
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-viral delivery of the CRISPR/Cas system: DNA versus RNA versus RNP†
Yi
Lin
a,
Ernst
Wagner
 *a and
Ulrich
Lächelt
*ab
*a and
Ulrich
Lächelt
*ab
aDepartment of Pharmacy and Center for NanoScience (CeNS), Ludwig-Maximilians-Universität München, Munich, Germany. E-mail: ernst.wagner@lmu.de; ulrich.laechelt@univie.ac.at
bDepartment of Pharmaceutical Sciences, University of Vienna, Vienna, Austria
First published on 25th January 2022
Abstract
Since its discovery, the CRISPR/Cas technology has rapidly become an essential tool in modern biomedical research. The opportunities to specifically modify and correct genomic DNA have also raised big hope for therapeutic applications by direct in vivo genome editing. In order to achieve the intended genome modifications, the functional unit of the CRISPR/Cas system finally has to be present in the nucleus of target cells. This can be achieved by delivery of different biomolecular Cas9 and gRNA formats: plasmid DNA (pDNA), RNA or Cas9 ribonucleoproteins (RNPs). While the initial research focussed on pDNA transfections, the currently most promising strategy for systemic non-viral in vivo delivery is based on RNA which has achieved remarkable results in the first clinical trials. RNP delivery receives much attention for ex vivo applications, but the translation to systemic in vivo genome editing in patients has not been reached so far. The article summarises the characteristics and differences of each format, provides an overview of the published delivery strategies and highlights recent examples of delivery systems including the status of clinical applications.
 Yi Lin | Yi Lin received his Master's degree in Pharmaceutics from National Engineering Research Center for Biomaterials (NERCB), Sichuan University, China in 2018. He is currently pursuing his Ph.D. at the Department of Pharmacy, Ludwig-Maximilians-Universität (LMU) München under the supervision of Prof. Ulrich Lächelt and Prof. Ernst Wagner. The current research of him focuses on the development of artificial oligoaminoamide peptides for Cas9/sgRNA ribonucleoprotein delivery and cancer therapy. |
 Ernst Wagner | Ernst Wagner is professor of Pharmaceutical Biotechnology at LMU and Center of Nanoscience since 2001. Previously he was Director Cancer Vaccines, Boehringer Ingelheim (first polymer-based gene therapy trial), group leader at IMP Vienna, postdoc at ETH Zurich and received a PhD in chemistry (TU Vienna) in 1985. He is Academician of European Academy of Sciences, member of College of Fellows of the Controlled Release Society, board of German Society for Gene Therapy and has authored 486 publications. |
 Ulrich Lächelt | Ulrich Lächelt is an assistant professor at the Deparment of Pharmaceutical Sciences of the University of Vienna since 2021. He studied pharmaceutics at the University of Heidelberg, received the degree of Doctor of Natural Sciences in the field of Pharmaceutical Biology at the LMU Munich in 2014 and was a group leader at the Chair of Pharmaceutical Biotechnology until his appointment at the University of Vienna. His research focuses on the intracellular delivery of biomolecules and the development of nanopharmaceuticals based on inorganic–organic hybrid materials. |
Introduction
The CRISPR/Cas9 system, which was initially discovered as a type of adaptive immune system in bacteria,1 revolutionized biomedical research due to the applicability as a programmable endonuclease.2 The genome editing tool is composed of the endonuclease Cas9 and a guide RNA (gRNA) which guides the protein to the target DNA sequence followed by a proto-spacer adjacent motif (PAM). The double-stranded DNA is unwound and a double-strand break (DSB) introduced 3–4 base-pairs upstream of the PAM sequence. The cellular machinery repairs DSBs in genomic DNA primarily via two distinct pathways (Fig. 1), non-homologous end joining (NHEJ) and homology directed repair (HDR).3,4 NHEJ represents the direct re-ligation of the cleaved DNA strands without involvement of a homologous DNA template. This error prone mechanism can lead to random insertions and deletions of nucleotides (indels) at the cleavage site which in part result in frameshift mutations and target gene knockout. In presence of a DNA template with homologous flanks, HDR mediates an accurate repair and the insertion of a defined DNA sequence, embedded between the homology arms at the cleavage site, can be realized. These strategies of using the cellular DSB repair machinery for introducing gene knock-outs or knock-ins are generally independent of the utilized endonuclease. However, the flexible control over the sequence recognition by simple variation of the gRNA component explains the uniqueness of CRISPR/Cas9 compared to other genome editors, such as transcription activator-like effector nucleases (TALENs) or zinc finger nucleases (ZFNs), which require individual protein design for each alteration of the target site.5 The functional unit of the CRISPR/Cas system is represented by the Cas9/gRNA ribonucleoprotein (RNP) complex which has to reach the nucleus of eukaryotic cells (eventually together with template DNA) for mediating the intended genomic modifications. To achieve this, different biomolecular formats can be selected and utilized for cellular delivery: plasmid DNA (pDNA) encoding for the Cas9 protein and the specific guide RNA (gRNA); a mixture of Cas9 mRNA and gRNA; or the pre-assembled RNP complex. Each strategy has its own features, prerequisites, advantages or drawbacks. This review presents approaches for the cellular delivery of the different biomolecular CRISPR/Cas9 formats and highlights their differences and characteristics.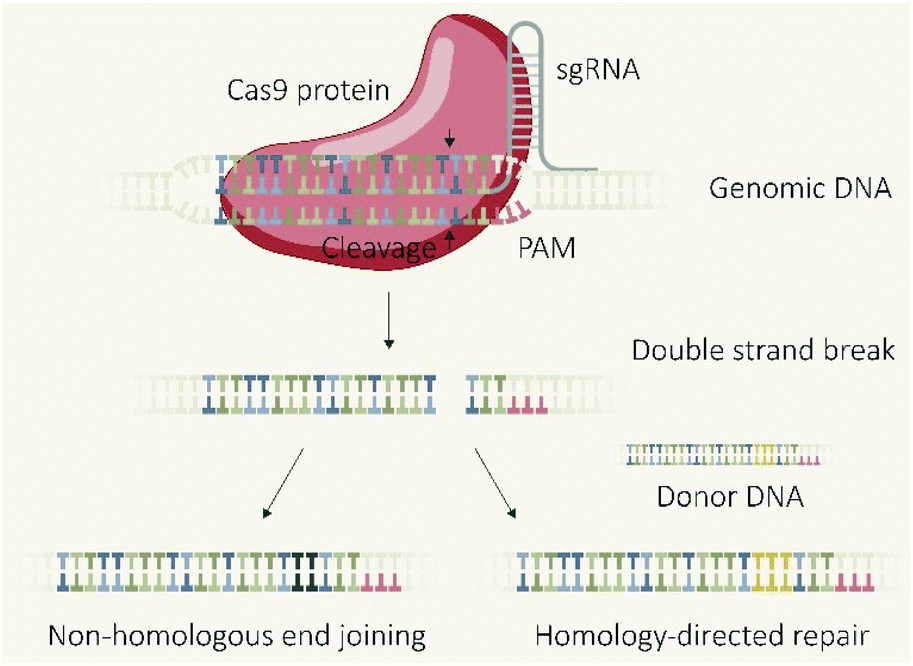 | ||
| Fig. 1 Mechanisms of CRISPR-Cas9 genome editing. | ||
Molecular basis of DNA, RNA and protein transfer
To achieve intended genome modifications with the CRISPR/Cas system, the RNP complex has to be present in the nucleus of target cells. In general, the artificial introduction of exogenous proteins into cells can be achieved by different strategies: direct cellular transport of the protein of interest, or deposition of genetic ‘blue prints’ which induce production of the foreign protein by the cellular machinery. As ‘blue prints’ based on the universal genetic code, DNA expression cassettes and mRNA can be used to trigger the protein production. Although both types of nucleic acids can achieve the same result, their application differs due to the involved biological mechanisms and individual characteristics. To understand the potential implications of different biomolecular formats on genome editing, the general characteristics and molecular basis of DNA, RNA and protein delivery are discussed first.DNA
Plasmid DNA (pDNA) is generally used as vector for non-viral DNA delivery, since the circular pDNA is not prone to degradation by exonucleases, can be complexed by common transfecting agents in more condensed form, is internalized into cells more efficiently and consequently achieves higher transgene expression than linear DNA constructs.6,7 The latter are mostly used, if a stable integration into the host genome is intended which preferentially occurs via homologous recombination of DNA with free ends, even though genome insertion can also occur with transferred pDNA at a rather low frequency. A technical advantage of pDNA is its facile and convenient production via amplification in bacteria. The other side of the coin is that bacterial backbone elements, such as a prokaryotic origin of replication, antibiotic resistance genes and unmethylated CpG motifs, can provoke immune reactions and short-lived transgene expression. To overcome this drawback, circular DNA constructs, only containing the expression cassette for the gene of interest (minicircle DNA), can be generated resulting in a smaller size, lower immunogenicity and longer persistence of transgene expression.8,9 Consistent with the flow of genetic information inside our cells, DNA first has to enter the nucleus for being transcribed into the corresponding mRNA, which is then translocated to the cytosol.RNA
By using mRNA as genetic ‘blue print’, the first steps of nuclear translocation and transcription can be omitted and the mRNA is directly translated by the ribosomes in the cytosol. As a result, mRNA transfer leads to a faster kinetic and earlier onset of protein expression in a higher number of cells compared to pDNA transfections. Single-cell analysis studies showed that the onset of protein expression occurs within a narrow time window a few hours after mRNA transfection, whereas pDNA transfections exhibit a higher variance of expression onset time in different cells, presumably due to the cell cycle dependence of nuclear translocation (Fig. 2).10 This also explains that the delivery of mRNA into non-dividing cells is generally more efficient, since the nuclear entry of pDNA is favored during the mitotic breakdown and reformation of the nuclear envelope.11,12 The reproducible and controllable nature of mRNA expression even enables approximation of intracellular pharmacokinetics represented by the area under the curve (AUC) of the intracellular protein product (Fig. 3).13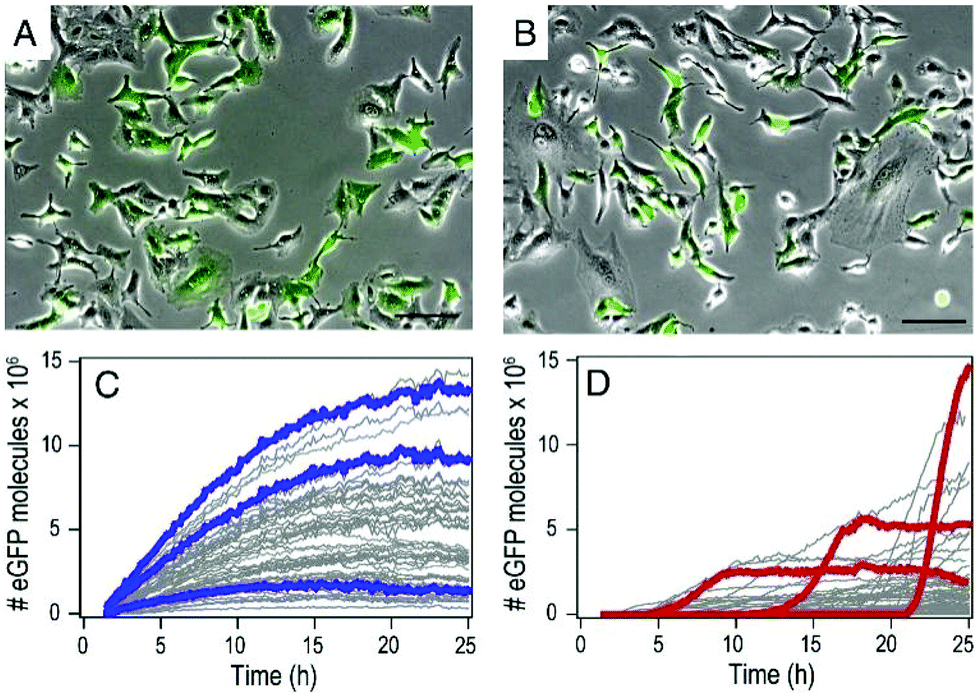 | ||
| Fig. 2 mRNA- and pDNA-mediated GFP expression kinetics in A549 cells. (A and B) Exemplary fluorescence microscopy images 25 h after transfection. (C and D) Representative GFP fluorescence time-courses after mRNA (C) and pDNA (D) transfections determined by single-cell analysis. mRNA transfections mediate early onset and continuous increase of GFP fluorescence, whereas pDNA transfections show less homogenously timed expression in different cells. Reproduced from ref. 10. Copyright (2014), with permission from Elsevier. | ||
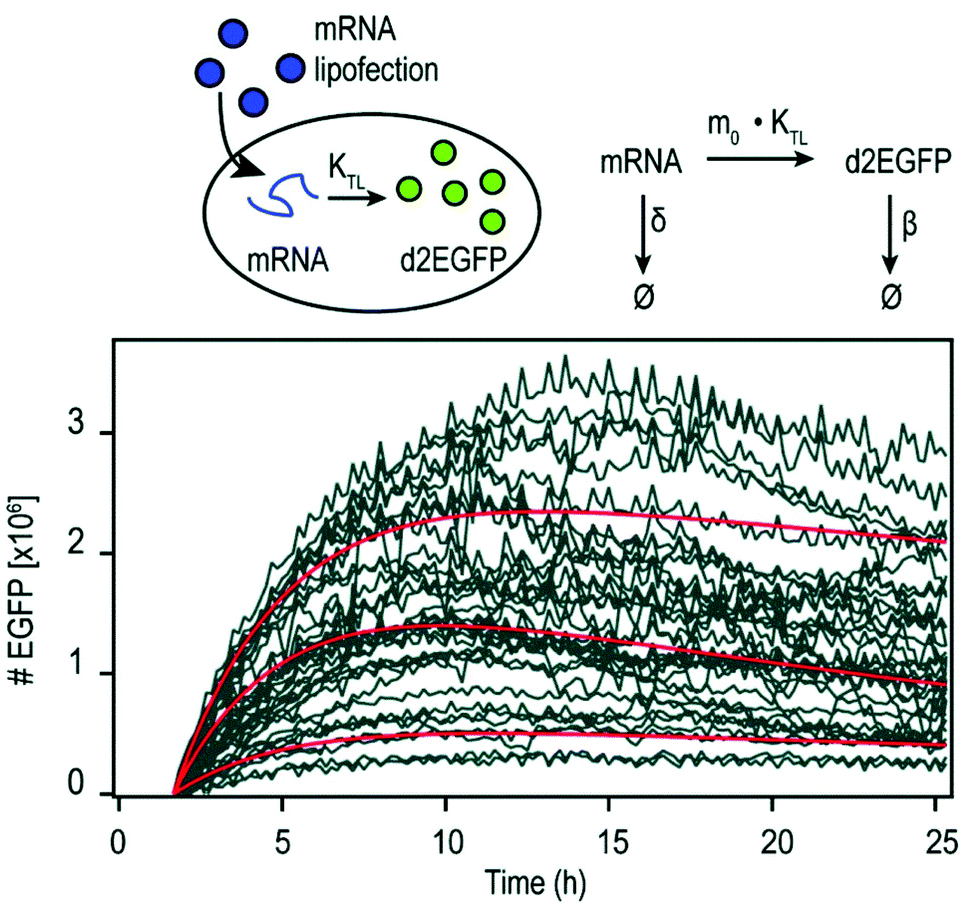 | ||
| Fig. 3 Pharmacokinetics of mRNA transfections. Top: Illustration of cellular mRNA lipofections and the reaction scheme describing underlying pharmacokinetic processes. Bottom: Exemplary time-courses of mRNA-mediated d2EGFP expression in A549 cells derived from micropattern-based single-cell arrays. The pharmacokinetics of intracellular protein production can be described by AUC = 0.48 × m0 × kTL × τmRNA × τProtien where AUC is the cumulative time-dose of the protein, m0 is the applied mRNA dose, kTL is the translation efficiency, and τ is the half-life of the mRNA or protein, respectively. Reproduced from ref. 13 under the terms of the Creative Commons CC BY-NC 3.0 License (2015). | ||
Already in the 1960s Smull et al., Vaheri and Pagano reported the use of histone, protamine sulfate and diethylaminoethyl dextran to enhance the infectiousness of viral RNA, which can be considered historical first nucleic acid transfections.14,15 In 1990, Wolff et al. published the first example of in vivo protein expression upon mRNA injection in mice.16
Although the utilization of mRNA as protein encoding nucleic acids has obvious advantages and first reports about RNA transfer in vitro and in vivo were known for decades, the research field required considerable time to establish suitable technologies and to show general feasibility of the approach. With the approval and extensive application of mRNA-based COVID-19 vaccines in 2021, the outstanding potential of mRNA transfer has finally been demonstrated impressively. Major hurdles for the development of mRNA-based therapeutics were the high innate immunogenicity of exogenous mRNA due to activation of Toll-like receptors, as well as a general low chemical stability and high nuclease sensitivity of RNA which are intrinsic properties of this particular type of nucleic acids.17,18 While DNA is the biological medium for long term storage of genetic information, RNA mainly serves as transient transmitter or regulator with a short half-life in biological fluids of only a few minutes. The group of Remaut investigated the decay kinetics of pDNA and mRNA and determined a half-life in biological samples of approximated 1–2 minutes for mRNA and 1–4 hours for pDNA (Fig. 4A).19 In fact, a high mRNA turnover rate, as a result of the interplay between regulated RNA synthesis and fast degradation, is essential to generate a dynamic transcriptome and keep cellular homeostasis. However, this makes clear, why the gene therapy research initially focused on the transfer of DNA, with higher stability and longer half-life, instead of RNA. This changed with the technological innovations and the development of chemically modified and codon-optimized mRNA with low immunogenicity, tuneable stability and high translatability.20,21 Heissig et al. used fluorescence lifetime imaging microscopy to assess the intracellular degradation of FRET-pair labelled short RNA oligonucleotides with different chemical modification patterns (phosphorothioate, 2′-O-Me, 2′-F).22 After transfection into HeLa cells, the intracellular integrity of the different oligonucleotides was monitored over time and a clear correlation with the extent of modification was confirmed (Fig. 4B).
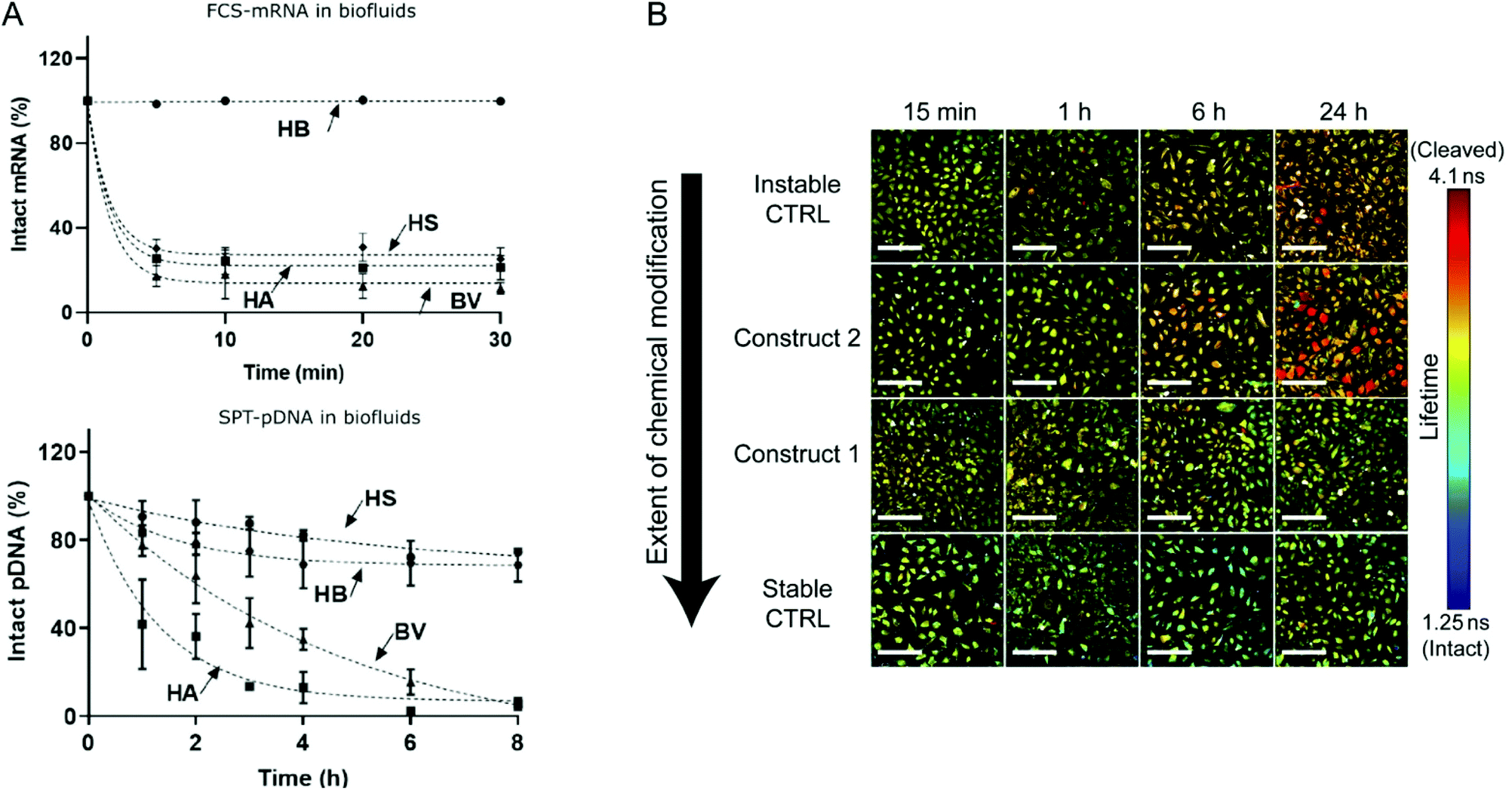 | ||
| Fig. 4 (A) Kinetics of mRNA and pDNA degradation in biological fluids. HEPES buffer (HB), human serum (HS), human ascites (HA), and bovine vitreous (BV) at 37° C. Top: mRNA decay as determined by fluorescence correlation spectroscopy. Bottom: Cy5-pDNA decay as determined by single particle tracking analysis. Reproduced with permission from ref. 19. Copyright 2020 Wiley-VCH GmbH. (B) Intracellular integrity of single stranded RNA molecules in dependence of chemical modifications visualized by fluorescence life time imaging. Reproduced with permission from ref. 22 under the terms of the Creative Commons CC BY 4.0 License (2017). | ||
Proteins
The direct delivery of proteins of interest represents an obvious strategy to achieve well controllable introduction into cells. Considering the intracellular pharmacokinetics, direct protein delivery achieves instantaneous availability without requirement of gene expression or mRNA translation and the duration of persistence only depends on the degradation of the protein. Protein degradation in eukaryotic cells mainly occurs via the ubiquitine-proteasome system or proteolysis in lysosomes and the half-lifes of different proteins vary significantly (Fig. 5). Therefore, the kinetic of intracellular availability can be expected to simplify to a dependency on the two parameters of protein dose and half-life, described by the exponential decay equation (Fig. 5A).23 | ||
| Fig. 5 Degradation of endogenous proteins. (A) Abundance changes of different endogenous proteins and (B) the distribution of protein half-lives. The kinetic of intracellular protein degradation can be described by P = P0·e−kt, where P is the abundance of the protein at each time point t, P0 is the initial protein availability and k is the degradation rate constant. Reproduced from ref. 23 under the terms of the Creative Commons CC BY 3.0 License (2016). | ||
Intracellular delivery
Despite the differences in the molecular biology of pDNA and mRNA, the two types of nucleic acids share high similarity in their chemical composition on the sub-molecular level and their physicochemical properties are dominated by the negatively charged phosphate backbones. With the exception of physical entrapment in liposomes or porous nanoparticles, co-precipitation and biomineralization strategies, most non-viral pDNA and mRNA delivery system are based on the electrostatic interaction with positively charged groups of the delivery systems. Cationic or cationizable oligo- and polymers, lipids and lipidoids have intensely been used for the transfer of pDNA and mRNA into cells. Also, the lipid nanoparticle (LNP) formulations of the COVID-19 mRNA vaccines contain cationizable lipids as essential component. Despite nucleic acid binding, these serve an additional essential purpose within the LNP delivery system: the amino-function of the lipid, with specific pKa around 6, is protonated in the acidic environment of endosomes, which triggers a conformational change, disruption of the endosomal membrane and release of the internalized material into the cytosol. This ‘endosomal escape’ is a generally critical step within the cellular delivery pathway and has to be addressed by all systems which are internalized via an endocytotic uptake mechanism. Also most polymeric transfecting agents rely on a specific protonation in the endosomal environment. Polyethylene imine (PEI), which is utilized for pDNA transfections since the 1990s, is characterized by a broad protonation range, including the endosomal pH, due to the highly repetitive diaminoethane motif.24 The discovery of the chemical principle and favourable consequences for cellular gene transfer, made PEI an archetype of polymeric transfecting agents and inspired scientists around the world to include PEI-like motifs into more defined and better tolerated delivery systems. By dissecting the chemical structure of PEI into shorter oligoamine segments, the relevance of specific protonation characteristics and the relation to biological activity became unveiled, while cytotoxicity was frequently reduced.25–31Early demonstrations of intracellular protein delivery by chemical carriers were reported in the 1970s based on liposomes.32,33 In contrast to nucleic acids which are, despite the existence of different types, relatively similar in their chemical composition and physico-chemical properties, proteins are an extremely diverse class of biomolecules. They vary in size, structural conformation, charge and hydrophilicity/hydrophobicity. Therefore, the development of generic strategies for protein delivery is sophisticated, and in many cases individual carrier design and optimization are required. Despite physical entrapment in liposomes, conjugation of cell penetrating peptides (CPPs) or protein transduction domains (PTDs)34–36 as well as nanomicelles,37–39 engineered nanocarriers40,41 and other strategies have been used for cellular protein delivery. Since classic nucleic acid transfecting agents, cationic polymers or lipids, generally only work with negatively charged proteins, a very flexible strategy is represented by charge-conversion. Here, the positively charged amino groups in proteins are reversibly converted into negatively charged functions, for instance via reaction with citraconic or cis-aconitic anhydride.42 The resulting protein derivatives with multiple negative charges are then susceptible for the electrostatic interaction and delivery by cationic transfecting agents. A similar aim is achieved by the genetic engineering of negatively charged protein analogs by fusion with supercharged proteins or peptide tags, which can enable electrostatic complexation by cationic delivery agents.43,44
Table 1 gives an overview over the different biomolecular formats for the introduction of exogenous proteins into cells and summarizes their properties and characteristics.
| pDNA | mRNA | Protein | |
|---|---|---|---|
| a The specified size limits of pDNA, mRNA and proteins are based on the considerations of: 1: minicircle DNA; 2: bacterial artificial chromosomes (BAC) as well as 3: the largest human protein titin and 4: the corresponding mRNA transcript. | |||
| Production | Bacterial amplification | In vitro transcription | Recombinant expression or isolation |
| Size | ≈ 2.000a1–300.000 bp![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a2 (≈ 1.200–185.000 kDa) a2 (≈ 1.200–185.000 kDa) |
≈ 500–100.000 nt a3 (≈ 160 kDa–32.000 kDa) | ≈ 50–30.000 aaa4 (≈ 5–4000 kDa) |
| Properties and appearance | Double-stranded DNA, circular, negatively charged, hydrophilic | Single-stranded RNA, linear, negatively charged, hydrophilic | Diverse (molecular weight, charge, isoelectric point, hydrophilicity, etc.) |
| Stability | Relative chemical stability, nucleolytic instability | High chemical and nucleolytic instability | Proteolytic instability, sensitivity to denaturation depending on individual protein |
| Kinetic | Variable onset of protein expression, cell cycle dependence, long duration of persistence possible | Fast and homogenous onset of protein expression, short duration of persistence | Ab initio availability of protein, minimal duration of persistence (dependent on protein half-life) |
| Individual problems | Risk of genome integration, potential immunogenicity due to bacterial sequences (not for minicircle DNA) | Chemical modifications required to increase stability and reduce immunogenicity | Potential immunogenicity, protease sensitivity, no generic delivery technologies for all kind of proteins |
| Delivery technologies | Ionic interaction with cationic polymers or lipids, encapsulation in or adsorption to inorganic nanoparticles | Conjugates (CPPs, polymers), encapsulation into micelles or liposomes, ionic interaction of negatively charged or charge-converted proteins with cationic polymers or lipids, encapsulation in or adsorption to inorganic nanoparticles | |
Physicochemical and physiological characteristics of different CRISPR/Cas9 formats
Cas9 plasmid DNA
Typical and well-established SpCas9 plasmids for transfection of mammalian cells are pX260 and pX330 from the Zhang laboratory. pX260 is an older CRISPR/Cas plasmid system, which contains three expression cassettes; one is used to drive SpCas9 nuclease expression and the other two are used to express the CRISPR RNA array and tracrRNA.45 At present, the most commonly used SpCas9 plasmid is pX330 that contains only two expression cassettes, a human codon-optimized SpCas9 and a chimeric single guide RNA. It has 8484 bp with 1.68 × 104 negative charges and a molecular weight of around 5.24 × 103 kDa. Generally, the pX330 vector can be digested by the restriction enzyme BbsI to insert a pair of annealed oligonucleotides that are designed based on the specific target site (20 bp) upstream of the NGG PAM sequence. Larger plasmid constructs containing an additional reporter gene (e.g. pX458 encoding GFP) to identify and enrich transfected cells, or a selection marker (e.g. pX459 encoding resistance to puromycin) to generate stable cell lines are also available.45Cas9 mRNA and sgRNA
Different from the two-in-one Cas9 plasmids, co-delivery of two individual components, Cas9 mRNA and sgRNA, is required in case of RNA-based strategies of the CRISPR/Cas system. Cas9 mRNA with the length of approximately 4500 nt is a linearized single-stranded RNA which is produced via in vitro transcription (IVT) followed by 5′ capping and 3′ poly-A modification.46,47 The 5′ capping of IVT mRNA mimics the natural eukaryotic mRNA which has a 7-methylguanosine (m7G) cap at the 5′ end.48 It can protect the mRNA against exonuclease degradation and assists in eukaryotic initiation factor (eIF) 4E recognition and binding during translation.49 The poly-A tail is a long chain of adenine nucleotides. It is added to the 3′ end of the mRNA transcript to increase its enzymatic stability, and it can act synergistically with the 5′ m7G cap to regulate translation efficiency as well as enhance protein expression.50,51 With regard to mRNA stabilization, introduction of phosphorothioates especially into the 5′ untranslated mRNA sequence has been shown to increase protein translation and to result in up to 22-fold enhanced protein expression.52 Generally, endocytosed IVT mRNA can be recognized by several endosomal innate immune receptors (TLR3, TLR7 and TLR8) and cytoplasmic proteins such as melanoma differentiation-associated protein 5 (MDA5) and retinoic acid inducible gene I protein (RIG-I), which may induce immune responses and inhibit the function of mRNA.17,53,54 The intrinsic immune activation activity of IVT mRNA is usually considered beneficial for immunotherapeutic vaccination, but a major disadvantage for non-immunotherapy-related applications since the induced immune responses can slow down the translation of mRNA and promote RNA degradation.53,54 Incorporation of chemically modified nucleotides, such as 2-thiouridine (s2U), 5-methyluridine (m5U), pseudouridine (Ψ), N1-methylpseudouridine (m1Ψ), 5-methylcytidine (m5C), 5-hydroxymethylcytosine (5hmC), into mRNA sequences are the mostly used strategies to overcome the high immunogenicity of IVT mRNA.20,55–57 Besides, some studies showed that lower immunogenicity can be achieved by sequential engineering and the use of high-performance liquid chromatography (HPLC)-purified mRNA.58,59Another key component of the RNA-based strategy is the sgRNA which has a smaller size of approximately 100 nt. sgRNA can either be generated by IVT or, due to the much shorter length, by solid-phase synthesis.60,61 IVT is a widely used approach for sgRNA production, since it is cost-effective, high-yielding and easy to implement. On the other hand, IVT sgRNA can be expected to face similar problems as other IVT generated RNA products, such as IVT mRNA, which could limit its applicability in vivo. Firstly, IVT RNA contains diverse variants with different lengths and structures which may cause variable and inconsistent editing efficiencies.53,62 Secondly, the stability of unmodified IVT RNA generally is not favourable for in vivo usage.63 Even if modified nucleotides are used for the IVT reaction, random incorporations of the nucleotides into the RNA sequence can still be a non-negligible issue.64 With regards to the CRISPR/Cas system, it has been reported that IVT sgRNA can induce immune responses and even cause cell apoptosis.65–67 In contrast to IVT sgRNA, chemically synthesized sgRNA is structurally well-defined and homogenous. Synthetic sgRNA which lacks a 5′-triphosphate group is less immunogenic and does not induce detectable immune response in many cell types.66–68 Moreover, the chemical oligonucleotide assembly enables site-specific incorporation of modified nucleotides during solid-phase synthesis. Various chemically modified nucleotides, including 2′-methyl (M), 2′-O-methyl-PS (MS), 2′-O-methyl-3′-thiophosphonoacetate (MSP), phosphorothioates, 2′-fluoro (F), 2′-O-methyl-3′-phosphonoacetate (MP), locked nucleic acids (LNA) and bridged nucleic acids (BNA), are developed to enhance the enzymatic stability, editing efficiency, specificity as well as to reduce the immunogenicity and off-target effects of sgRNA.69–77
Cas9/gRNA ribonucleoprotein (RNP)
Similar to the RNA strategy, protein-based delivery of the CRISPR/Cas system also requires two critical components in the formulation: the Cas9 nuclease and a guide RNA (gRNA). But in contrast to the mRNA-based strategy, which requires co-delivery of the discrete mRNA and sgRNA species, the Cas9 protein is first pre-complexed with gRNA to form the negatively charged Cas9/gRNA ribonucleoprotein (Cas9 RNP) complex, which can then be delivered as one single cargo entity. The typical SpCas9 protein comprises 1368 amino acids with a molecular weight of 158.4 kDa and a net positive charge of +22.44,78 The structure of the SpCas9 protein exhibits two lobes: an α-helix recognition lobe (REC) and a nuclease lobe (NUC).79 These two lobes are linked by the arginine-rich bridge helix (Arg) and the disordered linker (DL).79 The NUC lobe contains a RuvC domain and a HNH nuclease domain for cleavage of the target DNA, as well as a C-terminal domain (CTD) for recognizing the PAM sequence.79 The REC lobe consists of three α-helical domains and plays a vital role in recognition of DNA, regulation of conformational transition of the HNH nuclease domain and directing HNH to the cleavage site.79,80As gRNA component for Cas9 RNP assembly, two different variants can be utilized: (1) a crRNA:tracrRNA duplex, or (2) a single guide RNA (sgRNA). The two-component guide RNA represents the naturally occurring form of the bacterial CRISPR/Cas9 system and comprises a CRISPR RNA (crRNA) with a custom 20 nt targeting sequence and a trans-activating crRNA (tracrRNA), which is crucial for Cas9 recruitment.81 sgRNA is created by fusing the crRNA and tracrRNA sequences via a scaffold loop into a single RNA chimera, which became the most popular format for RNP-based CRISPR delivery. In the absence of gRNA, the Cas9 protein remains in an inactive conformation.82 When it is bound to gRNA, the REC lobe undergoes a conformational change which converts the inactive protein into the active RNP form.79,82 Since the binding of gRNA is highly associated with the functional domains of the Cas9 protein, the types and positions of modified nucleotides need to be carefully selected to avoid a potential interference with RNP formation. A very special feature of Cas9 RNPs, with high relevance for delivery, is the charge-conversion of the positively charged Cas9 protein upon binding of the negatively charged sgRNA component. The work by Zuris et al. on genetically engineered supercharged proteins demonstrated, that the sgRNA component in Cas9 RNPs already provides sufficient negative charge for the delivery with cationic lipids.44 Kuhn et al. reported the cellular delivery of Cas9 RNPs with oligoaminoamides, containing lipid-modifications and stabilizing tyrosine trimers,83 and determined by fluorescence correlation spectroscopy that the cationic oligomers only interact with fluorescently labelled Cas9, if the negatively charged sgRNA has been loaded.84 These results illustrate a significant characteristic of Cas9 RNPs which provides the basis for cellular delivery by cationic transfecting agents without requirement of protein derivatization or individual nanocarrier design.
Comparison of different formats of CRISPR/Cas9
Since each format has its intrinsically unique physicochemical and physiologic features, it is crucial to understand how these features affect the Cas9 action process and then select the best format for practical applications. The most direct distinction is the stability. Cas9 plasmid DNA (Cas9 pDNA) possesses the highest stability among the three formats. Chemically, DNA contains deoxyribose while RNA contains ribose. The absence of the single hydroxyl group in 2′ position of the pentose ring makes DNA more stable and less reactive than RNA.85 Moreover, Cas9 pDNA is a circular double-stranded DNA which is not vulnerable to exonucleases.86,87 Although Cas9 mRNA is more fragile than Cas9 pDNA, chemical modifications on the nucleotides can be applied to improve the stability as mentioned above.63,88Compared to pDNA and mRNA, which contain only a single type of biomolecules, Cas9 RNPs consist of protein and sgRNA and are susceptible to degradation by both proteases and RNases, hypothetically making it the least stable format. Besides, the Cas9 protein may not be resistant to organic solvents and is easily denatured. For example, denaturation of Cas9 RNP is observed in acidic citrate buffer at pH 4.0, which represent standard conditions for the formulations of nucleic acid loaded LNPs.89
Another obvious difference is the place of action. As discussed in the previous section, nuclear entry is an essential prerequisite for the transfer of pDNA. In contrast, Cas9 mRNA only requires delivery to the cytoplasm where the translation process may start immediately. However, in both cases, the RNP complex has to be assembled with gRNA after the Cas9 protein has been produced, which inevitably results in a certain latency between the intracellular availabilities of the protein and RNA components. In this regards, delivery of a priori formed functional RNPs is the most direct way for cellular genome editing, which generally is also considered the most efficient strategy.90 As a last step, the Cas9 RNP has to enter the nucleus to reach its target site and for this reasons nuclear localization signal (NLS)-fused Cas9 is commonly used.91 As discussed above, the duration of intracellular persistence depends on the stability of the genetic ‘blue prints’ and degradation of encoded protein. For genome editing applications, the CRISPR/Cas components are only transiently required until the intended genome modification has occurred; an additional presence in the cell generally is not favored, since the risk of off-target events can increase with the duration of presence in the cell. Therefore, a high control over the availability of the functional CRISPR/Cas system and a degradation or inactivation after the intended task has been fulfilled, are desirable. Due to the more rapid degradation within the target cells, Cas9 RNP and mRNA provide a shorter exposure time of the cellular genome to active Cas9 proteins and decrease the potential risk of off-target effects.92,93 In contrast, Cas9 pDNA can mediate longer gene expression that may increase off-target editing events.94
Independent of the biomolecular format, additional strategies to minimize mutations at unintended sites include shortening Cas9 half-life by tagging with degradation signals,95,96 reducing Cas9 activity by coupling to inhibitory domains,97 or selective activation of Cas9 variants by external triggers such as chemical reagents98,99 or light.100 In addition, other Cas9 variants have been designed to optimize the on-/off-target ratio, such as catalytically inactive Cas9 fused to the catalytic domain of the endonuclease FokI (dCas9-FokI),101 mutated variants with attenuated activity at mismatch targets, such as high-fidelity Cas9 (SpCas9-HF1),102 enhanced specificity Cas9 (eSpCas9),103 hyper-accurate Cas9 (HypaCas9),104 evoCas9,105 Sniper-Cas9106 and expanded PAM SpCas9 (xCas9).107 Nickases, generated by deactivation of the nucleolytic RuvC (Cas9-D10A) or HNH (Cas9-H840A) domain, induce single-strand nicks instead of double-strand breaks and therefore require a pair of gRNAs for gene disruption, which strongly increases the specificity.2,45,108
Apart from their stability and action features, safety is a major concern, especially considering potential in vivo applications. As stated above, random integration of Cas9 pDNA into the host genome may cause potentially harmful insertional mutagenesis as well as persistent Cas9 expression that can lead to off-target effects and MHC class I immune responses.92,109 In this regard, Cas9 RNP and mRNA appear as the safer choices for genome editing. Besides, the main type of immune response induced by these three formats is also different. Cas9 pDNA and mRNA mainly trigger innate immune responses by activating Toll-like receptors (TLRs), while Cas9 RNP can induce adaptive immune reactions.109–113 Recent studies have demonstrated that anti-Cas9 antibodies and T cells are pre-existing due to preceding bacterial infections.109,112,113 IgG antibodies against SpCas9 and anti-SpCas9 T cells were detected in 58% and 67% of human adults, respectively.109
Although Cas9 mRNA and RNP have certain advantages in terms of editing efficiency and safety, the costs are generally higher than for pDNA, since pDNA design, production and scale up are more feasible. In contrast, obtaining pure active Cas9 proteins is a time-consuming and complex process, which includes a series of critical steps, such as protein isolation from bacterial cultures, purification, suitable storage of the sensitive protein product etc.114 Notably, endotoxin contamination could occur during both pDNA and Cas9 protein preparation, while the cell-free mRNA production exhibits a much lower risk. Finally, the clinical translation of nucleic acid-based therapeutics is more mature than intracellular delivery of proteins, which is impressively demonstrated by the recent success of siRNA-based drugs and mRNA vaccines.115,116 To sum up this section, the individual CRISPR/Cas9 formats differ in their physicochemical properties, stability, action features, safety and production. Fig. 6 presents a summary of the distinction and individual characteristics. Notably, none of these formats can work without a suitable and efficient delivery technology. Therefore, the delivery strategies utilized for pDNA, mRNA and RNP-based CRISPR/Cas9 systems are reviewed in the following section.
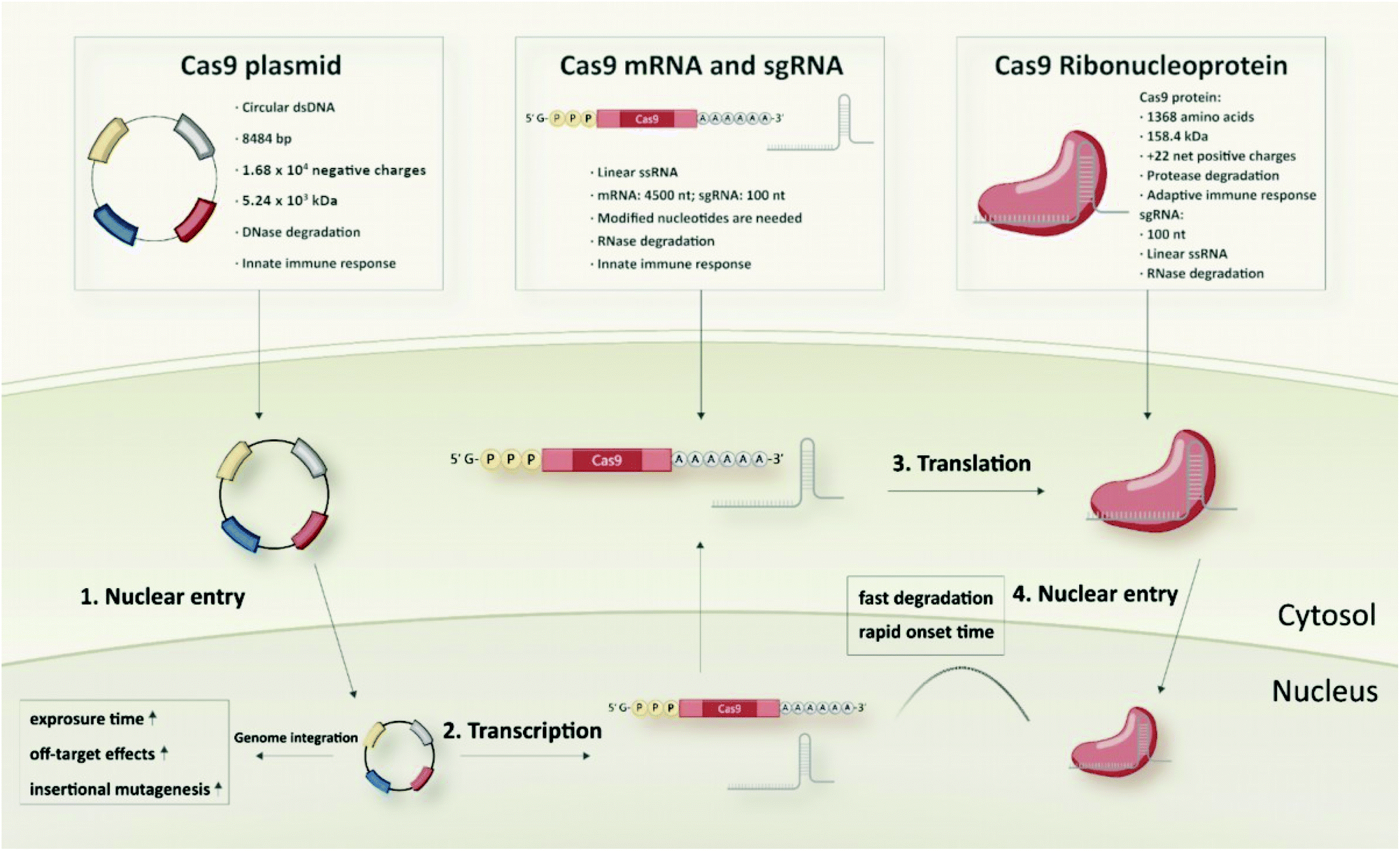 | ||
| Fig. 6 Comparison of different biomolecular CRISPR/Cas9 formats: Cas9 plasmid, Cas9 mRNA and sgRNA, and Cas9/sgRNA ribonucleoprotein. | ||
Non-viral delivery of different CRISPR/Cas9 formats
Up to now, various delivery strategies have been utilized for the delivery of the CRISPR/Cas9 system, which can be categorized into three general classes: (1) physical methods, (2) viral vectors, and (3) non-viral vectors.117 Several of these systems have already been utilized for ex vivo gene therapy approaches by applying genome editing to patient cells in clinical studies118 and technologies have been tested in direct in vivo applications, including systemic administration to patients.119 This article focusses on the non-viral vectors which are generated from diverse synthetic and bioderived materials. In general, each vector is designed for one specific Cas9 format, although there are a few examples that use polymers or nanoparticles for delivery of two or all three formats.120–123 Therefore, it is crucial for researchers to know the developed strategies for each format and which problems still remain. Given the different physicochemical and physiological features of Cas9 pDNA, mRNA and RNP, the materials and optimal ratio of each component in certain formulations will differ. In the following sections, the non-viral delivery systems of different CRISPR/Cas9 formats are discussed in detail, and specific examples of each class as well as its reachable target organs specified. In addition, a comprehensive overview over the different biomolecular formats and delivery strategies are provided in Table S1† (pDNA), Table S2† (RNA) and Table S3† (RNP).Delivery systems of Cas9 pDNA
In recent years, the FDA-approved polymer poly(lactic-co-glycolic acid) (PLGA) was frequently used for diverse drug delivery applications including nucleic acid delivery.131,132 Based on the success of cationic lipid-assisted PEG-b-PLGA nanoparticles (CLANs) for systemic siRNA delivery, Wang and colleagues developed a series of CLANs for cell type-specific delivery of Cas9 plasmids.133–137 For example, they encapsulated human CD68 promoter-containing Cas9 plasmids pM330 and pM458 into cationic BHEM-Chol lipid-assisted PEG5K-PLGA11K nanoparticles through a double emulsion method.136 The obtained CLANpM330 and CLANpM458 were efficiently taken up by T cells, B cells, neutrophils, monocytes and macrophages, but only expressed Cas9 protein in monocytes and macrophages due to the specific human CD68 promoter. Moreover, the functional system CLANpM330/sgNtn1 and CLANpM458/sgNtn1 induced Ntn1 deletion in monocytes and macrophages in vivo after intravenous injection. Interestingly, the target cell types of CLANs can be modulated by adjusting the mass fraction of PEG-PLGA and the ratio of the cationic lipid BHEM-Chol.137 A library of CLANs with various surface PEG densities and surface charges was established and a neutrophils targeting CLAN with lower PEG density and a higher surface charge was identified after screening in high-fat diet (HFD)-induced type 2 diabetes (T2D) mice. The optimized CLAN was able to deliver Cas9 pDNA to the neutrophils of the T2D mice and achieved disruption of the neutrophil elastase (NE) gene both in the liver and white adipose tissue, resulting in an improved glucose tolerance and insulin sensitivity. Besides, CLANs targeting B cells, chronic myeloid leukemia (CML) cells or dendritic cells (DCs) were generated.133–135 Yang et al. prepared lipid-polymer hybrid nanoparticles (LPHNs-cRGD) loaded with a plasmid pCas9/MGMT, targeting O6-methylguanine-DNA methyltransferase, based on lecithin, DSPE-PEG-cRGD, DSPE-PEG-biotin, PLGA and a nanoprecipitation method.138 The LPHNs were associated with microbubbles (MBs) composed of DPPC, DSPE-PEG-biotin and cholesterol via biotin–avidin interaction. Focused ultrasound (FUS) locally opened the BBB to facilitate the delivery of pCas9/MGMT into the brain in vivo, knockout MGMT in glioblastoma and to increase chemosensitivity towards temozolomide (TMZ) therapy. Alternatively to the use of lipids, coating of PLGA nanoparticles with chitosan (CS-PLGA NPs) was also reported.139In vitro knockout evaluation in HEK293T-GFP cells showed suppression of GFP expression up to 80% by CS-PLGA NPs.
To overcome the general toxicities of conventional transfection reagents, biodegradable polycations, such as poly(β-amino ester)s (PBAEs), were generated and used as gene delivery vehicles.140,141 Deng et al. designed a (1-(3-aminopropyl)-4-methylpiperazine) (AMP) modified PBAE (aPBAE) for delivering the CRISPR/Cas9 system to malignant tissues and knocking out cyclin-dependent kinase 5 (Cdk5) in vivo.142 In melanoma and breast cancer models, aPBAE/Cas9-Cdk5 significantly suppressed tumor growth and inhibited lung metastasis after intratumoral injection. To achieve higher transfection efficiency of aPBAE, Gao et al. utilized generation 0 PAMAM dendrimers (PAMAM-G0) as a branching unit of PAMAM-PBAE hyperbranched copolymers (hPPC) for Cas9 plasmid delivery.143 The study showed that hPPC1 with the highest degree of branching exhibited the strongest plasmid condensation capability and highest transfection efficiency compared with linear PBAE and the moderately branched hPPC2. The Green group synthesized a new class of bioreducible branched ester-amine quadpolymers (rBEAQs) for siRNA and DNA delivery by co-polymerizing acrylate monomers including the disulfide building block 2,2-disulfanediylbis(ethane-2,1-diyl) diacrylate in a one-pot Michael addition reaction.144 The best formulation of rBEAQs successfully co-delivered Cas9 pDNA and sgRNA into HEK293T-GFP cells and enabled 40% GFP knockout, which was the first time that co-delivery of Cas9 plasmid and sgRNA had been demonstrated for CRISPR-mediated genome editing. More recently, the same group designed a reporter system which enables differentiation between single cleavage edits and deletions mediated by two-fold cleavage edits for evaluating PBAE-based Cas9 pDNA and sgRNA co-delivery systems.145 The results demonstrated that a decrease of the total DNA dose significantly reduces two-fold cutting events but did not affect the extent of single cut editing.
Recently, it was reported that aminated poly(glycidyl methacrylate) (PGMA) could be a safe and effective gene vector.146 Zhang et al. reported the first example of CRISPR/Cas9-mediated Fbn1 gene editing in the aorta of adult mice using cholesterol (CHO)-terminated ethanolamine-aminated poly(glycidyl methacrylate) (PGEA) (Fig. 7A).147 The in vivo studies showed that the enrichment of the nanosystems in aortic tissues was improved by Angiotensin II (Ang II) infusion, and efficient Fbn1 gene disruption was observed in vascular smooth muscle cells (VSMCs) as well as the aorta. Nie et al. constructed a responsive charge-reversal carrier (Hep@PGEA/pCas9) which was composed of a negatively charged heparin core and a positively charged cyclodextrin-conjugated PGEA (CD-PGEA) shell to deliver Cas9 plasmids targeting the oncogene survivin for hepatocellular carcinoma therapy.148 In the reductive milieu of HCC cells with high glutathion concentration, the disulfide cross-linked heparin core disassembled to promote the release of Cas9 pDNA and mediate anti-tumor effects in an orthotopic HCC model. Moreover, the system further improved the therapeutic outcome of sorafenib, providing potential for a combination therapy of HCC.
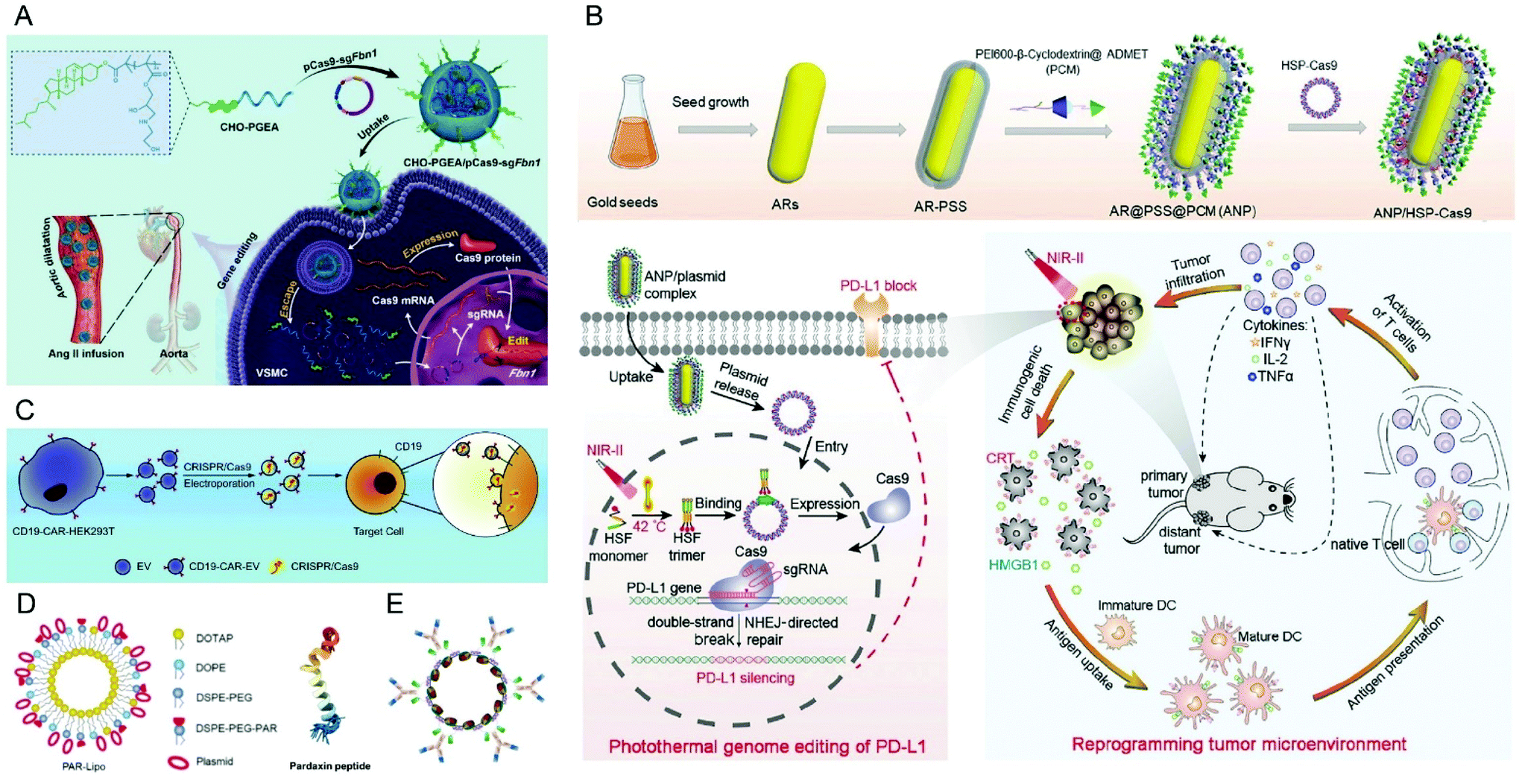 | ||
| Fig. 7 Representative delivery systems of Cas9 plasmids. (A) Cholesterol (CHO)-terminated ethanolamine-aminated poly(glycidyl methacrylate) (CHO-PGEA) for gene editing in the aorta. Reproduced with permission from ref. 147. Copyright 2019 John Wiley and Sons. (B) ANP/HSP-Cas9 plasmid complex for a photothermal genome-editing strategy and cancer immunotherapy. Reproduced with permission from ref. 165. Copyright 2021 John Wiley and Sons. (C) Tropism-facilitated delivery of Cas9 plasmids via CAR-EVs. Reproduced with permission from ref. 180. Copyright 2020 Elsevier. (D) Delivery of Cas9 plasmids through the non-lysosomal route using pardaxin peptide-modified liposomes. Reproduced with permission from ref. 161. Copyright 2020 John Wiley and Sons. (E) Antibody–chromatin complexes with Cas9 plasmids. Reproduced with permission from ref. 183. Copyright 2019 Oxford University Press. | ||
Apart from synthetic polymers, the natural cationic polysaccharide chitosan has a long tradition as transfecting agent and is still used for the creation of novel gene delivery systems.149 Recently, a chitosan-based stimuli responsive gene and drug co-delivery system was developed for HCC therapy.150 In this system, paclitaxel (PTX) and sgVEGFR2/Cas9 plasmid-preloaded chitosan was modified with β-galactose-carrying lactobionic acid (LA) to achieve asialoglycoprotein receptor (ASGPR) targeted delivery to HCC cells. In vivo knockout studies demonstrated 33.4% genome editing efficiency in tumor tissues and achieved synergistic therapeutic effects with PTX. To overcome the mucoadhesive properties of chitosan, Zhang et al. synthesized a series of PEGylated chitosan co-polymers by conjugating methoxy PEG (mPEG) to chitosan, and the obtained PEGylated chitosan was then evaluated for CRISPR/Cas9 plasmid delivery.151 An in vitro mucus model demonstrated that PEGylation of chitosan could improve the mucus-penetration of the nanocomplexes.
Xu's group developed a series of polycations for Cas9 plasmid delivery via a one-pot ring-opening reactions.152–154 By this strategy, an acid-responsive polycation (ARP) with numerous ortho ester linkages and hydroxyl groups was synthesized and subsequently fluorinated to produce ARP-F.152 The obtained ARP-F compacted the plasmid pCas9-survivin within stable nanoparticles and inhibited tumor growth in vivo by disrupting the survivin gene. The combination of the nanosystem with temozolomide demonstrated improved antitumoral effects due to an increased sensitivity towards the anticancer drug. Recently, Wang et al. fabricated PEGylated nanoparticles (P-HNPs) based on poly(γ-4-((2-(piperidin-1-yl)ethyl)aminomethyl)benzyl-L-glutamate) (PPABLG) for Cas9 plasmid and sgPlk1 delivery.155 The P-HNPs exhibited excellent colloidal stability and achieved 35% Plk1 knockout in HeLa tumors, resulting in 66.7% down-regulation of Plk1 protein levels, 71% tumor growth inhibition and 60% higher animal survival rate within 60 days. Self-assembled micelles consisting of quaternary ammonium-terminated poly(propylene oxide) (PPO-NMe3) and amphiphilic Pluronic F127 were designed by Lao et al. for Cas9 plasmid delivery.156 The optimized micelle system could efficiently delete the E7 oncogene in HeLa cells, which induced an inhibition of the downstream cancerous activity both in vitro and in vivo. More recently, Emami et al. developed biocompatible 4-arm polyrotaxane (PRX) nanoparticles and optimized them for genome editing. They found that 4-arm PRX modified with a redox-sensitive disulfide linker could improve intracellular pDNA release. The data also demonstrated that 4-arm PRX equipped with the cell penetrating peptide PipB and a peptide targeting the neural cell adhesion molecule (NCAM) successfully delivered Cas9 plasmids to muscle cells and enabled deletion of the DMD exons 45–55.157
Delivery systems of Cas9 mRNA and sgRNA
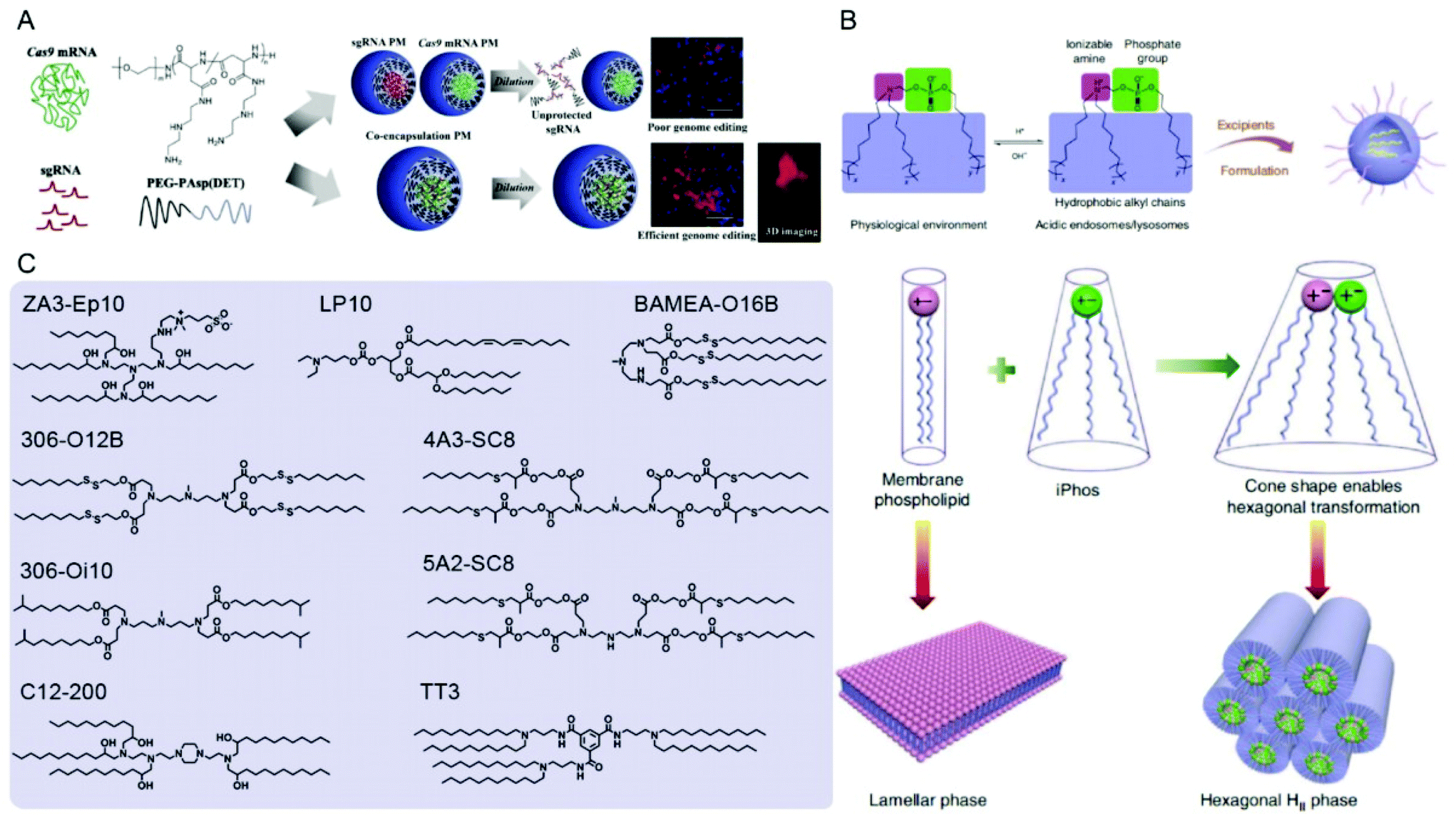 | ||
| Fig. 8 Representative delivery systems of Cas9 mRNA and sgRNA. (A) Poly(aspartamide)-based Cas9 mRNA/sgRNA polyplex micelles (PMs) for genome editing in the mouse brain. Reproduced with permission from ref. 187. Copyright 2021 Elsevier. (B) Multi-tailed ionizable phospholipids (iPhos) with enhanced hexagonal transformation capability at endosomal pH. Reproduced with permission from ref. 197. Copyright 2021 Springer Nature. (C) Representative lipids used for Cas9 mRNA and sgRNA delivery. | ||
In addition to SORT LNPs, other cell-selective or targeted LNPs were generated for specific applications. For instance, Peer and colleagues modified LNPs by coating them with anti-hEGFR to target human serous ovarian adenocarcinomas Ovcar8 (OV8) that highly express EGFR.199 Intraperitoneal injections of EGFR-targeted sgPlk1-LNPs into mice with disseminated peritoneal OV8 tumors achieved up to ∼80% gene editing in vivo, resulted in tumor growth inhibition and increase of survival by 80%. Tang et al. designed sialic acid-targeted phenylboronic acid (PBA)-derived LNPs which achieved much higher knockouts in HeLa cells than in non-cancer cells.200
The high potential of LNPs for RNA delivery in humans is not only demonstrated by the extensively used mRNA COVID-19 vaccines, but also the first clinical trial of systemic CRISPR-Cas9 in vivo gene editing, which is based on Cas9 mRNA and sgRNA.119 Gillmore et al. recently published interim results of the phase 1 clinical study. Six patients suffering from hereditary transthyretin amyloidosis (ATTR) were treated with a CRISPR LNP formulation targeting the transthyretin (TTR) gene to reduce the concentration of misfolded protein in serum. Single administrations resulted in 52% mean reduction of TTR serum levels at a dose of 0.1 mg kg−1 and 87% reduction at 0.3 mg kg−1. In a safety assessment period within 28 days after infusion only few and mild adverse reactions were observed. The study can be considered a milestone in the development of in vivo genome editing therapeutics and presumably is just the beginning of numerous developments in the future.
Delivery systems of Cas9 RNPs
In addition to PEI, cationic polypeptides such as polylysine and poly(aspartic acid) with cationic side-chain modifications were developed which generally feature higher biocompatibility and lower toxicity.205,206 Liu et al. reported a nano-Cas9 RNP system (nanoRNP) based on 2,5-dihydro-2,5-dioxofuran-3-acetic acid (CA)-bridged polylysine-g-poly(ethylene glycol) (PLys100-CA-mPEG77).205 The nanoRNP detached its mPEG shell in the acidic tumor microenvironment, facilitated cellular uptake of Cas9 and a combination of sgRNAs targeting STAT3 and RUNX1, which resulted in effective tumor growth inhibition in a heterogenous tumor model. Deng et al. utilized two copolymers to construct a tumor-targeted gene editing system for synergistic gene and photodynamic antitumoral effects (Fig. 9A).206 Firstly, nitrilotriacetic acid-disulfanediyldipropionate-polyethyleneglycol-b-polycaprolactone (NTA-SS-PEG-PCL) was assembled with the hydrophobic photosensitizer chlorin e6 (Ce6) into a micellar core. Then the chelate complexes of NTA with nickel ions on the surface of the micelles bound oligohistidine-tag containing Cas9 RNPs via coordinative interactions. Finally, iRGD-PEG-b-polyasparte-g-1,4-butanediamine [RGD-PEG-pAsp(DAB)] was added to neutralize the negative charge of the NPs and to provide tumor-targeting ability. The obtained T-CC-NPs escaped from lysosomes via NIR-induced production of reactive oxygen species (ROS) by Ce6 and released their Cas9/sgNrf2 cargo due to the reduction of disulfide bonds. The study indicated that Nrf2 deletion enhanced tumor cell sensitivity to ROS, leading to synergistic effects of PDT and Nrf2 gene knockout in vivo.
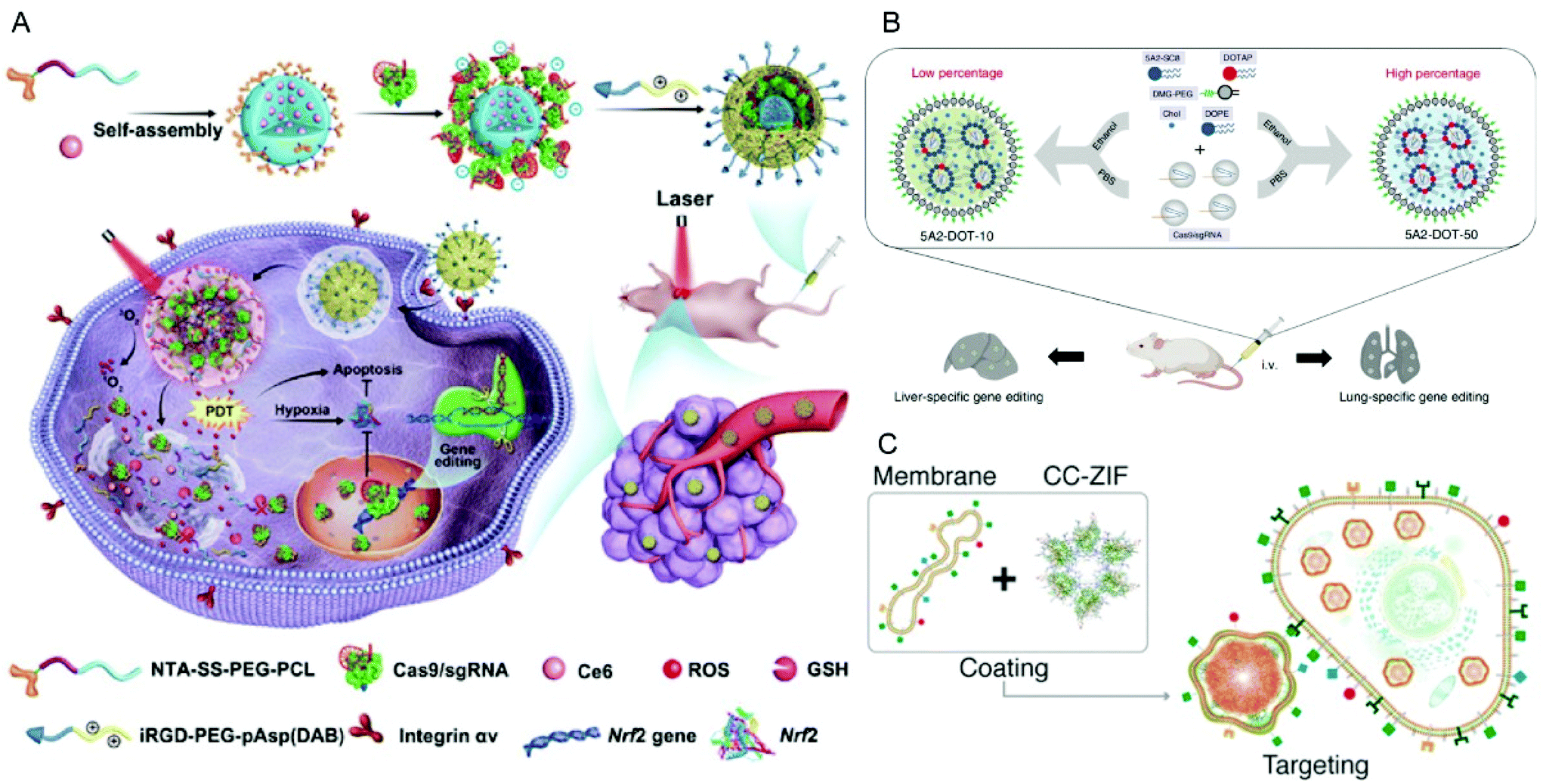 | ||
| Fig. 9 Representative delivery systems of Cas9 RNP. (A) Codelivery of Cas9 RNP and chlorin e6 for spatially controlled tumor-specific genome editing in vivo. Reproduced with permission from ref. 206. Copyright 2020 American Association for the Advancement of Science. (B) Tissue specific genome editing using DOTAP incorporated lipid nanoparticles. Reproduced with permission from ref. 89. Copyright 2020 Springer Nature. (C) Cell-type-specific Cas9 RNP delivery by cell membrane-coated ZIF-8. Reproduced with permission from ref. 222. Copyright 2020 American Chemical Society. | ||
As discussed in the previous pDNA part, PBAEs have been utilized as vectors for CRISPR-Cas9 plasmid delivery. In the context of protein delivery, classical PBAEs with highly positive charge may not be appropriate to encapsulate Cas9 proteins due to incompatibility with the surface charge density. Therefore, Green et al. synthesized a new class of PBAEs which contain both cationic and anionic functions via polymer end-capping with carboxylate ligands.207 The carboxylated PBAEs could self-assemble into nanoparticles with various types of proteins including Cas9 RNPs. The study demonstrated that a single intracranial injection of the nanoparticles carrying 3.5 pmol Cas9 RNP induced robust gene editing in an orthotopic murine glioma model. Likewise, Chen et al. synthesized a glutathione (GSH)-cleavable cross-linked polymer containing both cationic and anionic residues for better encapsulation of inhomogenously charged Cas9 RNPs.208 After local administration in vivo, the optimized nanoparticles enabled robust gene editing in retinal pigment epithelium (RPE) tissues and skeletal muscles. Recently, Liu et al. reported a phenylboronic acid-modified dendrimer that could also bind both negatively and positively charged proteins.209 The dendrimer effectively delivered various native proteins with different isoelectric points and sizes into cells. Particularly, it enabled efficient delivery of Cas9 RNPs into multiple cell lines and achieved excellent editing efficiency. Although numerous types of polymers were successfully synthesized and optimized for Cas9 RNP delivery, the question on structure–activity relationships is still hard to answer. For this purpose, Kumar et al. validated that combinatorial synthesis and high-throughput characterization methodologies coupled with machine learning enables the rapid discovery of potential polymers for Cas9 RNP delivery, which may accelerate the clinical translation of polymeric vectors in the future.210
In 2017, zeolitic imidazolate framework-8 (ZIF-8), a subclass of metal–organic frameworks (MOFs), was reported as the first example of MOF-based Cas9 RNP delivery by Khashab and colleagues.221 The CRISPR/Cas9 ZIF-8 (CC-ZIF) achieved 17% loading efficiency of Cas9 RNPs and 37% EGFP knockout in vitro. Moreover, they coated CC-ZIF with cancer cell membranes (C3-ZIFcell membrane type) to enable cell-type-specific delivery of Cas9 RNPs (Fig. 9C).222In vitro studies demonstrated higher repression of EGFP reporter expression when MCF-7 cells where treated with C3-ZIFMCF−7 compared to C3-ZIFHeLa, and in HeLa cells vice versa. In vivo fluorescence imaging confirmed that C3-ZIFMCF-7 could specifically accumulate in MCF-7 tumor sites. Recently, Yang et al. assembled imidazole-2-carboxaldehyde (2-ICA) and Zn2+ with Cas9 RNPs to generate an ATP-responsive delivery system (ZIF-90).223 The intracellular high concentration (10 mM) of ATP disassembled ZIF-90 due to the competitive coordination between Zn2+ and ATP, released Cas9 RNPs and induced up to 35% GFP knockout in HeLa cells. Liu et al. reported a novel approach for controlling hierarchical self-assembly of metal–organic cages (MOC) into supramolecular nanoparticles (SNPs) for Cas9 RNP delivery.224 The adamantane-functionalized Pd12L24 MOC (Ada-MOC) and β-cyclodextrin-conjugated PEI (PEI-βCD) assembled via host–guest interaction and encapsulated Cas9 RNPs. The obtained Cas9 SNPs achieved 40% knockout of GFP expression compared to non-treated cells.
As discussed in previous sections, mesoporous silica nanoparticles (MSNs) are widely used for delivery purposes, including the CRISPR/Cas9 system in nucleic acid or RNP form. However, the common technique of protein loading into MSNs is based on nonspecific physical absorption, which generally results in rather weak binding. To enhance the encapsulation efficiency of the protein in MSNs, Liu et al. developed a novel approach by modifying the surface of Cas9 protein with boronic acid and MSN scaffolds with amine groups.225 The dative bond formation between boronic acids and amines as well as electrostatic interactions increased the loading and intracellular delivery of Cas9 RNPs. Recently, Pan et al. constructed NIR-responsive upconversion nanoparticles (UCNPs) for Plk1 gene disruption in tumors.226 In this system, Cas9 RNPs were covalently anchored on the surface of UCNPs via a photocleavable 4-(hydroxymethyl)-3-nitrobenzoic acid (ONA) linker, and the formed UCNPs-Cas9 complexes were coated with PEI to facilitate endosomal escape after cellular internalization. Upon irradiation, UCNPs-Cas9@PEI converted 980 nm NIR light into local ultraviolet light which resulted in photo-responsive release of Cas9 RNPs and significant inhibition of tumor cell growth both in vitro and in vivo. In another study, Chen et al. used semiconducting copper sulfide (CuS) NPs as NIR-absorbing nanomaterials along with Cas9 RNPs, doxorubicin (DOX) and PEI to establish multifunctional NIR-triggered nanocomplexes for cancer combination therapy based on photothermal and chemotherapy.227 Besides, several 2D-nanomaterials were reported for Cas9 RNP delivery.228,229 Yue et al. developed a PEG and PEI dual-functionalized graphene oxide (GO) platform which enabled Cas9 RNP loading and CRISPR-mediated gene editing.228 Zhou et al. engineered a Cas9 protein with three NLS tags (Cas9N3) at the C terminus for a better loading onto black phosphorus nanosheets (BPs) via electrostatic interactions.229 The constructed Cas9N3-BPs entered the cells via both direct membrane penetration and endocytosis pathways, and the Cas9N3 with improved nuclear entry led to robust gene editing in vitro and in vivo.
 | ||
| Fig. 10 Representative delivery systems for Cas9 RNPs. (A) Blood–brain barrier-permeable dNP2-lipopeptide system for delivery of HypaCas9 RNPs. Reproduced with permission from ref. 236. Copyright 2019 American Chemical Society. (B) Extracellular nanovesicle-based Cas9 RNP delivery system. Reproduced with permission from ref. 241. Copyright 2020 Springer Nature. (C) Branched DNA-based delivery system for Cas9 RNP/antisense codelivery. Reproduced with permission from ref. 249. Copyright 2019 American Chemical Society. (D) Cas9 conjugate with trimeric N-acetyl galactosamine (GalNAc) ligands for asialoglycoprotein receptor-specific delivery. Reproduced with permission from ref. 251. Copyright 2018 American Chemical Society. (E) Lipo-oligoamino amides for Cas9/sgRNA RNP delivery. Reproduced with permission from ref. 84. Copyright 2020 American Chemical Society. Permission requests related to the material should be directed to the ACS. | ||
:3 ratio of aptamer/cholesterol increased the accumulation of EVs in HepG2 cells, human primary liver cancer-derived organoids and xenograft tumor models which could be utilized for WNT10B gene disruption with significant tumor growth suppression.
Apart from extracellular vesicles, virus-like particles were reported for Cas9 RNP delivery as another class of bio-derived vehicles. Mangeot et al. fabricated murine leukemia virus (MLV)-like particles to encapsulate Cas9 RNPs.245 The obtained nanoblades induced efficient genome editing in multiple primary cell lines and enabled genome editing in mouse embryos and livers. Doudna and colleagues demonstrated that HIV envelope glycoprotein-pseudotyped Cas9-VLPs could selectively edit genomes in CD4+ T cells.246
Current clinical trials of CRISPR/Cas
Although numerous delivery technologies and Cas9 variants were developed and scientists from industry as well as academia have put much efforts into the translation of these technologies, the clinical application of the CRISPR/Cas systems is still at an early stage. Generally, ex vivo and in vivo therapies can be discriminated, depending on the genome editing procedure occurring externally or internally of the human body. T cells, CAR-T cells, lymphocytes, hematopoietic stem and progenitor cells (HSPCs), induced pluripotent stem cells (iPSCs) and induced hepatic stem cells (iHSCs) are cell types of particular interest in current ex vivo applications.118 The native cells are isolated from patients, engineered by genomic modifications and then re-infused into the patients. In 2016, the first CRISPR clinical trial of PD-1 edited T cells in patients with advanced non-small-cell lung cancer (NCT02793856) started at the West China Hospital, Sichuan University. The researchers reported that T cells edited by nucleofection with Cas9 plasmids were generally safe for patients and off-target effects were observed at a low level.256 However, the desired responses were not observed in the patients, which may be due to insufficient T cell expansion, lack of antigen specificity or low editing efficiency. To improve this, several CRISPR-based ex vivo clinical trials using Cas9 mRNA or Cas9 RNPs as well as combinations with chimeric antigen receptor T cell (CAR-T) therapies are under investigation (NCT03166878, NCT03398967, NCT03545815, NCT03747965 and NCT04037566). Besides engineered T cells and CAR-T cells, modification of CCR5, BCL11A or mutant HBB in hematopoietic stem and progenitor cells (HSPCs) for treating hematologic diseases using CRISPR/Cas9 systems are in development.257 In contrast to ex vivo applications, where physical methods such as electroporation or nucleofection are feasible approaches to deliver the CRISPR/Cas9 systems, for in vivo applications, viral or non-viral vectors are generally required. The first in vivo administration of the CRISPR/Cas9 system to humans was conducted with EDIT-101, an AAV5 vector encoding for SaCas9 and two sgRNAs to mediate deletion of a mutated intronic sequence of the CPE290 gene (IVS26) after subretinal injections in patients suffering from Leber's Congenital Amaurosis Type 10.258 In the ongoing single ascending dose study, 18 participants with LCA10-IVS26 are treated with EDIT-101 (NCT03872479). As discussed above, lipid nanoparticles are used in the first systemically administered CRISPR therapy NTLA-2001 in patients with hereditary transthyretin amyloidosis with polyneuropathy (NCT04601051). An interim report of the ongoing phase 1 clinical study showed an impressive mean reduction of serum TTR protein by 87% in patients who received a single treatment of NTLA-2001 at a dose of 0.3 mg kg−1, which can be considered a first clinical demonstration of safe and efficient direct in vivo genome editing in humans.119Conclusions
Introduction of the CRISPR/Cas9 system into cells can be achieved by using different biomolecular formats. The delivery of DNA, mRNA and RNP introduces the components at different stages of the cellular flow of genetic information. This results in different kinetics and availability of the protein and RNA components required for assembly into the final Cas9/gRNA RNP complex. In the cases of DNA and RNA delivery, one or both CRISPR components are produced by the cellular machinery, which results in an unpreventable latency between the availabilities of gRNA and Cas9. RNP delivery represents the most direct strategy to introduce the pre-assembled and immediately functional CRISPR/Cas9 system into cells, which generally results in higher genome editing efficiency compared to the other biomolecular formats. RNPs are also more rapidly eliminated, since no genetic ‘blue-prints’ can provide additional supply after the complete dose has been applied. In this regard, a short persistence of the CRISPR/Cas system after the intended gene editing event has occured is generally desirable to prevent unintended off-target effects. In terms of immunogenicity, the different biomolecular formats also exhibit individual characteristics: while pDNA and mRNA mainly trigger innate immune responses, depending on the presence of bacterial backbone elements or nucleotide sequences, the Cas9 protein can provoke adaptive immune reactions. Due to the outstanding therapeutic potential of the CRISPR/Cas9 system, researchers around the world have worked on numerous delivery strategies based on polymeric, lipidic, inorganic, bioderived and other materials for each of the biomolecular formats. Table S1† (pDNA), Table S2† (RNA) and Table S3† (RNPs) give an overview over the research field, including those publications which are not described in the main part of the article. An interesting observation is the currently differing number of delivery strategies which can be found in the literature for the different biomolecular formats. A high number of pDNA based systems is not surprising, since plasmids were the first format being available for the utilisation of the CRISPR/Cas system and pDNA transfecting agents have a long tradition. Despite the obvious high clinical potential of RNA delivery, which has impressively been demonstrated by the mRNA COVID-19 vaccines and the first clinical trial of systemic CRISPR/Cas9 in vivo genome editing,119 the number of CRISPR RNA publications is the lowest. In contrast, a very high number of research articles on Cas9 RNP delivery demonstrates the high interest, and it is likely to become one preferred delivery strategy, at least for certain applications. This is attributed to the aspects, that Cas9/sgRNA RNPs exhibit several advantages, as described in the article. In addition, Cas9 RNPs represent a very special case where non-covalent ionic interactions enable robust encapsulation of the protein complexes and efficient delivery is possible similar to nucleic acid transfections. Based on these considerations and the characteristics of the different biomolecular formats, it is expected that especially the research on RNA and RNP delivery will continue to increase extensively with high impact on future therapies.Author contributions
Yi Lin: Investigation, writing – original draft. Ernst Wagner: Conceptualization, supervision, writing – original draft. Ulrich Lächelt: Conceptualization, supervision, writing – original draft.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank Joachim Rädler and Katrien Remaut for fruitful and inspiring discussions. The authors acknowledge support of their own related research by the UPGRADE (Unlocking Precision Gene Therapy) project that has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 825825. Y. L. appreciates the fellowship of the China Scholarship Council that supports his Ph.D. study at the LMU Munich, Germany.References
- F. J. M. Mojica, C. s. Díez-Villaseñor, J. García-Martínez and E. Soria, J. Mol. Evol., 2005, 60, 174–182 CrossRef CAS PubMed
.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef CAS PubMed
- E. Charpentier and J. A. Doudna, Nature, 2013, 495, 50–51 CrossRef CAS PubMed
- J. D. Sander and J. K. Joung, Nat. Biotechnol., 2014, 32, 347–355 CrossRef CAS PubMed
- T. Gaj, C. A. Gersbach and C. F. Barbas, Trends Biotechnol., 2013, 31, 397–405 CrossRef CAS PubMed
- C. Maucksch, B. Connor and C. Rudolph, Minicircle and Miniplasmid DNA Vectors, 2013, pp. 59–69, DOI:10.1002/9783527670420.ch5.
- K. Remaut, N. N. Sanders, F. Fayazpour, J. Demeester and S. C. De Smedt, J. Controlled Release, 2006, 115, 335–343 CrossRef CAS PubMed
- D. Kobelt, M. Schleef, M. Schmeer, J. Aumann, P. M. Schlag and W. Walther, Mol. Biotechnol., 2013, 53, 80–89 CrossRef CAS PubMed
- A. K. Levacic, S. Morys, S. Kempter, U. Lächelt and E. Wagner, Hum. Gene Ther., 2017, 28, 862–874 CrossRef CAS PubMed
- C. Leonhardt, G. Schwake, T. R. Stögbauer, S. Rappl, J.-T. Kuhr, T. S. Ligon and J. O. Rädler, Nanomedicine, 2014, 10, 679–688 CrossRef CAS PubMed
- S. Brunner, T. Sauer, S. Carotta, M. Cotten, M. Saltik and E. Wagner, Gene Ther., 2000, 7, 401–407 CrossRef CAS PubMed
- K. Remaut, N. Symens, B. Lucas, J. Demeester and S. C. De Smedt, J. Controlled Release, 2014, 179, 1–9 CrossRef CAS PubMed
- M. Ferizi, C. Leonhardt, C. Meggle, M. K. Aneja, C. Rudolph, C. Plank and J. O. Rädler, Lab Chip, 2015, 15, 3561–3571 RSC
- C. E. Smull, M. F. Mallette and E. H. Ludwig, Biochem. Biophys. Res. Commun., 1961, 5, 247–249 CrossRef
- A. Vaheri and J. S. Pagano, Virology, 1965, 27, 434–436 CrossRef CAS PubMed
- J. A. Wolff, R. W. Malone, P. Williams, W. Chong, G. Acsadi, A. Jani and P. L. Felgner, Science, 1990, 247, 1465–1468 CrossRef CAS PubMed
- J. Nelson, E. W. Sorensen, S. Mintri, A. E. Rabideau, W. Zheng, G. Besin, N. Khatwani, S. V. Su, E. J. Miracco, W. J. Issa, S. Hoge, M. G. Stanton and J. L. Joyal, Sci. Adv., 2020, 6, eaaz6893 CrossRef CAS PubMed
- D. Weissman, Expert Rev. Vaccines, 2015, 14, 265–281 CrossRef CAS PubMed
- H. Zhang, K. Rombouts, L. Raes, R. Xiong, S. C. De Smedt, K. Braeckmans and K. Remaut, Adv. Biosyst., 2020, 4, 2000057 CrossRef CAS PubMed
- M. S. D. Kormann, G. Hasenpusch, M. K. Aneja, G. Nica, A. W. Flemmer, S. Herber-Jonat, M. Huppmann, L. E. Mays, M. Illenyi, A. Schams, M. Griese, I. Bittmann, R. Handgretinger, D. Hartl, J. Rosenecker and C. Rudolph, Nat. Biotechnol., 2011, 29, 154–157 CrossRef CAS PubMed
- P. S. Kowalski, A. Rudra, L. Miao and D. G. Anderson, Mol. Ther., 2019, 27, 710–728 CrossRef CAS PubMed
- P. Heissig, W. Schrimpf, P. Hadwiger, E. Wagner and D. C. Lamb, PLoS One, 2017, 12, e0173401 CrossRef PubMed
- W. Chen, J. M. Smeekens and R. Wu, Chem. Sci., 2016, 7, 1393–1400 RSC
- O. Boussif, F. h. Lezoualc, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297 CrossRef CAS PubMed
- H. Uchida, K. Miyata, M. Oba, T. Ishii, T. Suma, K. Itaka, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2011, 133, 15524–15532 CrossRef CAS PubMed
- D. Schaffert, C. Troiber, E. E. Salcher, T. Fröhlich, I. Martin, N. Badgujar, C. Dohmen, D. Edinger, R. Kläger, G. Maiwald, K. Farkasova, S. Seeber, K. Jahn-Hofmann, P. Hadwiger and E. Wagner, Angew. Chem., Int. Ed., 2011, 50, 8986–8989 CrossRef CAS PubMed
- H. Uchida, K. Itaka, T. Nomoto, T. Ishii, T. Suma, M. Ikegami, K. Miyata, M. Oba, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2014, 136, 12396–12405 CrossRef CAS PubMed
- U. Lächelt, P. Kos, F. M. Mickler, A. Herrmann, E. E. Salcher, W. Rödl, N. Badgujar, C. Bräuchle and E. Wagner, Nanomedicine, 2014, 10, 35–44 CrossRef PubMed
- A. Jarzębińska, T. Pasewald, J. Lambrecht, O. Mykhaylyk, L. Kümmerling, P. Beck, G. Hasenpusch, C. Rudolph, C. Plank and C. Dohmen, Angew. Chem., Int. Ed., 2016, 55, 9591–9595 CrossRef PubMed
- P. Heller, D. Hobernik, U. Lächelt, M. Schinnerer, B. Weber, M. Schmidt, E. Wagner, M. Bros and M. Barz, J. Controlled Release, 2017, 258, 146–160 CrossRef CAS PubMed
- L. Beckert, L. Kostka, E. Kessel, A. Krhac Levacic, H. Kostkova, T. Etrych, U. Lächelt and E. Wagner, Eur. J. Pharm. Biopharm., 2016, 105, 85–96 CrossRef CAS PubMed
- G. Gregoriadis and R. A. Buckland, Nature, 1973, 244, 170–172 CrossRef CAS PubMed
- G. Weissmann, D. Bloomgarden, R. Kaplan, C. Cohen, S. Hoffstein, T. Collins, A. Gotlieb and D. Nagle, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 88 CrossRef CAS PubMed
- M. Becker-Hapak, S. S. McAllister and S. F. Dowdy, Methods, 2001, 24, 247–256 CrossRef CAS PubMed
- N. Nischan, H. D. Herce, F. Natale, N. Bohlke, N. Budisa, M. C. Cardoso and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2015, 54, 1950–1953 CrossRef CAS PubMed
- X. Liu, P. Zhang, W. Rödl, K. Maier, U. Lächelt and E. Wagner, Mol. Pharm., 2017, 14, 1439–1449 CrossRef CAS PubMed
- G. H. Gao, M. J. Park, Y. Li, G. H. Im, J.-H. Kim, H. N. Kim, J. W. Lee, P. Jeon, O. Y. Bang, J. H. Lee and D. S. Lee, Biomaterials, 2012, 33, 9157–9164 CrossRef CAS PubMed
- R. Tang, C. S. Kim, D. J. Solfiell, S. Rana, R. Mout, E. M. Velázquez-Delgado, A. Chompoosor, Y. Jeong, B. Yan, Z.-J. Zhu, C. Kim, J. A. Hardy and V. M. Rotello, ACS Nano, 2013, 7, 6667–6673 CrossRef CAS PubMed
- P. Zhang, B. Steinborn, U. Lächelt, S. Zahler and E. Wagner, Biomacromolecules, 2017, 18, 2509–2520 CrossRef CAS PubMed
- Y.-H. Lin, Y.-P. Chen, T.-P. Liu, F.-C. Chien, C.-M. Chou, C.-T. Chen and C.-Y. Mou, ACS Appl. Mater. Interfaces, 2016, 8, 17944–17954 CrossRef CAS PubMed
- J.-S. Lim, K. Lee, J.-N. Choi, Y.-K. Hwang, M.-Y. Yun, H.-J. Kim, Y. S. Won, S.-J. Kim, H. Kwon and S. Huh, Nanotechnology, 2012, 23, 085101 CrossRef PubMed
- Y. Lee, T. Ishii, H. Cabral, H. J. Kim, J.-H. Seo, N. Nishiyama, H. Oshima, K. Osada and K. Kataoka, Angew. Chem., Int. Ed., 2009, 48, 5309–5312 CrossRef CAS PubMed
- R. Mout, M. Ray, T. Tay, K. Sasaki, G. Y. Tonga and V. M. Rotello, ACS Nano, 2017, 11, 6416–6421 CrossRef CAS PubMed
- J. A. Zuris, D. B. Thompson, Y. Shu, J. P. Guilinger, J. L. Bessen, J. H. Hu, M. L. Maeder, J. K. Joung, Z.-Y. Chen and D. R. Liu, Nat. Biotechnol., 2015, 33, 73–80 CrossRef CAS PubMed
- L. Cong, F. A. Ran, D. Cox, S. Lin, R. Barretto, N. Habib, P. D. Hsu, X. Wu, W. Jiang, L. A. Marraffini and F. Zhang, Science, 2013, 339, 819–823 CrossRef CAS PubMed
- J. B. Miller, S. Zhang, P. Kos, H. Xiong, K. Zhou, S. S. Perelman, H. Zhu and D. J. Siegwart, Angew. Chem., 2017, 129, 1079–1083 CrossRef
- H. Kwon, M. Kim, Y. Seo, Y. S. Moon, H. J. Lee, K. Lee and H. Lee, Biomaterials, 2018, 156, 172–193 CrossRef CAS PubMed
- Y. Furuichi, Proc. Jpn. Acad., Ser. B, 2015, 91, 394–409 CrossRef CAS PubMed
- A. Roers, B. Hiller and V. Hornung, Immunity, 2016, 44, 739–754 CrossRef CAS PubMed
- D. J. Goss and F. E. Kleiman, Wiley Interdiscip. Rev.: RNA, 2013, 4, 167–179 CrossRef CAS PubMed
- A. Wadhwa, A. Aljabbari, A. Lokras, C. Foged and A. Thakur, Pharmaceutics, 2020, 12, 102 CrossRef CAS PubMed
- D. Kawaguchi, A. Kodama, N. Abe, K. Takebuchi, F. Hashiya, F. Tomoike, K. Nakamoto, Y. Kimura, Y. Shimizu and H. Abe, Angew. Chem., Int. Ed., 2020, 59, 17403–17407 CrossRef CAS PubMed
- U. Sahin, K. Karikó and Ö. Türeci, Nat. Rev. Drug Discovery, 2014, 13, 759–780 CrossRef CAS PubMed
- J. Devoldere, H. Dewitte, S. C. De Smedt and K. Remaut, Drug Discovery Today, 2016, 21, 11–25 CrossRef CAS PubMed
- K. Karikó, H. Muramatsu, F. A. Welsh, J. Ludwig, H. Kato, S. Akira and D. Weissman, Mol. Ther., 2008, 16, 1833–1840 CrossRef PubMed
- S. Vaidyanathan, K. T. Azizian, A. A. Haque, J. M. Henderson, A. Hendel, S. Shore, J. S. Antony, R. I. Hogrefe, M. S. Kormann, M. H. Porteus and A. P. McCaffrey, Mol. Ther. – Nucleic Acids, 2018, 12, 530–542 CrossRef CAS PubMed
- L. Warren, P. D. Manos, T. Ahfeldt, Y.-H. Loh, H. Li, F. Lau, W. Ebina, P. K. Mandal, Z. D. Smith, A. Meissner, G. Q. Daley, A. S. Brack, J. J. Collins, C. Cowan, T. M. Schlaeger and D. J. Rossi, Cell Stem Cell, 2010, 7, 618–630 CrossRef CAS PubMed
- A. Thess, S. Grund, B. L. Mui, M. J. Hope, P. Baumhof, M. Fotin-Mleczek and T. Schlake, Mol. Ther., 2015, 23, 1456–1464 CrossRef CAS PubMed
- E. Giuliani, C. Piovan, S. Bossi, S. Corna, C. Scavullo, M. Pema, T. Di Tomaso, P. Genovese, A. Lombardo, L. Naldini, C. Bordignon, G. P. Rizzardi and C. Bovolenta, Mol. Ther., 2016, 24, S53–S54 CrossRef
- S. A. Scaringe, Methods, 2001, 23, 206–217 CrossRef CAS PubMed
- D. J. Dellinger, Z. Timár, J. Myerson, A. B. Sierzchala, J. Turner, F. Ferreira, Z. n. Kupihár, G. Dellinger, K. W. Hill, J. A. Powell, J. R. Sampson and M. H. Caruthers, J. Am. Chem. Soc., 2011, 133, 11540–11556 CrossRef CAS PubMed
- N. Pardi, M. J. Hogan, F. W. Porter and D. Weissman, Nat. Rev. Drug Discovery, 2018, 17, 261–279 CrossRef CAS PubMed
- Q. Chen, Y. Zhang and H. Yin, Adv. Drug Delivery Rev., 2021, 168, 246–258 CrossRef CAS PubMed
- Y. Liu, E. Holmstrom, J. Zhang, P. Yu, J. Wang, M. A. Dyba, D. Chen, J. Ying, S. Lockett, D. J. Nesbitt, A. R. Ferré-D'Amaré, R. Sousa, J. R. Stagno and Y.-X. Wang, Nature, 2015, 522, 368–372 CrossRef CAS PubMed
- S. Kim, T. Koo, H.-G. Jee, H.-Y. Cho, G. Lee, D.-G. Lim, H. S. Shin and J.-S. Kim, Genome Res., 2018, 28, 367–373 CrossRef CAS PubMed
- W. Mu, N. Tang, C. Cheng, W. Sun, X. Wei and H. Wang, Protein Cell, 2019, 10, 461–465 CrossRef CAS PubMed
- B. Wienert, J. Shin, E. Zelin, K. Pestal and J. E. Corn, PLoS Biol., 2018, 16, e2005840 CrossRef PubMed
- D. Allen, M. Rosenberg and A. Hendel, Front. Genome Ed., 2021, 2, 617910 CrossRef PubMed
- S. M. A. Rahman, S. Seki, S. Obika, H. Yoshikawa, K. Miyashita and T. Imanishi, J. Am. Chem. Soc., 2008, 130, 4886–4896 CrossRef CAS PubMed
- M. Basila, M. L. Kelley and A. v. B. Smith, PLoS One, 2017, 12, e0188593 CrossRef PubMed
- J. D. Finn, A. R. Smith, M. C. Patel, L. Shaw, M. R. Youniss, J. van Heteren, T. Dirstine, C. Ciullo, R. Lescarbeau, J. Seitzer, R. R. Shah, A. Shah, D. Ling, J. Growe, M. Pink, E. Rohde, K. M. Wood, W. E. Salomon, W. F. Harrington, C. Dombrowski, W. R. Strapps, Y. Chang and D. V. Morrissey, Cell Rep., 2018, 22, 2227–2235 CrossRef CAS PubMed
- A. Hendel, R. O. Bak, J. T. Clark, A. B. Kennedy, D. E. Ryan, S. Roy, I. Steinfeld, B. D. Lunstad, R. J. Kaiser, A. B. Wilkens, R. Bacchetta, A. Tsalenko, D. Dellinger, L. Bruhn and M. H. Porteus, Nat. Biotechnol., 2015, 33, 985–989 CrossRef CAS PubMed
- K. S. Manning, A. N. Rao, M. Castro and T. A. Cooper, ACS Chem. Biol., 2017, 12, 2503–2509 CrossRef CAS PubMed
- A. Mir, J. F. Alterman, M. R. Hassler, A. J. Debacker, E. Hudgens, D. Echeverria, M. H. Brodsky, A. Khvorova, J. K. Watts and E. J. Sontheimer, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed
- M. Rahdar, M. A. McMahon, T. P. Prakash, E. E. Swayze, C. F. Bennett and D. W. Cleveland, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E7110–E7117 CrossRef CAS PubMed
- D. E. Ryan, D. Taussig, I. Steinfeld, S. M. Phadnis, B. D. Lunstad, M. Singh, X. Vuong, K. D. Okochi, R. McCaffrey, M. Olesiak, S. Roy, C. W. Yung, B. Curry, J. R. Sampson, L. Bruhn and D. J. Dellinger, Nucleic Acids Res., 2018, 46, 792–803 CrossRef CAS PubMed
- H. Yin, C.-Q. Song, S. Suresh, Q. Wu, S. Walsh, L. H. Rhym, E. Mintzer, M. F. Bolukbasi, L. J. Zhu, K. Kauffman, H. Mou, A. Oberholzer, J. Ding, S.-Y. Kwan, R. L. Bogorad, T. Zatsepin, V. Koteliansky, S. A. Wolfe, W. Xue, R. Langer and D. G. Anderson, Nat. Biotechnol., 2017, 35, 1179–1187 CrossRef CAS PubMed
- L. F. Ribeiro, L. F. Ribeiro, M. Q. Barreto and R. J. Ward, Int. J. Genomics, 2018, 2018, 1652567 Search PubMed
- F. Jiang and J. A. Doudna, Annu. Rev. Biophys., 2017, 46, 505–529 CrossRef CAS PubMed
- B. Sun, H. Chen and X. Gao, J. Controlled Release, 2021, 337, 698–717 CrossRef CAS PubMed
- J. A. Doudna and E. Charpentier, Science, 2014, 346, 1258096 CrossRef PubMed
- M. Jinek, F. Jiang, D. W. Taylor, S. H. Sternberg, E. Kaya, E. Ma, C. Anders, M. Hauer, K. Zhou, S. Lin, M. Kaplan, A. T. Iavarone, E. Charpentier, E. Nogales and J. A. Doudna, Science, 2014, 343, 1247997 CrossRef PubMed
- C. Troiber, D. Edinger, P. Kos, L. Schreiner, R. Kläger, A. Herrmann and E. Wagner, Biomaterials, 2013, 34, 1624–1633 CrossRef CAS PubMed
- J. Kuhn, Y. Lin, A. Krhac Levacic, N. Al Danaf, L. Peng, M. Höhn, D. C. Lamb, E. Wagner and U. Lächelt, Bioconjugate Chem., 2020, 31, 729–742, DOI:10.1021/acs.bioconjchem.9b00853
-
H. Lodish, A. Berk, S. L. Zipursky, P. Matsudaira, D. Baltimore and J. Darnell, Molecular Cell Biology, WH Freeman, 4th edn, 2000 Search PubMed
- A. V. Groll, Y. Levin, M. C. Barbosa and A. P. Ravazzolo, Biotechnol. Prog., 2006, 22, 1220–1224 CrossRef PubMed
- S. McLenachan, J. P. Sarsero and P. A. Ioannou, Genomics, 2007, 89, 708–720 CrossRef CAS PubMed
- S. H. Boo and Y. K. Kim, Exp. Mol. Med., 2020, 52, 400–408 CrossRef CAS PubMed
- T. Wei, Q. Cheng, Y.-L. Min, E. N. Olson and D. J. Siegwart, Nat. Commun., 2020, 11, 1–12 Search PubMed
- E. Kouranova, K. Forbes, G. Zhao, J. Warren, A. Bartels, Y. Wu and X. Cui, Hum. Gene Ther., 2016, 27, 464–475 CrossRef CAS PubMed
- G. Josipović, V. Zoldoš and A. Vojta, J. Biotechnol., 2019, 301, 18–23 CrossRef PubMed
- S. Kim, D. Kim, S. W. Cho, J. Kim and J.-S. Kim, Genome Res., 2014, 24, 1012–1019 CrossRef CAS PubMed
- C.-F. Xu, G.-J. Chen, Y.-L. Luo, Y. Zhang, G. Zhao, Z.-D. Lu, A. Czarna, Z. Gu and J. Wang, Adv. Drug Delivery Rev., 2021, 168, 3–29 CrossRef CAS PubMed
- X. Liang, J. Potter, S. Kumar, Y. Zou, R. Quintanilla, M. Sridharan, J. Carte, W. Chen, N. Roark, S. Ranganathan, N. Ravinder and J. D. Chesnut, J. Biotechnol., 2015, 208, 44–53 CrossRef CAS PubMed
- Z. Tu, W. Yang, S. Yan, A. Yin, J. Gao, X. Liu, Y. Zheng, J. Zheng, Z. Li, S. Yang, S. Li, X. Guo and X.-J. Li, Sci. Rep., 2017, 7, 42081 CrossRef CAS PubMed
- S. Yang, S. Li and X.-J. Li, Cell Rep., 2018, 25, 2653–2659 CrossRef CAS PubMed
- S. Aschenbrenner, S. M. Kallenberger, M. D. Hoffmann, A. Huck, R. Eils and D. Niopek, Sci. Adv., 2020, 6, eaay0187 CrossRef CAS PubMed
- B. Zetsche, S. E. Volz and F. Zhang, Nat. Biotechnol., 2015, 33, 139–142 CrossRef CAS PubMed
- K. M. Davis, V. Pattanayak, D. B. Thompson, J. A. Zuris and D. R. Liu, Nat. Chem. Biol., 2015, 11, 316–318 CrossRef CAS PubMed
- Y. Nihongaki, F. Kawano, T. Nakajima and M. Sato, Nat. Biotechnol., 2015, 33, 755–760 CrossRef CAS PubMed
- J. P. Guilinger, D. B. Thompson and D. R. Liu, Nat. Biotechnol., 2014, 32, 577–582 CrossRef CAS PubMed
- B. P. Kleinstiver, V. Pattanayak, M. S. Prew, S. Q. Tsai, N. T. Nguyen, Z. Zheng and J. K. Joung, Nature, 2016, 529, 490–495 CrossRef CAS PubMed
- I. M. Slaymaker, L. Gao, B. Zetsche, D. A. Scott, W. X. Yan and F. Zhang, Science, 2016, 351, 84–88 CrossRef CAS PubMed
- J. S. Chen, Y. S. Dagdas, B. P. Kleinstiver, M. M. Welch, A. A. Sousa, L. B. Harrington, S. H. Sternberg, J. K. Joung, A. Yildiz and J. A. Doudna, Nature, 2017, 550, 407–410 CrossRef CAS PubMed
- A. Casini, M. Olivieri, G. Petris, C. Montagna, G. Reginato, G. Maule, F. Lorenzin, D. Prandi, A. Romanel, F. Demichelis, A. Inga and A. Cereseto, Nat. Biotechnol., 2018, 36, 265–271 CrossRef CAS PubMed
- J. K. Lee, E. Jeong, J. Lee, M. Jung, E. Shin, Y.-h. Kim, K. Lee, I. Jung, D. Kim, S. Kim and J.-S. Kim, Nat. Commun., 2018, 9, 3048 CrossRef PubMed
- J. H. Hu, S. M. Miller, M. H. Geurts, W. Tang, L. Chen, N. Sun, C. M. Zeina, X. Gao, H. A. Rees, Z. Lin and D. R. Liu, Nature, 2018, 556, 57–63 CrossRef CAS PubMed
- F. A. Ran, P. D. Hsu, C.-Y. Lin, J. S. Gootenberg, S. Konermann, A. E. Trevino, D. A. Scott, A. Inoue, S. Matoba, Y. Zhang and F. Zhang, Cell, 2013, 154, 1380–1389 CrossRef CAS PubMed
- C. T. Charlesworth, P. S. Deshpande, D. P. Dever, J. Camarena, V. T. Lemgart, M. K. Cromer, C. A. Vakulskas, M. A. Collingwood, L. Zhang, N. M. Bode, M. A. Behlke, B. Dejene, B. Cieniewicz, R. Romano, B. J. Lesch, N. Gomez-Ospina, S. Mantri, M. Pavel-Dinu, K. I. Weinberg and M. H. Porteus, Nat. Med., 2019, 25, 249–254 CrossRef CAS PubMed
- H. Hemmi, O. Takeuchi, T. Kawai, T. Kaisho, S. Sato, H. Sanjo, M. Matsumoto, K. Hoshino, H. Wagner, K. Takeda and S. Akira, Nature, 2000, 408, 740–745 CrossRef CAS PubMed
- S. Vaidyanathan, K. T. Azizian, A. K. M. A. Haque, J. M. Henderson, A. Hendel, S. Shore, J. S. Antony, R. I. Hogrefe, M. S. D. Kormann, M. H. Porteus and A. P. McCaffrey, Mol. Ther. – Nucleic Acids, 2018, 12, 530–542 CrossRef CAS PubMed
- J. M. Crudele and J. S. Chamberlain, Nat. Commun., 2018, 9, 3497 CrossRef PubMed
- V. L. Simhadri, J. McGill, S. McMahon, J. Wang, H. Jiang and Z. E. Sauna, Mol. Ther. – Methods Clin. Dev., 2018, 10, 105–112 CrossRef CAS PubMed
- M. Behr, J. Zhou, B. Xu and H. Zhang, Acta Pharm. Sin. B, 2021, 11, 2150–2171 CrossRef CAS PubMed
- T. Coelho, D. Adams, A. Silva, P. Lozeron, P. N. Hawkins, T. Mant, J. Perez, J. Chiesa, S. Warrington, E. Tranter, M. Munisamy, R. Falzone, J. Harrop, J. Cehelsky, B. R. Bettencourt, M. Geissler, J. S. Butler, A. Sehgal, R. E. Meyers, Q. Chen, T. Borland, R. M. Hutabarat, V. A. Clausen, R. Alvarez, K. Fitzgerald, C. Gamba-Vitalo, S. V. Nochur, A. K. Vaishnaw, D. W. Y. Sah, J. A. Gollob and O. B. Suhr, N. Engl. J. Med., 2013, 369, 819–829 CrossRef CAS PubMed
- B. Z. Igyártó, S. Jacobsen and S. Ndeupen, Curr. Opin. Virol., 2021, 48, 65–72 CrossRef PubMed
- Z.-Y. He, K. Men, Z. Qin, Y. Yang, T. Xu and Y.-Q. Wei, Sci. China: Life Sci., 2017, 60, 458–467 CrossRef CAS PubMed
- Y. Li, Z. Glass, M. Huang, Z.-Y. Chen and Q. Xu, Biomaterials, 2020, 234, 119711 CrossRef CAS PubMed
- J. D. Gillmore, E. Gane, J. Taubel, J. Kao, M. Fontana, M. L. Maitland, J. Seitzer, D. O'Connell, K. R. Walsh, K. Wood, J. Phillips, Y. Xu, A. Amaral, A. P. Boyd, J. E. Cehelsky, M. D. McKee, A. Schiermeier, O. Harari, A. Murphy, C. A. Kyratsous, B. Zambrowicz, R. Soltys, D. E. Gutstein, J. Leonard, L. Sepp-Lorenzino and D. Lebwohl, N. Engl. J. Med., 2021, 385, 493–502 CrossRef CAS PubMed
- J. Guo, T. Wan, B. Li, Q. Pan, H. Xin, Y. Qiu and Y. Ping, ACS Cent. Sci., 2021, 7(6), 990–1000 CrossRef CAS PubMed
- J. Gong, H. X. Wang, Y. H. Lao, H. Hu, N. Vatan, J. Guo, T. C. Ho, D. Huang, M. Li, D. Shao and K. W. Leong, Adv. Mater., 2020, 32, 2003537 CrossRef CAS PubMed
- A. Noureddine, A. Maestas-Olguin, E. A. Saada, A. E. LaBauve, J. O. Agola, K. E. Baty, T. Howard, J. K. Sabo, C. R. S. Espinoza, J. A. Doudna, J. S. Schoeniger, K. S. Butler, O. A. Negrete, C. J. Brinker and R. E. Serda, Acta Biomater., 2020, 114, 358–368 CrossRef CAS PubMed
- Y. Wang, P. K. Shahi, X. Wang, R. Xie, Y. Zhao, M. Wu, S. Roge, B. R. Pattnaik and S. Gong, J. Controlled Release, 2021, 336, 296–309 CrossRef CAS PubMed
- N. Ryu, M.-A. Kim, D. Park, B. Lee, Y.-R. Kim, K.-H. Kim, J.-I. Baek, W. J. Kim, K.-Y. Lee and U.-K. Kim, Nanomedicine, 2018, 14, 2095–2102 CrossRef CAS PubMed
- C.-S. Cho, ISRN Mater. Sci., 2012, 2012, 798247 Search PubMed
- Z. Zhang, T. Wan, Y. Chen, Y. Chen, H. Sun, T. Cao, Z. Songyang, G. Tang, C. Wu, Y. Ping, F.-J. Xu and J. Huang, Macromol. Rapid Commun., 2019, 40, 1800068 CrossRef PubMed
- C. Liang, F. Li, L. Wang, Z.-K. Zhang, C. Wang, B. He, J. Li, Z. Chen, A. B. Shaikh, J. Liu, X. Wu, S. Peng, L. Dang, B. Guo, X. He, D. W. T. Au, C. Lu, H. Zhu, B.-T. Zhang, A. Lu and G. Zhang, Biomaterials, 2017, 147, 68–85 CrossRef CAS PubMed
- L. Li, L. Song, X. Liu, X. Yang, X. Li, T. He, N. Wang, S. Yang, C. Yu, T. Yin, Y. Wen, Z. He, X. Wei, W. Su, Q. Wu, S. Yao, C. Gong and Y. Wei, ACS Nano, 2017, 11, 95–111 CrossRef CAS PubMed
- L. Li, Z. Yang, S. Zhu, L. He, W. Fan, W. Tang, J. Zou, Z. Shen, M. Zhang, L. Tang, Y. Dai, G. Niu, S. Hu and X. Chen, Adv. Mater., 2019, 31, 1901187 CrossRef PubMed
- Y. Lyu, S. He, J. Li, Y. Jiang, H. Sun, Y. Miao and K. Pu, Angew. Chem., Int. Ed., 2019, 58, 18197–18201 CrossRef CAS PubMed
- X. Xu, K. Xie, X.-Q. Zhang, E. M. Pridgen, G. Y. Park, D. S. Cui, J. Shi, J. Wu, P. W. Kantoff, S. J. Lippard, R. Langer, G. C. Walker and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18638–18643 CrossRef CAS PubMed
- Y. Takashima, R. Saito, A. Nakajima, M. Oda, A. Kimura, T. Kanazawa and H. Okada, Int. J. Pharm., 2007, 343, 262–269 CrossRef CAS PubMed
- Y.-L. Luo, L.-F. Liang, Y.-J. Gan, J. Liu, Y. Zhang, Y.-N. Fan, G. Zhao, A. Czarna, Z.-D. Lu, X.-J. Du, S. Shen, C.-F. Xu, Z.-X. Lian and J. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 48259–48271 CrossRef CAS PubMed
- M. Li, Y.-N. Fan, Z.-Y. Chen, Y.-L. Luo, Y.-C. Wang, Z.-X. Lian, C.-F. Xu and J. Wang, Nano Res., 2018, 11, 6270–6282 CrossRef CAS
- Y. Liu, G. Zhao, C.-F. Xu, Y.-L. Luo, Z.-D. Lu and J. Wang, Biomater. Sci., 2018, 6, 1592–1603 RSC
- Y.-L. Luo, C.-F. Xu, H.-J. Li, Z.-T. Cao, J. Liu, J.-L. Wang, X.-J. Du, X.-Z. Yang, Z. Gu and J. Wang, ACS Nano, 2018, 12, 994–1005 CrossRef CAS PubMed
- Y. Liu, Z.-T. Cao, C.-F. Xu, Z.-D. Lu, Y.-L. Luo and J. Wang, Biomaterials, 2018, 172, 92–104 CrossRef CAS PubMed
- Q. Yang, Y. Zhou, J. Chen, N. Huang, Z. Wang and Y. Cheng, Int. J. Nanomed., 2021, 16, 185 CrossRef PubMed
- A. Srivastav, K. Gupta, D. Chakraborty, P. Dandekar and R. Jain, J. Pharm. Innovation, 2020, 1–14 CAS
- D. M. Lynn and R. Langer, J. Am. Chem. Soc., 2000, 122, 10761–10768 CrossRef CAS
- A. Akinc, D. M. Lynn, D. G. Anderson and R. Langer, J. Am. Chem. Soc., 2003, 125, 5316–5323 CrossRef CAS PubMed
- H. Deng, S. Tan, X. Gao, C. Zou, C. Xu, K. Tu, Q. Song, F. Fan, W. Huang and Z. Zhang, Acta Pharm. Sin. B, 2020, 10, 358–373 CrossRef CAS PubMed
- X. Gao, Z. Jin, X. Tan, C. Zhang, C. Zou, W. Zhang, J. Ding, B. C. Das, K. Severinov, I. I. Hitzeroth, P. R. Debata, D. He, X. Ma, X. Tian, Q. Gao, J. Wu, R. Tian, Z. Cui, W. Fan, Z. Huang, C. Cao, Y. Bao, S. Tan and Z. Hu, J. Controlled Release, 2020, 321, 654–668 CrossRef CAS PubMed
- Y. Rui, D. R. Wilson, K. Sanders and J. J. Green, ACS Appl. Mater. Interfaces, 2019, 11, 10472–10480 CrossRef CAS PubMed
- Y. Rui, M. Varanasi, S. Mendes, H. M. Yamagata, D. R. Wilson and J. J. Green, Mol. Ther. – Nucleic Acids, 2020, 20, 661–672 CrossRef CAS PubMed
- H. Gao, M. Elsabahy, E. V. Giger, D. Li, R. E. Prud'homme and J.-C. Leroux, Biomacromolecules, 2010, 11, 889–895 CrossRef CAS PubMed
- X. Zhang, C. Xu, S. Gao, P. Li, Y. Kong, T. Li, Y. Li, F. J. Xu and J. Du, Adv. Sci., 2019, 6, 1900386 CrossRef PubMed
- J.-J. Nie, Y. Liu, Y. Qi, N. Zhang, B. Yu, D.-F. Chen, M. Yang and F.-J. Xu, J. Controlled Release, 2021, 333, 362–373 CrossRef CAS PubMed
- D. Chuan, T. Jin, R. Fan, L. Zhou and G. Guo, Adv. Colloid Interface Sci., 2019, 268, 25–38 CrossRef PubMed
- B.-C. Zhang, P.-Y. Wu, J.-J. Zou, J.-L. Jiang, R.-R. Zhao, B.-Y. Luo, Y.-Q. Liao and J.-W. Shao, Chem. Eng. J., 2020, 393, 124688 CrossRef CAS
- H. Zhang, T. F. Bahamondez-Canas, Y. Zhang, J. Leal and H. D. Smyth, Mol. Pharm., 2018, 15, 4814–4826 CrossRef CAS PubMed
- Y. Qi, H. Song, H. Xiao, G. Cheng, B. Yu and F.-J. Xu, Small, 2018, 14, 1803061 CrossRef PubMed
- Y. Qi, Y. Liu, B. Yu, Y. Hu, N. Zhang, Y. Zheng, M. Yang and F. J. Xu, Adv. Sci., 2020, 7, 2001424 CrossRef CAS PubMed
- M. Shao, Y. Qi, D. Sui and F.-J. Xu, Biomater. Sci., 2021, 9, 7104–7114 RSC
- H.-X. Wang, Z. Song, Y.-H. Lao, X. Xu, J. Gong, D. Cheng, S. Chakraborty, J. S. Park, M. Li, D. Huang, L. Yin, J. Cheng and K. W. Leong, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4903–4908 CrossRef CAS PubMed
- Y.-H. Lao, M. Li, M. A. Gao, D. Shao, C.-W. Chi, D. Huang, S. Chakraborty, T.-C. Ho, W. Jiang, H.-X. Wang, S. Wang and K. W. Leong, Adv. Sci., 2018, 5, 1700540 CrossRef PubMed
- M. R. Emami, C. S. Young, Y. Ji, X. Liu, E. Mokhonova, A. D. Pyle, H. Meng and M. J. Spencer, Adv. Ther., 2019, 2, 1900061 CrossRef
- L. Zhang, P. Wang, Q. Feng, N. Wang, Z. Chen, Y. Huang, W. Zheng and X. Jiang, NPG Asia Mater., 2017, 9, e441–e441 CrossRef CAS
- C.-S. Wang, C.-H. Chang, T.-Y. Tzeng, A. M.-Y. Lin and Y.-L. Lo, Nanoscale Horiz., 2021, 6, 729–743 RSC
- M. Li, H. Xie, Y. Liu, C. Xia, X. Cun, Y. Long, X. Chen, M. Deng, R. Guo, Z. Zhang and Q. He, J. Controlled Release, 2019, 304, 204–215 CrossRef CAS PubMed
- H. Yin, X. Yuan, L. Luo, Y. Lu, B. Qin, J. Zhang, Y. Shi, C. Zhu, J. Yang, X. Li, M. Jiang, Z. Luo, X. Shan, D. Chen and J. You, Adv. Sci., 2020, 7, 1903381 CrossRef CAS PubMed
- C. Li, T. Yang, Y. Weng, M. Zhang, D. Zhao, S. Guo, B. Hu, W. Shao, X. Wang, A. Hussain, X. Liang and Y. Huang, Bioact. Mater., 2021, 9, 590–601 CrossRef PubMed
- D. Sun, Z. Sun, H. Jiang, A. M. Vaidya, R. Xin, N. R. Ayat, A. L. Schilb, P. L. Qiao, Z. Han, A. Naderi and Z.-R. Lu, Bioconjugate Chem., 2018, 30, 667–678 CrossRef PubMed
- Y. Chen, X. Chen, D. Wu, H. Xin, D. Chen, D. Li, H. Pan, C. Zhou and Y. Ping, Chem. Mater., 2020, 33, 81–91 CrossRef
- H. Tang, X. Xu, Y. Chen, H. Xin, T. Wan, B. Li, H. Pan, D. Li and Y. Ping, Adv. Mater., 2021, 33, 2006003 CrossRef CAS PubMed
- X. Chen, Y. Chen, H. Xin, T. Wan and Y. Ping, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2395–2405 CrossRef CAS PubMed
- Y. Tao, K. Yi, H. Hu, D. Shao and M. Li, J. Mater. Chem. B, 2021, 9, 94–100 RSC
- X. Xu, O. Koivisto, C. Liu, J. Zhou, M. Miihkinen, G. Jacquemet, D. Wang, J. M. Rosenholm, Y. Shu and H. Zhang, Adv. Ther., 2021, 4, 2000072 CrossRef CAS
- B.-C. Zhang, B.-Y. Luo, J.-J. Zou, P.-Y. Wu, J.-L. Jiang, J.-Q. Le, R.-R. Zhao, L. Chen and J.-W. Shao, ACS Appl. Mater. Interfaces, 2020, 12, 57362–57372 CrossRef CAS PubMed
- J. Shen, Z. Lu, J. Wang, Q. Hao, W. Ji, Y. Wu, H. Peng, R. Zhao, J. Yang, Y. Li, Z. Shi and X. Zhang, Adv. Mater., 2021, 2101993 CrossRef CAS PubMed
- A. Poddar, S. Pyreddy, F. Carraro, S. Dhakal, A. Rassell, M. R. Field, T. S. Reddy, P. Falcaro, C. M. Doherty and R. Shukla, Chem. Commun., 2020, 56, 15406–15409 RSC
- A. Hasanzadeh, F. Radmanesh, E. S. Hosseini, I. Hashemzadeh, J. Kiani, H. Nourizadeh, M. Naseri, Y. Fatahi, F. Chegini, Z. Madjd, A. Beyzavi, P. S. Kowalski and M. Karimi, Bioconjugate Chem., 2021, 32, 1875–1887 CrossRef CAS PubMed
- A. Hasanzadeh, F. Radmanesh, E. S. Hosseini, I. Hashemzadeh, J. Kiani, M. Naseri, H. Nourizadeh, Y. Fatahi, B. K. Y. Azar, B. G. Marani, A. Beyzavi, V. P. Mahabadi and M. Karimi, Nanomedicine, 2021, 16, 1673–1690 CrossRef CAS PubMed
- Y. Wu, J. Zheng, Q. Zeng, T. Zhang and D. Xing, Nano Res., 2020, 13, 2399–2406 CrossRef CAS
- X. Y. He, X. H. Ren, Y. Peng, J. P. Zhang, S. L. Ai, B. Y. Liu, C. Xu and S. X. Cheng, Adv. Mater., 2020, 32, 2000208 CrossRef CAS PubMed
- B.-Y. Liu, X.-Y. He, C. Xu, X.-H. Ren, R.-X. Zhuo and S.-X. Cheng, ACS Appl. Mater. Interfaces, 2019, 11, 23870–23879 CrossRef CAS PubMed
- B.-Y. Liu, X.-Y. He, C. Xu, L. Xu, S.-L. Ai, S.-X. Cheng and R.-X. Zhuo, Biomacromolecules, 2018, 19, 2957–2968 CrossRef CAS PubMed
- T. Briolay, T. Petithomme, M. Fouet, N. Nguyen-Pham, C. Blanquart and N. Boisgerault, Mol. Cancer, 2021, 20, 1–24 CrossRef PubMed
- S. M. Kim, Y. Yang, S. J. Oh, Y. Hong, M. Seo and M. Jang, J. Controlled Release, 2017, 266, 8–16 CrossRef CAS PubMed
- Q. Xu, Z. Zhang, L. Zhao, Y. Qin, H. Cai, Z. Geng, X. Zhu, W. Zhang, Y. Zhang, J. Tan, J. Wang and J. Zhou, J. Controlled Release, 2020, 326, 455–467 CrossRef CAS PubMed
- Y. Lin, J. Wu, W. Gu, Y. Huang, Z. Tong, L. Huang and J. Tan, Adv. Sci., 2018, 5, 1700611 CrossRef PubMed
- X.-H. Ren, X.-Y. He, B.-Y. Liu, C. Xu and S.-X. Cheng, ACS Appl. Bio Mater., 2020, 3, 7831–7839 CrossRef CAS PubMed
- T. Killian, A. Buntz, T. Herlet, H. Seul, O. Mundigl, G. Längst and U. Brinkmann, Nucleic Acids Res., 2019, 47, e55–e55 CrossRef CAS PubMed
- W.-J. Cheng, L.-C. Chen, H.-O. Ho, H.-L. Lin and M.-T. Sheu, Int. J. Nanomed., 2018, 13, 7079 CrossRef CAS PubMed
- Y. Zhang, S. Shen, G. Zhao, C.-F. Xu, H.-B. Zhang, Y.-L. Luo, Z.-T. Cao, J. Shi, Z.-B. Zhao, Z.-X. Lian and J. Wang, Biomaterials, 2019, 217, 119302 CrossRef CAS PubMed
- X. Li, Q. Feng, Z. Han and X. Jiang, Chin. J. Chem. Eng., 2021, 38, 216–220 CrossRef
- S. Abbasi, S. Uchida, K. Toh, T. A. Tockary, A. Dirisala, K. Hayashi, S. Fukushima and K. Kataoka, J. Controlled Release, 2021, 332, 260–268 CrossRef CAS PubMed
- X. Huang, R. Zheng, F. Ding, J. Yang, M. Xie, X. Liu, J. Li, J. Feng, X. Zhu and C. Zhang, ACS Mater. Lett., 2020, 2, 1509–1515 CrossRef CAS
- H. Yin, C.-Q. Song, J. R. Dorkin, L. J. Zhu, Y. Li, Q. Wu, A. Park, J. Yang, S. Suresh, A. Bizhanova, A. Gupta, M. F. Bolukbasi, S. Walsh, R. L. Bogorad, G. Gao, Z. Weng, Y. Dong, V. Koteliansky, S. A. Wolfe, R. Langer, W. Xue and D. G. Anderson, Nat. Biotechnol., 2016, 34, 328–333 CrossRef CAS PubMed
- C. Jiang, M. Mei, B. Li, X. Zhu, W. Zu, Y. Tian, Q. Wang, Y. Guo, Y. Dong and X. Tan, Cell Res., 2017, 27, 440–443 CrossRef CAS PubMed
- J. Liu, J. Chang, Y. Jiang, X. Meng, T. Sun, L. Mao, Q. Xu and M. Wang, Adv. Mater., 2019, 31, 1902575 CrossRef PubMed
- Y. Li, R. Jarvis, K. Zhu, Z. Glass, R. Ogurlu, P. Gao, P. Li, J. Chen, Y. Yu, Y. Yang and Q. Xu, Angew. Chem., 2020, 132, 15067–15074 CrossRef
- M. Qiu, Z. Glass, J. Chen, M. Haas, X. Jin, X. Zhao, X. Rui, Z. Ye, Y. Li, F. Zhang and Q. Xu, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(10), e2020401118 CrossRef CAS PubMed
- X. Zhao, Z. Glass, J. Chen, L. Yang, D. L. Kaplan and Q. Xu, Adv. Healthcare Mater., 2021, 10, 2000938 CrossRef CAS PubMed
- X. Zhang, B. Li, X. Luo, W. Zhao, J. Jiang, C. Zhang, M. Gao, X. Chen and Y. Dong, ACS Appl. Mater. Interfaces, 2017, 9, 25481–25487 CrossRef CAS PubMed
- Q. Cheng, T. Wei, L. Farbiak, L. T. Johnson, S. A. Dilliard and D. J. Siegwart, Nat. Nanotechnol., 2020, 15, 313–320 CrossRef CAS PubMed
- S. Liu, Q. Cheng, T. Wei, X. Yu, L. T. Johnson, L. Farbiak and D. J. Siegwart, Nat. Mater., 2021, 20, 701–710 CrossRef CAS PubMed
- L. Farbiak, Q. Cheng, T. Wei, E. Álvarez-Benedicto, L. T. Johnson, S. Lee and D. J. Siegwart, Adv. Mater., 2021, 2006619 CrossRef CAS PubMed
- D. Rosenblum, A. Gutkin, R. Kedmi, S. Ramishetti, N. Veiga, A. M. Jacobi, M. S. Schubert, D. Friedmann-Morvinski, Z. R. Cohen, M. A. Behlke, J. Lieberman and D. Peer, Sci. Adv., 2020, 6, eabc9450 CrossRef CAS PubMed
- Q. Tang, J. Liu, Y. Jiang, M. Zhang, L. Mao and M. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 46585–46590 CrossRef CAS PubMed
- Z. Chen, F. Liu, Y. Chen, J. Liu, X. Wang, A. T. Chen, G. Deng, H. Zhang, J. Liu, Z. Hong and J. Zhou, Adv. Funct. Mater., 2017, 27, 1703036 CrossRef PubMed
- T. Wan, Y. Chen, Q. Pan, X. Xu, Y. Kang, X. Gao, F. Huang, C. Wu and Y. Ping, J. Controlled Release, 2020, 322, 236–247 CrossRef CAS PubMed
- T. Wan, Q. Pan and Y. Ping, Sci. Adv., 2021, 7, eabe2888 CrossRef CAS PubMed
- Q. Ban, P. Yang, S.-J. Chou, L. Qiao, H. Xia, J. Xue, F. Wang, X. Xu, N. Sun, R. Y. Zhang, C. Zhang, A. Lee, W. Liu, T.-Y. Lin, Y.-L. Ko, P. Antovski, X. Zhang, S.-H. Chiou, C.-F. Lee, W. Hui, D. Liu, S. J. Jonas, P. S. Weiss and H.-R. Tseng, Small, 2021, 17, 2100546 CrossRef CAS PubMed
- Q. Liu, J. Cai, Y. Zheng, Y. Tan, Y. Wang, Z. Zhang, C. Zheng, Y. Zhao, C. Liu, Y. An, C. Jiang, L. Shi, C. Kang and Y. Liu, Nano Lett., 2019, 19, 7662–7672 CrossRef CAS PubMed
- S. Deng, X. Li, S. Liu, J. Chen, M. Li, S. Y. Chew, K. W. Leong and D. Cheng, Sci. Adv., 2020, 6, eabb4005 CrossRef CAS PubMed
- Y. Rui, D. R. Wilson, J. Choi, M. Varanasi, K. Sanders, J. Karlsson, M. Lim and J. J. Green, Sci. Adv., 2019, 5, eaay3255 CrossRef CAS PubMed
- G. Chen, A. A. Abdeen, Y. Wang, P. K. Shahi, S. Robertson, R. Xie, M. Suzuki, B. R. Pattnaik, K. Saha and S. Gong, Nat. Nanotechnol., 2019, 14, 974–980 CrossRef CAS PubMed
- C. Liu, T. Wan, H. Wang, S. Zhang, Y. Ping and Y. Cheng, Sci. Adv., 2019, 5, eaaw8922 CrossRef CAS PubMed
- R. Kumar, N. Le, Z. Tan, M. E. Brown, S. Jiang and T. M. Reineke, ACS Nano, 2020, 14, 17626–17639 CrossRef CAS PubMed
- T.-C. Ho, H. S. Kim, Y. Chen, Y. Li, M. W. LaMere, C. Chen, H. Wang, J. Gong, C. D. Palumbo, J. M. Ashton, H.-W. Kim, Q. Xu, M. W. Becker and K. W. Leong, Sci. Adv., 2021, 7, eabg3217 CrossRef CAS PubMed
- Y. A. Aksoy, B. Yang, W. Chen, T. Hung, R. P. Kuchel, N. W. Zammit, S. T. Grey, E. M. Goldys and W. Deng, ACS Appl. Mater. Interfaces, 2020, 12, 52433–52444 CrossRef CAS PubMed
- J.-Y. Ryu, E.-J. Won, H. A. R. Lee, J. H. Kim, E. Hui, H. P. Kim and T.-J. Yoon, Biomaterials, 2020, 232, 119736 CrossRef CAS PubMed
- K. Lee, M. Conboy, H. M. Park, F. Jiang, H. J. Kim, M. A. Dewitt, V. A. Mackley, K. Chang, A. Rao, C. Skinner, T. Shobha, M. Mehdipour, H. Liu, W.-c. Huang, F. Lan, N. L. Bray, S. Li, J. E. Corn, K. Kataoka, J. A. Doudna, I. Conboy and N. Murthy, Nat. Biomed. Eng., 2017, 1, 889–901 CrossRef CAS PubMed
- R. Shahbazi, G. Sghia-Hughes, J. L. Reid, S. Kubek, K. G. Haworth, O. Humbert, H.-P. Kiem and J. E. Adair, Nat. Mater., 2019, 18, 1124–1132 CrossRef CAS PubMed
- L. Zhang, L. Wang, Y. Xie, P. Wang, S. Deng, A. Qin, J. Zhang, X. Yu, W. Zheng and X. Jiang, Angew. Chem., Int. Ed., 2019, 58, 12404–12408 CrossRef CAS PubMed
- J. Wu, H. Peng, X. Lu, M. Lai, H. Zhang and X. C. Le, Angew. Chem., Int. Ed., 2021, 60, 11104–11109 CrossRef CAS PubMed
- Y.-W. Lee, R. Mout, D. C. Luther, Y. Liu, L. Castellanos-García, A. S. Burnside, M. Ray, G. Y. Tonga, J. Hardie, H. Nagaraj, R. Das, E. L. Phillips, T. Tay, R. W. Vachet and V. M. Rotello, Adv. Ther., 2019, 2, 1900041 CrossRef CAS PubMed
- X. Li, Y. Pan, C. Chen, Y. Gao, X. Liu, K. Yang, X. Luan, D. Zhou, F. Zeng, X. Han and Y. Song, Angew. Chem., Int. Ed., 2021, 60, 21200–21204 CrossRef CAS PubMed
- M. Hansen-Bruhn, B. E. F. de Ávila, M. Beltrán-Gastélum, J. Zhao, D. E. Ramírez-Herrera, P. Angsantikul, K. V. Gothelf, L. Zhang and J. Wang, Angew. Chem., Int. Ed., 2018, 57, 2657–2661 CrossRef CAS PubMed
- S. K. Alsaiari, S. Patil, M. Alyami, K. O. Alamoudi, F. A. Aleisa, J. S. Merzaban, M. Li and N. M. Khashab, J. Am. Chem. Soc., 2018, 140, 143–146 CrossRef CAS PubMed
- M. Z. Alyami, S. K. Alsaiari, Y. Li, S. S. Qutub, F. A. Aleisa, R. Sougrat, J. S. Merzaban and N. M. Khashab, J. Am. Chem. Soc., 2020, 142, 1715–1720 CrossRef CAS PubMed
- X. Yang, Q. Tang, Y. Jiang, M. Zhang, M. Wang and L. Mao, J. Am. Chem. Soc., 2019, 141, 3782–3786 CrossRef CAS PubMed
- J. Liu, T. Luo, Y. Xue, L. Mao, P. J. Stang and M. Wang, Angew. Chem., Int. Ed., 2021, 60, 5429–5435 CrossRef CAS PubMed
- B. Liu, W. Ejaz, S. Gong, M. Kurbanov, M. Canakci, F. Anson and S. Thayumanavan, Nano Lett., 2020, 20, 4014–4021 CrossRef CAS PubMed
- Y. Pan, J. Yang, X. Luan, X. Liu, X. Li, J. Yang, T. Huang, L. Sun, Y. Wang and Y. Lin, Sci. Adv., 2019, 5, eaav7199 CrossRef CAS PubMed
- C. Chen, Y. Ma, S. Du, Y. Wu, P. Shen, T. Yan, X. Li, Y. Song, Z. Zha and X. Han, Small, 2021, 17, 2101155 CrossRef CAS PubMed
- H. Yue, X. Zhou, M. Cheng and D. Xing, Nanoscale, 2018, 10, 1063–1071 RSC
- W. Zhou, H. Cui, L. Ying and X. F. Yu, Angew. Chem., 2018, 130, 10425–10429 CrossRef
- S. Ramakrishna, A.-B. K. Dad, J. Beloor, R. Gopalappa, S.-K. Lee and H. Kim, Genome Res., 2014, 24, 1020–1027 CrossRef CAS PubMed
- J. Yin, Q. Wang, S. Hou, L. Bao, W. Yao and X. Gao, J. Am. Chem. Soc., 2018, 140, 17234–17240 CrossRef CAS PubMed
- J. Yin, S. Hou, Q. Wang, L. Bao, D. Liu, Y. Yue, W. Yao and X. Gao, Bioconjugate Chem., 2019, 30, 898–906 CrossRef CAS PubMed
- S. M. Kim, S. C. Shin, E. E. Kim, S.-H. Kim, K. Park, S. J. Oh and M. Jang, ACS Nano, 2018, 12, 7750–7760 CrossRef CAS PubMed
- H. Park, J. Oh, G. Shim, B. Cho, Y. Chang, S. Kim, S. Baek, H. Kim, J. Shin, H. Choi, J. Yoo, J. Kim, W. Jun, M. Lee, C. J. Lengner, Y.-K. Oh and J. Kim, Nat. Neurosci., 2019, 22, 524–528 CrossRef CAS PubMed
- S. Krishnamurthy, C. Wohlford-Lenane, S. Kandimalla, G. Sartre, D. K. Meyerholz, V. Théberge, S. Hallée, A.-M. Duperré, T. Del'Guidice, J.-P. Lepetit-Stoffaes, X. Barbeau, D. Guay and P. B. McCray, Nat. Commun., 2019, 10, 4906 CrossRef PubMed
- T. T. Thach, D. H. Bae, N. H. Kim, E. S. Kang, B. S. Lee, K. Han, M. Kwak, H. Choi, J. Nam, T. Bae, M. Suh, J. K. Hur and Y. H. Kim, Small, 2019, 15, 1903172 CrossRef CAS PubMed
- P. K. Jain, J. H. Lo, S. Rananaware, M. Downing, A. Panda, M. Tai, S. Raghavan, H. E. Fleming and S. N. Bhatia, Nanoscale, 2019, 11, 21317–21323 RSC
- I. Lostalé-Seijo, I. Louzao, M. Juanes and J. Montenegro, Chem. Sci., 2017, 8, 7923–7931 RSC
- C. Montagna, G. Petris, A. Casini, G. Maule, G. M. Franceschini, I. Zanella, L. Conti, F. Arnoldi, O. R. Burrone, L. Zentilin, S. Zacchigna, M. Giacca and A. Cereseto, Mol. Ther. – Nucleic Acids, 2018, 12, 453–462 CrossRef CAS PubMed
- L. A. Campbell, L. M. Coke, C. T. Richie, L. V. Fortuno, A. Y. Park and B. K. Harvey, Mol. Ther., 2019, 27, 151–163 CrossRef CAS PubMed
- P. Gee, M. S. Y. Lung, Y. Okuzaki, N. Sasakawa, T. Iguchi, Y. Makita, H. Hozumi, Y. Miura, L. F. Yang, M. Iwasaki, X. H. Wang, M. A. Waller, N. Shirai, Y. O. Abe, Y. Fujita, K. Watanabe, A. Kagita, K. A. Iwabuchi, M. Yasuda, H. Xu, T. Noda, J. Komano, H. Sakurai, N. Inukai and A. Hotta, Nat. Commun., 2020, 11, 1334 CrossRef CAS PubMed
- X. Yao, P. Lyu, K. Yoo, M. K. Yadav, R. Singh, A. Atala and B. Lu, J. Extracell. Vesicles, 2021, 10, e12076 CAS
- Y. Ye, X. Zhang, F. Xie, B. Xu, P. Xie, T. Yang, Q. Shi, C.-Y. Zhang, Y. Zhang and J. Chen, Biomater. Sci., 2020, 8, 2966–2976 RSC
- J. Zhuang, J. Tan, C. Wu, J. Zhang, T. Liu, C. Fan, J. Li and Y. Zhang, Nucleic Acids Res., 2020, 48, 8870–8882 CrossRef CAS PubMed
- P. E. Mangeot, V. Risson, F. Fusil, A. Marnef, E. Laurent, J. Blin, V. Mournetas, E. Massouridès, T. J. Sohier, A. Corbin, F. Aubé, M. Teixeira, C. Pinset, L. Schaeffer, G. Legube, F.-L. Cosset, E. Verhoeyen, T. Ohlmann and E. P. Ricci, Nat. Commun., 2019, 10, 1–15 CrossRef PubMed
- J. R. Hamilton, C. A. Tsuchida, D. N. Nguyen, B. R. Shy, E. R. McGarrigle, C. R. S. Espinoza, D. Carr, F. Blaeschke, A. Marson and J. A. Doudna, Cell Rep., 2021, 35, 109207 CrossRef CAS PubMed
- W. Sun, W. Ji, J. M. Hall, Q. Hu, C. Wang, C. L. Beisel and Z. Gu, Angew. Chem., 2015, 127, 12197–12201 CrossRef
- F. Ding, X. Huang, X. Gao, M. Xie, G. Pan, Q. Li, J. Song, X. Zhu and C. Zhang, Nanoscale, 2019, 11, 17211–17215 RSC
- J. Liu, T. Wu, X. Lu, X. Wu, S. Liu, S. Zhao, X. Xu and B. Ding, J. Am. Chem. Soc., 2019, 141, 19032–19037 CrossRef CAS PubMed
- J. Shi, X. Yang, Y. Li, D. Wang, W. Liu, Z. Zhang, J. Liu and K. Zhang, Biomaterials, 2020, 256, 120221 CrossRef CAS PubMed
- R. Rouet, B. A. Thuma, M. D. Roy, N. G. Lintner, D. M. Rubitski, J. E. Finley, H. M. Wisniewska, R. Mendonsa, A. Hirsh, L. de Oñate, J. C. Barrón, T. J. McLellan, J. Bellenger, X. Feng, A. Varghese, B. A. Chrunyk, K. Borzilleri, K. D. Hesp, K. Zhou, N. Ma, M. Tu, R. Dullea, K. F. McClure, R. C. Wilson, S. Liras, V. Mascitti and J. A. Doudna, J. Am. Chem. Soc., 2018, 140, 6596–6603 CrossRef CAS PubMed
- X. He, Q. Long, Z. Zeng, L. Yang, Y. Tang and X. Feng, Adv. Funct. Mater., 2019, 29, 1906187 CrossRef CAS
- J. Qiao, W. Sun, S. Lin, R. Jin, L. Ma and Y. Liu, Chem. Commun., 2019, 55, 4707–4710 RSC
- X. Zhu, M.-M. Lv, J.-W. Liu, R.-Q. Yu and J.-H. Jiang, Chem. Commun., 2019, 55, 6511–6514 RSC
- X. Pan, X. Pei, H. Huang, N. Su, Z. Wu, Z. Wu and X. Qi, J. Controlled Release, 2021, 337, 686–697 CrossRef CAS PubMed
- Y. Lu, J. Xue, T. Deng, X. Zhou, K. Yu, L. Deng, M. Huang, X. Yi, M. Liang, Y. Wang, H. Shen, R. Tong, W. Wang, L. Li, J. Song, J. Li, X. Su, Z. Ding, Y. Gong, J. Zhu, Y. Wang, B. Zou, Y. Zhang, Y. Li, L. Zhou, Y. Liu, M. Yu, Y. Wang, X. Zhang, L. Yin, X. Xia, Y. Zeng, Q. Zhou, B. Ying, C. Chen, Y. Wei, W. Li and T. Mok, Nat. Med., 2020, 26, 732–740 CrossRef CAS PubMed
- E. González-Romero, C. Martínez-Valiente, C. García-Ruiz, R. P. Vázquez-Manrique, J. Cervera and A. Sanjuan-Pla, Haematologica, 2019, 104, 881 CrossRef PubMed
- News in Brief, Nat. Biotechnol., 2020, 38, 382–382 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1bm01658j |
| This journal is © The Royal Society of Chemistry 2022 |