
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent progress in self-healing polymers and hydrogels based on reversible dynamic B–O bonds: boronic/boronate esters, borax, and benzoxaborole
Seungwan
Cho
a,
Sung Yeon
Hwang
 *ab,
Dongyeop X.
Oh
*ab and
Jeyoung
Park
*ab
*ab,
Dongyeop X.
Oh
*ab and
Jeyoung
Park
*ab
aResearch Center for Bio-Based Chemistry, Korea Research Institute of Chemical Technology (KRICT), Ulsan, 44429, Republic of Korea. E-mail: crew75@krict.re.kr; dongyeop@krict.re.kr; jypark@krict.re.kr
bAdvanced Materials and Chemical Engineering, University of Science and Technology (UST), Daejeon 34113, Republic of Korea
First published on 18th May 2021
Abstract
Intrinsic self-healing polymeric materials are substances that relieve external stress and restore their original mechanical properties after extreme damage via dynamic covalent bonding in the polymeric structure or the reversible association of supramolecular motifs. Boronic ester-based dynamic covalent bonds formed between boronic acids and diols impart excellent self-healing properties to the hydrogels, organic gels, elastomers, and plastics depending on the characteristics of the corresponding polymer. In addition, these bonds induce internal reorganization in the chemical/physical structure in response to changes in the biological signals and biomaterials, such as hydrophilicity, pH, and the presence of glucose. This multi-responsiveness to stimuli as well as the self-healing, injectability, and biocompatibility of boronic ester-based polymers have led to several technological achievements with applicability in biomedical fields, including drug delivery, medical adhesion, bioimplants, and healthcare monitoring. This review provides an overview of various self-healing polymeric materials based on reversible B–O bonds, with a focus on boronic/boronate esters, borax, and benzoxaborole and their properties, responses to external stimuli, and applications.
 Seungwan Cho | Seungwan Cho received his BS and MS (2019) degrees from the Department of Polymer Science and Engineering at Pusan National University. Thereafter, he worked as a researcher in Research Center for Bio-Based Chemistry at KRICT for two years. His main area of research involves the synthesis and characterization of intrinsic self-healing polymers. |
 Sung Yeon Hwang | Sung Yeon Hwang received his PhD in 2011 from the Department of Fiber & Polymer Engineering at Hanyang University (South Korea) under the supervision of Prof. Seung Soon Im. He worked as a senior research engineer at the SKC Advanced Technology R&D Center for three years. Currently, he is a Director at the Research Center for Bio-Based Chemistry at KRICT, a Director of the Korean Bioplastics Association (KBPA), and an Associate Professor in Advanced Materials and Chemical Engineering at UST (South Korea). His research interests include self-healing and development of bioplastics for industrial applications and carbon neutrality. |
 Dongyeop X. Oh | Dongyeop X. Oh is a Senior Researcher at the Research Center for Bio-Based Chemistry at KRICT and an Associate Professor in Advanced Materials and Chemical Engineering at UST. He attended Hanyang University, in 2005, and received his BS and MS (2011) degrees in Polymer Science and Engineering. He investigated bioinspired advanced materials under the supervision of Prof. Dong Soo Hwang, and received PhD at POSTECH in South Korea. Upon finishing his doctorate, he began his professional career at KRICT in 2015. He seeks to integrate fundamental research with the development of sustainable and bio-renewable materials with advanced properties and functions. |
 Jeyoung Park | Jeyoung Park received his BS (Summa Cum Laude), MS, and PhD (2012) degrees from the Department of Chemistry at KAIST (South Korea) under the supervision of Prof. Sang Youl Kim, focused on controlled polymerization and self-assembly. After spending two years as an R&D researcher at SK Innovation Co., he is currently a Senior Researcher at the Research Center for Bio-Based Chemistry at KRICT and an Associate Professor in Advanced Materials and Chemical Engineering at UST. His research interests include self-healing and sustainable polymers used in industrial applications. Particular emphasis is placed on the design of novel polymer structures that are readily accessible. |
1. Introduction
Various materials constructed from polymers, metals, and ceramics are used in our daily lives. However, most of these materials require repair if damaged by external stimuli, which results in the deterioration or loss of their original performances if left unrepaired. As the need for constant replacement leads to environmental pollution and economic loss, research on self-healing materials that can allow extended life cycles via the autonomous repair of damage is necessary. Thus, self-healing materials have received considerable attention since the early 2000s. Many studies on self-healing polymeric materials have been conducted, but additional research is required to develop next-generation materials.1 Polymeric materials capable of self-healing have two main characteristics. First, the presence of a component within the material that enables self-healing, and second, a mobile phase in which the components that enable self-healing can migrate or diffuse to the damaged site. Thus, among the polymeric materials reported to date, studies on the self-healing of soft materials such as hydrogels and elastomers, in which the polymer chains tend to exhibit good mobility, are actively being conducted.2–7The self-healing properties of the polymeric materials can be divided into extrinsic and intrinsic self-healing, depending on whether the self-healing component is inserted into the polymer or is an original component in the polymer matrix (Fig. 1). Extrinsic self-healing materials cannot heal using their inherent properties. The components that enable healing, such as monomers, are dispersed in these materials in the form of capsules and the components inside the capsules are released upon damage.8 However, self-healing using this mechanism is not economically advantageous because it requires the use of expensive catalysts. Another disadvantage of this method is the difficulty in achieving repeated self-healing.
 | ||
| Fig. 1 Strategies used to prepare self-healing polymers. | ||
In the second category of intrinsic self-healing materials, healing is achieved through non-covalent or reversible dynamic covalent bonds in polymeric materials without the need for separate healing components, such as capsules. These materials show active non-covalent, supramolecular interactions, such as hydrogen bonding,9–13 dipole interactions,14 π–π interactions,15,16 metal complexation,17 and host–guest interactions.18 The mechanism of self-healing using supramolecular attractions is described as follows. When the non-covalent bond is broken by external shock, the force acts between the supramolecular chains present at the fracture site, resulting in self-assembly. Consequently, the broken network is rearranged and self-healing occurs at the fracture site.19 The mechanism of self-healing due to interactions between the supramolecules within the polymer is repetitive and allows relatively easy healing unlike extrinsic self-healing, which involves a limited number of healing cycles.
Repeated healing of polymers is also possible by intrinsic self-healing using reversible dynamic covalent bonds. Dynamic covalent bonding requires more time than self-healing via non-covalent bonding, but these materials possess good mechanical strength owing to their relatively stronger bond strengths.20–22 One of the representative strategies for achieving intrinsic self-healing polymeric materials using covalent bonds is the introduction of a Diels–Alder adduct into the material such that it can heal via the reversible formation and breakage of covalent bonds upon heating.23–25 The mechanism of self-healing using the Diels–Alder reaction is described as follows. In the damaged polymeric material, the Diels–Alder bonds break upon the application of heat and the chains become elastic at high temperatures. The elastic chains move to the fracture site to reform the Diels–Alder bonds upon a decrease in temperature; self-healing occurs as the network is re-formed. In addition to heat, light26 or radicals27,28 can be employed for the self-healing of polymers based on dynamic covalent bonding.
In addition to the Diels–Alder adducts, other examples of dynamic covalent bonds applicable to self-healing polymers include oxime,29,30 acylhydrazone,31,32 imine,33,34 disulfide,35–38 and boronic/boronate ester bonds. Boronic/boronate esters are formed via a combination of boronic acid and 1,2- or 1,3-diols, comprising the representative boron–oxygen (B–O) bonds. This review mainly focuses on self-healing polymers prepared using the bonds formed between boronic acid and diols, including the reversible B–O bonds. Reports on bulk polymers or elastomers containing boronic ester bonds, hydrogels with boronate ester bonds, and self-healing polymers containing borax or benzoxaborole compounds have been summarized, except for polymers with boroxine bonds prepared via the dehydration of boronic acid. Bulk self-healing polymer networks based on boroxines have been summarized in a recent review article by Sutti et al.39
2. Boronic acid and reversible esters
Boronic acid can act as a Lewis acid that can accept electron pairs (Fig. 2). Thus, it can form complexes with Lewis bases, such as hydroxide or electron-donating groups containing oxygen or nitrogen atoms. In an aqueous solution, boronic acid co-exists in its neutral state or is bound to a hydroxide ion, and its sp2 and sp3 hybrid orbitals adopt trigonal planar and tetragonal geometries, respectively. The relative equilibrium of these two forms is determined according to the Lewis acidity and the pKa of the boronic acid derivative. | ||
| Fig. 2 Dynamic equilibrium of boronic acid with 1,2- or 1,3-diols in an aqueous solution. | ||
The boronic ester and boronate ester are pentagonal or hexagonal rings in which boronic acid is bound to a cis-type 1,2- or 1,3-diol. These constitute the representative reversible dynamic covalent bonds that can impart self-healing properties to the polymers. These bonds can be reversibly formed or broken depending on the pH or aqueous media, which are sensitive to heat. Boronic acid can selectively bond to diols to form boronic esters or boronate esters, because of which boronic acid can be applied to sensors that can selectively detect the biomolecules including saccharides, such as glucose,40–42 or can be used as a component in the drug delivery systems43,44 and self-healing materials.
Boronic acid forms a boronate ester bond with 1,2- or 1,3-diols in an aqueous solution and a boronic ester bond with 1,2- or 1,3-diols in its bulk form or an organic solvent.39 In an aqueous solution, the neutral boronic ester constructed from boronic acid in its trigonal form is more unstable than it is before bonding to the diol owing to the ring strain, and is therefore easily hydrolyzed. Additionally, charged boronate esters generated from the tetragonal form of boronic acid are preferred over neutral boronate esters in an aqueous solution as these are relatively more stable.45 Boronic esters that do not undergo hydrolysis can also be formed, i.e., boronic esters that are relatively stable in their bulk form or an organic solvent in the absence of water. Thus, self-healing polymers containing boronic acid can be classified based on their presence in the bulk form or aqueous media.39,46 In this review, self-healing polymers prepared using boronic and boronate esters are discussed according to this classification.
Phenylboronic acid (PBA) derivatives are of significant importance because they have a good affinity for diol-containing biomolecules such as saccharides and peptidoglycans with high operational stability. Various substituents on the benzene ring affect their pKa and binding constants with diols.47,48 Electron-withdrawing substituents generally reduce the pKa of PBAs and preferably charge the boron atom negatively for gelation at a physiological pH in an aqueous solution.49 The structure–reactivity relationships should be considered when designing self-healing systems, particularly for biomedical applications.
Neutral boronic esters are difficult to form in aqueous solutions because these are susceptible to hydrolysis but can be generated when boronic acid and diol are combined in an organic solvent. In particular, bulk polymer materials capable of self-healing and reprocessing using boronic esters have recently been actively investigated and have generated considerable interest.39 Existing thermosets cannot melt or flow even at high temperatures due to cross-linking, but when a dynamic covalent bond is applied to a thermoset, it not only allows self-healing, but also reprocessability because of the associative and dissociative bonding mechanism. Among the recent studies, the Leibler group developed vitrimers,50 which are the thermosets that can be reprocessed using the reversible characteristics of boronic esters. Jing et al. introduced PBA as a cross-linking agent in novolac resin, which was used to produce a carbon fiber-reinforced polymer composite.51
Charged boronate esters are relatively stable when bound in aqueous solutions, particularly in alkaline aqueous solutions. Thus, charged boronate ester bonds have been extensively applied for self-healing in hydrogels, which mainly employ water as a solvent. Because of the pH-dependence of this bond, many charged boronate esters bound to the tetragonal boronic acid and diols are prevalent in aqueous solutions at high pH. At a low pH, many trigonal boronic acids are not bound to the diol. However, in the equilibrium reaction between boronic acid and ester, both boronic acid and diol have significant effects.47,52 Thus, to adjust the optimum pH to form boronate esters, the functional groups and pKa of both the diol and boronic acid must be considered.
3. Self-healing using neutral boronic ester bonds
The boronic ester bonds described in this chapter are mainly formed using a combination of boronic acids and cis-diols in an organic solvent or bulk rather than an aqueous solution. This type of bond is distinguishable from boroxine, a reversible bond that can be formed via the dehydration of boronic acid.53In the polymer networks containing boronic ester bonds, these bonds undergo facile bond exchange via associative or dissociative mechanism. There are three types of exchange reactions in boronic esters. The first is the metathesis reaction, in which two adjacent boronic esters are re-formed without the addition of water or free diol to form two different boronic esters. The second is the transesterification reaction, where the boronic ester reacts with the excess free diol. The two exchange reactions described above (metathesis and transesterification) are the reactions in which the boronic ester bonds are simultaneously broken and regenerated. As these reactions occur via an associative mechanism, a certain amount of bonding is maintained in the network such that the material has sufficient strength. The third exchange reaction is the hydrolysis/re-esterification reaction, which follows a dissociative mechanism, and proceeds in two stages. The boronic ester is first hydrolyzed and broken into its corresponding diol and boronic acid, and the decomposed diol and boronic acid undergo a re-esterification reaction with different boronic acid and diol. The polymer networks described in this chapter are capable of self-healing via these exchange reactions, but primarily by transesterification and hydrolysis/re-esterification.39
3.1 Combination of various boronic ester moieties and macromolecular structures
An early study on the synthesis of self-healing polymers containing boronic ester bonds and their application in linear polymers was published by the Lavigne group in 2005,54 in which poly(dioxaborolane) was synthesized (Fig. 3). To demonstrate the self-healing properties, the polymer was first decomposed via hydrolysis, the solvent was removed under reduced pressure, and the material was stored under vacuum. The resulting hydrolyzed material exhibited self-healing properties. Furthermore, the material that decomposed and self-healed via the above-mentioned process was confirmed to undergo healing based on the change in its relative molecular weight. This study reported the synthesis of a new polymer with good mechanical properties owing to the presence of reversible covalent bonds, which are stronger than supramolecular attractions. This polymer exhibited self-healing without the presence of any additional substances, such as catalysts, and had the capability to control the molecular weight, even after its synthesis. In particular, this study paved the way for the application of boronic esters to self-healing bulk polymeric materials.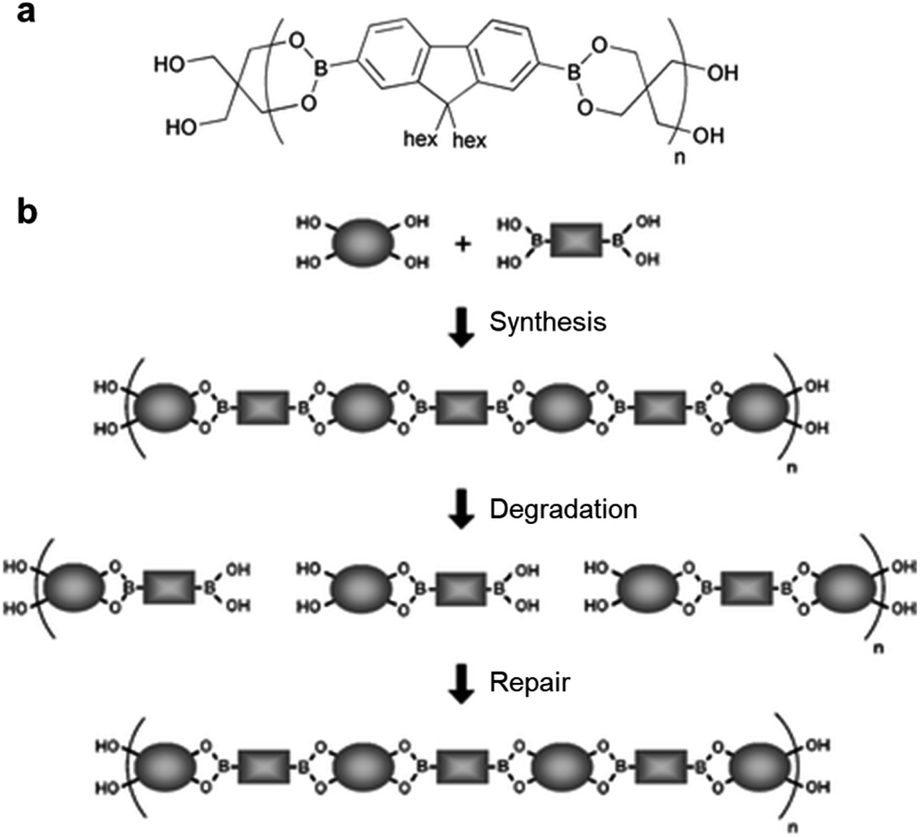 | ||
| Fig. 3 (a) Chemical structure of poly(dioxaborolane). (b) Schematic representation of the synthesis, hydrolytic degradation, and self-healing of poly(dioxaborolane). Reproduced from ref. 54 with permission from the RSC. | ||
In 2015, the Guan group studied transesterification, which is one of the exchange reactions mentioned above (Fig. 4).55 They initially investigated the rates of the exchange reactions in small molecules before studying the rate of the transesterification reactions in polymers in the presence of excess free diol. Two types of boronic esters were prepared, each containing PBA or o-(dimethylaminomethyl)PBA. The results showed that the latter exhibited a faster exchange reaction than the former because of adjacent nitrogen atoms in its structure. The effect of the functional groups on the exchange reaction rate in small molecules was similar to that observed in the bulk polymer. In the self-healing study, a higher healing rate and efficiency were observed when the o-aminophenylboronic group was introduced around the boronic ester bond than PBA. This study revealed the kinetics of the transesterification reaction between the free diols and boronic ester bonds in the presence of excess free diol and correlated the healing rate and efficiency with the functional groups present in the molecule. The possibility of regulating the exchange reaction rates and self-healing properties of the boronic esters upon introducing other functional groups into the molecule was also considered and implemented.
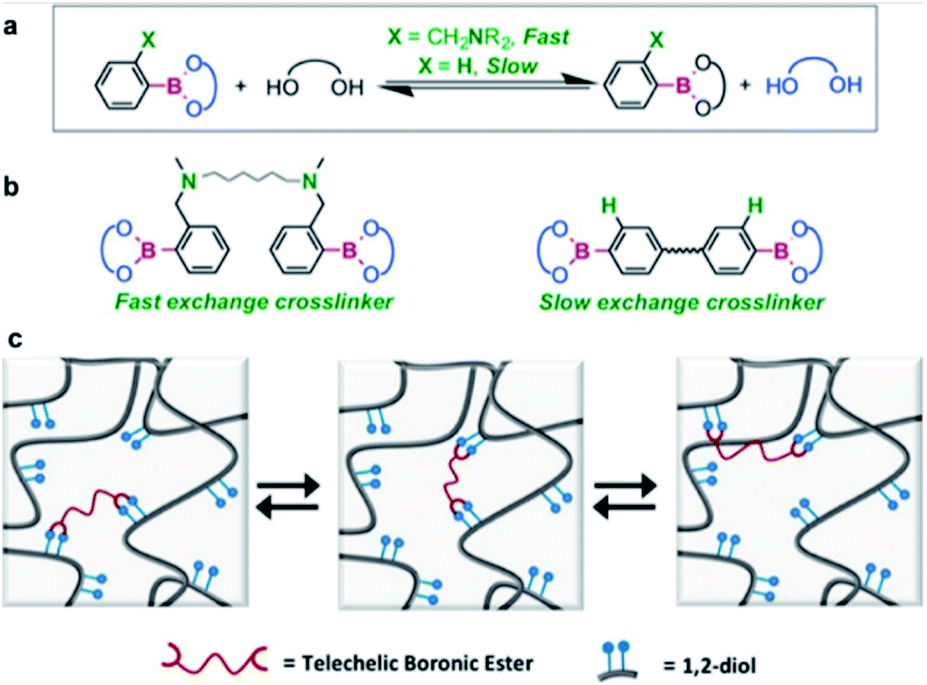 | ||
| Fig. 4 (a) Kinetics of the exchange reaction of boronic esters according to their neighboring functional groups. (b) Transesterification reaction using boronic ester as a cross-linking agent. (c) Schematic of the boronic ester exchange reaction. Reproduced from ref. 55 with permission from the ACS. | ||
The Sumerlin group synthesized a bulk polymer network that underwent an exchange reaction via hydrolysis/re-esterification. This material was capable of self-healing in high humidity or upon adding water under ambient conditions without requiring any external stimuli or catalysts, such as light and heat, which are required in self-healing bulk materials containing bonds in addition to the boronic ester bonds.56 This was the first study that showed that three-dimensional (3D) bulk polymer materials containing boronic ester bonds could self-heal at room temperature. A boronic ester diene monomer was prepared, and a polymer network containing boronic ester bonds was finally obtained via thiol–ene click chemistry using a thiol-containing monomer. The resulting polymer network had sufficient mobility for healing because its glass transition temperature was lower than the room temperature and exhibited stable hydrophobic properties in water despite the vulnerability of its existing boronic ester bonds to hydrolysis. Self-healing of cut specimens occurred simply by bringing the cut surfaces in contact at room temperature. This self-healing mechanism involved the hydrolysis and re-esterification of the boronic ester bonds present at the cut surface by water, allowing the healing to occur in multiple cut and repair experiments. Self-healing occurred even under high humidity (85% RH), but it was faster and higher healing efficiency was obtained only when a small amount of water was added. The fluidity of the boronic ester bonds present at the cut surface of the material increased because of the presence of water and self-healing occurred via re-esterification after hydrolysis.
As mentioned above, self-healing using boronic ester bonds is mainly possible via two mechanisms, transesterification or hydrolysis/re-esterification. In addition to the reports described above, the Sumerlin group conducted a study in 2018 that exploited both types of exchange reactions (transesterification and hydrolysis/re-esterification) simultaneously (Fig. 5).57 The amount of excess free diol was adjusted to investigate the self-healing effect via transesterification, and the humidity was controlled to examine the self-healing effect via hydrolysis/re-esterification. Using stress relaxation experiments, it was confirmed that high humidity and excess free diol led to a faster exchange reaction of the boronic ester bonds in the polymer network. Furthermore, when the self-healing efficiency was evaluated using tensile tests, the effect of the free diol was minimized under high-humidity conditions, and the healing efficiency was similar, regardless of the free diol content. Under low-humidity conditions, self-healing did not occur in the absence of free diol. The transesterification reaction showed a healing efficiency of 45% at the highest free diol content. In addition, the polymer network, which supplemented the stress relaxation and vulnerability toward creep behavior via additional cross-linking, showed high healing efficiency in three repetitive cut and heal experiments. This study revealed the exchange reaction mechanism that was more dominant in self-healing depending on the humidity and free diol content and presented the possibility of controlling the self-healing rate according to the ratio of humidity and free diol.
 | ||
| Fig. 5 (a) Cross-linked network of boronic ester prepared via photoinitiated thiol–ene click chemistry. Schematic of the boronic ester network exchange reaction in the presence of (b) free diol, (c) irreversible cross-linking, and (d) both free diol and irreversible cross-linking. Reproduced from ref. 57 with permission from the RSC. | ||
Polymeric materials that can undergo reprocessing and self-healing can be synthesized via transesterification or hydrolysis/re-esterification when boronic ester bonds are applied to the widely used materials such as rubbers or elastomers. These reprocessable and self-healing rubbers or elastomers are advantageous in terms of their environmental impact and resource conservation. The conventional preparation of a polymeric material cross-linked with sulfur using thermal and mechanical processing requires a considerable amount of energy. Additionally, the physical properties of the resulting materials deteriorate because of the inevitable decomposition of the polymeric chain during synthesis. To overcome these shortcomings, several studies have reported the application of boronic esters to commercial rubbers or elastomers to impart reprocessability as well as self-healing properties.
Polydimethylsiloxane (PDMS) elastomers are of commercial importance and significant interest in the field of polymers because of their excellent mechanical properties. In 2016, the Feng group synthesized a polysiloxane-based elastomer containing boronic ester bonds via a thiol–ene click reaction.58 This elastomer was prepared using poly[(mercaptopropyl) methylsiloxane] (PMMS) and 4-[(allyloxy)methyl]-2-(4-vinylphenyl)-1,3,2-dioxaborolan] (VPD) at room temperature. PMMS acted as a matrix in the elastomer, which was cross-linked with VPD. Cross-linking was achieved via a thiol–ene click reaction between the thiol group in PMMS and diene in VPD. The resulting elastomer was cut into two pieces and self-healing occurred without any external stimulation within 30 min once both pieces were brought into contact with each other (Fig. 6a). When the surface was treated with water, it healed faster (within 10 min). Thus, the self-healing properties of the polymer were attributed to the hydrolysis/re-esterification reaction of the boronic esters. In addition, to increase the mechanical strength and bond stability of the elastomer, a dual cross-linking silicone elastomer was prepared by introducing another irreversible bond via cross-linking along with the boronic ester bond. The dual cross-linked silicone elastomer was also capable of self-healing (Fig. 6b), which showed a high healing efficiency of ∼70% in the tensile tests. Furthermore, contact angle tests showed that the elastomer became more hydrophobic owing to the additional cross-linking via irreversible bonds, and the properties of the boronic esters that are vulnerable to hydrolysis could be compensated for.
 | ||
| Fig. 6 (a) Self-healing mechanism of a silicone elastomer cross-linked using VPD and a photograph of the cut elastomer at room temperature as well as its self-healing after 30 min. (b) Schematic of the self-healing mechanism of the dual cross-linked silicone elastomer. Reproduced from ref. 58 with permission from the Wiley. | ||
3.2 Potential industrial applications merited by mechanical robustness and reprocessability
In 2017, Leibler et al. introduced a study that improved the industrial applicability by designing boronic esters in commercial polymer structures, such as high-density polyethylene (HDPE), polystyrene (PS), and poly(methyl methacrylate) (PMMA) for the first time.50 Recyclable thermosets (called vitrimers) that have flowability when heat is applied were prepared (Fig. 7a). In other words, grounding processing and reprocessing several times through extrusion, compression, and injection molding was possible, similar to conventional thermoplastics. The experimental results show that the HDPE vitrimer did not deteriorate the properties during repeated processing, confirming the robustness of the dioxaborolane group (Fig. 7b). The vitrimer used in this study was able to overcome the limitations of the thermoplastics owing to its high resistance to environmental stress cracking (Fig. 7c). The PS vitrimer immersed in an ethanol/water solution (9/1 v/v) under load for 3 h was tested for flexural stress. It exhibited greater stability than conventional PS vitrimers. Even in pure water, no chemical degradation was observed for 3 months, which verified that the resistance of dioxaborolane-based vitrimers to hydrolysis is useful for industrial applications. | ||
| Fig. 7 (a) Synthetic scheme for various copolymers containing pendant dioxaborolanes (PS, PMMA, and HDPE). (b) Tensile properties of HDPE (gray column) and HDPE vitrimers (pristine: blue, reprocessed: light blue). (c) Environmental stress-cracking resistance of PS vitrimer. Reproduced from ref. 50 with permission from the AAAS. | ||
Jing et al. reported a study on the efficient recycling of carbon fiber-reinforced polymer (CFRP), which is widely used in large-scale industrial applications.51 By applying PBA to the novolac resin (NR) raw materials used in various fields, crosslinks by dynamic reversible boronate linkages were introduced (Fig. 8a). The prepared mixed system of NR and PBA (denoted as PBNR) had excellent processability, thermal stability, and mechanical properties. The alcoholysis nature of the boronate bonds showed that the PBNR and CF/PBNR complexes were fully recycled in ethanol at room temperature (Fig. 8b). The recovered resin solution and fibers could be reprocessed to produce new composites with almost the same mechanical properties as the original, and the recycled CF retained 95% of the tensile strength of the original fiber after the third recycling. The presented method provides new insights into the modification of existing engineering resins as functional sustainable materials. CF/PBNR composites are promising for applications in the automotive, aerospace, construction, adhesive, and electronic packaging industries. The strategies developed in this work can minimize the use of resources, waste generation, and related environmental pollution.
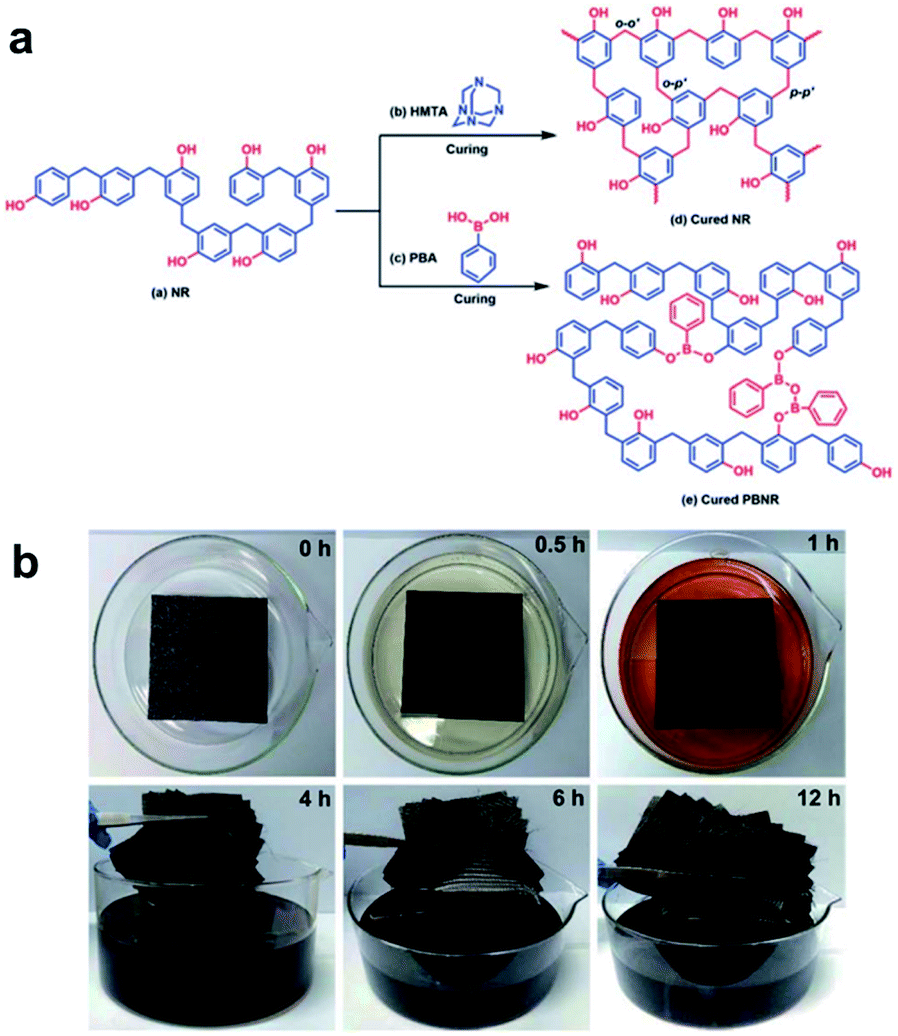 | ||
| Fig. 8 (a) Synthetic scheme for the cross-linked NR with hexamethylenetetramine and the reprocessable PBNR. (b) The time-lapsed degradation process of the CF/PBNR composite laminate in ethanol at room temperature. Reproduced from ref. 51 with permission from the RSC. | ||
Guo et al. studied the rubbers capable of self-healing and reprocessability upon the introduction of boronic ester bonds. In 2018, they developed a rubber cross-linked via boronic ester bonds prepared from styrene butadiene rubber (SBR), a widely used commercially available rubber.59 This rubber was synthesized using a thiol–ene click reaction between the double bonds in the pendant group of SBR and the thiol in 2,2′-(1,4-phenylene)-bis[4-mercaptan-1,3,2-dioxaborolane] (BDB) (Fig. 9a). The degree of cross-linking and mechanical properties was controlled by varying the BDB ratio. The resulting rubber was capable of self-healing via a transesterification reaction between the boronic ester bonds and allowed reprocessability. When the film was cut into two pieces, which were then brought into contact with each other to confirm the self-healing properties, the cut surface recovered within 1 h, and the recovered sample showed large elongation (Fig. 9b). In addition, when the healing efficiency was determined using tensile tests, the sample containing 3 phr BDB showed a recovery rate of ∼30% after 1 h at 80 °C and ∼90% after 24 h. This healing efficiency increased with increasing temperature, which further accelerated the boronic ester exchange reaction. In contrast, no dependence of the healing efficiency on the ratio of BDB was observed. This was attributed to the two contradicting effects. Although the cross-linking point at which the exchange reaction could be performed increased upon increasing the BDB ratio, the chain mobility decreased due to cross-linking. Moreover, the rubber could be reprocessed even after being cut into several pieces because of the boronic ester exchange reaction (Fig. 9c).
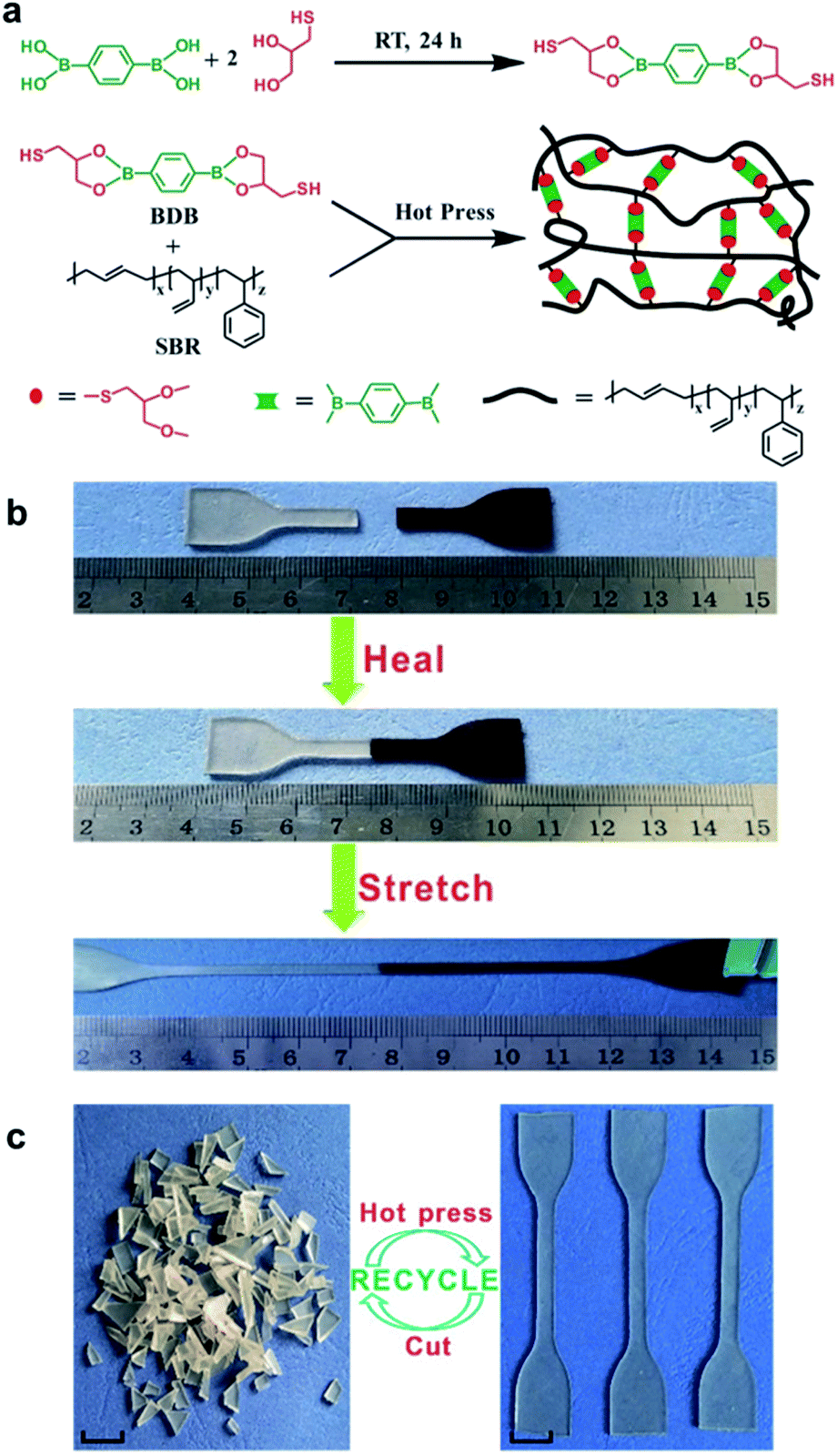 | ||
| Fig. 9 (a) Schematic of the synthesis of BDB and cross-linked SBR capable of self-healing. (b) Self-healing and stretching of SBR. (c) Photographs showing the reprocessability of SBR using heat. Reproduced from ref. 59 with permission from the ACS. | ||
The Guo group used natural rubber the following year to prepare a vitrimer, a mechanically robust elastomer with self-healing property and reprocessability.60 In this study, epoxidized natural rubber (ENR) was cross-linked with BDB. The cross-linked ENR network was denoted as BEx according to the concentration of BDB (x wt%) used. Stress relaxation was not possible in the networks cross-linked with a permanent bond other than the boronic ester bond. The BE series cross-linked using the boronic ester bonds were capable of stress relaxation via a transesterification reaction mechanism at temperatures of >160 °C. Because of these characteristics, the BE rubber network could be reprocessed at 160 °C. In addition, after cutting the samples into two pieces and healing for 1 h, a high degree of stretching was achievable. A comparison of the mechanical properties of BE5 healed at 80 °C for 24 h and those of the original sample before cutting using tensile tests revealed that the sample exhibited similar properties before and after cutting, and showed good healing efficiency. Water was added to the sample to confirm that the mechanism involved in self-healing was hydrolysis/re-esterification, but the hydrophobicity of the rubber matrix prevented water from penetrating the material and no hydrolysis occurred. Thus, self-healing in this sample occurred via the transesterification mechanism. Moreover, when ZnCl2 was added to the rubber to introduce a metal–ligand coordination bond, the mechanical properties enhanced without deteriorating the healing and reprocessing properties. This study proposed a strategy to fabricate elastomeric vitrimers with excellent self-healing properties and reprocessability with mechanical properties that could be employed in practical applications.
In 2020, Li et al. reported a study on the use of polyurethane networks containing catechol-derived boronic esters to prepare elastomeric vitrimers that are highly stretchable, healable, and adhesive (Fig. 10a).61 As an experiment to observe whether or not they could be used as an adhesive material, the elastomers were sandwiched between two pieces of glass substrates for the lap shear tests (adhering at 60 °C for 2 h and subsequently cooling to room temperature). The elastomers with free catechol groups showed stronger adhesion capabilities than those without residual catechol groups (Fig. 10b). For application in a strain sensor with the advantage of elastomer elasticity, an experiment of mixing silver flakes was performed. When the volume fraction of the silver was 23%, there was a linear relationship between the relative change in resistivity and the strain in the range of 30–60%. The electrical self-healing of the strain sensor was confirmed by cutting the sample with a blade and recovering its conductivity after bonding at room temperature for 1 h (Fig. 10c). However, the restoration of the mechanical strength of the silver composite was slower than that of the original elastomer matrix because of the limitations of the silver flakes on the mobility of the polymer chains and dynamic motif.
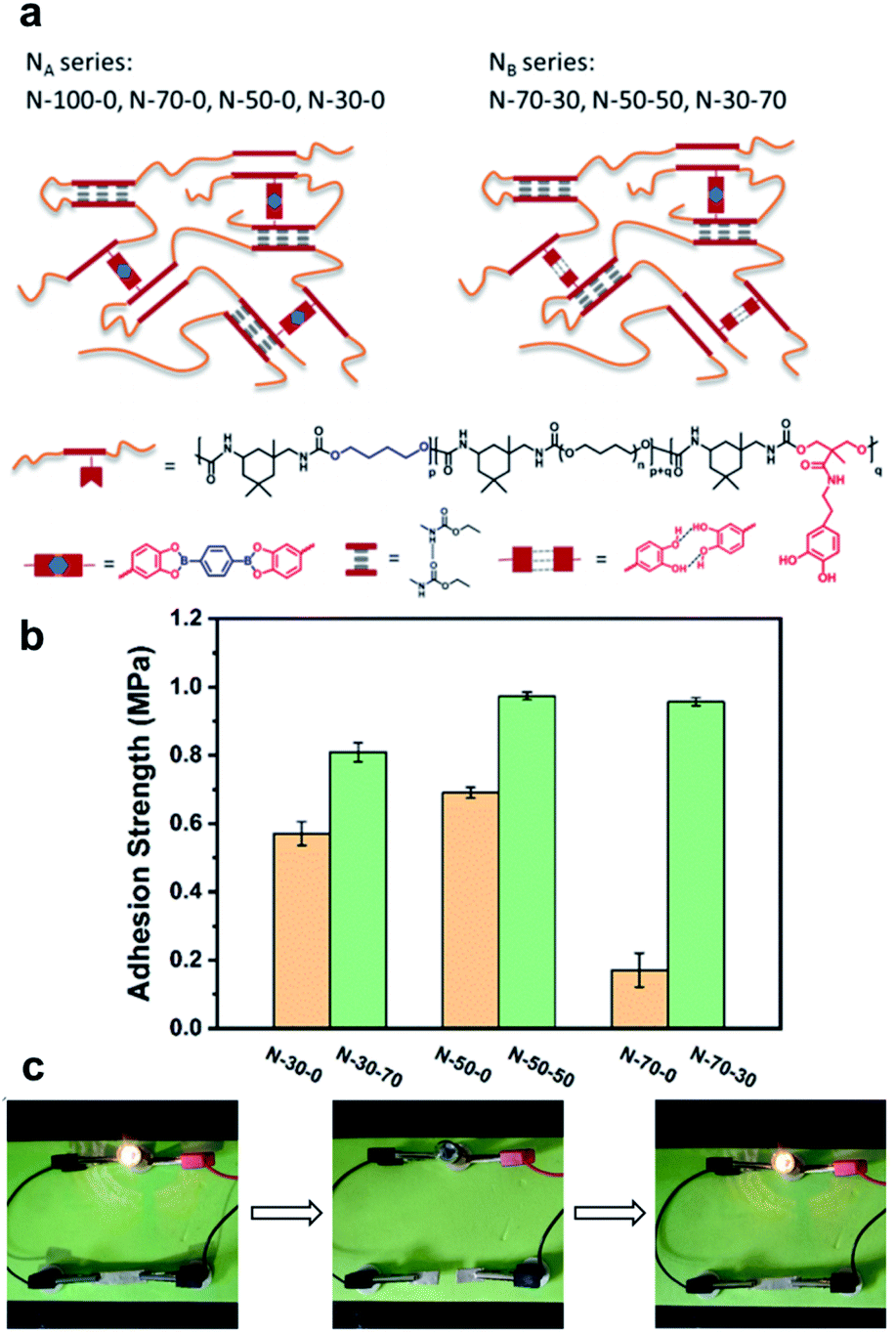 | ||
| Fig. 10 (a) Structures of elastomers with pendent catechol groups (NA: no free catechol and NB: free catechol). (b) The adhesion strength of the elastomers. (c) Demonstration of the healing process for the silver/elastomer composite with a light bulb. Reproduced from ref. 61 with permission from the ACS. | ||
Recently, in addition to the soft polymeric materials such as rubbers or elastomers, an interesting study was conducted to afford self-healing properties by employing boronic ester bonds in the organic crystals. In 2020, the Naumov group used 3-isopropoxyphenyl-boronic acid and catechol to synthesize organic crystals capable of self-healing via boronic ester bond formation (Fig. 11).62 When two pieces of this organic crystal were brought into contact with each other, self-healing occurred at the cut surface. The organic crystals showed ∼67% of their initial strength after the initial recovery step and showed 44% healing efficiency after five recoveries. Upon the application of tensile stress after the two pieces were brought into contact with each other for five seconds to determine if this self-healing was due to physical adhesion, coagulation by water, or non-covalent interactions such as static attraction, the two pieces did not heal over a short period of time and were immediately separated. Therefore, it was confirmed that self-healing was not due to secondary interactions. The mechanism of the exchange reaction through which the boronic ester bonds caused self-healing was also investigated. When water was added to the organic crystal sample, boronic ester bonds were hydrolyzed to generate boronic acid and catechol, but no re-esterification reaction occurred even after ball milling. In contrast, when free diol was present, a transesterification reaction occurred, which was confirmed using Fourier-transform infrared spectroscopy. In addition, 1H NMR spectroscopy was used to confirm that two adjacent boronic ester bonds re-formed two other boronic esters. These experiments confirmed that the hydrolysis/re-esterification mechanism did not significantly affect the self-healing of this sample, while self-healing was shown to occur via boronic ester transesterification and metathesis reactions. This study was the first report of an organic crystal with a considerably high healing efficiency. Through this study, the difference between hard and soft materials was mitigated, and the possibility of developing self-healing organic crystals was demonstrated.
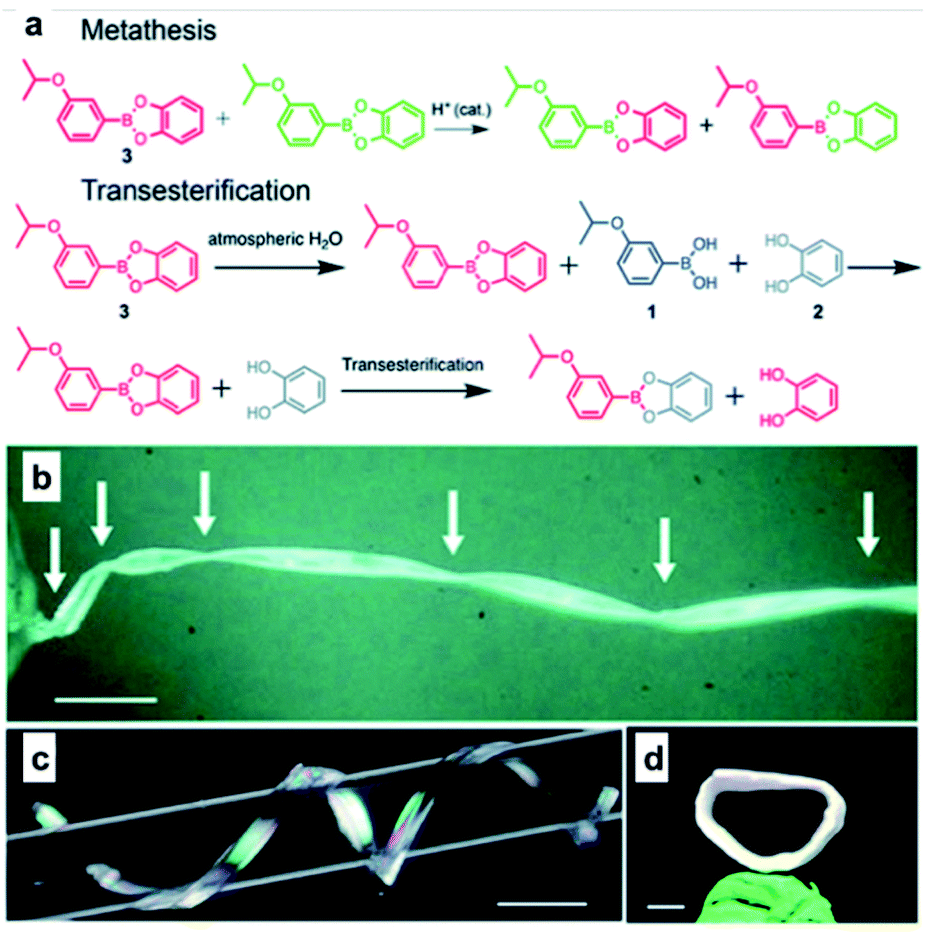 | ||
| Fig. 11 (a) Mechanism for metathesis and transesterification reactions applied to self-healing organic crystals. Crystals of 3 twisted by 1080° (b), coiled around a glass capillary (c), and those with their two ends joined together to form a crystal ring (d) (Scale bars: (b) 1 mm; (c) 2 mm; (d) 1 mm). Reproduced from ref. 62 with permission from the RSC. | ||
4. Self-healing using charged boronate ester bonds
As explained previously, boronic acid is a Lewis acid, and the trigonal planar sp2-hybridized boron atom changes into its tetragonal sp3-hybridized form when combined with a Lewis base, such as a hydroxide group to form a borate complex. Both forms of boronic acid are in equilibrium in an aqueous solution, and when a 1,2- or 1,3-diol is added, neutral and charged boronate esters are formed, respectively. The previous chapter summarized the self-healing polymers bearing neutral boronic esters reported to date, which are relatively unstable in aqueous solutions. In this chapter, studies on self-healing polymers containing charged boronate ester bonds reported in the literature are summarized as follows: Section 4.1 discusses the initial and representative self-healing hydrogels based on boronate esters, while Section 4.2 deals with biomedical application-oriented hydrogels. Section 4.3 introduces the study of additional self-healing moieties for reinforcing mechanical robustness or multi-responsiveness, while Section 4.4 discusses hydrogel studies using borax and benzoxaborole as charged self-healing motifs. Section 4.5 covers special studies on charged boronate ester-containing polymers, specifically bulk polymers or organogels, not hydrogels.4.1 Boronate esters applied in hydrogels
Hydrogels are polymeric materials produced by the retention of a large amount of water during the formation of a 3D cross-linked hydrophilic polymer network. Hydrogels are soft and flexible with biomimetic properties and good biocompatibility. Owing to these characteristics, hydrogels can be applied in various fields ranging from sensors, 3D printing inks, and actuators to various biomedical fields, such as tissue engineering and drug delivery.63–67 Self-healing properties are required for a more efficient application of the hydrogels in these fields. Hydrogels with self-healing properties are still being actively investigated for applications in biomedical fields,68,69 electronic materials, and e-skin.70–73 In particular, a mobile phase is essential for self-healing to allow the healing components to migrate to the damaged area; hydrogels are advantageous as these possess a suitable mobile phase because of the presence of water in their matrix.74–76 This section describes the hydrogels reported in the literature, which are prepared by employing boronate ester bonds (reversible covalent bonds) and exhibit self-healing properties. A polymer containing boronic acid forms a hydrogel via cross-linking with 1,2- or 1,3-diols in an aqueous solution at a pH similar to or higher than the pKa of boronic acid. Depending on the pH, boronic acid in the hydrogel can be separated from 1,2- or 1,3-diol or can be bound by the boronate ester bonds, with a dynamic equilibrium. Thus, boronate ester bonds are not rigid and can be flexible under their own weight.77 In addition, free boronic acid and diol formed via hydrolysis can interact with the adjacent free diol and boronic acid species to form new boronate ester bonds. Thus, the boronate ester bonds in the hydrogels can exhibit self-healing properties without external stimulation owing to this rearrangement mechanism.As mentioned earlier, the boronate ester bonds observed at equilibrium depend on the pH of the solution.78,79 A representative study on the equilibrium of boronate ester bonds in hydrogels was conducted by the Messersmith group in 2011. In this study, 1,3-benzenediboronic acid (BDBA) and 4-arm-poly(ethylene glycol) (PEG) catechol were used to synthesize a hydrogel sensitive to pH and capable of self-healing.79 Considering that the pKa of BDBA is 8.7 and that of catechol is 9.3, the experiment in this study was conducted using a solution of pH 9.0 to allow the formation of stable boronate ester bonds. The resulting hydrogel showed the formation of more boronate ester bonds, which were more stable when the pH was 9.0, i.e., under alkaline conditions. In contrast, as the boronate ester bond was unstable under acidic conditions, boronic acid and catechol dissociated and the gel was converted into a solution (Fig. 12a). In addition, when the hydrogel prepared at pH 9.0 was cut into two pieces and the cut surfaces were brought into contact with each other, the surface showed a rapid recovery owing to the dynamic equilibrium of the boronate ester bonds. The cut surface disappeared as it recovered within 30 s, and the recovered hydrogel could be stretched without further damage. This recovery occurred efficiently even after repeated cutting and healing cycles.
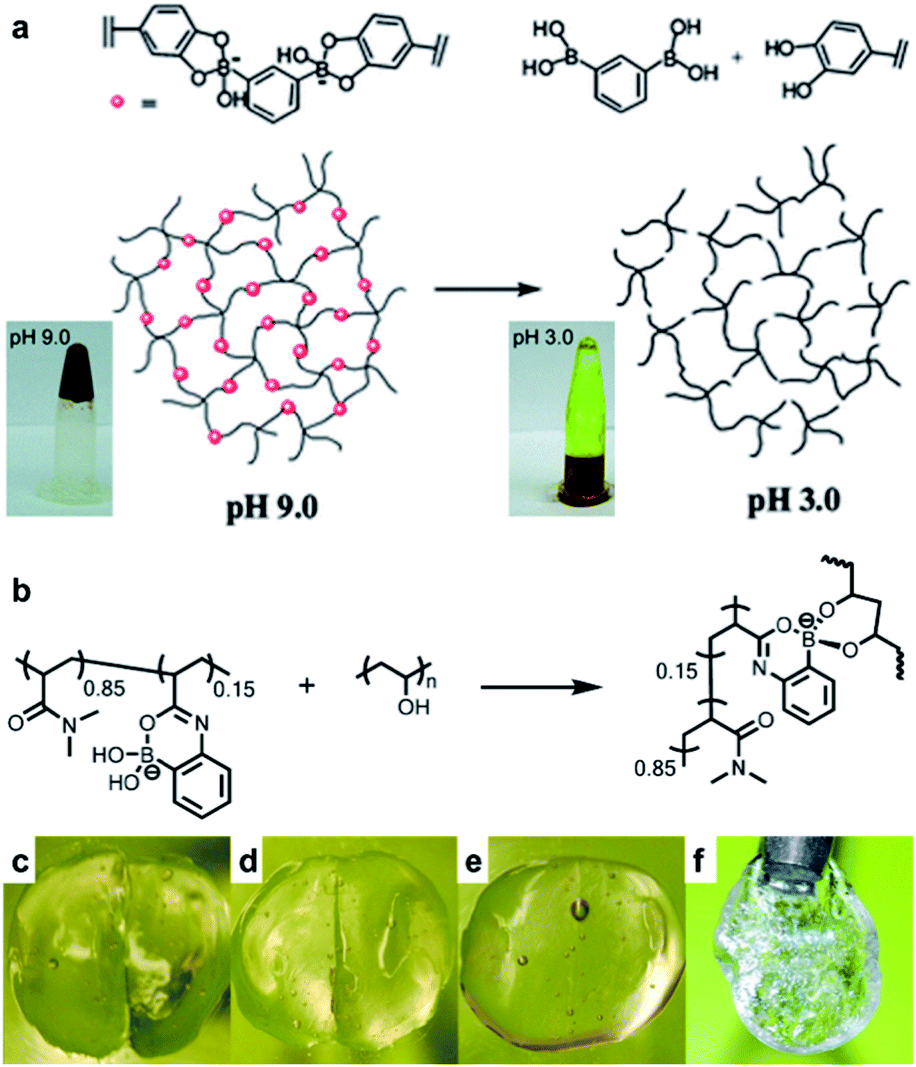 | ||
| Fig. 12 (a) Schematic representation of the pH response of a 4-arm-PEG catechol and BDBA-based hydrogel, and photographs of the changes observed in the hydrogel and its solution according to pH. Reproduced from ref. 79 with permission from the RSC. (b) Hydrogel formed at neutral pH using P(2APBA-co-DMA) (15 mol% 2APBA) and PVA. Photographs of the hydrogel formed using P(2APBA-co-DMA) (10 mol% 2APBA) and PVA (c) after being cut, (d) in contact immediately after being cut, (e) after healing for 60 min, and (f) healed gel suspended under its own weight. Reproduced from ref. 81 with permission from the ACS. | ||
As mentioned in a previous study, stable boronate ester bonds can be formed at a relatively high pH compared to those in vivo because the pKa values of most of the aryl boronic acids are in the range of 8–9.45 Therefore, for the application of a self-healing hydrogel containing boronate ester bonds in biomedical fields, it must be able to form and maintain stable bonds at pH of <8.80 In 2015, the Sumerlin group reported hydrogels capable of forming stable boronate ester bonds under acidic and neutral conditions.81 2-Acrylamidophenylboronic acid (2APBA) and N,N′-dimethylacrylamide (DMA) with hydrophilic properties and good biocompatibility were used as co-monomers to synthesize P(2APBA-co-DMA) and combined with poly(vinyl alcohol) (PVA) to afford a hydrogel (Fig. 12b). The resulting hydrogel maintained a stable gel state even under neutral and acidic pH conditions because the carbonyl oxygen of the acrylamide group and the boron atom of boronic acid formed a stable coordination complex. This hydrogel underwent a rapid exchange reaction, in which a new bond was formed upon the hydrolysis of boronate ester. In the cut and heal experiments, the cut disappeared within 60 min (Fig. 12c–f). This study showed that the boronate ester bonds could form and maintain stable bonds even at low pH via intramolecular coordination, thus suggesting a novel strategy for the synthesis of boronate ester hydrogels with self-healing properties for use over a wide range of pH conditions.
Boronate ester hydrogels are also injectable and self-healing owing to their dynamic bonding properties. Recently, the Park group developed a boronate ester hydrogel that was injectable with very high spinnability and capable of self-healing (Fig. 13).82 Prior to hydrogel synthesis, low-molecular-weight hyperbranched polyglycerol (LMPG) was synthesized. In the next step, in situ radical polymerization of 4-vinylphenyl boronic acid (VBA) and acrylamide was performed in an aqueous solution containing LMPG to prepare the final hydrogel. In this process, 1,2- or 1,3-diols present in LMPG and the boronic acid group in VBA formed a boronate ester bond, which played a key role in the cross-linking process. When the hydrogel was cut into two pieces and placed in contact with each other, self-healing occurred within 30 min, and the cut disappeared. The healing efficiency was determined by measuring the mechanical properties using a universal testing machine (UTM). The maximum load and toughness values of the sample that self-healed for 30 min were recovered to >97% of their initial values. Furthermore, this hydrogel could be stretched to >416 times its initial length, which was higher than that of slime, gum, and other highly stretchable hydrogels reported to date. In addition, when the hydrogel was injected using an 18G needle and stretched, water was removed, and it could be stretched into a fiber. This high stretchability could be attributed to the dynamic properties of the boronic acid ester bonds and high mobility owing to the low molecular weight of LMPG.
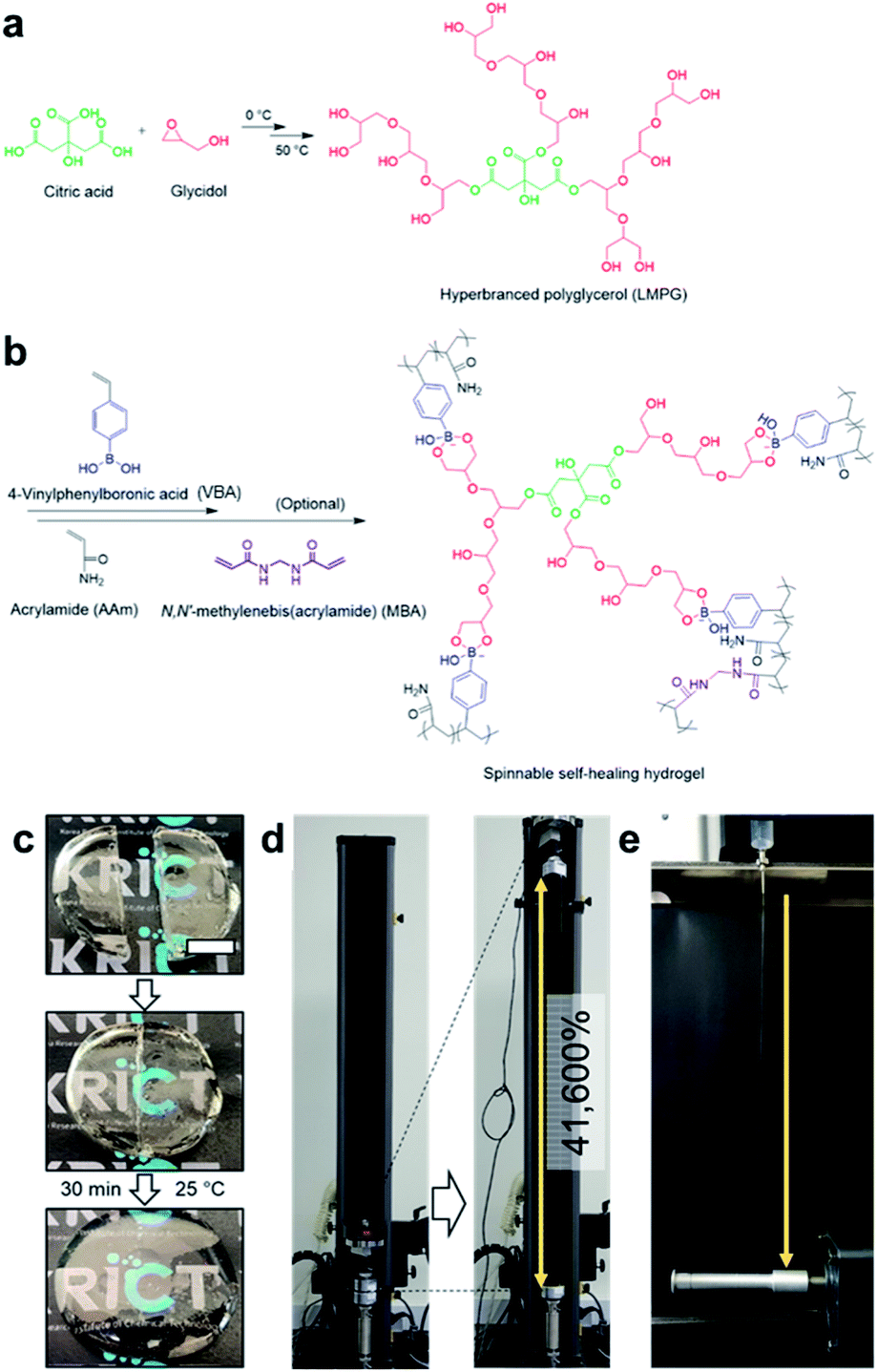 | ||
| Fig. 13 (a) Synthesis of LMPG using cationic ring-opening polymerization. (b) Preparation of a hydrogel using the radical polymerization of VBA and acrylamide. (c) Photographs of the self-healing process of the hydrogel, which self-heals after 30 min at room temperature. (d) Photographs before and after stretching the hydrogel ∼416 times its initial length using a UTM. (e) A photograph of the hydrogel spinning process. Reproduced from ref. 82 with permission from the Springer Nature. | ||
4.2 Self-healing boronate ester hydrogels used for biomedical applications
The B–C bond has a relatively low polarity and is therefore stable in aqueous systems. Recently, the incorporation of boronic acids into medicinal chemistry has steadily increased because the boronic acid moiety does not readily come off from organic compounds in physiological systems.83,84 In biomedical applications, the curing efficiency and mechanical properties of boronic acid-based self-healing materials can decrease in physiological systems because B–O bonds are unstable in aqueous systems. The pH of the system affects the equilibrium of boronic acid/esters in aqueous systems. This issue can be overcome by tuning the pKa of the boronic acid-based self-healing materials to the pH of the target biological system via molecular designing. Plants physiologically use the chemistry of boronic acid/ester pH tuning to control the association and dissociation of boronic acid/ester.85 In this regard, self-healing boronate ester hydrogels have mainly been studied for application in biomedical fields and are still receiving considerable research attention.86,87 The desired mechanical strength can be obtained relatively easily in boronate ester hydrogels by adjusting the ratio of the boronic acid and diol used or pH in the gel. Since boronate ester hydrogels have self-healing and shear-thinning properties due to the dynamic nature of the boronate ester bonds, they are injectable. In addition, these hydrogels are sensitive to glucose. Using these properties, boronate ester hydrogels can be used in tissue engineering and biomedical fields as they can be applied in drug delivery systems and cell cultures. For instance, boronic acid can form boronate esters in hydrogels, but competitively bonds with glucose as well as diols.88 Therefore, when glucose is added to boronate ester-containing hydrogels, the network of the gel collapses because the boronic acid bonds competitively with glucose. As the hydrogel encapsulating the drug enters the body and undergoes a competitive reaction with glucose, it causes the gel structure to collapse, and the drug encapsulated in the gel is released into the body. Thus, this reaction can be applied to drug delivery systems.89,90 In this section, we went beyond the synthesis and characterization of self-healing boronate esters and summarized studies on biocompatibility and biomedical applications, such as drug delivery and cell culture. For practical use, the safety profiles of B–O-based materials are significantly important; therefore, further studies on the short- and long-term effects on the human body, depending on the type and amount of self-healing polymers used, are required.In 2016, the Anderson group produced a PEG-based hydrogel with shear-thinning and self-healing properties, evaluated the biocompatibility of the resulting gel, and conducted a study to encapsulate a drug in the gel as well as its subsequent release.91 Initially, the NH2 group at the end of the 4-arm-PEG was modified using three types of boronic acid derivatives and glucose-like diols to synthesize PEG–FPBA (from fluorophenylboronic acid), PEG–PBA, PEG–APBA (from 2-formylphenylboronic acid), and PEG–diol (from D-glucolactone) (Fig. 14a and b). A total of nine hydrogels were prepared by forming boronate ester bonds in three solutions buffered at pH 6, 7, and 8. The boronic acid derivatives with high pKa values exhibit low mechanical strength at low pH, for example, PBA with the highest pKa value of 7.8 did not form a gel at pH 6. On the other hand, APBA, which has the lowest pKa value of 6.5–6.7, exhibited the strongest mechanical properties under all the pH conditions studied. This indicates that the pH and pKa of the boronic acid derivatives affect the degree of cross-linking of the boronate esters. In addition, the self-healing and shear-thinning properties of the hydrogels prepared at pH 7, which is similar to that found in vivo, were studied. After bringing the two pieces into contact with each other, the self-healing and stretching of the two pieces were visually confirmed to study the self-healing properties (Fig. 14c). The PEG–FPBA and PEG–PBA hydrogels exhibited shear-thinning properties. In particular, the PEG–FPBA hydrogel with a pKa value of 7.2, was injectable using a 21G needle (Fig. 14d). On the other hand, the PEG–APBA hydrogel with a pKa value of 6.5–6.7 displayed rigid and brittle properties similar to a typical cross-linked gel.
 | ||
| Fig. 14 (a) Schematic of the hydrogel synthesized using 4-arm-phenylboronic-acid-containing PEG and diol-containing PEG, and (b) their chemical structures and binding mechanism. (c) Self-healing properties (pH 7), (d) viscosity and shear-thinning behavior, (e) glucose concentration-dependent IgG release profile, and (f) cell viability at different time points (24–72 h) observed for the 10% w/v PEG–FPBA hydrogel. Reproduced from ref. 91 with permission from the Wiley. | ||
Three types of proteins were encapsulated in the injectable PEG–FPBA hydrogel to evaluate the delivery and release of the drug. The protein was efficiently encapsulated inside the gel without any significant changes in the properties of the hydrogel. The hydrogel encapsulating the protein was incubated in phosphate-buffered saline (PBS). Small proteins were rapidly released regardless of the size of the hydrogel mesh, whereas relatively large proteins were released depending on the mesh size. In addition, the release of proteins (immunoglobulin G, IgG) was promoted by the addition of glucose, as the hydrogel structure collapsed due to the binding of boronic acid to glucose (Fig. 14e). To apply this to the 3D cell culture scaffolds, the cells were dispersed in the hydrogel using an in situ method and then cultured for 72 h to evaluate the cytocompatibility of the cells. The dispersed cells showed relatively high cytocompatibility, and the cells were successfully cultured, even in the gel extracts. When the hydrogel was injected and implanted using an 18G needle, it showed good biocompatibility (Fig. 14f). In this study, the pKa of boronic acid used in the synthesis of self-healing and injectable boronate ester hydrogels and the mechanical properties of the gels according to the solution pH were compared. The biocompatibility was further demonstrated through encapsulation and cell culture experiments by employing the hydrogels, suggesting that hydrogels could be applied for drug delivery and tissue engineering.
In the same year, the Anderson group synthesized a polymer containing both boronic acid and glucose in one chain to obtain a hydrogel composed of a single-component polymer and studied the self-healing and drug release properties of the resulting gel.92 The single-component polymer was synthesized via radical polymerization of two types of monomers containing boronic acid and NH2 functional groups, and glucose was then introduced upon modification of the NH2 group to synthesize the final polymer. The hydrogel was formed in a 5 wt% aqueous solution when the composition of the boronic acid moiety in the polymer was increased from 10 to 60 wt% by adjusting the monomer ratio. Rhodamine B, which was inserted into the hydrogel as a model drug, was released when the hydrogel was immersed in a glucose solution. The higher the concentration of the glucose solution, greater was the amount of rhodamine B released. In this study, an injectable hydrogel was developed with self-healing and shear-thinning properties that could release the encapsulated drugs in response to glucose, exhibiting significant potential for biomedical applications.
In 2017, the Díaz group also prepared a single-component polymer hydrogel that was injectable, self-healing, suitable for living organisms, and capable of responding to various stimuli by modifying the alginate with boronic acid.93 The aqueous solution of alginate modified with boronic acid gelled under alkaline conditions without any additional diol (Fig. 15a). This was due to the formation of boronate ester bonds between the boronic acid moiety and diols present in the pyranose ring of the alginate. Gelation was possible at three aqueous concentrations of 3%, 4%, and 5% (w/v). The hydrogels prepared at 4% and 5% (w/v) concentrations were fragile and self-healed after damage; however, several hours were required to heal. In contrast, the hydrogel prepared at a concentration of 3% (w/v) was soft and stable upon cutting into two pieces that self-healed within a few minutes, thereby allowing stretching without further damage. A hole drilled in the middle of the hydrogel prepared at 3% (w/v) concentration was used to study its self-healing property, and this disappeared within 10 min. Artificial scratches on the hydrogel disappeared within 30 min when observed using an optical microscope. Rheological measurements showed that the storage modulus (G′) was greater than the loss modulus (G′′) upon the application of low strain (5%), but when high strain (500%) was applied, the gel ruptured, resulting in a greater loss modulus than the storage modulus. At a low strain of 5%, the storage modulus increased again, indicating self-healing of the gel (Fig. 15b). When a solution of pH 5 was added to the hydrogel prepared under alkaline conditions, the boronate ester bonds were broken, and the gel collapsed. The gel regained its stability upon the addition of NaOH solution, showing pH sensitivity. When the hydrogel was brought into contact with a fructose solution, the gel network collapsed because boronic acid interacted with the monosaccharide. Moreover, when HeLa cells were encapsulated in the gel, these showed good biocompatibility, as indicated by the high survival rates observed for the HeLa cells. This also suggested the possibility of biomedical applications owing to its injectability (Fig. 15c).
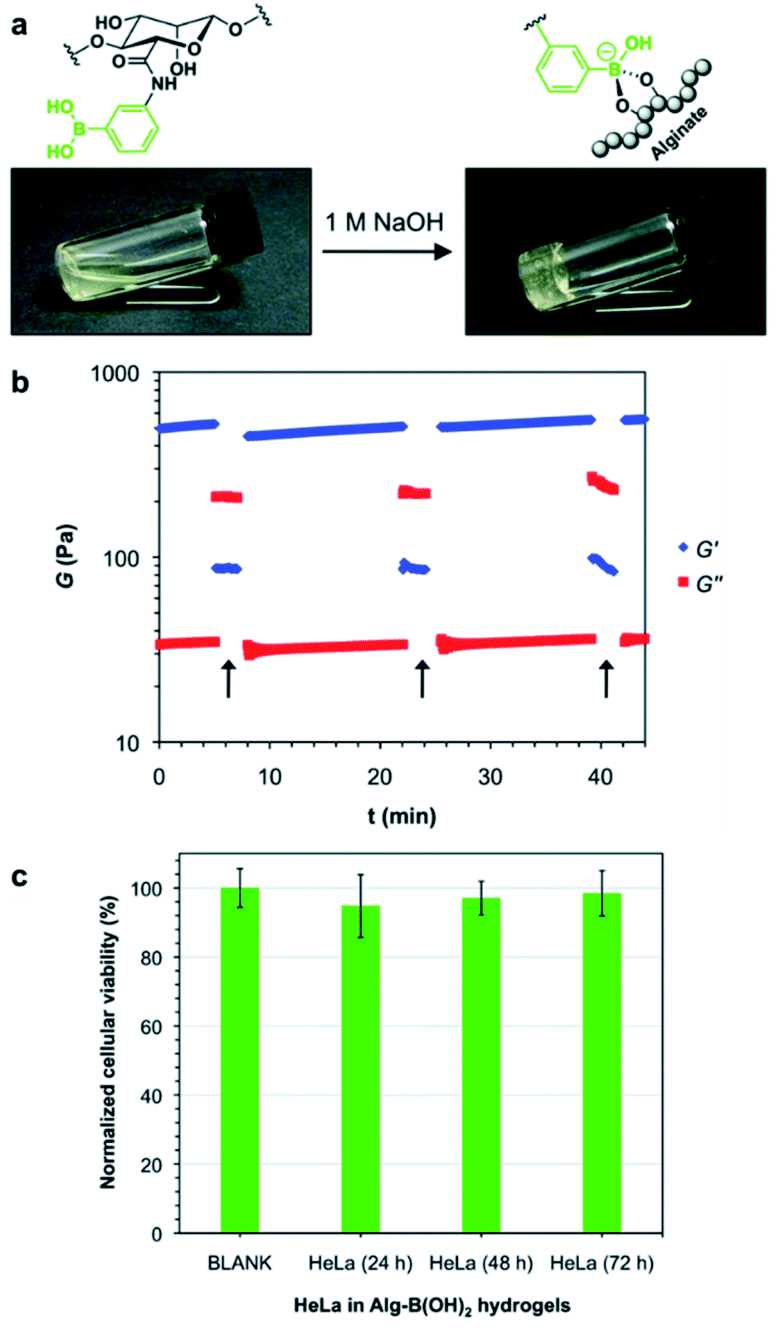 | ||
| Fig. 15 (a) Gelation of an aqueous solution of alginate modified with boronic acid upon increasing the pH. (b) Oscillatory rheological tests showing the thixotropy of the hydrogel. The arrows indicate the increase in the shear strain to 500%. (c) In vitro cytotoxicity of the hydrogel cultured with HeLa cells after 24, 48, and 72 h of incubation. Reproduced from ref. 93 with permission from the RSC. | ||
In 2018, the Lee group developed a hydrogel with various functions using a single polymer component consisting of alginate–boronic acid without the addition of inorganic nanoparticles, carbon materials, clay, or other polymers.94 The alginate–boronic acid-based single polymer was prepared by the introduction of a boronic acid group into the main chain of the alginate via a chemical reaction. The resulting alginate–boronic acid polymer formed a hydrogel at 2.3 wt% concentration in PBS solution of pH 7.4. The alginate–boronic acid hydrogel could be stretched 23 times longer than the hydrogels prepared using unmodified alginate or alginate–Ca2+ (Fig. 16a). In addition, owing to its shear-thinning properties, it was possible to inject the hydrogel in a one-dimensional manner, such as a fiber, using an 18G needle. A pulling experiment with alginate–boronic acid was conducted using atomic force microscopy to study the stretching properties of these materials at the molecular level. The force–distance graph showed a sawtooth pattern. This indicated that the bond between boronic acid and diol was broken by the applied force, and the resulting boronic acid and diol could reversibly reform the borate ester bond, thereby requiring another force to break this bond. As other boronic acids and diols could form bonds due to the polymer chain mobility in the hydrogel, the alginate–boronic acid hydrogels exhibited self-healing and reshaping properties. Consequently, after cutting and bringing the resulting pieces in contact with each other, the heart-shaped hydrogel healed within 5 min. In the step-strain test, the gel collapsed under high strain and then recovered its mechanical properties under low strain conditions. A higher healing efficiency was achieved when more boronic acid was substituted in the main chain of the alginate.
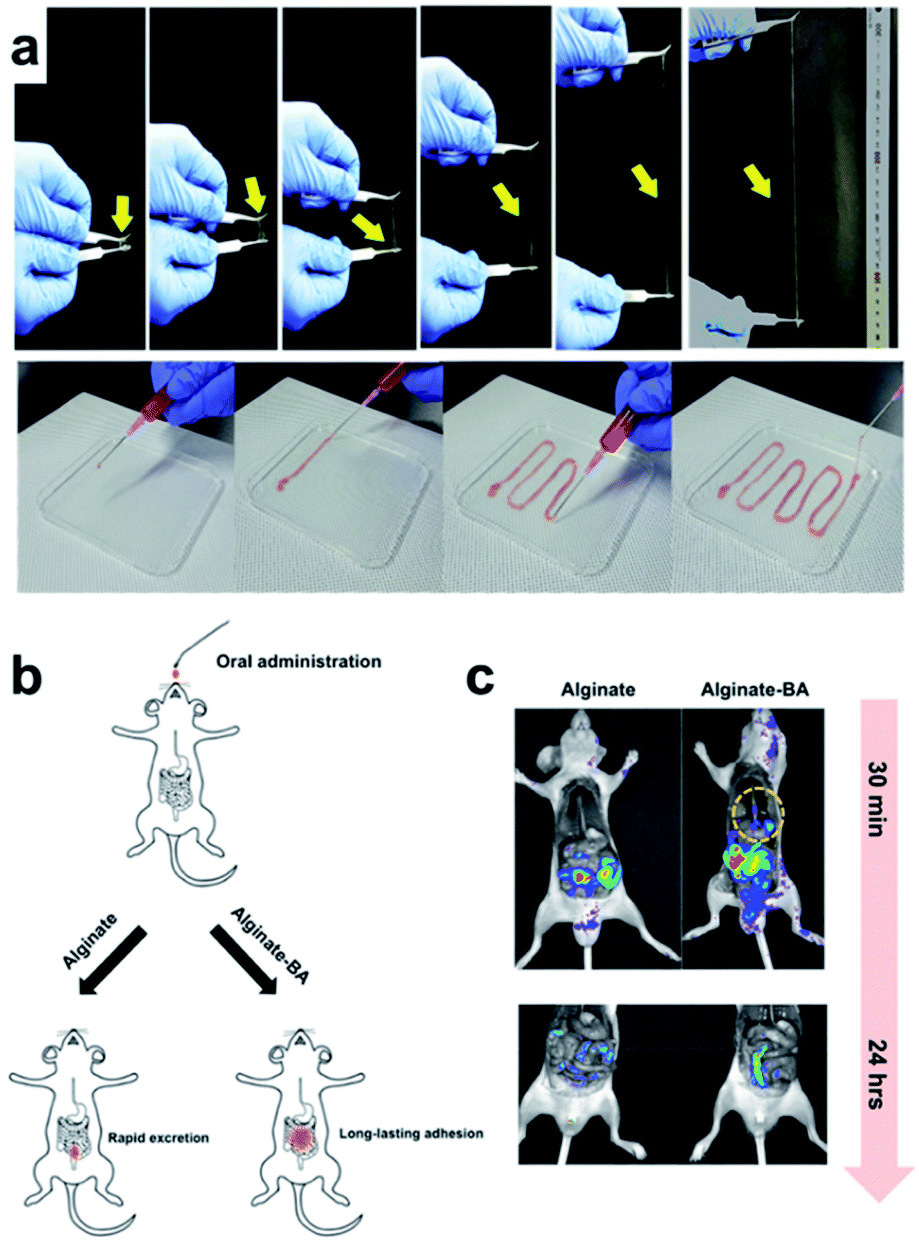 | ||
| Fig. 16 (a) Stretchability of the alginate–boronic acid hydrogel. (b) Schematic of its in vivo oral administration. (c) Time-dependent fluorescence image after the injection of alginate and alginate–boronic acid coated with Rho–dex. Reproduced from ref. 94 with permission from the ACS. | ||
Based on the frequency sweep experiment of the alginate–boronic acid hydrogels at pH values of 7.4 and 10, G′′ was higher at low frequencies while G′ was higher at high frequencies, and a crossover point was observed. Among these, G′ and G′′ were higher, and the gel behavior was improved because of the higher number of boronate ester bonds at pH 10 than at pH 7. Conversely, boronic acid and diol were completely bound at pH 12, and no crossover point was observed. This pH-dependent property was also observed in the sol–gel–sol transition upon the addition of a small amount of HCl or NaOH solution. In addition to the pH-dependence, the hydrogel showed a response to glucose, in which the storage moduli decreased after the addition of the glucose solution due to the interference of glucose with the cross-linking of the hydrogel. Moreover, the alginate–boronic acid hydrogel showed good potential as a pressure-sensitive adhesive because of its strong adhesive properties. Alginate–boronic acid and unmodified alginate were coated with a fluorescent substance (rhodamine B isothiocyanate–dextran (Rho–dex)) and injected into rats to determine the amount retained in the body along the gastrointestinal tract by fluorescence imaging (Fig. 16b and c). Alginate–boronic acid gelled in the esophagus and emitted fluorescence in the stomach, whereas alginate was mostly washed through the esophagus. Only Rho–dex was injected, and no fluorescence was observed as there was no mucoadhesion. The above-mentioned experiments suggested that alginate–boronic acid hydrogels with low toxicity, pH sensitivity, and viscosity could be used for oral delivery.
In 2018, the Zhang group conjugated PBA to M13 nanofibrous virus with a high aspect ratio (PBA–M13), and then prepared a hydrogel with PVA (Fig. 17).95 The pKa of PBA present in PBA–M13 was 7.7, which was lower than that of the aryl boronic acid (pKa = 9) due to the electron-withdrawing effect of the amide carbonyl group. At pH 7.4, the formation of a bond between PBA–M13 and the diol was confirmed using alizarin red S (ARS), a fluorescent dye containing a diol, and glucose. In addition, PBA-M13 showed liquid crystal properties with an uncharged hydrophobic nematic phase at a pH lower than the pKa and a negatively charged hydrophilic chiral nematic liquid crystal phase at a higher pH. A hydrogel was obtained by combining the PBA–M13 suspension with PVA solution at physiological pH. When two adjacent pieces of this hydrogel were brought into contact with one another, self-healing was observed as the cut disappeared after 1 h. A step-strain sweep was performed to quantitatively analyze the self-healing properties. At a high strain of 300%, G′′ was greater than G′, and the gel collapsed. At a low strain of 1%, G′ was greater than G′′, restoring the original gel state and G′/G′′ values. Moreover, the hydrogel was injectable using a 21G needle, and it was confirmed that the nanofibers inside the injected gel were oriented along the major axis using cryo-scanning electron microscopy (cryo-SEM). When ARS was injected into a part of the hydrogel, the ARS diffused inside the gel, forming a bond with PBA and the gel became red/orange after 1 h. Based on this principle, insulin was released more rapidly upon the injection of glucose into the hydrogel containing insulin in comparison to when glucose was not injected. This study showed that a boronate ester hydrogel could be applied to a wide range of biomaterials by preparing a hydrogel capable of injection, self-healing, glucose responsiveness, and internal structure control at physiological pH.
 | ||
| Fig. 17 (a) Preparation of PBA–M13. (b) Hydrogel prepared using PBA–M13 and PVA, and its self-healing behavior. (c) Fluorescence-on behavior upon the binding of ARS to PBA–M13 and its fluorescence-off behavior upon exchange with glucose. Reproduced from ref. 95 with permission from the ACS. | ||
Hydrogels are widely used in cell research. Decomposition of hydrogels assists cell proliferation and migration, but in hydrogels formed via covalent bonds, the structure collapses during its decomposition and may dissolve in a solvent. To compensate for this phenomenon, the Sumerlin and Kloxin groups applied dynamic boronate ester bonding to cell cultures in 2018.96 The hydrogel used in this study employed a monomer (2APBA)81 that could form stable boronate ester bonds under neutral conditions, which was developed by the same group in 2015 (Fig. 18a and b). Three hydrogels were prepared using three types of solutions, including PBS, a cell culture medium without serum, and a cell culture medium containing serum. Self-healing of the resulting hydrogel was confirmed using a hang test and rheological measurements. For the cell culture experiments, two model human cell lines (human breast cancer cells and human pulmonary fibroblasts) were encapsulated in the hydrogel. In the experiment, a solution containing a small amount of fibronectin was used to increase the cell adhesion by taking advantage of the interaction of boronic acid with the free diol in fibronectin. Both cell lines were encapsulated in the hydrogel and showed good viability even after seven days. After confirming the self-healing properties of the hydrogel and stability of the cells using this series of experiments, the cells were encapsulated into two hydrogels, and the two hydrogels were allowed to self-heal for 1 h in an incubator at 37 °C in 5% CO2 atmosphere. The proliferation of the two cell lines encapsulated in the self-healing hydrogel over one week was confirmed using confocal microscopy (Fig. 18c and d). This study showed that the hydrogel containing boronate ester bonds was capable of self-healing as well as the encapsulation of human cells, thus demonstrating the usefulness of self-healing materials in the preparation of co-culture media for cells. In addition, it allows more diverse biological applications.
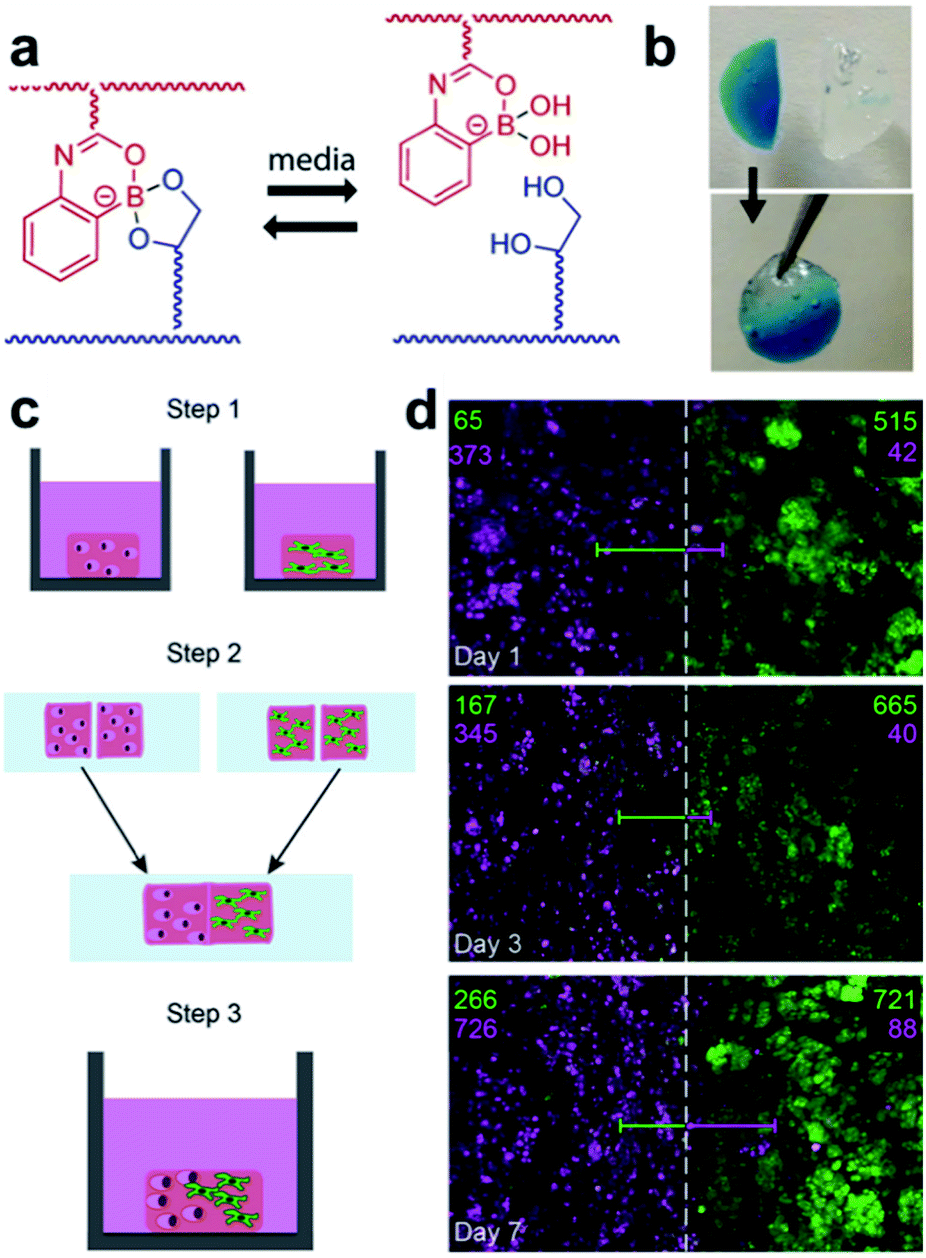 | ||
| Fig. 18 (a) Self-healing mechanism of the hydrogel composed of 2APBA and PVA. (b) Image of the hang test, (c) schematic of the cell co-culture experiment using the hydrogel, and (d) confocal z-stack image at the healed surface and cell quantification in the 1000 μm long region (number of fibroblasts (green) and breast cancer cells (pink) are shown). Reproduced from ref. 96 with permission from the ACS. | ||
The Das group synthesized a hydrogel with a unique structure using guanosine (G).97,98 Guanosine contains a 1,2-diol capable of forming a boronate ester bond with boronic acid, which can self-assemble into G-ribbons, G-quartets, and helical structures via hydrogen bonding. Among these, the G-quartet structure is more stable in the presence of metal ions, such as K+.99 In addition, the G-quartets can be stacked via π–π stacking interactions to form a larger G-quadruplex aggregate. Using these properties, a study was conducted in 2018 to apply the G-quadruplex hydrogel to 3D biomaterial printing.97 In this study, guanosine and three types of boronic acid, including PBA, 4-nitrophenyl boronic acid (4-NPBA), and 4-methoxyphenyl boronic acid (4-MPBA), were used to synthesize GPBA, 4-GNPBA, and 4-GMPBA, respectively. All synthesized hydrogels were self-assembled in the form of nanofibers, in which the G4-quartet structures were aggregated inside the gel (Fig. 19a–c), and the self-assembled structure was confirmed using circular dichroism (CD) and powder X-ray diffraction (PXRD). The high strengths of the GPBA and 4-GNPBA hydrogels were attributed to the high boronate ester conversion, whereas the 4-GMPBA gel was relatively weak due to the electron-donating effect of 4-MPBA, which was unsuitable for 3D printing. In addition, the GPBA hydrogel showed rapid self-healing in the strain sweep experiment, in which a high strain of 100% and a low strain of 0.1% were applied repeatedly. When the three pieces of the gel were brought into contact with one another, the cut surfaces disappeared after 30 min and formed a single gel. Moreover, the GPBA hydrogel showed good cell viability and uniform cell distribution when human dermal fibroblasts (HDFs) were used inside the gel, suggesting that this hydrogel could be applied as a 3D printing biomaterial (Fig. 19d–f).
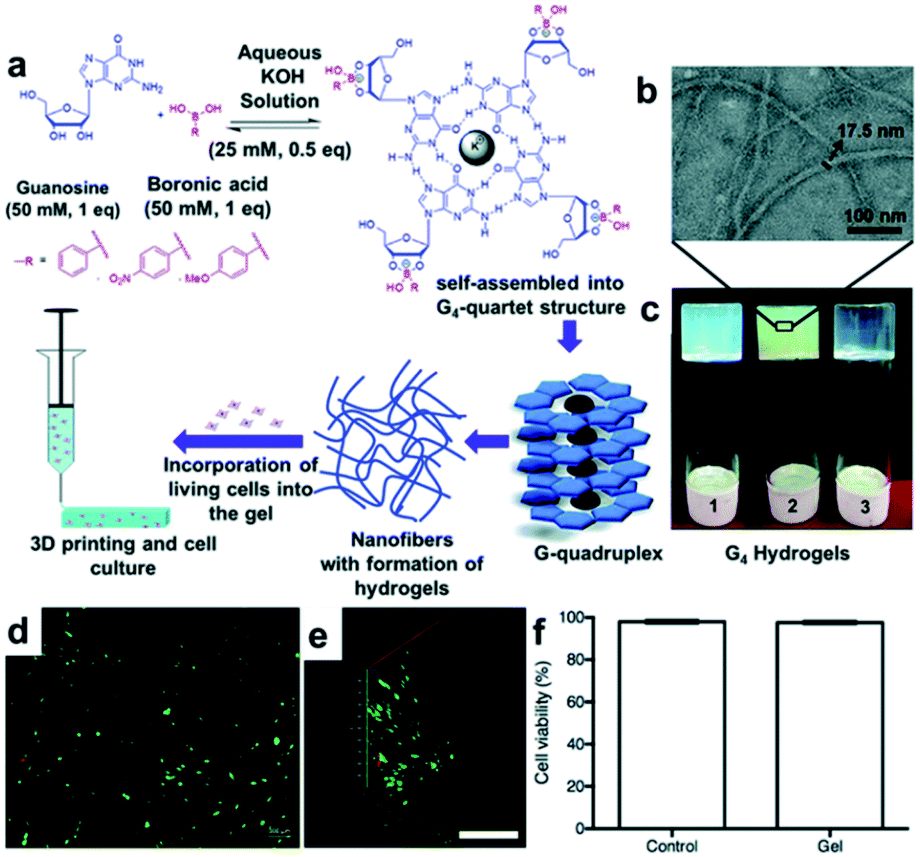 | ||
| Fig. 19 (a) Formation of G-quadruplex hydrogel via G-quartet self-assembly; schematic of its 3D printing and cell culture applications. (b) Transmission electron microscopy (TEM) image of 4-GNPBA and (c) photographs of GPBA (1), 4-GNPBA (2), and 4-GMPBA (3) hydrogels. (d) Fluorescence images of the live/dead cell assay of HDF cells in GBPA after 24 h of cell culture (scale bar = 100 μm). (e) Z-Stack fluorescence 3D image showing the adhered HDF cells inside the gel (scale bar = 500 μm). (f) Cell viability graph showing 98% viability in GPBA after 24 h of incubation. Reproduced from ref. 97 with permission from the RSC. | ||
In 2020, the same group developed a G-quadruplex hydrogel that slowly released the vitamins over 40 h at physiological pH and temperature using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of guanosine and naphthaleneboronic acid (1-NapBA).98 This hydrogel could maintain a stable structure through hydrogen bonding owing to the presence of K+, boronate ester bonding between guanosine and 1-NapBA, and π–π stacking interactions. Self-assembly into G-quartet and G-quadruplex structures in the presence of K+ was confirmed using a thioflavin T-binding assay, CD, PXRD, and morphological analyses. The hydrogel showed a gel state at a low deformation (strain) of 0.1–10% and a solution state under high strain (>10%). To confirm the self-healing properties, one dyed gel and one undyed gel were cut into five pieces, which were then brought into contact with each other for several minutes. As a result, the dyeing agent was dispersed throughout the gel, and the cut surface disappeared. In addition, the healed hydrogel supported its weight in the hang test. The hydrogel was injectable, could form several 3D shapes, and showed high biocompatibility in cell culture experiments using HeLa, MCF-7, and HEK293 cell lines. Furthermore, when vitamin B2 and B12 were encapsulated in the hydrogel and released in vivo, the fibril network structure of the G-quadruplex helped to slow down the release of vitamins. Therefore, the above-mentioned properties showed that the G-quadruplex gel used in this study was applicable to various biomedical fields, such as 3D bioprinting, biotransplant materials, and drug delivery.
:1 molar ratio of guanosine and naphthaleneboronic acid (1-NapBA).98 This hydrogel could maintain a stable structure through hydrogen bonding owing to the presence of K+, boronate ester bonding between guanosine and 1-NapBA, and π–π stacking interactions. Self-assembly into G-quartet and G-quadruplex structures in the presence of K+ was confirmed using a thioflavin T-binding assay, CD, PXRD, and morphological analyses. The hydrogel showed a gel state at a low deformation (strain) of 0.1–10% and a solution state under high strain (>10%). To confirm the self-healing properties, one dyed gel and one undyed gel were cut into five pieces, which were then brought into contact with each other for several minutes. As a result, the dyeing agent was dispersed throughout the gel, and the cut surface disappeared. In addition, the healed hydrogel supported its weight in the hang test. The hydrogel was injectable, could form several 3D shapes, and showed high biocompatibility in cell culture experiments using HeLa, MCF-7, and HEK293 cell lines. Furthermore, when vitamin B2 and B12 were encapsulated in the hydrogel and released in vivo, the fibril network structure of the G-quadruplex helped to slow down the release of vitamins. Therefore, the above-mentioned properties showed that the G-quadruplex gel used in this study was applicable to various biomedical fields, such as 3D bioprinting, biotransplant materials, and drug delivery.
4.3 Self-healing boronate ester hydrogels reinforced using dynamic covalent bonds or supramolecular attractions
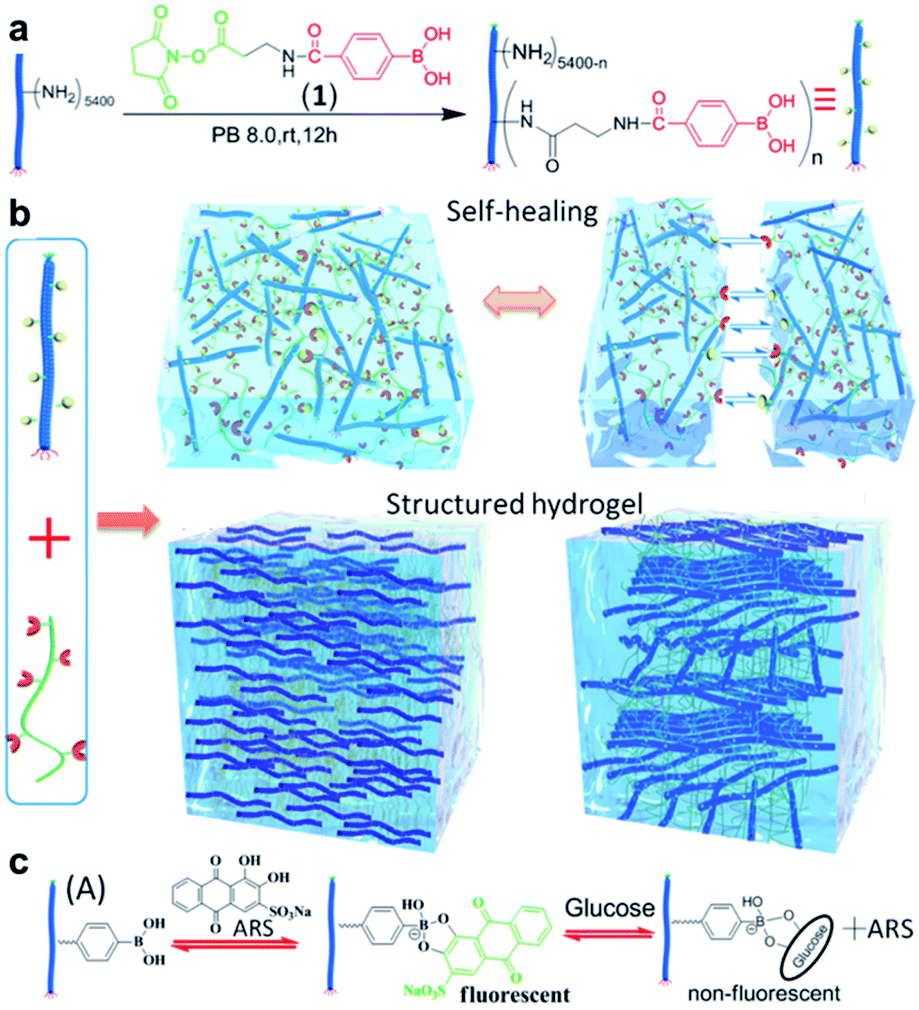
In Section 4.2, the previously reported hydrogels capable of self-healing mainly using boronate ester bonds as dynamic covalent bonds are summarized. However, some studies have reported the simultaneous application of two or more types of dynamic covalent bonds or a combination of dynamic covalent bonds and supramolecular interactions to a single polymer material to afford additional or improved electrical conductivity, shape memory properties, mechanical properties, stimulus sensitivity, or self-healing properties.100–103 This section describes studies based on boronate ester bonds in which additional dynamic covalent bonds or supramolecular forces are also applied.In 2014, the Chen group prepared a polymeric hydrogel capable of self-healing and shape memory using two interactions.104 In this study, the hydrogel composed of alginate grafted with PBA (Alg–PBA) and PVA showed self-healing capability and good deformability owing to the dynamic nature of the boronate ester bonds. In addition, the intermolecular cross-linking of the α-L-guluronate moiety in alginate and Ca+ species allowed the gel to stably maintain its temporary shape. In 2017, Zhang et al. prepared a hydrogel capable of 3D printing by introducing boronate ester and acylhydrazone bonds into an interpenetrating polymer network (IPN).105 The mechanical properties and gelation time of the resulting IPN hydrogel could be controlled using the polymer concentration and pH, such as the dissociation of the boronate ester bond with the presence of the reversible acylhydrazone bond at a low pH. The structure and self-healing properties of the hydrogel could also be adjusted using pH. In 2017, the Connal group synthesized a hydrogel capable of self-healing by employing the oxime bonds. After modification with boronic acid and the addition of tannic acid, the resulting hydrogel exhibited improved mechanical properties owing to the presence of boronate ester bonds.106
In 2017, the Zhang group studied a hydrogel capable of self-healing under ambient conditions by simultaneously introducing reversible boronate ester bonds and disulfide bonds into the hydrogels, which were also sensitive to pH, glucose, and redox reactions.107 In this study, a hydrogel was prepared by manipulating the catechol group present in the poly(acrylamide-co-dopamine methacrylamide) (P(AM–DOPMA)) chain, which could form a boronate ester bond with boronic acid. Bis(phenylboronic acid carbamoyl) cystamine (BPBAC) was used as the cross-linking agent for the main chain (Fig. 20a). The mechanical properties could be controlled by modifying the BPBAC ratio because BPBAC contained two boronic acid groups. In addition, this hydrogel was capable of a reversible gel–sol–gel phase transition that was responsive to glucose or pH. The phase also switched upon the addition of a redox reagent [dithiothreitol (DTT)] because of the disulfide group in BPBAC (Fig. 20b and c). To demonstrate multi-responsiveness to stimuli, such as pH, glucose, and redox, this hydrogel showed self-healing properties without any additional treatment or healing agent as two pieces of the P(AM–DOPMA) hydrogel adhered to one another after 1 min, and the cut surface disappeared after 5 min. This study provided a strategy to easily manufacture hydrogels that could be applied to drug delivery systems, medical adhesives, and sealants, while controlling the desired mechanical properties using commercially available reactants.
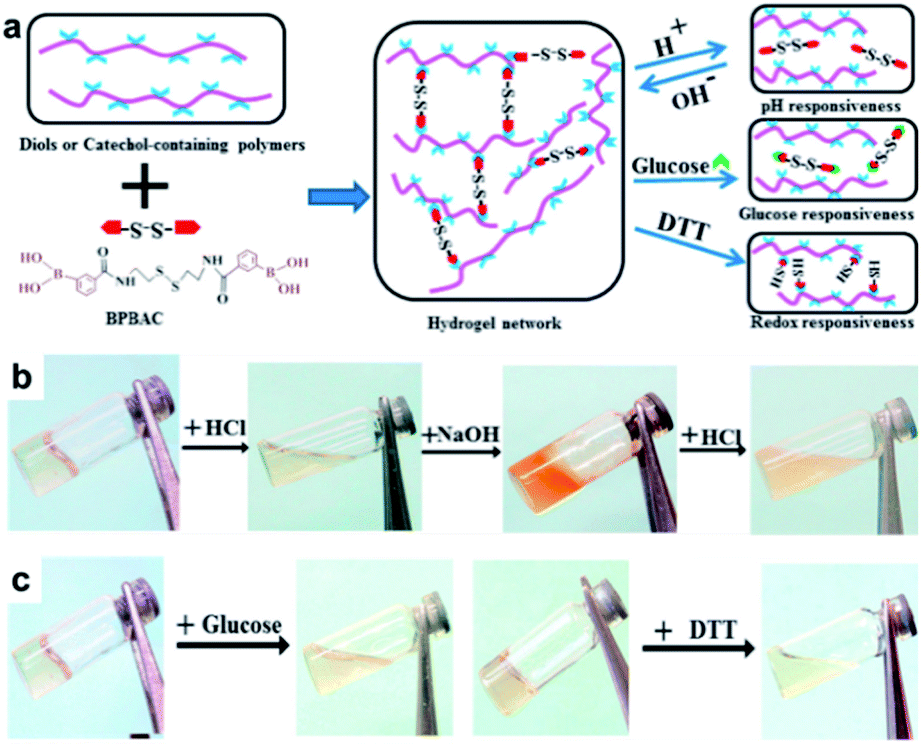 | ||
| Fig. 20 (a) Schematic for the formation of the BPBAC, PVA, and P(AM–DOPMA) hydrogel network and its multi-responsive properties. (b) pH-triggered gel–sol–gel phase transition and (c) response to glucose and redox stimuli. Reproduced from ref. 107 with permission from the ACS. | ||
In 2020, Tan et al. synthesized a hydrogel that simultaneously employed boronate ester and acylhydrazone bonds.108 The boronate ester bond was formed rapidly upon gelation, but the acylhydrazone bond was formed slowly. In addition, acylhydrazone contributed to the increased mechanical strength of the hydrogel owing to its relatively less dynamic nature. The mechanical behavior of the prepared hydrogel also changed depending on the water and polymer concentrations. The boronate ester bond significantly affected the mechanical strength at low concentrations, whereas the acylhydrazone bond contributed to >80% of the mechanical strength at high concentrations. To evaluate the self-healing properties of the hydrogel, the disk-shaped hydrogel was cut into two pieces, dyed, and subjected to the cut and heal experiment. The hydrogel composed of only boronate ester bonds showed excellent self-healing within 30 min, whereas the hydrogel comprising acylhydrazone bonds did not self-heal even after 24 h because the exchange reaction was hindered under weak alkaline conditions. Further experiments revealed that the hydrogel was sensitive to pH, fructose, and hydrazide. This study showed the individual effects of boronate ester and acylhydrazone bond on the mechanical behavior and self-healing properties of the hydrogels, respectively, providing a strategy for the preparation of hydrogels with desirable properties by controlling the composition of the bonds used.
4.4 Self-healing hydrogels containing B–O ester bonds with components other than boronic acid
The hydrogels discussed in Sections 4.2 and 4.3 employ boronic acid as a substance that bonds with 1,2- or 1,3-diols to form boronate ester bonds. In the self-healing hydrogels, dynamic B–O bonds can also be formed by combining diols with substances other than boronic acid. Substances that can be used for this purpose include borax and benzoxaborole. Borax is hydrolyzed in water to form boric acid and borate ions.109 The hydrolyzed anionic form of B(OH)4− can form B–O bonds with two diols in two steps. This bond was called boronate ester or diol–borate ester in a study that used borax to form B–O bonds; in this review, it is referred to as the boronate ester. Tetrahedral B–O bonds formed by the bonding of benzoxaborole with a diol are also called boronate esters and have been discussed herein.The first study described in this section used borax as a catalyst as well as a cross-linking agent. In 2015, Theato et al. synthesized a self-healing PEG-based hydrogel from commercial starting materials using a simple method (Fig. 21a).110 A cross-linkable polymer chain was prepared using the thiol–ene Michael polyaddition reaction between the diacrylate group at the end of the PEG chain and the thiol group at the end of DTT, which was catalyzed by borax to produce a PEG-based hydrogel. Borax acts as both a catalyst to assist the formation of the thioether bonds as well as a cross-linking agent to form boronate ester bonds upon reaction with the diol. Gelation was fast enough to occur between a time line of ∼40 s and 2 min depending on the concentration of borax used under ambient conditions. The resulting gel showed a G′ value of 104 Pa, which was similar to that observed for permanently cross-linked PEG hydrogels. When the hydrogel was cut into two pieces and brought into contact with each other, the cut surface disappeared within 30 min, and the gel recovered sufficient mechanical strength to support its weight. Self-healing was possible even during repeated cutting and healing experiments. Furthermore, the boronate ester bonds in this gel were sensitive to pH and heat. The bonds were broken upon heating or decreasing the pH, and formed again upon cooling and increasing the pH. This study was the first report employing borax as a catalyst to form a PEG-based hydrogel via the thiol–ene Michael polyaddition reaction, providing a new route to apply borax to gel sealants, biosensors, and regenerative medicine.
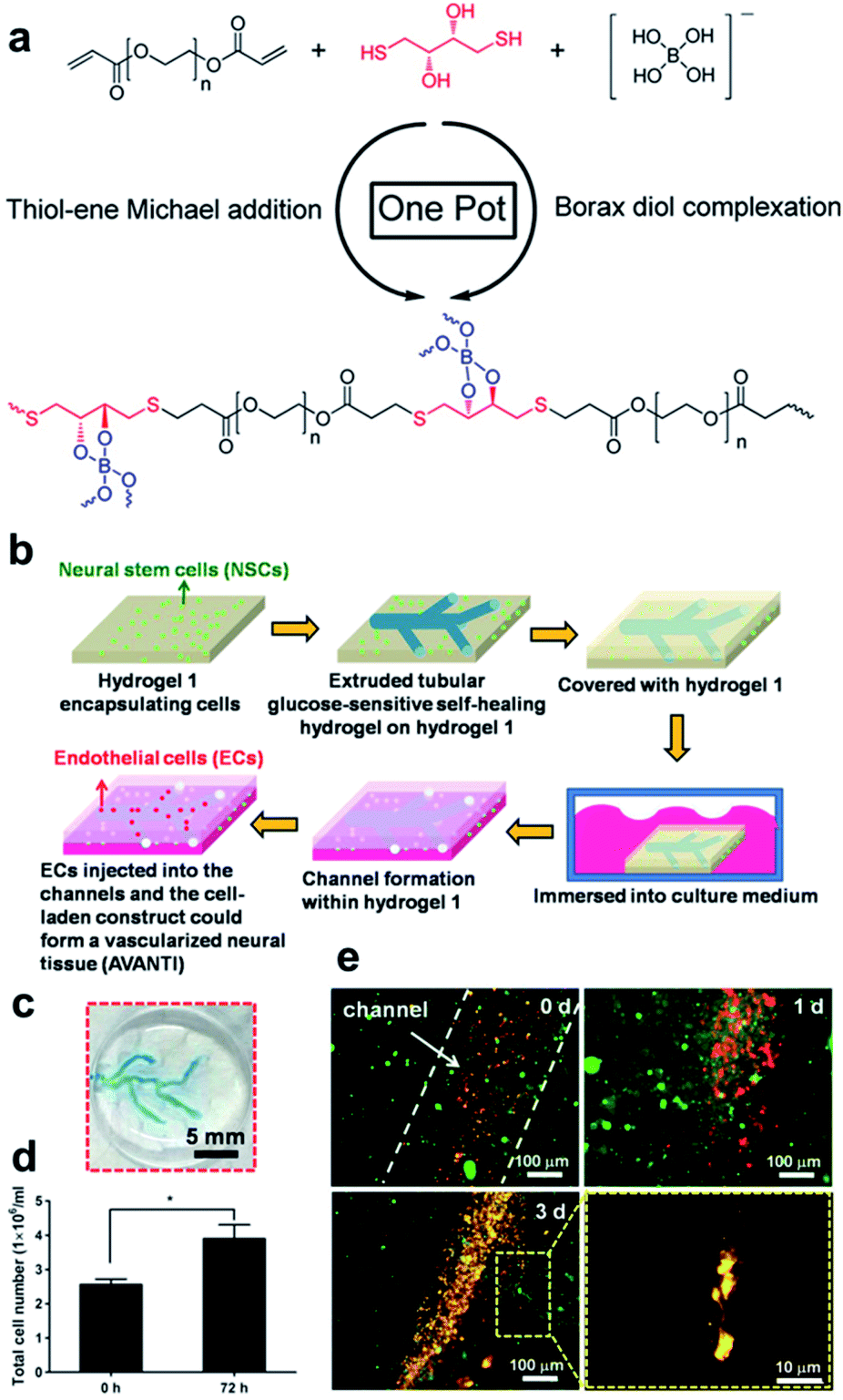 | ||
| Fig. 21 (a) Structural formula of PEG-hydrogel synthesized using one-pot reaction that employs borax as a catalyst and cross-linking agent in the thiol–ene Michael reaction. Reproduced from ref. 110 with permission from the ACS. (b) Schematic of the vascularization process using sacrificial and non-sacrificial hydrogels. (c) Appearance of a vascular network embedded in the non-sacrificial gel. (d) Number of cells in the construct determined after three days. (e) Morphologies of the NSCs and ECs encapsulated in the vascularized construct for three days, showing an EC lining at the early period as well as the migration of the ECs and precapillary formation after three days (red fluorescence: ECs; green fluorescence: NSCs). Reproduced from ref. 111 with permission from the Elsevier. | ||
In 2017, the Hsu group synthesized a hydrogel containing boronate ester bonds using borax as a catalyst via thiol–ene Michael reaction, which was applied in artificial bioimplants (Fig. 21b).111 Sacrificial and non-sacrificial hydrogels were used to create artificial blood vessels; sacrificial hydrogel was prepared via thiol–ene Michael reaction between PEG diacrylate and DTT, and the non-sacrificial hydrogel was composed of chitosan. Borax was added as a catalyst and cross-linking agent. The sacrificial hydrogel was injectable using a 26G needle and repetitive sol–gel transitions; thus, self-healing was shown to be possible in the step-strain tests. Properties such as injectability and self-healing allow facile designing of complex patterns, such as blood vessel structures (Fig. 21c). Therefore, the sacrificial hydrogel was injected into a non-sacrificial hydrogel encapsulated with neural stem cells (NSCs) in a pattern suitable for blood vessels, and a structure was constructed using the non-sacrificial hydrogel encapsulated with NSC. As boronic acid in the sacrificial hydrogel was sensitive to glucose, it melted to form a tube in the structure when placed in the culture medium. Vascularized neural tissue was prepared by the perfusion of endothelial cells (ECs) into this tube. In the prepared construct, EC proliferated along the tube and migrated to the non-sacrificial hydrogel, and cross-talk between EC and NSC promoted the differentiation of NSCs (Fig. 21d). Furthermore, EC formed a long-term vascular network, and NSCs formed neurodevelopmental tissues (Fig. 21e). Research using this boronate ester bond allowed easy designing of complex artificial blood vessels using a sacrificial hydrogel that was self-healing, injectable, and removed upon reaction with glucose, contributing toward the research on artificial blood vessels.
Borax can be used as a cross-linking agent for PVA, a simple polymer containing diols; thus, it has been widely used in the studies of PVA-borax hydrogel.112–114 Among the hydrogels reported to date containing B–O bonds obtained using borax, the mechanical strength or healing efficiency can also be improved by adding other substances. In 2016, Huang et al. synthesized a reinforced PVA–borax hydrogel by adding microfibrillated cellulose (MFC) as a nanofiller to further improve its mechanical properties.115 The production process consisted of a one-pot reaction, including ball milling and physical blending steps (Fig. 22). The MFC was well dispersed inside the hydrogel during ball milling, which was confirmed by analyzing the morphology before and after treatment using field-emission SEM and TEM. An appropriate duration for ball milling was 1–3 h; the fibers aggregated when this step was performed for longer time. The density of the fibers increased with an increase in the amount of added MFC, but MFC aggregation did not occur because both PVA and MFC are hydrophilic. Rheological analysis also showed results corresponding to those observed using the SEM. The amount of added MFC and duration of the ball milling step were closely related to mechanical strength. Addition of more MFC increased the mechanical strength and stiffness, but the optimum duration was 3 h; the physical properties declined beyond this time. To further confirm the self-healing properties of the hydrogel, two hydrogels were prepared, one of which was stained with rhodamine B. After the two hydrogels were cut in half and each piece was brought into contact with each other, self-healing occurred after 10 min at room temperature as the interface softened. The self-healing hydrogel could also be stretched by hand. Rheological analysis revealed that the gel collapsed at a high frequency (large amplitude) and recovered to its original gel state at low frequency (high amplitude). Moreover, the hydrogel was sensitive to pH as it showed a repetitive sol–gel phase transition depending on the pH. This study afforded a method for preparing biocompatible and non-toxic hydrogels that could be applied to many biomedical fields in an environmentally friendly and efficient manner using relatively inexpensive and readily available materials.
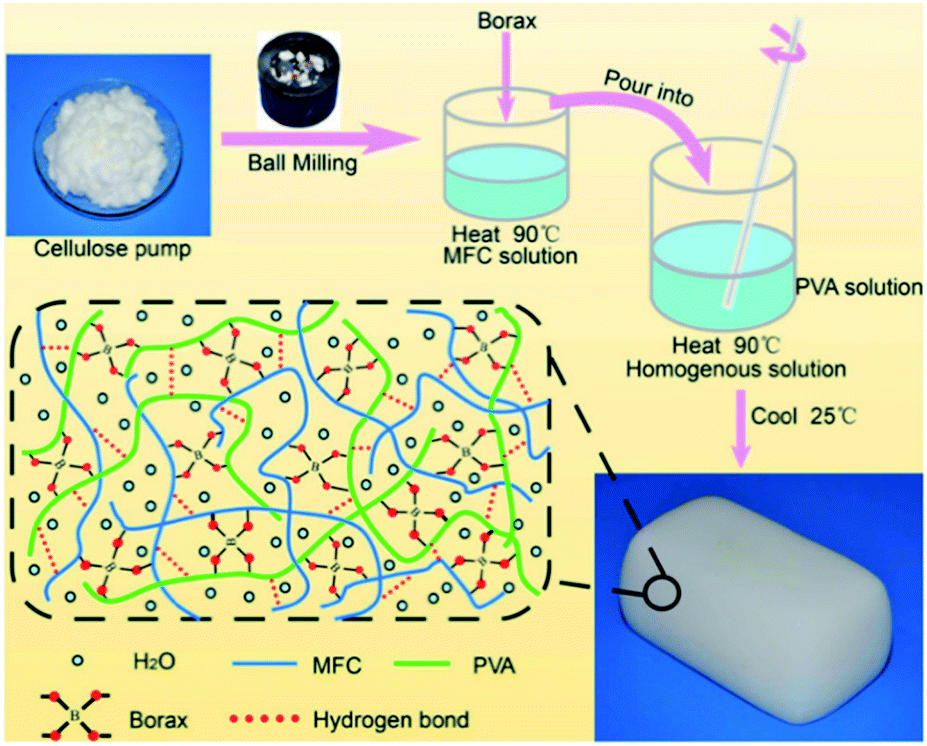 | ||
| Fig. 22 Schematic of the internal structure of a PVA–borax hydrogel enhanced with MFC prepared via one-pot tandem reaction. Reproduced from ref. 115 with permission from the ACS. | ||
In an investigation where cellulose was added to a PVA–borax hydrogel, Yang et al. introduced tannic acid-coated cellulose nanocrystals (TA@CNC) into the network of PVA and borax to obtain a nanocomposite hydrogel in 2019.116 First, PVA was dissolved in water at 98 °C and shaken at 90 °C after adding the TA@CNC and borax solution. This resulted in dynamic bonding between the hydroxyl group of PVA and boron of borax to form a PVA–borax hydrogel. In addition to the dynamic B–O bonds formed at this time, TA@CNC–PVA clusters were formed as a flexible PVA chain (shell) in the gel surrounding the hard TA@CNC (core). The resulting gel was capable of self-healing via dynamic bonding of the hydroxyl groups and showed improved healing properties and mechanical strength due to additional hydrogen bonds formed between TA@CNC and PVA. In particular, the stiffness and toughness improved without any deterioration in the extensibility due to TA@CNC. Additionally, strain-stiffening and fast stress relaxation were observed. Furthermore, when two disk-shaped gels stained with rhodamine B and methyl green were cut and brought into contact with each other, self-healing occurred, and the resulting healed hydrogel was capable of lifting a 200 g weight. The step-strain test showed that the structure of the gel collapsed, forming a solution under high strain (250 or 400%), but rapidly returned to its gel state under low strain (1%). The self-healing efficiency increased with an increase in the concentration of TA@CNC from 0 to 3 wt%. In contrast, the self-healing efficiency of the gel treated with glucose decreased significantly, indicating that the bonding between the diol and boron had a greater effect on healing than hydrogen bonding. As this hydrogel showed good adhesion to non-porous and porous materials, it could be used as a substitute for soft tissue (dynamically adhesive, strain-stiffening).
The Pan group applied a hydrogel comprising a bond between borax and a diol to a capacitor. In 2016, a polymer synthesized by grafting polyacrylic acid (PAA) onto PVA was applied to a capacitor (PVA-g-PAA),117 and in 2017, sodium alginate was used to prepare a hydrogel applicable to capacitors.118 The hydrogel was prepared by cross-linking sodium alginate modified with dopamine upon the addition of borax (Fig. 23). Potassium chloride (KCl) was added as an inorganic salt during gel formation, and the ionic conductivity increased upon increasing the amount of added salt and temperature. The salt also prevented freezing by interfering with the interactions between water and hydrophilic functional groups, resulting in high ionic conductivity even at −10 °C. The self-healing properties of the hydrogel electrolyte were also evaluated. After dyeing two hydrogels with different colors and cutting these into three pieces, the resulting six pieces were brought into contact with each other. Within 5 min, the gel sufficiently self-healed to support its own weight, and the interface disappeared upon microscopic observation. In addition, the healing efficiency and ionic conductivity of the sample healed once compared to those of the sample healed five times were similar and showed good recovery. To further investigate the self-healing mechanism, the cut pieces were treated with fructose and urea. When compared to the gel treated with urea, the gel treated with fructose was significantly less effective in healing than the original gel, indicating that self-healing occurred through a mechanism involving the binding of catechol and borate ester. When this hydrogel electrolyte was applied to a supercapacitor, it maintained a good charging capacity similar to the original charging capacity even after 10 damage/recovery cycles. Even at a low temperature of −10 °C, it exhibited a capacity of >80% of the charging capacity observed at room temperature. This study showed that the hydrogel electrolytes with self-healing properties and cold resistance could be applied to energy storage devices, such as electronic materials that can be worn or carried, smart clothing, and flexible robots.
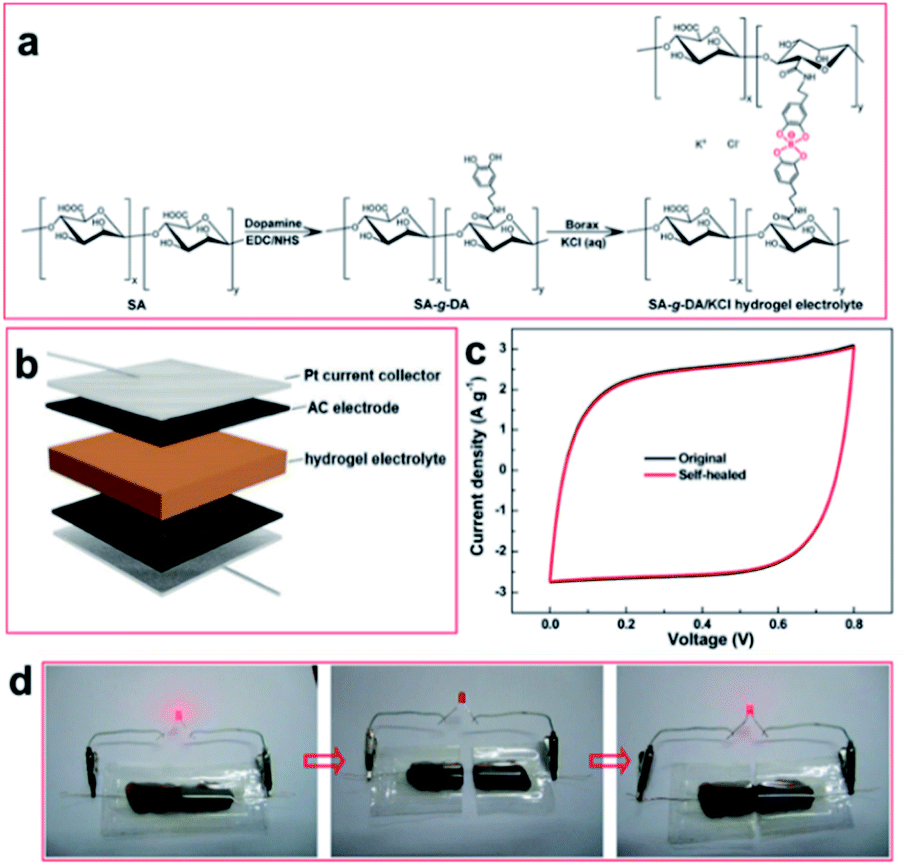 | ||
| Fig. 23 (a) Synthesis of a hydrogel using sodium alginate (SA) modified with dopamine, borax, and KCl. (b) Illustration of the assembly of a capacitor and (c) cyclic voltammograms obtained before/after self-healing at 100 mV s−1. (d) Four capacitors connected in series and used to light an LED via cut/heal process. Reproduced from ref. 118 with permission from the ACS. | ||
In 2017, Lee et al. developed a piezoresistive strain sensor that was highly stretchable and self-healing.119 The hydrogel was synthesized by adding PVA and borax to a single-wall carbon nanotube (SWCNT) solution (Fig. 24a). When the hydrogel was cut and the pieces were brought into contact with each other at room temperature, partial healing within 30 s and complete healing after 60 s were observed. Most of its initial conductivity was recovered within 3.2 s, and the conductivity recovery rate was 98 ± 0.8% over five repeated experiments (Fig. 24b and c). The recovery of its conductivity was also confirmed in a circuit connected to an LED. The LED turned off when the hydrogel was cut and turned back on upon self-healing. The hydrogel with SWCNTs acted as a strain sensor that could withstand very high elastic strain of up to 1000% and afforded a high gauge factor, i.e., sensitivity, which is capable of detecting human motions, such as strain, flexion, and twist forces, with a repetitive and excellent response (Fig. 24d and e). This study suggested that the developed self-healing sensor could be used to detect human movement upon its incorporation into gloves and clothing, or upon attachment to the skin, which implied that self-healing sensors could also be applied to electronic devices.
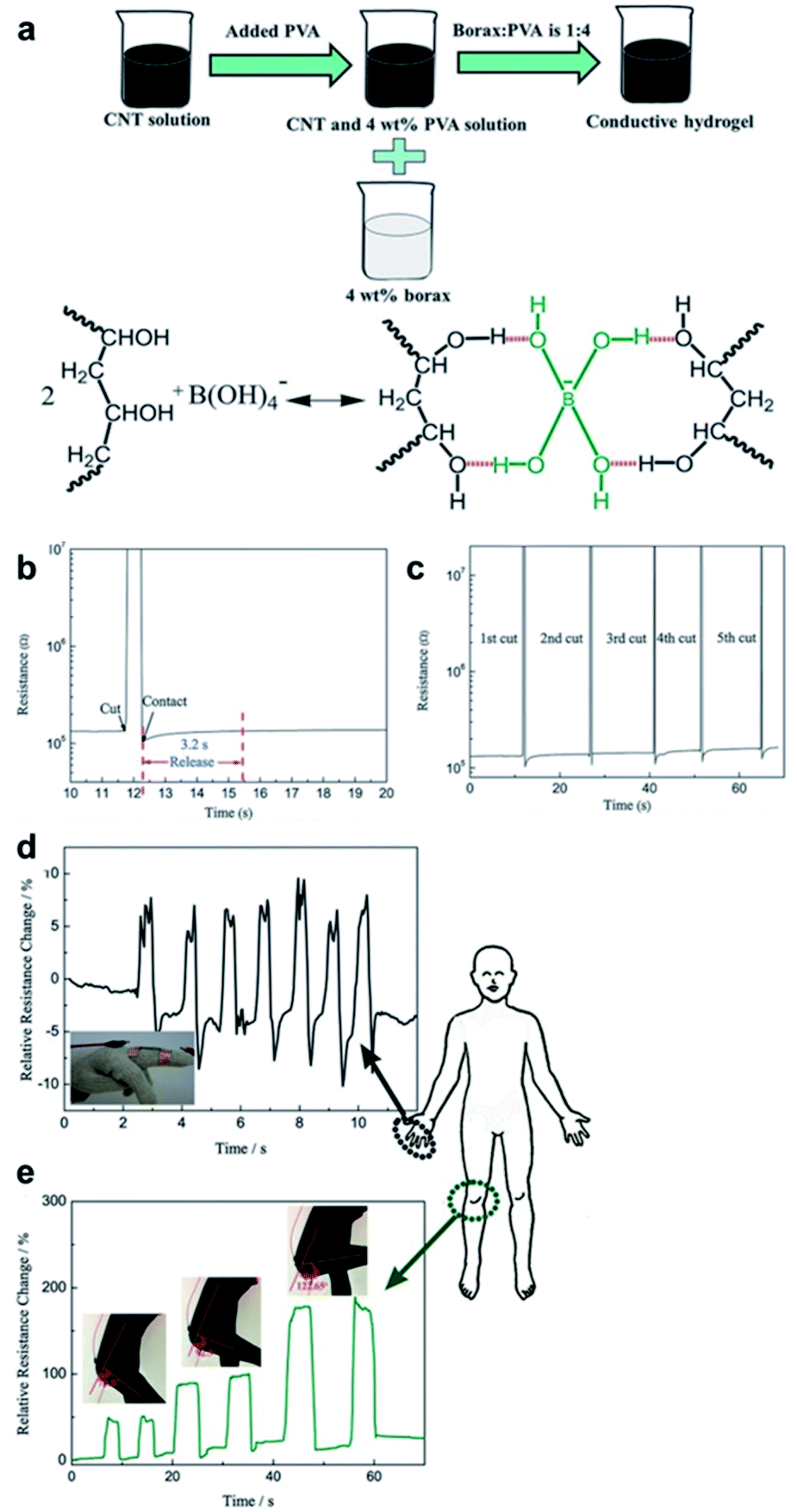 | ||
| Fig. 24 (a) Manufacturing method used to prepare PVA–borax hydrogel with electrical conductivity via the introduction of SWCNT. (b and c) Resistance changes observed during electrical healing under ambient conditions using (b) time-lapsed and (c) cut and heal cycles performed at the same location. (d and e) Monitoring of human motions in real-time. Relative resistance changes depending on (d) the bending and release of the index finger and (e) bending of knee at different angles. Reproduced from ref. 119 with permission from the Wiley. | ||
Hydrogels also have high biocompatibility and skin-like properties, allowing the easy delivery of drugs by their attachment to the skin in the form of patches. In 2020, Park et al. developed a malleable and stretchable hydrogel patch using PVA, polyhydroxyethyl methacrylate (PHEMA), and borax.120 The manufacturing process used to prepare the hydrogel was as follows. First, the in situ polymerization of HEMA monomers in PVA solution resulted in the formation of PHEMA micelles via self-assembly owing to its hydrophobic backbone. Thereafter, borax was added, and the hydrogel was formed via cross-linking between borax and PVA rather than PHEMA (Fig. 25). When the PHEMA content was 57–27 wt%, the micelles were formed with an even distribution. The hydrogels prepared using a PHEMA content of 43 wt% exhibited better ductility, extremely low tensile strength and modulus, and better healing efficiency than those without PHEMA because of the plasticizing role of the PHEMA particles in addition to the reversible PVA–borax bonding. This is because PHEMA micelles do not chemically bond to borax and move freely in the PVA matrix, absorbing mechanical shock. Moreover, the release of the PHEMA micelles was confirmed by dropping an aqueous solution on the hydrogel to examine its potential use as a patch. The particles escaped from the hydrogel due to the concentration gradient, allowing the use of the hydrogel in drug delivery applications. PHEMA was released slowly because with an increase in pH, the cross-linking between borax and PVA increased, whereas the PHEMA micelles were rapidly released in an aqueous solution at low pH (pH 2). Furthermore, when the release of Nile red (NR), a hydrophobic fluorescent substance, from the hydrogel was examined, PHEMA was shown to aid the release of NR to the exterior of the hydrogel, which was also applied to porcine skin. The hydrogel showed minimal in vivo cytotoxicity and skin irritation. This study provided a new strategy for the development of excellent drug delivery patches in the field of medicine and cosmetics.
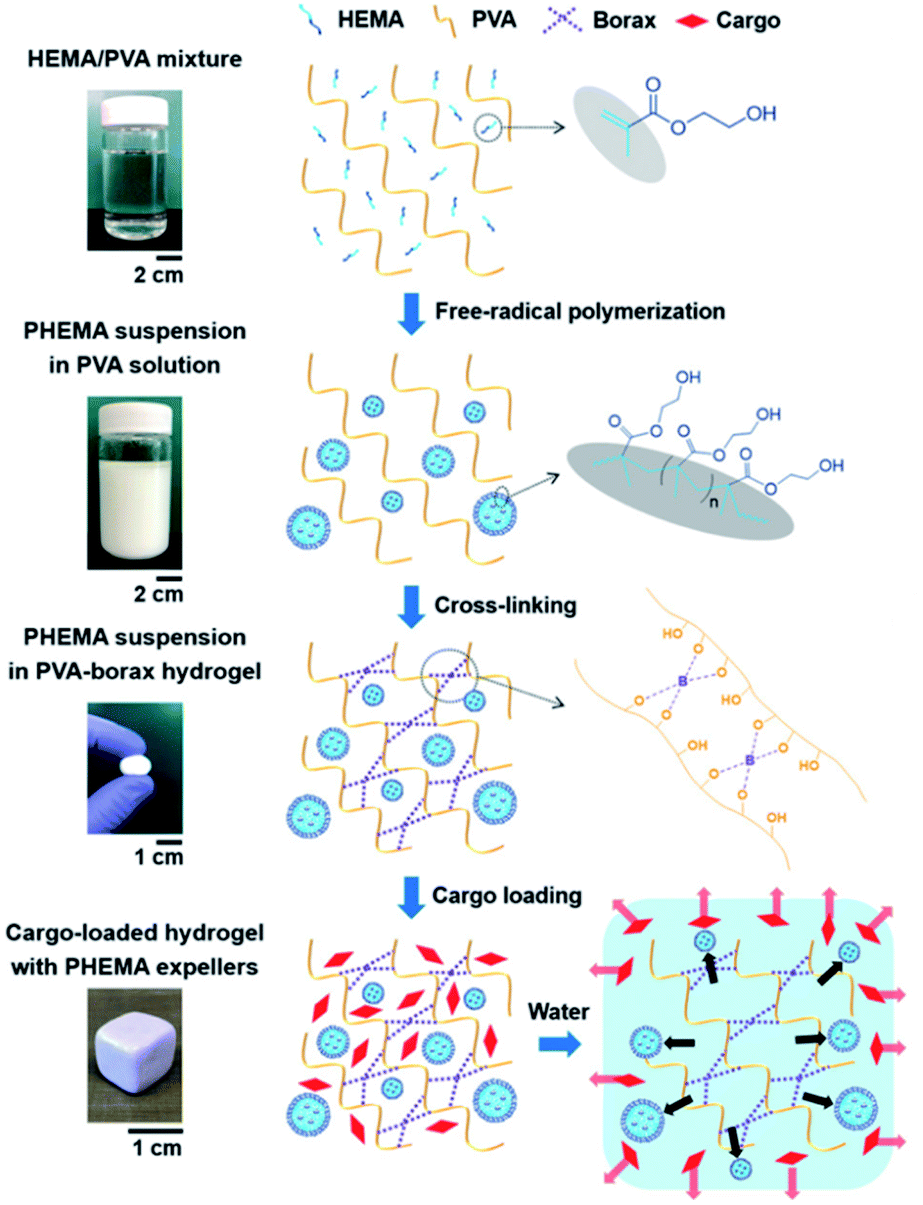 | ||
| Fig. 25 Schematic of the drug release model and manufacturing method used to prepare a PVA–borax hydrogel embedding PHEMA in the micelles. Reproduced from ref. 120 with permission from the Wiley. | ||
In 2018, the Narain group synthesized a hydrogel bearing a benzoxaborole group, which could be applied in the biomedical field. Benzoxaborole has a pKa of 7.2, which is close to the physiological pH and reacts with diols to form a reversible B–O bond.121 They prepared a hydrogel using a biocompatible 2-methacryloyloxyethyl phosphorylcholine (MPC)-based zwitterionic polymer with sugar or catechol as the diol.122,123 In this study, two MPC copolymers were used, including poly(2-methacryloyloxyethyl phosphorylcholine-st-5-methacrylamido-1,2-benzoxaborole) (PMB) containing a benzoxaborole functional group and poly(2-methacryloyloxyethyl phosphorylcholine-st-2-gluconamidoethyl methacrylamide) (PMG) containing a sugar moiety (Fig. 26a). The PMB copolymer had a single fixed molar composition of 15 mol% benzoxaborole, and the PMG copolymer included three molar compositions of 20, 50, or 80 mol% sugar. After dissolving the PMB copolymer and three types of PMG copolymers in three buffer solutions (pH 7.4, 8.4, and 9.4), a gel was formed within 15 s upon combining the copolymer solutions of PMB and PMG. SEM analysis revealed that all nine hydrogels had interconnected structures, and the dynamic rheological measurements for each gel showed that the mechanical properties could be adjusted according to the sugar content and pH. In particular, the hydrogel was more elastic and exhibited better mechanical properties when prepared with a higher sugar content at high pH (Fig. 26b and c).
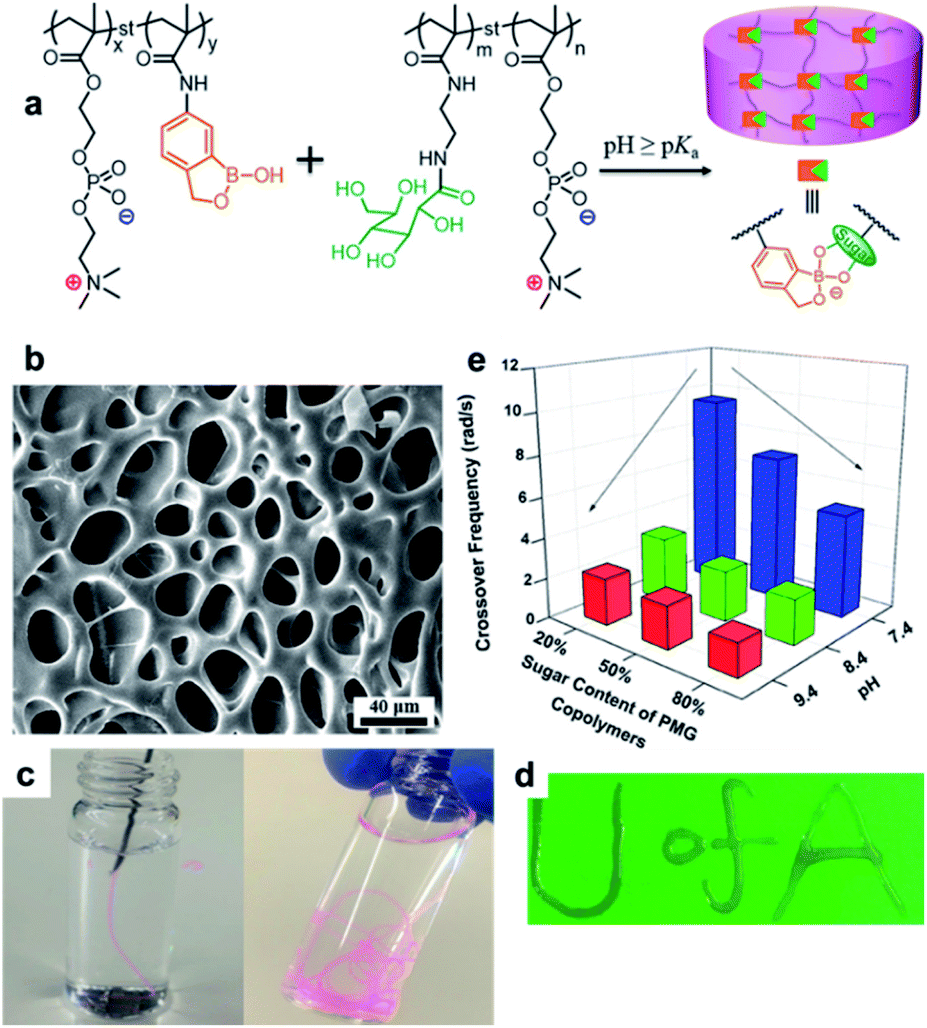 | ||
| Fig. 26 (a) Synthesis of PMBG hydrogel using zwitterionic copolymers (PMB and PMG) bearing benzoxaborole and sugar pendant, respectively. (b) SEM image of the PMBG hydrogel. (c and d) Injectable properties of PMBG-50%-7.4. (e) Crossover frequency data. Reproduced from ref. 122 with permission from the ACS. | ||
In addition, self-healing was possible via the rearrangement of the interactions between the benzoxaborole and sugar moieties. The self-healing properties were evaluated using the hydrogel (PMBG-50%-7.4) prepared with a buffer solution of PMB and 50 mol% sugar. The step-strain test showed that G′ became smaller than G′′ when a large strain of 200% was applied. However, when a low strain of 1% was applied, the recovery rate was close to 95% of the initial G′ value. Recovery was possible in repeated experiments and could be confirmed visually. Moreover, when three pieces of the hydrogel were brought into contact with each other without external stimulation, recovery occurred within ∼20 s, and the resulting hydrogel was able to support its weight. In addition, a gel–sol–gel phase transition occurred in PMBG-50%-7.4, owing to its pH-dependent properties, and the existing bonds could be broken due to the addition of competitive molecules containing diols, such as fructose. The hydrogel could also be used as a 3D bioprinting material because of its injectability or as a drug delivery system that could release a drug when the bonding in the hydrogel was broken under weakly acidic pH conditions (pH ∼ 6.8) in vivo owing to its pH and sugar sensitivity (Fig. 26d and e). Furthermore, it could be applied to various biomedical fields owing to its biocompatibility, as shown via in vitro cytotoxicity tests.
In a similar study, Narain et al. synthesized an injectable self-healing hydrogel in 2019, which was sensitive to various stimuli (temperature, pH, and sugar).124 Initially, an ABA-type terpolymer was prepared using a hydrophilic glycopolymer at both ends and a temperature-sensitive polymer in the middle of the chain. When the resulting ABA-type polymer was combined with a polymer containing a benzoxaborole group, the diol in the glycopolymer and benzoxaborole formed a benzoxaborole–diol covalent bond, thereby generating the hydrogel. Self-healing occurred within 1 h when two pieces of the resulting hydrogel coated with rhodamine B and methylene blue dyes were brought into contact with each other. In addition to its self-healing properties, the hydrogel also showed injectability due to its shear-thinning, repetitive gel–sol–gel transition depending on the pH, excellent fructose binding ability, and temperature-dependent mechanical properties. This study described a hydrogel that could respond to more than three stimuli, and was injectable and self-healing, allowing its potential application in biomedical fields because of the relatively low pKa of benzoxaborole (pKa = 7.2).
4.5 Self-healing in polymer networks other than hydrogels
In addition to the hydrogels containing water, studies have been conducted on self-healing polymers using boronate ester bonds. This section describes self-healing using boronate ester bonds in bulk polymer networks or organogels in addition to the hydrogels.In 2018, the Yoshie group investigated a bulk polymer network containing tetrahedral boronate ester bonds, which was capable of self-healing under ambient conditions.125 This polymer network was synthesized using poly(dopamine acrylamide-co-n-butyl acrylate)[P(DA-co-BA)], p-phenyldiboronic acid (PDBA), and triethylamine to form a polymer film (Fig. 27a). The ratio of DA to PDBA was 10:3 or 10:5, indicating that catechol formed a boronate ester bond at 60% or 100%, respectively. This bond was reversible and afforded sufficient cross-linking to exhibit good mechanical properties because the formation of the bond occurred faster than hydrolysis in the presence of water. In this study, the self-healing efficiency was defined as the recovery of its toughness during tensile tests. The polymer film with a cross-linking point of 60% did not self-heal sufficiently at 30% RH. However, it recovered ∼30% of its initial toughness after seven days at 55% RH and recovered almost all of its initial toughness after three days at 75% RH. This was possible because the boronate ester bond formation was promoted by the diffusion of water molecules in the polymer matrix. In contrast, in the polymer film with a cross-linking point of 100%, the healing efficiency was relatively low because of the disturbance in the chain mobility. In addition, application of pressure in the presence of heat or moisture after completely cutting the polymer film confirmed that the film was reprocessable.
 | ||
| Fig. 27 (a) Self-healing mechanism of PDBA under humid conditions. Reproduced from ref. 125 with permission from the RSC. (b) Synthesis of organogel using PVA, FPBA, and TDH. (c) Photographs of the gels combined into a single gel after 30 s via self-healing and its stretchability after 5 min. Reproduced from ref. 127 with permission from the RSC. | ||
Boronic acid can form boronate ester in aqueous alkaline solutions. However, boronate ester bonds can also be formed by employing B–N bonds between boron and nitrogen in the organogels in organic solvents other than aqueous solutions. In 2014, the Severin group studied organogels, in which boronate ester was employed using B–N bonding.126 In 2019, Zheng et al. developed an organogel capable of rapid self-healing without requiring a separate catalyst or external stimulation via bonding between boronate ester and acylhydrazone.127 In this study, PVA, 4-formylphenylboronic acid (FPBA), and tartaric acid dihydrazide (TDH) were used under alkaline conditions to synthesize a gel in situ (Fig. 27b). The resulting gel was capable of self-healing due to dynamic covalent bonding, and in particular, the adhesive properties of PVA assisted in the bonding of the cut surface. When the gel was cut into two pieces in the cut and heal test and brought into contact with each other for 30 s, the pieces could support their own weight. These could also stretch up to four times their original length after 5 min (Fig. 27c). Optical microscopy revealed that the cut surface nearly disappeared after 30 min, and the tensile stress almost recovered after 10 min, which was similar to that observed for the original gel. In addition to the self-healing properties, the chirality of TDH was applied to this gel. When this organogel was converted into a xerogel, it exhibited the selective absorption of organic dyes, particularly methylene blue, via π–π interactions.
5. Conclusions and outlook
Self-healing has received considerable attention in recent years because of its economic and environmental advantages as it can effectively extend the life of materials, allowing significant technological progress. In this review, various polymeric materials capable of self-healing via reversible B–O bonding, such as boronic/boronate esters, borax, and benzoxaborole, were summarized, with a focus on dynamic covalent bonding between boronic esters and diols.In the bulk phases such as elastomers and plastics, boronic ester bonds exhibit self-healing properties through metathesis, transesterification, or hydrolysis/re-esterification, enabling the recovery of their mechanical strength and reprocessability upon damage. Studies have mainly focused on the chemical modifications of boronic acid derivatives and free diols, as well as moisture variation, which affect the mechanical properties of the materials. In the hydrogels, boronate esters are existed with high binding constants. In addition to their self-healing properties, these hydrogels change their physical/chemical shape in response to the changes in pH or the presence of external diol/boronic acids, such as glucose, exhibiting their potential for use in drug delivery, medical adhesion, and biomedical applications. To synthesize a hydrogel that can be practically applied at physiological pH, a 2APBA or amide carbonyl-boronate ester-containing structure with an adjacent electron-withdrawing group was designed. Injectability due to shear thinning was exhibited in some cases, and the hydrogel was simultaneously applied to self-healing and cell culture, affording a more diverse biological application. This hydrogel showed a synergistic improvement in its mechanical properties with the presence of an additional self-healing motif (dynamic covalent bonds and supramolecular forces). Reversible B–O bonds have also been applied to various boron-based compounds, such as borax and benzoxaborole.
Tables 1 and 2 summarize the self-healing motif, polymer matrix, mechanical, self-healing, and reprocessing properties of the literature introduced in this review. Hydrogels with unreported tensile properties were summarized as storage moduli at 1 rad s−1 or 1% strain. It is noteworthy that in cases where the tensile strength was less than 5 MPa, self-healing was possible at room temperature, but it was greatly affected by moisture. When the mechanical strength increased to >9 MPa, a healing temperature of >80 °C for >24 h was required. As reported by Leibler et al., self-healing occurred best at 20–50 MPa of HDPE/PS/PMMA containing dioxaborolane that can be reprocessed at >180 °C.50 These conventional plastics were evaluated to increase the industrial applicability because they were less influenced by moisture. In their bulk state, the reported boronic ester-based polymers can self-heal at room temperature under high humidity. The maximum tensile strength is 4.4 MPa, as reported by Sumerlin et al.56 In contrast, the tensile strength of elastomers prepared from other room temperature self-healing motifs has been reported to be relatively high with a range of 6.5–40 MPa.35,128–130 Yang et al. reported the best case for the hydrogel, which was at a tensile strength of 0.1 MPa (∼15 kPa at room temperature, ∼20 wt% of solid content), performed by a combination of borax/PVA/tannic acid.116 The mechanical properties of the B–O-based hydrogels are judged to be much inferior to those of other self-healing motifs-based hydrogels, with a tensile strength range of 0.5–8 MPa.131–134 However, it is expected that further developments and applications will be possible when new approaches improve the insufficient mechanical properties of the B–O-based hydrogels by finding innovative macromolecular engineering methods with the best B–O motif combination based on the thermal stability of the B–O bonds at high temperature and the advantages of multi-stimuli responsiveness.
| Dynamic bond | System | Tensile properties | Healing conditions | Healing efficiencyc | Remarks | Ref. | |
|---|---|---|---|---|---|---|---|
| σ (MPa) | ε b (%) | ||||||
| a Ultimate tensile strength. b Elongation at break. c Estimated tensile toughness. d Room temperature. e Healing efficiency is calculated by peak stress. | |||||||
| Boronic ester | Bulk | ∼1.5 | ∼90 | 3 days at 85% humidity (rtd) | >60%e | Reprocessing in 85% humidity | 57 |
| ∼3 | ∼550 | 16 h at 50 °C | >95% | Reprocessing at 80 °C | 55 | ||
| ∼4.4 | ∼58 | 3 days at 85% humidity (rt) | >85% | — | 56 | ||
| Bulk (polyurethane) | ∼0.45 | ∼900 | 1 h at 25 °C | 94% | Reprocessing at 60 °C in <30% humidity; adhesion | 61 | |
| Bulk (PDMS) | 1.28 | ∼0.85 | Water layered on the cut surface (1 day at rt) | 64%e | — | 58 | |
| Bulk (SBR) | 1.94 | 262 | 24 h at 80 °C | ∼90% | Reprocessing at 160 °C | 59 | |
| Bulk (ENR) | 9.55 | 648 | 24 h at 80 °C | ∼100% | Reprocessing at 160 °C | 60 | |
| Bulk (HDPE/PS/PMMA) | ∼20–50 | ∼700 | — | — | Reprocessing at >180 °C | 50 | |
| Boronate ester | Bulk | 2.1 | 450 | No wait at 75% humidity (rt) | 96% | Reprocessing at 60 °C or 75% humidity | 125 |
| Borax | Hydrogel (catechol) | ∼0.004 | 300 | 20 min at rt | ∼100% | — | 118 |
| Hydrogel (PVA) | ∼0.005 | 3330 | 1 min at 25 °C | 74% | — | 120 | |
| ∼0.006 | ∼650 | 20 min at rt | ∼100%e | — | 117 | ||
| Boronate ester/acylhydrazone | Hydrogel (PBA/PVA) | ∼0.055 | ∼115 | — | — | — | 108 |
| Borax | Hydrogel (PVA/tannic acid) | 0.10 | 916 | 60 s at 25 °C | 92% | Adhesion | 116 |
| Boronate ester | Organogel (PVA in DMSO) | 0.005 | 250 | 10 min at 25 °C | 94% | — | 127 |
| Dynamic bond | System | Storage modulusa (kPa) | Temp.; content; pH | Ref. | |
|---|---|---|---|---|---|
| a Storage modulus at 1 Hz (or 1% strain). b Storage modulus at 0.05% strain. c After self-healing, the original storage modulus was almost restored. d 1 M NaOH (75 μL) per mL was estimated to have a pH of ∼12.9. | |||||
| Boronate ester | PBA/PVA | 0.15 | 25 °C; 2.5 wt% | pH 7.4 | 95 |
| Boronate ester/metal–ligand | 1 | 25 °C; ∼1.5 wt% | >pH 8.5 | 104 | |
| Boronate ester/acylhydrazone | ∼4 | rt; ∼20 wt% | pH 8.6 | 108 | |
| Boronate ester | PBA/glucose | ∼0.1 | 5 wt% | Water | 92 |
| PBA/alginate | 0.32 | rt; 2.3 wt% | pH 7.4 | 94 | |
| 0.63 | 25 °C; 3 wt% | ∼pH 12.9d | 93 | ||
| PBA/maltose | ∼1 | 1.5 wt% | pH 7.4 | 80 | |
| Boronate ester/disulfide | PBA/catechol | ∼1 | 25 °C; 10 wt% | pH 8.0 | 107 |
| Boronate ester/acylhydrazone | 5 | 25 °C; 10 wt% | pH 7 | 105 | |
| Boronate ester/oxime | PBA/tannic acid | ∼9 | 25 °C; ∼8 wt% | pH 8.14 | 106 |
| Boronate ester | 2APBA/PVA | ∼0.7 | 10 wt% | pH 4.0 | 81 |
| BDBA/catechol | ∼0.8 | 20 °C; 15 wt% | pH 9.0 | 79 | |
| PBA/guanosine | 0.9 | 25 °C; ∼2.5 wt% | pH 7.6 | 97 | |
| NapBA/guanosine | 1.5 | 25 °C; ∼2.5 wt% | pH 7.4 | 98 | |
| 2APBA/PVA | 2 | rt; 10 wt% | pH 7.4 | 96 | |
| FPBA/glucose | ∼20b | 37 °C; 10 wt% | pH 7.0 | 91 | |
| Borax | Catechol | ∼1.5c | rt; ∼2.5 wt% | Water | 118 |
| PVA | ∼2.3 | 30 °C; ∼7 wt% | Water | 115 | |
| ∼3 | rt; ∼4 wt% | Water | 119 | ||
| ∼6 | 25 °C; 17.5 wt% | Water | 120 | ||
| DTT | ∼10 | 25 °C; 12.5 wt% | pH 7.4 | 110 | |
| ∼10 | 25 °C; ∼12.5 wt% | pH 7 | 111 | ||
| PVA/tannic acid | ∼15 | rt; ∼20 wt% | Water | 116 | |
| Benzoxaborole | Catechol | ∼0.1 | 25 °C; 10 wt% | pH 7.4 | 123 |
| Galactose | ∼0.25 | 25 °C; 10 wt% | pH 8.4 | 124 | |
| Sugar | ∼1 | 25 °C; 10 wt% | pH 7.4 | 122 | |
| Boronate ester | Organogel (PVA in DMSO) | ∼1c | 25 °C; 5 wt% | pH 8 | 127 |
Considering the chemical and physical properties of the boronate ester-based self-healing polymeric materials reported to date, future research is expected to diversify practically in the following direction: (1) overcoming the concurrent limits of the mechanical performance while maintaining the self-healing abilities by incorporating novel macromolecular architectures and optimizing the substituents of boronic acid/diol-derivatives and (2) using new materials that are less affected by moisture or water, i.e., those that do not deteriorate the physical properties in a long-term environment in actual use. In addition, the next hydrogels are expected to include in vitro practical applications in transdermal drug delivery patches, medical adhesives, and healthcare monitoring sensors, as well as in vivo applications such as injectable drug administration and tissue regeneration. Such biomedical applications must be accompanied by toxicity studies on their biocompatibility and safety to contribute to the next generation of biomedical applications.
Author contributions
S. Cho performed the formal analysis and wrote the original draft. S. Y. Hwang, D. X. Oh, and J. Park performed the conceptualization, supervision, and writing – review & editing of the manuscript. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by KRICT (Core Project; SS2142-10) and the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2018R1C1B6000966).Notes and references
- B. S. Sumerlin, Science, 2018, 362, 150–151 Search PubMed
.
- S. Terryn, J. Brancart, D. Lefeber, G. Van Assche and B. Vanderborght, Sci. Robot., 2017, 2, eaan4268 Search PubMed
- T. P. Huynh, P. Sonar and H. Haick, Adv. Mater., 2017, 29, 1604973 Search PubMed
- W. Wang, R. Narain and H. Zeng, Front. Chem., 2018, 6, 497 Search PubMed
- J. Xu, G. Wang, Y. Wu, X. Ren and G. Gao, ACS Appl. Mater. Interfaces, 2019, 11, 25613–25623 Search PubMed
- S. Talebian, M. Mehrali, N. Taebnia, C. P. Pennisi, F. B. Kadumudi, J. Foroughi, M. Hasany, M. Nikkhah, M. Akbari, G. Orive and A. Dolatshahi-Pirouz, Adv. Sci., 2019, 6, 1801664 Search PubMed
- R. Sanka, B. Krishnakumar, Y. Leterrier, S. Pandey, S. Rana and V. Michaud, Front. Mater., 2019, 6, 137 Search PubMed
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 Search PubMed
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. Folmer, J. K. Hirschberg, R. F. Lange, J. K. Lowe and E. Meijer, Science, 1997, 278, 1601–1604 Search PubMed
- A. Faghihnejad, K. E. Feldman, J. Yu, M. V. Tirrell, J. N. Israelachvili, C. J. Hawker, E. J. Kramer and H. Zeng, Adv. Funct. Mater., 2014, 24, 2322–2333 Search PubMed
- J. H. Yoon, S.-M. Kim, Y. Eom, J. M. Koo, H.-W. Cho, T. J. Lee, K. G. Lee, H. J. Park, Y. K. Kim, H.-J. Yoo, S. Y. Hwang, J. Park and B. G. Choi, ACS Appl. Mater. Interfaces, 2019, 11, 46165–46175 Search PubMed
- J. H. Yoon, S.-M. Kim, H. J. Park, Y. K. Kim, D. X. Oh, H.-W. Cho, K. G. Lee, S. Y. Hwang, J. Park and B. G. Choi, Biosens. Bioelectron., 2020, 150, 111946 Search PubMed
- S.-M. Kim, S.-A. Park, S. Y. Hwang, E. S. Kim, J. Jegal, C. Im, H. Jeon, D. X. Oh and J. Park, Polymers, 2017, 9, 663 Search PubMed
- Y. Cao, T. G. Morrissey, E. Acome, S. I. Allec, B. M. Wong, C. Keplinger and C. Wang, Adv. Mater., 2017, 29, 1605099 Search PubMed
- S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. Harris, W. Hayes, M. E. Mackay and S. J. Rowan, Chem. Commun., 2009, 6717–6719 Search PubMed
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 Search PubMed
- S. Bode, R. K. Bose, S. Matthes, M. Ehrhardt, A. Seifert, F. H. Schacher, R. M. Paulus, S. Stumpf, B. Sandmann, J. Vitz, A. Winter, S. Hoeppener, S. J. Garcia, S. Spange, S. van der Zwaag, M. D. Hager and U. S. Schubert, Polym. Chem., 2013, 4, 4966–4973 Search PubMed
- J.-B. Hou, X.-Q. Zhang, D. Wu, J.-F. Feng, D. Ke, B.-J. Li and S. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 12105–12113 Search PubMed
- F. Herbst, D. Döhler, P. Michael and W. H. Binder, Macromol. Rapid Commun., 2013, 34, 203–220 Search PubMed
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467 Search PubMed
- J. Wu, L. H. Cai and D. A. Weitz, Adv. Mater., 2017, 29, 1702616 Search PubMed
- J. Chen, F. Li, Y. Luo, Y. Shi, X. Ma, M. Zhang, D. Boukhvalov and Z. Luo, J. Mater. Chem. A, 2019, 7, 15207–15214 Search PubMed
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 Search PubMed
- Y.-L. Liu and T.-W. Chuo, Polym. Chem., 2013, 4, 2194–2205 Search PubMed
- Y. Y. Jo, A. S. Lee, K.-Y. Baek, H. Lee and S. S. Hwang, Polymer, 2017, 108, 58–65 Search PubMed
- T. Hughes, G. P. Simon and K. Saito, ACS Appl. Mater. Interfaces, 2019, 11, 19429–19443 Search PubMed
- C. e. Yuan, M. Z. Rong, M. Q. Zhang, Z. P. Zhang and Y. C. Yuan, Chem. Mater., 2011, 23, 5076–5081 Search PubMed
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 Search PubMed
- S. Mukherjee, M. R. Hill and B. S. Sumerlin, Soft Matter, 2015, 11, 6152–6161 Search PubMed
- N. Kuhl, M. Abend, S. Bode, U. S. Schubert and M. D. Hager, J. Appl. Polym. Sci., 2016, 133, 44168 Search PubMed
- G. Xiao, Y. Wang, H. Zhang, L. Chen and S. Fu, Carbohydr. Polym., 2019, 218, 68–77 Search PubMed
- C. Sun, H. Jia, K. Lei, D. Zhu, Y. Gao, Z. Zheng and X. Wang, Polymer, 2019, 160, 246–253 Search PubMed
- A. Chao, I. Negulescu and D. Zhang, Macromolecules, 2016, 49, 6277–6284 Search PubMed
- P. Wang, L. Yang, B. Dai, Z. Yang, S. Guo, G. Gao, L. Xu, M. Sun, K. Yao and J. Zhu, Eur. Polym. J., 2020, 123, 109382 Search PubMed
- S. M. Kim, H. Jeon, S. H. Shin, S. A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 Search PubMed
- X. Li, R. Yu, Y. He, Y. Zhang, X. Yang, X. Zhao and W. Huang, ACS Macro Lett., 2019, 8, 1511–1516 Search PubMed
- S. Choi, Y. Eom, S. M. Kim, D. W. Jeong, J. Han, J. M. Koo, S. Y. Hwang, J. Park and D. X. Oh, Adv. Mater., 2020, 32, 1907064 Search PubMed
- Y. Eom, S.-M. Kim, M. Lee, H. Jeon, J. Park, E. S. Lee, S. Y. Hwang, J. Park and D. X. Oh, Nat. Commun., 2021, 12, 621 Search PubMed
- A. P. Bapat, B. S. Sumerlin and A. Sutti, Mater. Horiz., 2020, 7, 694–714 Search PubMed
- S. D. Bull, M. G. Davidson, J. M. Van den Elsen, J. S. Fossey, A. T. A. Jenkins, Y.-B. Jiang, Y. Kubo, F. Marken, K. Sakurai, J. Zhao and T. D. James, Acc. Chem. Res., 2013, 46, 312–326 Search PubMed
- X.-t. Zhang, G.-j. Liu, Z.-w. Ning and G.-w. Xing, Carbohydr. Res., 2017, 452, 129–148 Search PubMed
- G. Fang, H. Wang, Z. Bian, J. Sun, A. Liu, H. Fang, B. Liu, Q. Yao and Z. Wu, RSC Adv., 2018, 8, 29400–29427 Search PubMed
- H. Gaballa and P. Theato, Biomacromolecules, 2019, 20, 871–881 Search PubMed
- H. Gaballa, J. Shang, S. Meier and P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 422–431 Search PubMed
- G. Vancoillie and R. Hoogenboom, Polym. Chem., 2016, 7, 5484–5495 Search PubMed
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 Search PubMed
- W. L. A. Brooks, C. C. Deng and B. S. Sumerlin, ACS Omega, 2018, 3, 17863–17870 Search PubMed
- Y. Suzuki, D. Kusuyama, T. Sugaya, S. Iwatsuki, M. Inamo, H. D. Takagi and K. Ishihara, J. Org. Chem., 2020, 85, 5255–5264 Search PubMed
- C. Zhang, M. D. Losego and P. V. Braun, Chem. Mater., 2013, 25, 3239–3250 Search PubMed
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 Search PubMed
- S. Wang, X. Xing, X. Zhang, X. Wang and X. Jing, J. Mater. Chem. A, 2018, 6, 10868–10878 Search PubMed
- B. Preinerstorfer, M. Lämmerhofer and W. Lindner, J. Sep. Sci., 2009, 32, 1673–1685 Search PubMed
- Y. Qin, C. Cui and F. Jäkle, Macromolecules, 2007, 40, 1413–1420 Search PubMed
- W. Niu, C. O'Sullivan, B. M. Rambo, M. D. Smith and J. J. Lavigne, Chem. Commun., 2005, 4342–4344 Search PubMed
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 Search PubMed
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098–2106 Search PubMed
- J. J. Cash, T. Kubo, D. J. Dobbins and B. S. Sumerlin, Polym. Chem., 2018, 9, 2011–2020 Search PubMed
- Y. Zuo, Z. Gou, C. Zhang and S. Feng, Macromol. Rapid Commun., 2016, 37, 1052–1059 Search PubMed
- Y. Chen, Z. Tang, X. Zhang, Y. Liu, S. Wu and B. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 24224–24231 Search PubMed
- Y. Chen, Z. Tang, Y. Liu, S. Wu and B. Guo, Macromolecules, 2019, 52, 3805–3812 Search PubMed
- Y. Yang, F.-S. Du and Z.-C. Li, ACS Appl. Polym. Mater., 2020, 2, 5630–5640 Search PubMed
- P. Commins, M. B. Al-Handawi, D. P. Karothu, G. Raj and P. Naumov, Chem. Sci., 2020, 11, 2606–2613 Search PubMed
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121 Search PubMed
- E. Caló and V. V. Khutoryanskiy, Eur. Polym. J., 2015, 65, 252–267 Search PubMed
- S. Cascone and G. Lamberti, Int. J. Pharm., 2020, 573, 118803 Search PubMed
- D. Chimene, R. Kaunas and A. K. Gaharwar, Adv. Mater., 2020, 32, 1902026 Search PubMed
- J. Fu and M. in het Panhuis, J. Mater. Chem. B, 2019, 7, 1523–1525 Search PubMed
- Q. Li, C. Liu, J. Wen, Y. Wu, Y. Shan and J. Liao, Chin. Chem. Lett., 2017, 28, 1857–1874 Search PubMed
- S. Uman, A. Dhand and J. A. Burdick, J. Appl. Polym. Sci., 2020, 137, 48668 Search PubMed
- M. A. Darabi, A. Khosrozadeh, R. Mbeleck, Y. Liu, Q. Chang, J. Jiang, J. Cai, Q. Wang, G. Luo and M. Xing, Adv. Mater., 2017, 29, 1700533 Search PubMed
- S.-H. Shin, W. Lee, S.-M. Kim, M. Lee, J. M. Koo, S. Y. Hwang, D. X. Oh and J. Park, Chem. Eng. J., 2019, 371, 452–460 Search PubMed
- S.-H. Shin, S.-M. Kim, H. Jeon, S. Y. Hwang, D. X. Oh and J. Park, ACS Appl. Polym. Mater., 2020, 2, 5352–5357 Search PubMed
- Z. Deng, H. Wang, P. X. Ma and B. Guo, Nanoscale, 2020, 12, 1224–1246 Search PubMed
- Z. Wei, J. H. Yang, J. Zhou, F. Xu, M. Zrínyi, P. H. Dussault, Y. Osada and Y. M. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 Search PubMed
- D. L. Taylor and M. in het Panhuis, Adv. Mater., 2016, 28, 9060–9093 Search PubMed
- Y. Liu and S.-h. Hsu, Front. Chem., 2018, 6, 449 Search PubMed
- W. L. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 Search PubMed
- L. Wang, M. Liu, C. Gao, L. Ma and D. Cui, React. Funct. Polym., 2010, 70, 159–167 Search PubMed
- L. He, D. E. Fullenkamp, J. G. Rivera and P. B. Messersmith, Chem. Commun., 2011, 47, 7497–7499 Search PubMed
- D. Tarus, E. Hachet, L. Messager, B. Catargi, V. Ravaine and R. Auzély-Velty, Macromol. Rapid Commun., 2014, 35, 2089–2095 Search PubMed
- C. C. Deng, W. L. Brooks, K. A. Abboud and B. S. Sumerlin, ACS Macro Lett., 2015, 4, 220–224 Search PubMed
- H.-S. Yang, S. Cho, Y. Eom, S.-A. Park, S. Y. Hwang, H. Jeon, D. X. Oh and J. Park, Macromol. Res., 2021, 29, 140–148 Search PubMed
- J. Plescia and N. Moitessier, Eur. J. Med. Chem., 2020, 195, 112270 Search PubMed
- H. S. Ban and H. Nakamura, Chem. Rec., 2015, 15, 616–635 Search PubMed
- A. Yoshinari and J. Takano, Front. Plant Sci., 2017, 8, 1951 Search PubMed
- B. Marco-Dufort and M. Tibbitt, Mater. Today Chem., 2019, 12, 16–33 Search PubMed
- Y. Guan and Y. Zhang, Chem. Soc. Rev., 2013, 42, 8106–8121 Search PubMed
- A. Kikuchi, K. Suzuki, O. Okabayashi, H. Hoshino, K. Kataoka, Y. Sakurai and T. Okano, Anal. Chem., 1996, 68, 823–828 Search PubMed
- A. Matsumoto, T. Ishii, J. Nishida, H. Matsumoto, K. Kataoka and Y. Miyahara, Angew. Chem., Int. Ed., 2012, 51, 2124–2128 Search PubMed
- T. Yang, R. Ji, X.-X. Deng, F.-S. Du and Z.-C. Li, Soft Matter, 2014, 10, 2671–2678 Search PubMed
- V. Yesilyurt, M. J. Webber, E. A. Appel, C. Godwin, R. Langer and D. G. Anderson, Adv. Mater., 2016, 28, 86–91 Search PubMed
- Y. Dong, W. Wang, O. Veiseh, E. A. Appel, K. Xue, M. J. Webber, B. C. Tang, X.-W. Yang, G. C. Weir, R. Langer and D. G. Anderson, Langmuir, 2016, 32, 8743–8747 Search PubMed
- A. Pettignano, S. Grijalvo, M. Haering, R. Eritja, N. Tanchoux, F. Quignard and D. D. Díaz, Chem. Commun., 2017, 53, 3350–3353 Search PubMed
- S. H. Hong, S. Kim, J. P. Park, M. Shin, K. Kim, J. H. Ryu and H. Lee, Biomacromolecules, 2018, 19, 2053–2061 Search PubMed
- X. Zhi, C. Zheng, J. Xiong, J. Li, C. Zhao, L. Shi and Z. Zhang, Langmuir, 2018, 34, 12914–12923 Search PubMed
- M. E. Smithmyer, C. C. Deng, S. E. Cassel, P. J. LeValley, B. S. Sumerlin and A. M. Kloxin, ACS Macro Lett., 2018, 7, 1105–1110 Search PubMed
- A. Biswas, S. Malferrari, D. M. Kalaskar and A. K. Das, Chem. Commun., 2018, 54, 1778–1781 Search PubMed
- T. Ghosh, A. Biswas, P. K. Gavel and A. K. Das, Langmuir, 2020, 36, 1574–1584 Search PubMed
- G. M. Peters, L. P. Skala, T. N. Plank, H. Oh, G. Manjunatha Reddy, A. Marsh, S. P. Brown, S. R. Raghavan and J. T. Davis, J. Am. Chem. Soc., 2015, 137, 5819–5827 Search PubMed
- F. Yu, X. Cao, J. Du, G. Wang and X. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 24023–24031 Search PubMed
- Z. Wei, J. H. Yang, Z. Q. Liu, F. Xu, J. X. Zhou, M. Zrínyi, Y. Osada and Y. M. Chen, Adv. Funct. Mater., 2015, 25, 1352–1359 Search PubMed
- Z. Jiang, A. Bhaskaran, H. M. Aitken, I. C. Shackleford and L. A. Connal, Macromol. Rapid Commun., 2019, 40, 1900038 Search PubMed
- S. Mukherjee, W. L. Brooks, Y. Dai and B. S. Sumerlin, Polym. Chem., 2016, 7, 1971–1978 Search PubMed
- H. Meng, P. Xiao, J. Gu, X. Wen, J. Xu, C. Zhao, J. Zhang and T. Chen, Chem. Commun., 2014, 50, 12277–12280 Search PubMed
- Y. Wang, H. Yu, H. Yang, X. Hao, Q. Tang and X. Zhang, Macromol. Chem. Phys., 2017, 218, 1700348 Search PubMed
- J. Collins, M. Nadgorny, Z. Xiao and L. A. Connal, Macromol. Rapid Commun., 2017, 38, 1600760 Search PubMed
- R. Guo, Q. Su, J. Zhang, A. Dong, C. Lin and J. Zhang, Biomacromolecules, 2017, 18, 1356–1364 Search PubMed
- Y. Liu, Y. Liu, Q. Wang, Y. Han, H. Chen and Y. Tan, Polymers, 2020, 12, 487 Search PubMed
- R. K. Schultz and R. R. Myers, Macromolecules, 1969, 2, 281–285 Search PubMed
- L. He, D. Szopinski, Y. Wu, G. A. Luinstra and P. Theato, ACS Macro Lett., 2015, 4, 673–678 Search PubMed
- T.-C. Tseng, F.-Y. Hsieh, P. Theato, Y. Wei and S.-h. Hsu, Biomaterials, 2017, 133, 20–28 Search PubMed
- G. Keita, A. Ricard, R. Audebert, E. Pezron and L. Leibler, Polymer, 1995, 36, 49–54 Search PubMed
- H.-L. Lin, Y.-F. Liu, T. L. Yu, W.-H. Liu and S.-P. Rwei, Polymer, 2005, 46, 5541–5549 Search PubMed
- J. Han, T. Lei and Q. Wu, Carbohydr. Polym., 2014, 102, 306–316 Search PubMed
- B. Lu, F. Lin, X. Jiang, J. Cheng, Q. Lu, J. Song, C. Chen and B. Huang, ACS Sustainable Chem. Eng., 2017, 5, 948–956 Search PubMed
- C. Shao, L. Meng, M. Wang, C. Cui, B. Wang, C.-R. Han, F. Xu and J. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 5885–5895 Search PubMed
- Z. Wang, F. Tao and Q. Pan, J. Mater. Chem. A, 2016, 4, 17732–17739 Search PubMed
- F. Tao, L. Qin, Z. Wang and Q. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 15541–15548 Search PubMed
- G. Cai, J. Wang, K. Qian, J. Chen, S. Li and P. S. Lee, Adv. Sci., 2017, 4, 1600190 Search PubMed
- S. H. Shin, Y. Eom, E. S. Lee, S. Y. Hwang, D. X. Oh and J. Park, Adv. Healthcare Mater., 2020, 9, 2000876 Search PubMed
- H. Kim, Y. J. Kang, S. Kang and K. T. Kim, J. Am. Chem. Soc., 2012, 134, 4030–4033 Search PubMed
- Y. Chen, W. Wang, D. Wu, M. Nagao, D. G. Hall, T. Thundat and R. Narain, Biomacromolecules, 2018, 19, 596–605 Search PubMed
- Y. Chen, D. Diaz-Dussan, D. Wu, W. Wang, Y.-Y. Peng, A. B. Asha, D. G. Hall, K. Ishihara and R. Narain, ACS Macro Lett., 2018, 7, 904–908 Search PubMed
- Y. Chen, Z. Tan, W. Wang, Y.-Y. Peng and R. Narain, Biomacromolecules, 2018, 20, 1028–1035 Search PubMed
- C. Kim, H. Ejima and N. Yoshie, J. Mater. Chem. A, 2018, 6, 19643–19652 Search PubMed
- N. Luisier, K. Schenk and K. Severin, Chem. Commun., 2014, 50, 10233–10236 Search PubMed
- S. Ren, P. Sun, A. Wu, N. Sun, L. Sun, B. Dong and L. Zheng, New J. Chem., 2019, 43, 7701–7707 Search PubMed
- H. Wang, Y. Yang, M. Nishiura, Y. Higaki, A. Takahara and Z. Hou, J. Am. Chem. Soc., 2019, 141, 3249–3257 Search PubMed
- L. Zhang, Z. Liu, X. Wu, Q. Guan, S. Chen, L. Sun, Y. Guo, S. Wang, J. Song, E. M. Jeffries, C. He, F.-L. Qing, X. Bao and Z. You, Adv. Mater., 2019, 31, 1901402 Search PubMed
- Z. Shi, J. Kang and L. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 23484–23493 Search PubMed
- H. Jiang, G. Zhang, X. Feng, H. Liu, F. Li, M. Wang and H. Li, Compos. Sci. Technol., 2017, 140, 54–62 Search PubMed
- M. Guo, Y. Wu, S. Xue, Y. Xia, X. Yang, Y. Dzenis, Z. Li, W. Lei, A. T. Smith and L. Sun, J. Mater. Chem. A, 2019, 7, 25969–25977 Search PubMed
- T. Yuan, X. Cui, X. Liu, X. Qu and J. Sun, Macromolecules, 2019, 52, 3141–3149 Search PubMed
- T. Liu, S. Zou, C. Hang, J. Li, X. Di, X. Li, Q. Wu, F. Wang and P. Sun, Polym. Chem., 2020, 11, 1906–1918 Search PubMed
| This journal is © The Royal Society of Chemistry 2021 |