
Emerging elemental two-dimensional materials for energy applications
Zongxiao
Wu†
,
Junlei
Qi†
,
Wenbin
Wang
,
Zhiyuan
Zeng
 and
Qiyuan
He
*
and
Qiyuan
He
*
Department of Materials Science and Engineering, City University of Hong Kong, Hong Kong SAR, China. E-mail: qiyuanhe@cityu.edu.hk
First published on 25th June 2021
Abstract
Elemental two-dimensional (2D) materials possess distinct properties and superior performances across a multitude of fundamental and practical research fields. Although a tremendous number of earlier studies focus on graphene and its derivatives, other emerging elemental 2D materials, such as borophene, silicene, black phosphorene, antimonene, tellurene, bismuthene and arsenene, are attracting increasing research interest in electronics and optoelectronics, as well as various energy storage and conversion applications, owing to their unique structural, electrochemical and electronic properties. In particular, emerging elemental 2D materials often possess large surface areas, high theoretical capacity, structural anisotropicity, high carrier mobility and tunable bandgaps, making them promising candidates in many energy storage and conversion technologies. Recently, we have witnessed remarkable progress in the preparation, characterization, and application of many emerging elemental 2D materials. However, the challenging yet vastly different synthetic strategies and resulting physicochemical properties, along with complex theoretical and experimental results, make it difficult to navigate through their applications in specific research fields. In this perspective, we summarize the progress of emerging elemental 2D materials in terms of their preparation methods, properties and figures of merit in energy storage and catalytic applications. Furthermore, we present our insight into the challenges and opportunities, which would hopefully shed light on the great potential of this ever-expanding field.
 Qiyuan He | Dr Qiyuan He is currently an Assistant Professor in the Department of Materials Science & Engineering at the City University of Hong Kong. He obtained his PhD Degree from Nanyang Technological University in Singapore in 2013. He then joined the University of California, Los Angeles as a Postdoctoral Fellow before returning to Nanyang Technological University as a Research Fellow in 2016. Dr He's research scope is highly interdisciplinary, focusing on the fundamentals of semiconductor interfaces and their applications in nanoelectronics, iontronics, chemical/biological sensors, catalysis, and on-chip electrochemistry, and of course, Star Wars. |
1. Introduction
The discovery of graphene has sparked tremendous interest in two-dimensional (2D) materials in both fundamental and practical research for well over a decade, owing to their many unique physical and chemical properties.1,2 In particular, the atomic-thin morphology, large surface area and layered structure make them ideal candidates for energy devices. Looking beyond graphene, researchers have also explored a variety of 2D materials, such as hexagonal boron nitride (h-BN)3 and transition metal dichalcogenides (TMDs).4 Among the enormous 2D materials family, elemental 2D materials are one unique type of elemental material with 2D layered or sheet-like structures, often referred to as Xenes, demonstrating promising prospects in electronic and energy fields.5–7 Compared with TMDs, which usually exhibit indirect bandgaps, many elemental 2D materials possess superior mobility (tellurene, black arsenene, etc.) and direct band gaps (BP, black arsenene, etc.). While reviews of elemental 2D materials in electronics could be found previously,8–11 in this perspective, we will elaborate on the recent advances of elemental 2D materials in the fields of energy storage and catalysis.Until now, over a dozen elemental 2D materials have been experimentally realized, with more still in theoretical predictions.12–14 An overview of all the theoretically and experimentally explored elemental 2D materials is shown in Table 1. Due to the vast different synthetic strategies and properties,5 we resolve to use the most straightforward categorization according to element groups. For group 13 elements, the monolayer structures of two elements, boron and gallium, have been successfully synthesized and termed borophene and gallenene,15–30 respectively. Due to the carbon-neighbor position of boron in the periodic table of elements, borophene is expected to possess some properties similar to those of graphene,16 such as high capacity, and electronic and ionic conductivity, indicating prospects for use in metal ion batteries as an anode material. Similarly, with atomic structures analogous to graphene, silicene (2D silicon), germanene (2D germanium), stanene (2D tin) and plumbene (2D lead) in group 14 have also been experimentally explored. Tunable bandgaps with various surface engineering strategies are their most attractive properties, offering a fascinating prospect of discrete engineering applications.31
|
In group 15, black phosphorene (denoted as BP) remains the most explored elemental 2D material besides graphene. Previously, 2D black phosphorus was commonly denoted as phosphorene or simply black phosphorus. However, novel allotropes, such as violet phosphorene32 and blue phosphorene,33 have recently been synthesized. Thereby, we use the term BP to represent black phosphorene. Few-layer BP exhibits significantly higher carrier mobility and larger theoretical capacity for metal-ion batteries than other 2D materials.34 Additionally, its tunable bandgap and abundant active sites are favorable for catalytic applications.35 However, the inferior stability of BP limits its practical application, which remains an important barrier to overcome. Furthermore, other elemental 2D materials in groups 15 and 16 with analogous lattice structures to BP, i.e., arsenene (2D As), antimonene (2D Sb), bismuthene (2D Bi), and tellurene (2D Te), exhibiting tunable band gaps, high carrier mobility, high theoretical capacity, and more importantly, excellent environmental stability, are being actively pursued theoretically and experimentally.36 They provide a wide range of potential candidates for high-performance electrode materials with good stability in energy storage and conversion fields.
In this perspective, we highlight the ever-expanding family of elemental 2D materials and their intriguing applications in energy storage, including batteries and supercapacitors, and energy conversion, including electrocatalysis, photocatalysis and solar cells. Firstly, we provide an overview of the basic properties of emerging elemental 2D materials, followed by a comprehensive summary of their synthetic methods. Next, their figures of merit in specific fields of energy storage and conversion are discussed. Finally, we share our perspective on the challenges and opportunities in this developing research field and hope to shed some light on the fundamental exploration and future application.
2. Structures and properties
2.1 Elemental 2D materials of group 13
Both borophene (2D boron) and gallenene (2D gallium) show high anisotropy due to the different arrangements of atoms along two different lattice directions. Many different phases of borophene, such as 2-Pmmn, β12, χ3, and honeycomb (Fig. 1a–d), have been synthesized and relatively comprehensively studied.15,20,26 For example, the 2-Pmmn phase holds a bulked structure with slight corrugation along the a direction (zigzag direction). But along the b direction (armchair direction), the corrugations of this structure are removed. 2-Pmmn borophene is predicted to have great anisotropy in the electronic band structure, exhibiting metallic properties along the armchair direction while possessing a large bandgap in the zigzag direction.15 The excellent electron and ion transport along the armchair direction and the high capacity make borophene an attractive material in ion batteries. In contrast to the 2-Pmmn phase, other obtained structures (β12, χ3, and the honeycomb) are all planar, which can be attributed to the weak interaction with the underlying substrate. As shown in Fig. 1b and c, the sheet structures of β12 and χ3 are similar, with the main difference being the hexagonal boron array for the β12 phase and the narrow zigzag boron array for the χ3 phase.20 In particular, honeycomb borophene shows a V-shape dI/dV curve via scanning tunneling spectroscopy (STS) near the Fermi energy, suggesting the Dirac band structures similar to graphene.26 Additionally, the χ6 and γ-B28 phases of borophene have been successfully synthesized with study on their properties still in progress.28 | ||
Fig. 1 Crystal structures of elemental 2D materials. (a) 2-Pmmn, (b) β12, (c) χ3, and (d) honeycomb borophene.14,25,36 (a) Reproduced with permission,14 American Association for the Advancement of Science (AAAS). (b) and (c) Reproduced with permission,25 Springer Nature. (d) Reproduced with permission,36 Elsevier. (e) Planar and (f) buckled gallenene. Reproduced with permission,38 AAAS. (g)–(j) Theoretical buckled hexagonal structures of group 14 elemental materials. Reproduced with permission,40 Wiley-VCH. (g) Flat; (h) chair; (i) boat and (j) washboard. (k) Black phosphorene. Reproduced with permission,53 Wiley-VCH. (l) Antimonene. Reproduced with permission,62 American Chemical Society. (m) Bismuthene. Reproduced with permission,68 Wiley-VCH. (n) Grey arsenene. Reproduced with permission,76 American Chemical Society. (o) Black arsenene. Reproduced with permission,78 Wiley-VCH. (p) and (q) Tellurene from the [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 20] direction (p) and front view (q). Reproduced with permission,81 Springer Nature. 20] direction (p) and front view (q). Reproduced with permission,81 Springer Nature. | ||
Two stable 2D anisotropies of gallenene have been recently synthesized, which, again, show both buckled and planar configurations (as shown in Fig. 1e and f), respectively.17,52 Due to the finite density of states at the Fermi energy, both allotropes of gallenene show metallic characteristics, however, with different band structures. The bands of buckled configuration are highly dispersive with a large bandwidth, suggesting lighter charge carriers and higher mobilities.
2.2 Elemental 2D materials of group 14
In contrast to graphene, most elemental 2D materials in group 14 (silicene,37 germanene,38 stanene,39 and plumbene40) show buckled honeycomb structures, resulting from the mixed hybridization of sp3 and sp2, except for the recent discovery of a planar stanene polymorph.53,54 The buckling positions vary in the hexagonal buckled structures, termed chair, boat, and washboard, respectively.55,56Fig. 1g–j illustrates the flat and buckled hexagonal structure. The most stable structures for each element were also predicted, i.e., triple-layer MoS2 geometry for stanene, large honeycomb dumbbell arrangements for silicene and germanene, and high-buckled 2D phases for plumbene.57,58 In addition, theoretical studies have predicted many “non-graphene” 2D carbons. Among them, the two most theoretically stable allotropes are phagraphene (consisting of 5-6-7 carbon rings) and penta-graphene (consisting entirely of pentagons).41,42 Like 2D materials in group 13, buckled 2D materials in group 14 possess Dirac cone semimetal band structures.59,60 For example, the band structures of various graphene allotropes are predicted to range from metallic61,62 to wide bandgap semiconductors,63,64 resulting from the different degrees of sp2 and sp3 hybridization. For instance, penta-graphene may possess an intrinsic quasi-direct band gap of 3.25 eV,42 which is close to that of ZnO and GaN, while phagraphene is metallic.652.3 Elemental 2D materials of group 15
The 2D forms of group 15 elements are similarly termed analogous to graphene, namely phosphorene (2D BP), arsenene (2D As), antimonene (2D Sb), and bismuthene (2D Bi), which exhibit different crystal orientations and band structures.Black phosphorene (BP) possesses a side-centered layered orthorhombic lattice structure, the space group of which is classified as Cmca. For each P atom in the crystal lattice, there are three nearest neighbor P atoms. The puckered atom layers can be regarded as two parallel sub-layers within the plane (Fig. 1k).43 And P atoms form two kinds of geometries (zigzag-like and armchair-like) along different directions, which lead to strong anisotropic properties. Also, the interlayer spacing of BP is about 0.53 nm,66 larger than that of graphene (0.33 nm).67 Such large interlayer spacing results from the puckered structure, as well as the AB Bernal stacking of two phosphorene layers in the unit cell.68 Few-layer BP shows predominant p-type behavior, and is one among the few examples of p-type 2D materials along with WSe2 and tellurene.69 It maintains direct bandgaps, which can be tuned by its thickness from 0.3 eV (bulk) to 2 eV (monolayer).70,71 Few-layer BP possesses high theoretical carrier mobility, i.e., 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–26000 cm2 V−1 s−1 for monolayer BP.70 However, BP faces severe chemical degradation under ambient conditions. Tremendous efforts have been made to understand the exact mechanism of BP degradation from theoretical and experimental respects. With spectroscopic investigation, it was found that the oxidation process of BP is most likely directly related to the oxygen concentration and light density.72 Additionally, Wang et al. found that O2 spontaneously dissociates on the surface of few-layer BP under ambient conditions and consequently initializes the oxidation process.73 In contrast to popular belief, H2O would not directly interact with BP until it has been already oxidized. Most recently, researchers also successfully synthesized violet phosphorene32 and blue phosphorene,33 which are still at the primary stage of investigation.
000–26000 cm2 V−1 s−1 for monolayer BP.70 However, BP faces severe chemical degradation under ambient conditions. Tremendous efforts have been made to understand the exact mechanism of BP degradation from theoretical and experimental respects. With spectroscopic investigation, it was found that the oxidation process of BP is most likely directly related to the oxygen concentration and light density.72 Additionally, Wang et al. found that O2 spontaneously dissociates on the surface of few-layer BP under ambient conditions and consequently initializes the oxidation process.73 In contrast to popular belief, H2O would not directly interact with BP until it has been already oxidized. Most recently, researchers also successfully synthesized violet phosphorene32 and blue phosphorene,33 which are still at the primary stage of investigation.
For antimonene, β-phase antimonene is found to be the most thermodynamically stable phase. According to transmission electron microscope (TEM) characterization and calculations, β-antimonene exhibits a buckled hexagonal atomic structure with two atoms per unit cell and an interlayer distance of 3.73 Å (ref. 74) (Fig. 1l). Based on theoretical calculation results, β-antimony remains metallic until the thickness is reduced to two layers, and the monolayer antimonene exhibits a relatively large bandgap of 2.28 eV.19 Experimentally, β-antimonene obtained by liquid-phase exfoliation shows thickness-dependent bandgaps between 0.8 eV (5–7 nm) and 1.44 eV (0.3–1 nm).44 Moreover, unlike BP, β-antimonene shows good stability under ambient conditions. Verified by optical microscopy and Raman characterization, monolayer antimonene on a PdTe2 substrate prepared via MBE showed no change in optical microscopy and Raman spectra after annealing at 380 K for 10 minutes before the oxidation begins.75 Similarly, few-layer antimonene flakes prepared by chemical vapor deposition could remain stable for 10 minutes below 250 °C and show no change in Raman spectra for three months under ambient conditions at room temperature.76
Bismuthene is another relatively stable elemental 2D material of group 15. Analogous to antimonene, few-layer bismuthene also crystallizes with a buckled layered structure, which is the most thermodynamically stable phase among all its allotropies.77 Monolayer bismuthene exhibits a 2D hexagonal lattice with a washboard structure (Fig. 1m).45,78 The interlayer spacing of monolayer bismuthene is 3.9 Å,45 which is smaller than that of BP, still generally favorable for ion intercalation in the electrochemical process. Although bulk bismuth remains metallic, few-layer bismuthene is predicted to be an n-type 2D semiconductor with direct bandgaps. Similar to BP, the bandgap of bismuthene is expected to be thickness dependent from 0 V (bulk) to 0.55 eV (monolayer).78 Moreover, bismuthene may possess exceptionally high carrier mobility, ranging from 5.7 × 106 (bulk) to 20000 cm2 V−1 s−1 (0.1–2 μm thin films), which is superior to that of few-layer BP.79
Arsenene has two thermodynamically stable phases, gray arsenene with a buckled lattice structure (Fig. 1n) and black arsenene with a puckered lattice structure (Fig. 1o). Both demonstrate better chemical stability than BP.46,80 Gray arsenene has the P3m1 space group81 and exhibits a lattice constant of 3.61 Å.82 The atomic layers are stacked via ABC arrangement.83,84 On the other hand, black arsenene is analogous to BP with a puckered structure.85 It exhibits the Pmna space group81 and an interlayer distance of 5.46 Å.86 Both grey and black arsenene possess thickness-dependent indirect bandgaps. The bandgaps of monolayer grey and black arsenene are expected to be 1.63 and 0.83 eV, respectively,81 which are yet to be experimentally confirmed. Besides, grey arsenene and black arsenene are expected to have moderate carrier mobilities of 527.40 cm2 V−1 s−1 (ref. 87) and 176.82 cm2 V−1 s−1,88 respectively.
2.4 Elemental 2D materials of group 16
There are two phases of tellurene, α and β, with the α-phase being more thermodynamically stable. α-phase tellurene shows a trigonal crystal lattice similar to their bulk counterpart, in which Te atoms form individual helical chains and then spiral along axes parallel to the [0001] direction at the center and corners of the hexagonal elementary cell89 (Fig. 1p and q). Each Te atom has two nearest atoms and is bonded via a covalent bond. Both the monolayer tellurene film MBE-grown on graphene layers90 and free-standing few-layer tellurene nanosheets prepared through a wet-chemistry method89 exhibit lattice constants consistent with the α-phase with trigonal configuration. On the other hand, the monolayer tellurene film MBE-grown on highly oriented pyrolytic graphite (HOPG) substrates has a crystal structure consistent with the β-phase.47 The substrate, as well as the growth kinetics, is believed to play a critical role in the atomic structure of 2D tellurene, which necessitates more efforts in promoting the theoretical understanding and the experimental investigation of 2D tellurene.Many recent studies aimed to explore the electric band structure of tellurene. Qiao et al. reported that bilayer tellurene possessed an indirect bandgap of 1.17 eV in theoretical calculations. The bandgap will reduce with increasing layer numbers, reaching 0.31 eV for bulk tellurium.91 Experimentally, monolayer tellurene grown on graphene is a p-type semiconductor with a bandgap of 0.92 eV, which is determined by dI/dV spectra from STS.90 Tellurene also demonstrates high carrier mobility ranging from hundreds to thousands of cm2 V−1 s−1, depending on the thickness and synthetic methods.89,92 In addition, the solution-grown tellurene possesses excellent chemical stability. Such tellurene-based field-effect transistors maintain the performance in air for more than two months.93
Other than tellurene, the successful synthesis of 2D selenene in group 16 is also reported recently.48 However, there is little research on aspects of energy applications of selenene, which calls for further studies.
Above all, elemental 2D materials exhibit rich atomic structures, such as puckered structures for BP and black arsenene, and buckled structures for β-antimonene and grey arsenene. Moreover, different elemental 2D materials possess various electronic characteristics, including carrier mobility and tunable bandgap scope. Specifically, some elemental 2D materials (BP, arsenene, etc.) exhibit a direct band gap and high mobility, and demonstrate excellent application potential. In conclusion, the various atomic structures combined with the electronic characteristics of elemental 2D materials, facilitate their extensive applications in a variety of energy storage and conversion areas.
3. Synthetic approaches
Despite the significant synthetic challenges, a variety of reliable fabrication strategies have been developed to prepare elemental 2D materials with controlled crystal structures and morphologies. These novel synthetic methods can be classified into two main strategies, top-down methods (mechanical cleavage, liquid-phase exfoliation, and electrochemical exfoliation) and bottom-up methods (chemical vapor deposition and molecular beam epitaxy). In top-down methods, 2D materials are usually obtained by various exfoliation processes that break the weak van der Waals interaction between atomic layers. In comparison, bottom-up methods refer to the direct synthesis of monolayer to few-layer 2D materials through different chemical reactions in the gas or liquid phases.3.1 Top-down methods
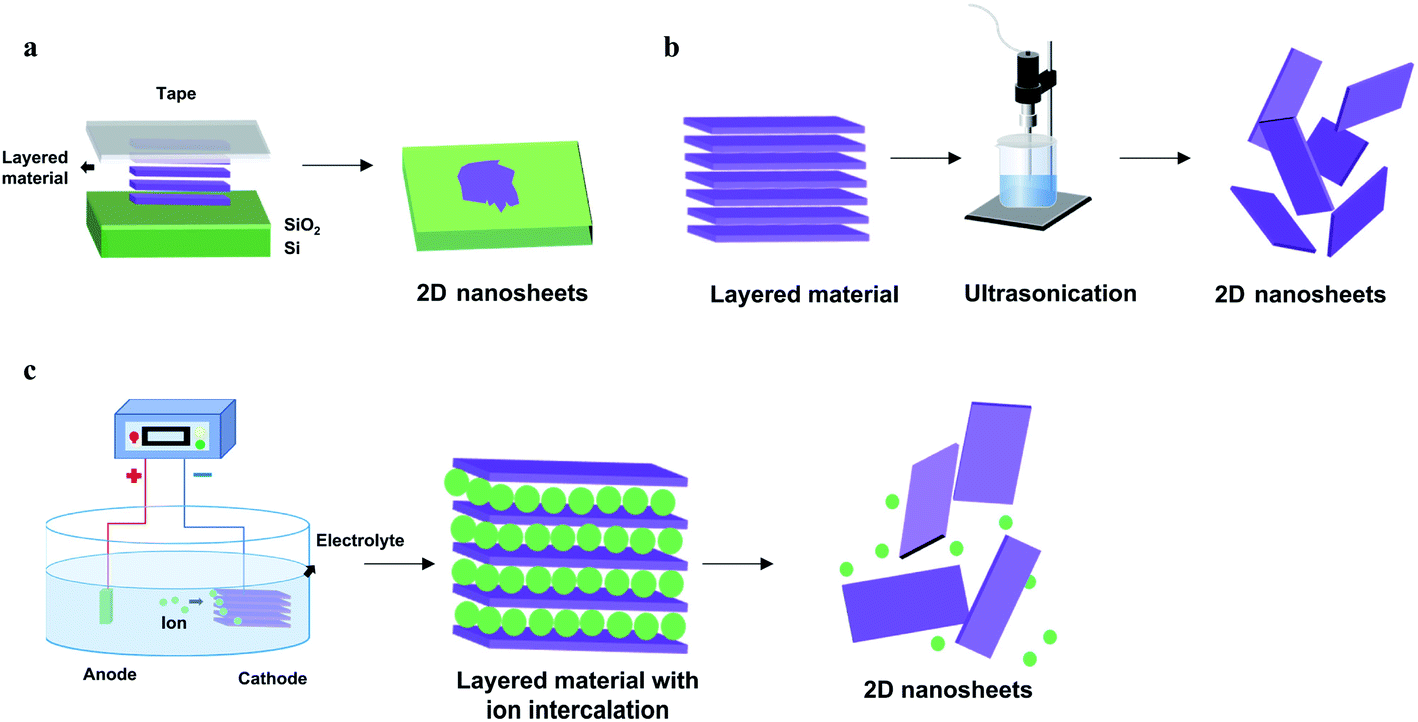 | ||
| Fig. 2 Schematic illustration of top-down preparation methods of elemental 2D materials. (a) Mechanical exfoliation. (b) Liquid-phase exfoliation. (c) Electrochemical exfoliation. | ||
Notably, numerous modifications have emerged in recent studies to tackle these problems. For instance, a modified mechanical exfoliation method has been reported, capable of producing a wide range of 2D materials with a large lateral size.98 It is achieved by adopting oxygen plasma treatment to remove ambient absorbents on the substrate prior to the exfoliation and additional heat treatment during the exfoliation to enhance the contact between bulk layered materials and the substrate. In addition, metal-assisted mechanical exfoliation has been developed to synthesize high-quality BP flakes efficiently.99 With the assistance of the deposited gold/silver layer, the adhesive force between metal layers and bulk layered materials is significantly enhanced, resulting in enhanced production and increased product sizes.
In general, top-down approaches rely on the facile exfoliation process and are relatively cost-effective, however still facing various limitations. Mechanical exfoliation is severely restricted by its low yield, while the products obtained by liquid exfoliation often exhibit a small lateral size and a large distribution of thickness. Alternatively, electrochemical exfoliation is more efficient, highly controllable and mass productive. However, it is not applicable for all elemental 2D materials due to the difference in source crystals and the intercalation mechanism.
3.2 Bottom-up methods
Bottom-up methods refer to the direct synthesis of nanomaterials from precursors by chemical reactions, which generally produce high-quality 2D elemental materials in scale. In particular, due to the large energy and bonding configuration difference between bulk and 2D layered materials in groups 13 and 14, it is extremely difficult to exfoliate 2D layered structures from the bulk sources directly.116 Bottom-up approaches are, therefore, favored.As shown in Fig. 3a, the first successful synthesis of borophene was reported using MBE growth on a Ag(111) substrate.15 In this experiment, a mixture of two borophene phases was observed, which were identified as striped and homogeneous phases, respectively. Almost at the same time, Feng et al. reported the synthesis of borophene by MBE in a parallel study.20 When the substrate was changed to Au, besides the growth of few-layer nanosheets, borophene islands with a triangular structure and honeycomb lattice were also observed.55 Interestingly, during the growth, borophene island discrete nanoclusters were initially observed before the formation of larger nanosheets with increasing boron coverage, resulting from the increasing boron dose. It is worth mentioning that elemental 2D materials with a honeycomb structure (for instance, graphene, silicene, germanene, and stanine) are favored in many applications due to the Dirac fermions and other exotic properties hosted by the highest lattice hole density among the typical lattice structures.119,120 However, the energetical instability caused by electron deficiency of boron makes it challenging to synthesize a honeycomb structure, which is finally obtained on an Al(111) substrate using MBE under UHV conditions.26 The small size (from several to hundreds of nanometers) of the obtained borophene on Ag, Al, or Au surfaces limits its detailed property characterization and applications.15,26 To this end, Wu et al. synthesized large-area borophene with a domain size of 10–100 μm2 by diffusion of boron into copper, followed by repeated resurfacing and recrystallization processes.29,30 Recently, Ir(111) was also used as the epitaxial substrate to produce large-area borophene.28 Unlike earlier reported studies, the borophene formed a single phase (χ6) in a wide range of experimental conditions.
 | ||
| Fig. 3 Schematic illustration of bottom-up preparation methods of elemental 2D materials. (a) Diagram of molecular beam epitaxy (MBE) growth of Borophene on the Ag(111) substrate. Reproduced with permission,14 AAAS. (b) Diagram of the homemade two-zone furnace for the chemical vapor deposition (CVD) growth of the borophene thin film on Cu foil.17 (c) Schematic diagram of the prepared borophene from the top view in (b).17 (b) and (c) Reproduced with permission,17 Wiley-VCH. (d) Morphology evolution from 1D Te structures to 2D tellurene in the hydrothermal synthesis of tellurene. Reproduced with permission,81 Springer Nature. | ||
In group 14, silicene has been successfully synthesized by epitaxial growth on different substrates such as Ag(111),56,121–123 Ag(110),124 MoS2,125 ZrBr2(0001),126 Ir(111),127 and Au(110)128 at different temperatures and deposition rates. Due to the 3/4 ratio of lattice constants between Ag and Si, silver is the most appropriate substrate for the epitaxial growth of silicene among other substrates.129,130 Many potential substrates to grow silicene have been predicted by theoretical calculations, such as aluminum nitride,131,132 chalcogenide-layered compounds,133 ZnS,132 CaF2,134 and gallium dichalcogenides,135 which are yet to be experimentally realized.
Germanene by MBE has been successfully fabricated on Au(111),136 Al(111),137 Pt(111),138,139 and Cu(111).140 Recently, a novel segregation method to synthesize large-scale germanene has been developed using a Ag(111) thin film epitaxially grown on as the substrate. Monolayer and multilayer stanene nanosheets were firstly obtained on a Bi2Te3 substrate by MBE, and the atomic and electronic structures of stanene were experimentally determined, which is consistent with the calculation results.141 Later, MBE growth of stanene was demonstrated on Sb(111),142 Ag(111),39 and Cu(111)39 substrates. Interestingly, it has been found in density-functional theory (DFT) calculations.39 that various epilayer structures grown on Ag(111) might spontaneously recover to become a perfect stanene structure after departing from the substrate.
0} facet of the nucleated seeds, thereby leading to the formation of the 1D structure. As the growth continues, PVP is insufficient to cover the crystal plane, hence significantly enhances the growth of {100} surfaces along the {1![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 10} direction, which contributes to the formation of the 2D morphology of tellurene. In addition, few-layer hexagonal antimonene nanosheets have been prepared via a facile colloidal synthesis method.150 The XRD characterization of the final product confirmed the identity of the rhombohedral phase (β-antimonene) without impurities. Bismuthene has also been synthesized via the hydrothermal method by reduction of the precursor Bi2O2CO3.153 Moreover, a one-step wet-chemistry method has been developed to prepare bismuthene with an average thickness of 0.6 nm,151 based on the simple reduction of BiCl3 by NaBH4.
10} direction, which contributes to the formation of the 2D morphology of tellurene. In addition, few-layer hexagonal antimonene nanosheets have been prepared via a facile colloidal synthesis method.150 The XRD characterization of the final product confirmed the identity of the rhombohedral phase (β-antimonene) without impurities. Bismuthene has also been synthesized via the hydrothermal method by reduction of the precursor Bi2O2CO3.153 Moreover, a one-step wet-chemistry method has been developed to prepare bismuthene with an average thickness of 0.6 nm,151 based on the simple reduction of BiCl3 by NaBH4.
In “bottom-up” methods, elemental 2D materials are constructed from atomic or molecular precursors. Compared with “top-down” methods, “bottom-up” methods can break the requirements of layered source crystals and achieve reasonable yield. However, “bottom-up” methods such as the commonly used MBE and CVD, heavily rely on the chemical bonds/interactions between precursors and substrates, and therefore are usually limited to the lattice symmetry and parameters. Further development calls for more exploration into the combination of precursors, substrates and deposition conditions. In addition, most wet-chemistry methods are only acceptable for specific nanoscale materials with uncontrollable sizes. It is particularly difficult to obtain elemental 2D materials in a monolayer via wet-chemistry approaches. And the surfactants resulting from the synthesis process are undesirable in the consequent applications. Overall, it is still challenging to find general, effective, high-yield and low-cost methods for the controllable synthesis of 2D materials.
4. Applications in energy storage
4.1 Batteries
The majority of applications of elemental 2D materials in energy storage fields are utilization as electrode materials for batteries, including Li-ion batteries (LIB), Na-ion batteries (SIB), Li–sulfur batteries (LSB), and other next-generation batteries. | ||
| Fig. 4 Schematic diagram of structure evolution of elemental 2D materials during the operation of a Li-ion battery and Na-ion battery. (a) Schematics of structural evolution of bismuthene to Na3Bi via NaBi during electrochemical sodiation. Reproduced with permission,68 Wiley-VCH. (b) Structures of lithiated silicene. Reproduced with permission,153 Elsevier. (c) Top and side views of the fully relaxed structures of LixAs and NaxAs. Reproduced with permission,154 Royal Society of Chemistry. | ||
The diffusion behavior of Li in few-layer BP has received the most attention.43,157 Notably, few-layer BP shows a puckered lattice structure and a large interlayer distance of 0.53 nm, providing a suitable interlayer channel for the ion insertion/extraction.162 BP can be intercalated with three Li ions at the end of the lithiation stage to form the Li3P phase and thereby possesses a high theoretical volume of 2596 mA h g−1. It has a multistep lithiation process: BP → LiP → Li2P → Li3P. Additionally, theoretical results demonstrate a strong diffusion anisotropy, with a lower energy barrier (0.08 eV) along the zigzag direction, compared with the high energy barrier (0.68 eV) along the armchair direction.157 The shallow energy barrier along the zigzag direction implies ultrahigh Li diffusivity, which is 102 times faster than that on MoS2 or 104 times faster than that on graphene. Such highly anticipated anisotropy has been experimentally realized.163
The potential of borophene as an anode material for metal-ion batteries is also investigated via first-principles calculations. At full lithium insertion forming Li0.75B, borophene possesses a high theoretical specific capacity of 1860 mA h g−1.158,164 Similar to BP, borophene also exhibits a strong Li diffusion anisotropy, i.e., the energy barrier for Li diffusion is only 2.6 meV along the furrow direction of corrugated borophene, much lower than that of the perpendicular direction (325.1 meV).158
In addition, the sodiation/desodiation mechanism of antimonene is researched based on DFT calculations, along with experimental characterization, including in situ synchrotron X-ray diffraction (XRD) and ex situ selected-area electron diffraction (SAED).74,165 Antimonene has a theoretical capacity of 660 mA h g−1 for SIB, with reversible crystalline phase evolution (Sb → NaSb → Na3Sb) during the ion insertion/extraction process, which is verified by the experimental characterization. Additionally, antimonene showed anisotropic volume expansion along the a/b plane in the Na ion intercalation process.74 This can be explained by the lower Na-ion diffusion barrier of 0.14 eV, enabling fast sodiation reaction kinetics and excellent cyclic performance for SIB.
Similarly, first-principles calculations are implemented to explore few-layer bismuthene in LIB and SIB.166 The calculated results demonstrated that bismuthene possessed high theoretical storage capacities of 2276 mA h g−1 and 2149 mA h g−1 for Li and Na, respectively. The sodiation storage mechanism in bismuthene is also experimentally verified by in situ TEM with a multistep crystal phase evolution (Fig. 4a), Bi → NaBi → Na3Bi, accompanied by a large anisotropic volume expansion of 142% occurring mainly along the z-axis.45
Furthermore, silicene, germanene and stanene are predicted to possess theoretical capacities of 954 mA h g−1, 369 mA h g−1 and 226 mA h g−1 for both Li or Na ions storage, respectively (Fig. 4b).160 In addition, theoretical results of monolayer arsenene as the anode for (Li/Na/Mg)-ion batteries, as shown in Fig. 4c, demonstrates that monolayer arsenene possesses a high theoretical capacity of 1609.77 mA h g−1 for Li, 1073.18 mA h g−1 for Na, and 1430.91 mA h g−1 for Mg.161
Inspired by the progress in theoretical calculations and the rapid development of synthetic methods, extensive experimental studies have been carried out to determine the potential of emerging elemental 2D materials as an anode in LIB and SIB.
Due to the high theoretical volume and good ion diffusivity, BP is the most researched elemental 2D material for use as a battery anode and probably one of the most promising ones, demonstrating high capacity up to ∼2000 mA h g−1,68 which is far beyond those of other 2D anode materials for LIB or SIB, such as MXene.167 Still, the main drawback of BP in the electrochemical process involves the huge volume change and the destruction of the crystal structure, which lead to lower coulombic efficiency (≈8%) and unsatisfactory reversible specific capacity in the following cycles.168 To deal with these problems, researchers have made extensive efforts to composite BP with other functional materials to enhance its mechanical strength. In one good example, few-layer BP nanosheets were sandwiched between graphene layers to form a composite SIB anode, exhibiting an ultrahigh specific capacity of 2440 mA h g−1 at a current density of 0.05 A g−1 along with good cycling performance.163 The graphene layers not only enhance the electrical conductivity but also provide mechanical support to accommodate the sizeable anisotropic expansion of phosphorene layers along the y and z axial directions, which is further verified by in situ TEM characterization. Similarly, BP–graphene (BP–G) hybrid paper was fabricated by vacuum filtration of a mixed dispersion containing graphene and BP, demonstrating a high electrical conductivity(about 1000 S cm−1) and excellent flexibility.169 As a result, the LIB showed a high specific capacity of 501 mA h g−1, good rate capability, and prolonged cycling performance with a capacity retention of 80.2% at a current density of 500 mA g−1 after 500 cycles.
Beyond BP, few-layer antimonene nanosheets prepared via the LPE method have also been used as anode materials in Li-ion batteries.165 As illustrated in Fig. 5a and b, antimonene nanosheets exhibited better cycling performance and rate capability than bulk antimony and antimony nanoparticles derived from similar LPE process. Based on the first-principles calculation, antimonene exhibited a minimal Li+ ion diffusion barrier of 0.25 eV along the in-plane channel, whereas the Li-ion diffusion across layers required a larger energy barrier of 1.14 eV. Thereby, antimonene with a large lateral size facilitated the ion insertion process and enhanced Li-ion storage compared with bulk antimony and antimony nanoparticles. Besides, ultrathin bismuthene nanosheets were prepared with a similar LPE method and used as an anode for SIB.159 Notably, the time-sequences of high-resolution transmission electron microscope (HRTEM) images in Fig. 5c indicate that the intercalation of Na ions does not collapse the crystal structure of 2D bismuthene. Moreover, after four cycles in the in situ TEM study, bismuthene nanosheets showed a small lateral expansion of around 13% and exhibited a superior cyclic ability after 200 cycles compared with the bulk counterpart.
 | ||
| Fig. 5 (a) and (b) the electrochemical performance of antimonene in Li-ion battery.158 (a) Schematic diagram of the Li ion diffusion barrier in antimonene. (b) Cycling performances of the anode made of bulk β-phase antimony, antimonene nanosheets, and antimony nanoparticles in a Li-ion battery. (a) and (b) Reproduced with permission,158 Royal Society of Chemistry. (c) (i)–(ix) are time-sequences HR-TEM images of bismuthene nanosheets during the sodiation process in a Na-ion battery. Scale bar: 10 nm. Reproduced with permission,152 American Chemical Society. | ||
To overcome this polysulfide dissolution, a novel BP functionalized separator was utilized in LSB, showing much enhanced specific capacity and cycling performance compared to the commercial polypropylene separator.171 According to first-principles calculations, BP exhibits a high binding energy with various lithium polysulfide/sulfide species and thereby provides strong adsorption of lithium polysulfide/sulfide species to suppress the “shuttle effect”. Hence, the functionalized separator can effectively reduce material loss and improve the electrochemical performance of LSB.
In another theoretical study, the potential of borophene to serve as a good host of cathode materials for LSB172,173 was investigated. The puckered lattice structure is expected to contribute to an ultrahigh adsorption ability towards lithium polysulfides. However, it also suggested that the lithium polysulfides could decompose on borophene with no energy barrier, leading to the separation of lithium and sulfur and therefore undesirable sulfur loss during charge–discharge cycles.173 In contrast, defective borophene demonstrates moderate adsorption energy toward lithium polysulfides, which can not only efficiently anchor lithium polysulfides to suppress the “shuttle effect”, but also avoid the dissolution of sulfur materials.
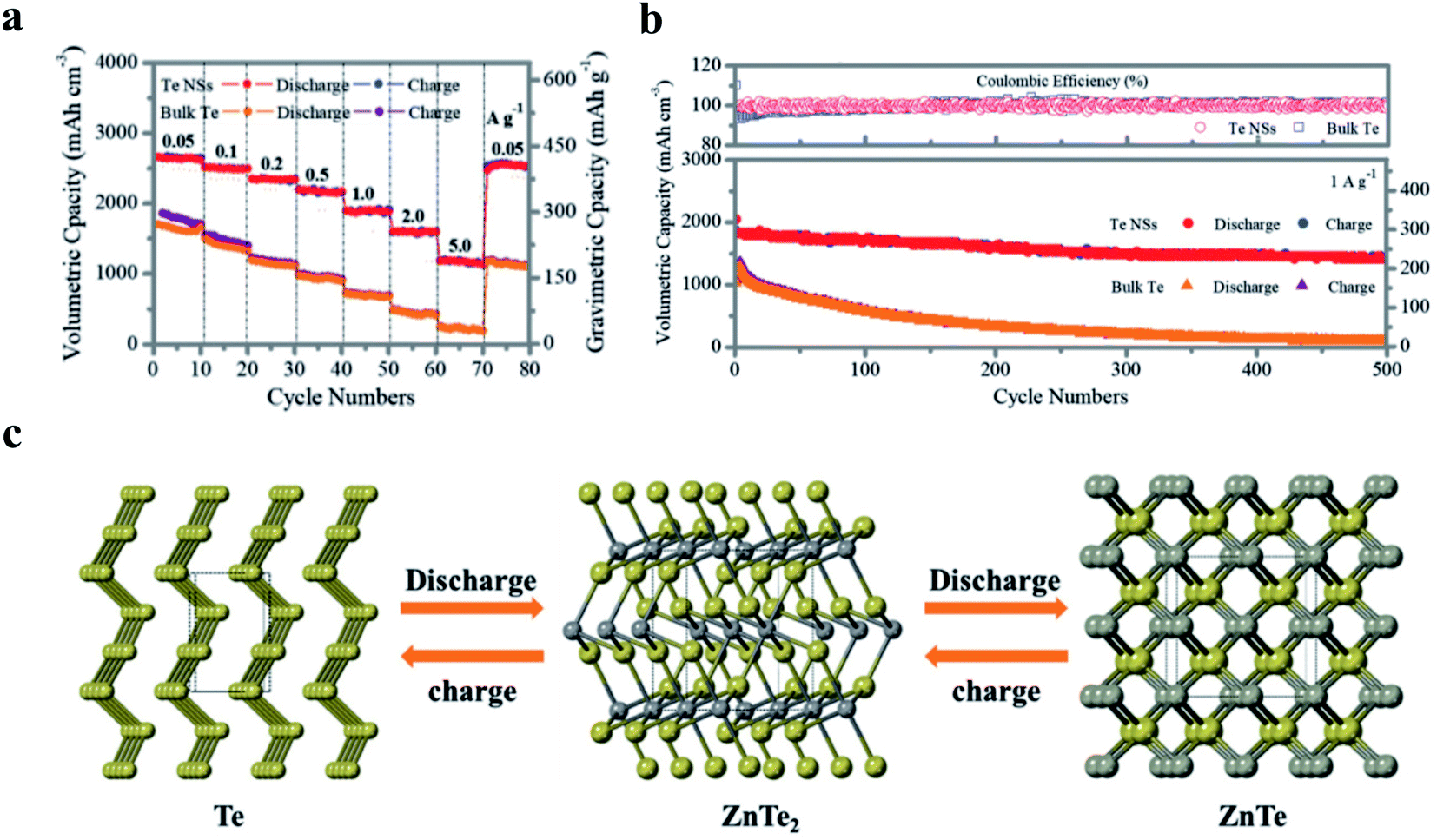 | ||
| Fig. 6 Tellurene as an anode for high-performance Zn–Te batteries. (a) Rate performance based on different current densities from 0.05 to 5 A g−1. (b) Cycling performance and coulombic efficiency of Te NSs and bulk Te at 1 A g−1. (c) Schematic representation for phase transformation during the discharging process (yellow: Te atoms, gray: Zn atoms). Reproduced with permission,167 Wiley-VCH. | ||
4.2 Supercapacitor
Supercapacitors are an electrochemical energy storage device that can provide higher power density than batteries as well as higher energy density than conventional dielectric capacitors.175 The large surface area, large interlayer spacing and high carrier mobility make elemental 2D materials promising electrode materials for supercapacitors. For example, a free-standing polypyrrole (PPy)/BP film was prepared via a facile electrodeposition method and served as a supercapacitor electrode.176 BP nanosheets efficiently suppress the disordered packing of PPy during electrodeposition, thereby facilitating ion diffusion and electron transport as well as enhancing stability during the electrochemical processes. The hybrid electrode exhibits a high capacitive value of 497.5 F g−1 and good cycling performance. A highly flexible supercapacitor was assembled based on the PPy/BP composite film, showing great potential for a variety of electronics. In this work, thin BP nanosheets (fewer than five layers), via the LPE method, are hybridized with graphene nanosheets to form the BP/graphene film through vacuum filtration, which was further transformed into an oxidized free-standing black phosphorus (oBP)/reduced graphene oxide (rGO) hybrid film (oBP/rGO, Fig. 7a and b) via controlled ozonation and subsequent thermal treatment.177 As is shown in Fig. 7c, the free-standing BP/rGO hybrid film exhibited enhanced electrochemical performance in a supercapacitor compared with BP and graphene, with a high specific capacitance of 478 F g−1 at 1 A g−1 (four times greater than that of black phosphorus) and good rate capability. The specific capacitance of the BP/rGO hybrid film is higher than those of electrodes based on other 2D materials, e.g., conductive 2D titanium carbide, which possesses a specific capacitance of 245 F g−1 at a scan rate of 2 mV s−1.178 Based on DFT calculations and experimental characterization, such as in situ Raman spectroelectrochemical spectra studies, it is evident that the molecular-level redox-active P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O sites obtained from controlled ozone-driven oxidation provide more active sites for proton accumulation and lead to the high specific capacitance. Additionally, hybridizing BP with electronically conductive rGO results in a high electronic conductivity, which is favorable to the rate capability.
O sites obtained from controlled ozone-driven oxidation provide more active sites for proton accumulation and lead to the high specific capacitance. Additionally, hybridizing BP with electronically conductive rGO results in a high electronic conductivity, which is favorable to the rate capability.
 | ||
| Fig. 7 Supercapacitor based on the hybrid structure of oxidized black phosphorus (oBP) and a reduced graphene oxide film (oBP/rGO).170 (a) Illustration of the schematic model of the oBP/rGO hybrid film. (b) Cross-sectional-view HR-TEM image of the free-standing oBP/rGO hybrid film, showing the heterointerface of the oBP/rGO hybrid film. Scale bar, 1 nm. (c) Rate capability of 2D BP, rGO, oBP/GO and free-standing oBG/rGO (foBG) at various current densities from 1 to 50 A g−1. 2D BP: black phosphorene; rGO: reduced graphene oxide; oBP/GO: oBP and graphene oxide hybrid film; oBP/rGO: oBP and reduced graphene oxide hybrid film. Reproduced with permission,170 Springer Nature. | ||
Besides, antimonene also serves as a promising electrode material for supercapacitors.179 Few-layer antimonene demonstrates remarkable electrochemical performance in supercapacitors with a capacitance of 1578 F g−1, which implied high charge storage capacity and could be attributed to 2D layered atomic structure as well as the high intrinsic pseudocapacitive activity of antimonene.180 However, although the high capacitance of antimonene was confirmed by follow-up research studies,181,182 the mechanism for the high intrinsic pseudocapacitive activity of antimonene requires further investigation. The assembled device shows very competitive performance with energy and power densities of 20 mW h kg−1 and 4.8 kW kg−1, respectively. Additionally, antimonene was also hybridized with other active materials or conductive substrates to enhance electrochemical performance. For example, an antimonene/3D-Ni foam hybrid electrode demonstrates an ultrahigh specific capacity of 1618.41 mA h g−1 (∼6854.45 F g−1) at a low scan rate of 5 mV s−1.182 Recently, the novel VS4/antimonene composite electrode also exhibited excellent electrochemical performance for supercapacitors.183 The hybridization of VS4 with highly electroactive few-layer antimonene (4–6 layers) significantly increased the number of active surface sites owing to the synergistic combination of two highly pseudocapacitive materials. The VS4/antimonene showed a specific capacitance of 1447 F g−1 at 6 A g−1 and good rate capability, that it retains 782.1 F g−1 at a large current density of 30 A g−1. And the VS4/antimonene‖VS4/antimonene symmetry supercapacitor demonstrated high specific capacitance (539.4 F g−1@3 A g−1), excellent cycling stability (174% after 10000 cycles), and high energy and power density (93.5 W h kg−1 and 1705 W kg−1).
5. Applications in electrocatalysis
Recently, there have been rapid theoretical and experimental developments of elemental 2D materials in electrocatalytic reactions, including the hydrogen evolution reaction (HER), oxygen evolution reaction (OER), oxygen reduction reaction (ORR), carbon dioxide reduction (CO2RR), and N2 reduction reaction (NRR).5.1 Water splitting
Water splitting involves the hydrogen evolution reaction (HER) at the cathode and the oxygen evolution reaction (OER) at the anode, respectively, in an electrolytic cell. Borophene,184,185 antimonene,104,186 black arsenene187 and BP188 have been reported as electrocatalysts for the electrocatalytic HER or OER.BP has been considered to be a promising electrocatalyst for water splitting. However, plain BP exhibits instability and limited electrochemical performance, which is possibly caused by either a low density of catalytic sites or a limited electrical conductivity.188 Thereby, composite materials based on BP and other nanomaterials are commonly used as high-performance electrocatalysts for water splitting. For example, BP was prepared by LPE and formed a heterostructure with N-doped graphene (NG) by sonication to serve as an electrocatalyst for overall water splitting.188 The heterostructure electrocatalyst exhibited good electrochemical performance with a low cell voltage of 1.54 V at 10 mA cm−2 for overall water splitting in 1 M KOH solution, outperforming the integrated Pt/C@RuO2 couple (1.60 V). It is believed that directional interfacial electron transfer induced by different Fermi levels between BP and NG not only enriched the electron density, but also optimized H adsorption/desorption to promote the HER, as well as enhanced the favorable formation of key OER intermediates (OOH*) to improve the OER. In addition, few-layer antimonene nanosheets were prepared by LPE and utilized as bifunctional electrocatalysts for the HER and OER.186 Both the HER and OER catalytic performance of few-layer antimonene nanosheets without any treatment showed great potential in the application of overall water splitting under alkaline conditions. Antimonene nanosheets have demonstrated an overpotential of 666 mV to achieve the current density of 1 mA cm−2 and a relatively large Tafel slope of 217.2 mV dec−1 in 1 M KOH solution in the HER. Also, it showed a moderate OER performance with a Tafel slope of 261.3 mV dec−1 and long-term stability in alkaline KOH solution for bifunctional water splitting, i.e., both the HER and OER.
Furthermore, researchers have also forecasted the electrocatalysis performance of some elemental 2D materials via theoretical calculations. The metallic band structure and the multiple active sites of borophene make it promising in the HER.20 The borophene electrocatalyst is expected to show an ultralow overpotential due to the small free energy of hydrogen adsorption (ΔGH) of the β12, α, and β1 phases of borophene. Additionally, as reported by Feng and coworkers, pure black arsenene is unfavorable for the HER process due to the extremely weak binding strength of the hydrogen atom on its surface. However, the embedded O atoms at the surface of arsenene are expected to increase the binding strength of H to an appropriate degree and greatly improve the catalytic performance of black arsenene for the HER.187
Although some elemental 2D materials are inherently unfavorable for catalysis, they demonstrate potential as supporting substrates in some cases, which can be attributed to many of their unique characteristics.189,190 Like most 2D materials, elemental 2D materials provide high surface areas, which are beneficial to the uniform dispersion of nanomaterials on the substrates. Additionally, elemental 2D materials often possess a high carrier concentration or large carrier mobility, which significantly facilitate charge transport in composite catalysts. Moreover, the various synergistic effects between substrates and nanomaterials on them could effectively enhance electrocatalytic activity. For example, a modified LPE process was reported to fabricate few-layer antimonene nanosheets while simultaneously growing RuS2 nanodots on the antimonene during the LPE process, as shown in Fig. 8a.189 The Ru–S–Sb/antimonene composite exhibits moderate HER performance with a low overpotential of 153 mV at a current density of 10 mA cm−2 (Fig. 8b) and a small Tafel slope of 118 mV dec−1 in 1.0 M phosphate-buffered saline (Fig. 8c). In addition, the catalytic performance of various single transition metal (TM) atoms supported on borophene with vacancies (denoted as the TM-BH system) has been investigated for the electrocatalytic ORR and OER.190 Among them, Fe supported on borophene (Fe-BH) and Rh supported on borophene (Rh-BH) are two promising ORR catalysts with overpotentials of 430 mV and 470 mV, respectively. Furthermore, the Rh-BH also shows impressive OER performance with the overpotential of 240 mV, which is smaller than that of commercial RuO2 by near 370 mV. Shi et al. also developed an electrocatalyst based on cobalt or Co3O4 nanoparticles decorated on BP nanosheets, exhibiting a low overpotential and excellent stability in the OER in alkaline media.191
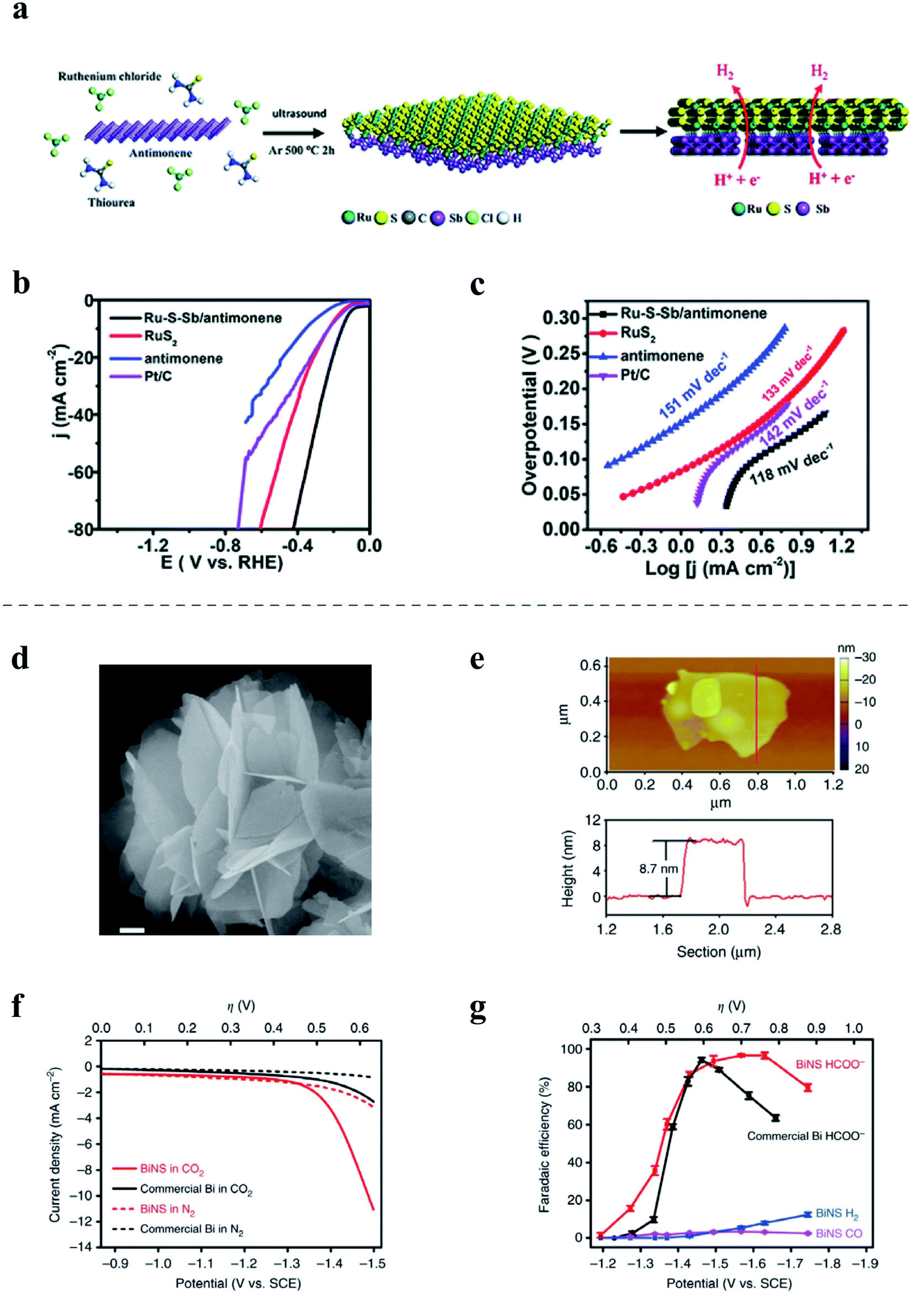 | ||
| Fig. 8 (a–c) Preparation diagram and electrochemical performance of Ru––S––Sb/antimonene180 for the HER: (a) schematic of the preparation of Ru–S–Sb/antimonene; (b) polarization curves at 5 mV s−1 in the electrolyte; (c) Tafel plots of Ru–S–Sb/antimonene, RuS nanodots, antimonene nanosheets and commercial Pt/C. (d–g) Characterization and electrochemical performance of free-standing bismuthene nanosheets144 in the CO2RR: (d) SEM image of bismuthene nanosheets, scale bar, 200 nm; (e) AFM image and the corresponding height profile of the bismuthene nanosheet; (f) polarization curves of bismuthene nanosheets (BiNS) and a commercial Bi nanopowder in N2- or CO2-saturated 0.5 M NaHCO3; (g) potential-dependent faradaic efficiencies of HCOO−, CO, and H2 on BiNS in comparison with the faradaic efficiency of HCOO− on the commercial Bi nanopowder. (a–c) Reproduced with permission,180 Royal Society of Chemistry. (d–g) Reproduced with permission,144 Springer Nature. | ||
5.2 CO2 reduction reaction (CO2RR) and N2 reduction reaction (NRR)
Elemental 2D material-based electrocatalysts have attracted numberous research interest for the CO2RR and NRR. For instance, the active orbital and electrons of the zigzag and diff-zigzag type edges of few-layer BP enable selective electrocatalysis of N2 to NH3,192 achieving a high ammonia yield of 31.37 μg h−1 mg−1cat. Further study on few-layer antimonene electrocatalysts has reported an impressive NH3 yield of 180.4 μg h−1 mg−1cat and faradaic efficiency of 11.6% when tested in 0.1 M KOH under ambient conditions.193 In addition, bismuthene electrocatalysts also demonstrated efficient NH3 production.194,195The Li group reported the synthesis of free-standing bismuthene nanosheets (Fig. 8d and e) with a thickness of ∼8.7 nm via in situ topotactic transformation of bismuth oxyiodide and used as a electrocatalyst in the CO2RR. As is illustrated in Fig. 8f and g, bismuthene nanosheets demonstrated a cathodic current onset at approximately −1.3 V versus SCE in CO2-saturated 0.5 M NaHCO3 solution,196 which increased to 11 mA cm−2 at −1.5 V versus SCE. And the initial faradaic efficiency was 16% at ∼−1.3 V, which quickly rose to >95% at −1.5 V and maintained close to 100% until reaching −1.7 V. Bismuthene nanosheets showed good performance with high selectivity (∼100% for formate formation) and FEHCOO− (up to 98%) at an overpotential of −580 mV as well as good cycling performance and an ultrasmall onset overpotential of <90 mV. Additionally, based on DFT calculations, borophene was also predicted as a promising catalyst for the CO2RR due to its great capture capacity of CO2,197,198 although yet to be experimentally proven. Additionally, borophene has also been theoretically studied as the supporting substrate for Cu atomic chains as an electrocatalyst for the CO2RR.199 Based on first-principles calculations, the unique chain structures of Cu atoms played a key role in lowering the overpotential of the CO2RR, implying great catalytic abilities of Cu-supported borophene for CO2RR. Furthermore, silicene on Ag(111) was predicted to show good catalytic activity for CO2 hydrogenation, which could be selectively controlled by the number of layers.200 Based on the binding energy with the reaction intermediates, the supported silicene monolayer is expected to be favorable to form carbon monoxide, formic acid, and formaldehyde, while few-layer silicene is expected to form methanol and methane.
6. Applications in photocatalysis
Photocatalysis directly converts solar energy to chemical energy,201,202 which could be an effective approach to overcome the intermittent nature of solar illumination in renewable energy production. The large surface area as well as the tunable bandgap, endows elemental 2D materials with great potential to serve as efficient photocatalysts. Recently, many theoretical calculation results have predicted potential interest for some emerging elemental 2D semiconductors as photocatalysts, including BP,203 blue phosphorene,204,205 violet phosphorene,206 antimonene,207,208 and arsenene.209–211 However, the experimental realization is still limited, with most of the advances focusing on BP and its composite.6.1 Photocatalytic hydrogen production
Few-layer BP is one of the most promising candidates for photocatalytic water splitting among elemental 2D materials, owing to its suitable conduction band (CB) position for hydrogen production and tunable bandgaps that can accommodate a wide range of photon absorption from ultraviolet to near-infrared. However, the practical application of BP in photocatalysis is primarily limited by the low stability and rapid recombination of photoinduced charge carriers, possibly related to its relatively narrow bandgap.212 Constructing semiconductor heterostructures of elemental 2D materials and semiconductors is an effective approach to suppress charge carrier recombination, accelerate carrier mobility, and consequently enhance photocatalytic performance.213–216 For example, the Z-scheme heterostructure strategy is often adopted to construct preferable band alignment to reduce the recombination of photoinduced charge carriers.217 When the photocatalytic reaction happens in a Z-scheme band alignment, the photogenerated electrons in one semiconductor with lower reduction ability recombine with the photogenerated holes in the other semiconductor with a lower oxidation ability. Therefore, the photogenerated electrons in a semiconductor with high reduction ability and photogenerated holes with high oxidation ability can avoid recombination and participate in the photocatalytic reaction. As illustrated in Fig. 9a, a Z-scheme 2D/2D heterojunction of BP/Bi2WO6 was prepared by sonication treatment of the mixture solution of BP and monolayer Bi2WO6 nanosheets and used as a photocatalyst in both photocatalytic water splitting and NO removal.218 The heterostructure photocatalyst exhibits a high H2 evolution rate of 21042 μmol g−1, which is 9.15 times higher than that of pristine monolayer Bi2WO6 and also higher than that of other previously reported 2D photocatalysts, such as WS2/CdS heterostructures, which demonstrated a photocatalytic HER performance of ∼4200 μmol g−1 h−1.219 Similarly, the NO removal ratio is up to 67%, which is as high as 2.6 times that of pure monolayer Bi2WO6. 5,5-dimethyl-1-pyrroline-N-oxide (DMPO)-electron spin resonance spectroscopy (ESR) was utilized to track active radicals during the photodegradation process and proved the Z-scheme charge transfer mechanism. The electrons in the CB of Bi2WO6 combine with the holes in the valence band (VB) of BP. Consequently, the photoinduced electrons remain in the CB of BP to produce H2 while the holes remain in the VB of Bi2WO6 to oxidize NO, as illustrated in Fig. 9b.
 | ||
| Fig. 9 (a and b) Photocatalytic NO removal based on the composite film of BP/monolayer Bi2WO6:209 (a) schematic illustration of the synthesis of the BP/monolayer Bi2WO6 heterojunction; (b) mechanism of photocatalytic NO removal and water splitting based on the BP/monolayer Bi2WO6 heterojunction under visible-light irradiation, showing the various redox potentials versus RHE. (c–e) Photocatalytic H2 evolution based on atomic cobalt species (ACS) functionalized tellurene nanosheets (Te NS-ACS):210 (c) illustration of the synthesis of Te NS-ACS; (d) the photocatalytic H2 evolution activities of pure tellurene nanosheets (Te NS) with and without ACS functionalization and Eosin Y (EY) as a light absorber under visible light; (e) mechanism of the photocatalytic H2 evolution based on Te NS-ACS. (a and b) Reproduced with permission,209 Wiley-VCH. (c–e) Reproduced with permission,210 Wiley-VCH. | ||
Furthermore, recent studies also show the potential of tellurene in photocatalytic H2 generation. As shown in Fig. 9c, tellurene nanosheets were functionalized with single atomic cobalt species (ACS) and used as a catalyst for photocatalytic H2 production.220 The ACS functionalized tellurene nanosheets (denoted as Te NS-ACS) demonstrated a H2 evolution rate of 130.8 μmol h−1 under visible light irradiation (Fig. 9d), with Eosin Y (EY) as a light absorber and triethanolamine (TEOA) as the sacrificial agent. The boosted electrochemical performance could be attributed to coordination between Te nanosheets and atomic cobalt, which altered the electronica structure and introduced intermediate energy states. As displayed in Fig. 9e, the intermediate energy states could act as trap sites to accommodate the photogenerated electrons, promoting charge separation and transfer. Qiu and coworkers prepared ultrathin 2D tellurene nanosheets (2–5 nm) via the LPE method and fabricated a tellurene/CdS heterostructure as a high-performance photocatalyst for photocatalytic hydrogen evolution.221 The tellurene/CdS heterostructure exhibits an enhanced hydrogen production rate of 234.4 μmol h−1, which is 7.8 times higher than that of bare CdS (30.2 μmol h−1). Remarkably, the heterostructure photocatalyst maintained a stable photocatalytic H2 production for more than 10 h. DFT calculation and time-resolved photoluminescence decay spectra spectroscopy suggested that the photoinduced electrons transfer from CdS to tellurene to produce H2 rapidly. The superior photocatalytic performance could be attributed to the low electron–hole recombination, massive surface active sites and high charge mobility of tellurene.
6.2 Photocatalytic CO2 reduction reaction and the N2 reduction reaction
Photocatalytic reduction of CO2 and N2 provides a valuable path to effectively mitigate the greenhouse effect as well as produce useful chemical fuels or high-value-adding chemicals. Elemental 2D materials have been studied as high-performance photocatalysts for the CO2RR and NRR. For example, the BP-based composite has demonstrated excellent performance as a photocatalyst in photocatalytic CO2RR.222 The LPE-derived BP could uniformly self-assemble with the cubic perovskite nanocrystal CsPbBr3 to form a heterostructure photocatalyst (Fig. 10a). As illustrated in Fig. 10b and c, the hybrid photocatalyst possessed good cycling performance and enhanced performance with high conversion rates of CO2 to CO and CH4 at 44.7 and 10.7 μmol g−1 h−1, compared with the pure CsPbBr3 nanocrystals or BP nanosheets. The high performance is ascribed to the efficient charge separation at the CsPbBr3/BP interface that significantly reduced the recombination of photoinduced electrons and holes. Also, the introduction of BP provided more active sites, which facilitated the charge transfer kinetics of CO2 reduction. In another work, the hydroxyl group was utilized to modify monolayer BP to enhance the electrochemical ability for the CO2RR.223 Isopropanol solution was added as a solvent for ultrasonication to add hydroxy functional groups to BP to enhance its stability. The hydroxyl-modified BP showed stable photocatalysis to reduce CO2 to CO under visible light, with an impressive CO production rate of 112.6 μmol h−1 g−1. | ||
| Fig. 10 Photocatalytic CO2RR based on the heterostructure of the cubic perovskite nanocrystal (CsPbBr3) assembled on BP (CsPbBr3/BP):212 (a) schematic illustration of the synthesis of CsPbBr3/BP. (b) The production of CH4 (blue) and CO (red) in ethyl acetate with 0.4 vol% H2O solvents after 3 h based on CsPbBr3/BP with different weight ratios of BP. (c) Cycle stability of photocatalytic CO2 reduction under light irradiation based on CsPbBr3/BP. Reproduced with permission,212 Elsevier. | ||
Interestingly, the aforementioned Te NS-ACS composite could also serve as a photocatalyst for efficient photocatalytic CO2RR producing 104.5 μmol of CO within two hours.220 The functionalization of ACS introduces numerous intermediate energy states, trapping photoinduced electrons, which further suppresses the recombination of photogenerated carriers and contributes to high photocatalytic performance.
Most recently, elemental 2D materials have also found their use in photocatalytic nitrogen fixation. For example, the 2D BP/CdS hybrid is reported as an efficient photocatalyst for the NRR,224 showing a photocurrent density of 13.58 μA cm−2, which is much higher than that of pristine CdS (6.79 μA cm−2). H2 production was negligible, which demonstrates the high selectivity of CdS/2D BP photocatalyst for N2 reduction. The DFT calculations revealed that BP nanosheets could extract electrons from adjacent CdS, which suppresses photoinduced electron–hole recombination and promotes photocatalytic performance.
6.3 Photocatalytic applications in environmental fields
Elemental 2D materials also show potential as metal-free photocatalysts for photoinduced H2O2 production and water purification. A heterostructure photocatalyst based on the hybrid of BP and graphitic carbon nitride (g-C3N4) was reported recently.225 The composite exhibited excellent photocatalytic performance for light-induced superoxide radical (˙O2−) evolution and consequently showed an elevated ability in rhodamine B (RhB) degradation. The degradation rates of RhB after 15 min photoreaction for 10% BP/C3N4, C3N4, and BP nanosheets were 98%, 55%, and 2%, respectively. The enhanced performance of BP/g-C3N4 could be attributed to the visible light adsorption of BP and high charge separation efficiency at the heterojunction interface. Remarkably, the photocatalytic H2O2 generation rate of BP/g-C3N4 reached 3463 μmol h−1 g−1, which was about 14.5 times higher than that of bulk g-C3N4.7. Application in solar cells
Solar cells, which can turn unlimited solar energy into electric energy, have been developed and utilized practically since the 1970s. The most recent development in nanomaterials, especially 2D materials, provides enormous opportunities for high-performance solar cells beyond conventional bulk materials. Notably, some elemental 2D materials with high carrier mobility and tunable band gaps are considered to be promising candidates for solar cells.Many theoretical studies have investigated the potential of elemental 2D materials in solar cells.226 For example, Hu et al. constructed the heterostructure consisting of hydrogen atom or fluorine atom-modified phosphorene nanoflakes and studied its optoelectronic properties.227 Based on calculation result, the heterojunction is predicted to exhibit a high energy conversion efficiency of 20%, making them suitable for high efficiency and low-cost solar cells. Additionally, tellurene possesses a suitable band gap, high carrier mobility and strong light absorption, which make it promising for solar cells.228 By using first-principles density functional theory (DFT) calculations, Gao et al. demonstrated that the heterojunction constructed from halogen-modified zigzag tellurene possesses desirable type 2 band alignments, small band offsets and reduced band gaps for enhanced sunlight absorption, resulting in a high power-conversion efficiency (PCE) of 22.6% in heterojunction solar cells.
Experimental investigation of elemental 2D materials for solar cells has also been carried out recently. LPE-derived antimonene quantum dots were applied as effective photoactive materials in solid-state solar cells.229 Antimonene has good optical absorption due to its approximately 1.5 eV band gap, which is close to ideal for a solar absorber, along with high light stability. The fabricated solar cells exhibited an encouraging PCE of 3.07% in the forward and reverse scan modes (without employing any interfacial optimization) and good stability with 90% of the initial PCE maintained after 1000 hours of operation. Besides, monolayer antimonene nanosheets are ideal for the hole extraction layer in planar inverted perovskite solar cells.230 And it can effectively improve the PCE of perovskite solar cells from 17.58% to 20.05%, due to fast hole extraction and efficient hole transfer at the perovskite/hole transport layer interface.
The various applications of elemental 2D materials in energy fields are summarized in Table 2. From Table 2, one can see that though phosphorene, bismuthene and antimonene are well studied in the energy storage and conversation fields to date; research on materials such as gallinene and plumbene is still in its infancy stage. Furthermore, most of the research of moderately well-studied elemental 2D materials (arsenene, germanene, and stanene) heavily lean on battery applications, indicating extensive opportunities to explore other energy applications.
| Emerging elemental 2D materials | Typical materials | |||
|---|---|---|---|---|
| a LIB is the Li-ion battery; SIB is the Na-ion battery; LSB is the Li–sulfur battery; HER is the hydrogen evolution reaction, OER is the oxygen evolution reaction; CO2RR is the CO2 reduction reaction, and NRR is the nitrogen reduction reaction. Th. is short for theory and Exp. is short for experiment. | ||||
| Batteries (mA h−1 g−1) | LIB | Th. | B: 1860,158 Si: 954,160 Ge: 369,160 Sn: 226,160 As: 1609 (ref. 161) | Graphite: 372,231 MoS2: 334,232 Ti3C2: 320 (ref. 233) |
| Exp. | BP: 920@0.1 A g−1,163 Sb: 584@0.5C (ref. 165) | MoS2: 912@1C (ref. 234) | ||
| SIB | Th. | As: 1073 (ref. 161) | MoS2: 670 (ref. 235) | |
| Exp. | BP: 2440@0.05 A g−1,169 Sb: 642@0.1C,74 Bi: 381@0.2 mA cm−2,45 I: 250@0.1 A g−1 (ref. 51) | Graphite: 284@20 mA g−1,236 MXene: 267@0.1 A g−1,237 MoS2: from 190 to 85 after 100 cycle238 | ||
| Others | BP and borophene show potential as a good host of the cathode material for LSB.171,173 Zn–Te battery exhibits a high capacity of 419 mA h−1 g−1@0.05 A g−1 (ref. 174) | |||
| Supercapacitor (F g−1) | BP: 478@1 A g−1,177 Sb: 1578@14 A g−1 (ref. 180) | MoS2: 100@1 mV s−1,239 Ti3C2: 245@2 mV s−1 (ref. 240) | ||
| Electrocatalysis (mV vs. RHE)@10 mA cm−2 | HER | BP: 290 in 1 M KOH,241 Sb: 153 in buffer189 | 2H-MoS2: 208 in 0.5 M H2SO4,242 1T-MoS2: 252 in 0.5 M H2SO4,243 WSe2: 245 in 0.5 M H2SO4 (ref. 244) | |
| OER | B: 240 (Th.),190 BP: 430 in 1 M NaOH,245 Sb: 1.56 V in 1 M KOH (ref. 186) | MoS2: 450 in 0.5 M H2SO4,246 Co3O4: 318 in 1 M NaOH (ref. 247) | ||
| ORR | B: 470 (Th.)190 | |||
| NRR | BP: faradaic efficiency of 5.0%@−0.7 V in 0.01 M HCl (ref. 192) | |||
| CO2RR | Bi: faradaic efficiency >95%@−1.5 V (ref. 196) | |||
| Photocatalysis (Exp.) | BP: H2 evolution rate: 21042 μmol h−1 g−1;218 NO removal ratio: 67%;218 CO2RR: CO and CH4 evolution rate of 44.7 and 10.7 μmol g−1 h−1;222 NRR: NH3 evolution rate of 57.64 μmol L−1 h−1 (ref. 224) |
MoS2: H2 evolution rate: 1472 μmol h−1 g−1,248 1T-MoS2: H2 evolution rate: 26000 μmol h−1 g−1 (ref. 249) |
||
| Te: H2 evolution rate: 130.8 μmol h−1;220 CO2RR: 104.5 μmol CO (2 h)220 | ||||
| Solar cell (power conversion efficiency) | Th. | Si: 29%,226 Te: 22.6%,228 BP: 20% (ref. 227) | Bulk GaAs: ∼30% (ref. 250) | |
| Exp. | Sb: 3.07% (ref. 229) | MoS2: 5.23% (ref. 251) | ||
8. Summary and outlook
In the past decade, significant progress has been made in the theoretical and experimental exploration of elemental 2D materials, which immensely expand the scope of the ever-growing 2D materials family. Elemental 2D materials possess many unique properties, including large specific areas, abundant active sites, tunable band gaps and high carrier mobility, showing potential in various energy fields, including batteries, electrocatalysis and photocatalysis. The synthetic difficulties, along with the stability issues, have severely limited the practical applications of elemental 2D materials. While the continuous progress targeting the synthetic challenges has been summarized in previous sections, researchers have recently made great efforts to stabilize emerging elemental 2D materials to expand their applications scope and enhance the performance. For example, BP, as one of the most promising, has been modified in various ways to achieve commendable stability in batteries, electrocatalysis and photocatalysis. Additionally, some elemental 2D materials in groups 15 and 16 (including antimonene, tellurene, arsenene, etc.) exhibit higher mobility than TMD and higher stability than BP, and are more likely to shine in practical applications in the near future.252 For example, tellurene, which can be synthesized on a large scale via the facile hydrothermal method, demonstrates great potential in high-performance cathodes for Zn–Te batteries, as well as in other energy and electronic applications, such as solar cells, photodetectors and gas sensors.93 In addition, restricted by the synthetic approaches, the applications of many emerging elemental 2D materials in groups 13 and 14 still require further investigation.Although immerse advances have been made in the realization and practical application of elemental 2D materials, the rapidly expanding elemental 2D materials are still an emerging area that faces plenty of difficulties. Several critical issues and possible solutions are discussed below.
First of all, we recognize the incredible challenges in the synthesis of elemental 2D materials with high yield and sizeable lateral dimensions. In one respect, some elemental materials, such as silicene and borophene, are impossible to obtain via direct exfoliation of bulky sources due to the strong sp3 hybridized bonds. Synthesis of these elemental 2D materials currently relies on MBE, which is not only demanding in terms of the equipment, reaction conditions and substrates, but is also impractical for large-scale applications. In the other respect, traditional widely applied exfoliation methods and other bottom-up methods are also limited by either the crystal quality or product lateral size. To this end, more efforts in developing novel synthetic methods towards high-quality and high-yield production of elemental 2D materials remain the priority in this growing field.
Second, significant knowledge gaps between the theoretical studies and experimental results still challenge the further development of elemental 2D materials for energy applications in terms of structures, properties, and operational mechanisms. First of all, although great progress has been achieved in expanding the elemental 2D material family experimentally, the synthesis of many previously predicted structures, such as aluminene (2D aluminum),49 indiene (2D indium),50 few-layer graphdiyne,253 penta-graphene64 and green phosphorene,254 still necessitates more effort. Additionally, there are some crucial discrepancies about the band structures of elemental 2D materials between the predicted and experiment results, such as black arsenene and tellurene. Moreover, despite the fact that abundant theoretical results have revealed a variety of potential energy applications for various elemental 2D materials, experimental realization calls for further attempts. For example, borophene is considered to be a promising electrode material for Li–S batteries. However, it has not been experimentally verified yet until now.
Third, the heterostructures of elemental 2D materials and other nanomaterials ranging from 2D materials to 0D nanoparticles could significantly expand the capability of elemental 2D materials. The heterostructure strategy could provide more new properties inaccessible to the individual components due to various synergistic effects. For example, the aforementioned Ru–S–Sb/antimonene189 and CsPbBr3/black222 heterostructures have shown improved catalytic performance compared with the individual components. Specifically, 2D van der Waals heterojunctions (vdWHs) have invoked substantial interest due to their ability to tailor the electronic properties of 2D structures through interfacial coupling,255 which could effectively optimize performance in energy applications. For example, the BP g-C3N4 2D/2D van der Waals heterostructure reduced photoinduced carrier recombination and exhibited enhanced photocatalysis performance compared to the pure BP nanosheets or g-C3N4 nanosheets.256
Fourth, the achievement in the synthetic effort of emerging elemental 2D materials calls for further exploration in novel energy applications beyond electrochemistry. The large surface area, tunable band structures and diverse interfacial properties could shine in alternative energy conversion technologies such as solar-thermal clean water production219,220 or electrokinetic power generation.221,222
Last but not least, the development of novel characterization techniques could offer new perspectives in the investigation of emerging nanomaterials, including elemental 2D materials, which is especially beneficial for the mechanism study in energy applications. Recently, a variety of advanced characterization techniques have been incorporated in the investigation of energy storage and photo-/electro-catalysts, such as in situ Raman spectroscopy,257,258in situ XRD spectra,259 cryogenic transmission electron microscopy,260,261in situ transmission electron microscopy,262,263 and many more. Remarkably, the recently developed on-chip electrochemical approach4,264,265 is particularly suitable for the electrochemical applications of 2D electrocatalysts with a limited size, yield and stability, as well as diverse crystal phases,266,267 which could significantly expand the exploration of elemental 2D materials and their heterostructures in energy applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Q. H. acknowledges the support from the Start-Up Grant (project no. 7200656 and 9610482) and Grants (project no. 7005468) from the City University of Hong Kong.References
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed
.
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G.-H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed
- K. Zhang, Y. Feng, F. Wang, Z. Yang and J. Wang, J. Mater. Chem. C, 2017, 5, 11992–12022 RSC
- Y. He, Q. He, L. Wang, C. Zhu, P. Golani, A. D. Handoko, X. Yu, C. Gao, M. Ding, X. Wang, F. Liu, Q. Zeng, P. Yu, S. Guo, B. I. Yakobson, L. Wang, Z. W. Seh, Z. Zhang, M. Wu, Q. J. Wang, H. Zhang and Z. Liu, Nat. Mater., 2019, 18, 1098–1104 CrossRef CAS PubMed
- A. J. Mannix, B. Kiraly, M. C. Hersam and N. P. Guisinger, Nat. Rev. Chem., 2017, 1, 0014 CrossRef CAS
- W. Tao, N. Kong, X. Ji, Y. Zhang, A. Sharma, J. Ouyang, B. Qi, J. Wang, N. Xie, C. Kang, H. Zhang, O. C. Farokhzad and J. S. Kim, Chem. Soc. Rev., 2019, 48, 2891–2912 RSC
- N. R. Glavin, R. Rao, V. Varshney, E. Bianco, A. Apte, A. Roy, E. Ringe and P. M. Ajayan, Adv. Mater., 2020, 32, 1904302 CrossRef CAS PubMed
- Z. Lin, C. Wang and Y. Chai, Small, 2020, 16, 2003319 CrossRef CAS PubMed
- Z. Meng, J. Zhuang, X. Xu, W. Hao, S. X. Dou and Y. Du, Adv. Mater. Interfaces, 2018, 5, 1800749 CrossRef
- M. Qiu, Z. T. Sun, D. K. Sang, X. G. Han, H. Zhang and C. M. Niu, Nanoscale, 2017, 9, 13384–13403 RSC
- Y. Xu, Z. Shi, X. Shi, K. Zhang and H. Zhang, Nanoscale, 2019, 11, 14491–14527 RSC
- T. Wang, H. Wang, Z. Kou, W. Liang, X. Luo, F. Verpoort, Y.-J. Zeng and H. Zhang, Adv. Funct. Mater., 2020, 30, 2002885 CrossRef CAS
- M. Z. Mohamad Nasir and M. Pumera, TrAC, Trends Anal. Chem., 2019, 121, 115696 CrossRef CAS
- N. Antonatos, H. Ghodrati and Z. Sofer, Appl. Mater. Today, 2020, 18, 100502 CrossRef
- A. J. Mannix, X.-F. Zhou, B. Kiraly, J. D. Wood, D. Alducin, B. D. Myers, X. Liu, B. L. Fisher, U. Santiago, J. R. Guest, M. J. Yacaman, A. Ponce, A. R. Oganov, M. C. Hersam and N. P. Guisinger, Science, 2015, 350, 1513–1516 CrossRef CAS PubMed
-
B. Feng, L. Chen and K. Wu, in 2D Boron: Boraphene, Borophene, Boronene, 2021, ch. 3, pp. 51–72, DOI:10.1007/978-3-030-49999-0_3
- M.-L. Tao, Y.-B. Tu, K. Sun, Y.-L. Wang, Z.-B. Xie, L. Liu, M.-X. Shi and J.-Z. Wang, 2D Mater., 2018, 5, 035009 CrossRef
- G. Tai, T. Hu, Y. Zhou, X. Wang, J. Kong, T. Zeng, Y. You and Q. Wang, Angew. Chem., Int. Ed., 2015, 127, 15693–15697 CrossRef
- S. Zhang, Z. Yan, Y. Li, Z. Chen and H. Zeng, Angew. Chem., Int. Ed., 2015, 54, 3112–3115 CrossRef CAS PubMed
- B. Feng, J. Zhang, Q. Zhong, W. Li, S. Li, H. Li, P. Cheng, S. Meng, L. Chen and K. Wu, Nat. Chem., 2016, 8, 563–568 CrossRef CAS PubMed
- S. Xu, Y. Zhao, J. Liao, X. Yang and H. Xu, Nano Res., 2016, 9, 2616–2622 CrossRef CAS
- Q. Zhong, L. Kong, J. Gou, W. Li, S. Sheng, S. Yang, P. Cheng, H. Li, K. Wu and L. Chen, Phys. Rev. Mater., 2017, 1, 021001 CrossRef
- Q. Zhong, J. Zhang, P. Cheng, B. Feng, W. Li, S. Sheng, H. Li, S. Meng, L. Chen and K. Wu, J. Phys.: Condens. Matter, 2017, 29, 095002 CrossRef PubMed
- G. P. Campbell, A. J. Mannix, J. D. Emery, T.-L. Lee, N. P. Guisinger, M. C. Hersam and M. J. Bedzyk, Nano Lett., 2018, 18, 2816–2821 CrossRef CAS PubMed
- X. Ji, N. Kong, J. Wang, W. Li, Y. Xiao, S. T. Gan, Y. Zhang, Y. Li, X. Song and Q. Xiong, Adv. Mater., 2018, 30, 1803031 CrossRef PubMed
- W. Li, L. Kong, C. Chen, J. Gou, S. Sheng, W. Zhang, H. Li, L. Chen, P. Cheng and K. Wu, Sci. Bull., 2018, 63, 282–286 CrossRef CAS
- B. Kiraly, X. Liu, L. Wang, Z. Zhang, A. J. Mannix, B. L. Fisher, B. I. Yakobson, M. C. Hersam and N. P. Guisinger, ACS Nano, 2019, 13, 3816–3822 CrossRef CAS PubMed
- N. A. Vinogradov, A. Lyalin, T. Taketsugu, A. S. Vinogradov and A. Preobrajenski, ACS Nano, 2019, 13, 14511–14518 CrossRef CAS PubMed
- R. Wu, I. K. Drozdov, S. Eltinge, P. Zahl, S. Ismail-Beigi, I. Božović and A. Gozar, Nat. Nanotechnol., 2019, 14, 44–49 CrossRef CAS PubMed
- R. Wu, A. Gozar and I. Božović, npj Quantum Mater., 2019, 4, 40 CrossRef
- H. Liu, J. Gao and J. Zhao, J. Phys. Chem. C, 2013, 117, 10353–10359 CrossRef CAS
- L. Zhang, H. Huang, B. Zhang, M. Gu, D. Zhao, X. Zhao, L. Li, J. Zhou, K. Wu, Y. Cheng and J. Zhang, Angew. Chem., Int. Ed., 2020, 59, 1074–1080 CrossRef CAS PubMed
- W. Zhang, H. Enriquez, Y. Tong, A. Bendounan, A. Kara, A. P. Seitsonen, A. J. Mayne, G. Dujardin and H. Oughaddou, Small, 2018, 14, 1804066 CrossRef PubMed
- A. E. Del Rio Castillo, V. Pellegrini, H. Sun, J. Buha, D. A. Dinh, E. Lago, A. Ansaldo, A. Capasso, L. Manna and F. Bonaccorso, Chem. Mater., 2018, 30, 506–516 CrossRef CAS
- R. Feng, W. Lei, G. Liu and M. Liu, Adv. Mater., 2018, 30, 1804770 CrossRef PubMed
- W. Huang, C. Li, L. Gao, Y. Zhang, Y. Wang, Z. N. Huang, T. Chen, L. Hu and H. Zhang, J. Mater. Chem. C, 2020, 8, 1172–1197 RSC
- M. J. Cherukara, B. Narayanan, H. Chan and S. K. Sankaranarayanan, Nanoscale, 2017, 9, 10186–10192 RSC
- J. Yuhara, H. Shimazu, K. Ito, A. Ohta, M. Araidai, M. Kurosawa, M. Nakatake and G. Le Lay, ACS Nano, 2018, 12, 11632–11637 CrossRef CAS PubMed
- J. Yuhara, Y. Fujii, K. Nishino, N. Isobe, M. Nakatake, L. Xian, A. Rubio and G. Le Lay, 2D Mater., 2018, 5, 025002 CrossRef
- J. Yuhara, B. He, N. Matsunami, M. Nakatake and G. Le Lay, Adv. Mater., 2019, 31, 1901017 CrossRef PubMed
- Z. Wang, X.-F. Zhou, X. Zhang, Q. Zhu, H. Dong, M. Zhao and A. R. Oganov, Nano Lett., 2015, 15, 6182–6186 CrossRef CAS PubMed
- S. Zhang, J. Zhou, Q. Wang, X. Chen, Y. Kawazoe and P. Jena, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2372–2377 CrossRef CAS PubMed
- Y. Zhang, L. Wang, H. Xu, J. Cao, D. Chen and W. Han, Adv. Funct. Mater., 2020, 30, 1909372 CrossRef CAS
- X. Wang, J. He, B. Zhou, Y. Zhang, J. Wu, R. Hu, L. Liu, J. Song and J. Qu, Angew. Chem., Int. Ed., 2018, 130, 8804–8809 CrossRef
- J. Zhou, J. Chen, M. Chen, J. Wang, X. Liu, B. Wei, Z. Wang, J. Li, L. Gu, Q. Zhang, H. Wang and L. Guo, Adv. Mater., 2019, 31, 1807874 CrossRef PubMed
- S. M. Beladi-Mousavi, A. M. Pourrahimi, Z. Sofer and M. Pumera, Adv. Funct. Mater., 2019, 29, 1807004 Search PubMed
- J. Chen, Y. Dai, Y. Ma, X. Dai, W. Ho and M. Xie, Nanoscale, 2017, 9, 15945–15948 RSC
- J. Qin, G. Qiu, J. Jian, H. Zhou, L. Yang, A. Charnas, D. Y. Zemlyanov, C.-Y. Xu, X. Xu, W. Wu, H. Wang and P. D. Ye, ACS Nano, 2017, 11, 10222–10229 CrossRef CAS PubMed
- J. Yuan, N. Yu, K. Xue and X. Miao, Appl. Surf. Sci., 2017, 409, 85–90 CrossRef CAS
- D. Singh, S. K. Gupta, I. Lukačević and Y. Sonvane, RSC Adv., 2016, 6, 8006–8014 RSC
- M. Qian, Z. Xu, Z. Wang, B. Wei, H. Wang, S. Hu, L.-M. Liu and L. Guo, Adv. Mater., 2020, 32, 2004835 CrossRef CAS PubMed
- V. Kochat, A. Samanta, Y. Zhang, S. Bhowmick, P. Manimunda, S. A. S. Asif, A. S. Stender, R. Vajtai, A. K. Singh, C. S. Tiwary and P. M. Ajayan, Sci. Adv., 2018, 4, e1701373 CrossRef PubMed
- A. Molle, J. Goldberger, M. Houssa, Y. Xu, S.-C. Zhang and D. J. N. m. Akinwande, Nat. Mater., 2017, 16, 163–169 CrossRef CAS PubMed
- J. Deng, B. Xia, X. Ma, H. Chen, H. Shan, X. Zhai, B. Li, A. Zhao, Y. Xu and W. Duan, Nat. Mater., 2018, 17, 1081–1086 CrossRef CAS PubMed
- S. Balendhran, S. Walia, H. Nili, S. Sriram and M. Bhaskaran, Small, 2015, 11, 640–652 CrossRef CAS PubMed
- P. Vogt, P. De Padova, C. Quaresima, J. Avila, E. Frantzeskakis, M. C. Asensio, A. Resta, B. Ealet and G. Le Lay, Phys. Rev. Lett., 2012, 108, 155501 CrossRef PubMed
- P. Rivero, J.-A. Yan, V. M. García-Suárez, J. Ferrer and S. Barraza-Lopez, Phys. Rev. B, 2014, 90, 241408 CrossRef CAS
- F. Matusalem, M. Marques, L. K. Teles and F. Bechstedt, Phys. Rev. B, 2015, 92, 045436 CrossRef
- D. Wang, L. Chen, X. Wang, G. Cui and P. Zhang, Phys. Chem. Chem. Phys., 2015, 17, 26979–26987 RSC
- J. C. Garcia, D. B. De Lima, L. V. Assali and J. F. Justo, Phys. Chem. Chem. Phys., 2011, 115, 13242–13246 CrossRef CAS
- X. Fan, J. Li and G. Chen, RSC Adv., 2017, 7, 17417–17426 RSC
- G. Long, Y. Zhou, M. Jin, B. Kan, Y. Zhao, A. Gray-Weale, D.-e. Jiang, Y. Chen and Q. Zhang, Carbon, 2015, 95, 1033–1038 CrossRef CAS
- J.-W. Jiang, J. Leng, J. Li, Z. Guo, T. Chang, X. Guo and T. Zhang, Carbon, 2017, 118, 370–375 CrossRef CAS
- Y. Aierken, O. Leenaerts and F. M. Peeters, Phys. Rev. B, 2016, 94, 155410 CrossRef
- D. Wu, S. Wang, J. Yuan, B. Yang and H. Chen, Phys. Chem. Chem. Phys., 2017, 19, 11771–11777 RSC
- X. Wang, A. M. Jones, K. L. Seyler, V. Tran, Y. Jia, H. Zhao, H. Wang, L. Yang, X. Xu and F. Xia, Nat. Nanotechnol., 2015, 10, 517–521 CrossRef CAS PubMed
- C. Zhang, X. Peng, Z. Guo, C. Cai, Z. Chen, D. Wexler, S. Li and H. Liu, Carbon, 2012, 50, 1897–1903 CrossRef CAS
- J. Pang, A. Bachmatiuk, Y. Yin, B. Trzebicka, L. Zhao, L. Fu, R. G. Mendes, T. Gemming, Z. Liu and M. H. Rummeli, Adv. Energy Mater., 2018, 8, 1702093 CrossRef
- Q. He, Y. Liu, C. Tan, W. Zhai, G.-h. Nam and H. Zhang, ACS Nano, 2019, 13, 12294–12300 CrossRef CAS PubMed
- J. Qiao, X. Kong, Z.-X. Hu, F. Yang and W. Ji, Nat. Commun., 2014, 5, 4475 CrossRef CAS PubMed
- X. Zhu, T. Zhang, Z. Sun, H. Chen, J. Guan, X. Chen, H. Ji, P. Du and S. Yang, Adv. Mater., 2017, 29, 1605776 CrossRef PubMed
- A. Favron, E. Gaufrès, F. Fossard, A.-L. Phaneuf-L'Heureux, N. Y. W. Tang, P. L. Lévesque, A. Loiseau, R. Leonelli, S. Francoeur and R. Martel, Nat. Mater., 2015, 14, 826–832 CrossRef CAS PubMed
- G. Wang, W. J. Slough, R. Pandey and S. P. Karna, 2D Mater., 2016, 3, 025011 CrossRef
- W. Tian, S. Zhang, C. Huo, D. Zhu, Q. Li, L. Wang, X. Ren, L. Xie, S. Guo, P. K. Chu, H. Zeng and K. Huo, ACS Nano, 2018, 12, 1887–1893 CrossRef CAS PubMed
- X. Wu, Y. Shao, H. Liu, Z. Feng, Y.-L. Wang, J.-T. Sun, C. Liu, J.-O. Wang, Z.-L. Liu, S.-Y. Zhu, Y.-Q. Wang, S.-X. Du, Y.-G. Shi, K. Ibrahim and H.-J. Gao, Adv. Mater., 2017, 29, 1605407 CrossRef PubMed
- Q. Wu and Y. J. Song, Chem. Commun., 2018, 54, 9671–9674 RSC
- S. Zhang, M. Xie, F. Li, Z. Yan, Y. Li, E. Kan, W. Liu, Z. Chen and H. Zeng, Angew. Chem., Int. Ed., 2016, 128, 1698–1701 CrossRef
- T. Chai, X. Li, T. Feng, P. Guo, Y. Song, Y. Chen and H. Zhang, Nanoscale, 2018, 10, 17617–17622 RSC
- E. S. Walker, S. R. Na, D. Jung, S. D. March, J.-S. Kim, T. Trivedi, W. Li, L. Tao, M. L. Lee, K. M. Liechti, D. Akinwande and S. R. Bank, Nano Lett., 2016, 16, 6931–6938 CrossRef CAS PubMed
- M. Zhong, Q. Xia, L. Pan, Y. Liu, Y. Chen, H.-X. Deng, J. Li and Z. Wei, Adv. Funct. Mater., 2018, 28, 1802581 CrossRef
- C. Kamal and M. Ezawa, Phys. Rev. B, 2015, 91, 085423 CrossRef
- P. Vishnoi, M. Mazumder, S. K. Pati and C. N. R. Rao, New J. Chem., 2018, 42, 14091–14095 RSC
- Y.-p. Wang, C.-w. Zhang, W.-x. Ji, R.-w. Zhang, P. Li, P.-j. Wang, M.-j. Ren, X.-l. Chen and M. Yuan, J. Phys. D: Appl. Phys., 2016, 49, 055305 CrossRef
- Y. Hu, Z.-H. Qi, J. Lu, R. Chen, M. Zou, T. Chen, W. Zhang, Y. Wang, X. Xue, J. Ma and Z. Jin, Chem. Mater., 2019, 31, 4524–4535 CrossRef CAS
- J. Carrete, L. J. Gallego and N. Mingo, J. Phys. Chem. Lett., 2017, 8, 1375–1380 CrossRef CAS PubMed
- Y. Chen, C. Chen, R. Kealhofer, H. Liu, Z. Yuan, L. Jiang, J. Suh, J. Park, C. Ko, H. S. Choe, J. Avila, M. Zhong, Z. Wei, J. Li, S. Li, H. Gao, Y. Liu, J. Analytis, Q. Xia, M. C. Asensio and J. Wu, Adv. Mater., 2018, 30, 1800754 CrossRef PubMed
- D. Guo, B. Shao, C. Li and Y. Ma, Superlattices Microstruct., 2016, 100, 324–334 CrossRef CAS
- B. Peng, H. Zhang, H. Shao, K. Xu, G. Ni, J. Li, H. Zhu and C. M. Soukoulis, J. Mater. Chem. A, 2018, 6, 2018–2033 RSC
- Y. Wang, G. Qiu, R. Wang, S. Huang, Q. Wang, Y. Liu, Y. Du, W. A. Goddard, M. J. Kim, X. Xu, P. D. Ye and W. Wu, Nat. Electron., 2018, 1, 228–236 CrossRef
- X. Huang, J. Guan, Z. Lin, B. Liu, S. Xing, W. Wang and J. Guo, Nano Lett., 2017, 17, 4619–4623 CrossRef CAS PubMed
- J. Qiao, Y. Pan, F. Yang, C. Wang, Y. Chai and W. Ji, Sci. Bull., 2018, 63, 159–168 CrossRef CAS
- C. Shen, Y. Liu, J. Wu, C. Xu, D. Cui, Z. Li, Q. Liu, Y. Li, Y. Wang, X. Cao, H. Kumazoe, F. Shimojo, A. Krishnamoorthy, R. K. Kalia, A. Nakano, P. D. Vashishta, M. R. Amer, A. N. Abbas, H. Wang, W. Wu and C. Zhou, ACS Nano, 2020, 14, 303–310 CrossRef CAS PubMed
- W. Wu, G. Qiu, Y. Wang, R. Wang and P. Ye, Chem. Soc. Rev., 2018, 47, 7203–7212 RSC
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth and S. Roth, Nature, 2007, 446, 60–63 CrossRef CAS PubMed
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372–377 CrossRef CAS PubMed
- P. Ares, F. Aguilar-Galindo, D. Rodríguez-San-Miguel, D. A. Aldave, S. Díaz-Tendero, M. Alcamí, F. Martín, J. Gómez-Herrero and F. Zamora, Adv. Mater., 2016, 28, 6332–6336 CrossRef CAS PubMed
- C. Sabater, D. Gosálbez-Martínez, J. Fernández-Rossier, J. G. Rodrigo, C. Untiedt and J. J. Palacios, Phys. Rev. Lett., 2013, 110, 176802 CrossRef CAS PubMed
- Y. Huang, E. Sutter, N. N. Shi, J. Zheng, T. Yang, D. Englund, H.-J. Gao and P. Sutter, ACS Nano, 2015, 9, 10612–10620 CrossRef CAS PubMed
- L. Guan, B. Xing, X. Niu, D. Wang, Y. Yu, S. Zhang, X. Yan, Y. Wang and J. Sha, Chem. Commun., 2018, 54, 595–598 RSC
- P. Yasaei, B. Kumar, T. Foroozan, C. Wang, M. Asadi, D. Tuschel, J. E. Indacochea, R. F. Klie and A. Salehi-Khojin, Adv. Mater., 2015, 27, 1887–1892 CrossRef CAS PubMed
- W. Zheng, J. Lee, Z.-W. Gao, Y. Li, S. Lin, S. P. Lau and L. Y. S. Lee, Adv. Energy Mater., 2020, 10, 1903490 CrossRef CAS
- J. Kang, J. D. Wood, S. A. Wells, J.-H. Lee, X. Liu, K.-S. Chen and M. C. Hersam, ACS Nano, 2015, 9, 3596–3604 CrossRef CAS PubMed
- A. O'Neill, U. Khan, P. N. Nirmalraj, J. Boland and J. N. Coleman, J. Phys. Chem. C, 2011, 115, 5422–5428 CrossRef
- C. Gibaja, M. Assebban, I. Torres, M. Fickert, R. Sanchis-Gual, I. Brotons, W. S. Paz, J. J. Palacios, E. G. Michel, G. Abellán and F. Zamora, J. Mater. Chem. A, 2019, 7, 22475–22486 RSC
- F. Liu, C. Wang, X. Sui, M. A. Riaz, M. Xu, L. Wei and Y. Chen, Carbon Energy, 2019, 1, 173–199 CrossRef
- N. Liu, P. Kim, J. H. Kim, J. H. Ye, S. Kim and C. J. Lee, ACS Nano, 2014, 8, 6902–6910 CrossRef CAS PubMed
- C. Shen, T. Cheng, C. Liu, L. Huang, M. Cao, G. Song, D. Wang, B. Lu, J. Wang, C. Qin, X. Huang, P. Peng, X. Li and Y. Wu, J. Mater. Chem. A, 2020, 8, 453–460 RSC
- J. Li, P. Song, J. Zhao, K. Vaklinova, X. Zhao, Z. Li, Z. Qiu, Z. Wang, L. Lin, M. Zhao, T. S. Herng, Y. Zuo, W. Jonhson, W. Yu, X. Hai, P. Lyu, H. Xu, H. Yang, C. Chen, S. J. Pennycook, J. Ding, J. Teng, A. H. Castro Neto, K. S. Novoselov and J. Lu, Nat. Mater., 2021, 20, 181–187 CrossRef CAS PubMed
- C. Wang, Q. He, U. Halim, Y. Liu, E. Zhu, Z. Lin, H. Xiao, X. Duan, Z. Feng, R. Cheng, N. O. Weiss, G. Ye, Y.-C. Huang, H. Wu, H.-C. Cheng, I. Shakir, L. Liao, X. Chen, W. A. Goddard III, Y. Huang and X. Duan, Nature, 2018, 555, 231–236 CrossRef CAS PubMed
- A. Ambrosi, Z. Sofer and M. Pumera, Angew. Chem., Int. Ed., 2017, 56, 10443–10445 CrossRef CAS PubMed
- M. B. Erande, S. R. Suryawanshi, M. A. More and D. J. Late, Eur. J. Inorg. Chem., 2015, 2015, 3102–3107 CrossRef CAS
- Z. Huang, H. Hou, Y. Zhang, C. Wang, X. Qiu and X. Ji, Adv. Mater., 2017, 29, 1702372 CrossRef PubMed
- Z. Zeng, Z. Yin, X. Huang, H. Li, Q. He, G. Lu, F. Boey and H. Zhang, Angew. Chem., Int. Ed., 2011, 123, 11289–11293 CrossRef
- X. Tang, L. Hu, T. Fan, L. Zhang, L. Zhu, H. Li, H. Liu, J. Liang, K. Wang, Z. Li, S. Ruan, Y. Zhang, D. Fan, W. Chen, Y.-J. Zeng and H. Zhang, Adv. Funct. Mater., 2019, 29, 1808746 CrossRef
- Y. Yang, S. Leng and W. Shi, Electrochem. Commun., 2021, 126, 107025 CrossRef CAS
- A. Lherbier, A. R. Botello-Méndez and J.-C. Charlier, 2D Mater., 2016, 3, 045006 CrossRef
- T. Niu, W. Zhou, D. Zhou, X. Hu, S. Zhang, K. Zhang, M. Zhou, H. Fuchs and H. Zeng, Adv. Mater., 2019, 31, 1902606 CrossRef PubMed
- T. Paulauskas, F. G. Sen, C. Sun, P. Longo, Y. Zhang, S. W. Hla, M. K. Y. Chan, M. J. Kim and R. F. Klie, Nanoscale, 2019, 11, 14698–14706 RSC
- Z. Y. Meng, T. C. Lang, S. Wessel, F. F. Assaad and A. Muramatsu, Nature, 2010, 464, 847–851 CrossRef CAS PubMed
- A. M. Attaran, S. Abdol-Manafi, M. Javanbakht and M. Enhessari, J. Nanostruct. Chem., 2016, 6, 121–128 CrossRef
- K. Kawahara, T. Shirasawa, R. Arafune, C.-L. Lin, T. Takahashi, M. Kawai and N. J. S. s. Takagi, Surf. Sci., 2014, 623, 25–28 CrossRef CAS
- N. Takagi, C.-L. Lin, K. Kawahara, E. Minamitani, N. Tsukahara, M. Kawai and R. J. P. i. S. S. Arafune, Appl. Surf. Sci., 2015, 90, 1–20 CAS
- B. Feng, Z. Ding, S. Meng, Y. Yao, X. He, P. Cheng, L. Chen and K. Wu, Nano Lett., 2012, 12, 3507–3511 CrossRef CAS PubMed
- P. De Padova, C. Quaresima, C. Ottaviani, P. M. Sheverdyaeva, P. Moras, C. Carbone, D. Topwal, B. Olivieri, A. Kara and H. J. A. P. L. Oughaddou, Appl. Phys. Lett., 2010, 96, 261905 CrossRef
- D. Chiappe, E. Scalise, E. Cinquanta, C. Grazianetti, B. van den Broek, M. Fanciulli, M. Houssa and A. Molle, Adv. Mater., 2014, 26, 2096–2101 CrossRef CAS PubMed
- A. Fleurence, R. Friedlein, T. Ozaki, H. Kawai, Y. Wang and Y. Yamada-Takamura, Phys. Rev. Lett., 2012, 108, 245501 CrossRef PubMed
- L. Meng, Y. Wang, L. Zhang, S. Du, R. Wu, L. Li, Y. Zhang, G. Li, H. Zhou and W. A. Hofer, Nano Lett., 2013, 13, 685–690 CrossRef CAS PubMed
- H. Enriquez, A. Mayne, A. Kara, S. Vizzini, S. Roth, B. Lalmi, A. P. Seitsonen, B. Aufray, T. Greber, R. Belkhou, G. Dujardin and H. Oughaddou, Appl. Phys. Lett., 2012, 101, 021605 CrossRef
- V. A. Lubarda, Mech. Mater., 2003, 35, 53–68 CrossRef
- K. T. Butler, P. E. Vullum, A. M. Muggerud, E. Cabrera and J. H. Harding, Phys. Rev. B, 2011, 83, 235307 CrossRef
- M. Houssa, G. Pourtois, V. V. Afanas'ev and A. Stesmans, Appl. Phys. Lett., 2010, 97, 112106 CrossRef
- M. Houssa, G. Pourtois, M. M. Heyns, V. V. Afanas'ev and A. Stesmans, J. Electrochem. Soc., 2011, 158, H107 CrossRef CAS
- E. Scalise, M. Houssa, E. Cinquanta, C. Grazianetti, B. van den Broek, G. Pourtois, A. Stesmans, M. Fanciulli and A. Molle, 2D Mater., 2014, 1, 011010 CrossRef CAS
- S. Kokott, P. Pflugradt, L. Matthes and F. Bechstedt, J. Phys.: Condens. Matter, 2014, 26, 185002 CrossRef CAS PubMed
- Y. Ding and Y. Wang, Appl. Phys. Lett., 2013, 103, 043114 CrossRef
- M. E. Dávila, L. Xian, S. Cahangirov, A. Rubio and G. Le Lay, New J. Phys., 2014, 16, 095002 CrossRef
- M. Derivaz, D. Dentel, R. Stephan, M.-C. Hanf, A. Mehdaoui, P. Sonnet and C. Pirri, Nano Lett., 2015, 15, 2510–2516 CrossRef CAS PubMed
- L. Li, S.-z. Lu, J. Pan, Z. Qin, Y.-q. Wang, Y. Wang, G.-y. Cao, S. Du and H.-J. Gao, Adv. Mater., 2014, 26, 4820–4824 CrossRef CAS PubMed
- A. Acun, L. Zhang, P. Bampoulis, M. Farmanbar, A. van Houselt, A. N. Rudenko, M. Lingenfelder, G. Brocks, B. Poelsema, M. I. Katsnelson and H. J. W. Zandvliet, J. Phys.: Condens. Matter, 2015, 27, 443002 CrossRef CAS PubMed
- Z. Qin, J. Pan, S. Lu, Y. Shao, Y. Wang, S. Du, H.-J. Gao and G. Cao, Adv. Mater., 2017, 29, 1606046 CrossRef PubMed
- F.-f. Zhu, W.-j. Chen, Y. Xu, C.-l. Gao, D.-d. Guan, C.-h. Liu, D. Qian, S.-C. Zhang and J.-f. Jia, Nat. Mater., 2015, 14, 1020–1025 CrossRef CAS PubMed
- J. Gou, L. Kong, H. Li, Q. Zhong, W. Li, P. Cheng, L. Chen and K. Wu, Phys. Rev. Mater., 2017, 1, 054004 CrossRef
- A. T. Hoang, K. Qu, X. Chen and J.-H. Ahn, Nanoscale, 2021, 13, 615–633 RSC
- M. Huang, B. Deng, F. Dong, L. Zhang, Z. Zhang and P. Chen, Small Methods, 2021, 5, 2001213 CrossRef
- C. Li, Y. Wu, B. Deng, Y. Xie, Q. Guo, S. Yuan, X. Chen, M. Bhuiyan, Z. Wu, K. Watanabe, T. Taniguchi, H. Wang, J. J. Cha, M. Snure, Y. Fei and F. Xia, Adv. Mater., 2018, 30, 1703748 CrossRef PubMed
- H. Xu, X. Han, Z. Li, W. Liu, X. Li, J. Wu, Z. Guo and H. Liu, Adv. Mater. Interfaces, 2018, 5, 1801048 CrossRef
- X. Zhang, J. Jiang, A. A. Suleiman, B. Jin, X. Hu, X. Zhou and T. Zhai, Adv. Funct. Mater., 2019, 29, 1906585 CrossRef CAS
- G. Yang and S.-J. Park, Materials, 2019, 12, 1177 CrossRef CAS PubMed
- M. Amani, C. Tan, G. Zhang, C. Zhao, J. Bullock, X. Song, H. Kim, V. R. Shrestha, Y. Gao and K. Crozier, ACS Nano, 2018, 12, 7253–7263 CrossRef CAS PubMed
- L. Peng, S. Ye, J. Song and J. Qu, Angew. Chem., Int. Ed., 2019, 58, 9891–9896 CrossRef CAS PubMed
- F. Yang, A. O. Elnabawy, R. Schimmenti, P. Song, J. Wang, Z. Peng, S. Yao, R. Deng, S. Song, Y. Lin, M. Mavrikakis and W. Xu, Nat. Commun., 2020, 11, 1088 CrossRef CAS PubMed
- Y. Wang, S. Yao, P. Liao, S. Jin, Q. Wang, M. J. Kim, G. J. Cheng and W. Wu, Adv. Mater., 2020, 32, 2002342 CrossRef CAS PubMed
- H. Xu, S. Yang and B. Li, J. Mater. Chem. A, 2020, 8, 149–157 RSC
- M. A. Ledina, N. Bui, X. Liang, Y. G. Kim, J. Jung, B. Perdue, C. Tsang, J. Drnec, F. Carlà, M. P. Soriaga, T. J. Reber and J. L. Stickney, J. Electrochem. Soc., 2017, 164, D469–D477 CrossRef CAS
- Z.-Q. Shi, H. Li, Q.-Q. Yuan, Y.-H. Song, Y.-Y. Lv, W. Shi, Z.-Y. Jia, L. Gao, Y.-B. Chen, W. Zhu and S.-C. Li, Adv. Mater., 2019, 31, 1806130 CrossRef PubMed
- S. Khatun, A. Banerjee and A. J. Pal, Nanoscale, 2019, 11, 3591–3598 RSC
- W. Li, Y. Yang, G. Zhang and Y.-W. Zhang, Nano Lett., 2015, 15, 1691–1697 CrossRef CAS PubMed
- H. R. Jiang, Z. Lu, M. C. Wu, F. Ciucci and T. S. Zhao, Nano Energy, 2016, 23, 97–104 CrossRef CAS
- Y. Huang, C. Zhu, S. Zhang, X. Hu, K. Zhang, W. Zhou, S. Guo, F. Xu and H. Zeng, Nano Lett., 2019, 19, 1118–1123 CrossRef PubMed
- B. Mortazavi, A. Dianat, G. Cuniberti and T. Rabczuk, Electrochim. Acta, 2016, 213, 865–870 CrossRef CAS
- H. Benzidi, M. Lakhal, M. Garara, M. Abdellaoui, A. Benyoussef, A. El kenz and O. Mounkachi, Phys. Chem. Chem. Phys., 2019, 21, 19951–19962 RSC
- L.-Q. Sun, M.-J. Li, K. Sun, S.-H. Yu, R.-S. Wang and H.-M. Xie, J. Phys. Chem. C, 2012, 116, 14772–14779 CrossRef CAS
- J. Sun, H.-W. Lee, M. Pasta, H. Yuan, G. Zheng, Y. Sun, Y. Li and Y. Cui, Nat. Nanotechnol., 2015, 10, 980–985 CrossRef CAS PubMed
- Z. Zhang, S. N. Shirodkar, Y. Yang and B. I. Yakobson, Angew. Chem., Int. Ed., 2017, 129, 15623–15628 CrossRef
- Y. Gao, W. Tian, C. Huo, K. Zhang, S. Guo, S. Zhang, X. Song, L. Jiang, K. Huo and H. Zeng, J. Mater. Chem. A, 2019, 7, 3238–3243 RSC
- M. I. Khan, G. Nadeem, A. Majid and M. Shakil, Mater. Sci. Eng., B, 2021, 266, 115061 CrossRef CAS
- Y. Dong, H. Shi and Z.-S. Wu, Adv. Funct. Mater., 2020, 30, 2000706 CrossRef CAS
- H. Liu, K. Hu, D. Yan, R. Chen, Y. Zou, H. Liu and S. Wang, Adv. Mater., 2018, 30, 1800295 CrossRef PubMed
- L. Chen, G. Zhou, Z. Liu, X. Ma, J. Chen, Z. Zhang, X. Ma, F. Li, H.-M. Cheng and W. Ren, Adv. Mater., 2016, 28, 510–517 CrossRef CAS PubMed
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed
- J. Sun, Y. Sun, M. Pasta, G. Zhou, Y. Li, W. Liu, F. Xiong and Y. Cui, Adv. Mater., 2016, 28, 9797–9803 CrossRef CAS PubMed
- F. Li and J. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 42836–42844 CrossRef CAS PubMed
- H. R. Jiang, W. Shyy, M. Liu, Y. X. Ren and T. S. Zhao, J. Mater. Chem. A, 2018, 6, 2107–2114 RSC
- Z. Chen, Q. Yang, F. Mo, N. Li, G. Liang, X. Li, Z. Huang, D. Wang, W. Huang, J. Fan and C. Zhi, Adv. Mater., 2020, 32, 2001469 CrossRef CAS PubMed
- W. Raza, F. Ali, N. Raza, Y. Luo, K.-H. Kim, J. Yang, S. Kumar, A. Mehmood and E. E. Kwon, Nano Energy, 2018, 52, 441–473 CrossRef CAS
- S. Luo, J. Zhao, J. Zou, Z. He, C. Xu, F. Liu, Y. Huang, L. Dong, L. Wang and H. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 3538–3548 CrossRef CAS PubMed
- P. Nakhanivej, X. Yu, S. K. Park, S. Kim, J.-Y. Hong, H. J. Kim, W. Lee, J. Y. Hwang, J. E. Yang, C. Wolverton, J. Kong, M. Chhowalla and H. S. Park, Nat. Mater., 2019, 18, 156–162 CrossRef CAS PubMed
- M. Ghidiu, M. R. Lukatskaya, M.-Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CrossRef CAS PubMed
- J. Yu, J. Zhou, P. Yao, H. Xie, M. Zhang, M. Ji, H. Liu, Q. Liu, C. Zhu and J. Xu, Adv. Energy Mater., 2019, 9, 1902462 CrossRef CAS
- E. Martínez-Periñán, M. P. Down, C. Gibaja, E. Lorenzo, F. Zamora and C. E. Banks, Adv. Energy Mater., 2018, 8, 1702606 CrossRef
- M. Mohamed Ismail, J. Vigneshwaran, S. Arunbalaji, D. Mani, M. Arivanandhan, S. P. Jose and R. Jayavel, Dalton Trans., 2020, 49, 13717–13725 RSC
- V. K. Mariappan, K. Krishnamoorthy, P. Pazhamalai, S. Natarajan, S. Sahoo, S. S. Nardekar and S.-J. Kim, Nano Energy, 2020, 77, 105248 CrossRef CAS
- S. Singal, A. Joshi, G. Singh and R. K. Sharma, J. Power Sources, 2020, 475, 228669 CrossRef CAS
- S. H. Mir, S. Chakraborty, P. C. Jha, J. Wärnå, H. Soni, P. K. Jha and R. Ahuja, Appl. Phys. Lett., 2016, 109, 053903 CrossRef
- L. Shi, C. Ling, Y. Ouyang and J. Wang, Nanoscale, 2017, 9, 533–537 RSC
- X. Ren, Z. Li, H. Qiao, W. Liang, H. Liu, F. Zhang, X. Qi, Y. Liu, Z. Huang, D. Zhang, J. Li, J. Zhong and H. Zhang, ACS Appl. Energy Mater., 2019, 2, 4774–4781 CrossRef CAS
- S. Shen, Y. Gan, X.-x. Xue, J. Wei, L.-m. Tang, K. Chen and Y. Feng, Appl. Phys. Express, 2019, 12, 075502 CrossRef
- Z. Yuan, J. Li, M. Yang, Z. Fang, J. Jian, D. Yu, X. Chen and L. Dai, J. Am. Chem. Soc., 2019, 141, 4972–4979 CrossRef CAS PubMed
- Y. Li, J. Chen, J. Huang, Y. Hou, L. Lei, W. Lin, Y. Lian, X. Zhonghua, H.-H. Yang and Z. Wen, Chem. Commun., 2019, 55, 10884–10887 RSC
- Y. Singh, S. Back and Y. Jung, Phys. Chem. Chem. Phys., 2018, 20, 21095–21104 RSC
- F. Shi, Z. Geng, K. Huang, Q. Liang, Y. Zhang, Y. Sun, J. Cao and S. Feng, Adv. Sci., 2018, 5, 1800575 CrossRef PubMed
- L. Zhang, L.-X. Ding, G.-F. Chen, X. Yang and H. Wang, Angew. Chem.,
Int. Ed., 2019, 58, 2612–2616 CrossRef CAS PubMed
- M. Bat-Erdene, G. Xu, M. Batmunkh, A. S. R. Bati, J. J. White, M. J. Nine, D. Losic, Y. Chen, Y. Wang, T. Ma and J. G. Shapter, J. Mater. Chem. A, 2020, 8, 4735–4739 RSC
- L. Li, C. Tang, B. Xia, H. Jin, Y. Zheng and S.-Z. Qiao, ACS Catal., 2019, 9, 2902–2908 CrossRef CAS
- R. Zhang, L. Ji, W. Kong, H. Wang, R. Zhao, H. Chen, T. Li, B. Li, Y. Luo and X. Sun, Chem. Commun., 2019, 55, 5263–5266 RSC
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu, Y. Li and Y. Li, Nat. Commun., 2018, 9, 1320 CrossRef PubMed
- Q. Sun, Z. Li, D. J. Searles, Y. Chen, G. Lu and A. Du, J. Am. Chem. Soc., 2013, 135, 8246–8253 CrossRef CAS PubMed
- X. Tan, H. A. Tahini and S. C. Smith, ACS Appl. Mater. Interfaces, 2017, 9(23), 19825–19830 CrossRef CAS PubMed
- H. Shen, Y. Li and Q. Sun, Nanoscale, 2018, 10, 11064–11071 RSC
- S. Zhou, W. Pei, J. Zhao and A. Du, Nanoscale, 2019, 11, 7734–7743 RSC
- B. Luo, G. Liu and L. Wang, Nanoscale, 2016, 8, 6904–6920 RSC
- A. K. Singh, K. Mathew, H. L. Zhuang and R. G. Hennig, J. Phys. Chem. Lett., 2015, 6, 1087–1098 CrossRef CAS PubMed
- J. Yan, P. Verma, Y. Kuwahara, K. Mori and H. Yamashita, Small Methods, 2018, 2, 1800212 CrossRef
- A. Maibam, D. Chakraborty, K. Joshi and S. Krishnamurty, New J. Chem., 2021, 45, 3570–3580 RSC
- B.-J. Wang, X.-H. Li, R. Zhao, X.-L. Cai, W.-Y. Yu, W.-B. Li, Z.-S. Liu, L.-W. Zhang and S.-H. Ke, J. Mater. Chem. A, 2018, 6, 8923–8929 RSC
- Y.-L. Lu, S. Dong, W. Zhou, S. Dai, B. Zhou, H. Zhao and P. Wu, Phys. Chem. Chem. Phys., 2018, 20, 11967–11975 RSC
- A. Kokabi and S. B. Touski, Phys. E, 2020, 124, 114336 CrossRef CAS
- J. Barrio, C. Gibaja, J. Tzadikov, M. Shalom and F. Zamora, Adv. Sustainable Syst., 2019, 3, 1800138 CrossRef
- X.-H. Li, B.-J. Wang, X.-L. Cai, W.-Y. Yu, L.-W. Zhang, G.-D. Wang and S.-H. Ke, RSC Adv., 2017, 7, 44394–44400 RSC
- X. Niu, Y. Li, Q. Zhou, H. Shu and J. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 42856–42861 CrossRef CAS PubMed
- K. Ren, W. Tang, M. Sun, Y. Cai, Y. Cheng and G. Zhang, Nanoscale, 2020, 12, 17281–17289 RSC
- J. Chu, X. Han, Z. Yu, Y. Du, B. Song and P. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 20404–20411 CrossRef CAS PubMed
- Y. Gong, J. Shen, Y. Zhu, W. Yan, J. Zhu, L. Hou, D. Xie and C. Li, Appl. Surf. Sci., 2021, 545, 149012 CrossRef CAS
- X. Ji, Y. Kang, T. Fan, Q. Xiong, S. Zhang, W. Tao and H. Zhang, J. Mater. Chem. A, 2020, 8, 323–333 RSC
- X. Wang, B. Zhou, Y. Zhang, L. Liu, J. Song, R. Hu and J. Qu, J. Alloys Compd., 2018, 769, 316–324 CrossRef CAS
- M. Zhu, M. Fujitsuka, L. Zeng, M. Liu and T. Majima, Appl. Catal., B, 2019, 256, 117864 CrossRef CAS
- J. Low, C. Jiang, B. Cheng, S. Wageh, A. A. Al-Ghamdi and J. Yu, Small Methods, 2017, 1, 1700080 CrossRef
- J. Hu, D. Chen, Z. Mo, N. Li, Q. Xu, H. Li, J. He, H. Xu and J. Lu, Angew. Chem., Int. Ed., 2019, 58, 2073–2077 CrossRef CAS PubMed
- X. Zong, J. Han, G. Ma, H. Yan, G. Wu and C. Li, J. Phys. Chem. C, 2011, 115, 12202–12208 CrossRef CAS
- L. Shi, X. Ren, Q. Wang, Y. Li, F. Ichihara, H. Zhang, Y. Izumi, L. Ren, W. Zhou, Y. Yang and J. Ye, Small, 2020, 16, 2002356 CrossRef CAS PubMed
- B. Qiu, C. Wang, J. Wang, Z. Lin, N. Zhang, L. Cai, X. Tao and Y. Chai, Mater. Today Energy, 2021, 21, 100720 CrossRef CAS
- X. Wang, J. He, J. Li, G. Lu, F. Dong, T. Majima and M. Zhu, Appl. Catal., B, 2020, 277, 119230 CrossRef CAS
- X. Zhu, S. Huang, Q. Yu, Y. She, J. Yang, G. Zhou, Q. Li, X. She, J. Deng, H. Li and H. Xu, Appl. Catal., B, 2020, 269, 118760 CrossRef CAS
- Z.-K. Shen, Y.-J. Yuan, P. Wang, W. Bai, L. Pei, S. Wu, Z.-T. Yu and Z. Zou, ACS Appl. Mater. Interfaces, 2020, 12, 17343–17352 CrossRef CAS PubMed
- M. Wen, J. Wang, R. Tong, D. Liu, H. Huang, Y. Yu, Z.-K. Zhou, P. K. Chu and X.-F. Yu, Adv. Sci., 2019, 6, 1801321 CrossRef PubMed
- J. Lv, M. Xu, S. Lin, X. Shao, X. Zhang, Y. Liu, Y. Wang, Z. Chen and Y. Ma, Nano Energy, 2018, 51, 489–495 CrossRef CAS
- W. Hu, L. Lin, C. Yang, J. Dai and J. Yang, Nano Lett., 2016, 16, 1675–1682 CrossRef CAS PubMed
- Y. Gao, K. Wu, W. Hu and J. Yang, Phys. Chem. Chem. Phys., 2020, 22, 28414–28422 RSC
- C. Zhang, Y. Li, P. Zhang, M. Qiu, X. Jiang and H. Zhang, Sol. Energy Mater. Sol. Cells, 2019, 189, 11–20 CrossRef CAS
- F. Zhang, J. He, Y. Xiang, K. Zheng, B. Xue, S. Ye, X. Peng, Y. Hao, J. Lian and P. Zeng, Adv. Mater., 2018, 30, 1803244 CrossRef PubMed
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed
- H. Shu, F. Li, C. Hu, P. Liang, D. Cao and X. Chen, Nanoscale, 2016, 8, 2918–2926 RSC
- Q. Tang, Z. Zhou and P. Shen, J. Am. Chem. Soc., 2012, 134, 16909–16916 CrossRef CAS PubMed
- H. Hwang, H. Kim and J. Cho, Nano Lett., 2011, 11, 4826–4830 CrossRef CAS PubMed
- X.-L. Fan, Y. Yang, P. Xiao and W.-M. Lau, J. Mater. Chem. A, 2014, 2, 20545–20551 RSC
- Y. Wen, K. He, Y. Zhu, F. Han, Y. Xu, I. Matsuda, Y. Ishii, J. Cumings and C. Wang, Nat. Commun., 2014, 5, 1–10 Search PubMed
- Y. Wu, P. Nie, J. Wang, H. Dou and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 39610–39617 CrossRef CAS PubMed
- L. David, R. Bhandavat and G. Singh, ACS Nano, 2014, 8, 1759–1770 CrossRef CAS PubMed
- J. M. Soon and K. P. Loh, Electrochem. Solid-State Lett., 2007, 10, A250 CrossRef CAS
- M. Ghidiu, M. R. Lukatskaya, M.-Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CrossRef CAS PubMed
- L. Shao, H. Sun, L. Miao, X. Chen, M. Han, J. Sun, S. Liu, L. Li, F. Cheng and J. Chen, J. Mater. Chem. A, 2018, 6, 2494–2499 RSC
- Y. He, Q. He, L. Wang, C. Zhu, P. Golani, A. D. Handoko, X. Yu, C. Gao, M. Ding and X. Wang, Nat. Mater., 2019, 18, 1098–1104 CrossRef CAS PubMed
- S. Venkateshwaran and S. M. Senthil Kumar, ACS Sustainable Chem. Eng., 2019, 7, 2008–2017 CrossRef CAS
- Y. Sun, X. Zhang, B. Mao and M. Cao, Chem. Commun., 2016, 52, 14266–14269 RSC
- R. Prasannachandran, T. V. Vineesh, A. Anil, B. M. Krishna and M. M. Shaijumon, ACS Nano, 2018, 12, 11511–11519 CrossRef CAS PubMed
- J. Wu, M. Liu, K. Chatterjee, K. P. Hackenberg, J. Shen, X. Zou, Y. Yan, J. Gu, Y. Yang and J. J. A. M. I. Lou, Adv. Mater. Interfaces, 2016, 3, 1500669 CrossRef
- R. Wei, M. Fang, G. Dong, C. Lan, L. Shu, H. Zhang, X. Bu and J. C. Ho, ACS Appl. Mater. Interfaces, 2018, 10, 7079–7086 CrossRef CAS PubMed
- J. Chen, X. J. Wu, L. Yin, B. Li, X. Hong, Z. Fan, B. Chen, C. Xue and H. Zhang, Angew. Chem., Int. Ed., 2015, 127, 1226–1230 CrossRef
- U. Maitra, U. Gupta, M. De, R. Datta, A. Govindaraj and C. Rao, Angew. Chem., Int. Ed., 2013, 52, 13057–13061 CrossRef CAS PubMed
- S. Eyderman and S. John, Sci. Rep., 2016, 6, 28303 CrossRef PubMed
- M.-L. Tsai, S.-H. Su, J.-K. Chang, D.-S. Tsai, C.-H. Chen, C.-I. Wu, L.-J. Li, L.-J. Chen and J.-H. He, ACS Nano, 2014, 8, 8317–8322 CrossRef CAS PubMed
- B. Li, C. Lai, G. Zeng, D. Huang, L. Qin, M. Zhang, M. Cheng, X. Liu, H. Yi, C. Zhou, F. Huang, S. Liu and Y. Fu, Small, 2019, 15, 1804565 CrossRef PubMed
- Y. Zhang, P. Huang, J. Guo, R. Shi, W. Huang, Z. Shi, L. Wu, F. Zhang, L. Gao, C. Li, X. Zhang, J. Xu and H. Zhang, Adv. Mater., 2020, 32, 2001082 CrossRef CAS PubMed
- T. Kaewmaraya, L. Ngamwongwan, P. Moontragoon, W. Jarernboon, D. Singh, R. Ahuja, A. Karton and T. Hussain, J. Hazard. Mater., 2021, 401, 123340 CrossRef CAS PubMed
- Y. Liu, Y. Huang and X. Duan, Nature, 2019, 567, 323–333 CrossRef CAS PubMed
- T. Wu, Y. Ma, Z. Qu, J. Fan, Q. Li, P. Shi, Q. Xu and Y. Min, ACS Appl. Mater. Interfaces, 2019, 11, 5136–5145 CrossRef CAS PubMed
- Y. Deng, A. D. Handoko, Y. Du, S. Xi and B. S. Yeo, ACS Catal., 2016, 6, 2473–2481 CrossRef CAS
- Y.-F. Huang, P. J. Kooyman and M. T. M. Koper, Nat. Commun., 2016, 7, 12440 CrossRef CAS PubMed
- W. Sheng, S. Kattel, S. Yao, B. Yan, Z. Liang, C. J. Hawxhurst, Q. Wu and J. G. Chen, Energy Environ. Sci., 2017, 10, 1180–1185 RSC
- Y. Li, Y. Li and Y. Cui, Chem, 2018, 4, 2250–2252 CAS
- Y. Li, W. Huang, Y. Li, W. Chiu and Y. Cui, ACS Nano, 2020, 14, 9263–9276 CrossRef CAS PubMed
- Z.-L. Xu, J.-Q. Huang, W. G. Chong, X. Qin, X. Wang, L. Zhou and J.-K. Kim, Adv. Energy Mater., 2017, 7, 1602078 CrossRef
- H. Huang, J. Wang, X. Yang, R. Hu, J. Liu, L. Zhang and M. Zhu, Angew. Chem., Int. Ed., 2020, 132, 14612–14618 CrossRef
- M. Ding, Q. He, G. Wang, H.-C. Cheng, Y. Huang and X. Duan, Nat. Commun., 2015, 6, 7867 CrossRef CAS PubMed
- H. Yang, Q. He, Y. Liu, H. Li, H. Zhang and T. Zhai, Chem. Soc. Rev., 2020, 49, 2916–2936 RSC
- H. Li, X. Zhou, W. Zhai, S. Lu, J. Liang, Z. He, H. Long, T. Xiong, H. Sun, Q. He, Z. Fan and H. Zhang, Adv. Energy Mater., 2020, 10, 2002019 CrossRef CAS
- Y. Chen, Z. Lai, X. Zhang, Z. Fan, Q. He, C. Tan and H. Zhang, Nat. Rev. Chem., 2020, 4, 243–256 CrossRef CAS
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |