
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect amidation of metallaaromatics: access to N-functionalized osmapentalynes via a 1,5-bromoamidated intermediate†
Hongjian
Wang
 a,
Yonghong
Ruan
a,
Yu-Mei
Lin
*a and
Haiping
Xia
*ab
a,
Yonghong
Ruan
a,
Yu-Mei
Lin
*a and
Haiping
Xia
*ab
aState Key Laboratory of Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: linyum@xmu.edu.cn; hpxia@xmu.edu.cn
bShenzhen Grubbs Institute, Department of Chemistry, Southern University of Science and Technology, Shenzhen 518055, China
First published on 7th April 2021
Abstract
The direct C–H amidation or imidation of metallaaromatics with N-bromoamides or imides has been achieved under mild conditions and leads to the formation of a family of N-functionalized metallapentalyne derivatives. A unique 1,5-bromoamidated species has been identified, and can be viewed as a σH-adduct intermediate in a nucleophilic aromatic substitution. The 1,5-addition of both electrophilic and nucleophilic moieties into the metallaaromatic framework demonstrates a novel pathway in contrast to the typical radical process of arene C–H amidation involving N-haloamide reagents.
Introduction
The direct C–H amidation of arenes provides an efficient and economical approach to construction of valuable C–N bonds avoiding the use of prefunctionalized aromatic components.1 Various catalysts and effective amidation reagents have been explored and have led to improvements in the scope of substrates and functional group tolerance.2 Activated N–X amide reagents, such as N-haloimides or N-acyloxyimides, typically undergo homolytic cleavage to generate a nitrogen-centered radical, which can undergo N-aryl bond formation.3,4 The reactions usually proceed through photoredox catalysis,4e,4f and are potential alternatives to the transition metal mediated catalysis assisted by directing groups.Transition-metal-containing metallaaromatic compounds are analogues of the corresponding organic aromatic compounds,5 and have attracted growing interest.6–13 Two main strategies have been developed for the construction of the N-aryl bond in metallaaromatic compounds. Electrophilic substitution reaction of metallaaromatic compounds is a convenient route to obtain nitro-substituted derivatives,14 which are useful for further N-functionalization.15 For example, Wright et al. synthesized amido-substituted and imido-substituted fused-ring metallabenzenes starting from nitro-substituted iridabenzofurans via multiple consecutive reactions.15 As an alternative, by pre-installation of a functional group on the metallacycle, intramolecular nucleophilic aromatic substitutions and cine-substitution reactions can be realized to access N-functionalization metallaaromatics.16 Despite these impressive advances, approaches to amido-substituted metallaaromatic usually require multistep pathways or prefunctionalized starting materials. Direct C–H amidation has emerged as a step- and atom-economical alternative, but is elusive and its successful completion is challenging.
Herein we report the direct amidation of metallaaromatics with N-bromoamides and N-bromoimides, respectively. A planar Möbius metallaaromatic, osmapentalyne, was employed as a model substrate and a series of N-functionalized metallapentalyne derivatives have been synthesized under mild conditions at room temperature (rt) (Scheme 1). The identified 1,5-bromoamidation metallacyclic intermediate suggests that the reaction proceeds a unique pathway involving electrophilic addition and nucleophilic attack. This is quite different from the typical radical process of organic arene C–H amidation involving N-haloamide reagents.
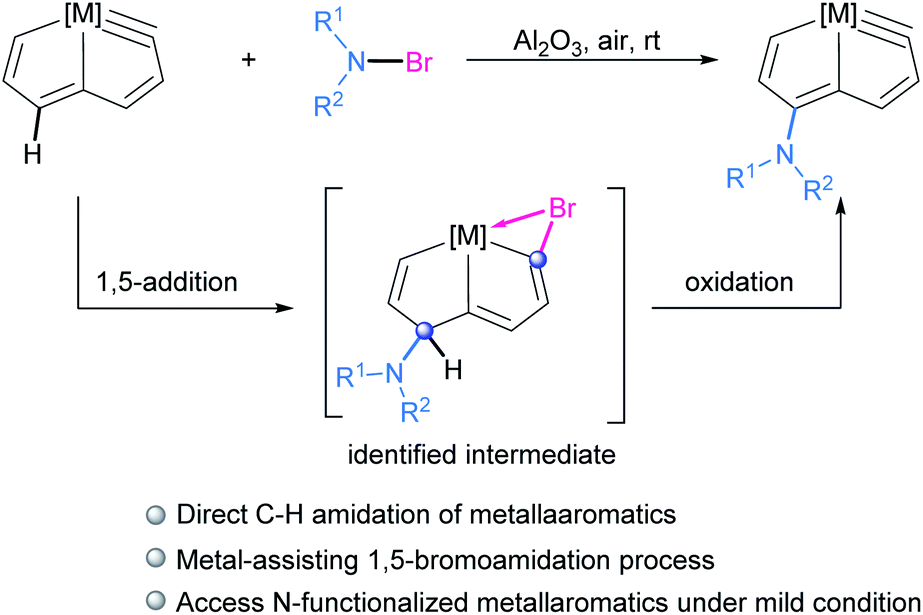 | ||
| Scheme 1 Direct amidation of metallaaromatics via 1,5-bromoamidated intermediate. | ||
Results and discussion
Synthesis and characterization of osmapentalyne 1
We have previously reported some interesting metallaaromatics, i.e. metallapentalynes containing a metal–carbon triple bond in a five-membered ring, which exhibit the rare planar Möbius aromaticity.8b,17 In order to investigate the direct amidation strategy to obtain its N-functionalized derivatives, we envisioned that metallapentalyne with relatively electron-deficient groups would participate in the substitution of hydrogen by the amidating reagent. Accordingly, we designed and synthesized the tri-substituted osmapentalyne 1. Details of the synthesis and characterization are in the ESI (see ESI, p. S4†). As shown in Fig. 1, the metallacycle contains an osmium shared by two fused five-membered rings. The eight atoms (Os1, C1–C7) are coplanar as reflected by the mean deviation of 0.010 Å from the least squares plane and the sum of angles in the fused five-membered rings, which are both 540.0°. The coplanarity as well as the Os–C and C–C bond distances in the fused five membered ring indicates it is a delocalized metallabicyclic compound, which resembles our previously reported aromatic osmapentalynes.8b,17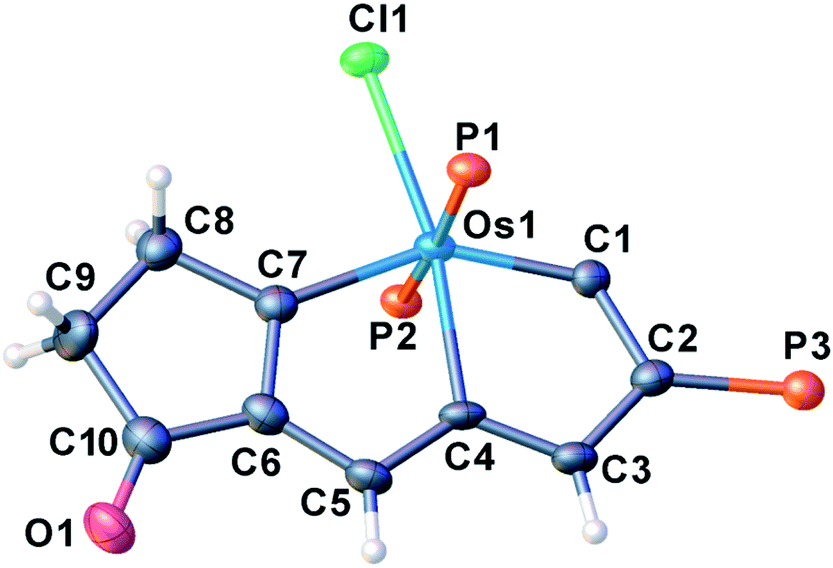 | ||
| Fig. 1 X-ray crystal structure of the cation of 1 (thermal ellipsoids are set at the 50% probability level and the phenyl groups in the PPh3 moieties are omitted for clarity). Selected bond lengths [Å] and angles [°]: Os1–C1: 1.858(3), Os1–C4: 2.106(3), Os1–C7: 2.045(3), C1–C2: 1.398(4), C2–C3: 1.398(5), C3–C4: 1.398(5), C4–C5: 1.397(5), C5–C6: 1.410(5), C6–C7: 1.396(5), C4–Os1–C1: 72.31(13), C7–Os1–C4: 75.01(13). | ||
Direct C–H amidation or imidation of osmapentalyne 1: procedure, scope and characterization of products
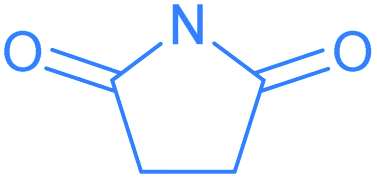
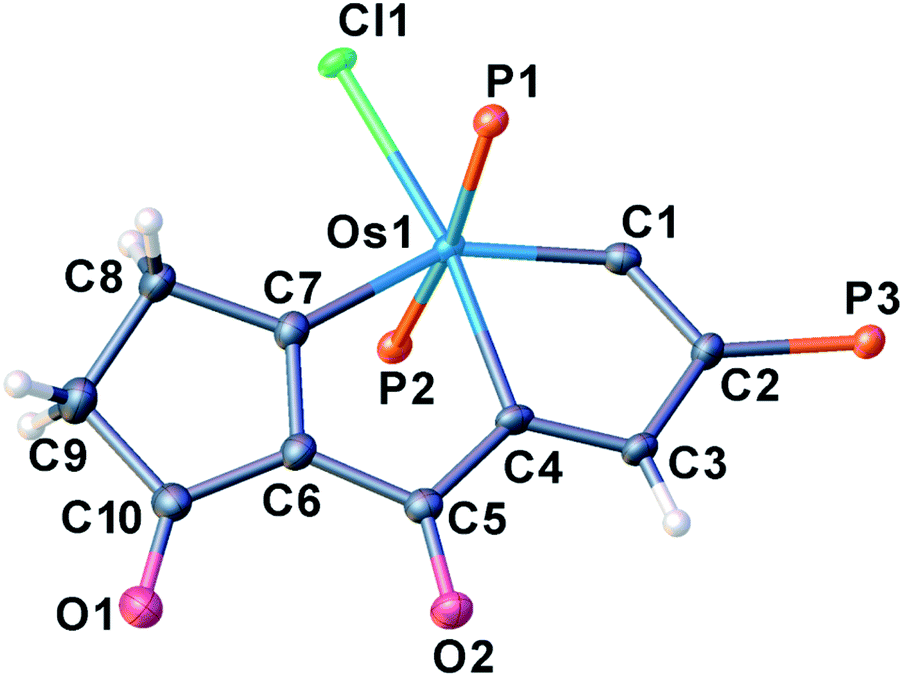
We investigated the reactions of osmapentalyne 1 with various amidating reagents (Table 1). When 1 was reacted with N-bromophthalimide (NBP) in the presence of aluminium oxide in air at rt for 12 h, complex 2a was formed in 92% yield. The product 2a can also be obtained in 81% yield when the reaction mixture was treated with CH3COONa instead of Al2O3 (see ESI, p. S5†). Treatment of compound 1 with N-bromosuccinimide (NBS) under similar conditions led to the formation of 2b in 90% yield. Complexes 2a and 2b were characterized by X-ray crystallographic analysis. As shown in Fig. 2, the metallacyclic skeleton of 2a is similar to that of 1, with the exception of the phthalimido substitute attached to C5. The length of the C5–N1 bond (1.430(3) Å) indicates its single-bond character, and is similar to the reported N-aryl single bond (1.464(4) Å) in the imide-substituted metallaaromatics.15 The dihedral angle between the metallabicyclic unit and the imide group is 62.40°. The Os1–C1 bond length (1.842(2) Å) suggests that it maintains the Os![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond character. The C–C bond lengths (1.388–1.414 Å) in the fused five-membered rings are intermediate between the lengths of C–C and C
C triple bond character. The C–C bond lengths (1.388–1.414 Å) in the fused five-membered rings are intermediate between the lengths of C–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and show no significant bond-length alternation. The planarity and the bond lengths in the fused five membered rings indicate it is a delocalized metallabicyclic compound. All these parameters are consistent with 2a being a C5-phthalimido-substituted osmapentalyne derivative.
C bonds and show no significant bond-length alternation. The planarity and the bond lengths in the fused five membered rings indicate it is a delocalized metallabicyclic compound. All these parameters are consistent with 2a being a C5-phthalimido-substituted osmapentalyne derivative.



 | ||
| Fig. 2 X-ray crystal structure of the cation of 2a (thermal ellipsoids are set at the 50% probability level and the phenyl groups in the PPh3 moieties are omitted for clarity). Selected bond lengths [Å] and angles [°]: Os1–C1: 1.842(2), Os1–C4: 2.111(2), Os1–C7: 2.044(2), C1–C2: 1.404(3), C2–C3: 1.404(3), C3–C4: 1.414(3), C4–C5: 1.397(3), C5–C6: 1.397(3), C6–C7: 1.388(3), C5–N1: 1.430(3), C4–Os1–C1: 72.32(9), C7–Os1–C4: 75.89(9). | ||
The solid-state structure of 2a is fully supported by NMR spectroscopy. In the 1H NMR spectrum, the characteristic signal of H3 (δ = 8.30 ppm) appears in the aromatic region. Carbon C5 bearing the phthalimido substitute has a peak at δ = 140.3 ppm. In the 31P{1H} NMR spectrum of 2a, only one singlet (δ = −0.18 ppm) is observed for the two PPh3 ligands, indicating that the two phosphorus atoms are equivalent.
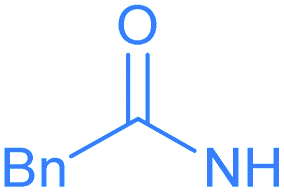
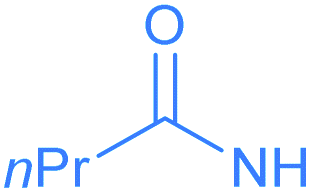
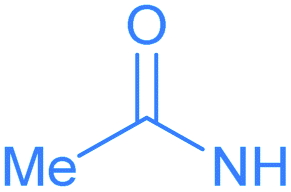
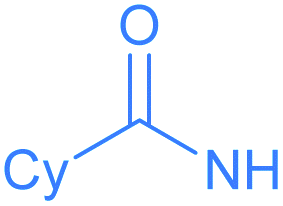
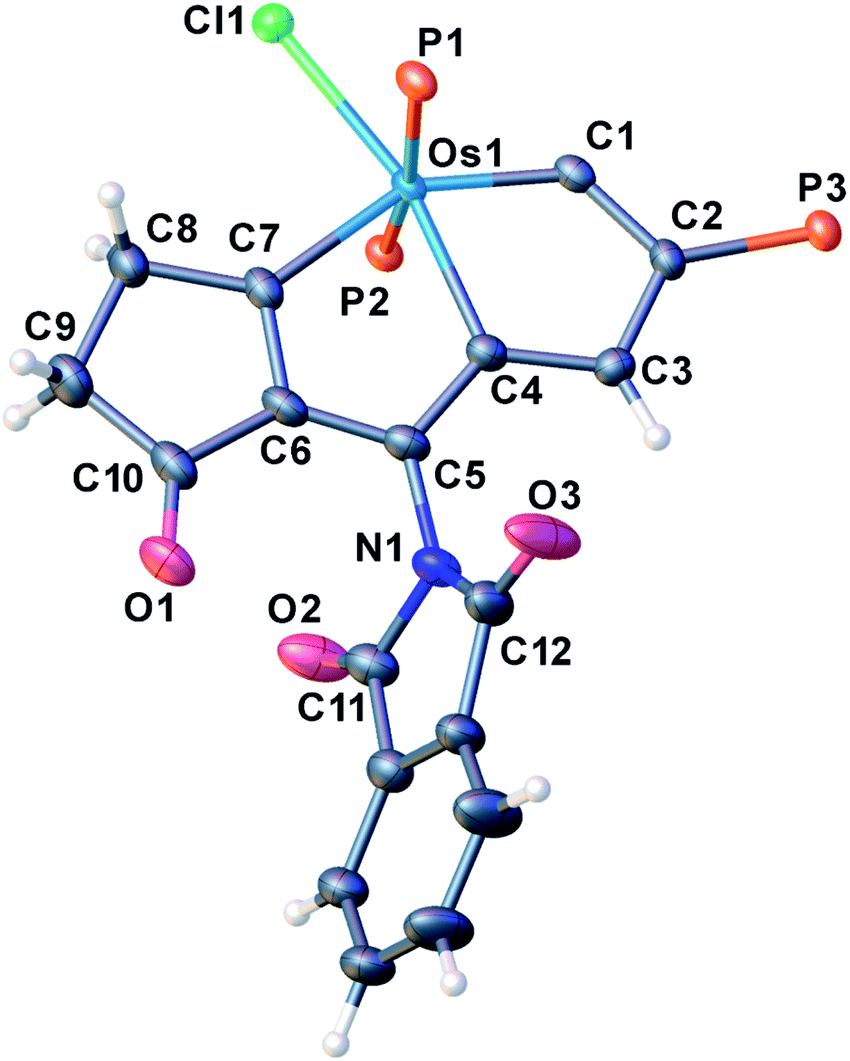
Encouraged by the effective imidation of osmapentalyne with NBP and NBS, we then examined other amidating reagents. N-Bromocarboxamides substituted with different groups were subjected to the standard reaction conditions. The methyl, phenyl, benzyl, n-propyl and cyclohexyl groups were all well-tolerated, yielding the corresponding products in moderate to excellent yields (71–93%, 2c–2g) (Table 1). Complexes 2c, 2d and 2e have been fully characterized by X-ray crystallographic analysis and all the structures were further supported by NMR spectroscopy and high-resolution mass spectrometry (HRMS) (see ESI p. S44–S53†).
The structural parameters of the fused rings in compound 2c resemble those in 2a (Fig. 3). The acetamide group attached at C5 position is indicated by the N1–C5 bond length (1.387(11) Å), which is close to that of the reported N-aryl single bond (1.391(4) Å) in osmabenzoxazolone.16c The intramolecular hydrogen bonds N1H⋯O1 (2.107 Å) and H3⋯O2 (2.105 Å) were also observed in the acetamide-substituted osmapentalyne.
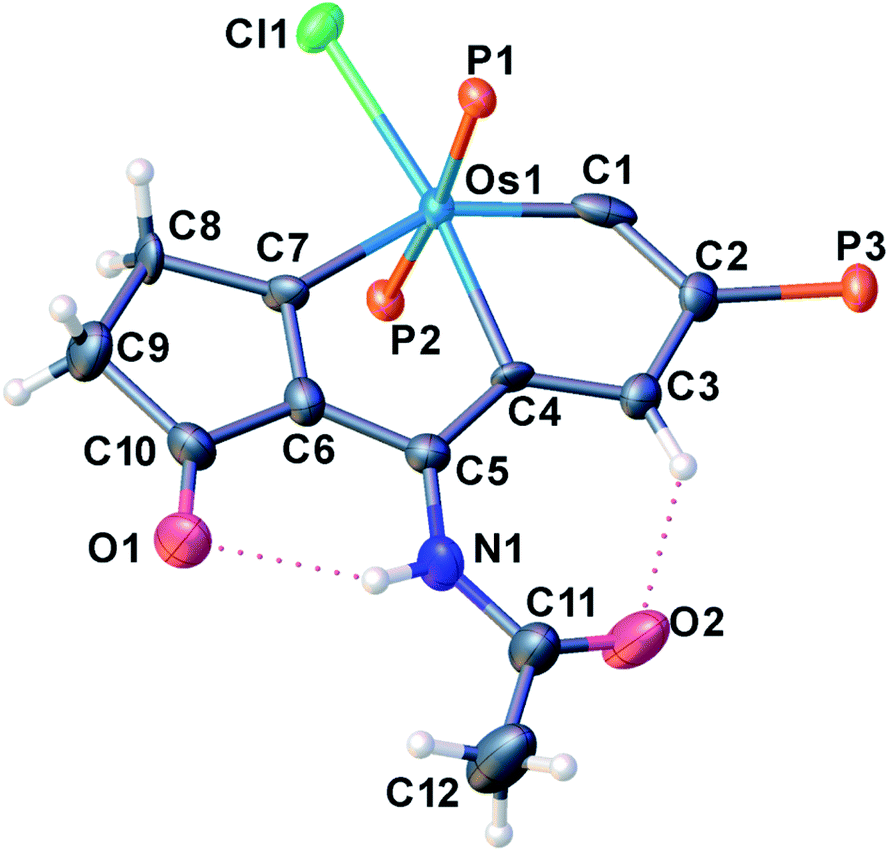 | ||
| Fig. 3 X-ray crystal structure of the cation of 2c (thermal ellipsoids are set at the 50% probability level and the phenyl groups in the PPh3 moieties are omitted for clarity). Selected bond lengths [Å] and angles [°]: Os1–C1: 1.818(10), Os1–C4: 2.146(7), Os1–C7: 2.057(8), C1–C2: 1.411(12), C2–C3: 1.415(11), C3–C4: 1.394(10), C4–C5: 1.411(11), C5–C6: 1.427(11), C6–C7: 1.356(11), C5–N1: 1.387(11), N1H⋯O1: 2.107, H3⋯O2: 2.105 C4–Os1–C1: 72.6(3), C7–Os1–C4: 75.7(3). | ||
Investigation of the mechanism: identification of key intermediates and control experiments
Control experiments were performed to elucidate the mechanism of the reaction (Table 2). It is well known that NBS or NBP can generally participate either in radical or ionic pathways depending on the cleavage of the N–Br bond.18 When the reaction was conducted in the dark, no influence on the reaction efficiency and yield was observed (entry 2). Addition of the radical scavenger BHT (2,6-di-tert-butyl-4-methylphenol) to the reaction mixture under the standard conditions had no obvious effect on the product yield (entry 3). This result suggests that a radical pathway is unlikely.|
|
|||
|---|---|---|---|
| Entry |
|
Additive | Yieldb (%) |
| a Reaction conditions: 1 (0.20 mmol) with NXS (0.60 mmol) and Al2O3 (5.0 mmol) in CH2Cl2 (10.0 mL) for 12 h at rt in air. b Isolated yield. c In the dark. d The reaction was carried out in the presence of BHT (1.0 mmol). e Only a mixture of unidentified species was detected. [Os] = OsCl(PPh3)2. | |||
| 1 | Br | — | 90 |
| 2c | Br | — | 90 |
| 3d | Br | BHT | 88 |
| 4 | I | — | 0 |
| 5 | Cl | — | 0e |
| 6 | OH | — | 0 |
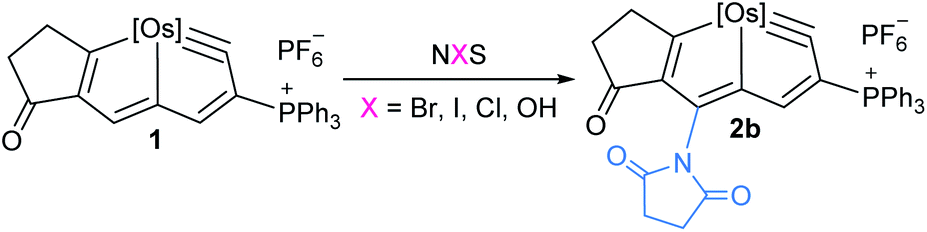

The in situ NMR showed that compound 3a can be identified from the reaction of 1 with NBP within 15 min (Scheme 2). HRMS spectrometry showed a dominant peak at m/z = 1380.1864, consistent with the adduct of cationic 1 and NBP [C72H55BrClNO3OsP3]+ (1380.1855) (see ESI p. S56†). The structure can be inferred from the NMR spectra, which indicates that it is a bromoamidation product with bromide and nitrogen moieties at C1 and C5 positions, respectively. The 1H NMR spectrum of 3a displays a signal for the H5 proton at δ = 6.12 ppm determined by 1H–13C heteronuclear single quantum coherence (HSQC) spectroscopy, which is significantly shifted to a higher field when compared to that of 1 (δ = 9.46 ppm). The signal from C5 (δ = 57.5 ppm) is dramatically shifted upfield when compared to the corresponding resonance observed in 1 (δ = 147.0 ppm) and 2a (δ = 140.3 ppm). The values strongly support the sp3 hybridization state of C5. In addition, the H3 proton signal (δ = 6.22 ppm) is also moved upfield compared with that of 1 (δ = 8.11 ppm) and previously reported metallapentalenes (ca. 7–9 ppm),19 but is comparable with those observed in osmacyclopentadiene derivatives (δ = 6.41 ppm).20 Notably, the 13C{1H} NMR and DEPT-135 spectra confirmed the C1 as a quaternary carbon at δ = 198.3 ppm. The chemical shift is upfield compared with those found for related osmacyclopentadienes (ca. δ = 218–235 ppm) due to the bromide attached at C1.20,21 The resonances of the two carbonyl carbons in the substituted imide group were observed at δ = 169.7 and 166.2 ppm, respectively. The 31P{1H} NMR spectrum shows signals from the two phosphorus atoms at δ = −22.68 and −21.11 ppm, respectively. These data all suggest that 3a is a dearomatization product derived from the 1,5-addition of NBP to 1 (Table 3).
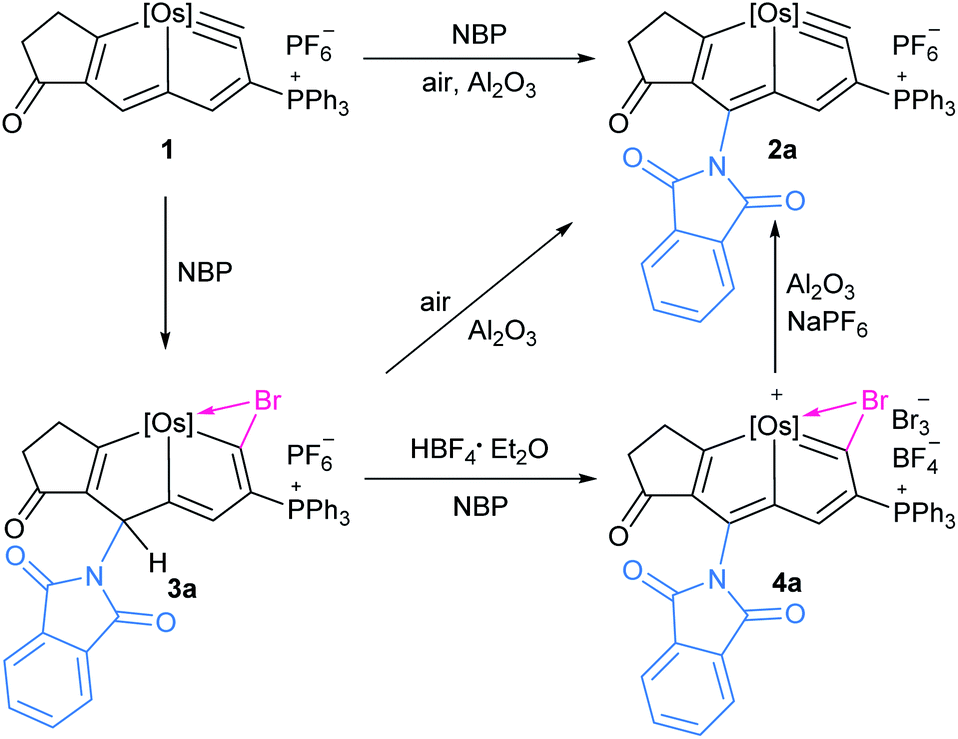 | ||
| Scheme 2 Trapping intermediates 3a and 4a in the formation of 2a from 1, [Os] = OsCl(PPh3)2. | ||
| Compound | δ(1H) (ppm) | δ(13C) (ppm) | ||
|---|---|---|---|---|
| H3 | H5 | C1 | C5 | |
| 1 | 8.11 | 9.46 | 324.4 | 147.0 |
| 2a | 8.30 | — | 327.1 | 140.3 |
| 3a | 6.22 | 6.12 | 198.3 | 57.5 |
| 4a | 8.13 | — | 270.7 | 151.0 |
The reaction of 1 with NBS could also give rise to the analogous 1,5-addition product 3b. Complex 3a and 3b could be recognized as the intermediate σH-adduct in nucleophilic aromatic substitution reactions.22 Strong nucleophiles such as RO−, H−, NH2− have participated in the formation of the isolated σH-adducts.23 It is challenging however for weak nucleophiles such as an imide anion to attack the aromatic ring. The isolation of a σH-adduct involving imide anion is limited to extremely electron-deficient aromatics.24
Haloamidation of an unsaturated bond using an electrophilic halogen source such as NBS is an important and well documented synthetic route to vicinal haloamide compounds.25 In contrast, the reactions of amide reagents toward arenes or heteroarenes usually result in either imidation or halogenation.26 In our case, the unique feature of osmapentalyne permits the bromine addition to the OsC carbyne, promoting the unusual 1,5-bromoamidation of metallaaromatics. This provides an economical route to access difunctionalized metallacycles.
Treatment of 3a with excessive amounts of NBP as an oxidant in the presence of HBF4·Et2O, led to the formation of the oxidation product 4a (Scheme 2). The solid-state structure of 4a was characterized by X-ray single-crystal diffraction (Fig. 4). The bond lengths of Os1–C1 (1.949(5) Å) and C1–Br1 (1.896(5) Å) as well as Br1–C1–Os1 bond angle (90.2(2)°) are comparable to those of the reported metallabromirenium compound (1.935 (11) Å, 1.872(10) Å and Br1–C7–Os1 95.8(5)°, respectively).27 The Os1–Br1 bond length (2.7241(5) Å) was slightly shorter than that of the reported metallabromirenium (2.8254(12) Å), demonstrating the stronger interaction between the bromide atom and the metal center. The other metal–carbon bonds, Os1–C4 (2.146(5) Å) and Os1–C7 (2.023(5) Å) and the C–C bond lengths (1.366–1.425 Å) are within the range observed for 2a and other osmapentalenes,19a suggesting the rearomatization of 4a. The NMR data are consistent with the crystal structure. In particular, the signal from C1 appears at δ = 270.7 ppm, suggesting the carbene character of C1. This differs from the Os–C single bond observed in 3a (198.3 ppm). Signals from the remaining ring carbon atoms are observed at δ = 159.8 (C2), 125.4 (C3), 174.1 (C4), 151.0 (C5), 164.3 (C6) and 220.2 (C7) ppm, respectively. The 1H NMR spectrum of 4a also shows the signal for H3 at δ = 8.13 ppm in the aromatic region. The singlet peak of the two PPh3 ligands at δ = −5.11 ppm in the 31P{1H} NMR spectrum of 4a reflects the reconstruction of a molecular mirror plane.
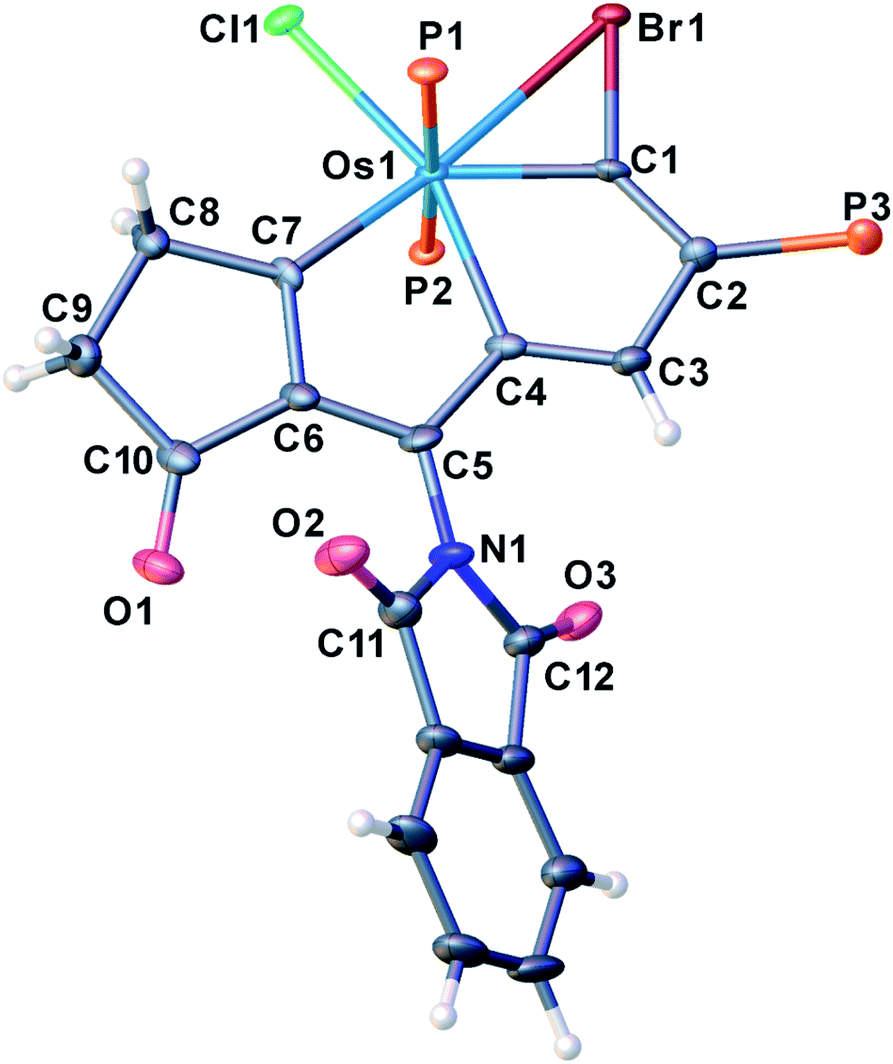 | ||
| Fig. 4 X-ray crystal structure of the cation of 4a (thermal ellipsoids are set at the 50% probability level and the phenyl groups in the PPh3 moieties are omitted for clarity). Selected bond lengths [Å] and angles [°]: Os1–C1: 1.949(5), Os1–C4: 2.146(5), Os1–C7: 2.023(5), Os1–Br1: 2.7241(5), C1–Br1: 1.896(5), C1–C2: 1.366(7), C2–C3: 1.425(7), C3–C4: 1.377(7), C4–C5: 1.414(7), C5–C6: 1.393(7), C6–C7: 1.389(7), C5–N1: 1.379(7), C4–Os1–C1: 68.38(19), C7–Os1–C4: 74.12(19), Os1–C1–Br1: 90.2(2), Br–Os1–C1: 44.10(13), Os1–Br1–C1: 45.67(15). | ||
Experimentally, N-halogen imides other than NBS were used in the reactions (Table 2). No reaction was observed with N-iodosuccinimide (NIS), which might only reflect the poor reactivity of N-iodoimides (entry 4). When NCS was used in the reaction, a mixture of unidentified species was produced (entry 5). The smaller atomic size of the chlorine atom is known to make it harder to sustain the unsaturated three membered ring due to the imperfect orbital overlap.27 In addition, upon replacement of NBS by N-hydroxysuccinimide (NHS), no desired product was observed (entry 6). The bromide cation appears to play a vital role in the reaction.
Elimination of the bromine cation from 4a could generate the osmapentalyne 2a in high yield upon the addition of Al2O3 under an argon atmosphere in the presence of NaPF6 (Scheme 2). In this context, a reasonable mechanism was proposed for the direct amidation reaction (Scheme 3). Initially, the addition of a bromine cation derived from the N-bromoamide to the metal–carbon triple bond produces an extremely electron-deficient cationic metallabromirenium A. Subsequently, nucleophilic addition of imide anion afforded the stabilized intermediate σH-adduct 3a, and this is followed by elimination of hydride ion by oxidation to accomplish the SNAr reaction giving the metallaaromatic product 4a. Elimination of the bromine cation by Al2O3 affords the final amido-substituted product 2a.
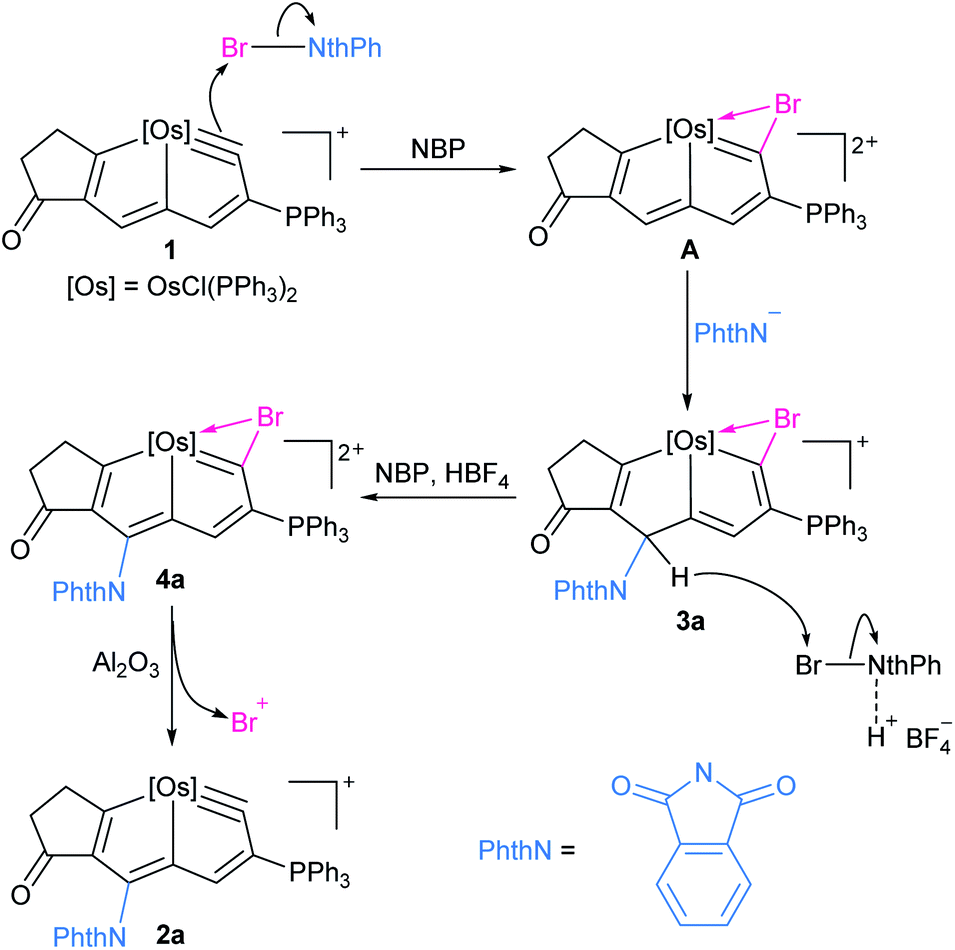 | ||
| Scheme 3 Proposed mechanism for the direct amidation of metallaaromatics, [Os] = OsCl(PPh3)2. | ||
While 1 was reacted with NBP in the presence of wet acetic acid, and then the mixture was treated by aluminium oxide, compound 5 can be isolated in 68% yield (Scheme 4). The X-ray crystallographic analysis confirms it is a new metallacyclic skeleton containing a five-membered metallacycloallene (Fig. 5). Notably, carbonylation at C5 position in 5 was observed rather than N-functionalization as in 2. The proposed mechanism for the formation of 5 showed nucleophilic attack of H2O to metallabromirenium A afforded intermediate B, which would be oxidized into carbonyl complex C. The cationic C is detected by HRMS spectrometry (m/z = 1249.1461, [C64H50BrClO2OsP3]+ calculated m/z = 1249.1483) (see ESI p. S62†). The ready elimination of the bromine cation afforded the product 5 containing a metal-vinylidene moiety (see ESI p. S19†). It is presumably that the nucleophilicity of NBP in the presence of the acetic acid is dramatically reduced, thus the C5 position of 1 was prior to being attacked by H2O. The strategy provides a convenient route to access extraordinary metallacycles with a high degree of skeletal complexity.
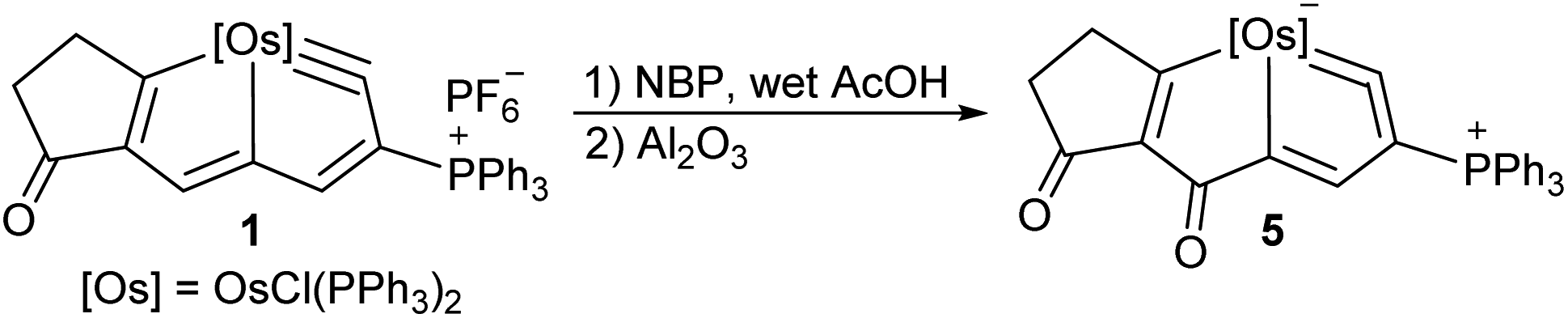 | ||
| Scheme 4 Formation of 5 by reaction of 1 with H2O, [Os] = OsCl(PPh3)2. | ||
 | ||
| Fig. 5 X-ray crystal structure of 5 (thermal ellipsoids are set at the 50% probability level and the phenyl groups in the PPh3 moieties are omitted for clarity). Selected bond lengths [Å] and angles [°]: Os1–C4: 2.111(3), Os1–C1: 1.870(3), Os1–C7: 2.059(3), O2–C5: 1.250(3), O1–C10: 1.223(4), C1–C2: 1.374(4), C3–C2: 1.450(4), C4–C3: 1.366(4), C4–C5: 1.465(4), C6–C5: 1.449(4), C6–C7: 1.382(4), C7–C8: 1.512(4), C8–C9: 1.530(4), C10–C9: 1.519(4), C6–C10: 1.464(4), C2–C1–Os1: 130.0(2), C1–C2–C3: 107.3(2), O2–C5–C4: 124.3(3), O1–C10–C6: 129.1(3). | ||
Further, transformation of imide product 2a was performed by treatment of 2a with hydrazine hydrate, afforded amino-substituted osmapentalyne 6 in high yield (90%) (Scheme 5). Amino-substituted metallaromatics exhibit rich reactivities towards many reagents.15 Complex 6 represents the first amino-substituted metallapentalyne, which is useful for further functionalization of these unusual metallacyclic compounds.
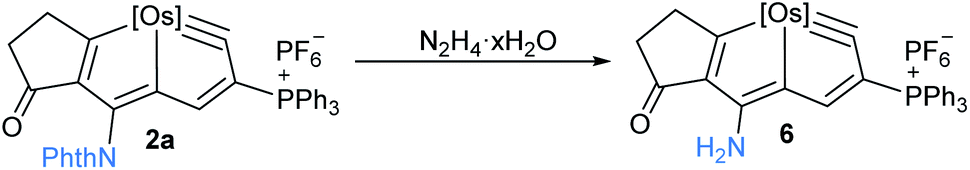 | ||
| Scheme 5 Transformation of complex 2a to amino-substituted osmapentalyne 6, [Os] = OsCl(PPh3)2. | ||
Thermal stability and spectroscopic properties
The solid state of amidation products 2a–2g can be stored in air at room temperature at least for two weeks. They also exhibit good thermal stability even when heated at 100 °C in air for 5 h (see ESI p. S20†). The UV-Vis absorption spectra of the precursor 1 and its derivatives 2a–2g were investigated. As shown in Fig. 6, compound 1 displays an absorption maximum at λmax = 460 nm. The absorption bands for 2a and 2b, bearing electron-withdrawing imide groups were observed at approximately λmax = 472 nm, slightly red-shifted compared to that of 1, while 2c–2g with electron-donating amide groups at around λmax = 450 nm was slightly blue-shifted. The results show that amidation of metallaaromatic compounds is a feasible methodology with which to subtly tune their photophysical properties.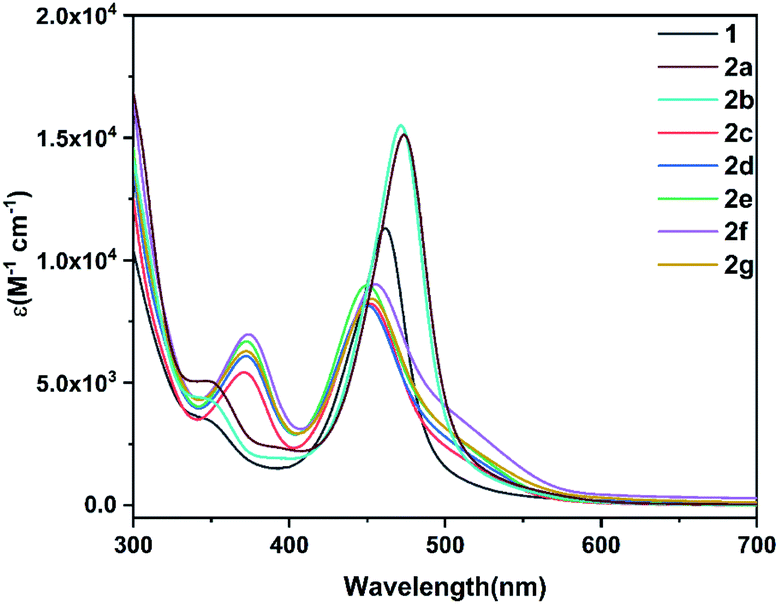 | ||
| Fig. 6 UV-Vis spectra of osmapentalyne 1 and its N-functionalized derivatives 2a–2g (CH2Cl2, rt, 5.0 × 10−5 M). | ||
Conclusions
We have developed a direct and efficient amidation of metallaaromatics, leading to N-functionalized osmapentalynes. The successful isolation of the key intermediates, the 1,5-bromoamidation and metallabromirenium species, confirms that the reaction is a three-step process that first involves electrophilic addition, followed by nucleophilic attack and then oxidation. This study provides insight into a strategy to construct functionalized metallaaromatics and significantly facilitates further exploration of their properties and potential applications.Author contributions
H. X. and Y.-M. L. conceived the project. H. W. performed the experiments. H. W., Y.-M. L., and Y. R. analyzed and interpreted the experimental data. Y.-M. L. and J. W. drafted the paper. All of the authors discussed the results and contributed to the preparation of the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. U1705254, 22071206, 21931002 and 21671164).Notes and references
- For reviews, see: (a) J. Bariwal and E. Van der Eycken, Chem. Soc. Rev., 2013, 42, 9283–9303 RSC; (b) M.-L. Louillat and F. W. Patureau, Chem. Soc. Rev., 2014, 43, 901–910 RSC; (c) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040–1052 CrossRef CAS PubMed; (d) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed; (e) R. K. Rit, M. Shankar and A. K. Sahoo, Org. Biomol. Chem., 2017, 15, 1282–1293 RSC.
- For recent examples, see: (a) X. Hu, G. Zhang, L. Nie, T. Kong and A. Lei, Nat. Commun., 2019, 10, 5467 CrossRef PubMed; (b) H. J. Kim, J. Kim, S. H. Cho and S. Chang, J. Am. Chem. Soc., 2011, 133, 16382–16385 CrossRef CAS PubMed; (c) A. A. Kantak, S. Potavathri, R. A. Barham, K. M. Romano and B. DeBoef, J. Am. Chem. Soc., 2011, 133, 19960–19965 CrossRef CAS PubMed; (d) R. Shrestha, P. Mukherjee, Y. Tan, Z. C. Litman and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 8480–8483 CrossRef CAS PubMed; (e) K. Foo, E. Sella, I. Thomé, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5279–5282 CrossRef CAS PubMed; (f) T. Yamaguchi, E. Yamaguchi and A. Itoh, Org. Lett., 2017, 19, 1282–1285 CrossRef CAS PubMed; (g) K. Sun, Y. Li and Q. Zhang, Sci. China: Chem., 2015, 58, 1354–1358 CrossRef CAS.
- (a) R. A. Lidgett, E. R. Lynch and E. B. McCall, J. Chem. Soc., 1965, 3754–3759 RSC; (b) J. C. Day, M. G. Katsaros, W. D. Kocher, A. E. Scott and P. S. Skell, J. Am. Chem. Soc., 1978, 100, 1950–1951 CrossRef CAS; (c) L. Stella, Angew. Chem., Int. Ed. Engl., 1983, 22, 337–350 CrossRef; (d) U. Lüning and P. S. Skell, Tetrahedron, 1985, 41, 4289–4302 CrossRef.
- (a) T. Xiong and Q. Zhang, Chem. Soc. Rev., 2016, 45, 3069–3087 RSC; (b) M. D. Kärkäs, ACS Catal., 2017, 7, 4999–5022 CrossRef; (c) L. J. Allen, P. J. Cabrera, M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2014, 136, 5607–5610 CrossRef CAS PubMed; (d) H. Kim, T. Kim, D. G. Lee, S. W. Roh and C. Lee, Chem. Commun., 2014, 50, 9273–9276 RSC; (e) S. C. Cosgrove, J. M. C. Plane and S. P. Marsden, Chem. Sci., 2018, 9, 6647–6652 RSC; (f) L. Song, L. Zhang, S. Luo and J.-P. Cheng, Chem.–Eur. J., 2014, 20, 14231–14234 CrossRef CAS PubMed; (g) T. Kuribara, M. Nakajima and T. Nemoto, Org. Lett., 2020, 22, 2235–2239 CrossRef CAS PubMed.
- For reviews, see: (a) D. Chen, Y. Hua and H. Xia, Chem. Rev., 2020, 120, 12994–13086 CrossRef CAS PubMed; (b) B. J. Frogley and L. J. Wright, Chem.–Eur. J., 2018, 24, 2025–2038 CrossRef CAS PubMed; (c) W. Ma, C. Yu, T. Chen, L. Xu, W.-X. Zhang and Z. Xi, Chem. Soc. Rev., 2017, 46, 1160–1192 RSC; (d) G. Jia, Acc. Chem. Res., 2004, 37, 479–486 CrossRef CAS PubMed; (e) J. R. Bleeke, Acc. Chem. Res., 2007, 40, 1035–1047 CrossRef CAS PubMed; (f) L. J. Wright, Dalton Trans., 2006, 1821–1827 RSC; (g) C. W. Landorf and M. M. Haley, Angew. Chem., Int. Ed., 2006, 45, 3914–3936 CrossRef CAS PubMed; (h) C. Zhu and H. Xia, Acc. Chem. Res., 2018, 51, 1691–1700 CrossRef CAS PubMed.
- (a) M. A. Esteruelas, A. B. Masamunt, M. Oliván, E. Oñate and M. Valencia, J. Am. Chem. Soc., 2008, 130, 11612–11613 CrossRef CAS PubMed; (b) T. Bolaño, R. Castarlenas, M. A. Esteruelas and E. Oñate, J. Am. Chem. Soc., 2006, 128, 3965–3973 CrossRef PubMed; (c) P. Barrio, M. A. Esteruelas and E. Oñate, J. Am. Chem. Soc., 2004, 126, 1946–1947 CrossRef CAS PubMed.
- (a) J. R. Bleeke, P. Putprasert, T. Thananatthanachon and N. P. Rath, Organometallics, 2008, 27, 5744–5747 CrossRef CAS PubMed; (b) V. Jacob, C. W. Landorf, L. N. Zakharov, T. J. R. Weakley and M. M. Haley, Organometallics, 2009, 28, 5183–5190 CrossRef CAS.
- (a) B. J. Frogley and L. J. Wright, Angew. Chem., Int. Ed., 2017, 56, 143–147 CrossRef CAS PubMed; (b) C. Zhu, S. Li, M. Luo, X. Zhou, Y. Niu, M. Lin, J. Zhu, Z. Cao, X. Lu, T. Wen, Z. Xie, P. V. Schleyer and H. Xia, Nat. Chem., 2013, 5, 698–703 CrossRef CAS PubMed; (c) W. Ruan, T.-F. Leung, C. Shi, K. H. Lee, H. H. Y. Sung, I. D. Williams, Z. Lin and G. Jia, Chem. Sci., 2018, 9, 5994–5998 RSC; (d) J. Chen, K.-H. Lee, H. H. Y. Sung, I. D. Williams, Z. Lin and G. Jia, Angew. Chem., Int. Ed., 2016, 55, 7194–7198 CrossRef CAS PubMed.
- (a) Z. Huang, Y. Zheng, W.-X. Zhang, S. Ye, L. Deng and Z. Xi, Angew. Chem., Int. Ed., 2020, 59, 14394–14398 CrossRef CAS PubMed; (b) C. Yu, M. Zhong, Y. Zhang, J. Wei, W. Ma, W.-X. Zhang, S. Ye and Z. Xi, Angew. Chem., Int. Ed., 2020, 59, 19048–19053 CrossRef CAS PubMed; (c) Y. Zheng, C.-S. Cao, W. Ma, T. Chen, B. Wu, C. Yu, Z. Huang, J. Yin, H.-S. Hu, J. Li, W.-X. Zhang and Z. Xi, J. Am. Chem. Soc., 2020, 142, 10705–10714 CrossRef CAS PubMed.
- (a) Á. Vivancos, M. Paneque, M. L. Poveda and E. Álvarez, Angew. Chem., Int. Ed., 2013, 52, 10068–10071 CrossRef PubMed; (b) P. Lara, M. Paneque, M. L. Poveda, L. L. Santos, J. E. Valpuesta, E. Carmona, S. Moncho, G. Ujaque, A. Lledos, E. Alvarez and K. Mereiter, Chem.–Eur. J., 2009, 15, 9034–9045 CrossRef CAS PubMed; (c) M. Paneque, M. L. Poveda, N. Rendón and K. Mereiter, J. Am. Chem. Soc., 2004, 126, 1610–1611 CrossRef CAS PubMed.
- (a) M. Kuno, K. Suzuki, T. Nakamura and M. Yamashita, Organometallics, 2020, 39, 4139–4142 CrossRef CAS; (b) T. Nakamura, K. Suzuki and M. Yamashita, J. Am. Chem. Soc., 2017, 139, 17763–17766 CrossRef CAS PubMed; (c) T. Nakamura, K. Suzuki and M. Yamashita, J. Am. Chem. Soc., 2014, 136, 9276–9279 CrossRef CAS PubMed; (d) A. J. Ashe III, T. R. Diephouse and M. Y. Elsheikh, J. Am. Chem. Soc., 1982, 104, 5693–5699 CrossRef; (e) A. J. Ashe III, Organometallics, 2009, 28, 4236–4248 CrossRef.
- (a) M. Saito, M. Sakaguchi, T. Tajima, K. Ishimura, S. Nagase and M. Hada, Science, 2010, 328, 339–342 CrossRef CAS PubMed; (b) M. Nakada, T. Kuwabara, S. Furukawa, M. Hada, M. Minoura and M. Saito, Chem. Sci., 2017, 8, 3092–3097 RSC; (c) J. B. Chen, V. G. Young and R. J. Angelici, J. Am. Chem. Soc., 1995, 117, 6362–6363 CrossRef CAS.
- (a) V. C. Arias-Coronado, R. Pereira-Cameselle, A. Ozcelik, M. Talavera, A. Peña-Gallego, J. L. Alonso-Gómez and S. Bolaño, Chem.–Eur. J., 2019, 25, 13496–13499 CrossRef CAS PubMed; (b) M. Talavera, A. Peña-Gallego, J. L. Alonso-Gómez and S. Bolaño, Chem. Commun., 2018, 54, 10974–10976 RSC; (c) C. S. Chin, H. Lee and M.-S. Eum, Organometallics, 2005, 24, 4849–4852 CrossRef CAS; (d) C. S. Chin and H. Lee, Chem.–Eur. J., 2004, 10, 4518–4522 CrossRef CAS PubMed.
- (a) W. Y. Hung, B. Liu, W. Shou, T. B. Wen, C. Shi, H. H. Sung, I. D. Williams, Z. Lin and G. Jia, J. Am. Chem. Soc., 2011, 133, 18350–18360 CrossRef CAS PubMed; (b) C. E. F. Rickard, W. R. Roper, S. D. Woodgate and L. J. Wright, Angew. Chem., Int. Ed., 2000, 39, 750–752 CrossRef CAS; (c) B. J. Frogley, A. F. Dalebrook and L. J. Wright, Organometallics, 2016, 35, 400–409 CrossRef CAS.
- B. J. Frogley, L. C. Perera and L. J. Wright, Chem.–Eur. J., 2018, 24, 4304–4309 CrossRef CAS PubMed.
- (a) T. Wang, S. Li, H. Zhang, R. Lin, F. Han, Y. Lin, T. B. Wen and H. Xia, Angew. Chem., Int. Ed., 2009, 48, 6453–6456 CrossRef CAS PubMed; (b) T. Wang, H. Zhang, F. Han, L. Long, Z. Lin and H. Xia, Chem.–Eur. J., 2013, 19, 10982–10991 CrossRef CAS PubMed; (c) F. Han, T. Wang, J. Li, H. Zhang and H. Xia, Chem.–Eur. J., 2014, 20, 4363–4372 CrossRef CAS PubMed.
- (a) C. Zhu, Y. Yang, J. Wu, M. Luo, J. Fan, J. Zhu and H. Xia, Angew. Chem., Int. Ed., 2015, 54, 7189–7192 CrossRef CAS PubMed; (b) Q. Zhuo, J. Lin, Y. Hua, X. Zhou, Y. Shao, S. Chen, Z. Chen, J. Zhu, H. Zhang and H. Xia, Nat. Commun., 2017, 8, 1912 CrossRef PubMed; (c) S. Chen, L. Liu, X. Gao, Y. Hua, L. Peng, Y. Zhang, L. Yang, Y. Tan, F. He and H. Xia, Nat. Commun., 2020, 11, 4651 CrossRef CAS PubMed.
- (a) Z. Wang, L. Lin, P. Zhou, X. Liu and X. Feng, Chem. Commun., 2017, 53, 3462–3465 RSC; (b) Y. Cai, X. Liu, Y. Hui, J. Jiang, W. Wang, W. Chen, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2010, 49, 6160–6164 CrossRef CAS PubMed; (c) Y.-Y. Yeung, X. Gao and E. J. Corey, J. Am. Chem. Soc., 2006, 128, 9644–9645 CrossRef CAS PubMed; (d) W. Z. Yu, F. Chen, Y. A. Cheng and Y.-Y. Yeung, J. Org. Chem., 2015, 80, 2815–2821 CrossRef CAS PubMed.
- (a) C. Zhu, C. Yang, Y. Wang, G. Lin, Y. Yang, X. Wang, J. Zhu, X. Chen, X. Lu, G. Liu and H. Xia, Sci. Adv., 2016, 2, e1601031 CrossRef PubMed; (b) C. Zhu, Y. Yang, M. Luo, C. Yang, J. Wu, L. Chen, G. Liu, T. Wen, J. Zhu and H. Xia, Angew. Chem., Int. Ed., 2015, 54, 6181–6185 CrossRef CAS PubMed.
- M. Luo, Z. Deng, Y. Ruan, Y. Cai, K. Zhuo, H. Zhang and H. Xia, Organometallics, 2019, 38, 3053–3059 CrossRef CAS.
- J. Chen, Z.-A. Huang, Y. Hua, H. Zhang and H. Xia, Organometallics, 2015, 34, 340–347 CrossRef CAS.
-
(a) M. M
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) kosza and K. Wojciechowski, Chem. Rev., 2004, 104, 2631–2666 CrossRef PubMed;
(b) M. Mąkosza, Chem. Soc. Rev., 2010, 39, 2855–2868 RSC;
(c) V. N. Charushin and O. N. Chupakhin, Mendeleev Commun., 2007, 17, 249–254 CrossRef CAS;
(d) O. N. Chupakhin and V. N. Charushin, Tetrahedron Lett., 2016, 57, 2665–2672 CrossRef CAS.
kosza and K. Wojciechowski, Chem. Rev., 2004, 104, 2631–2666 CrossRef PubMed;
(b) M. Mąkosza, Chem. Soc. Rev., 2010, 39, 2855–2868 RSC;
(c) V. N. Charushin and O. N. Chupakhin, Mendeleev Commun., 2007, 17, 249–254 CrossRef CAS;
(d) O. N. Chupakhin and V. N. Charushin, Tetrahedron Lett., 2016, 57, 2665–2672 CrossRef CAS. - (a) G. R. Clark, L. A. Ferguson, A. E. McIntosh, T. Sohnel and L. J. Wright, J. Am. Chem. Soc., 2010, 132, 13443–13452 CrossRef CAS PubMed; (b) F. Kloss, V. Krchnak, A. Krchnakova, S. Schieferdecker, J. Dreisbach, V. Krone, U. Mollmann, M. Hoelscher and M. J. Miller, Angew. Chem., Int. Ed., 2017, 56, 2187–2191 CrossRef CAS PubMed; (c) T. Lemek, M. Makosza, D. S. Stephenson and H. Mayr, Angew. Chem., Int. Ed., 2003, 42, 2793–2795 CrossRef CAS PubMed; (d) H. C. Van der Plas, V. N. Charushin and B. Van Veldhuizen, J. Org. Chem., 1983, 48, 1354–1357 CrossRef CAS.
- (a) I. V. Borovlev, O. P. Demidov, N. A. Kurnosova, G. A. Amangasieva and E. K. Avakyan, Chem. Heterocycl. Compd., 2015, 51, 170–175 CrossRef CAS; (b) M. K. Stern and B. K. Cheng, J. Org. Chem., 1993, 58, 6883–6888 CrossRef CAS.
- (a) A. Alix, C. Lalli, P. Retailleau and G. Masson, J. Am. Chem. Soc., 2012, 134, 10389–10392 CrossRef CAS PubMed; (b) L. Song, S. Luo and J.-P. Cheng, Org. Lett., 2013, 15, 5702–5705 CrossRef CAS PubMed; (c) M. Li, H. Yuan, B. Zhao, F. Liang and J. Zhang, Chem. Commun., 2014, 50, 2360–2363 RSC.
- Imidation: see ref. 4. Halogenation: (a) G. K. S. Prakash, T. Mathew, D. Hoole, P. M. Esteves, Q. Wang, G. Rasul and G. A. Olah, J. Am. Chem. Soc., 2004, 126, 15770–15776 CrossRef CAS PubMed; (b) F. Mo, J. M. Yan, D. Qiu, F. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 2028–2032 CrossRef CAS PubMed; (c) Y. Nishii, M. Ikeda, Y. Hayashi, S. Kawauchi and M. Miura, J. Am. Chem. Soc., 2020, 142, 1621–1629 CrossRef CAS PubMed; (d) S. Duan, J. Turk, J. Speigle, J. Corbin, J. Masnovi and R. J. Baker, J. Org. Chem., 2000, 65, 3005–3009 CrossRef CAS PubMed; (e) M. De Rosa, G. Cabrera Nieto and F. Ferrer Gago, J. Org. Chem., 1989, 54, 5347–5350 CrossRef CAS.
- M. Luo, C. Zhu, L. Chen, H. Zhang and H. Xia, Chem. Sci., 2016, 7, 1815–1818 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization details of new compounds, crystallographic details, NMR spectra. CCDC 2053993 (S), 2054261 (1), 2054330 (2a), 2054286 (2b), 2054272 (2c), 2054274 (2d), 2054285 (2e), 2054269 (4a) and 2069503 (5). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01571k |
| This journal is © The Royal Society of Chemistry 2021 |