
Materials and system design for direct electrochemical CO2 conversion in capture media
Shuzhen
Zhang†
a,
Celia
Chen†
a,
Kangkang
Li
 b,
Hai
Yu
b and
Fengwang
Li
*a
b,
Hai
Yu
b and
Fengwang
Li
*a
aSchool of Chemical and Biomolecular Engineering, The University of Sydney Nano Institute, The University of Sydney, NSW 2006, Australia. E-mail: fengwang.li@sydney.edu.au
bCSIRO Energy, 10 Murray Dwyer Circuit, Mayfield West, NSW 2304, Australia
First published on 10th June 2021
Abstract
The electrochemical CO2 reduction reaction (eCO2RR) has been regarded as a promising means to store renewable electricity in the form of value-added chemicals or fuels. However, most of the present eCO2RR studies focus on the conversion of pure CO2. The CO2 valorisation chain – from CO2 capture to the eCO2RR – requires significant energy and capital inputs in each of the capture, purification, conversion, and product separation steps. The integration of upstream CO2 capture and downstream electrochemical conversion by direct electrolysis of capture media, such as amine and carbonate salts, offers a potential solution to energy- and cost-efficient utilisation of CO2. In this perspective, we first summarise the present advances in the direct eCO2RR from CO2-capture media. We then focus on potential development directions of materials and systems that boost the process to a phase of high selectivity towards valuable products (e.g., syngas, ethylene, and ethanol). We conclude by highlighting the major challenges and emerging opportunities in the area of integrated electrochemical CO2 utilisation systems.
 Fengwang Li | Fengwang Li is a Lecturer and DECRA Fellow in the School of Chemical and Biomolecular Engineering at the University of Sydney (USYD). His research aims for a “greener”, carbon-neutral future relying on electrochemical energy. Dr Li completed his bachelor's and master's degrees in Chemistry at Renmin University of China and obtained his PhD in Chemistry from Monash University in 2017. Before joining USYD, Dr Li completed his postdoctoral research at the University of Toronto, where he focused on developing catalysts and systems for the conversion of carbon dioxide to fuels and chemicals. |
Introduction
The renewable energy-driven electrochemical CO2 reduction reaction (eCO2RR) towards value-added feedstocks and fuels is a promising solution to develop carbon-neutral energy supply and distribution systems. The considerable recent growth of this field is not only driven by the continuously falling price of renewable electricity, but also the fact that the eCO2RR processes are on the path towards commercially relevant levels.1 However, most of the present eCO2RR studies are based on pure CO2 as a feedstock while little attention has been paid to the entire CO2 valorisation process.In the context of CO2 utilisation that is aimed at carbon emission reduction, CO2 has to be firstly captured using carbon-capture technologies from various CO2 sources, such as point sources (e.g., flue gas from a coal-fired power plant), air and oceanwater.2–4 Commercially relevant amine-based carbon-capture processes require heat to drive the regeneration of CO2 from the capture media, which is the most costly step in the whole process chain: a typical amine scrubbing process costs around US$50 to $150 per tonne of CO2 captured, with over 60% of the cost associated with the regeneration and compression steps.5–7 Meanwhile, the heat for such capture solution regeneration usually comes from fossil fuels, inevitably bringing extra carbon emissions, which is against the scheme of a carbon neutral future.
Process intensification that integrates CO2 capture and utilisation has shown potential to reduce the costs associated with separated carbon capture, storage and utilisation.8 Value-added chemical production, such as production of organic carbonates, has been demonstrated in integrated CO2 capture from power stations, combustion or flue gas, followed directly by chemical CO2 conversion.9–11 Electricity-driven electrochemical CO2 conversion enables the employment of electrical energy – potentially from renewable sources – instead of thermal energy to regenerate and subsequently convert CO2 from capture media. This also allows the captured CO2 to be converted in situ to value-added products (e.g., syngas). This reaction pathway offers a promising solution to simplify the entire CO2 valorisation chain and reduce the overall energy requirements and costs of the CO2 utilisation process.12
To date, CO2 capture and electrochemical conversion are mostly studied independently with only a few attempts at integrated CO2 capture and conversion. This perspective summarises recent key studies regarding the integration of CO2 capture and eCO2RR, and, on this basis, proposes a series of potential design principles and strategies for future development of such integrated CO2 utilisation techniques. The perspective further discusses several techno-economic concepts that could serve as guidelines for future scale-up and concludes with major challenges and opportunities in this emerging field of research.
Materials and systems for CO2-capture-medium reduction
CO2 capture
The use of CO2 capture solvents as electrolytes, as high as several molar in concentration, circumvents the mass transfer limit of CO2 that would otherwise be encountered in aqueous electrolytes where the CO2 solubility is typically around 30 mM.12 There are various choices of CO2 capture media, from polymer membranes to organic and inorganic liquid adsorbents,13 and the mostly widely used ones are amine-based and alkaline absorption solvents, the focus of this perspective.Amine-based CO2 capture is the most mature and commonly used capture means for industrial CO2 sources.6 It usually consists of two steps (Fig. 1): the amine first reacts chemically with CO2 to form carbamate via reaction (1); in the subsequent stage, the carbamate is regenerated by heating at approximately 100–120 °C.14
| 2RNH2(aq) + CO2(g) → RNHCOO(aq)− + RNH3(aq)+ | (1) |
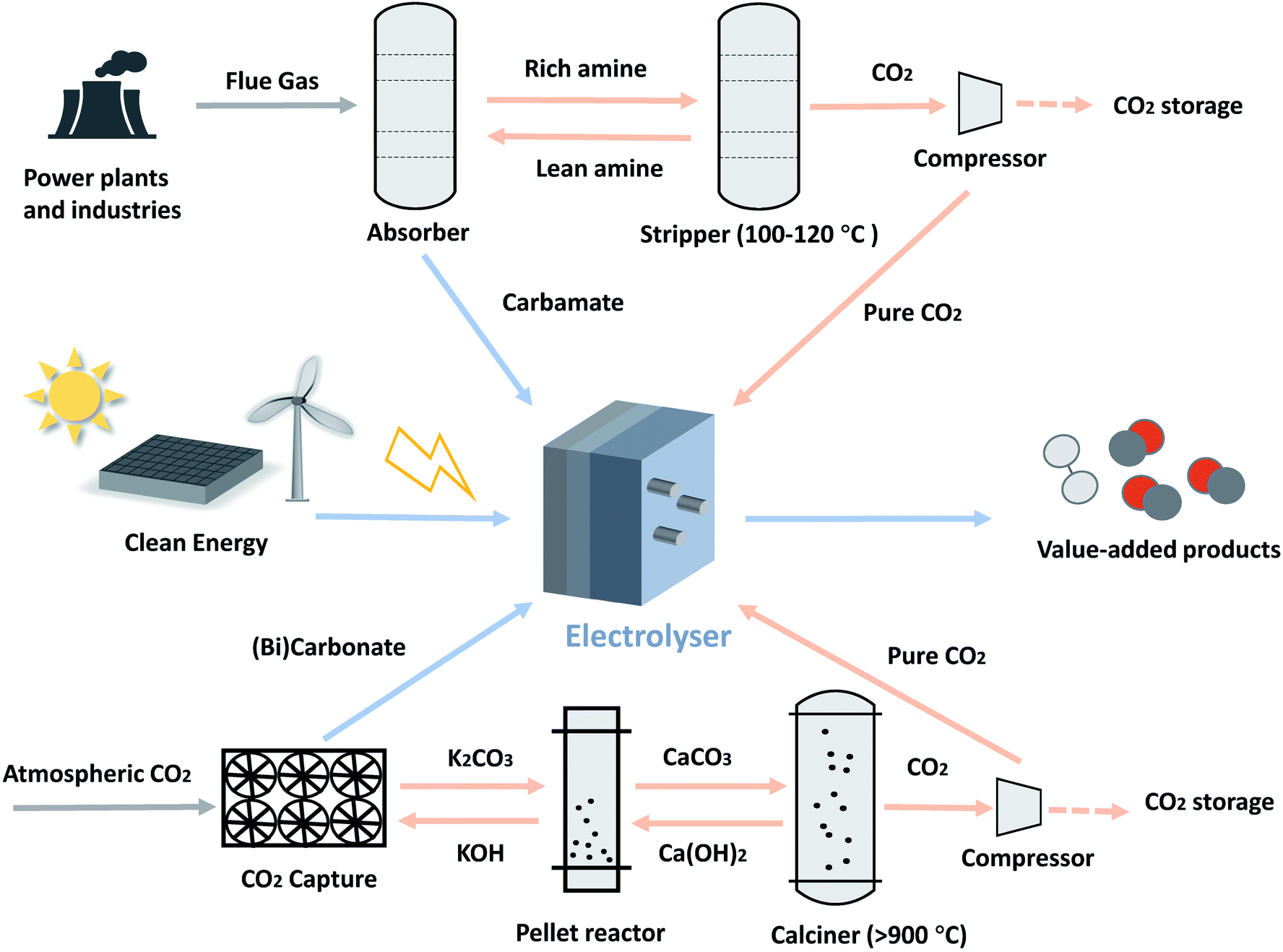 | ||
| Fig. 1 Simplified process flow diagrams for direct capture media electrolysis and gaseous conventional CO2 electrolysis pathway. The blue line indicates the integration utilisation pathway; the orange line indicates the traditional CO2 utilisation pathway. | ||
For a typical chemical absorption based direct air capture process, CO2 in the atmosphere is captured by a hydroxide solution to form carbonate and/or bicarbonate via reactions (2) and (3), and then regenerated from CaCO3 through the high-temperature calcination step (>900 °C).4,15 This pathway requires carbonate salt palletisation, calcination, and compression to produce pressurised CO2 (Fig. 1).
| CO2(g) + 2KOH(aq) → K2CO3(aq) + H2O(l) | (2) |
| CO2(g) + K2CO3(aq) + H2O(l) → 2KHCO3(aq) | (3) |
Amine based CO2 conversion
In 2017, Chen et al. reported electrochemical CO2 conversion using a range of electrodeposited metallic electrodes (e.g., Pb, Cu, In; Fig. 2a–c) in a CO2 saturated 30% (w/w) monoethanolamine (MEA) aqueous solution.16 Compared with the smooth counterpart, the porous electrode showed improved CO2RR selectivity, which the authors attributed to an increase of local pH and thus suppression of the competing hydrogen evolution reaction (HER) caused by the consumption of protons inside the pores. The authors further found a drastic improvement of CO2RR selectivity when a surfactant, cetyltrimethylammonium bromide (CTAB), was added into the reaction medium (Fig. 2d), and ascribed this enhancement to the adsorption of CTAB to inhibit the HER.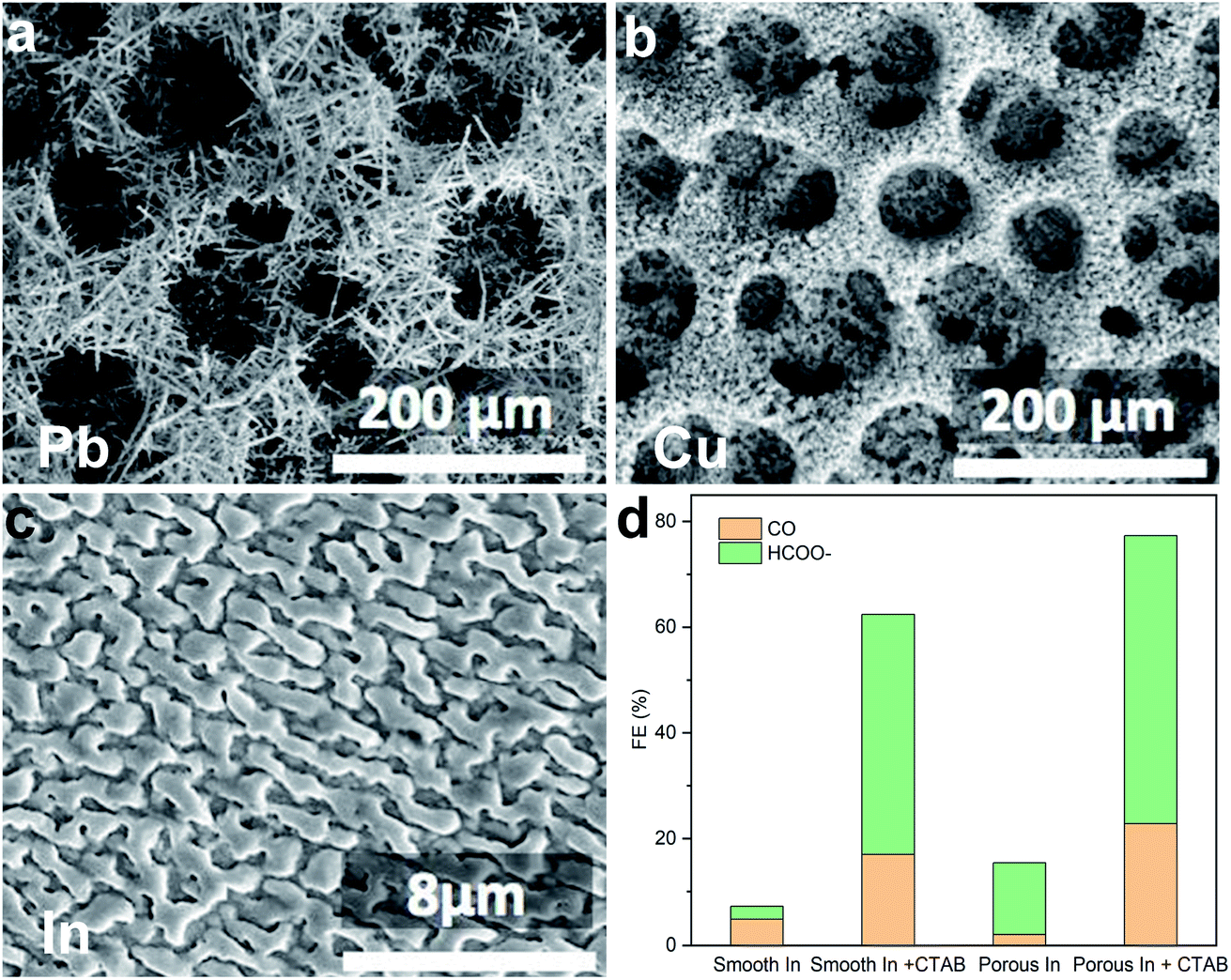 | ||
| Fig. 2 (a–c) Scanning electron microscopy (SEM) images of the porous structure Pb, Cu, and In electrodes. (d) Faradaic efficiency of CO and formate on In electrodes. Reprinted from ref. 16 with permission. Copyright 2017 Wiley-VCH. | ||
In this work, the authors argued – based on the control experiments where using MEA aqueous solution without free molecular CO2 resulted in H2 as the dominant product – that the CO and formate came from dissolved CO2 in MEA solution instead of carbamate.
Very recently, Goetheer et al. studied an integration platform of CO2 capture media electrolysis system using a mixture of 2-amino-2-methyl-1-propanol (AMP) and propylene carbonate (PC) solution as the electrolyte.17 The direct electrolysis of captured CO2 in this mixed medium produced various products, such as formate, glycolate, oxalate, and CO, with a formate faradaic efficiency (FE) up to 50%. Increasing the temperature from 20 to 75 °C steered selectivity to CO with an FE up to 45%. The authors believed that carbamate/bicarbonate species were not themselves directly reduced; instead, with increasing temperature, it was the desorbed CO2 from the carbamate species that underwent electrochemical reduction.
Kratz et al. evaluated several commercially available amine compounds including MEA, ethylenediamine (EDA), and decylamine (DCA) on glassy carbon and Cu electrodes, and found that EDA, owing to its dual capturing sites, showed the best efficiency compared with the others. The authors discovered that the carbamate electrolysis towards CO was significantly improved when using Cu as the catalyst material, with a FE of 58% in EDA. In comparison, the CO FE was 2.3% on glassy carbon. Cu also significantly improved the current density up to 18.4 mA cm−2 (vs. 0.63 mA cm−2 on glassy carbon) at −0.76 V vs. the reversible hydrogen electrode (RHE).18
One challenge for direct carbamate reduction is that the electrostatic repulsion between the negatively charged carbamate and negative potential imposed on the cathode makes it difficult for the carbamate to approach the electrode for the reduction to take place. To address this, Sargent and co-workers tailored the electrochemical double layer so that the chemisorbed CO2 in the form of carbamate can be directly reduced. With the aid of an alkali cation and accelerated mass transport by further system design (temperature and concentration), they demonstrated a direct carbamate conversion to CO with an FE of 72% at 50 mA cm−2.19
(Bi)carbonate-based CO2 conversion
The direct utilisation of bicarbonate (HCO3−) or carbonate (CO32−) as a source of carbon is compelling as they are the major carbon species in commonly used neutral and weak basic solutions with a saturated concentration (e.g., 3.3 M for KHCO3) an order of magnitude higher than that of dissolved CO2, offering a much higher achievable reduction current. Oceanwater, which accounts for absorption of ∼40% of anthropogenic CO2 since the industrial revolution, can potentially be the tank of such carbon sources as the current oceanwater CO2 capture relies on the CO2/bicarbonate equilibrium.20–23Direct bicarbonate electroreduction dates back to 1983 when Hori reported an H-cell architecture with a Hg electrode that produced formate from the reduction of a 1.0 M NaHCO3 solution without a CO2 feed but at a small partial current density (<1 mA cm−2).24 In 2015, Kanan and co-workers reported that a Pd catalyst in an H-cell was able to reduce 2.8 M KHCO3 into formate at a current density of 3.2 mA cm−2.25 These early studies showed the feasibility of direct electrochemical reduction of bicarbonate, but they are limited by low current density and product selectivity (only formate reported).
In 2019, Berlinguette and colleagues introduced a flow cell system that converted a model carbon-capture solution (3 M KHCO3) into CO. Using a bipolar membrane (BPM) as the ion-exchange membrane, a high H+ flux is transported from the anode side to the cathode side, where HCO3− reacts with these protons to generate local high-concentration CO2 at the catalyst/electrolyte interface. The product observed other than formate was CO, with FEs of 81% at 25 mA cm−2 and 37% at 100 mA cm−2.12 This product selectivity is reasonable as Ag, which favours production of CO in the eCO2RR, was used as the electrode material and the species that was reduced on the electrode was essentially CO2 instead of bicarbonate anions.
The same group further used a free-standing porous Ag electrode to improve the selectivity and stability of the conversion.26 The FE of CO reached 78% at 100 mA cm−2, and the performance loss after 80 hours of operation was less than 3%. These performances obtained in bicarbonate electrolysis were comparable to the CO2-fed CO2RR in gas-diffusion-based flow cells. It was believed that the performance improvement came from innovations in electrode materials. First, the addition of Ag nanowires significantly increased the electrochemical surface area (ECSA) of the Ag electrode and increased the abundance of exposed corner and edge active sites that promote chemisorption of both reactants and key intermediates. Additionally, the high length-to-diameter ratio, afforded by nanowire decoration, provided excess pores and channels for the transport of CO2 and electrolyte that resulted in a faster reaction rate. In the same year, they reported that a similar flow cell system with a bismuth-coated carbon (Bi/C) cathode produced formate with a FE of 64% at a current density of 100 mA cm−2, which was also comparable with CO2-fed electrolysers.15
To reduce device voltage and thereby energy input, the hydrogen oxidation reaction was used to replace the oxygen evolution reaction (OER) at the anode.27 As a result, the electrolysis of bicarbonate to CO reached a commercially relevant current density of 500 mA cm−2 at a full-cell voltage of 2.2 V. In comparison, the full-cell voltage of a similar cell configuration that operates the OER and bicarbonate reduction was 4.4 V to maintain a current density of 100 mA cm−2.
It is worth noting that, in a typical alkaline CO2 absorption system, CO2 is converted to carbonate. A further reaction step is required to convert it to bicarbonate, requiring additional energy.28 Thus, it is more valuable for direct carbonate electrolysis. On this front, the Sargent group reported a similar BPM strategy to locally generate CO2 from carbonate for the production of syngas.29 Syngas (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2:CO ratio) was generated at a current density of 150 mA cm−2 with an energy efficiency of 35%, achieving 100% carbon utilisation across the system. The carbonate-to-syngas system operated stably for 145 h.
:1 H2:CO ratio) was generated at a current density of 150 mA cm−2 with an energy efficiency of 35%, achieving 100% carbon utilisation across the system. The carbonate-to-syngas system operated stably for 145 h.
Future directions and considerations
Catalytic materials design
One of the greatest challenges in carbamate electrolysis is to break the C–N bond in carbamate to achieve a high local CO2 concentration or to facilitate the direct carbamate electrolysis. Ongoing experiences in catalyst design and mechanistic studies aimed at lowering the regeneration energy based on the current thermal regeneration mode can be borrowed into the carbamate electrolysis. For example, solid acid catalysts (e.g., ZrO2 and Ag2O/Ag2CO3) have been intensively investigated, which potentially reduce the CO2-amine regeneration energy requirement, as the acidic site facilitates weakening of the C–N bond. The metal atom (Lewis acid) and protons (Brønsted acid) can attach to the carbamate to grab its lone pair of electrons, subsequently changing the configuration of N atoms from sp2 to sp3 and weakening the C–N bond by stretching. As the C–N bond in carbamate is weakened, it could be broken with less energy and CO2 can be released more easily.30,31Presently, direct capture media electrolysis products are limited to C1 products (i.e., CO and formate; Table 1). Progress on the transformation of CO2 into multicarbon (C2+) products has been sought after. However, the challenges are obvious: the formation of C2+ products from the eCO2RR relies on the formation of the C–C bond.32 Cu provides excellent C–C coupling ability and produces C2+ chemicals in gas-fed eCO2RR systems.33 However, there is currently no report with appreciable selectivity towards C2+ products from direct electrolysis of carbamate or (bi)carbonate. On the other side, even in the gas-fed eCO2RR, the long-term stability of Cu electrodes faces considerable challenges. This presents a conundrum in the presence of corrosive amines in carbamate electrolysis. The strategies to suppress the corrosion of electrocatalysts could be potential solutions to achieve activity and stability of the Cu electrode.34,35
| Materials | Reaction media | Applied potential | Products | FE (%) | j (mA cm−2) | Ref. |
|---|---|---|---|---|---|---|
| a Full-cell potential. b Partial current density. | ||||||
| Porous In | 30% (w/w) MEA aqueous solution containing 0.1% (w/w) of CTAB | −0.8 V vs. RHE | CO | 22.8 | n.a. | 16 |
| HCOO− | 54.5 | |||||
| Porous Bi | HCOO− | 36 | ||||
| Porous Pb | HCOO− | 60.8 | ||||
| Porous Ag | CO | 38.2 | ||||
| Pb coil | 2 M 2-amino-2-methyl-1-propanol and 0.7 M tetraethylammonium chloride in propylene carbonate solution | −2.5 V vs. Ag/AgCl | HCOO− | 50 | 25 | 17 |
| Au foil | 1 M 2-amino-2-methyl-1-propanol and 0.7 M tetraethylammonium chloride in propylene carbonate solution | −1.6 V vs. Ag/AgCl | CO | 45 | 10 | |
| Glassy carbon | Pure EDA | −0.76 V vs. RHE | CO | 2.3 | 0.63 | 18 |
| Cu | Pure EDA | −0.76 V vs. RHE | CO | 58 | 18.4 | |
| Ag nanoparticle | 30 wt% MEA and 2 M KCl | ∼−0.75 V vs. RHE | CO | 72 | 50 | 19 |
| Hg | 1 M NaHCO3 | n.a. | HCOO− | n.a. | <1b | 24 |
| Pd | 2.8 M KHCO3 | −0.35 V vs. RHE | HCOO− | 88 | 3.45b | 25 |
| Ag | 3 M KHCO3 | −3.5 Va | CO | 37 | 100 | 12 |
| Porous Ag | 3 M KHCO3 | −3.5 ± 0.1 Va | CO | 78 | 100 | 26 |
| Bi on carbon | 3 M KHCO3 | −4 Va | HCOO− | 64 | 100 | 15 |
| Ag foam | 3 M KHCO3 | −2.2 Va | CO | 15 | 500 | 27 |
| Ag | 1 M K2CO3 | −3 Va | CO | 28 | 100 | 29 |
Recently, supported organometallics, metal–organic frameworks (MOFs), and single-atom catalysts are starting to show promising rates in the eCO2RR.36 Also, high FE towards C2+ products at high current density (>200 mA cm−2) was reported in neutral electrolytes through molecular tuning on heterogeneous electrocatalysts.37 These novel catalyst design means may be a promising solution to achieve C2+ product formation in the capture medium reduction systems.
Mechanisms underpinning the capture medium electrolysis are still unclear, and the lack of such mechanistic guidelines retards the development of effective catalyst materials. An in-depth understanding of the dynamic environment (catalytically active site, diffusion and adsorption of reactants, identity of intermediates, and desorption of products) at the catalyst surface by in situ studies are desirable. In situ/operando spectroscopies (e.g., operando X-ray absorption spectroscopy and operando Raman spectroscopy) coupled with computational modelling may provide powerful toolkits. Such efforts of fundamental mechanistic studies would guide the design of novel catalysts and optimise reaction conditions.
Electrode structure design
The direct capture medium electrolysis system is different from the gas-fed electrolysers. In the gas-fed system, the formation of a three-phase interface (CO2/catalyst/electrolyte) at the gas-diffusion electrode (GDE) is critical to achieve high reactivity and stability. However, in the capture medium electrolysis, CO2 comes from the liquid phase rather than the gas phase and thus a gas-diffusion layer (GDL) that facilitates CO2 transport is not a necessity. Therefore, optimisation for gas-fed flow cell systems may not be transferrable to the design of electrode structures for capture medium electrolysis.Berlinguette and colleagues found that the hydrophobic GDL components (Fig. 3) decreased in situ CO2 generation from bicarbonate and thus reduced the formation of CO.38 This observation suggests that GDL used in liquid-fed electrolysers is expected to efficiently transport the liquid-phase streams, which is opposite to the hydrophobic requirement in gas-fed CO2 electrolysers to prevent excessive accumulation of liquid (so called ‘flooding’). New strategies in tailoring electrocatalyst distribution, hydrophobicity, and ion transport characteristics of the GDEs are desired to achieve a long stability and efficient conversion for the direct capture medium electrolysis.
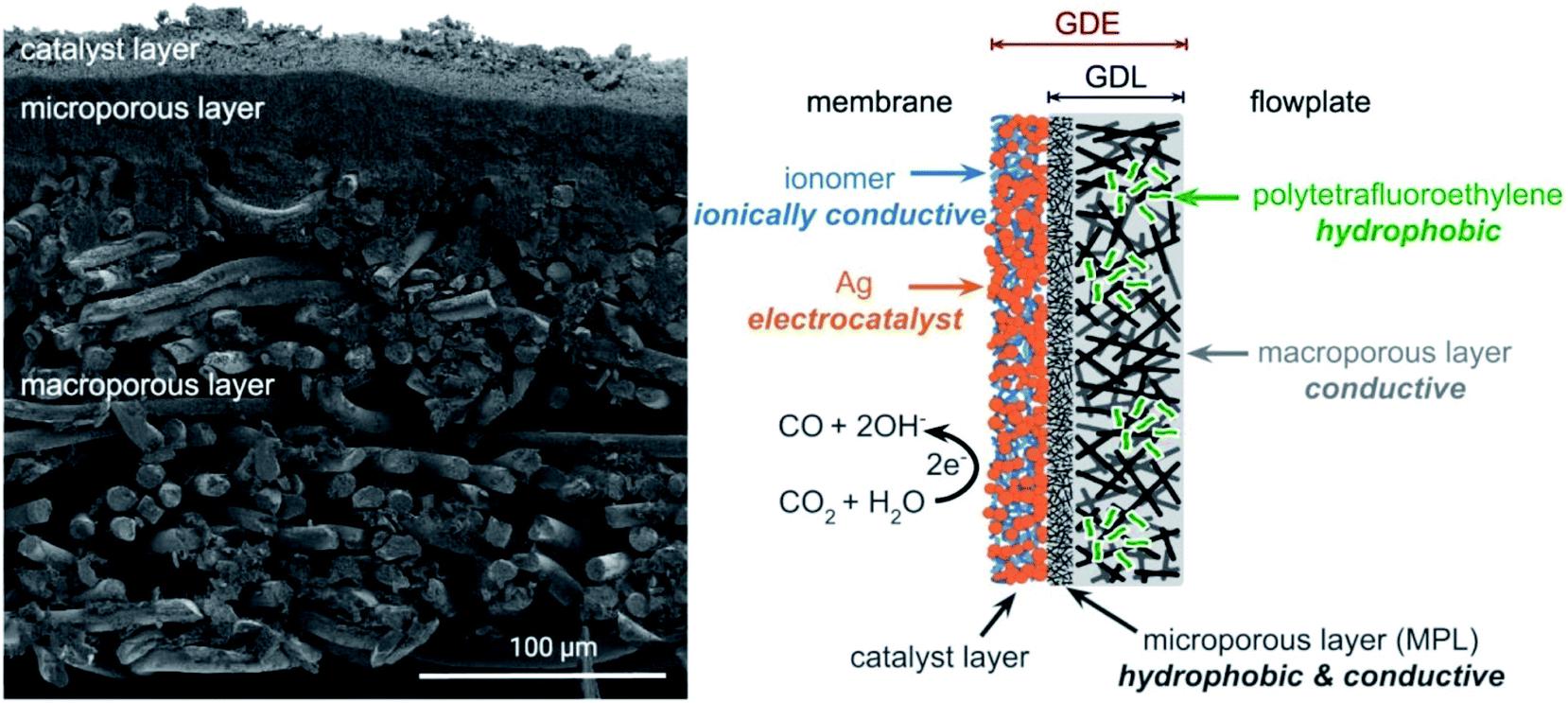 | ||
| Fig. 3 Cross-sectional SEM micrograph and schematic diagram of a GDE. Reprinted from ref. 38 with permission. Copyright 2020 American Chemical Society. | ||
Considerations in the reaction environment
There is a large family of CO2-capture media, especially the amine-based ones.39 Amine design or using a mixed amine system as the electrolyte could be another solution to accelerate breaking the C–N bond. Several factors should be taken into account along this research path. An increase in the number and bulkiness of the substituent in amines tends to lower regeneration energy as the C–N bond becomes weaker, but this comes at the expense of lower absorption rate.40 Meanwhile, different from aqueous electrolytes, amines normally have a high viscosity. Using amines with high concentration or high viscosity as electrolytes for direct electrolysis may lead to a decrease in reaction rate and efficiency due to mass transfer limitations.41Trace impurities, such as SOx and NOx, are common in point source emissions.42 The influence of these impurities should be considered carefully. Jiao et al. studied the impact of SO2 and various NOx (including NO, NO2, and N2O) on CO2 electrolysis with various catalysts in gas-fed flow cells.43,44 Their results indicated that the presence of SO2 and NOx reduces the efficiency of CO2 electrolysis due to the preferential reduction of these impurities. In the case of Ag and Sn, the effect of the SO2 impurity is reversible. In contrast, a selectivity shift to formate was observed on the Cu catalyst with the suppression of C2+ products, indicating that Cu is highly sensitive to SO2 impurities. The impact of NOx impurities is reversible. It is expected that the challenges of these impurities, especially SOx, are present in the direct electrolysis of carbon capture media and thus a pre-electrolysis purification is possibly a necessity. The CO2 obtained from direct air capture technology may be more suitable as the CO2 sources are cleaner than fossil sources.43,44
As discussed in the preceding section, it is challenging for the negatively charged carbamate and (bi)carbonate to approach the cathode due to electrostatic repulsion. Tuning ion species in the electrical double layer19 or using alternative amines hold promise to overcome this fundamental challenge.
Considerations in techno-economics
A techno-economic assessment is useful to establish performance targets and offers guidance for future technology development. Compared with the existing, separated process of CO2 capture and electrochemical conversion means, direct electrolysis has considerable advantages. Besides bypassing the costly capture media regeneration and gas compression steps, another significant advantage of the direct electrolysis in improving economic viability is that it reduces capital and maintenance costs of the overall CO2 capture and utilisation process. Taking the typical amine absorption process as an example, the current process (Fig. 1) requires a two-section process including an amine absorption and a stripping tower while the direct electrolysis does not require the stripping process. This has potential to reduce the capital and equipment maintenance costs associated with the stripping process. Jens et al. reported an assessment of cost-saving by integrating CO2 capture with utilisation and concluded that, with a feed upstream containing >30 mol% CO2, 46% savings in energy demand can be achieved compared to a process without integration.45 Gao et al. found that, although the higher operating voltages of the integrated electrolysis system cause higher energy and operating expenses than those for the gas-fed alternative, it reduces significantly the capital costs since several units, such as the stripper, are no longer required.46The eCO2RR generates a number of possible products; aiming at the most economically viable products would bring advantage to improve the market competitiveness. A techno-economic assessment showed that, under the current gas-fed eCO2RR conditions, CO and formic acid are the most economically viable products with the net present values (NPVs) of US$13.5 million and US$39.4 million, respectively.47 Both products have been reported by the current direct capture medium process. Among the products, syngas (a mixture of CO and H2), as a valuable intermediate used for manufacture of chemicals and fuels with a considerable global market size, should be highlighted as one of the most feasible targets for the direct capture medium electrolysis.48 One advantage of the direct capture medium electrolysis over the gas-fed eCO2RR process is that the single-pass conversion efficiency could be much higher (if not 100% due to the small amount of unreacted CO2 locally generated) as the gaseous product would not be otherwise diluted by the large amount of unreacted CO2.
A complete energy and economic analysis are favoured to look at the entire CO2 capture and valorisation chain. This may start from different sources of CO2, each step in the valorisation process, and the end product applications, market size, and energy supply systems involved.
Conclusion and outlook
The attempts of direct electrolysis of carbon capture media have made some considerable progress. Industrial-level current densities (≥100 mA cm−2) have been achieved through advances in materials and system design (Table 1). Besides, the direct (bi)carbonate electrolysis offers a promising way to solve the ‘carbonate formation’ issue which troubles the low-temperature gaseous CO2 electrolysis in neutral and alkaline electrolytes.49Despite these recent advances, the direct electrolysis of both carbamate and (bi)carbonate is limited by low CO2 availability at the interface of electrode/electrolyte. Further advances towards one-step reduction require strategies to ease the cleavage of the C–N bond and to increase the accessibility of negatively charged carbon-containing species. Physical chemistry characterisation of bond formation and cleavage and the adsorption and desorption on the electrode surface may shed light on mechanisms underpinning new materials design strategies. Additionally, the corrosive nature of amines to metals, the most widely used electrode materials for electrocatalysis, presents a potential challenge in maintaining electrolyser lifetime and catalyst stability. The evaluation of the impact of such corrosion on the durability and robustness of systems should be taken into consideration.
To summarise, previous studies have proven the feasibility of direct electrolysis and show considerable economic benefits relative to a separated CO2 valorisation system. Further advances in targeted strategies for catalyst materials, electrolyser and system designs, and mechanistic understanding, are warranted.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. Z. is grateful to CSIRO Energy for a top-up scholarship. F. L. is grateful for his Australian Research Council Discovery Early Career Researcher Award (project number DE200100477) funded by the Australian Government.References
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef.
- B. D. Patterson, F. Mo, A. Borgschulte, M. Hillestad, F. Joos, T. Kristiansen, S. Sunde and J. A. Van Bokhoven, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 12212–12219 CrossRef CAS PubMed.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo and L. A. Hackett, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- M. Ramdin, T. W. de Loos and T. J. Vlugt, Ind. Eng. Chem. Res., 2012, 51, 8149–8177 CrossRef CAS.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- K. Li, W. Leigh, P. Feron, H. Yu and M. Tade, Appl. Energy, 2016, 165, 648–659 CrossRef CAS.
- W. A. Smith, T. Burdyny, D. A. Vermaas and H. Geerlings, Joule, 2019, 3, 1822–1834 CrossRef CAS.
- A. Barthel, Y. Saih, M. Gimenez, J. D. Pelletier, F. E. Kühn, V. D'Elia and J.-M. Basset, Green Chem., 2016, 18, 3116–3123 RSC.
- I. S. Metcalfe, M. North, R. Pasquale and A. Thursfield, Energy Environ. Sci., 2010, 3, 212–215 RSC.
- A. Chapman, C. Keyworth, M. Kember, A. Lennox and C. Williams, ACS Catal., 2015, 5, 1581–1588 CrossRef CAS.
- T. Li, E. W. Lees, M. Goldman, D. A. Salvatore, D. M. Weekes and C. P. Berlinguette, Joule, 2019, 3, 1487–1497 CrossRef CAS.
- B. Li, Y. Duan, D. Luebke and B. Morreale, Appl. Energy, 2013, 102, 1439–1447 CrossRef CAS.
- C. Sun and P. K. Dutta, Ind. Eng. Chem. Res., 2016, 55, 6276–6283 CrossRef CAS.
- T. Li, E. W. Lees, Z. Zhang and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2624–2630 CrossRef CAS.
- L. Chen, F. Li, Y. Zhang, C. L. Bentley, M. Horne, A. M. Bond and J. Zhang, ChemSusChem, 2017, 10, 4109–4118 CrossRef CAS PubMed.
- E. Pérez-Gallent, C. Vankani, C. Sánchez-Martínez, A. Anastasopol and E. Goetheer, Ind. Eng. Chem. Res., 2021, 60, 4269–4278 CrossRef.
- M. Abdinejad, Z. Mirza, X.-A. Zhang and H.-B. Kraatz, ACS Sustainable Chem. Eng., 2020, 8, 1715–1720 CrossRef CAS.
- G. Lee, Y. C. Li, J.-Y. Kim, T. Peng, D.-H. Nam, A. Sedighian Rasouli, F. Li, M. Luo, A. H. Ip, Y.-C. Joo and E. H. Sargent, Nat. Energy, 2021, 6, 46–53 CrossRef CAS.
- C.-F. de Lannoy, M. D. Eisaman, A. Jose, S. D. Karnitz, R. W. DeVaul, K. Hannun and J. L. Rivest, Int. J. Greenhouse Gas Control, 2018, 70, 243–253 CrossRef CAS.
- M. D. Eisaman, J. L. Rivest, S. D. Karnitz, C.-F. de Lannoy, A. Jose, R. W. DeVaul and K. Hannun, Int. J. Greenhouse Gas Control, 2018, 70, 254–261 CrossRef CAS.
- T. DeVries, Global Biogeochem. Cycles, 2014, 28, 631–647 CrossRef CAS.
- I. A. Digdaya, I. Sullivan, M. Lin, L. Han, W.-H. Cheng, H. A. Atwater and C. Xiang, Nat. Commun., 2020, 11, 4412 CrossRef PubMed.
- Y. Hori and S. Suzuki, J. Electrochem. Soc., 1983, 130, 2387 CrossRef CAS.
- X. Min and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4701–4708 CrossRef CAS PubMed.
- Z. Zhang, E. W. Lees, F. Habibzadeh, D. A. Salvatore, S. Ren, G. Simpson, D. G. Wheeler, A. Liu and C. P. Berlinguette, ChemRxiv, 2021 DOI:10.26434/chemrxiv.12891071.v3.
- Z. Zhang, E. Lees, S. Ren, A. Huang and C. Berlinguette, ChemRxiv, 2021 DOI:10.26434/chemrxiv.13665074.v1.
- A. J. Welch, E. Dunn, J. S. DuChene and H. A. Atwater, ACS Energy Lett., 2020, 5, 940–945 CrossRef CAS.
- Y. C. Li, G. Lee, T. Yuan, Y. Wang, D.-H. Nam, Z. Wang, F. P. García de Arquer, Y. Lum, C.-T. Dinh, O. Voznyy and E. H. Sargent, ACS Energy Lett., 2019, 4, 1427–1431 CrossRef CAS.
- U. H. Bhatti, S. Nam, S. Park and I. H. Baek, ACS Sustainable Chem. Eng., 2018, 6, 12079–12087 CrossRef CAS.
- U. H. Bhatti, D. Sivanesan, S. Nam, S. Y. Park and I. H. Baek, ACS Sustainable Chem. Eng., 2019, 7, 10234–10240 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. Stephens, K. Chan and C. Hahn, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS.
- X. Li, S. Fu, W. Zhang, S. Ke, W. Song and J. Fang, Sci. Adv., 2020, eabd1580 CrossRef CAS PubMed.
- J. Peng, B. Chen, Z. Wang, J. Guo, B. Wu, S. Hao, Q. Zhang, L. Gu, Q. Zhou and Z. Liu, Nature, 2020, 586, 390–394 CrossRef CAS PubMed.
- R. I. Masel, Z. Liu, H. Yang, J. J. Kaczur, D. Carrillo, S. Ren, D. Salvatore and C. P. Berlinguette, Nat. Nanotechnol., 2021, 16, 118–128 CrossRef CAS.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z.-Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C.-S. Tan, D.-H. Nam, R. Quintero-Bermudez, T.-T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- E. W. Lees, M. Goldman, A. G. Fink, D. J. Dvorak, D. A. Salvatore, Z. Zhang, N. W. Loo and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2165–2173 CrossRef CAS.
- W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu and J. Wang, Chem. Soc. Rev., 2020, 49, 8584–8686 RSC.
- F. A. Chowdhury, H. Okabe, H. Yamada, M. Onoda and Y. Fujioka, Energy Procedia, 2011, 4, 201–208 CrossRef CAS.
- E. Pérez-Gallent, C. Vankani, C. Sánchez-Martínez, A. Anastasopol and E. Goetheer, Ind. Eng. Chem. Res., 2020, 60, 4269–4278 CrossRef.
- J. Zhang, P. Xiao, G. Li and P. A. Webley, Energy Procedia, 2009, 1, 1115–1122 CrossRef CAS.
- W. Luc, B. H. Ko, S. Kattel, S. Li, D. Su, J. G. Chen and F. Jiao, J. Am. Chem. Soc., 2019, 141, 9902–9909 CrossRef CAS.
- B. H. Ko, B. Hasa, H. Shin, E. Jeng, S. Overa, W. Chen and F. Jiao, Nat. Commun., 2020, 11, 5856 CrossRef CAS PubMed.
- C. M. Jens, L. Müller, K. Leonhard and A. Bardow, ACS Sustainable Chem. Eng., 2019, 7, 12270–12280 CAS.
- N. Gao, C. Quiroz-Arita, L. A. Diaz and T. E. Lister, J. CO2 Util., 2021, 43, 101365 CrossRef CAS.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Nat. Catal., 2018, 1, 32–39 CrossRef CAS.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |