
Design of biodegradable and biocompatible conjugated polymers for bioelectronics
Joshua
Tropp
 *a and
Jonathan
Rivnay
*b
*a and
Jonathan
Rivnay
*b
aDepartment of Biomedical Engineering, Center for Advanced Regenerative Engineering, and Simpson Querrey Institute, Northwestern University, Evanston, Illinois 60208, USA. E-mail: joshua.tropp@northwestern.edu
bDepartment of Biomedical Engineering, Center for Advanced Regenerative Engineering, and Simpson Querrey Institute, Northwestern University, Evanston, Illinois 60208, USA. E-mail: jrivnay@northwestern.edu
First published on 22nd September 2021
Abstract
The emerging field of bioelectronics leverages the optoelectronic properties of synthetic materials to interface with living systems. The convergence of modern electronics with biology has offered lifesaving medical treatments, with applications related to drug delivery, regenerative engineering, and continuous biosignal monitoring for healthcare on the horizon. This next generation of bioelectronic technologies requires an intimate biointerface, necessitating electroactive materials which are both mechanically and physiochemically compatible. Organic systems such as conjugated polymers offer an alternative design space for electroactive materials that are mechanically compatible (flexible, stretchable, conformal) and chemically tunable through various well-established synthetic methods and can therefore be tailored for integration with biological systems. Currently, conjugated polymers utilized for bioelectronic applications consist of prominent high-performing materials emerging from adjacent organic electronic communities with slight chemical modifications, and are therefore generally not well-suited for the entire lifecycle of a biomaterial. While early investigations have demonstrated the potential of such conjugated polymers as semiconductors and conductors in vivo, their limited biodegradability and long-term biocompatibility have slowed widespread adoption and clinical translation. To aid in the development of the next generation of bioelectronic materials, this review details various synthetic strategies to endow a conjugated material with degradability and biocompatibility. Prominent examples of conjugated materials are used to illustrate design principles, current limitations, and future directions towards such electroactive materials. The main factors that need to be considered for the rational design of biodegradable and biocompatible conjugated polymers for bioelectronic applications are highlighted, with future directions emphasized.
 Joshua Tropp | Dr Joshua Tropp is a Postdoctoral Associate in the Biomedical Engineering Department at Northwestern University. Joshua earned his PhD in Polymer Science and Engineering in 2020 from the University of Southern Mississippi, where he focused on the design and synthesis of pi-conjugated polymers for various sensing applications. In the Rivnay Lab, his current studies focus on the development of novel materials for biosensing, regenerative engineering, and other bioelectronic applications. |
 Jonathan Rivnay | Dr Jonathan Rivnay earned his BSc in 2006 from Cornell University (Ithaca, NY). He then moved to Stanford University (Stanford, CA) where he earned a MSc and PhD in Materials Science and Engineering studying the structure and electronic transport properties of organic electronic materials. In 2012, he joined the Department of Bioelectronics at the Ecole des Mines de Saint-Etienne in France as a Marie Curie post-doctoral fellow, working on conducting polymer-based devices for bioelectronics. Jonathan spent 2015–2016 as a member of the research staff in the Printed Electronics group at the Palo Alto Research Center (Palo Alto, CA) before joining the Department of Biomedical Engineering at Northwestern University in 2017. He is a recipient of an NSF CAREER award, ONR Young Investigator award, and has been named an Alfred P. Sloan Research Fellow, and MRS Outstanding Early Career Investigator. |
1. Introduction
Organic bioelectronics encompasses areas of study that electronically couple organic (semi)conductors with living systems. Today, this convergence of modern electronics with biology offers lifesaving technologies and tools to understand the complexities of life. Implantable electronic devices have been successfully implemented for well-established medical treatments—pacemakers, cochlear implants, and both deep-brain and vagus nerve stimulation.1 The next generation of bioelectronic technologies offer solutions related to drug delivery, regenerative engineering, and continuous healthcare monitoring, among others, further enriching the human condition in the process.2Key to these technologies are active materials which interface electronics with biological tissue. Current platforms rely on inorganic materials as conductors and semiconductors, which demonstrate chemical and mechanical instabilities with tissue, and therefore limit the compatibility of bioelectronic devices within the body. Organic systems such as conjugated polymers (CPs) offer an alternative design space for electroactive materials that are flexible and chemically tunable through various well-established synthetic methods, and can therefore be tailored for integration with biological systems.3 Early demonstrations have adapted prominent, high-performing materials emerging from the organic thin-film transistor (OTFT), photovoltaic (OPV), and electrochromic communities for in vivo applications. As these chemical modifications are made on previously designed materials post hoc, CPs utilized for bioelectronic applications today are generally not well-suited for the entire lifecycle of both temporary and permanent implantable technologies. Just as the complete lifecycle of consumer electronics and their components were not considered, leading to the growing problem of e-waste in the environment, the fate of organic electronic materials within the human body has been neglected. The resulting (bio)e-waste (i.e. degraded and/or intact implanted components) and chemical incompatibility of established organic materials requires a next generation of CPs rationally designed for in vivo bioelectronic applications. Beyond the necessary electrical performance for the intended device, the material may require controlled transience and/or long-term biocompatibility within the targeted biological environment; technical definitions for such features are outlined in Table 1.4,5 Transience of a material allows disappearance through absorption or degradation within the body through safe, biologically benign end products. Transient bioelectronic platforms prevent invasive secondary extraction surgeries of implanted devices, while also reducing (bio)e-waste within the body. While biocompatibility is a relatively ambiguous behavior, it generally refers to the ability of a biomaterial to perform its desired function without causing deleterious effects on the biological system. While all materials elicit a host response, it is the nature of the response which is the defining mark of the material compatibility. Biocompatible materials enable intimate integration with cells and tissue without eliciting reactive inflammatory responses – critical for both short term (i.e. transient) and long-term implantation.
| Term | Definition |
|---|---|
| a Indicates terminology defined through IUPAC recommendations. Other terms were influenced by other authoritative sources.4,5 | |
| —Biomaterial specific terms— | |
| Biointerface | The region of contact between a biomolecule, cell, biological tissue, or living organism with another biomaterial. The biotic/abiotic interface between an engineered material/device and a living system |
| Biocompatiblea | The ability of a material to be in contact with a living system without producing an adverse effect |
| Biodegradablea | The ability of a material to be broken down into innocuous components by microorganisms (enzymatic processes resulting from cellular action) |
| Bioabsorbable | The ability of a material to be degraded or dissolved and subsequently metabolized or assimilated within an organism |
| Bioresorbablea | The ability of a compound or device to be totally eliminated or assimilated through natural pathways. For polymeric materials, degradation is necessary prior to bioresorption. This is the preferred term for in vivo processes ending with total elimination of implant material and degradation of its byproducts with no residual side-effects |
| Transience | The ability of a compound or device to dissolve or decompose via hydrolysis or composting in diverse types of environments within a systematically programmed manner. All components can be resorbed or metabolized by the body through safe and complete degradation into biologically benign end products |
| – Passive | Transience begins immediately upon device/material deployment |
| – Active | Transience does not initiate until the device/material is exposed to a stimulus |
| —Organic electronic material related terms— | |
| Charge carrier | A mobile electron, hole, or ion by which electric charge passes through a material |
| Mobility (μ)a | Measure of the ease at which charge carriers can move through a material. Measured in cm2 (V s)−1 |
| Electrical conductivity (σ) | Measure of the ease at which electric current can flow through a material. It is the electric current density divided by the electric field strength. Measured in S cm−1 |
| Ionic conductivity (σionic) | Measure of the ease at which an ion can pass through a material. It is usually coupled to the polymer segmental motion and is therefore dominated by motion in the amorphous regions of conjugated polymers |
| Intrinsically conductive | Materials which demonstrate electrical, magnetic, and optical properties typical of metals and semiconductors without the addition of an external additive or dopant |
| Dopanta | Charge-transfer agent used to generate, by oxidation or reduction, positive or negative charges in a conducting polymer |
Although several organic materials demonstrating transience and/or biocompatibility have been recently reported, the majority of such disclosures have focused on device performance, manufacturing techniques, and demonstrations ex vivo. This is probably due to the stringent requirements contemporary applications have placed on materials properties, requiring a new lens with which organic materials need to be designed. In this review, therefore, we focus on different chemical strategies and concepts to achieve transience and biocompatibility in organic bioelectronic materials for in vivo applications. The outlined design principles may facilitate the widespread implementation of these materials and expedite the clinical adoption of the technologies they enable. For extensive reviews regarding the general application of organic electronic materials in biomedical applications, we direct the reader to prior accounts.1,3,6–10 For comprehensive reviews regarding the degradation and biocompatibility of other device components beyond the active material (substrate, dielectric, conductor), we direct the reader elsewhere.11,12
2. Organic electronic materials for in vivo degradation
Of the strict requirements placed on material design for successful short-term in vivo operation (days to months),13 biodegradability/bioresorbability of device components (substrate, conductor, semiconductor, dielectric, and encapsulant) is critical to avoid invasive periodic renewal/extraction surgeries and infections from long-term exposure. Beyond the desire for general transience, material design is further complicated due to the need for tunable and triggered degradation, which is highly dependent on the application and biological process(es) of interest. For example, devices for accelerated wound healing necessitate stable and consistent operation for beyond 4–6 weeks, while short-term monitoring applications may only require a few days.14 Most transient devices are passive, utilizing degradable or biofluid soluble components protected by a temporary encapsulant. Stable operation occurs until dissolution of the encapsulation layer, followed by the degradation of the device within a short period. Since the initial report of water-soluble transient devices in 2009,15 extensive studies have explored routes to triggered and kinetically tunable degradation through internal and external physicochemical stimuli;13,14 through a particular focus on encapsulation material design. While prior biological applications (logic, circuits, sensing, etc.) utilizing CPs focused on their semiconductive properties, contemporary bioelectronic technologies leverage various active and passive (surface chemistry, morphology, ion/molecular delivery, thermal activity, potential, etc.) properties, requiring a direct and intimate biointerface (Table 1) and thus preclude the use of encapsulants.3,8 Therefore, there is a need for organic semiconductors and conductors with stable performance and controlled degradation in biological environments for successful implementation in vivo.2.1 Entirely conjugated materials
Compared to inorganic materials, organics offer soft, low-cost, scalable, processible, and synthetically tunable alternatives to achieve controlled degradation and biocompatibility (see Section 3). The sensitivity of degradation kinetics in common inorganic systems (Si NM, a-Si, Si–Ge, Ge, ZnO) to pH, temperature, surface roughness/morphology, and ionic concentration,16,17 may be dramatically reduced in organics through material design.18 Targeted organic materials should consist of components that can be safely reabsorbed or metabolized by the body and completely degrade into biologically benign end products after successful operation in a temporary bioelectronic device.19 One logical source of inspiration for biodegradable/bioresorbable chemical motifs comes from naturally occurring conjugated small molecules such as p-type beta (β)-carotene and n-type indigo, which have been successfully utilized in ingestible electronics applications (Fig. 1a).20 Synthetic chromophores derived from natural anthraquinone, as well as perylene diimide, and 5,5′-bis-(7-dodecyl-9H-fluoren-2-yl)-2,2′-bithiophene (DDFTTF) have also been used as semiconductors in transient devices with modest mobilities.20,21 Polymers and oligomers derived from these natural and synthetic aromatic systems offer distinct advantages for bioelectronics applications compared to small molecules such as: tunable electronic structure, good processability, high charge-carrier transport (Table 1), among others.22 Due to their availability and electrical conductivity upon intrinsic or external doping, derivatives of polypyrrole (PPy), polyaniline (PANi), polythiophene (PTh), and poly(3,4-ethylenedioxythiophene) (PEDOT) have been thoroughly investigated for biointerfacing (Fig. 1b).3 However, the same double (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and triple (C
C) and triple (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) bonds enabling conductivity, have strong bond dissociation energies and provide rigidity, limiting the biodegradability of CPs and therefore their in vivo efficacy and clinical translation within temporary implantable technologies. Early examples avoided degradability through the use of sparingly water-soluble conjugated polyelectrolytes (CPEs) which would erode over time.23,24 These functionalized “bioerodable” CPs demonstrated high cellular internalization, low cytotoxic profiles, and were hypothesized to be eliminated from the body through renal filtration (<40 kDa).25 However, in vivo studies are rarely performed to ascertain the biological fate and biocompatibility of conjugated polymeric materials (see Section 3).
C) bonds enabling conductivity, have strong bond dissociation energies and provide rigidity, limiting the biodegradability of CPs and therefore their in vivo efficacy and clinical translation within temporary implantable technologies. Early examples avoided degradability through the use of sparingly water-soluble conjugated polyelectrolytes (CPEs) which would erode over time.23,24 These functionalized “bioerodable” CPs demonstrated high cellular internalization, low cytotoxic profiles, and were hypothesized to be eliminated from the body through renal filtration (<40 kDa).25 However, in vivo studies are rarely performed to ascertain the biological fate and biocompatibility of conjugated polymeric materials (see Section 3).
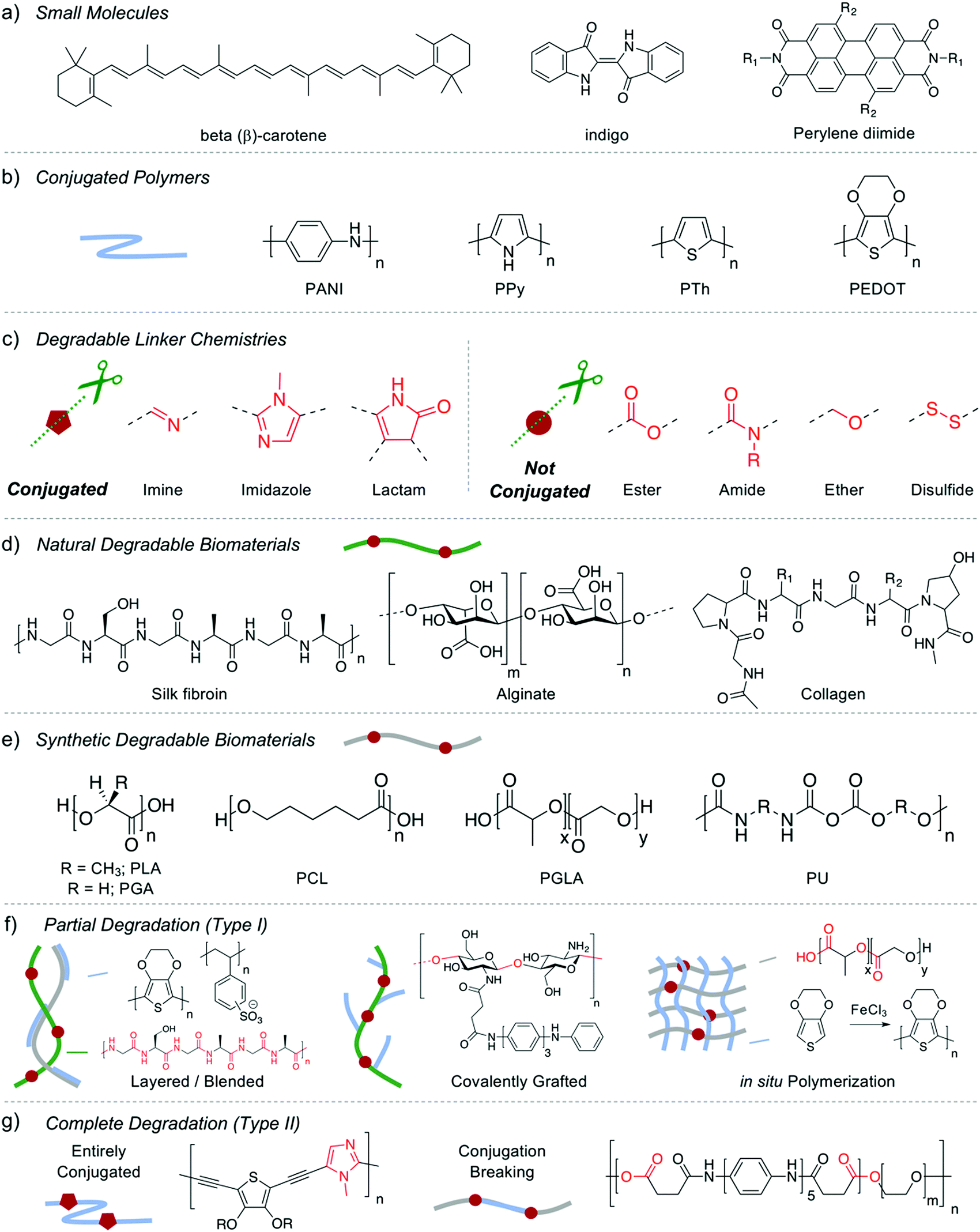 | ||
| Fig. 1 Chemical structures of (a) small molecules, (b) common conjugated polymers, (c) degradable linkers, (d) natural biomaterials, (e) synthetic biomaterials, and representative materials utilized in bioelectronic applications with (f) type I and (g) type II degradation. | ||
Organic materials can be designed with cleavable chemical motifs (Fig. 1c), which allow partial or complete degradation into smaller biologically benign products which can be processed through phagocytosis, metabolization, bioabsorption, or excretion. Scission can afford biodegradation via disintegration without full chemical breakdown (type I, Fig. 1f) or molecular cleavage of the entire material into monomeric and oligomeric components (type II, Fig. 1g).11 Biodegradable type I materials can be obtained simply through the introduction of the organic material as a conductive filler within a biodegradable polymer. Natural biopolymers including polysaccharides (cellulose, chitin/chitosan, alginate) and proteins (silk, collagen, gelatin) have been widely employed for enzymatically degradable materials for bioelectronics (Fig. 1d), and also offer good biocompatibility, tunable morphologies, soft mechanical properties, natural abundance, and multiple sites for chemical modification.26 Synthetic polymers such as polylactide (PLA), polyglycolide (PGA), polycaprolactone (PCL), poly(lactide-co-glycolide) (PLGA), and polyurethane (PU) and been utilized to similar affect (Fig. 1e).18,27 Conductive polymers can be blended, covalently grafted, or polymerized within the synthetic or natural biopolymer to afford degradable conductive composites for implantable devices. This strategy is limited, however, as only the biopolymer degrades and therefore the conjugated polymer is deposited within the body once the device has been disintegrated. The relatively high loading of CP filler required for adequate conductivity can make this strategy toxic for in vivo applications. Recent work has focused on creative material design to overcome the potential toxicity of nondegradable CPs through incorporation of cleavable chemical motifs within the CP, instead of the synthetic or natural degradable polymer matrix (Fig. 1f).
Fully disintegrable (type II) organic semiconductors can be afforded by utilizing degradable or reversible dynamic covalent linkages within each repeat unit (Fig. 1c). Seminal work in this area has demonstrated that Schiff base chemistry can be utilized as hydrolyzable linkages within the polymer backbone to afford high-performance transient and biocompatible semiconductors.28 A polymer based on diketopyrrolopyrrole (DPP), a lactam based heterocycle, was synthesized through a polycondensation with p-phenylenediamine, to afford a conjugated organic semiconductor (PDPP-PD) with hole mobilities as high as 0.34 cm2 V−1 s−1 for use in an OTFT (Fig. 2a). Each repeat unit was held together by imine (−CN−) linkages which were stable under neutral and basic-pH conditions while also maintaining conjugation throughout the organic semiconductor. Under mildly acidic conditions (pH = 4.6) the imine linkages hydrolyzed, disintegrating the material, which was monitored by UV-vis spectroscopy and NMR studies. Devices fabricated with PDPP-PD demonstrated stable operation for several days in DI water, and full disintegration after 30 days in acidic buffer (Fig. 2b). The same group built upon this strategy to develop stretchable and degradable organic semiconductors through nanoconfinement within a biodegradable polycaprolactone elastomer (E-PCL, Fig. 2c).29 The stretchable urethane-based polymer (E-PCL) features polycaprolactone, a common synthetic biodegradable polymer (Fig. 1e), allowing for both components of the composite to be degradable. Side chain engineering of the PDPP-PD polymer, as well as blending ratio optimization, was used to afford nanoconfined semiconducting fibril aggregates within the elastomeric E-PCL matrix (Fig. 2d). Both components of the composite displayed degradation in acidic aqueous solutions and preliminary in vitro cell culture experiments suggested high viability (>99.5%, Fig. 2e). The reported platform offers an exciting opportunity to develop a vast array of degradable conjugated polymers for transient electronics through the polymerization of various (hetero)aromatic monomers and oligomers through imine linkages. Future work must focus on the in vivo compatibility/toxicity of the monomeric byproducts released upon the disintegration of the semiconductor to ensure safe usage in implantable devices (see Section 3). The system, as currently designed, requires low-pH for degradation, limiting transience to acidic environments such as the digestive system. This limitation can be overcome through the use of triggerable release of acids, such as the thermal rupture of methanesulfonic acid (MSA) droplets,30 as well as other cascade or combination triggering events used in the field.14 Alternative conjugated linkage chemistries would expand the toolbox available to synthetic chemists to access degradable organic semiconductors, however, there is inherent resistance of most charge-conducting chemistries to hydrolytic cleavage. Alternative dynamic C–C bonds may offer avenues to stimuli-responsive conjugated linkage chemistries through Diels–Alder cycloaddition, Knoevenagel condensation, and various other reversible reactions.31,32 Similarly, degradable heterocycles could be employed as linkers within CPs. Recently functional imidazole derivatives have been utilized in thiophene and diketopyrrolopyrrole based conjugated polymer nanoparticles for biomedical imaging, with reactive oxygen-induced degradation.33,34 The incorporation of similar imidazole derivatives in electroactive rather than fluorescent CPs, may offer type II organic semiconductors which oxidatively disintegrate for transient electronics.
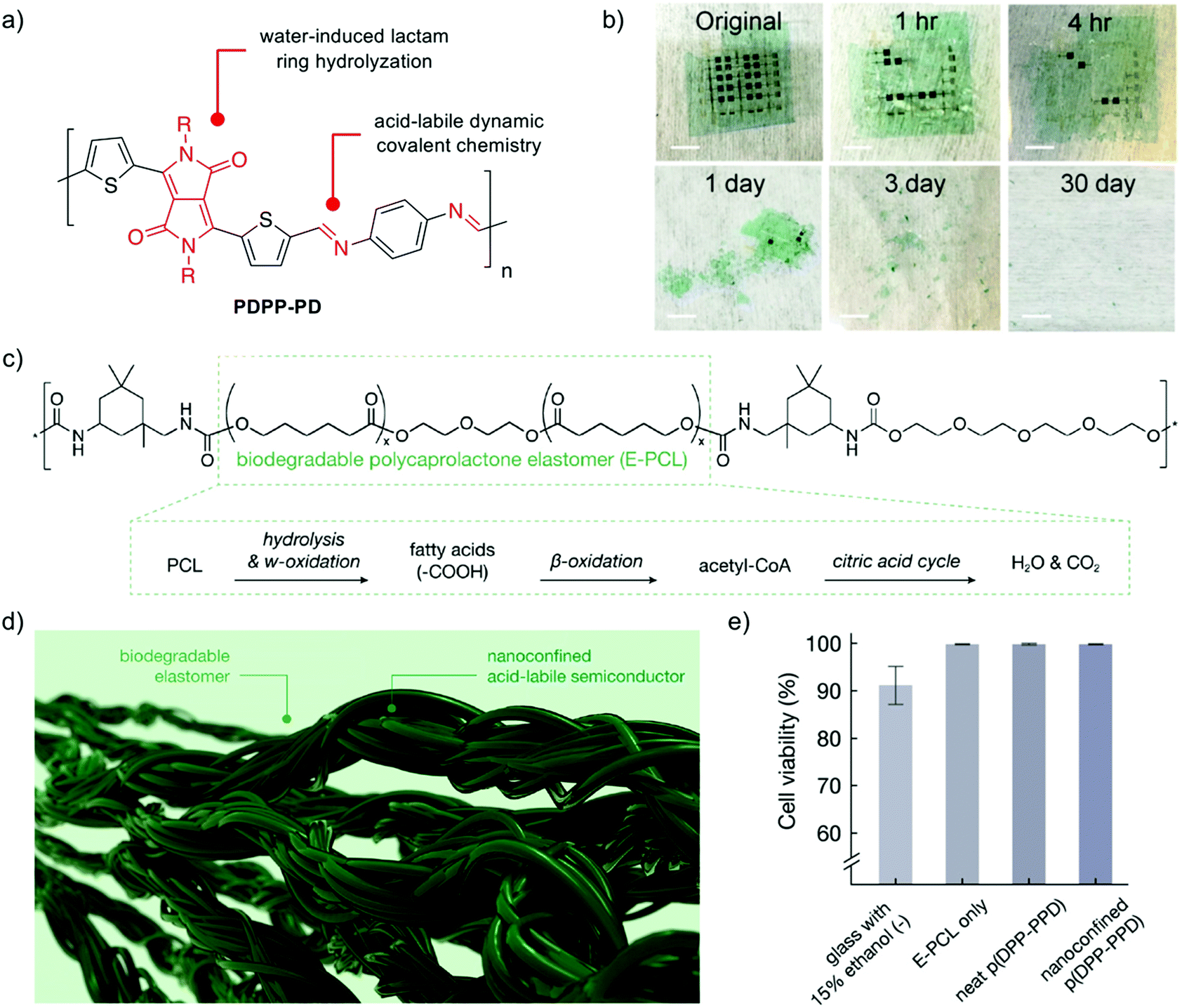 | ||
| Fig. 2 (a) Chemical structure of PDPP-PD. (b) Photographs of PDPP-PD-based device at various stages of disintegration in acidic media. Images adapted with permission from ref. 28. Copyright 2017 National Academy of Sciences. (c) Chemical structure of the biodegradable elastomer E-PCL, and the reported degradation pathway. (d) Illustration of nanoconfined acid-labile semiconductor fibers embedded within a biodegradable elastomer. (e) Cell viability for glass with ethanol (negative control), E-PCL, PDPP-PD, and nanoconfined PDPP-PD. Reprinted with permission from ref. 29. Copyright 2019 American Chemical Society. | ||
2.2 Materials with conjugation breaking
As degradable fully conjugated polymers are rare, conjugated oligomers have been widely explored as alternatives for bioelectronic applications.18,27 Unlike CPs which tend to have batch-to-batch variation in molecular weight distribution by virtue of step-growth polymerization kinetics, oligomers are well-defined, while also maintaining similar electroactivity and redox behavior, making them ideal for well-controlled investigations. Biodegradable materials are developed through the incorporation of (semi)conductive oligomers within the backbone of synthetic biodegradable materials (Fig. 1e) via cleavable linkages (Fig. 1c). It has been hypothesized that short oligomers (<10 monomers) can be consumed by macrophages, therefore rendering these materials both electroactive and entirely degradable. A more detailed discussion of these claims is presented in Section 3.1. The first reported linear material with this design utilized a pyrrole–thiophene–pyrrole oligomer which was tethered via degradable ester linkages to short aliphatic blocks (Fig. 3a).35 The conductive block-copolymer demonstrated complete degradation via esterase after two weeks, and in vivo biocompatibility as investigated through subcutaneous implantation into rats for 14 and 29 days. This method has been extended to oligomers of thiophene (Fig. 3b),36 aniline,37,38 and various polymer architectures including; star-shaped, hyperbranched, crosslinked, graft, and hydrogel-based degradable systems.27 The majority of such reports have focused on aniline oligomers due to relative ease with which well defined, hetero-functionalized systems can be synthesized, for subsequent copolymerization or initiation of ring-opening polymerization (ROP).39,40 The electroactive and degradable nature of oligomeric aniline-based block copolymers have been utilized for tissue engineering,41 with examples including the enhancement of myogenic differentiation,42 osteogenic differentiation,43 as well as myelin gene expression and neurotrophin secretion of Schwann cells.44 However, recent reports have demonstrated aniline-based materials have strong protonation-dependent conductivity and deleterious effects on cells when exposed for long periods (see Section 3.1), highlighting the need for alternative biocompatible and electrically stable conjugated oligomers.45 Further progress in this area will rely on advancements in synthetic methods to access alternative electroactive and biocompatible oligomers. Recently, a novel method for the controlled and scalable synthesis of well-defined oligomers of 3,4-ethylenedioxythiophene (EDOT) was reported, affording hetero-bifunctional oligo-EDOT materials with high conductivity and chemical stability for bioelectronic applications.46 This synthetic strategy was recently adopted to utilize oligoEDOTs as macroinitiators for the polymerization of PCL to access electroactive, degradable, and biocompatible ABA block copolymers for neural tissue engineering (Fig. 3c).47 The oligoEDOT-PCL constructs demonstrated sufficient solubility and processability for solvent electrospinning, allowing for the fabrication of fibrous membranes for tissue engineering (Fig. 3d). To investigate the potential of the oligoEDOT platform for neural tissue engineering, neural stem cells (NSCs) were cultured on tetraEDOT-PCL films, and a pulsed direct current (DC) was applied. An increase in mean NSC neurite length was observed following stimulation of the tetraEDOT-PCL films (142.1 ± 10.4 μm) compared to the unstimulated films (111.4 ± 8.7 μm) (Fig. 3e). Neurite branching also increased upon stimulation (Fig. 3f), suggesting EDOT-based scaffolds offer an exciting new material platform for tissue engineering. Future work could benefit from adopting design motifs, complex architectures, methods, and applications demonstrated with prior aniline-based block copolymers and applying them to oligoEDOT systems. Scalable synthetic methods that enable access to alternative hetero-functionalized conjugated oligomers will define the future directions of these platforms.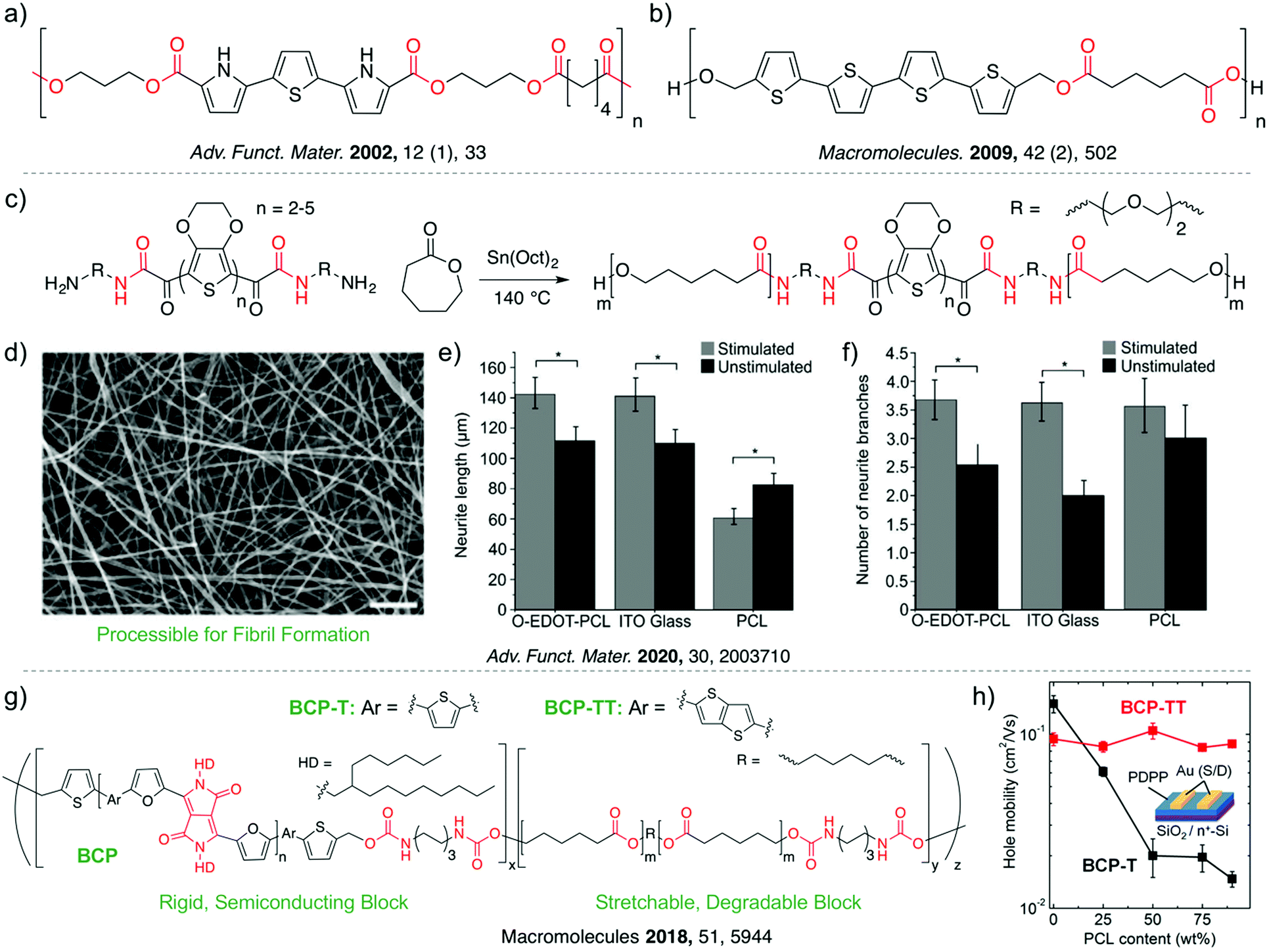 | ||
| Fig. 3 Chemical structure of biodegradable, electrically conductive polymers based on oligomers of (a) pyrrole–thiophene–pyrrole, and (b) quaterthiophene. (c) OligoEDOT-PCL synthesis by ring opening polymerization. (d) SEM image of oligoEDOT-PCL scaffold created using solution electrospinning. Scale bar: 20 μM. (e) Mean neurite length of NSCs (neural stem cells) on substrates with or without electrical stimulation. (f) Neurite branching of NSCs on substrates with or without electrical stimulation. Reprinted with permission from ref. 47. Copyright 2020 John Wiley and Sons. (g) Chemical structure of degradable block copolymers with diisocyanate linkers. (h) Schematic illustration of OFET structure and relationship between charge-carrier mobility and PCL content. Reprinted with permission from ref. 49. Copyright 2018 American Chemical Society. | ||
Similar degradable materials can also be accessed through the linkage of conjugated polymers, rather than oligomers, with biodegradable segments. Segmented conjugated copolymers can offer high mobility unlike analogous oligomer-based systems, as sufficiently long CP segments can interconnect aggregates with locally efficient intermolecular charge transport.48 Recently a stretchable and degradable (type I) material was developed based on a semiconducting furan flanked diketopyrrolopyrrole (DPP) block, and insulating degradable PCL blocks (Fig. 3g).49 Conjugated prepolymer blocks with molecular weights ranging between 19.1 kg mol−1 < Mn < 25.1 kg mol−1 were polymerized and end-capped with 2-thiophenemethanol groups. Independently synthesized PCL soft blocks were copolymerized at varying ratios with the 2-thiophenemethanol-capped DPP blocks using hexamethylene diisocyanate to form urethane linkages. The PCL blocks provided stretchability (>100%) and complete degradation in less than 3 days in 0.5 M NaOH and 12 weeks in phosphate buffered saline. Remarkably, the relatively high hole mobility of ∼0.1 cm2 V−1 s−1 was independent of up to 90 wt% of PCL block incorporation (Fig. 3h). Similar multiblock copolymer systems may provide an alternative route to oligomer-based copolymer systems with significantly enhanced conductivity, however the DPP semiconducting block does not completely degrade or absorb in vivo. Future work could extend this approach to type I degradable conjugated materials (see Section 2.1) to afford stretchable, biocompatible, and fully degradable materials.
3. Chemically biocompatible organic materials
Biocompatibility generally refers to the ability of a biomaterial or device to perform its desired function without eliciting undesirable host responses from the biological system. The ambiguous and contextual nature of this material property makes it challenging to identify chemical motifs which support biocompatibility. The variables that confound biocompatibility analysis of a material include, but are not limited to chemical structure, concentration, supporting materials and additives, duration of use, degradation byproducts, mechanical properties, and the intended biological environment of interest. As such translatable design guidelines to achieve general compatibility of organic electronic materials remain nascent. The standard method for assessing biocompatibility for materials intended for implantable devices is the International Organization for Standardization (ISO) 10993: Biological evaluation of medical devices. To evaluate the suitability of a novel material for a medical device, ISO 10993 requires extensive chemical analysis (structure and purity) for screening and initial assessment for biological safety. It is then required to test the material or device through a series of in vitro tests per ISO 10993 standards for a particular application for screening before in vivo testing and clinical studies. The required tests are predicated on a decision tree that considers the intended device application, the nature of body contact, and the contact duration. All systems require evaluation for cytotoxicity, sensitization, and irritation/intracutaneous reactivity; the approved battery of tests for these effects is partially outlined in Table 2. For further details regarding tests specific to other biological effects, we refer the reader to ISO 10993.| Test | Explanation | Ref.a |
|---|---|---|
| a Representative examples of the corresponding biocompatibility test being performed on conjugated polymers. | ||
| —In vitro cytotoxicity— | ||
| MTT assay | Colorimetric assay for assessing cell metabolic activity as an indicator of cell viability, proliferation, and cytotoxicity (in vitro) | 56 and 57 |
| Agar (disk) diffusion assay | Visual test for assessing cytotoxicity and antimicrobial activity through the comparison of zones of inhibition (in vitro) | 62 |
| MEM elution assay | Colorimetric test for assessing cytotoxicity of a material as it affects L929 cells after treatment (in vitro) | — |
| Neutral red uptake assay | Colorimetric test for assessing cytotoxicity of a material as it affects BALB/c 3T3 cells (in vitro). Commonly used to predict in vivo rodent LD50 starting doses for acute oral systemic toxicity | — |
| XTT test | Colorimetric test for assessing cytotoxicity of a material as it affects mitochondrial enzymes of L929 cells (in vitro) | 70 |
| —Irritation and skin sensitization— | ||
| Local lymph node assay (LLNA) | Skin sensitization is assessed through the material induced growth of lymphocytes which is measured through radiolabeling, bioluminescence, or immunoassay (in vivo, preferred method) | — |
| Guinea pig assays | Skin sensitization is assessed through the allergic reaction of test animals when intradermally exposed to the material. Can be performed with or without an adjuvant (in vivo) | 56 and 63 |
| Animal intracutaneous test | Irritation caused by an implanted material is assessed after injection into the back of a rabbit. A numerical grading system is used to assess intradermal reactions (in vivo, rabbits preferred) | 56 |
| Skin irritation test | Irritation is assessed through the chemical-induced cell damage and subsequent inflammatory cascade in reconstructed human epidermis (RhE) tissues after topical application of the material (in vitro) | 63 |
Care must also be taken not to infer in vivo biocompatibility from in vitro analyses. While a recent update of the ISO 10993 standards stresses in vitro testing, with in vivo testing to only be carried out when absolutely necessary, responses to the material may dramatically differ between the two environments. Even in the most well-designed in vitro experiments, critical interactions such as inflammatory and immune responses cannot be predictably replicated. Biocompatibility of materials and their degradation products should be thoroughly investigated in vivo, the intended environment for the bioelectronic device, rather than assumed from in vitro analyses when possible.50 With these methods and challenges considered, the in vivo biocompatibility of organic electronic materials is poorly understood, as such analyses have rarely been performed in a systematic manner for CP-based materials. To aid the reader, representative examples of these tests being correctly applied to CP-based materials are provided in Table 2. While not explicitly required for material biocompatibility determination by the ISO 10993 guidelines, assessments regarding material influence on other relevant cellular processes such as oxidative stress and autophagy should be considered when possible.105,106 The remainder of this section will highlight literature reports relating chemical features of organic electronic materials to biocompatibility assessments. The chosen examples highlight key design principles and areas for future investigation. For extensive reviews regarding other methods toward biocompatibility, such as mechanical deformation, we direct the reader to prior accounts.51,52 When operating electrochemical devices, the formation of reactive side-products may also reduce biocompatibility; the reader is directed elsewhere for such considerations.107
3.1 Biocompatibility of prominent bioelectronic materials
From the dozens of CP systems developed since the 1970s, only a select few have been seriously considered as electroactive biomaterials (Fig. 1b).3 Broad statements about the biocompatibility of these materials are challenging as the presence of unreacted monomer, oligomeric chains, dopant ions, and other potential contaminants such as solvent or catalyst may lead to the toxicity of a particular batch of polymer.2,53,54 Common monomers have been found to be moderately toxic as determined by the median lethal dose (oral/subcutaneous) for pyrrole (LD50 = 137/98 mg kg−1), aniline (LD50 = 250/1400 mg kg−1), and EDOT (LD50 = 615/894 mg kg−1), emphasizing the need for leachable removal and extensive chemical analysis of each batch before implementation. The two most commonly utilized CPs for biomedical applications are PPy and PANI as composites, blends, and pure films/extracts.6,7,55 Toxicity testing has demonstrated good overall biological performance for PPy extracts within both in vitro culture models and in vivo animal models in accordance with the ISO 10993 standards.7,56–59 The biocompatibility of PANI is more controversial as several reports of chronic inflammation have caused reservations,60–62 despite a plethora of examples demonstrating good in vitro and in vivo biocompatibility.7,55,63 Recently, a direct comparison of PPy and PANI biocompatibility was performed under the same conditions with cytotoxicity and embryotoxicity investigated using the ISO 10993 standards.57 Similar biocompatibility of the two materials was confirmed, and the authors suggested prior observations of inflammation may have risen from impurities or doping state. While less common, PEDOT and its derivatives/composites have also demonstrated biocompatibility in vitro and in vivo.64–67 PEDOT has been utilized as an alternative to PPy due to its improved stability, and PANI due to its improved conductivity, processability, and routes for synthetic modification.7,68–70 Biopolymer composites of these materials have been shown to improve biocompatibility while also acting as dopants, and improving electrochemical stability, conductivity, morphology, adhesion, and processability.6,26,69,71 Conjugated materials have been successfully adsorbed, entrapped, electrospun, polymerized in situ, and covalently modified with synthetic and natural biopolymers to achieve biocompatible electroactive composites. While in-depth biocompatibility studies of these composites have been promising, very little is known about their long-term toxicity in vivo, adding barriers for application within permanent implantable technologies (i.e. pacemakers, cochlear implants, neural protheses, etc.).72 Recently, PEDOT-based coatings such as Amplicoat® have been successfully commercialized with ISO 10993 biocompatibility for chronic applications (electrophysiology mapping, neuromodulation, and cardiac rhythm management).73 Perhaps the biggest limitation of CP-based materials and related composites for temporary implantable technologies is their inherent inability to degrade or be cleared through the body, leading to the possibility of long-term toxicity (see Section 2).Claims have been made in the literature that water-soluble conjugated polymers below a molecular weight of ∼40–50 kDa are appropriate for renal filtration and are therefore bioerodable.23,24,74 This is a misleading claim and should not be assumed, as renal clearance is related to the hydrodynamic radius of the polymer relative to glomerular capillary pores within the kidney.25 As such, factors beyond molecular weight such as side chain composition, molecular architecture, and attached biomacromolecules can influence renal clearance. While systematic studies on aliphatic polymers for drug delivery applications have been performed to correlate effective molecular size to renal clearance,25,75 such investigations have not been reported for CPs. Concerns regarding the long-term compatibility of CPs have led to the extensive use of conjugated oligomer-based block copolymers (see Section 2.2). These copolymer films and hydrogels contain electroactive oligomeric units, generally oligoaniline,40 covalently bound to biocompatible polymers through degradable linkages (Fig. 4b), and have been predominantly utilized in tissue engineering applications.18,27,41,45 While many reports have highlighted the biocompatibility of the copolymer scaffolds in vitro and in vivo, few specifically investigate the toxicity of the oligomeric byproducts after degradation. As it is nearly impossible to reproduce the conditions that materials encounter when implanted in vivo in the laboratory, it is a significant challenge to predict the exact chemical structure of the degradation byproducts and assess toxicity. Several investigations have evaluated the toxicity of various isolated aniline oligomers in order to relate structure (size and functionality) to cytotoxicity (Fig. 4a).76–78 When examining the cellular responses to aniline oligomers with systematic variation in length, the trimer demonstrated the highest toxicity when compared to the dimer and tetramer in both mouse embryonic fibroblasts (NIH-3T3) and human adenocarcinomic alveolar basal epithelial cells (A549). While the sequence of toxicity for the remaining oligomers varied between the two cell lines, the substantial difference in molecular weight between the dimer and the tetramer did not lead to a significant difference in cell viability (Fig. 4a(i)).78 These results demonstrate the nontrivial chain length dependence of conjugated oligomers to their toxicity, which seems to be obfuscated by end groups and cell type. Subsequent studies further investigated the effects of different end-functional groups on aniline oligomer toxicity using a glucose terminated tetramer, and pentamers with both amine and carboxyl groups, respectively.77 Significant differences in cellular responses within both human cervical carcinoma (HeLa) and A549 cell lines were observed, with the glucose terminated tetramer demonstrating the highest toxicity in both types of cells. It was hypothesized that differences in cellular responses could be correlated to water solubility, which was controlled by end group functionality (Fig. 4a(ii)). A comparison between the two studies support this hypothesis as, within the same A549 cell line, the tetramer with a single glucose group was more toxic than that with a single amine, where glucose provided more solubilizing power in aqueous environments. Similarly, the pentamer with two amine groups was less toxic than the trimer with the same functionality, where a smaller oligomer would be expected to have a greater proportion of polar functionalities to impart greater aqueous solubility. As glucose is a ubiquitous biomolecule, further investigations should be taken before concluding the relative role solubility plays in oligomer toxicity. Taken together, these studies suggest that the toxicity of aniline oligomers is complex but can be tailored by the modification of different end groups. These studies also provide evidence of general compatibility of these oligomers in vivo through the lack of generated reactive oxygen species and hemocompatibility evaluation. Further investigations are necessary to understand the structural dependence of oligomer biocompatibility for aniline and other classes of conjugated structures. A focus on the impact of end group chemistries on toxicity is important as these groups remain after molecular cleavage of common oligomer-based block copolymers, and therefore dictate cellular response. Structure–property relationships should be made with caution, as end group chemistries may interfere in various metabolic processes and therefore augment cytotoxicity.
 | ||
| Fig. 4 (a) Factors that influence the cellular response to isolated aniline oligomers. These include (i) molecular weight, (ii) functionality, and (iii) the cellular environment. (b) Generalized structure of a conducting copolymer consisting of electroactive conjugated oligomers and blocks of biocompatible synthetic polymers. Structures of the most prevalent conjugated oligomers incorporated (i) within the backbone and (ii) as grafts/side chains of electroactive biomaterials. Details of biocompatibility assessments upon partial or complete degradation of materials consisting of each oligomer are annotated. | ||
While investigations of isolated oligomers provide a path toward realizing structure–function–property relationships, it is a challenge to predict the exact chemical structure of the byproducts that will be released upon cleavage of the block copolymer. Therefore, a more precise method involves the degradation of the material with subsequent toxicity assessment in vitro and in vivo, an approach seldom performed.79–85 The electroactive material can be introduced in vitro as a water-soluble extract or a solid, as well as in vivo through subcutaneous implantation—followed by common assays to investigate viability/cytotoxicity (Table 2), cell proliferation, and gene expression. Fig. 4b highlights toxicity/viability investigations of the copolymers, where the biocompatibility of the material over time generally acts as a surrogate for that of the degraded oligomeric byproducts, which are slowly released through ester hydrolysis. As previously mentioned in Section 2.2, original toxicity investigations of degraded electroactive block copolymers incorporating pyrrole–thiophene–pyrrole in vivo and quaterthiophene in vitro suggested long-term compatibility (Fig. 4b).35,36 Aniline oligomers have been more widely utilized,40,41 and have been incorporated into the backbone (Fig. 4b(i)) or as grafts (Fig. 4b(ii)) within various polymer architectures.27 While modification of the copolymer composition, topology, oligomer length, and aniline loading have demonstrated substantial variations in conductivity, mechanical properties, and morphology, favorable short-term biocompatibility has been consistently observed. The long-term compatibility of these materials is less apparent, as the slow kinetics of material degradation does not allow significant oligomeric byproduct to be released within the time frame commonly used to assess biocompatibility in vitro (1–8 days). Therefore assessments, as currently performed, do not provide an accurate understanding of the chronic toxicity of degraded aniline oligomers. While investigations in vivo do suggest long-term compatibility of these materials, deleterious effects have been observed at high aniline loadings, limiting the achievable conductivity (10−7 to 10−4).40 The reliance on ester linkages also precludes the controlled degradation of these materials, limiting their effectiveness in vivo. Future progress in this field could be aided by active linkage chemistries (see Section 2.1) and alternative conductive oligomers such as EDOT (see Section 2.2). For extensive reviews regarding electroactive oligomer-based materials and their application, we direct the reader to prior accounts.27,40,41
3.2 Chemical compatibility through functionalization
Backbone design provides an important, but incomplete, role in determining the macroscopic properties, such as biocompatibility, of conjugated materials. Chemical functionalization through side chain engineering provides a far-reaching method to manipulate material properties. Side chain design has been widely utilized to tune solution processability and as an effective strategy to regulate interchain interactions of polymers, and therefore the packing/film morphology to enhance performance of OPVs, OTFTs, organic light-emitting diodes (OLEDs), and organic photodetectors (OPDs), among others.86,87 Beyond regulating optoelectronic properties such as charge carrier mobilities, side chains may also impart latent reactivity,88 ionic conductivity,22 molecular recognition,89 and various modalities for optical/electronic sensing.90 While side chain engineering has been used as a tool to modulate material behavior with respect to cells,91,92 such investigations of bioelectronic materials are in their infancy. Such attempts at biointerfacing have focused on the covalent attachment of biomolecules to CPs deposited at the surfaces of electrodes for electrochemical biosensing applications. Elegant approaches to incorporate biomolecules to CP surfaces have been demonstrated through an array of high-yielding chemistries, a complete survey of which is outside of the scope of this review.69,70,91,93A primary concern of implanted materials and their corresponding devices for long-term applications is the nonspecific adsorption of biomolecules and cells which can obscure the biointerface. The introduction of synthetic, rather than biological, side chains has been offered as a solution to reduce nonspecific adsorption in bioelectronic systems, adopting well-established design guidelines from the polymer chemistry community.94 Such antifouling behavior is tailored through the incorporation of large biocompatible synthetic polymer (Fig. 1e) grafts,91,95 or short hydrophilic (oligoether, ionic, zwitterionic) side chains92 onto the CP backbone. Grafting approaches have provided advantageous chemical, physical, and mechanical properties, however much of the field is not specifically targeted for biointerface applications. As such biocompatibility investigations of grafted CPs remain rare.96–99 Short hydrophilic side chains have been utilized to control antifouling and antimicrobial properties to greater effect,100,101 however only a handful have been assessed for in vitro biocompatibility, with in vivo investigations virtually absent. A major challenge in determining biocompatibility guidelines through side chain engineering, is the overwhelming use of electropolymerization rather than solution phase synthetic protocols, complicating structural analysis. As many of the required techniques to determine structure–function–property relationships take place in solution (i.e. nuclear magnetic resonance spectroscopy, size-exclusion chromatography, etc.), complete polymer characterization for biocompatibility is non-trivial. Future work could leverage parallel derivatization techniques, as is common in the adjacent field of organic electronics,86,89 to expand the chemical diversity of side chains of CPs for biointerfacing applications. Such assessments should leverage international standards for determining biocompatibility (Table 2), as to provide comparable analysis towards the rational development of design guidelines.
4. Conclusions
Organic electroactive biomaterials offer an alternative design space toward new and exciting implantable bioelectronic devices. The next generation of implantable devices related to drug delivery, regenerative engineering, and continuous healthcare monitoring require an intimate biointerface with the organic electronic material, creating significant challenges for contemporary CPs. The prevailing paradigm for materials design focuses on electrical performance rather than other important figures of merit for in vivo devices such as degradability and biocompatibility. This lack of foresight has crippled progress in the field, and the associated externalities such as (bio)e-waste and chronic toxicity have delayed translation into clinical settings. The same carbon–carbon double (CC) and triple (CC) bonds enabling conductivity in CPs are also resistant to hydrolytic and enzymatic degradation, requiring creative solutions to access transient materials. The introduction of labile units within the polymer backbone to connect conjugated oligomers, blocks, and biopolymers affords a wide variety of tunable degradable organic electronic biomaterials. Growth in this field will closely follow novel synthetic methods and the introduction of alternative cleavable linkage chemistries. Active, rather than passive, chemistries that allow chain scission to occur within the desired period and environment are particularly important to expand the toolbox available for material design. The compatibility of these materials and their corresponding degradation byproducts must be assessed when considering a material for in vivo applications to avoid chronic toxicity and inflammatory responses. Designing biocompatible organic electronic materials has been a significant challenge due to the lack of standardized characterization within the literature, dramatic differences between each system (powder, film, composite, blend, network, etc.), and the profound effect of leachable toxicants remaining from synthesis. Side-by-side in-house ISO 10993 compliant comparative toxicity analyses of various materials would reduce inter-laboratory variations in assay performance and experimental set-up. While rarely performed,57,102 such studies can aid in the development of material design guidelines, particularly when paired with systematic chemical modification (backbone and side chain). Investigations could rapidly aid in the development of such structure–function–property relationships through modular synthetic approaches such as click chemistry.103,104 In addition, in vivo verification of in vitro results will be necessary before significant strides can be made towards clinical translation. The required battery of tests for biovalidation necessitates that such materials can be synthesized at scale, a consideration that should be made during the design process. Rational design which focuses on the entire lifecycle and chemical compatibility of the material, will pave the way for the next generation of high-performing implantable devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. T. was primarily supported by an ONR YIP (SP0056955). J. R. gratefully acknowledges support from the Alfred P. Sloan Foundation (FG-2019-12046).References
- E. Zeglio, A. L. Rutz, T. E. Winkler, G. G. Malliaras and A. Herland, Adv. Mater., 2019, 31, 1806712 CrossRef PubMed
.
- R. A. Green, N. H. Lovell, G. G. Wallace and L. A. Poole-Warren, Biomaterials, 2008, 29, 3393–3399 CrossRef CAS PubMed
- S. Inal, J. Rivnay, A.-O. Suiu, G. G. Malliaras and I. McCulloch, Acc. Chem. Res., 2018, 51, 1368–1376 CrossRef CAS PubMed
- Y. Liu, Y. Zheng and B. Hayes, Sci. China Mater., 2017, 60, 377–391 CrossRef CAS
- M. Vert, Y. Doi, K.-H. Hellwich, M. Hess, P. Hodge, P. Kubisa, M. Rinaudo and F. Schué, Pure Appl. Chem., 2012, 84, 377–410 CAS
- R. Balint, N. J. Cassidy and S. H. Cartmell, Acta Biomater., 2014, 10, 2341–2353 CrossRef CAS PubMed
- N. K. Guimard, N. Gomez and C. E. Schmidt, Prog. Polym. Sci., 2007, 32, 876–921 CrossRef CAS
- A. J. Petty, R. L. Keate, B. Jiang, G. A. Ameer and J. Rivnay, Chem. Mater., 2020, 32, 4095–4115 CrossRef CAS
- S. G. Higgins, A. Lo Fiego, I. Patrick, A. Creamer and M. M. Stevens, Adv. Mater. Technol., 2020, 5, 2000384 CrossRef CAS
- D. Ohayon and S. Inal, Adv. Mater., 2020, 32, 2001439 CrossRef CAS PubMed
- V. R. Feig, H. Tran and Z. Bao, ACS Cent. Sci., 2018, 4, 337–348 CrossRef CAS PubMed
- J. A. Chiong, H. Tran, Y. Lin, Y. Zheng and Z. Bao, Adv. Sci., 2021, 2101233 CrossRef CAS PubMed
- R. Singh, M. J. Bathaei, E. Istif and L. Beker, Adv. Healthcare Mater., 2020, 9, 2000790 CrossRef CAS PubMed
- G. Lee, Y. S. Choi, H.-J. Yoon and J. A. Rogers, Matter, 2020, 3, 1031–1052 CrossRef
- D.-H. Kim, Y.-S. Kim, J. Amsden, B. Panilaitis, D. L. Kaplan, F. G. Omenetto, M. R. Zakin and J. A. Rogers, Appl. Phys. Lett., 2009, 95, 133701 CrossRef PubMed
- S.-W. Hwang, C. H. Lee, H. Cheng, J.-W. Jeong, S.-K. Kang, J.-H. Kim, J. Shin, J. Yang, Z. Liu, G. A. Ameer, Y. Huang and J. A. Rogers, Nano Lett., 2015, 15, 2801–2808 CrossRef CAS PubMed
- Y. S. Choi, J. Koo and J. Rogers, MRS Bull., 2020, 45, 103–112 CrossRef
- K. Liu and B. Liu, Biomacromolecules, 2018, 19, 1783–1803 CrossRef PubMed
- W. B. Han, J. H. Lee, J.-W. Shin and S.-W. Hwang, Adv. Mater., 2020, 32, 2002211 CrossRef CAS PubMed
- M. Irimia-Vladu, P. A. Troshin, M. Reisinger, L. Shmygleva, Y. Kanbur, G. Schwabegger, M. Bodea, R. Schwödiauer, A. Mumyatov, J. W. Fergus, V. F. Razumov, H. Sitter, N. S. Sariciftci and S. Bauer, Adv. Funct. Mater., 2010, 20, 4069–4076 CrossRef CAS
- C. J. Bettinger and Z. Bao, Adv. Mater., 2010, 22, 651–655 CrossRef CAS PubMed
- B. D. Paulsen, K. Tybrandt, E. Stavrinidou and J. Rivnay, Nat. Mater., 2020, 19, 13–26 CrossRef CAS PubMed
- D. Mawad, K. Gilmore, P. Molino, K. Wagner, P. Wagner, D. L. Officer and G. G. Wallace, J. Mater. Chem., 2011, 21, 5555–5560 RSC
- A. N. Zelikin, D. M. Lynn, J. Farhadi, I. Martin, V. Shastri and R. Langer, Angew. Chem., Int. Ed., 2002, 41, 141–144 CrossRef CAS PubMed
- M. E. Fox, F. C. Szoka and J. M. J. Fréchet, Acc. Chem. Res., 2009, 42, 1141–1151 CrossRef CAS PubMed
- C. Wang, T. Yokota and T. Someya, Chem. Rev., 2021, 121, 2109–2146, DOI:10.1021/acs.chemrev.0c00897
- B. Guo, L. Glavas and A.-C. Albertsson, Prog. Polym. Sci., 2013, 38, 1263–1286 CrossRef CAS
- T. Lei, M. Guan, J. Liu, H.-C. Lin, R. Pfattner, L. Shaw, A. F. McGuire, T.-C. Huang, L. Shao, K.-T. Cheng, J. B. H. Tok and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5107 CrossRef CAS PubMed
- H. Tran, V. R. Feig, K. Liu, H.-C. Wu, R. Chen, J. Xu, K. Deisseroth and Z. Bao, ACS Cent. Sci., 2019, 5, 1884–1891 CrossRef CAS PubMed
- C. W. Park, S.-K. Kang, H. L. Hernandez, J. A. Kaitz, D. S. Wie, J. Shin, O. P. Lee, N. R. Sottos, J. S. Moore, J. A. Rogers and S. R. White, Adv. Mater., 2015, 27, 3783–3788 CrossRef CAS PubMed
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC
- S. Huang, X. Kong, Y. Xiong, X. Zhang, H. Chen, W. Jiang, Y. Niu, W. Xu and C. Ren, Eur. Polym. J., 2020, 141, 110094 CrossRef CAS
- T. Repenko, A. Rix, S. Ludwanowski, D. Go, F. Kiessling, W. Lederle and A. J. C. Kuehne, Nat. Commun., 2017, 8, 470 CrossRef PubMed
- J. Liu, S. Wang, X. Cai, S. Zhou and B. Liu, Chem. Commun., 2018, 54, 2518–2521 RSC
- T. J. Rivers, T. W. Hudson and C. E. Schmidt, Adv. Funct. Mater., 2002, 12, 33–37 CrossRef CAS
- N. K. E. Guimard, J. L. Sessler and C. E. Schmidt, Macromolecules, 2009, 42, 502–511 CrossRef CAS
- L. Huang, X. Zhuang, J. Hu, L. Lang, P. Zhang, Y. Wang, X. Chen, Y. Wei and X. Jing, Biomacromolecules, 2008, 9, 850–858 CrossRef CAS PubMed
- L. Huang, J. Hu, L. Lang, X. Wang, P. Zhang, X. Jing, X. Wang, X. Chen, P. I. Lelkes, A. G. MacDiarmid and Y. Wei, Biomaterials, 2007, 28, 1741–1751 CrossRef CAS PubMed
- Z. Wei and C. F. J. Faul, Macromol. Rapid Commun., 2008, 29, 280–292 CrossRef CAS
- P. Zarrintaj, B. Bakhshandeh, M. R. Saeb, F. Sefat, I. Rezaeian, M. R. Ganjali, S. Ramakrishna and M. Mozafari, Acta Biomater., 2018, 72, 16–34 CrossRef CAS PubMed
- B. Guo and P. X. Ma, Biomacromolecules, 2018, 19, 1764–1782 CrossRef CAS PubMed
- J. Chen, R. Dong, J. Ge, B. Guo and P. X. Ma, ACS Appl. Mater. Interfaces, 2015, 7, 28273–28285 CrossRef CAS
- L. Li, M. Yu, P. X. Ma and B. Guo, J. Mater. Chem. B, 2016, 4, 471–481 RSC
- Y. Wu, L. Wang, B. Guo, Y. Shao and P. X. Ma, Biomaterials, 2016, 87, 18–31 CrossRef CAS
- A. da Silva and S. Córdoba de Torresi, Front. Mater., 2019, 6, 98 CrossRef
- C. D. Spicer, M. A. Booth, D. Mawad, A. Armgarth, C. B. Nielsen and M. M. Stevens, Chem, 2017, 2, 125–138 CAS
- K. I. Ritzau-Reid, C. D. Spicer, A. Gelmi, C. L. Grigsby, J. F. Ponder Jr, V. Bemmer, A. Creamer, R. Vilar, A. Serio and M. M. Stevens, Adv. Funct. Mater., 2020, 30, 2003710 CrossRef CAS
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed
- F. Sugiyama, A. T. Kleinschmidt, L. V. Kayser, M. A. Alkhadra, J. M. H. Wan, A. S. C. Chiang, D. Rodriquez, S. E. Root, S. Savagatrup and D. J. Lipomi, Macromolecules, 2018, 51, 5944–5949 CrossRef CAS
- J. M. Anderson, Regener. Biomater., 2016, 3, 73–77 CrossRef CAS PubMed
- B. V. Khau, A. D. Scholz and E. Reichmanis, J. Mater. Chem. C, 2020, 8, 15067–15078 RSC
- S. E. Root, S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Chem. Rev., 2017, 117, 6467–6499 CrossRef CAS PubMed
- V. Kašpárková, P. Humpolíček, J. Stejskal, Z. Capáková, P. Bober, K. Skopalová and M. Lehocký, Polymers, 2019, 11, 362 CrossRef
- J. M. Fonner, L. Forciniti, H. Nguyen, J. D. Byrne, Y.-F. Kou, J. Syeda-Nawaz and C. E. Schmidt, Biomed. Mater., 2008, 3, 034124 CrossRef PubMed
- T. H. Qazi, R. Rai and A. R. Boccaccini, Biomaterials, 2014, 35, 9068–9086 CrossRef CAS PubMed
- X. Wang, X. Gu, C. Yuan, S. Chen, P. Zhang, T. Zhang, J. Yao, F. Chen and G. Chen, J. Biomed. Mater. Res., Part A, 2004, 68, 411–422 CrossRef PubMed
- P. Humpolíček, V. Kašpárková, J. Pacherník, J. Stejskal, P. Bober, Z. Capáková, K. A. Radaszkiewicz, I. Junkar and M. Lehocký, Mater. Sci. Eng., C, 2018, 91, 303–310 CrossRef PubMed
- D. D. Ateh, H. A. Navsaria and P. Vadgama, J. R. Soc., Interface, 2006, 3, 741–752 CrossRef CAS PubMed
- A. Ramanaviciene, A. Kausaite, S. Tautkus and A. Ramanavicius, J. Pharm. Pharmacol., 2007, 59, 311–315 CrossRef CAS PubMed
- C. H. Wang, Y. Q. Dong, K. Sengothi, K. L. Tan and E. T. Kang, Synth. Met., 1999, 102, 1313–1314 CrossRef CAS
- A. Borriello, V. Guarino, L. Schiavo, M. A. Alvarez-Perez and L. Ambrosio, J. Mater. Sci.: Mater. Med., 2011, 22, 1053–1062 CrossRef CAS PubMed
- N. Bagheri, M. Mansour Lakouraj, S. R. Nabavi, H. Tashakkorian and M. Mohseni, RSC Adv., 2020, 10, 25290–25304 RSC
- P. Humpolicek, V. Kasparkova, P. Saha and J. Stejskal, Synth. Met., 2012, 162, 722–727 CrossRef CAS
- M. Asplund, E. Thaning, J. Lundberg, A. C. Sandberg-Nordqvist, B. Kostyszyn, O. Inganäs and H. von Holst, Biomed. Mater., 2009, 4, 045009 CrossRef CAS PubMed
- S. M. Richardson-Burns, J. L. Hendricks, B. Foster, L. K. Povlich, D.-H. Kim and D. C. Martin, Biomaterials, 2007, 28, 1539–1552 CrossRef CAS PubMed
- R. F. Vreeland, C. W. Atcherley, W. S. Russell, J. Y. Xie, D. Lu, N. D. Laude, F. Porreca and M. L. Heien, Anal. Chem., 2015, 87, 2600–2607 CrossRef CAS PubMed
- S.-C. Luo, E. Mohamed Ali, N. C. Tansil, H.-H. Yu, S. Gao, E. A. B. Kantchev and J. Y. Ying, Langmuir, 2008, 24, 8071–8077 CrossRef CAS PubMed
- C. Boehler, Z. Aqrawe and M. Asplund, Bioelectron. Med., 2019, 2, 89–99 CrossRef
- M. J. Donahue, A. Sanchez-Sanchez, S. Inal, J. Qu, R. M. Owens, D. Mecerreyes, G. G. Malliaras and D. C. Martin, Mater. Sci. Eng., R, 2020, 140, 100546 CrossRef
- B. Wu, B. Cao, I. M. Taylor, K. Woeppel and X. T. Cui, Front. Chem., 2019, 7, 178 CrossRef PubMed
- T. Nezakati, A. Seifalian, A. Tan and A. M. Seifalian, Chem. Rev., 2018, 118, 6766–6843 CrossRef CAS PubMed
-
A. Sanchez-Sanchez, I. del Agua, G. G. Malliaras and D. Mecerreyes, in Smart Polymers and their Applications, ed. M. R. Aguilar and J. San Román, Woodhead Publishing, 2nd edn, 2019, pp. 191–218 DOI:10.1016/B978-0-08-102416-4.00006-5
- S. Narayan, L. Stoica, A. Liess and A. Reisinger, Presented in part at the 2021 Design of Medical Devices Conference, Minneapolis, MN, USA, 2021.
- J. G. Hardy, J. Y. Lee and C. E. Schmidt, Curr. Opin. Biotechnol, 2013, 24, 847–854 CrossRef CAS PubMed
- M. J. Knauf, D. P. Bell, P. Hirtzer, Z. P. Luo, J. D. Young and N. V. Katre, J. Biol. Chem., 1988, 263, 15064–15070 CrossRef CAS
- M. R. Gizdavic-Nikolaidis, J. Bennett, Z. Zujovic, S. Swift and G. A. Bowmaker, Synth. Met., 2012, 162, 1114–1119 CrossRef CAS
- H. Qi, M. Liu, L. Xu, L. Feng, L. Tao, Y. Ji, X. Zhang and Y. Wei, Toxicol. Res., 2013, 2, 427–433 CrossRef CAS
- X. Zhang, H. Qi, S. Wang, L. Feng, Y. Ji, L. Tao, S. Li and Y. Wei, Toxicol. Res., 2012, 1, 201–205 CrossRef CAS
- T. Hu, Y. Wu, X. Zhao, L. Wang, L. Bi, P. X. Ma and B. Guo, Chem. Eng. J., 2019, 366, 208–222 CrossRef CAS
- J. Qu, X. Zhao, Y. Liang, Y. Xu, P. X. Ma and B. Guo, Chem. Eng. J., 2019, 362, 548–560 CrossRef CAS
- H. Cui, L. Cui, P. Zhang, Y. Huang, Y. Wei and X. Chen, Macromol. Biosci., 2014, 14, 440–450 CrossRef CAS PubMed
- H. Cui, Y. Liu, Y. Cheng, Z. Zhang, P. Zhang, X. Chen and Y. Wei, Biomacromolecules, 2014, 15, 1115–1123 CrossRef CAS PubMed
- H. Cui, J. Shao, Y. Wang, P. Zhang, X. Chen and Y. Wei, Biomacromolecules, 2013, 14, 1904–1912 CrossRef CAS PubMed
- R. Dong, X. Zhao, B. Guo and P. X. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 17138–17150 CrossRef CAS PubMed
- E. Jin, Z. Zhang, H. Lian, X. Chen, C. Xiao, X. Zhuang and X. Chen, Eur. Polym. J., 2017, 88, 67–74 CrossRef CAS
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS
- Y. Yang, Z. Liu, G. Zhang, X. Zhang and D. Zhang, Adv. Mater., 2019, 31, 1903104 CrossRef CAS
- J. Freudenberg, D. Jänsch, F. Hinkel and U. H. F. Bunz, Chem. Rev., 2018, 118, 5598–5689 CrossRef CAS PubMed
- R. Zhan and B. Liu, Chem. Rec., 2016, 16, 1715–1740 CrossRef CAS PubMed
- G. Anantha-Iyengar, K. Shanmugasundaram, M. Nallal, K.-P. Lee, M. J. Whitcombe, D. Lakshmi and G. Sai-Anand, Prog. Polym. Sci., 2019, 88, 1–129 CrossRef CAS
- A. J. Hackett, J. Malmström and J. Travas-Sejdic, Prog. Polym. Sci., 2017, 70, 18–33 CrossRef CAS
- J.-G. Wu, J.-H. Chen, K.-T. Liu and S.-C. Luo, ACS Appl. Mater. Interfaces, 2019, 11, 21294–21307 CrossRef CAS PubMed
- D. Mantione, I. Del Agua, A. Sanchez-Sanchez and D. Mecerreyes, Polymers, 2017, 9, 354 CrossRef
- Z. K. Zander and M. L. Becker, ACS Macro Lett., 2018, 7, 16–25 CrossRef CAS
- P. Baek, L. Voorhaar, D. Barker and J. Travas-Sejdic, Acc. Chem. Res., 2018, 51, 1581–1589 CrossRef CAS PubMed
- A. C. da Silva, A. T. S. Semeano, A. H. B. Dourado, H. Ulrich and S. I. Cordoba de Torresi, ACS Omega, 2018, 3, 5593–5604 CrossRef CAS PubMed
- B. G. Molina, L. Cianga, A.-D. Bendrea, I. Cianga, C. Alemán and E. Armelin, Polym. Chem., 2019, 10, 5010–5022 RSC
- S. Maione, G. Fabregat, L. J. del Valle, A.-D. Bendrea, L. Cianga, I. Cianga, F. Estrany and C. Alemán, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 239–252 CrossRef CAS
- H. Zhao, B. Zhu, S.-C. Luo, H.-A. Lin, A. Nakao, Y. Yamashita and H.-H. Yu, ACS Appl. Mater. Interfaces, 2013, 5, 4536–4543 CrossRef CAS PubMed
- B. Cao, Q. Tang, L. Li, C.-J. Lee, H. Wang, Y. Zhang, H. Castaneda and G. Cheng, Chem. Sci., 2015, 6, 782–788 RSC
- H. Zhao, B. Zhu, J. Sekine, S.-C. Luo and H.-H. Yu, ACS Appl. Mater. Interfaces, 2012, 4, 680–686 CrossRef CAS PubMed
- Z. Capáková, K. A. Radaszkiewicz, U. Acharya, T. H. Truong, J. Pacherník, P. Bober, V. Kašpárková, J. Stejskal, J. Pfleger, M. Lehocký and P. Humpolíček, Mater. Sci. Eng., C, 2020, 113, 110986 CrossRef PubMed
- A. K. Williams, J. Tropp, E. R. Crater, N. Eedugurala and J. D. Azoulay, ACS Appl. Polym. Mater., 2019, 1, 309–314 CrossRef CAS
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed
- M. Katerji, M. Filippova and P. Duerksen-Hughes, Oxid. Med. Cell. Longevity, 2019, 2019, 1279250 Search PubMed
- D. J. Klionsky,
et al.
, Autophagy, 2021, 17, 1–382 CrossRef PubMed
- A. Giovannitti, R. B. Rashid, Q. Thiburce, B. D. Paulsen, C. Cendra, K. Thorley, D. Moia, J. T. Mefford, D. Hanifi, D. Weiyuan, M. Moser, A. Salleo, J. Nelson, I. McCulloch and J. Rivnay, Adv. Mater., 2020, 32, 1908047 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2021 |