DOI:
10.1039/D1SC01375K
(Review Article)
Chem. Sci., 2021,
12, 6800-6819
Recent advances in single atom catalysts for the electrochemical carbon dioxide reduction reaction
Received
9th March 2021
, Accepted 7th April 2021
First published on 26th April 2021
Abstract
The electrochemical carbon dioxide reduction reaction (CO2RR) offers a promising solution to mitigate carbon emission and at the same time generate valuable carbonaceous chemicals/fuels. Single atom catalysts (SACs) are encouraging to catalyze the electrochemical CO2RR due to the tunable electronic structure of the central metal atoms, which can regulate the adsorption energy of reactants and reaction intermediates. Moreover, SACs form a bridge between homogeneous and heterogeneous catalysts, providing an ideal platform to explore the reaction mechanism of electrochemical reactions. In this review, we first discuss the strategies for promoting the CO2RR performance, including suppression of the hydrogen evolution reaction (HER), generation of C1 products and formation of C2+ products. Then, we summarize the recent developments in regulating the structure of SACs toward the CO2RR based on the above aspects. Finally, several issues regarding the development of SACs for the CO2RR are raised and possible solutions are provided.
 Jincheng Zhang | Jincheng Zhang is currently a PhD student at Nanyang Technological University. His research interest focuses on coordination engineering of single-atom catalysts. |
 Hongbin Yang | Hongbin Yang is a Professor at Suzhou University of Science and Technology. He received his B.S. (1998) from Lanzhou University and PhD (2008) from Fudan University. His active research interests include electrocatalysis, photo-electrochemistry and carbon-based catalysts. |
 Bin Liu | Bin Liu received his B.Eng. (1st Class Honors) and M.Eng. degrees in Chemical Engineering from the National University of Singapore and obtained his PhD degree in Chemical Engineering from the University of Minnesota in 2011. Thereafter, he moved to the University of California, Berkeley, and worked as a postdoctoral researcher in the Department of Chemistry from 2011 to 2012 before joining the School of Chemical and Biomedical Engineering at Nanyang Technological University as an Assistant Professor in 2012. He is now an Associate Professor at NTU. His main research interests are electrocatalysis, photovoltaics and photoelectrochemistry. |
1. Introduction
Excessive emission of CO2 resulting from overconsumption of fossil fuels causes the green-house effect. The CO2RR not only provides a solution to reduce CO2 concentration in the atmosphere, but also is able to simultaneously produce valuable carbonaceous chemicals/fuels.1,2 Electrocatalytic reduction of CO2 driven by renewable electricity is environmentally friendly, and has become a hotspot in the field of CO2 conversion. The CO2RR involves very complex reactions, coupled with multiple proton and electron transfer steps,3 which can convert CO2 into carbon monoxide (CO), methane (CH4), formic acid (HCOOH), methanol (CH3OH), ethylene (C2H4), ethanol (C2H5OH), etc:| | | CO2 + 2H+ + 2e− → HCOOH | (1) |
| | | CO2 + 2H+ + 2e− → CO + H2O | (2) |
| | | CO2 + 6H+ + 6e− → CH3OH + H2O | (3) |
| | | CO2 + 8H+ + 8e− → CH4 + 2H2O | (4) |
| | | 2CO2 + 12H+ + 12e− → C2H4 + 4H2O | (5) |
| | | 2CO2 + 12H+ + 12e− → C2H5OH + 3H2O | (6) |
Many catalysts have been explored to break the robust C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, such as sulfides,4–6 phosphides,7,8 oxides,9–13 and so on. However, the performance of the CO2RR is still unsatisfactory, showing low activity and poor product selectivity,14,15 especially for liquid C2+ products. Therefore, developing highly active catalysts with good selectivity and stability is crucial for promoting this clean energy conversion technology.16–18
O bond, such as sulfides,4–6 phosphides,7,8 oxides,9–13 and so on. However, the performance of the CO2RR is still unsatisfactory, showing low activity and poor product selectivity,14,15 especially for liquid C2+ products. Therefore, developing highly active catalysts with good selectivity and stability is crucial for promoting this clean energy conversion technology.16–18
Since Qiao et al.19 developed the Pt1/FeOx single atom catalyst (SAC) in 2011, SACs have received considerable attention. SACs usually consist of a metal center coordinated with non-metal atoms. Because most of the metal sites can be exposed at the catalyst's surface and they possess a unique electronic structure, SACs show high intrinsic catalytic activity.20–23 Additionally, SACs are ideal models for elucidating the structure–activity relationship because of their homogeneous atomically-dispersed active sites.24–27 Furthermore, coupled with in situ characterization techniques, the structural evolution of active sites in SACs can be probed, which helps to further design next-generation catalysts.28,29 Recently, the application of SACs achieved significant advancement in the field of electrochemical reactions.7,30 In the electrochemical CO2RR, SACs exhibit encouraging performance in terms of catalytic activity, product selectivity, and stability.31–33Fig. 1 summarizes the reported elements in SACs for the electrochemical CO2RR.
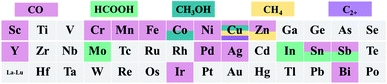 |
| | Fig. 1 The reported elements in SACs for the electrochemical CO2RR. | |
The metal elements used to construct SACs for the electrochemical CO2RR include three types, transition metal (Mo, Fe, Co, Ni, Cu, and Zn), noble metal (Pd, Ag and Ir) and p-block (Sn, Sb and Bi) elements (Fig. 1). Most of the reported SACs electrochemically reduce CO2 to CO, especially TM-SACs. However, it is also reported that Co, Cu, Mo, Sn and Sb SACs can reduce CO2 to liquid products (e.g., HCOOH, CH3OH, etc.). As shown in Fig. 1, for the same metal SAC, the reported major CO2RR products are different, for example, Co-SAC can reduce CO2 to CO or CH3OH, and Sn and Sb-SAC can reduce CO2 to CO or HCOOH. The determining factor for product selectivity is still under controversy. Therefore, to clearly elucidate the structure–activity relationship in the CO2RR and further develop SACs with higher product selectivity, especially for C2+ products, a systematic review of previous studies of SACs in the CO2RR is urgently needed. Fig. 2 shows the development of SAC-based electrocatalysts for the CO2RR.
 |
| | Fig. 2 The development of SAC-based electrocatalysts for the CO2 RR. | |
In this review, the strategies to enhance the CO2RR performance are first outlined, including the suppression of the HER, and the generation of C1 products and C2+ products. Then, recent advances in promoting CO2RR performance using SACs are summarized. Finally, some challenges in this field are proposed and possible solutions are provided at the end.
2. Strategies to improve the CO2RR
2.1. Suppression of the HER
The HER is the main side reaction competing with the CO2RR. The HER can proceed via two mechanisms, the Volmer–Heyrovsky and the Volmer–Tafel mechanism.34–36 In the Volmer step, a proton from the electrolyte reacts with an electron to form an adsorbed hydrogen on the catalyst surface (H*). If the second step is the Heyrovsky reaction, the H* will directly react with a proton and another electron to generate H2. If the Tafel reaction happens, another proton combines with another electron to form a second H*, which will couple with the first H* to generate H2.| | | H* + H+ + e− → H2 Heyrovsky | (8) |
The Tafel route requires two adjacent active sites to adsorb two H*, which is difficult in the case of SACs. So, the Heyrovsky mechanism is more plausible for the HER on SACs. Previous studies have shown that the activation energy of the Heyrovsky step is typically two times that of the Tafel step. Consequently, the HER can be considerably suppressed on SACs compared to their nanoparticle counterparts. A similar principle is also applicable to the CO2RR, where the Heyrovsky mechanism is more plausible.
| | | CO2 + H+ + e− → COOH* | (10) |
| | | COOH* + H* → CO + H2O Tafel | (11) |
| | | COOH* + H+ + e− → CO + H2O Heyrovsky | (12) |
The difference between the thermodynamic limiting potentials (UL(CO2) – UL(H2)) is a descriptor to indicate the selectivity of the CO2RR (UL(CO2) and UL(H2) are the thermodynamic limiting potentials for the CO2RR and HER, respectively). The larger the value, the higher the selectivity of CO.
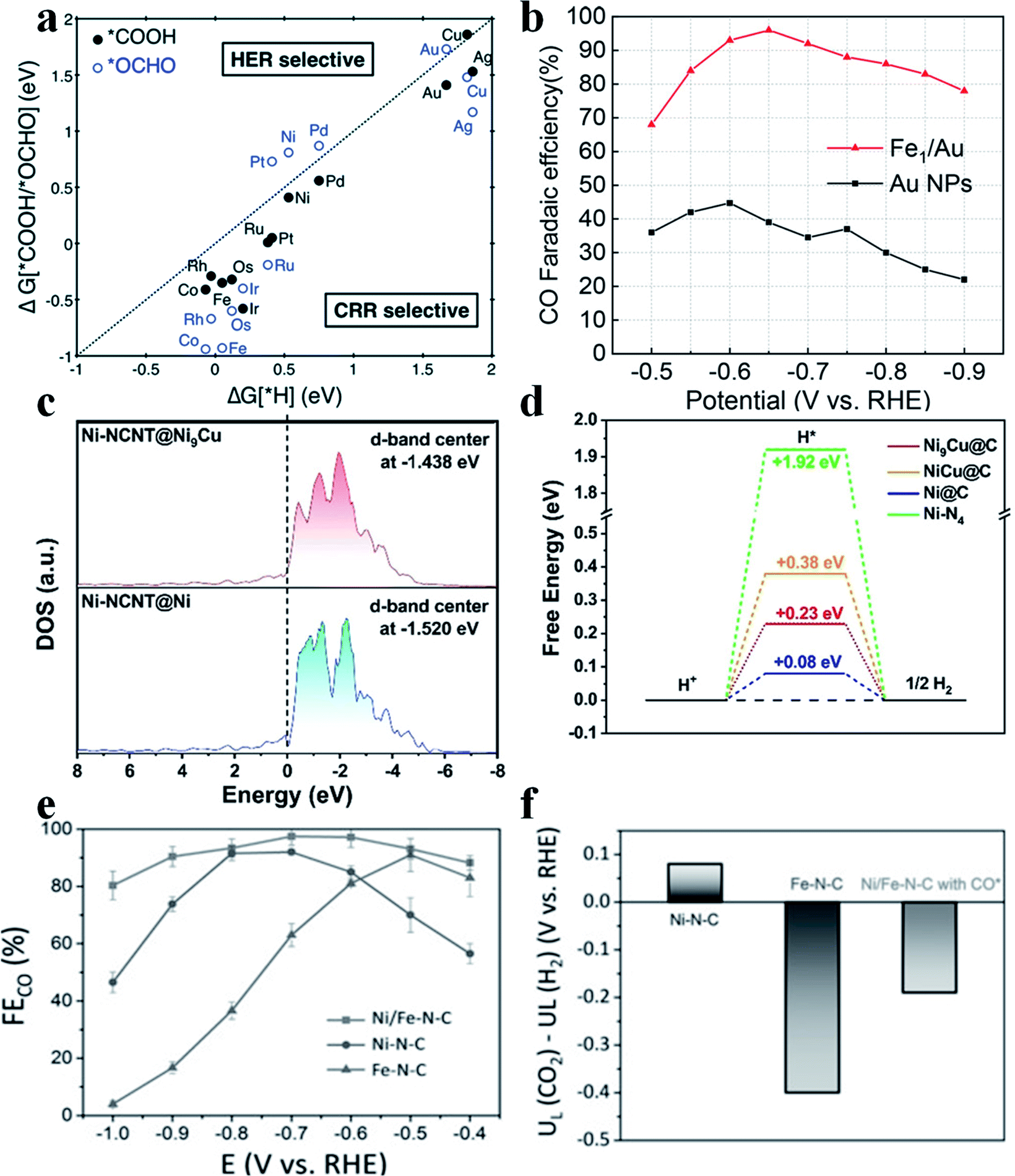
The HER is the main side reaction competing with the CO2RR, which needs to be suppressed so as to improve the selectivity of the CO2RR. Jung et al. calculated the adsorption energy of H (ΔG(H*)), *COOH and *OCHO (ΔG[*COOH/*OCHO]) and found that most SACs possess a ΔG(H*) larger than the ΔG[*COOH/*OCHO], suggesting that the CO2RR was more favorable than the HER on most SACs (Fig. 3a).7 Although the HER is intrinsically unfavorable on SACs, the selectivity towards CO2RR products still needs to be further improved. Yao et al. reported that by introducing Fe atoms into Au nanoparticles (Au NPs), the HER could be effectively suppressed, which greatly enhanced the CO faradaic efficiency (FE) in the CO2RR.37 Furthermore, the introduced Fe atoms bound with O atoms in CO2, while the nearby Au atoms bound with C atoms in CO2. Such a synergistic effect boosted the performance of Fe/Au (Fig. 3b). Other metal elements have also been studied to constrain the HER in SACs. Liu et al. introduced Cu into Ni SAC to form Ni(I)-NCNT@Ni9Cu to tune the adsorption energy of H*. Electrochemical results showed that Ni(I)-NCNT@Ni9Cu delivered a specific current density of −32.87 mA cm−2 and a TOF of 1962 h−1 at an overpotential of 620 mV for generating CO with 97% FECO. DFT calculations found that the d-band center was upshifted from −1.520 eV to −1.438 eV after the introduction of Cu, which strengthened the adsorption of H* and thus further impeded the HER (Fig. 3c and d).38 The HER dominates at high overpotentials in single Fe atom catalysts. Zhao et al. introduced single Ni atoms into Fe-SAC and found that such a dual atom center could efficiently regulate the adsorption energy of H*39 to suppress the HER and thus promote FECO (Fig. 3e and f).
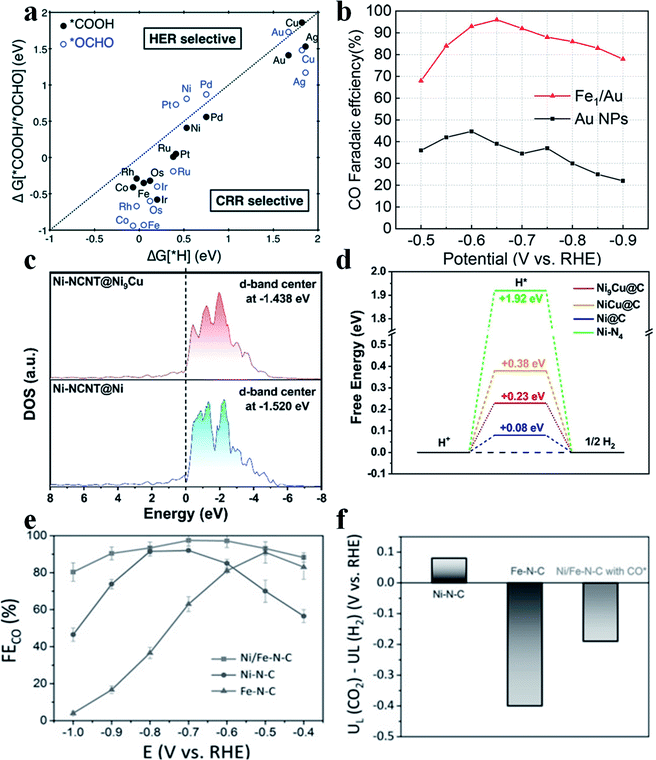 |
| | Fig. 3 (a) Free energy change of the first protonation step in the CO2RR and HER on various SACs. Catalysts below the dotted parity line are CO2RR selective. Reproduced with permission from ref. 7. Copyright (2017) The Royal Society of Chemistry. (b) CO faradaic efficiency of Fe1/Au, compared with Au NPs, at different applied cathodic potentials. Reproduced with permission from ref. 37. Copyright (2020) American Chemical Society. (c) Calculated DOS for NiI-NCNT@Ni and NiI-NCNT@Ni9Cu. (d) The calculated free-energy diagram of the HER on the (111) surface of Ni@C, Ni9Cu@C, NiCu@C, and Ni–N4 sites. Reproduced with permission from ref. 38. Copyright (2020) Wiley-VCH. (e) FECO of Ni/Fe-N-C, Ni-N-C and Fe-N-C at various applied potentials. (f) Difference in limiting potentials for the CO2RR and HER for different catalysts. Reproduced with permission from ref. 39. Copyright (2019) Wiley-VCH. | |
Another issue in the electrochemical CO2RR is that the solubility of CO2 in aqueous electrolyte is typically low (34 mM),40 and the reactant diffusion limitation restricts the reaction rate of the CO2RR particularly at high overpotentials, while the HER becomes more competitive. To tackle this challenge, many strategies have been developed, such as increasing the device pressure,41 lowering the reaction temperature,42 adopting a flow-cell device,43 and using a solid electrolyte.44
2.2. Generation of C1 products
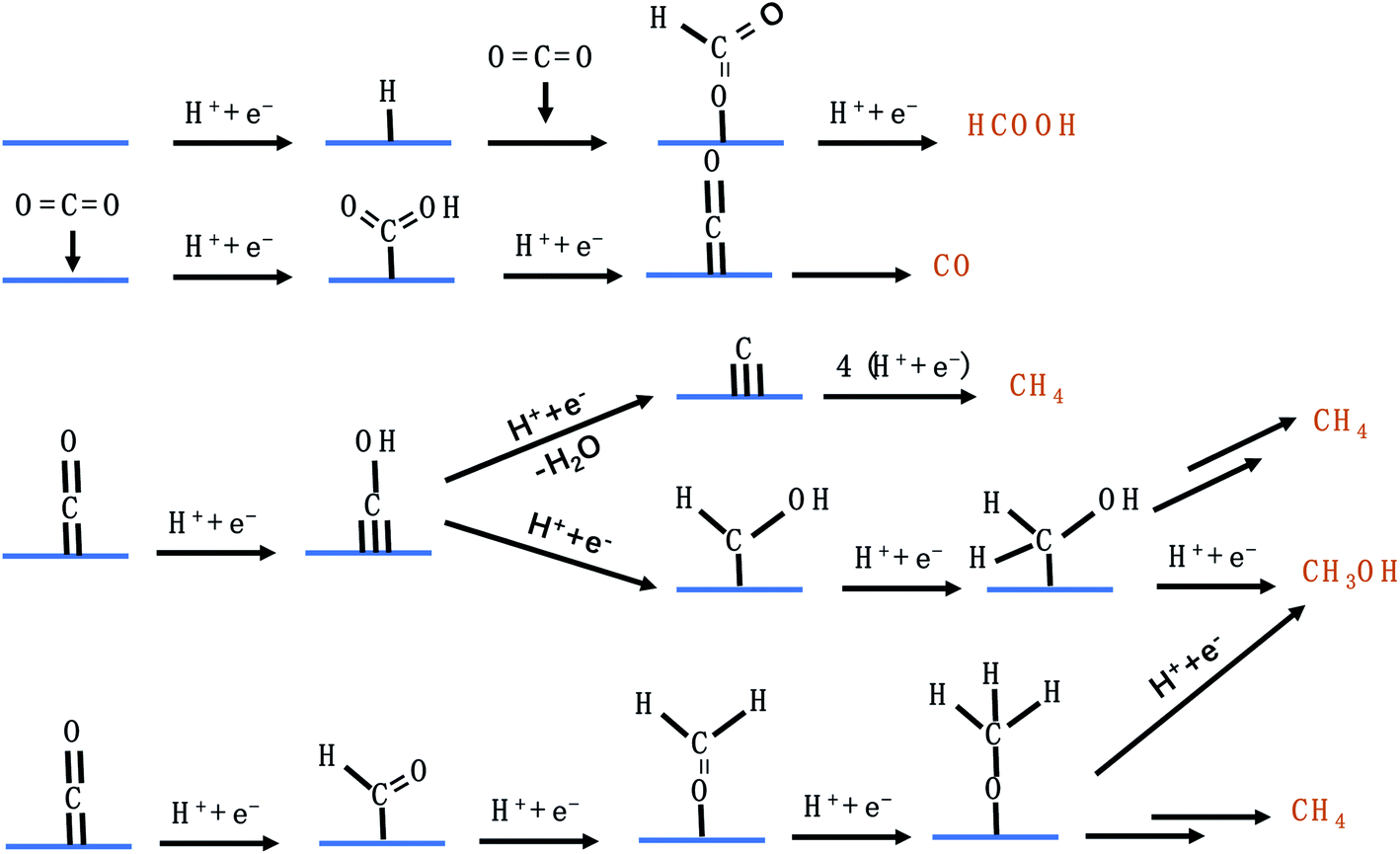
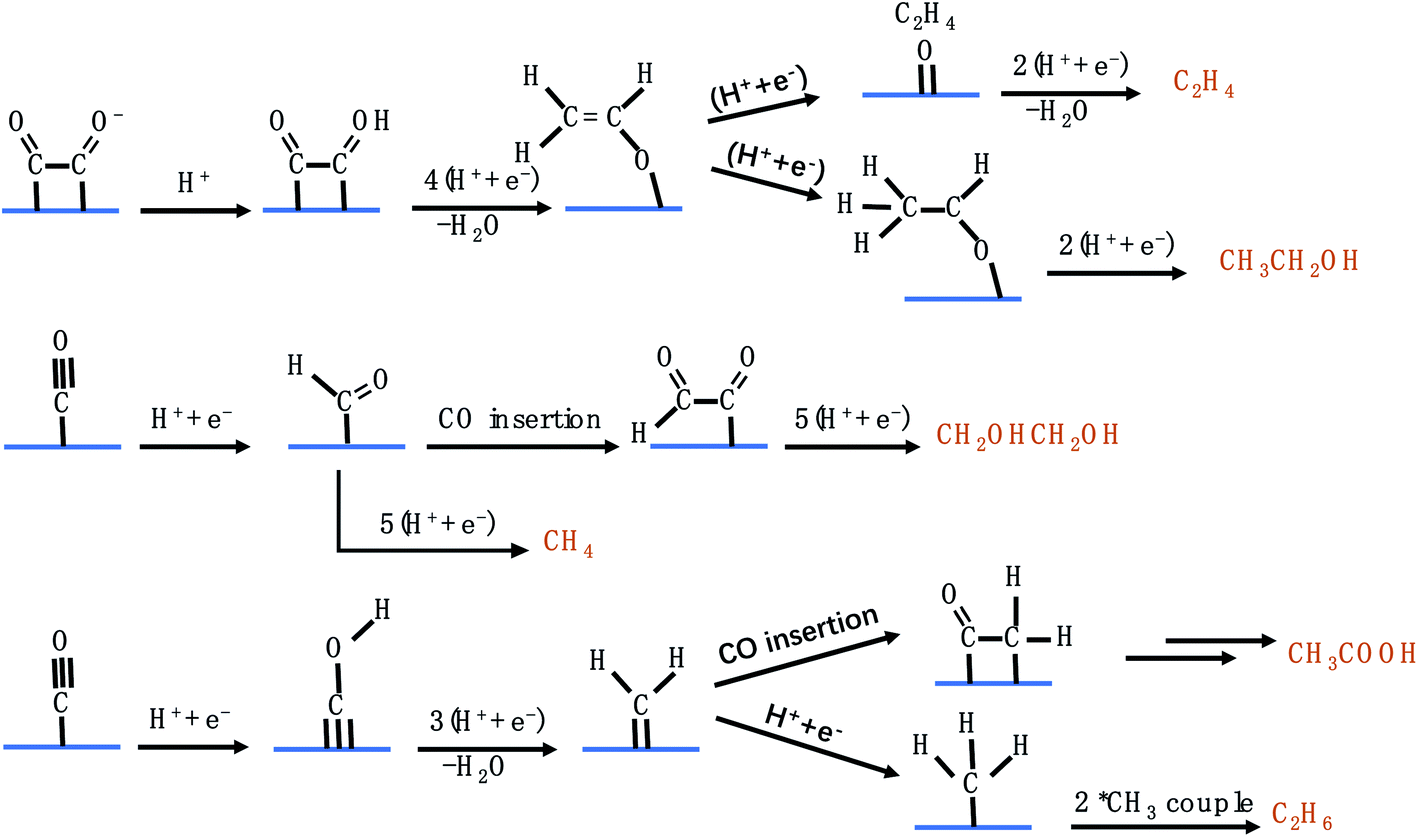
C1 products from the CO2RR include CO, HCOO−, CH3OH, and CH4. Fig. 4 lists the possible pathways for the C1 products. To reduce CO2 to CO, *COOH is first generated on the catalyst's surface via a proton coupled electron transfer (PCET) step, followed by *CO formation through dehydrogenation of *COOH, which is finally desorbed from the catalyst to release CO.45 If the catalyst adsorbs *CO very strongly, the catalytically active sites will be poisoned and the HER becomes dominant. If the catalyst adsorbs *CO loosely, *CO will be desorbed from the catalyst surface to generate CO. Only when the catalytic surface adsorbs *CO neither too strongly nor too weakly, further reduction reaction can take place. HCOO− is formed via the *OCHO intermediate, which has a medium adsorption energy.46 *OCHO can be bound to the catalyst surface via one or two oxygen atoms. As there are no neighbouring active sites, *OCHO is more likely to bind to the SAC via only one O atom as shown in Fig. 4. Both *CHO and *COH pathways are able to produce CH4, forming *CHOH and then *CH2OH and finally branching into two routes (CH3OH and CH4).47–51 *C is formed via dehydration of *COH and constitutes another pathway to generate CH4. Carbon deposition covered on the copper catalyst after the CO2RR confirmed the existence of *C. Furthermore, *CH3O formed via protonation of *CHO is an important intermediate for CH4 and CH3OH.50,52–56 It is worth mentioning that the energy barriers for both pathways (*CH2OH and *CH3O) to produce CH3OH are high, making CH3OH generation rather difficult.
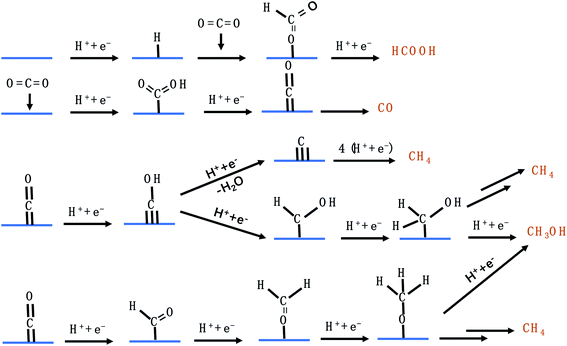 |
| | Fig. 4 The possible CO2RR pathways for C1 products. | |
2.3. Generation of C2 products
C2 products from the CO2RR include C2H4, C2H5OH, C2H6, CH3COOH, C2H6O2, etc. Several protonation and electron transfer steps engage in the formation of C2 products, making the reactions very sophisticated. As many possible pathways are capable of generating the same product, this leads to controversy about the reaction mechanisms.
Fig. 5 shows the possible CO2RR pathways for C2 products. In the first route, CH2CHO* acts as an important intermediate to produce C2H4 and CH3CH2OH,57 which can branch into two pathways. One transforms to *C2H4O, which dissociates and releases C2H4. The other forms CH3CHO* which is further reduced to generate CH3CH2OH. It is noteworthy that the calculated energy barrier for CH3CHO* → CH3CH2OH is 0.2 eV, which is higher than that for CH2CHO* → *O + C2H4, explaining the higher selectivity for C2H4 than for CH3CH2OH among different C2 products.58–62 In the second route, starting from *CO, CH2OHCH2OH can be formed through CO insertion into *CHO coupled with several protonation and electron transfer steps. *CHO can also undergo another pathway to produce CH4. The pathway towards CH4 production has a lower energy barrier than that towards CH2OHCH2OH production, disclosing that very little CH2OHCH2OH can be detected. In the third route, *CH2, originating from *CO, serves as an important intermediate to produce CH3COOH and C2H6.63–66 As discussed above, the C2 pathways involve two close active sites to adsorb intermediates, which are severely limited in SACs. Consequently, very few groups have reported the generation of C2 products on SACs via the CO2RR.
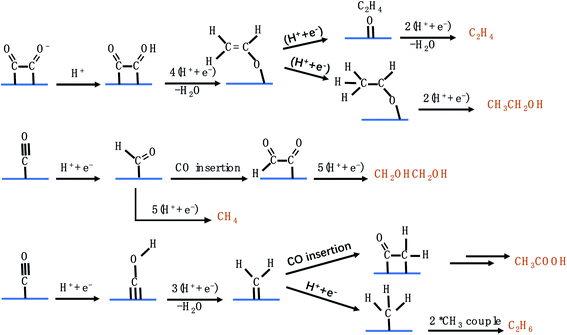 |
| | Fig. 5 The possible CO2RR pathways for C2 products. | |
3. SACs for the CO2RR
3.1. Advantages of SACs
Compared with traditional catalysts, the metal single atom sites in SACs locate at the catalyst's surface. Such an attribute makes SACs possess maximum atom utilization efficiency and thus exhibit excellent catalytic performance. Furthermore, the metal centers in SACs can be influenced by the near-range coordination environment and long-range interactions. Therefore, the electronic structure of SACs is tunable, which helps to improve the selectivity of the CO2RR. Also, SACs are ideal models for exploring the structure–activity relationship because of their homogeneous atomically-dispersed metal active centers.
3.2. CO2 to CO
Various strategies have been developed to promote the catalytic performance of SACs for the CO2RR to CO, including regulation of the center metal atom and manipulation of the coordination environment of the center metal atom. Recently, great achievements have been made in the production of CO from CO2 on SACs. But, for commercial applications, further improvements in intrinsic catalytic activity and stability are needed. In this section, the developed strategies to promote the production of CO on SACs via the CO2RR are summarized.
3.2.1. Modulation of the metal atomic center for boosting CO2 reduction to CO.
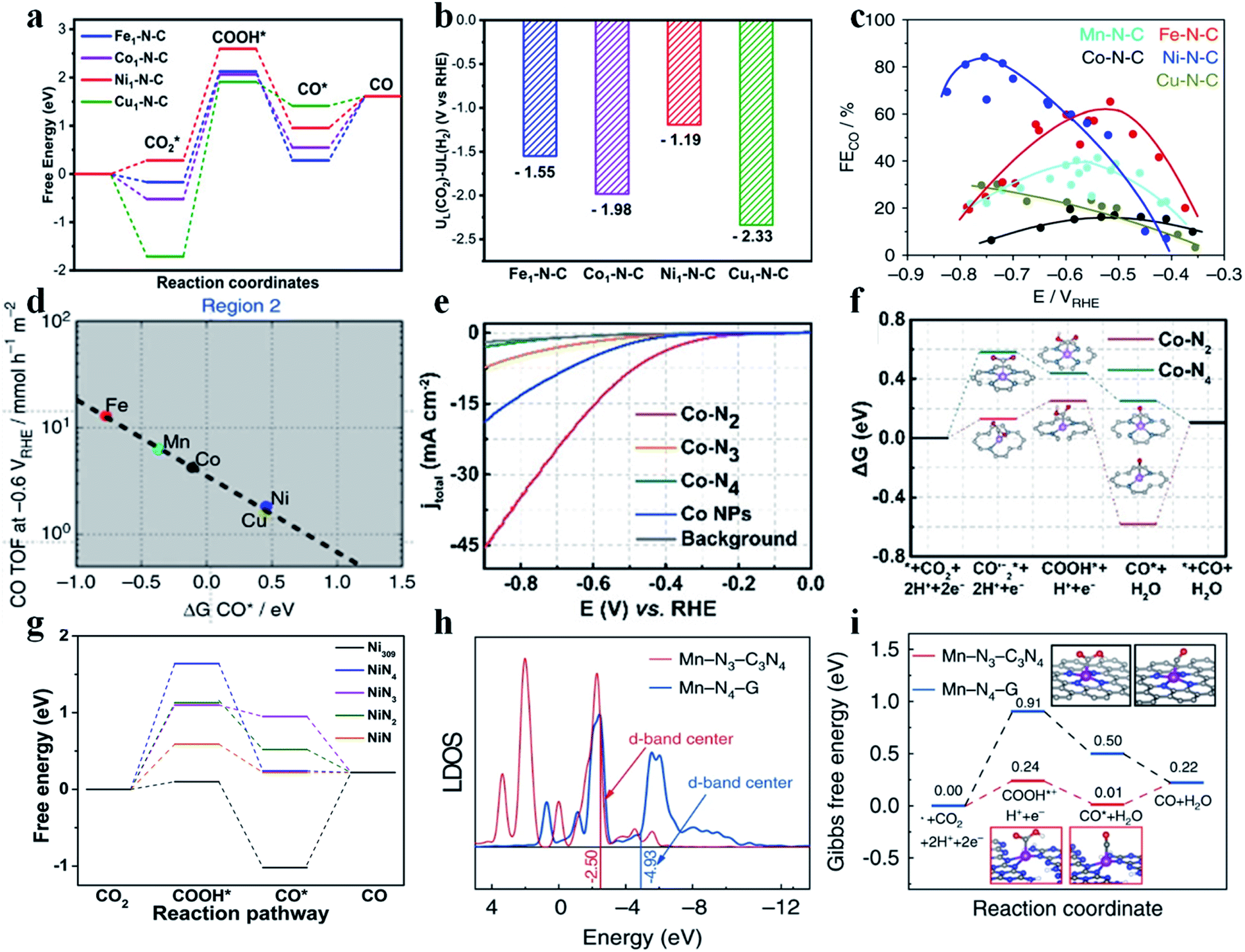
SACs with different metal centers possess different d-band centers, which determine their catalytic performance in the CO2RR. Typically, the higher the d-band center is, the stronger the SAC binds with the intermediates. Jiang et al. synthesized various SACs implanted in N-doped carbon (M1-N-C; M = Fe, Co, Ni, and Cu) by calcining metal–organic frameworks (MOF).67 Aberration-corrected high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images showed sparsely distributed bright dots, and Fourier transformed extended X-ray absorption fine structure (FT-EXAFS) spectra confirmed the absence of M–M bonds in all SACs, indicating that the metal atoms were atomically dispersed. In the N K-edge near-edge X-ray absorption fine structure (NEXAFS) spectra, a peak was found broadened and split into a doublet (a1 and a2), revealing that the metal atoms were stabilized by pyridinic N. EXAFS fitting results further showed that the metal centers in all SACs were coordinated with four pyridinic N atoms. Besides, all SACs possessed a similar N content and metal loading amount (about 4.94 at% and 1.75 wt%, respectively). Electrochemical results exhibited that Ni1-N-C had the best activity and selectivity towards CO production, followed by Fe1-N-C, Co1-N-C, and Cu1-N-C. Specifically, the TOF and CO partial current density of Ni1-N-C reached 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 315 h−1 and 27 mA cm−2, respectively, at −0.8 V vs. RHE. Ni1-N-C displayed a much smaller Tafel slope (98 mV per decade) than the other SACs (104 mV per decade for Fe1-N-C, 142 mV per decade for Co1-N-C, and 118 mV per decade for Cu1-N-C). Additionally, Ni1-N-C also showed long-term durability at −0.8 V vs. RHE for 10 h without obvious attenuation in terms of current density and FECO. Density functional theory (DFT) calculations were performed to explain the differences among these SACs. As stated above, all SACs have similar structural parameters except for the metal centers, and offer an ideal model to study the role of metal atomic centers in CO2RR performance. The calculated free energy diagram revealed that the formation of *COOH was the rate-determining step (RDS) for all SACs. Ni1-N-C and Fe1-N-C had a similar energy barrier for *COOH formation, lower than that of Co1-N-C and Cu1-N-C. Besides, the energy barrier for CO desorption on Ni1-N-C was lower than that on Fe1-N-C, suggesting favorable CO release from Ni1-N-C (Fig. 6a). The UL(CO2) – UL(H2) values for Ni1-N-C, Fe1-N-C, Co1-N-C and Cu1-N-C are −1.19 eV, −1.55 eV, −1.98 eV, and −2.33 eV, respectively. The most positive value of UL(CO2) – UL(H2) for Ni1-N-C supported the conclusion that Ni1-N-C had the highest selectivity towards CO production (Fig. 6b). Peter et al. also prepared a series of transition metal SACs (Fe-N-C, Co-N-C, Ni-N-C, Cu-N-C, and Mn-N-C) and studied their CO2RR performance.68 The results demonstrated that Fe-N-C produced the most CO at low overpotentials, while Ni-N-C produced the most CO at high overpotentials (Fig. 6c). Fe-N-C, Mn-N-C, and Co-N-C all bind to CO* very strongly such that desorption of CO becomes the RDS for these SACs. Ni-N-C and Cu-N-C display a weak binding to *COOH and thus they require higher overpotentials to initiate the CO2RR. Interestingly, a small amount of CH4 was detected in the CO2RR catalyzed on Fe-N-C and Mn-N-C due to their strong adsorption towards CO*, indicating their potential for producing CH4 (Fig. 6d). Other studies also attained a similar consensus that the Ni SAC possessed the highest activity and selectivity for the CO2RR to CO among different SACs.69–73
315 h−1 and 27 mA cm−2, respectively, at −0.8 V vs. RHE. Ni1-N-C displayed a much smaller Tafel slope (98 mV per decade) than the other SACs (104 mV per decade for Fe1-N-C, 142 mV per decade for Co1-N-C, and 118 mV per decade for Cu1-N-C). Additionally, Ni1-N-C also showed long-term durability at −0.8 V vs. RHE for 10 h without obvious attenuation in terms of current density and FECO. Density functional theory (DFT) calculations were performed to explain the differences among these SACs. As stated above, all SACs have similar structural parameters except for the metal centers, and offer an ideal model to study the role of metal atomic centers in CO2RR performance. The calculated free energy diagram revealed that the formation of *COOH was the rate-determining step (RDS) for all SACs. Ni1-N-C and Fe1-N-C had a similar energy barrier for *COOH formation, lower than that of Co1-N-C and Cu1-N-C. Besides, the energy barrier for CO desorption on Ni1-N-C was lower than that on Fe1-N-C, suggesting favorable CO release from Ni1-N-C (Fig. 6a). The UL(CO2) – UL(H2) values for Ni1-N-C, Fe1-N-C, Co1-N-C and Cu1-N-C are −1.19 eV, −1.55 eV, −1.98 eV, and −2.33 eV, respectively. The most positive value of UL(CO2) – UL(H2) for Ni1-N-C supported the conclusion that Ni1-N-C had the highest selectivity towards CO production (Fig. 6b). Peter et al. also prepared a series of transition metal SACs (Fe-N-C, Co-N-C, Ni-N-C, Cu-N-C, and Mn-N-C) and studied their CO2RR performance.68 The results demonstrated that Fe-N-C produced the most CO at low overpotentials, while Ni-N-C produced the most CO at high overpotentials (Fig. 6c). Fe-N-C, Mn-N-C, and Co-N-C all bind to CO* very strongly such that desorption of CO becomes the RDS for these SACs. Ni-N-C and Cu-N-C display a weak binding to *COOH and thus they require higher overpotentials to initiate the CO2RR. Interestingly, a small amount of CH4 was detected in the CO2RR catalyzed on Fe-N-C and Mn-N-C due to their strong adsorption towards CO*, indicating their potential for producing CH4 (Fig. 6d). Other studies also attained a similar consensus that the Ni SAC possessed the highest activity and selectivity for the CO2RR to CO among different SACs.69–73
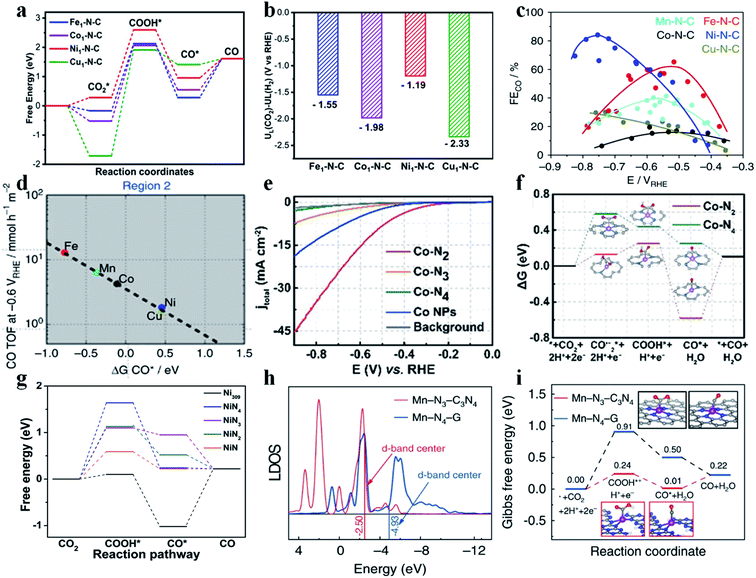 |
| | Fig. 6 (a) Calculated free energy diagram of CO2 reduction to CO and (b) the values of UL(CO2) – UL(H2) for various M1-N-C catalysts. Reproduced with permission from ref. 67. Copyright (2020) Wiley-VCH. (c) FECO for various SACs at different potentials. (d) TOFCO at −0.6 V vs. RHE as a function of CO free adsorption energy. Reproduced with permission from ref. 68. Copyright (2017) Springer Nature. (e) LSV curves of Co–N2, Co–N3, Co–N4, and Co NPs and pure carbon paper as the background. (f) Calculated Gibbs free energy diagram for CO2 electroreduction to CO on Co–N2 and Co–N4. Reproduced with permission from ref. 74. Copyright (2018) Wiley-VCH. (g) Free energy diagram for conversion of CO2 to CO on a Ni-SAC and Ni309 cluster at U = 0 V vs. RHE. Reproduced with permission from ref. 76. Copyright (2019) Elsevier. (h) Local density of states (LDOS) of Mn in Mn-N3-C3N4 and Mn-N4-G. (i) Calculated Gibbs free energy diagram for the CO2RR over Mn-N3-C3N4 and Mn-N4-G. Reproduced with permission from ref. 77. Copyright (2020) Springer Nature. | |
3.2.2. Effect of coordination number on CO2 reduction to CO.
Typically, in SACs, the metal atomic center is coordinated with four non-metal atoms. However, this structure does not always give the best CO2RR activity. Regulating the coordination number in SACs offers an effective strategy to tune the adsorption energy of reaction intermediates and thus their catalytic performance. Generally, the lower the coordination number, the stronger the adsorption of intermediates on SACs. Li et al. prepared Co–N2, Co–N3, and Co–N4 by calcining Co/Zn zeolitic imidazolate frameworks (ZIFs) at different temperatures.74 Since N species became volatile at high temperature, Co–N2, Co–N3, and Co–N4 could be obtained at 1000 °C, 900 °C, and 800 °C. The atomically dispersed Co atoms could be directly observed by HAADF-STEM. EXAFS confirmed the existence of Co–N bonds (at around 1.4 Å) and the absence of Co–Co bonds. The intensity of the Co–N peak for Co–N2 was found much lower than that for Co–N3 and Co–N4, indicating lower coordination around the Co center in Co–N2. EXAFS fitting gave the Co–N coordination number in these Co-SACs of 2.2, 3.1, and 4.1, respectively. Electrochemical measurements showed that Co–N2 had the highest activity to reduce CO2 to CO (Fig. 6e). Co–N2, Co–N3, and Co–N4 have similar structural parameters except the Co–N coordination number, and provide an ideal platform to elucidate the influence of coordination number on CO2RR performance. DFT calculations disclosed that the endergonic CO2˙−* formation was the RDS for these Co-SACs. Owing to the higher d-band center of Co–N2 (−0.81 eV) than that of Co–N4 (−1.06 eV), Co–N2 had a lower energy barrier than Co–N4 to form COO˙−*, explaining the higher CO2RR activity on Co–N2 (Fig. 6f). Additionally, the authors further measured the OH− adsorption strength to simulate the binding affinity of COO˙−* as COO˙−* was unstable in the electrolyte. Co–N2 showed a more negative OH− adsorption peak, verifying its stronger adsorption to COO˙−*. Considering the higher catalytic activity of Co–N4 towards the HER,75 the enhanced CO2RR performance of Co–N2 could also be related to the increase in UL(CO2) – UL(H2). This work indicated that lower coordination number in SACs would enhance the adsorption of CO2RR intermediates. Li et al. performed DFT calculations to study Ni SACs with different coordination numbers (NiNx; x = 1–4) toward catalyzing the CO2RR.76 The results show that the RDS for all studied NiNx SACs is the formation of COOH* – an endergonic step. NiN has the lowest coordination number and thus possesses the smallest energy barrier to generate the COOH* intermediate, justifying its highest CO2RR activity (Fig. 6g). Zhang et al. synthesized Mn-N3 and Mn-N4 catalysts and measured their CO2RR performance. The Mn-N3 catalyst delivered a 98.8% CO FE with a CO partial current density of 14 mA cm−2 at an overpotential of 0.44 V. In situ X-ray absorption spectroscopy showed that Mn-N3 facilitated the formation of COOH*, which is the RDS. DFT calculation gave an energy barrier of 0.24 eV for Mn-N3 and 0.91 eV for Mn-N4 (Fig. 6i). Local density of states (LDOS) revealed that the d-band centers of Mn-N3 and Mn-N4 were located at −2.50 eV and −4.93 eV, respectively (Fig. 6h).77 Other researchers also reached a similar conclusion that the lower coordination number in SACs indeed strengthened the adsorption of CO2RR intermediates.78–83
3.2.3. Effect of the coordinated nitrogen type on CO2 reduction to CO.
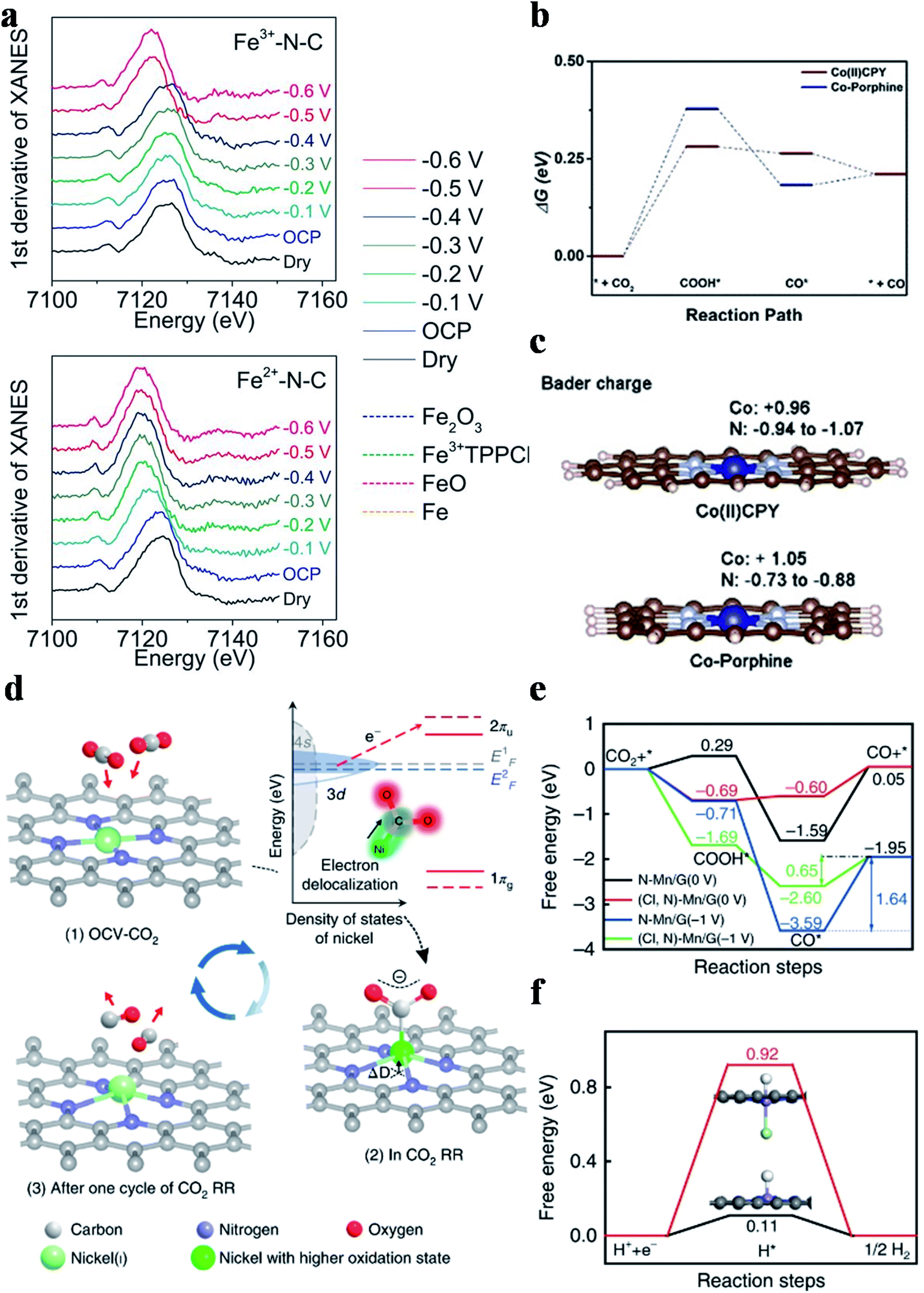
Four types of nitrogen atoms exist in SACs, namely pyridinic, pyrrolic, graphitic and oxidized N. The metal centers in SACs could be bonded with pyridinic N or pyrrolic N. The d-band center for pyrrolic-N coordinated metal sites is lower than that for pyridinic-N coordinated metal sites and thus pyrrolic-N coordinated metal sites bind to the CO2RR intermediates more weakly. Hu et al. prepared Fe3+-N-C coordinated with pyrrolic N and Fe2+-N-C coordinated with pyridinic N using different pyrolysis precursors.84 Fe3+-N-C could reduce CO2 to CO with a CO partial current density of 94 mA cm−2 at an overpotential of 340 mV. No catalytic performance decay was observed in 12 hours of continuous reaction. Operando X-ray absorption spectroscopy verified the true active sites of Fe3+ ions coordinated with pyrrolic N atoms and the +3 oxidation state of Fe was maintained during the reaction (Fig. 7a). Furthermore, electrochemical measurements indicated that the faster CO2 activation and weaker CO adsorption were responsible for the superior catalytic activity of Fe3+-N-C. Other research groups also regarded M-pyrrolic N as the active center for the CO2RR.85,86 Wang et al. constructed Co(II)CPY (Co atoms coordinated with four pyridinic N atoms) and Co-porphine (Co atoms coordinated with four pyrrolic N atoms) and found that Co(II)CPY showed better CO2RR activity than Co-porphine.87 DFT calculations suggested that the formation of COOH* was the RDS, in which Co(II)CPY had a lower energy barrier (Fig. 7b). Bader charge analysis showed that the Co atoms in Co(II)CPY could get more electrons from the ligand compared with the case in Co-porphine (Fig. 7c). Projected density of states (PDOS) demonstrated that the dz2 orbital of Co(II)CPY was closer to the Fermi level before the reaction and had a larger overlap with the 2p orbital of COOH after the reaction, substantiating the enhancement of COOH* adsorption in Co(II)CPY. Other research studies also reached the same conclusion that M-pyridinic N species served as the reaction sites in catalyzing the CO2RR.72,73,88–91 Since the controversy about which type of M–N species is the active site(s) in the CO2RR still remains, more attention has to be paid on this research hotspot.
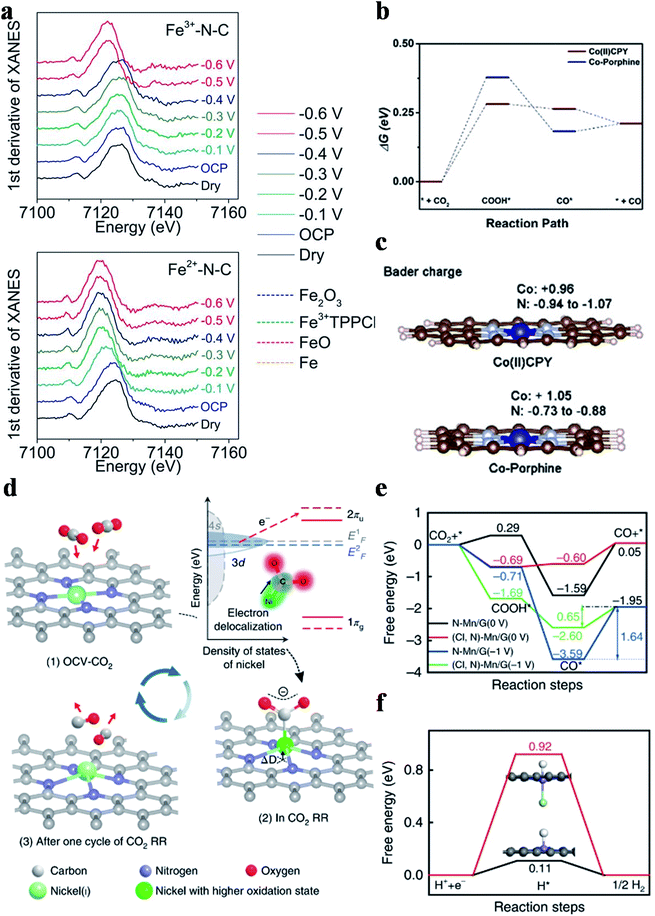 |
| | Fig. 7 (a) The first derivative of the XANES spectra for Fe3+-N-C and Fe2+-N-C as dry powder (black) and on a glassy carbon electrode at the open circuit potential (OCP) (blue), −0.1 V (light blue), −0.2 V (green), −0.3 V (dark green), −0.4 V (dark blue), −0.5 V (red), and −0.6 V (pink) versus RHE, with Fe2O3 (blue dashed), Fe3+TPPCl (green dashed), FeO (pink dashed), and Fe foil (orange dashed) references. Reproduced with permission from ref. 84. Copyright (2019) the American Association for the Advancement of Science. (b) Free energy profiles of Co(II)CPY/graphene and Co-porphine/graphene. (c) Bader charge of Co and N atoms. Reproduced with permission from ref. 87. Copyright (2020) Wiley-VCH. (d) Structural evolution of the active sites in electrochemical CO2 reduction. Reproduced with permission from ref. 92. Copyright (2018) Springer Nature. (e) Calculated free energy of the CO2RR. (f) Calculated free energy of hydrogen adsorption. Reproduced with permission from ref. 93. Copyright (2019) Springer Nature. | |
3.2.4. Effect of heteroatoms on CO2 reduction to CO.
Heteroatom doping could induce electron transfer between the metal atomic center and the surrounding non-metal atoms, leading to the distinct electronic structure and hence the electrocatalytic performance. Liu et al. synthesized a S doped Ni-SAC (Ni–N3S) and Ni–N4 and studied their CO2RR mechanisms.92 The CO2RR onset potential of Ni–N3S was 100 mV lower than that of Ni–N4. Operando XAS showed that the valence state of Ni atoms in Ni SACs was +1, serving as the true active sites. Moreover, the Ni+ sites were identified as having strong interactions with CO2, where electrons were transferred from Ni+ to the C 2p orbital in CO2 to generate CO2δ−, facilitating the CO2 activation and further reduction (Fig. 7d). Consequently, the catalyst achieved a specific current of 350 A gcatalyst−1 and a TOF of 14800 h−1 at an overpotential of 0.61 V with 97% FECO. The authors also proposed the activation process of CO2 on the Ni(I) active center and its structural evolution during the CO2RR (Fig. 7d). Cl doping in a Mn-SAC was reported by Zhang et al. to improve the CO2RR activity. Mn-SACs usually displayed poor selectivity towards CO production because of their too strong adsorption for CO*, making CO release rather difficult. After introducing Cl into the Mn-SAC ((Cl, N)-Mn/G), the selectivity towards CO was significantly increased from 17% to 97% with the partial current density for CO production exhibiting an 11-fold increase. PDOS revealed that the d-band center of (Cl, N)-Mn/G was lower than that of MnN4, weakening the CO* adsorption and thus improving the CO selectivity. The free energy diagram further supported this conclusion. Specifically, the energy barrier of the RDS (desorption of CO*) decreased from 1.64 eV for MnN4 to 0.65 eV for (Cl, N)-Mn/G (Fig. 7e). Moreover, the energy barrier for H2 generation increased from 0.11 eV for MnN4 to 0.92 eV for (Cl, N)-Mn/G, implying that the HER was largely suppressed (Fig. 7f).93
3.2.5. Effect of dual metal atoms on CO2 reduction to CO.
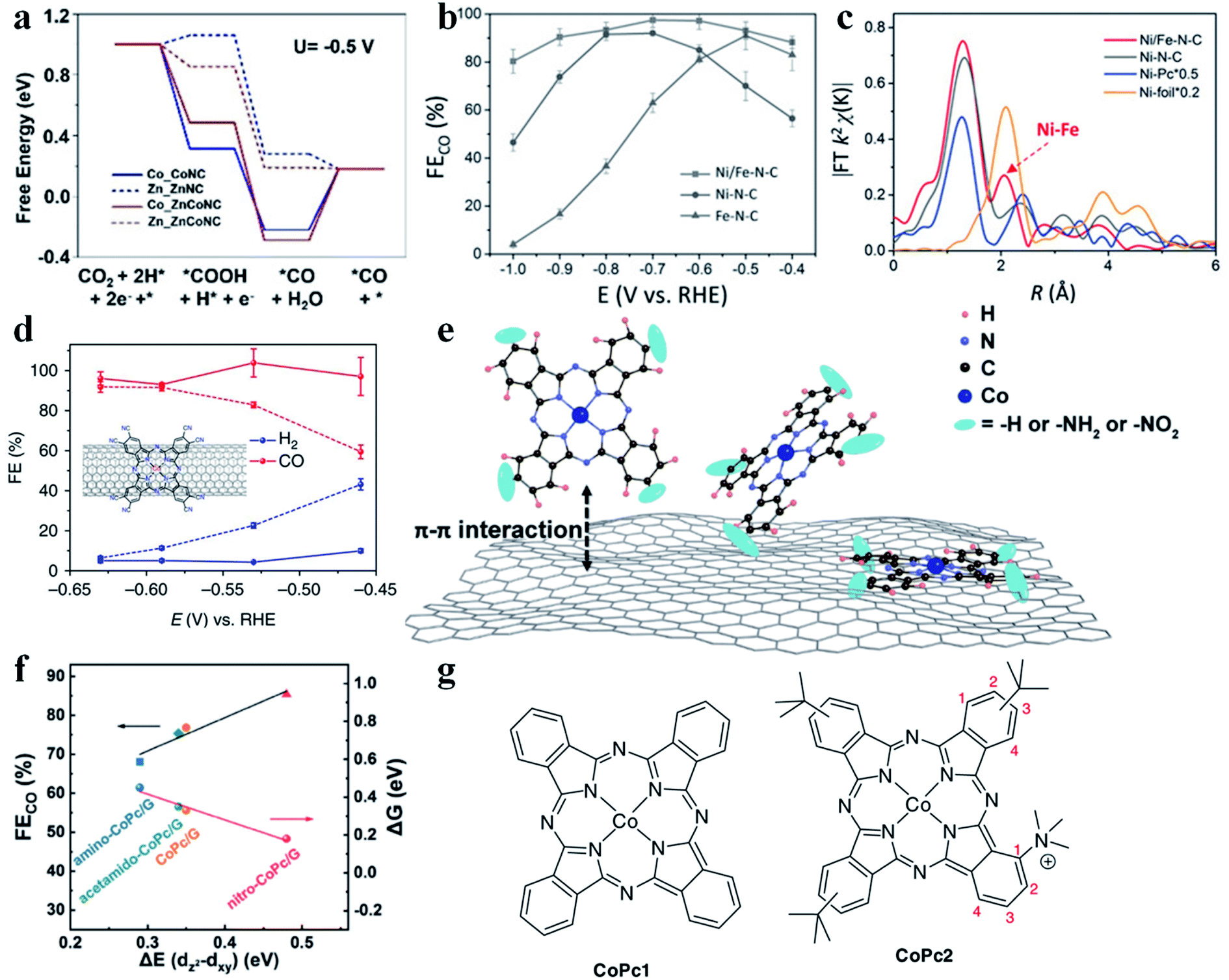
Although much progress has been made in the CO2RR to produce CO using SACs, their performance could be further improved through constructing dual metal atomic sites. Gong et al. demonstrated an example of neighboring Zn and Co monomers (ZnCoNC) for enhancing CO production in the CO2RR.94 ZnCoNC displayed a CO FE of 93.2% at −0.5 V vs. RHE in 30 h. XANES confirmed the interaction between Zn and Co. According to the free energy diagram, after introducing Co into the Zn-SAC, the formation of COOH* (the RDS for the Zn-SAC) changed from endergonic to exergonic (Fig. 8a), which enhanced the adsorption of COOH* and thus promoted CO production. Zhao et al. synthesized a Ni–Fe dual-metal-atom catalyst (NiFe-N-C).95 Electrochemical study disclosed that NiFe-N-C possessed higher activity and selectivity than Ni-N-C and Fe-N-C in the CO2RR (Fig. 8b and c). The calculated free energy diagram showed that the energy barrier for COOH* formation (RDS) was much lower in NiFe-N-C than in Ni-N-C. Therefore, Fe doping could strengthen the binding affinity of Ni atoms for COOH*. It is worth mentioning that two mechanisms are possible for enhancing the CO2RR performance in dual metal atom catalysts. One is that only one metal site acts as the active center, and the introduction of the second metal atom regulates the electronic structure of the first metal site. The other is that both metal sites serve as the active centers for adsorbing reaction intermediates and engage in the reaction. To date, the reported examples belong to the former mechanism. If the reaction could follow the latter mechanism on dual metal atom catalysts, the scaling relationship might be broken, which provides another possibility to tune the CO2RR performance.
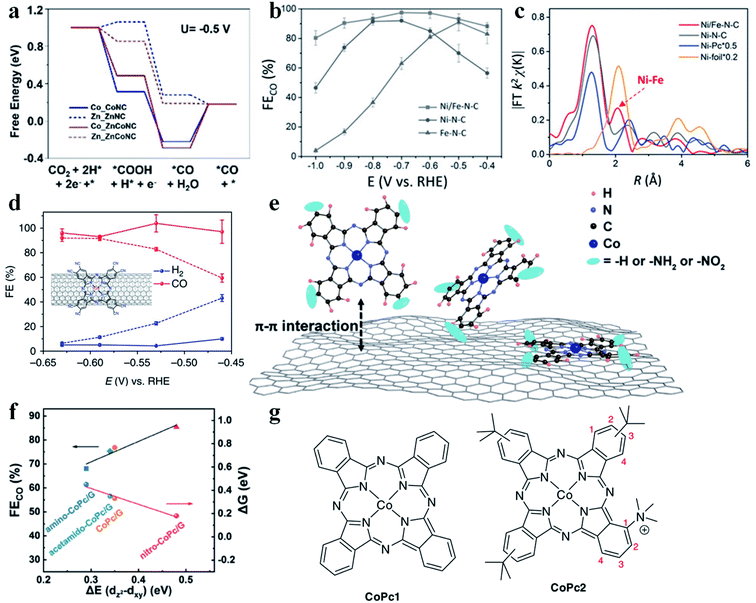 |
| | Fig. 8 (a) Calculated free energy for CO2 reduction to CO. Reproduced with permission from ref. 94. Copyright (2020) Wiley-VCH. (b) FE of CO for various SACs. (c) Fourier transformation of the EXAFS spectra at R space. Reproduced with permission from ref. 95. Copyright (2019) Wiley-VCH. (d) Faradaic efficiencies of reduction products at different potentials for CoPc-CN/CNT (solid line) in comparison with CoPc/CNT (dotted line). The inset in (d) shows the molecular structure of CoPc-CN, which is anchored on a CNT. The average values and error bars in (e) are based on six measurements during three reaction runs (two product analysis measurements were performed in each run). The error bars represent standard deviation of six measurements. The data are all iR corrected. Reproduced with permission from ref. 97. Copyright (2017) Springer Nature. (e) Schematic illustration showing the construction of single cobalt atom catalysts for the CO2RR. (f) The linear relationship between the CO faradaic efficiency/Gibbs free energy of CO2 reduction to the *CO2− intermediate and the energy difference ΔE(dz2 − dxy). Reproduced with permission from ref. 96. Copyright (2020) Springer. (g) CoPc1 (left) bears no substituents, and CoPc2 (right) bears one trimethyl ammonium group at position 1 of the isoindole subunits, and three tert-butyl groups (position 2 or 3) of the other subunits. Reproduced with permission from ref. 98. Copyright (2019) Springer Nature. | |
3.2.6. Effect of surrounding functional groups on CO2 reduction to CO.
The charge state of the metal atomic center determines its intrinsic catalytic activity. Therefore, introducing functional groups (electron-withdrawing or electron-donating) into SACs offers an effective strategy to manipulate their CO2RR performance. Huang et al. prepared CoPc/G, amino functionalized CoPc/G (amino-CoPc/G), and nitro functionalized CoPc/G (nitro-CoPc/G) and studied the effect of electron-withdrawing/electron-donating groups in SACs on the CO2RR activity (Fig. 8e).96 Electrochemical measurements showed that nitro-CoPc/G possessed the highest activity and selectivity for generating CO. DFT calculations revealed that formation of *CO2− was the RDS, where nitro-CoPc/G had the lowest energy barrier. After the introduction of the electron-withdrawing nitro group, the electron density around the Co site decreased. Also, the dz2 orbital was upshifted and became less affected by other lower energy level orbitals, like dxy. Another important descriptor was also found, that is, the larger the energy difference between the dz2 and dxy orbital, the smaller the energy barrier to form *CO2− (Fig. 8f). Wang et al. introduced an electron-withdrawing cyano group into CoPc/CNT (CoPc-CN/CNT) and found that both activity and CO selectivity increased obviously (Fig. 8d).97 The electron-withdrawing cyano group could prompt the generation of Co(I), which was regarded as the active site for the CO2RR. The enhanced Co(II)/Co(I) redox transition at more positive potentials in CoPc-CN/CNT supported this proposal. Moreover, the cyano group also reduced the affinity of the Co site for CO*, facilitating the release of CO. In addition to the nitro group and cyano group, a trimethyl ammonium group (CoPc2) was also introduced into CoPc1 (Fig. 8g).98 After such a modification, the CoPc2 showed high selectivity towards CO production, while the CO partial current density increased substantially.
3.3. HCOOH
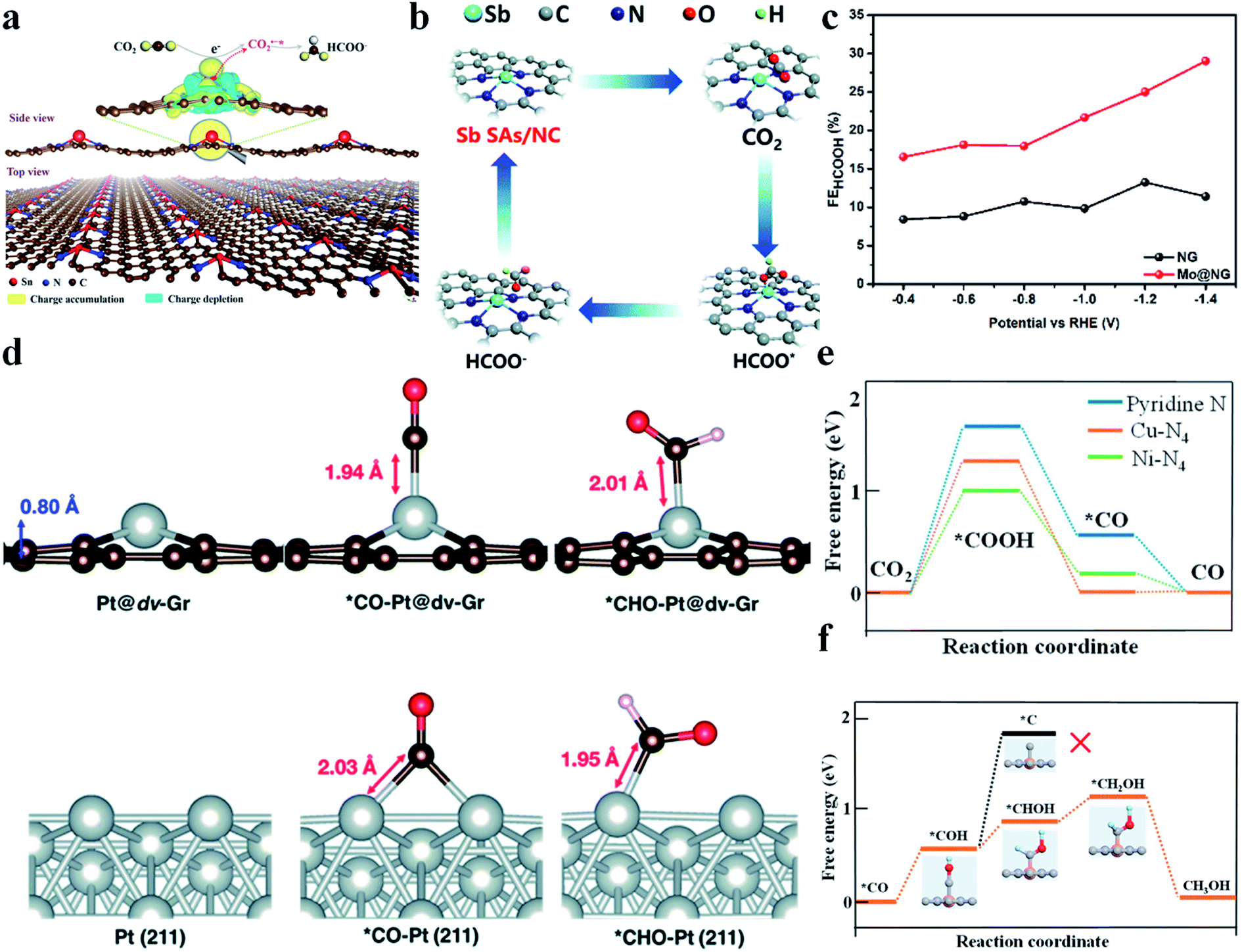
It is reported that SACs with a p-block metal (Sn, Sb) center are able to catalyze the CO2RR to produce formic acid or formate. Xie et al. synthesized SnN2C2 and found that it could reduce CO2 to formate at a very low onset overpotential of 60 mV and with a high TOF of 11930 h−1 at −1.14 V vs. RHE.99 Moreover, its activity did not decay during 200 h of continuous reaction. DFT calculations revealed that electrons were transferred from Sn atoms to graphene, making Sn atoms positively charged. Fourier transformed infrared spectroscopy (FTIR) spectra and Gibbs free energy calculations confirmed that Snδ+ could facilitate CO2 activation and protonation through stabilizing CO2˙−* and HCOO−* (Fig. 9a). Wei et al. constructed a NiSn atomic pair (NiSn-APC) and also achieved a good performance in the CO2RR to formate.100 NiSn-APC produced HCOOH with a high TOF of 4752 h−1 and formate productivity of 36.7 mol h−1 gSn−1 at −1.02 V vs. RHE. In situ attenuated total reflection-infrared (ATR-IR) spectra and theoretical calculations confirmed the electron redistribution on Sn, which is induced by the adjacent Ni atoms. Moreover, ΔG (*HCOOH) (−0.05 eV) was much smaller than ΔG (*COOH) (1.36 eV), explaining its high selectivity towards formate production. Sb–N4 (Sb SA/NC) reported by Li's group also displayed a good CO2RR performance.101 Sb SA/NC produced formate with a FE of 94% at −0.8 V vs. RHE. In situ XAFS and DFT calculations verified that the positively charged Sbδ+-N4 was the origin of the high catalytic activity. Sb SA/NC possessed a ΔG (HCOO*) (0.21 eV) smaller than ΔG (COOH*) (1.00 eV) and ΔG (H*) (0.81 eV), implying that formate production was more favorable than CO and H2 (Fig. 9b). Xie et al. reported that Mo atoms supported on N doped graphene (Mo@NG) could also efficiently reduce CO2 to produce HCOOH. Mo@NG generated HCOOH with a high FE, twice as that of NG, and achieved a formate yield of 747 mmol gcatalyst−1·h−1 (Fig. 9c).102
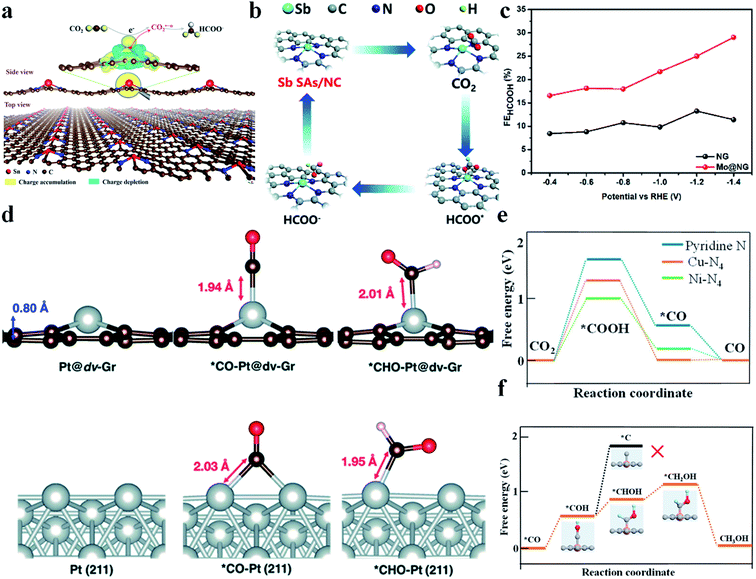 |
| | Fig. 9 (a) Plot showing 3D differential charge density of single Snδ+ atoms on N-doped graphene and the schematic illustration of CO2 electroreduction into formate. The yellow and blue isosurfaces correspond to the increase and depletion in the number of electrons, respectively. Reproduced with permission from ref. 99. Copyright (2019) Wiley-VCH. (b) Proposed CO2RR mechanism on Sb SA/NC. Reproduced with permission from ref. 101. Copyright (2020) The Royal Society of Chemistry. (c) Comparison of formate faradaic efficiency between NG and Mo@NG. Reproduced with permission from ref. 102. Copyright (2019) Elsevier. (d) Optimized geometries of Pt@dv-Gr and Pt (211) before and after adsorption of *CO and *CHO. Reproduced with permission from ref. 7. Copyright (2017) The Royal Society of Chemistry. (e) Free energy diagram of CO2 reduction to CO on pyridine N, Ni–N4, and Cu–N4. (f) Free energies for conversion of *CO to CH3OH on Cu–N4. Orange, gray, dark blue, red, and light blue spheres stand for Cu, C, N, O, and H atoms, respectively. Reproduced with permission from ref. 105. Copyright (2019) American Chemical Society. | |
3.4. CH3OH
CH3OH is a high value-added product in the CO2RR. However, as discussed above, both CO2RR pathways to generate CH3OH (*CH2OH and *CH3O) possess very high energy barriers and thus its production remains a big challenge. Wang et al. immobilized cobalt phthalocyanine (CoPc) onto carbon nanotubes (CNTs), denoted as CoPc/CNT.103 CoPc/CNT was found to be able to reduce CO2 to CH3OH with a FE of 44% and a partial current density greater than 10 mA cm−2 at −0.94 V vs. RHE. But the catalytic activity of CoPc/CNT quickly decayed after 1 h of electrochemical reaction. To enhance the stability, an amino functional group was introduced into CoPc/CNT to form CoPc-NH2/CNT, whose stability was greatly improved. In addition to experiments, theoretical studies have also been conducted to prompt the discovery of potential catalysts to reduce CO2 to CH3OH. Cai et al. anchored Co, Rh, and Ir atoms onto porphyrin-like graphene.104 First, the structural stability of the SACs was confirmed from the strong hybridization between metal d-orbitals and N 2p-orbitals. Then, the energy barriers for the CO2RR to CH3OH were calculated. CoN4/graphene was found most promising among the three studied catalysts for CH3OH generation with a RDS energy barrier of only 0.59 eV. Jung et al. conducted DFT calculations to predict a series of transition metal SACs on graphene with single and double vacancies.7 They figured out that Ni and Pt SACs with double vacancies (Ni@dv-Gr, Pt@dv-Gr) were promising to reduce CO2 to CH3OH. Taking Pt@dv-Gr as an example, CO* was adsorbed on one Pt atom via the top-on adsorption configuration, while bulk Pt adsorbed CO* via two Pt atoms adopting the bridge adsorption mode. As a result, bulk Pt adsorbed CO* so strongly that the transformation of CO* to *CHO – an important intermediate for CH3OH formation – became rather difficult. In contrast, Pt@dv-Gr had a much smaller energy barrier for this step and held the premise for CH3OH production (Fig. 9d). He et al. developed a simple method to prepare a Cu-SAC on through-hole carbon nanofibers (Cu SAs/TCNFs).105 The Cu SAs/TCNFs (Cu–N4) could generate methanol with a FE of 44%. DFT calculations found that the energy barrier for COOH* formation was much larger than that for CHOH* formation on Cu–N4, which effectively suppressed the CO production. Moreover, Cu–N4 could adsorb CO* more strongly than Ni–N4, which allowed CO* to be further reduced (Fig. 9e). Additionally, the energy barrier to form C* from COH*, an important step towards CH4, was calculated to be 1.88 eV, which is much larger than that to form CH3OH (Fig. 9f). Therefore, CH3OH was more favorable than CH4 in the CO2RR. In summary, the mechanism of the CO2RR to form CH3OH on SACs was nearly completely based on DFT calculation. The lack of understanding of the structural characteristics poses a big challenge for designing efficient SACs for reducing CO2 to CH3OH.
3.5. CH4
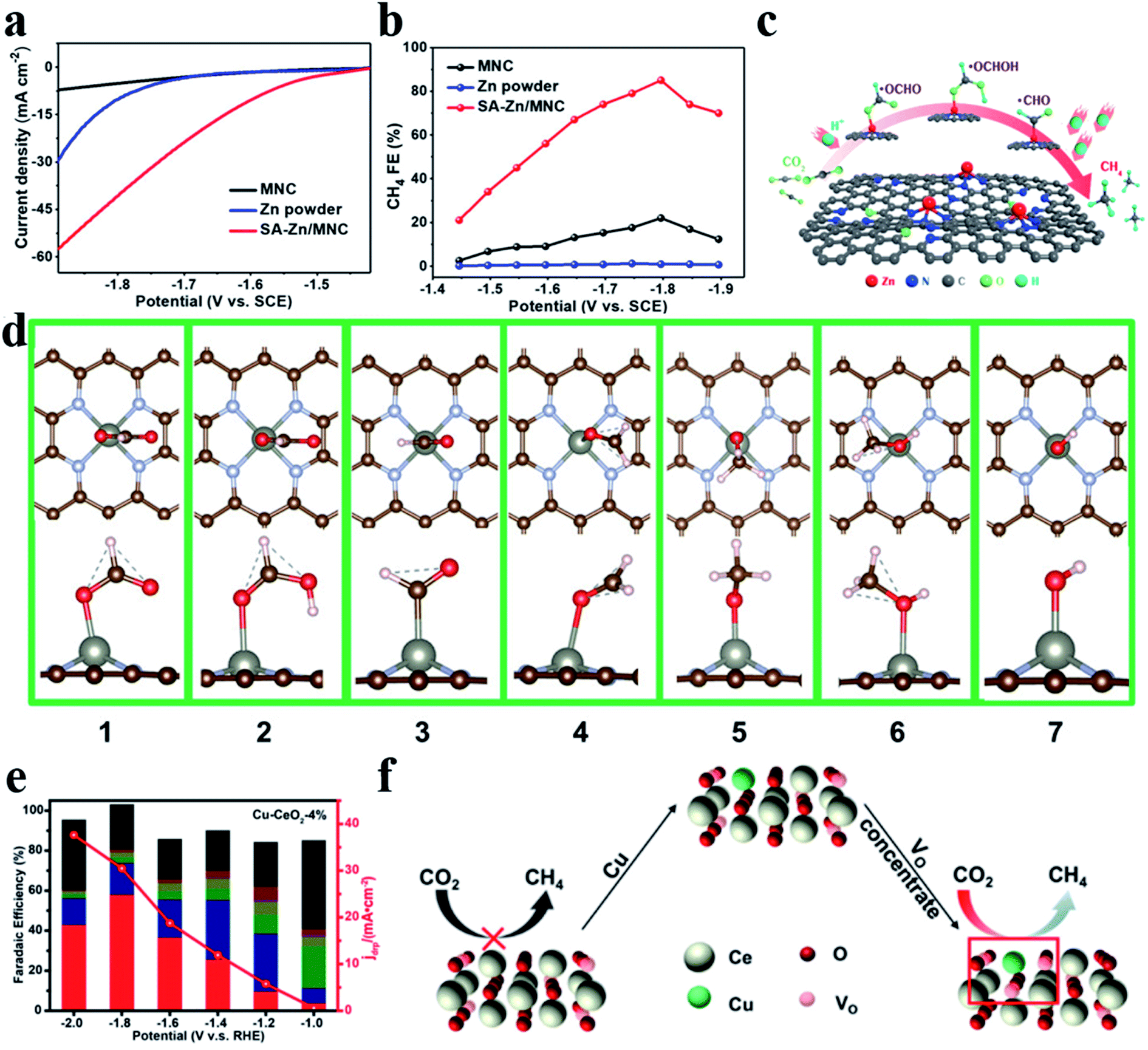
CH4 is another important product in the CO2RR, which undergoes an 8e− transfer process. Strong adsorption affinity of metal sites for the CO* intermediate is required for generating CH4. Xin et al. reported Zn–N4 that could stably reduce CO2 to CH4 with a FE of 85% and a partial current density of −31.8 mA cm−2 at −1.8 V vs. the saturated calomel electrode (SCE) (Fig. 10a–c).106 DFT calculations showed that Zn atoms tended to bond with O atoms to form *OCHO (ΔG (*OCHO) = 0.46 eV) rather than with C atoms to form COOH* (ΔG (*COOH) = 1.2 eV) (Fig. 10d). Furthermore, surface-enhanced infrared absorption spectroscopy enabled direct observation of the existence of *OCH3 and *OCH2. Zheng et al. deposited Cu atoms on CeO2 nanorods (Cu-CeO2), where Cu atoms were stabilized by three oxygen vacancies. Cu-CeO2 could reduce CO2 into CH4 with a FE of 58% (Fig. 10e and f).107 Theoretical calculations revealed that the synergistic effect between atomic dispersion of Cu and multiple surrounding oxygen vacancies as well as the contribution from the CeO2 framework expedited CO2 adsorption and activation.
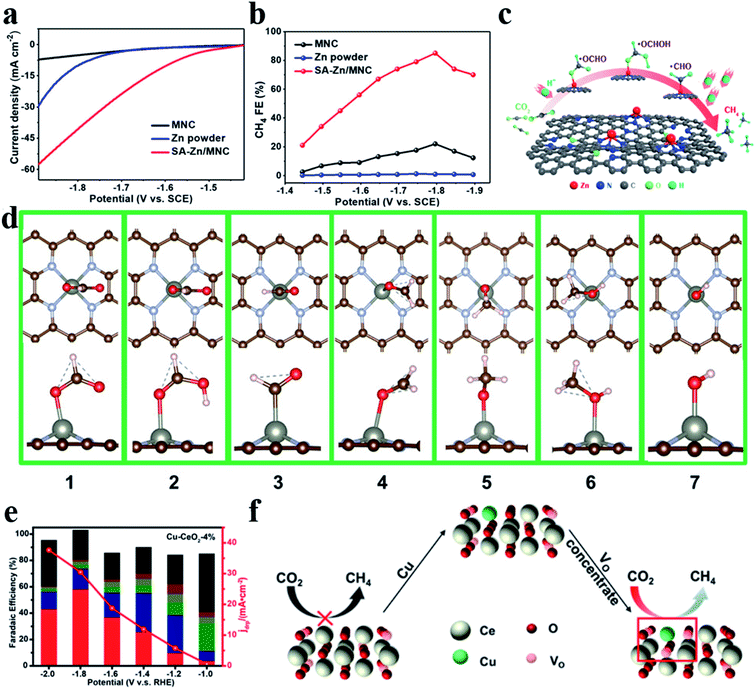 |
| | Fig. 10 (a) Polarization curves for MNC, Zn powder, and SA-Zn/MNC. (b) FEs for the electrochemical CO2RR to CH4 at different applied potentials. (c) CO2RR pathway for single Zn atoms to produce CH4. (d) The most stable structure. The red and pink balls represent O and H atoms, respectively. Reproduced with permission from ref. 106. Copyright (2020) American Chemical Society. (e) Faradaic efficiencies (bars, left y-axis) and the deep reduction product current density (jdrp, red curve, right y-axis) of Cu-CeO2-4%. The red column represents CH4. (f) Effect of oxygen vacancies on the CO2RR. Reproduced with permission from ref. 107. Copyright (2018) American Chemical Society. | |
3.6. C2+
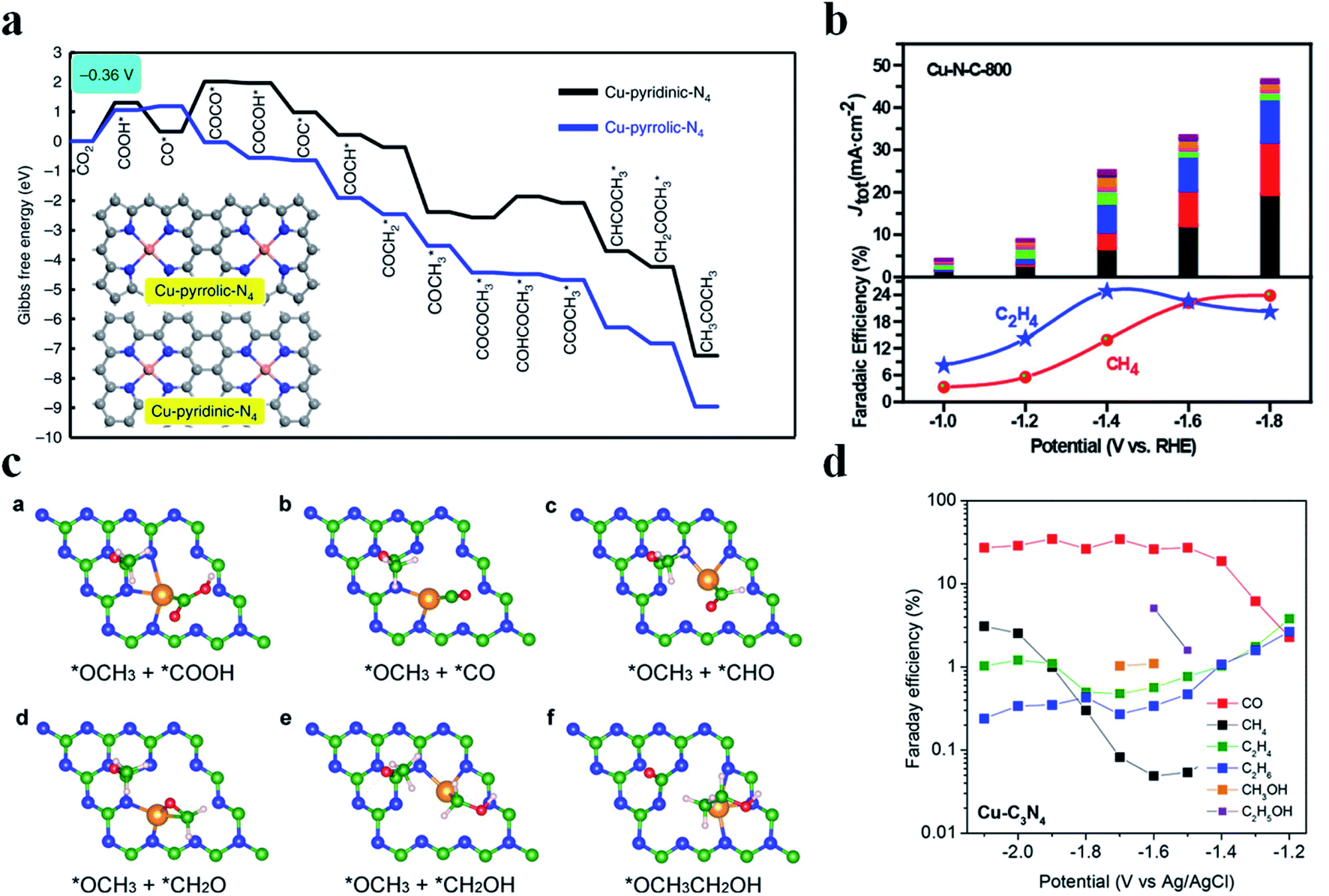
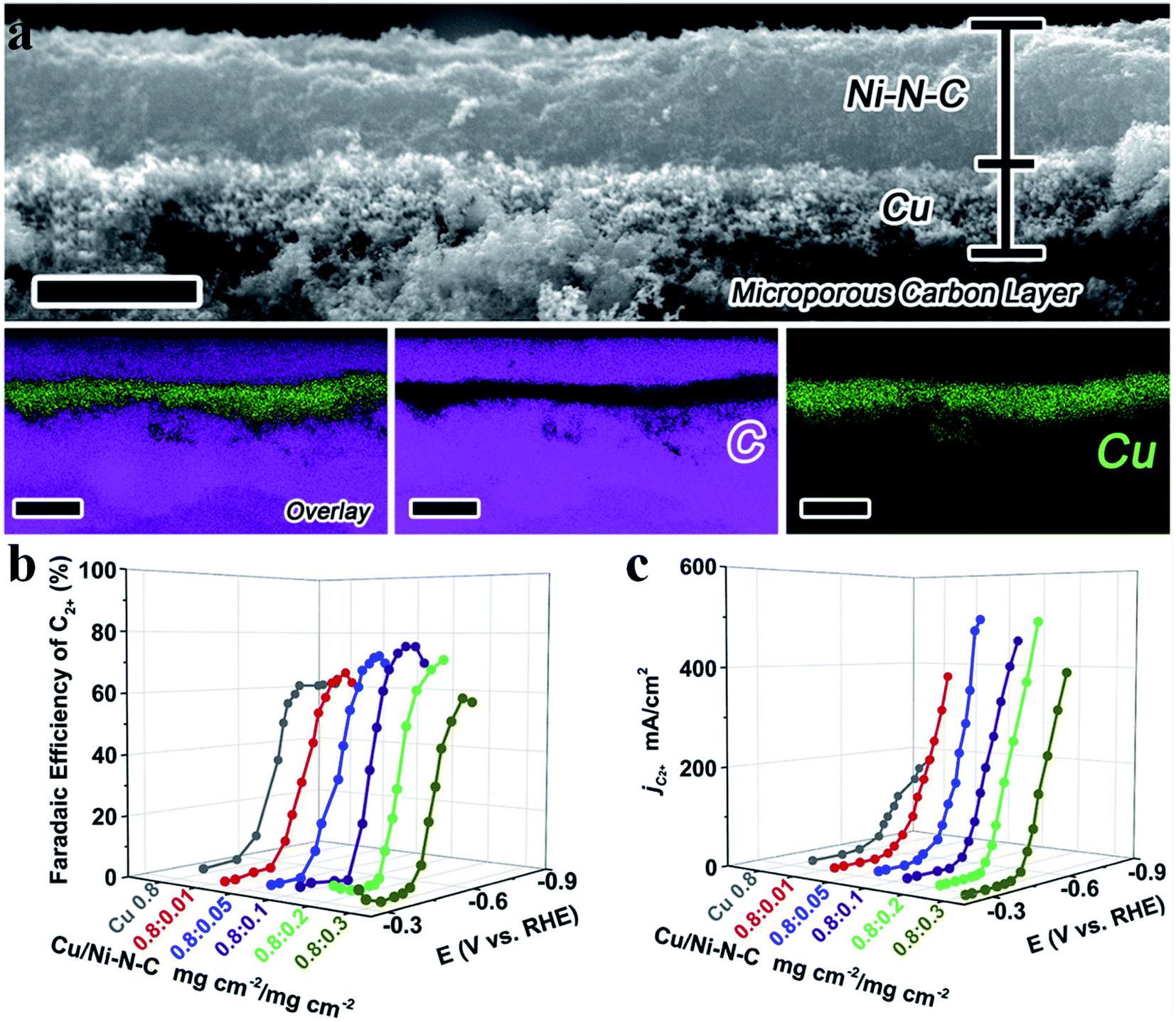
Generation of C2+ products in the CO2RR requires C–C coupling, which possesses a high energy barrier and is difficult to take place in SACs. Cu nanoparticles have been shown as an excellent catalyst to reduce CO2 to form C2+ products, while this is not the case for Cu SACs due to the lack of neighbouring active sites to allow for C–C coupling. Fontecave et al. reported that a Cu SAC (Cu0.5NC) could reduce CO2 to ethanol with a FE of 43% at −1.2 V vs. RHE in 0.1 M CsHCO3 solution.108 A higher FEethanol (∼66%) could be achieved using CO as the feed gas, suggesting that CO* might be the important intermediate to form ethanol. Operando XAS analysis showed that the initial isolated Cu atoms would aggregate and form Cu NPs during the CO2RR, acting as the true active sites. Interestingly, Cu atoms could be recovered after the reaction, indicating that such a transition was reversible. Xu et al. also observed a similar phenomenon where Cu atoms transformed into Cun clusters (n = 3 and 4) during the electrochemical reaction and then returned back to their initial state after the reaction.109 Chen et al. designed a Cu SAC loaded on N-doped porous carbon (Cu-SA/NPC) to reduce CO2 into C2+ products.110 Cu-SA/NPC produced acetone with a FE of 36.7% and a yield of 336.1 μg h−1 at −0.36 V vs. RHE. Theoretical calculations pointed out that Cu coordinated with four pyrrolic N atoms was the true active center, which decreased the energy barrier of CO2 activation and C–C coupling. Also, the most possible pathway to form acetone was proposed: CO2 → COOH* → CO* → COCO* → COCOH* → COC* → COCH* → COCH2* → COCH3* → COCOCH3* → COHCOCH3* → CCOCH3* → CHCOCH3* → CH2COCH3* → CH3COCH3 (Fig. 11a). Zheng et al. regulated the population of Cu–Nx sites on NC (Cu-N-C) by controlling the pyrolysis temperature (800 °C and 900 °C), where Cu-N-C-800 contained 4.9 mol% metal atoms and Cu-N-C-900 had 2.4 mol% metal atoms. Adjacent Cu–N2 sites existed among the densely distributed Cu-Nx in Cu-N-C-800, enabling C–C coupling for C2H4 formation with a FE of 24.8% at −1.4 V vs. RHE (Fig. 11b).111 DFT calculations disclosed that Cu–N2 bound with CO* more strongly (−1.17 eV) than Cu–N4 (−0.19 eV), which was favorable for CO* to participate in the further reaction. Moreover, the adjacent Cu–N2 sites possessed a much lower energy barrier for C–C coupling (0.14 eV) than the isolated Cu–N2 (1.38 eV). Cu-N-C-900 with fewer neighboring Cu–N2 sites generated much less C2H4, substantiating the important role of the adjacent Cu–N2 sites in C–C coupling. Qiao et al. came up with a novel dual active center mechanism to produce C2+ products in the CO2RR, where Cu atoms bound with reaction intermediates having carbon as the anchoring atom (*COOH, *CO, and *CHO) and C atoms on C3N4 bound with reaction intermediates having oxygen as the anchoring atom (*OCH2, *OCH3, *O, and *OH) (Fig. 11c).112 The detection of various C2+ products validated the reaction mechanism, though the selectivity of C2+ products was low (Fig. 11d).
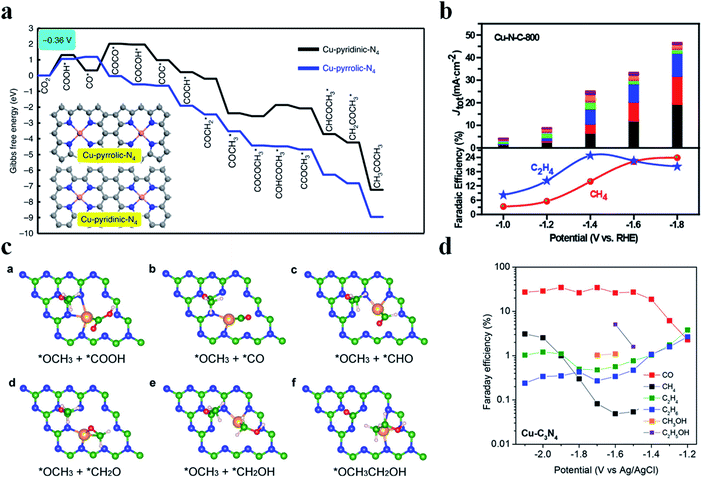 |
| | Fig. 11 (a) Free energy diagrams calculated at a potential of −0.36 V vs. RHE for CO2 reduction to CH3COCH3 on Cu-pyridinic-N4 and Cu-pyrrolic-N4 sites of Cu-SA/NPC (the computational models are included in the figure). Reproduced with permission from ref. 110. Copyright (2020) Springer Nature. (b) CO2 electroreduction performance of the Cu-N-C-800 catalyst. Top panel: current densities, with colors indicating different products as shown at the bottom. Bottom panel: faradaic efficiencies of C2H4 and CH4. Reproduced with permission from ref. 111. Copyright (2020) American Chemical Society. (c) Key reaction intermediates for CH3CH2OH generation on Cu–C3N4. (d) The measured faradaic efficiencies of various products on Cu–C3N4 at different overpotentials. Reproduced with permission from ref. 112. Copyright (2017) American Chemical Society. | |
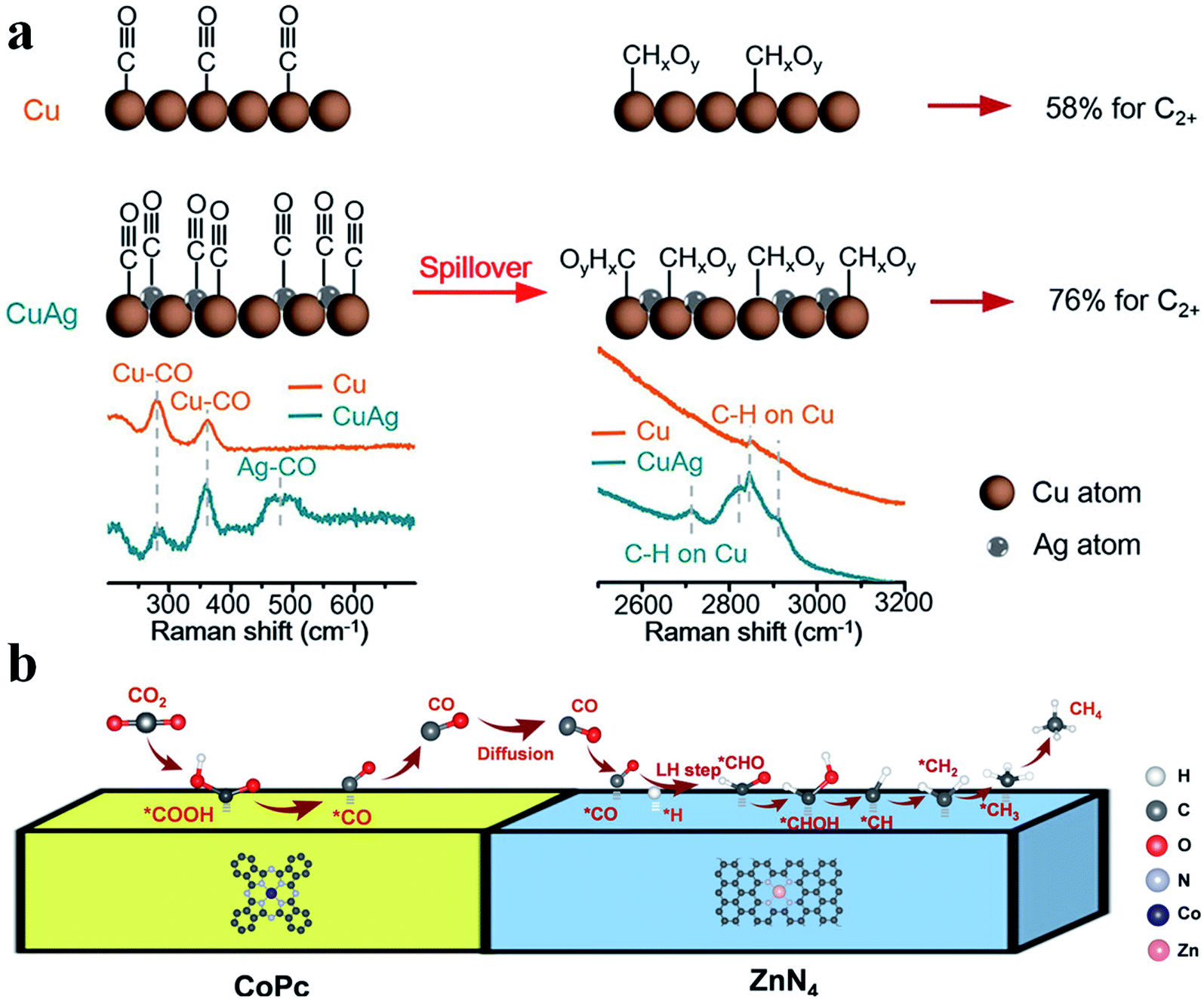
Tandem catalysis provides another efficient strategy to produce C2+ products via the CO2RR, in which plenty of CO* is first generated on one type of active site and it diffuses to the other type of active site, and then gets further reduced to produce C2+ products. Grätzel et al. fabricated a Cu2O–Ag tandem catalyst. CO* was first produced on the Ag sites and then underwent C–C coupling on the Cu sites to generate C2H4. Operando Raman spectroscopy allowed direct observation of the enhanced coverage of CH-containing intermediates (Fig. 12a).113 Consequently, the partial current density of C2H4 reached −18.1 mA cm−2 at −1.05 V vs. RHE on the CuAg catalyst, while it was −8.5 mA cm−2 on the Cu catalyst. A similar tandem catalyst was demonstrated using Ni SAC as the CO generator.114 By optimizing the loading amount of Ni-N-C, the Cu/Ni-N-C tandem electrode could reduce CO2 into C2+ products at a high production efficiency (80% FE) and rate (over 500 mA cm−2) (Fig. 13a–c).115 Therefore, the tandem catalysis strategy not only efficiently promoted the C2+ product faradaic efficiency, but also substantially enhanced the reaction rate. Wang et al. reported that CoPc@Zn-N-C was able to reduce CO2 to CH4via a two-step tandem reaction.116 CO was first generated on CoPc, and then it was further reduced to CH4 on the Zn–N4 site. DFT calculations disclosed that CoPc not only engaged in the first step to produce CO but also took part in the next step, where it increased the availability of H* over nearby N sites in Zn–N4 and thus benefited CH4 formation (Fig. 12b). The above studies highlighted that multiple sites are essential for multi-electron product formation in the CO2RR, in which the multiple sites can be adjacent single atom sites, or metal atoms and coordination atoms.
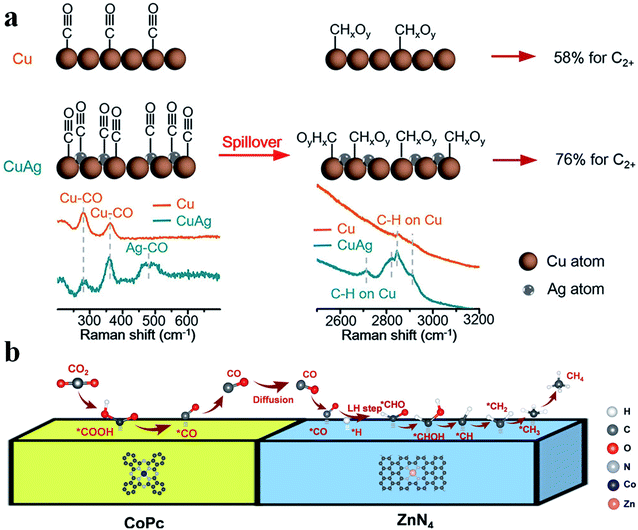 |
| | Fig. 12 (a) CO2RR mechanism for producing C2+ products on Cu and CuAg catalysts. Reproduced with permission from ref. 113. Copyright (2019) American Chemical Society. (b) A proposed reaction mechanism of the CO2RR to CH4 over CoPc@Zn-N-C. Reproduced with permission from ref. 116. Copyright (2020) Wiley-VCH. | |
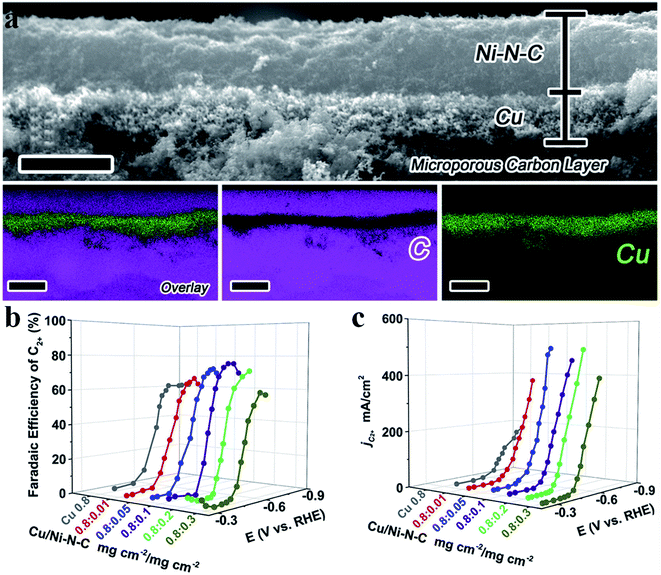 |
| | Fig. 13 (a) A cross-sectional SEM image and EDS elemental mapping of a Cu/Ni-N-C tandem electrode with two catalyst layers. Scale bars: 4 μm for the SEM image and 5 μm for the elemental mapping. (b) FEC2+ for the Cu/Ni-N-C tandem electrode with Ni-N-C loading varying from 0.01 to 0.3 mg cm−2. (c) Partial current density of the Cu/Ni-N-C tandem electrode with Ni-N-C loading varying from 0.01 to 0.3 mg cm−2. Reproduced with permission from ref. 115. Copyright (2020) Elsevier. | |
The mechanism of the CO2RR to C2+ products on Cu-based catalysts is still controversial due to the multiple electron transfer and dynamic structural transformation in the reaction. The above-mentioned challenges are also present for Cu-N-C SACs. A few studies reported that C2+ could be produced by Cu-N-C SACs due to the strong adsorption of CO2 reduction intermediates on Cu atoms, which is favorable for the C–C coupling. For example, some groups showed that C2+ formation was catalyzed by small Cu nanoparticles or clusters in Cu-N-C SACs, which came from aggregation of Cu single atoms under negative bias. But others reported that Cu single atoms were stable in the CO2RR, and the formation of the C2+ products was on the dual active sites (Cu and N in Cu-N-C SACs). Based on the reported results, the stability of Cu-N-C SACs in the CO2RR seems to be related to the synthesis method and electrochemical reaction environment, which is directly correlated to the binding strength of Cu single atoms with the substrate. Therefore, it is crucial to develop novel strategies to enhance the binding strength between the central metal atom and the substrate for further studying the reaction mechanism of the CO2RR to C2+ products on SACs. Right now, besides the Cu-N-C SAC, there are no other reported SACs capable of reducing CO2 to form C2+ products. As Zn and Co could strongly adsorb CO2RR intermediates, combining Zn and Co together to develop a Zn and Co diatomic catalyst might facilitate the reduction of CO2 to form C2+ products.
SACs supported on metals belong to single-atom alloys and some reviews have already summarized their progress.117,118 Here, we focus on SACs supported on carbon-based substrates. Besides carbon, other substrates have also been used to support metal single atoms. For example, Li et al. deposited Ag single atoms on MnO2 and found that Ag single atoms in Ag1/MnO2 had a higher electronic density close to the Fermi level than Ag nanoparticles, which enabled them to reduce CO2 to CO with a FE of 95.7% at −0.85 V vs. RHE.119 Sun et al. reported that Sn single atoms supported on oxygen-deficient CuO could reduce CO2 to ethylene with a FE of 48.5%, resulting from the lowered CO dimerization energy due to Sn doping.120 The CO2RR is limited by the scaling relationship. Hybridizing d-orbitals of transition metals with p-orbitals of main group elements or metal carbides or nitrides offers a promising strategy to break the scaling relationship. Jung et al. inserted a series of single metal atoms into the surface defect sites of TiC, denoted as M@d-TiC,49 and Ir@d-TiC was found to display a very low overpotential of only −0.09 V to reduce CO2 to produce CH4. pDOS revealed that the lack of sigma-type bonding interaction between *CO and single Ir atoms in Ir@d-TiC resulted in an appreciable decrease in the limiting potential. Inspired by these studies, substrates beyond carbon deserve more attention to be studied.
4. Summary and outlook
To sum up, in this review, we introduced the theoretical considerations to guide the design of CO2RR catalysts in terms of HER suppression, generation of C1 products and formation of C2+ products, which were later supported by experiments (Table 1 summarizes the products beyond CO using SACs in the CO2RR). Although much progress has been made in this field, many challenges still remain. In the following we list the main issues and offer our solutions to address these obstacles.
Table 1 Products beyond CO using SACs in the CO2RR
| SACs |
Structures |
Products |
Ref. |
| Snδ+ |
SnN2C2 |
HCOOH |
99
|
| Sb SA/NC |
SbN4 |
HCOOH |
101
|
| NiSn-APC |
N4–Ni–Sn–N4 |
HCOOH |
100
|
| Mo-NG |
MoN4 |
HCOOH |
102
|
| CoPc/CNT |
CoN4 |
CH3OH |
103
|
| CuSA/TCNFs |
CuN4 |
CH3OH |
105
|
| SA-Zn/MNC |
ZnN4 |
CH4 |
106
|
| CoPc@Zn-N-C |
ZnN4 |
CH4 |
116
|
| Cu–CeO2 |
Cu–O3 |
CH4 |
107
|
| Cu0.5NC |
CuN4 |
C2H5OH |
108
|
| Cu/C-0.4 |
CuO4 |
C2H5OH |
109
|
| Cu-N-C-800(900) |
Cu–N2(N4) |
C2H4&CH4 |
111
|
| Cu-SA/NPC |
CuN4 |
CH3COCH3 |
110
|
(1) Most of the SACs are prepared at high temperatures (>800 °C), which makes the synthesis uncontrollable.121–123 As a result, the construction of SACs with a precise coordination environment is difficult to realize. Up to now, the coordination number is solely determined by EXAFS. However, EXAFS can only give the average structure124–126 of SACs instead of the local structure. The high similarity of the M–C, M–N, and M–O bonds makes it hard to exactly figure out the coordination structure and the coordination number. Also, even if SACs possess the same coordination number, many possible models exist. For example, for the M-SAC with a coordination number of 3, the structure can be M–N3, M–N3–C, or M–N3–V. Moreover, these species can be located in plane or at the edges. The above uncertainty leads to the divergence in the effect of coordination number. Without a definite structure, it is impossible for DFT calculation to simulate the reaction mechanism.127–130 Preparing SACs at low temperature or designing immobilized molecular catalysts is able to ensure a well-defined structure.131–135 Additionally, more advanced characterization techniques are needed to be developed to better study the structure of SACs.136–139
(2) The structure of SAC does not always remain constant during the electrochemical reaction. For example, Cu single atoms could be transformed into Cu clusters during the CO2RR. Whether SACs undergo structural transitions and identification of the actual active site(s) in SACs are still the conundrums. In situ characterization provides a platform to monitor the structural evolution of SACs during the reaction.140–144 Time-resolved in situ measurements should be developed to figure out the true active sites and obtain an overall picture of the reaction mechanism.
(3) Multiple protonation and electron transfer steps are involved in the CO2RR, making the reaction mechanism very complicated.145–148 For the C2+ products, lots of possible pathways for C–C coupling are proposed but cannot be confirmed by the current characterization techniques.149 Also, the distribution of C2+ products is broad, and the selectivity is rather poor. Furthermore, it is still unclear whether protonation and electron transfer happen simultaneously (PCET mechanism).150–154 Without a thorough understanding of the reaction mechanism, designing and searching for highly active and selective catalysts would be challenging. Constructing SACs with a well-defined structure and coupling in situ techniques together with the rapid developing computational methods are conducive to elucidating the underlying reaction mechanisms155,156 (Fig. 14).
 |
| | Fig. 14
In situ measurements and theoretical calculations prompt the development of SACs. | |
(4) Most of the SACs are in the form of powder, and show unsatisfactory performance in real devices.157–161 Particularly, the solubility of CO2 is low in aqueous solution (34 mM) and the CO2 mass diffusion limitation dominates the CO2RR particularly at high current densities. Hybridizing SACs with a gas diffusion support or designing monolithic SAC electrodes has the potential to tackle this challenge. Moreover, flow cells and membrane electrode assembly cells have been developed rapidly, which assists in further improving CO2RR performance to meet the industry requirement.162–167
Author contributions
J. Z., H. B. Y., and B. L. conceived the topic and structure of the article. All authors reviewed and contributed to this paper.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the fund from the Singapore Ministry of Education Academic Research Fund (AcRF) Tier 1: RG4/20, Tier 2: MOET2EP10120-0002, Agency for Science, Technology and Research (A*Star) IRG: A20E5c0080, Jiangsu Specially-Appointed Professor Program, Interdisciplinary Project of Yangzhou University (yzuxk202013) and National Natural Science Foundation of China (No. 22075195).
References
- Z. Song, L. Zhang, K. Doyle-Davis, X. Fu, J. L. Luo and X. Sun, Adv. Energy Mater., 2020, 10, 2001561 CrossRef CAS.
- J. Su, R. Ge, Y. Dong, F. Hao and L. Chen, J. Mater. Chem. A, 2018, 6, 14025–14042 RSC.
- F. Zhang, H. Zhang and Z. Liu, Curr. Opin. Green Sust. Chem., 2019, 16, 77–84 Search PubMed.
- Y. Chen, K. Chen, J. Fu, A. Yamaguchi, H. Li, H. Pan, J. Hu, M. Miyauchi and M. Liu, Nano Mater. Sci., 2020, 2, 235–247 CrossRef.
- S. Piontek, K. Junge Puring, D. Siegmund, M. Smialkowski, I. Sinev, D. Tetzlaff, B. R. Cuenya and U.-P. Apfel, Chem. Sci., 2019, 10, 1075–1081 RSC.
- Z. Chen, X. Zhang, M. Jiao, K. Mou, X. Zhang and L. Liu, Adv. Energy Mater., 2020, 10, 1903664 CrossRef CAS.
- S. Back, J. Lim, N. Y. Kim, Y. H. Kim and Y. Jung, Chem. Sci., 2017, 8, 1090–1096 RSC.
- S. Mou, T. Wu, J. Xie, Y. Zhang, L. Ji, H. Huang, T. Wang, Y. Luo, X. Xiong and B. Tang, Adv. Mater., 2019, 31, 1903499 CrossRef PubMed.
- Q. Gong, P. Ding, M. Xu, X. Zhu, M. Wang, J. Deng, Q. Ma, N. Han, Y. Zhu and J. Lu, Nat. Commun., 2019, 10, 1–10 CrossRef CAS.
- J. Kim, W. Choi, J. W. Park, C. Kim, M. Kim and H. Song, J. Am. Chem. Soc., 2019, 141, 6986–6994 CrossRef CAS PubMed.
- J. Y. Choi, C. K. Lim, B. Park, M. Kim, A. Jamal and H. Song, J. Mater. Chem. A, 2019, 7, 15068–15072 RSC.
- P. Deng, H. Wang, R. Qi, J. Zhu, S. Chen, F. Yang, L. Zhou, K. Qi, H. Liu and B. Y. Xia, ACS Catal., 2019, 10, 743–750 CrossRef.
- L. R. L. Ting, R. García-Muelas, A. J. Martín, F. L. Veenstra, S. T. J. Chen, Y. Peng, E. Y. X. Per, S. Pablo-García, N. López and J. Pérez-Ramírez, Angew. Chem., 2020, 132, 21258–21265 CrossRef.
- R. Matheu, E. Gutierrez-Puebla, M. Á. Monge, C. S. Diercks, J. Kang, M. S. Prévot, X. Pei, N. Hanikel, B. Zhang and P. Yang, J. Am. Chem. Soc., 2019, 141, 17081–17085 CrossRef CAS.
- H. Zhang, X. Chang, J. G. Chen, W. A. Goddard, B. Xu, M.-J. Cheng and Q. Lu, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- C. Kim, F. Dionigi, V. Beermann, X. Wang, T. Möller and P. Strasser, Adv. Mater., 2019, 31, 1805617 CrossRef PubMed.
- N. Martić, C. Reller, C. Macauley, M. Löffler, B. Schmid, D. Reinisch, E. Volkova, A. Maltenberger, A. Rucki and K. J. Mayrhofer, Adv. Energy Mater., 2019, 9, 1901228 CrossRef.
- N. Wanninayake, Q. Ai, R. Zhou, M. A. Hoque, S. Herrell, M. I. Guzman, C. Risko and D. Y. Kim, Carbon, 2020, 157, 408–419 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- L. Wang, W. Zhang, S. Wang, Z. Gao, Z. Luo, X. Wang, R. Zeng, A. Li, H. Li and M. Wang, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- G. X. Pei, X. Y. Liu, X. Yang, L. Zhang, A. Wang, L. Li, H. Wang, X. Wang and T. Zhang, ACS Catal., 2017, 7, 1491–1500 CrossRef CAS.
- C. Zhu, S. Fu, J. Song, Q. Shi, D. Su, M. H. Engelhard, X. Li, D. Xiao, D. Li and L. Estevez, Small, 2017, 13, 1603407 CrossRef PubMed.
- L. Jiao, H. Yan, Y. Wu, W. Gu, C. Zhu, D. Du and Y. Lin, Angew. Chem., 2020, 132, 2585–2596 CrossRef.
- B. Han, R. Lang, B. Qiao, A. Wang and T. Zhang, Chin. J. Catal., 2017, 38, 1498–1507 CrossRef CAS.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- X. Su, X.-F. Yang, Y. Huang, B. Liu and T. Zhang, Acc. Chem. Res., 2018, 52, 656–664 CrossRef PubMed.
- X. Li, X. Yang, J. Zhang, Y. Huang and B. Liu, ACS Catal., 2019, 9, 2521–2531 CrossRef CAS.
- X. Li, H. Y. Wang, H. Yang, W. Cai, S. Liu and B. Liu, Small Methods, 2018, 2, 1700395 CrossRef.
- F. Lü, H. Bao, Y. Mi, Y. Liu, J. Sun, X. Peng, Y. Qiu, L. Zhuo, X. Liu and J. Luo, Sustainable Energy Fuels, 2020, 4, 1012–1028 RSC.
- J. Lin, A. Wang, B. Qiao, X. Liu, X. Yang, X. Wang, J. Liang, J. Li, J. Liu and T. Zhang, J. Am. Chem. Soc., 2013, 135, 15314–15317 CrossRef CAS.
- B. Zhang, H. Asakura, J. Zhang, J. Zhang, S. De and N. Yan, Angew. Chem., 2016, 128, 8459–8463 CrossRef.
- F. Yang, P. Song, X. Liu, B. Mei, W. Xing, Z. Jiang, L. Gu and W. Xu, Angew. Chem., Int. Ed., 2018, 57, 12303–12307 CrossRef CAS PubMed.
- J. M. Bockris and E. Potter, J. Electrochem. Soc., 1952, 99, 169 CrossRef CAS.
- P. C. Vesborg, B. Seger and I. Chorkendorff, J. Phys. Chem. Lett., 2015, 6, 951–957 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- X. Shen, X. Liu, S. Wang, T. Chen, W. Zhang, L. Cao, T. Ding, Y. Lin, D. Liu, L. Wang, W. Zhang and T. Yao, Nano Lett., 2021, 21, 686–692 CrossRef CAS PubMed.
- T. Zhang, X. Han, H. Yang, A. Han, E. Hu, Y. Li, X. Q. Yang, L. Wang, J. Liu and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 12055–12061 CrossRef CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- R. M. Enick and S. M. Klara, Chem. Eng. Commun., 1990, 90, 23–33 CrossRef CAS.
- P. Singh, D. W. Brilman and M. J. Groeneveld, Int. J. Greenhouse Gas Control, 2011, 5, 61–68 CrossRef CAS.
- L. W. Diamond and N. N. Akinfiev, Fluid Phase Equilib., 2003, 208, 265–290 CrossRef CAS.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed.
- S. Yao, S. Hosohara, Y. Shimizu, N. Miura, H. Futata and N. Yamazoe, Chem. Lett., 1991, 20, 2069–2072 CrossRef.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS PubMed.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- Y. Hori, K. Kikuchi and S. Suzuki, Chem. Lett., 1985, 14, 1695–1698 CrossRef.
- W. Li, M. Seredych, E. Rodríguez-Castellón and T. J. Bandosz, ChemSusChem, 2016, 9, 606–616 CrossRef CAS PubMed.
- S. Back and Y. Jung, ACS Energy Lett., 2017, 2, 969–975 CrossRef CAS.
- C. Ao, B. Feng, S. Qian, L. Wang, W. Zhao, Y. Zhai and L. Zhang, J. CO2 Util., 2020, 36, 116–123 CrossRef CAS.
- D. Y. Shin, J. S. Won, J. A. Kwon, M.-S. Kim and D.-H. Lim, Comput. Theor. Chem., 2017, 1120, 84–90 CrossRef CAS.
- M. Le, M. Ren, Z. Zhang, P. T. Sprunger, R. L. Kurtz and J. C. Flake, J. Electrochem. Soc., 2011, 158, E45 CrossRef CAS.
- Y. Kotb, S.-E. K. Fateen, J. Albo and I. Ismail, J. Electrochem. Soc., 2017, 164, E391 CrossRef CAS.
- M. Tang, H. Shen, H. Xie and Q. Sun, ChemPhysChem, 2020, 21, 779–784 CrossRef CAS PubMed.
- L. Ji, L. Li, X. Ji, Y. Zhang, S. Mou, T. Wu, Q. Liu, B. Li, X. Zhu and Y. Luo, Angew. Chem., 2020, 132, 768–772 CrossRef.
- E. Boutin, M. Wang, J. C. Lin, M. Mesnage, D. Mendoza, B. Lassalle-Kaiser, C. Hahn, T. F. Jaramillo and M. Robert, Angew. Chem., Int. Ed., 2019, 58, 16172–16176 CrossRef CAS PubMed.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS.
- S. Kusama, T. Saito, H. Hashiba, A. Sakai and S. Yotsuhashi, ACS Catal., 2017, 7, 8382–8385 CrossRef CAS.
- N. Altaf, S. Liang, R. Iqbal, M. Hayat, T. R. Reina and Q. Wang, J. CO2 Util., 2020, 40, 101205 CrossRef CAS.
- H. Ning, X. Wang, W. Wang, Q. Mao, Z. Yang, Q. Zhao, Y. Song and M. Wu, Carbon, 2019, 146, 218–223 CrossRef CAS.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, Chem. Commun., 1988, 17–19 RSC.
- D. W. DeWulf, T. Jin and A. J. Bard, J. Electrochem. Soc., 1989, 136, 1686 CrossRef CAS.
- C. Genovese, M. E. Schuster, E. K. Gibson, D. Gianolio, V. Posligua, R. Grau-Crespo, G. Cibin, P. P. Wells, D. Garai and V. Solokha, Nat. Commun., 2018, 9, 1–12 CrossRef CAS PubMed.
- R. De, S. Gonglach, S. Paul, M. Haas, S. Sreejith, P. Gerschel, U. P. Apfel, T. H. Vuong, J. Rabeah and S. Roy, Angew. Chem., 2020, 132, 10614–10621 CrossRef.
- G. Feng, W. Chen, B. Wang, Y. Song, G. Li, J. Fang, W. Wei and Y. Sun, Chem.–Asian J., 2018, 13, 1992–2008 CrossRef CAS PubMed.
- C. Chen, J. F. K. Kotyk and S. W. Sheehan, Chem, 2018, 4, 2571–2586 CAS.
- L. Jiao, W. Yang, G. Wan, R. Zhang, X. Zheng, H. Zhou, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 20589–20595 CrossRef CAS PubMed.
- W. Ju, A. Bagger, G. P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef PubMed.
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng, G. Li, Q. Hu, X. Ren, Q. Zhang and J. Liu, Nat. Commun., 2020, 11, 1–8 Search PubMed.
- S. Liu, H. B. Yang, S. F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren and Z. Kuang, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS PubMed.
- Q. He, D. Liu, J. H. Lee, Y. Liu, Z. Xie, S. Hwang, S. Kattel, L. Song and J. G. Chen, Angew. Chem., Int. Ed., 2020, 59, 3033–3037 CrossRef CAS PubMed.
- Y. Cheng, S. Zhao, B. Johannessen, J. P. Veder, M. Saunders, M. R. Rowles, M. Cheng, C. Liu, M. F. Chisholm and R. De Marco, Adv. Mater., 2018, 30, 1706287 CrossRef PubMed.
- P. Lu, Y. Yang, J. Yao, M. Wang, S. Dipazir, M. Yuan, J. Zhang, X. Wang, Z. Xie and G. Zhang, Appl. Catal., B, 2019, 241, 113–119 CrossRef CAS.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed.
- L. Zhang, J. Xiao, H. Wang and M. Shao, ACS Catal., 2017, 7, 7855–7865 CrossRef CAS.
- C. Zhao, Y. Wang, Z. Li, W. Chen, Q. Xu, D. He, D. Xi, Q. Zhang, T. Yuan, Y. Qu, J. Yang, F. Zhou, Z. Yang, X. Wang, J. Wang, J. Luo, Y. Li, H. Duan, Y. Wu and Y. Li, Joule, 2019, 3, 584–594 CrossRef CAS.
- J. Feng, H. Gao, L. Zheng, Z. Chen, S. Zeng, C. Jiang, H. Dong, L. Liu, S. Zhang and X. Zhang, Nat. Commun., 2020, 11, 4341 CrossRef PubMed.
- J. Zhang, H. Yang and B. Liu, Adv. Energy Mater., 2021, 11, 2002473 CrossRef CAS.
- G. Gao, S. Bottle and A. Du, Catal. Sci. Technol., 2018, 8, 996–1001 RSC.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- Y. Wang, G. Jia, X. Cui, X. Zhao, Q. Zhang, L. Gu, L. Zheng, L.-H. Li, Q. Wu and D. J. Singh, Chem, 2021, 7, 436–449 CAS.
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Nano Res., 2020, 13, 1842–1855 CrossRef CAS.
- Y. Lin, P. Liu, E. Velasco, G. Yao, Z. Tian, L. Zhang and L. Chen, Adv. Mater., 2019, 31, 1808193 CrossRef.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang and X. Meng, Angew. Chem., 2016, 128, 14522–14526 CrossRef.
- D. Yang, H. Yu, T. He, S. Zuo, X. Liu, H. Yang, B. Ni, H. Li, L. Gu and D. Wang, Nat. Commun., 2019, 10, 1–10 CrossRef.
- L. Sun, Z. Huang, V. Reddu, T. Su, A. C. Fisher and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 17104–17109 CrossRef CAS.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty and D. Su, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- J. Liu, X. Kong, L. Zheng, X. Guo, X. Liu and J. Shui, ACS Nano, 2020, 14, 1093–1101 CrossRef CAS PubMed.
- N. Wang, Z. Liu, J. Ma, J. Liu, P. Zhou, Y. Chao, C. Ma, X. Bo, J. Liu and Y. Hei, ACS Sustainable Chem. Eng., 2020, 8, 13813–13822 CrossRef CAS.
- Y. Cheng, S. Yang, S. P. Jiang and S. Wang, Small Methods, 2019, 3, 1800440 CrossRef.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai and R. Chen, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- B. Zhang, J. Zhang, J. Shi, D. Tan, L. Liu, F. Zhang, C. Lu, Z. Su, X. Tan and X. Cheng, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- W. Zhu, L. Zhang, S. Liu, A. Li, X. Yuan, C. Hu, G. Zhang, W. Deng, K. Zang, J. Luo, Y. Zhu, M. Gu, Z. J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2020, 59, 12664–12668 CrossRef CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS.
- X. Ren, S. Liu, H. Li, J. Ding, L. Liu, Z. Kuang, L. Li, H. Yang, F. Bai, Y. Huang, T. Zhang and B. Liu, Sci. China: Chem., 2020, 63, 1727–1733 CrossRef CAS.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Nat. Commun., 2017, 8, 14675 CrossRef PubMed.
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Işci and C. P. Berlinguette, Nat. Commun., 2019, 10, 1–8 CrossRef.
- X. Zu, X. Li, W. Liu, Y. Sun, J. Xu, T. Yao, W. Yan, S. Gao, C. Wang, S. Wei and Y. Xie, Adv. Mater., 2019, 31, e1808135 CrossRef PubMed.
- W. Xie, H. Li, G. Cui, J. Li, Y. Song, S. Li, X. Zhang, J. Y. Lee, M. Shao and M. Wei, Angew. Chem., Int. Ed., 2020, 60, 7382–7388 CrossRef PubMed.
- Z. Jiang, T. Wang, J. Pei, H. Shang, D. Zhou, H. Li, J. Dong, Y. Wang, R. Cao, Z. Zhuang, W. Chen, D. Wang, J. Zhang and Y. Li, Energy Environ. Sci., 2020, 13, 2856–2863 RSC.
- P. Huang, M. Cheng, H. Zhang, M. Zuo, C. Xiao and Y. Xie, Nano Energy, 2019, 61, 428–434 CrossRef CAS.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS.
- Z. Wang, J. Zhao and Q. Cai, Phys. Chem. Chem. Phys., 2017, 19, 23113–23121 RSC.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed.
- L. Han, S. Song, M. Liu, S. Yao, Z. Liang, H. Cheng, Z. Ren, W. Liu, R. Lin, G. Qi, X. Liu, Q. Wu, J. Luo and H. L. Xin, J. Am. Chem. Soc., 2020, 142, 12563–12567 CrossRef CAS PubMed.
- Y. Wang, Z. Chen, P. Han, Y. Du, Z. Gu, X. Xu and G. Zheng, ACS Catal., 2018, 8, 7113–7119 CrossRef CAS.
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvao Tizei, A. Zitolo, F. Jaouen, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean, R. E. Winans, D.-J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 2455 CrossRef CAS PubMed.
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T.-K. Sham, C. Yang, Y. Ji, L. Qian, X. Xu and G. Zheng, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS.
- Y. Jiao, Y. Zheng, P. Chen, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2017, 139, 18093–18100 CrossRef CAS.
- J. Gao, H. Zhang, X. Guo, J. Luo, S. M. Zakeeruddin, D. Ren and M. Grätzel, J. Am. Chem. Soc., 2019, 141, 18704–18714 CrossRef CAS PubMed.
- X. Wang, J. F. de Araújo, W. Ju, A. Bagger, H. Schmies, S. Kühl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS PubMed.
- X. She, T. Zhang, Z. Li, H. Li, H. Xu and J. Wu, Cell Rep. Phys. Sci., 2020, 1, 100051 CrossRef.
- L. Lin, T. Liu, J. Xiao, H. Li, P. Wei, D. Gao, B. Nan, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2020, 59, 22408–22413 CrossRef CAS.
- T. Zhang, A. G. Walsh, J. Yu and P. Zhang, Chem. Soc. Rev., 2021, 50, 569–588 RSC.
- R. T. Hannagan, G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Chem. Rev., 2020, 120, 12044–12088 CrossRef CAS PubMed.
- N. Zhang, X. Zhang, L. Tao, P. Jiang, C. Ye, R. Lin, Z. Huang, A. Li, D. Pang and H. Yan, Angew. Chem., Int. Ed., 2021, 60, 6170–6176 CrossRef CAS PubMed.
- Y. Jiang, C. Choi, S. Hong, S. Chu, T.-S. Wu, Y.-L. Soo, L. Hao, Y. Jung and Z. Sun, Cell Rep. Phys. Sci., 2021, 2, 100356 CrossRef.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang and H. Sun, Nat. Catal., 2018, 1, 63–72 CrossRef CAS.
- M. Xiao, J. Zhu, L. Ma, Z. Jin, J. Ge, X. Deng, Y. Hou, Q. He, J. Li and Q. Jia, ACS Catal., 2018, 8, 2824–2832 CrossRef CAS.
- H. Yang, L. Shang, Q. Zhang, R. Shi, G. I. Waterhouse, L. Gu and T. Zhang, Nat. Commun., 2019, 10, 1–9 CrossRef.
- F. De Groot, Chem. Rev., 2001, 101, 1779–1808 CrossRef CAS.
- J. Yano and V. K. Yachandra, Photosynth. Res., 2009, 102, 241 CrossRef CAS PubMed.
-
D. Koningsberger and R. Prins, X-ray absorption: principles, applications, techniques of EXAFS, SEXAFS, and XANES, 1988 Search PubMed.
- Y. Gao, Z. Cai, X. Wu, Z. Lv, P. Wu and C. Cai, ACS Catal., 2018, 8, 10364–10374 CrossRef CAS.
- Z. Lu, P. Lv, Y. Liang, D. Ma, Y. Zhang, W. Zhang, X. Yang and Z. Yang, Phys. Chem. Chem. Phys., 2016, 18, 21865–21870 RSC.
- Y. Feng, Q. Wan, H. Xiong, S. Zhou, X. Chen, X. I. Pereira Hernandez, Y. Wang, S. Lin, A. K. Datye and H. Guo, J. Phys. Chem. C, 2018, 122, 22460–22468 CrossRef CAS.
- L. Qin, Y. Q. Cui, T. L. Deng, F. H. Wei and X. F. Zhang, ChemPhysChem, 2018, 19, 3346–3349 CrossRef CAS PubMed.
- R. Brimblecombe, A. Koo, G. C. Dismukes, G. F. Swiegers and L. Spiccia, J. Am. Chem. Soc., 2010, 132, 2892–2894 CrossRef CAS PubMed.
- F. Li, B. Zhang, X. Li, Y. Jiang, L. Chen, Y. Li and L. Sun, Angew. Chem., 2011, 123, 12484–12487 CrossRef.
- S. W. Sheehan, J. M. Thomsen, U. Hintermair, R. H. Crabtree, G. W. Brudvig and C. A. Schmuttenmaer, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- P. Garrido-Barros, C. Gimbert-Suriñach, R. Matheu, X. Sala and A. Llobet, Chem. Soc. Rev., 2017, 46, 6088–6098 RSC.
- B. C. Martindale, G. A. Hutton, C. A. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018–6025 CrossRef CAS PubMed.
- J. Liu, B. R. Bunes, L. Zang and C. Wang, Environ. Chem. Lett., 2018, 16, 477–505 CrossRef CAS.
- A. Vasileff, Y. Zhu, X. Zhi, Y. Zhao, L. Ge, H. M. Chen, Y. Zheng and S. Z. Qiao, Angew. Chem., 2020, 59, 19649–19653 CrossRef CAS PubMed.
- C. Xu, A. Vasileff, Y. Zheng and S. Z. Qiao, Adv. Mater. Interfaces, 2021, 8, 2001904 CrossRef CAS.
- C. Xu, X. Zhi, A. Vasileff, D. Wang, B. Jin, Y. Jiao, Y. Zheng and S.-Z. Qiao, Small Structures, 2021, 2, 2000058 CrossRef.
- S. Mukerjee, T. Thurston, N. Jisrawi, X. Yang, J. McBreen, M. Daroux and X. Xing, J. Electrochem. Soc., 1998, 145, 466 CrossRef CAS.
- S. Cheong, J. Watt, B. Ingham, M. F. Toney and R. D. Tilley, J. Am. Chem. Soc., 2009, 131, 14590–14595 CrossRef CAS PubMed.
- F. Hui, C. Li, Y. Chen, C. Wang, J. Huang, A. Li, W. Li, J. Zou and X. Han, Nano Res., 2020, 13, 3019–3024 CrossRef CAS.
- W. Yang, Y. Kim, P. K. Liu, M. Sahimi and T. T. Tsotsis, Chem. Eng. Sci., 2002, 57, 2945–2953 CrossRef CAS.
- K. Rhodes, R. Meisner, Y. Kim, N. Dudney and C. Daniel, J. Electrochem. Soc., 2011, 158, A890 CrossRef CAS.
- M. Asadi, B. Kumar, A. Behranginia, B. A. Rosen, A. Baskin, N. Repnin, D. Pisasale, P. Phillips, W. Zhu and R. Haasch, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- X. Liu, J. Xiao, H. Peng, X. Hong, K. Chan and J. K. Nørskov, Nat. Commun., 2017, 8, 1–7 CrossRef PubMed.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., 2015, 127, 5268–5271 CrossRef.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- A. D. Handoko, F. Wei, B. S. Yeo and Z. W. Seh, Nat. Catal., 2018, 1, 922–934 CrossRef CAS.
- B. Mondal, J. Song, F. Neese and S. Ye, Curr. Opin. Chem. Biol., 2015, 25, 103–109 CrossRef CAS.
- L. R. L. Ting and B. S. Yeo, Curr. Opin. Electrochem., 2018, 8, 126–134 CrossRef CAS.
- S. Ringe, C. G. Morales-Guio, L. D. Chen, M. Fields, T. F. Jaramillo, C. Hahn and K. Chan, Nat. Commun., 2020, 11, 1–11 Search PubMed.
- A. Bachmeier and F. Armstrong, Curr. Opin. Chem. Biol., 2015, 25, 141–151 CrossRef CAS.
- X. Li, L. Liu, X. Ren, J. Gao, Y. Huang and B. Liu, Sci. Adv., 2020, 6, eabb6833 CrossRef CAS PubMed.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS.
- J. Zhang, H. Yang, J. Gao, S. Xi, W. Cai, J. Zhang, P. Cui and B. Liu, Carbon Energy, 2020, 2, 276–282 CrossRef CAS.
- J. Gao, H. bin Yang, X. Huang, S.-F. Hung, W. Cai, C. Jia, S. Miao, H. M. Chen, X. Yang and Y. Huang, Chem, 2020, 6, 658–674 CAS.
- J. Gao and B. Liu, ACS Mater. Lett., 2020, 2, 1008–1024 CrossRef CAS.
- L. Huang, J. Chen, L. Gan, J. Wang and S. Dong, Sci. Adv., 2019, 5, eaav5490 CrossRef CAS PubMed.
- S. Liang, C. Hao and Y. Shi, ChemCatChem, 2015, 7, 2559–2567 CrossRef CAS.
- T. Frey and M. Linardi, Electrochim. Acta, 2004, 50, 99–105 CrossRef CAS.
-
R. H. Barton, P. R. Gibb, J. A. Ronne and H. H. Voss, Membrane Electrode Assembly for an Electrochemical Fuel Cell and a Method of Making an Improved Membrane Electrode Assembly, US Pat., 6057054, 2000 Search PubMed.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Energy Environ. Sci., 2019, 12, 1950–1968 RSC.
- J. K. Jang, H. S. Moon, I. S. Chang and B. H. Kim, J. Microbiol. Biotechnol., 2005, 15, 438–441 Search PubMed.
- W. Sun, W. Zhang, H. Su, P. Leung, L. Xing, L. Xu, C. Yang and Q. Xu, Int. J. Hydrogen Energy, 2019, 44, 32231–32239 CrossRef CAS.
- S.-F. Hung, Pure Appl. Chem., 2020, 92, 1937–1951 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a