., 2019, 58, 7823–7828 CrossRef CAS PubMed
- J. Wang, J.-G. Wang, H. Liu, C. Wei and F. Kang, J. Mater. Chem. A, 2019, 7, 13727–13735 RSC
- L. Ma, S. Chen, C. Long, X. Li, Y. Zhao, Z. Liu, Z. Huang, B. Dong, J. A. Zapien and C. Zhi, Adv. Energy Mater., 2019, 9, 1902446 CrossRef CAS
- R. Trócoli and F. La Mantia, ChemSusChem, 2015, 8, 481–485 CrossRef PubMed
- Y. Yang, Y. Tang, G. Fang, L. Shan, J. Guo, W. Zhang, C. Wang, L. Wang, J. Zhou and S. Liang, Energy Environ. Sci., 2018, 11, 3157–3162 RSC
- C. Xia, J. Guo, Y. Lei, H. Liang, C. Zhao and H. N. Alshareef, Adv. Mater., 2018, 30, 1705580 CrossRef PubMed
- N. Zhang, Y. Dong, M. Jia, X. Bian, Y. Wang, M. Qiu, J. Xu, Y. Liu, L. Jiao and F. Cheng, ACS Energy Lett., 2018, 3, 1366–1372 CrossRef CAS
- P. Hu, T. Zhu, X. Wang, X. Wei, M. Yan, J. Li, W. Luo, W. Yang, W. Zhang, L. Zhou, Z. Zhou and L. Mai, Nano Lett., 2018, 18, 1758–1763 CrossRef CAS PubMed
- A. Khayum M, M. Ghosh, V. Vijayakumar, A. Halder, M. Nurhuda, S. Kumar, M. Addicoat, S. Kurungot and R. Banerjee, Chem. Sci., 2019, 10, 8889–8894 RSC
- Z. Guo, Y. Ma, X. Dong, J. Huang, Y. Wang and Y. Xia, Angew. Chem., Int. Ed., 2018, 57, 11737–11741 CrossRef CAS PubMed
- W. Yang, X. Du, J. Zhao, Z. Chen, J. Li, J. Xie, Y. Zhang, Z. Cui, Q. Kong, Z. Zhao, C. Wang, Q. Zhang and G. Cui, Joule, 2020, 4, 1557–1574 CrossRef CAS
- Q. Zhao, W. Huang, Z. Luo, L. Liu, Y. Lu, Y. Li, L. Li, J. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, eaao1761 CrossRef PubMed
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 CrossRef CAS
- L. Ma, S. Chen, X. Li, A. Chen, B. Dong and C. Zhi, Angew. Chem., Int. Ed., 2020, 59, 23836–23844 CrossRef CAS PubMed
- S.-B. Wang, Q. Ran, R.-Q. Yao, H. Shi, Z. Wen, M. Zhao, X.-Y. Lang and Q. Jiang, Nat. Commun., 2020, 11, 1634 CrossRef PubMed
- Q. Zhang, J. Luan, L. Fu, S. Wu, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 15841–15847 CrossRef CAS PubMed
- Z. Kang, C. Wu, L. Dong, W. Liu, J. Mou, J. Zhang, Z. Chang, B. Jiang, G. Wang, F. Kang and C. Xu, ACS Sustainable Chem. Eng., 2019, 7, 3364–3371 CrossRef CAS
- Y. Zeng, X. Zhang, R. Qin, X. Liu, P. Fang, D. Zheng, Y. Tong and X. Lu, Adv. Mater., 2019, 31, 1903675 CrossRef PubMed
- Q. Yang, Y. Guo, B. Yan, C. Wang, Z. Liu, Z. Huang, Y. Wang, Y. Li, H. Li, L. Song, J. Fan and C. Zhi, Adv. Mater., 2020, 32, 2001755 CrossRef CAS PubMed
- X. Xie, S. Liang, J. Gao, S. Guo, J. Guo, C. Wang, G. Xu, X. Wu, G. Chen and J. Zhou, Energy Environ. Sci., 2020, 13, 503–510 RSC
- H. Yang, Z. Chang, Y. Qiao, H. Deng, X. Mu, P. He and H. Zhou, Angew. Chem., Int. Ed., 2020, 59, 9377–9381 CrossRef CAS PubMed
- J. Hao, X. Li, S. Zhang, F. Yang, X. Zeng, S. Zhang, G. Bo, C. Wang and Z. Guo, Adv. Funct. Mater., 2020, 30, 2001263 CrossRef CAS
- Y. Dong, S. Di, F. Zhang, X. Bian, Y. Wang, J. Xu, L. Wang, F. Cheng and N. Zhang, J. Mater. Chem. A, 2020, 8, 3252–3261 RSC
- A. Naveed, H. Yang, J. Yang, Y. Nuli and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 2760–2764 CrossRef CAS PubMed
- C. Zhang, J. Holoubek, X. Wu, A. Daniyar, L. Zhu, C. Chen, D. P. Leonard, I. A. Rodríguez-Pérez, J.-X. Jiang, C. Fang and X. Ji, Chem. Commun., 2018, 54, 14097–14099 RSC
- R. Qin, Y. Wang, M. Zhang, Y. Wang, S. Ding, A. Song, H. Yi, L. Yang, Y. Song, Y. Cui, J. Liu, Z. Wang, S. Li, Q. Zhao and F. Pan, Nano Energy, 2021, 80, 105478 CrossRef CAS
- L. Cao, D. Li, E. Hu, J. Xu, T. Deng, L. Ma, Y. Wang, X.-Q. Yang and C. Wang, J. Am. Chem. Soc., 2020, 142, 21404–21409 CrossRef PubMed
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef PubMed
- H. Qiu, X. Du, J. Zhao, Y. Wang, J. Ju, Z. Chen, Z. Hu, D. Yan, X. Zhou and G. Cui, Nat. Commun., 2019, 10, 5374 CrossRef PubMed
- J. Hao, L. Yuan, C. Ye, D. Chao, K. Davey, Z. Guo and S. Qiao, Angew. Chem., Int. Ed., 2021, 60, 7366–7375 CrossRef PubMed
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef PubMed
- J.-B. Brubach, A. Mermet, A. Filabozzi, A. Gerschel, D. Lairez, M. P. Krafft and P. Roy, J. Mater. Chem. B, 2001, 105, 430–435 Search PubMed
- J. Zhao, J. Zhang, W. Yang, B. Chen, Z. Zhao, H. Qiu, S. Dong, X. Zhou, G. Cui and L. Chen, Nano Energy, 2019, 57, 625–634 CrossRef
- N. Chang, T. Li, R. Li, S. Wang, Y. Yin, H. Zhang and X. Li, Energy Environ. Sci., 2020, 13, 3527–3535 RSC
- F. Wang, O. Borodin, M. S. Ding, M. Gobet, J. Vatamanu, X. Fan, T. Gao, N. Eidson, Y. Liang, W. Sun, S. Greenbaum, K. Xu and C. Wang, Joule, 2018, 2, 927–937 CrossRef
- O. Borodin, M. Olguin, P. Ganesh, P. R. C. Kent, J. L. Allen and W. A. Henderson, Phys. Chem. Chem. Phys., 2016, 18, 164–175 RSC
- L. Suo, O. Borodin, Y. Wang, X. Rong, W. Sun, X. Fan, S. Xu, M. A. Schroeder, A. V. Cresce, F. Wang, C. Yang, Y.-S. Hu, K. Xu and C. Wang, Adv. Energy Mater., 2017, 7, 1701189 CrossRef
- Y. Dong, M. Jia, Y. Wang, J. Xu, Y. Liu, L. Jiao and N. Zhang, ACS Appl. Energy Mater., 2020, 3, 11183–11192 CrossRef
- X. Chen, X. Shen, T.-Z. Hou, R. Zhang, H.-J. Peng and Q. Zhang, Chem, 2020, 6, 2242–2256 Search PubMed
- Z. Hou, M. Dong, Y. Xiong, X. Zhang, Y. Zhu and Y. Qian, Adv. Energy Mater., 2020, 10, 1903665 CrossRef
- N. Zhang, M. Jia, Y. Dong, Y. Wang, J. Xu, Y. Liu, L. Jiao and F. Cheng, Adv. Funct. Mater., 2019, 29, 1807331 CrossRef
Footnotes |
| † Dedicated to the 100th anniversary of Hebei University. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06734b |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |