
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High capacity ammonia adsorption in a robust metal–organic framework mediated by reversible host–guest interactions†
Lixia
Guo
 a,
Xue
Han
a,
Yujie
Ma
a,
Jiangnan
Li
a,
Wanpeng
Lu
a,
Weiyao
Li
a,
Daniel
Lee
b,
Ivan
da Silva
c,
Yongqiang
Cheng
d,
Svemir
Rudić
c,
Pascal
Manuel
c,
Mark D.
Frogley
e,
Anibal J.
Ramirez-Cuesta
d,
Martin
Schröder
*a and
Sihai
Yang
*a
a,
Xue
Han
a,
Yujie
Ma
a,
Jiangnan
Li
a,
Wanpeng
Lu
a,
Weiyao
Li
a,
Daniel
Lee
b,
Ivan
da Silva
c,
Yongqiang
Cheng
d,
Svemir
Rudić
c,
Pascal
Manuel
c,
Mark D.
Frogley
e,
Anibal J.
Ramirez-Cuesta
d,
Martin
Schröder
*a and
Sihai
Yang
*a
aDepartment of Chemistry, University of Manchester, Manchester, M13 9PL, UK. E-mail: M.Schroder@manchester.ac.uk; Sihai.Yang@manchester.ac.uk
bDepartment of Chemical Engineering and Analytical Science, University of Manchester, Manchester, M13 9PL, UK
cISIS Facility, STFC Rutherford Appleton Laboratory, Oxfordshire, OX11 0QX, UK
dNeutron Scattering Division, Neutron Sciences Directorate, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
eDiamond Light Source, Harwell Science and Innovation Campus, Oxfordshire, OX11 0DE, UK
First published on 13th April 2022
Abstract
To understand the exceptional adsorption of ammonia (NH3) in MFM-300(Sc) (19.5 mmol g−1 at 273 K and 1 bar without hysteresis), we report a systematic investigation of the mechanism of adsorption by a combination of in situ neutron powder diffraction, inelastic neutron scattering, synchrotron infrared microspectroscopy, and solid-state 45Sc NMR spectroscopy. These complementary techniques reveal the formation of reversible host–guest supramolecular interactions, which explains directly the observed excellent reversibility of this material over 90 adsorption–desorption cycles.
Annual global production of ammonia (NH3) is around 170 million tonnes reflecting its role as a major feedstock for agriculture and industry.1 The high hydrogen content (17.8 wt %) and hydrogen volume density (105 kg m−3) of NH3 make it a desirable carbon-free hydrogen carrier, and NH3 is therefore regarded as a surrogate for the H2 economy. However, the corrosive and toxic nature of NH3 makes the development of stable storage materials with high and reversible uptakes extremely challenging. Conventional sorbent materials such as zeolites,2 activated carbons,3 and organic polymers4 have been investigated for the storage of NH3, but show low and often irreversible uptakes. Metal–organic framework (MOF) materials have been postulated as promising candidates for gas storage due to their high surface areas and versatile pore structures.5 As opposed to conventional adsorbents, the affinities of MOF materials to a target gas can be tailored by grafting the pore interior with functional groups to anchor the gas through coordination, hydrogen bonding, electrostatic interactions, acid–base interactions or π···π stacking.5–7 A large number of MOFs with functional groups (e.g. –COOH,8 –OH9) and open metal sites10 have been reported to impart enhanced affinity to gas molecules. Several state-of-the-art MOFs, such as MOF-177,11 M2Cl2BBTA [BBTA = 1H,5H-benzo(1,2-d:4,5-d′)bistriazole; M = Co, Mn],12 M2Cl2(BTDD) {BTDD = bis(1H-1,2,3-triazolo[4,5-b],[4′,5′-i])dibenzo[1,4]dioxin; M = Mn, Co, Ni and Cu}13 as well as MFM-300(M) (M = Al, Fe, V, Cr, In),6,14 have been investigated for NH3 adsorption. However, due to the reactive and corrosive nature of NH3, many MOF systems showed structural degradation and/or significant loss of uptake after consecutive cycles owing to irreversible host–guest binding. So far, only a very limited number of MOFs exhibit reversible NH3 sorption over multiple cycles.6,8,13–16 Unravelling the molecular details of host–guest interactions is of critical importance if new efficient NH3 storage systems are to be developed. This is however highly challenging, not least because hydrogen atoms are invisible in X-ray diffraction experiments and NH3 molecules can act as a rapid rotor even in solid state.
The mechanism of adsorption of NH3 in MFM-300(Sc) was examined systematically using gas isotherms, breakthrough experiments, in situ solid-state nuclear magnetic resonance (ssNMR) spectroscopy, synchrotron infrared microspectroscopy, neutron powder diffraction (NPD) and inelastic neutron scattering (INS) techniques, coupled with DFT modelling. Distinct new insights have been gained into the mechanism of adsorption compared with a recent report based on theoretical and infrared spectroscopic studies of this system.17 Importantly, we found the exceptional NH3 uptake (19.5 mmol g−1 at 273 K and 1 bar) by MFM-300(Sc) was mediated by reversible host–guest and guest–guest hydrogen bond interactions. The moderate strength of the host–guest interaction in MFM-300(Sc) leads to excellent adsorption reversibility and stability with full retention of the capacity over 90 cycles.
MFM-300(Sc) shows a three-dimensional framework containing [ScO4(OH)2] octahedra which are connected via the cis-μ2-OH groups into infinite chains, and further coordinated by the BPTC4− ligand (H4BPTC = biphenyl-3,3,5,5-tetracarboxylic acid) (Fig. S1, ESI†).18 Desolvated MFM-300(Sc) shows a Brunauer–Emmett–Teller (BET) surface area of 1390 m2 g−1 and a pore volume of 0.48 cm3 g−1 (Fig. S2, ESI†). MFM-300(Sc) exhibits high thermal stability up to 500 °C under N2 (Fig. S3, ESI†) and high chemical stability in aqueous solutions of pH of 7–12 as well as in various organic solvents (Fig. S4, ESI†).
Adsorption isotherms of NH3 for MFM-300(Sc) were measured at 273–313 K, where an exceptional uptake of 19.5 mmol g−1 was recorded at 273 K and 1.0 bar (Fig. 1a), reducing to 13.5 mmol g−1 at 298 K. MFM-300(Sc) shows the highest NH3 uptake among the MFM-300(M) family6,14 primarily due to its large pore size and pore volume allowing the accommodation of additional NH3 molecules in the pore. MFM-300(Sc) shows an NH3 uptake of 13.5 mmol g−1 at 298 K and 1.0 bar, comparing favourably with state-of-the-art materials (Fig. 1d and Table S6, ESI†). The uptake of NH3 in MFM-300(Sc) between 273 and 313 K decreases gradually with increasing temperature, consistent with an exothermic adsorption mechanism.19 The isosteric heat of adsorption (Qst) for NH3 in MFM-300(Sc) decreases from 60 to 30 kJ mol−1 with increasing loading of NH3 from 1 to 10 mmol g−1 (Fig. S7, ESI†), confirming the presence of moderate adsorbate–adsorbent binding interaction. The repeated isotherm of NH3 at 273 K using regenerated MFM-300(Sc) shows no loss in capacity with full retention of its porosity (Fig. S6, ESI†). 90 consecutive cycles of adsorption–desorption were conducted at 298 K and confirmed excellent reversibility and stability of adsorption (Fig. 1b), with retention of the crystal structure of MFM-300(Sc) as confirmed by powder X-ray diffraction (PXRD) (Fig. S5, ESI†). The ability of MFM-300(Sc) to capture NH3 at low concentrations (1000 ppm) was confirmed by dynamic breakthrough experiments at 298 K with a dynamic uptake of 1.65 mmol g−1, consistent with that measured by isotherms (1.74 mmol g−1 at 10 mbar, equivalent to 1000 ppm; Fig. 1c). With an exceptional adsorption capacity and excellent regenerability, MFM-300(Sc) represents a promising candidate for applications in NH3 storage and transport.
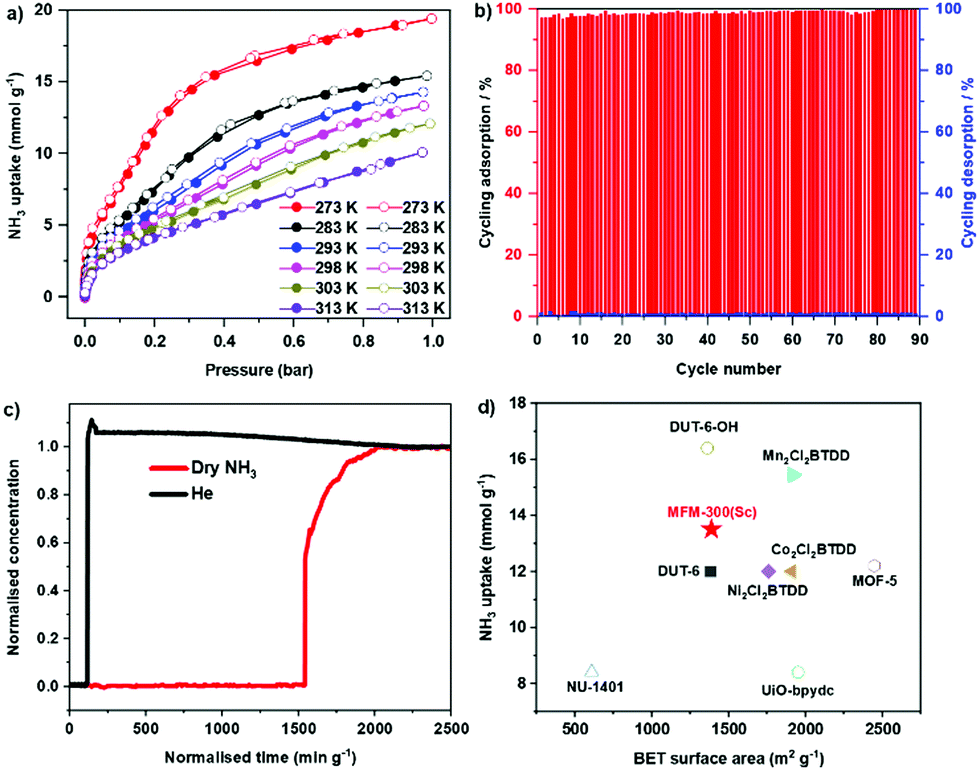 | ||
| Fig. 1 (a) Adsorption isotherms for NH3 in MFM-300(Sc) at 273–313 K (adsorption: solid symbols; desorption: open symbols). (b) 90 cycles of adsorption–desorption of NH3 in MFM-300(Sc) under pressure-swing conditions. (c) Breakthrough curve for NH3 (1000 ppm diluted in He) through a fixed-bed packed with MFM-300(Sc) at 298 K and 1.0 bar. (d) Comparison of NH3 uptake at 1 bar and 298 K for selected materials plotted against their surface areas (solid symbols: reversible sorption; hollow symbols: irreversible sorption; full details are given in the supplementary information). | ||
In situ NPD data of MFM-300(Sc) as a function of ND3 loading were collected and Rietveld refinements revealed the preferential binding sites for ND3 (Fig. 2). Interestingly, the NH3-induced rearrangement of metal–ligand (Sc–O) bonds via insertion of NH3 molecules into the MOF upon ND3 binding as predicted by a DFT study17 was not observed here. For MFM-300(Sc)·(ND3)1.25, {[Sc2(L)(OD0.6H0.4)2]·(ND2.05H0.95)1.25}, only one binding site was found, interacting primarily with the bridging μ2-OH groups at the four corners of its square-shaped channel [Obridge–H⋯ND3 = 1.96(1) Å] (Fig. 2a and b). At the higher loading of MFM-300(Sc)·(ND3)2.6, {[Sc2(L)(OD0.75H0.25)2]·(ND2.42H0.58)2.6}, two distinct binding sites were identified (Fig. 2c and d). Site I is fully occupied by ND3 molecules (1 ND3/Sc), with hydrogen bonding between the μ2-OH groups and the ND3 molecule [Obridge–H⋯ND3 = 1.93(1) Å]. This is complemented by additional electrostatic interactions [ND3⋯aromatic rings = 3.13(1) Å], and hydrogen bonding [ND3⋯Oligand = 3.24(1) Å]. Site II (0.3 ND3/Sc) exhibited hydrogen bonding with the ND3 at site I [2.30(3) Å and 2.24(2) Å], propagating along the 1D channel to form a cooperative {ND3}∞ network. Similar to other MFM-300 analogues,6,14 hydrogen/deuterium site exchange was also observed between the adsorbed ND3 molecules and the μ2-OH group for MFM-300(Sc).
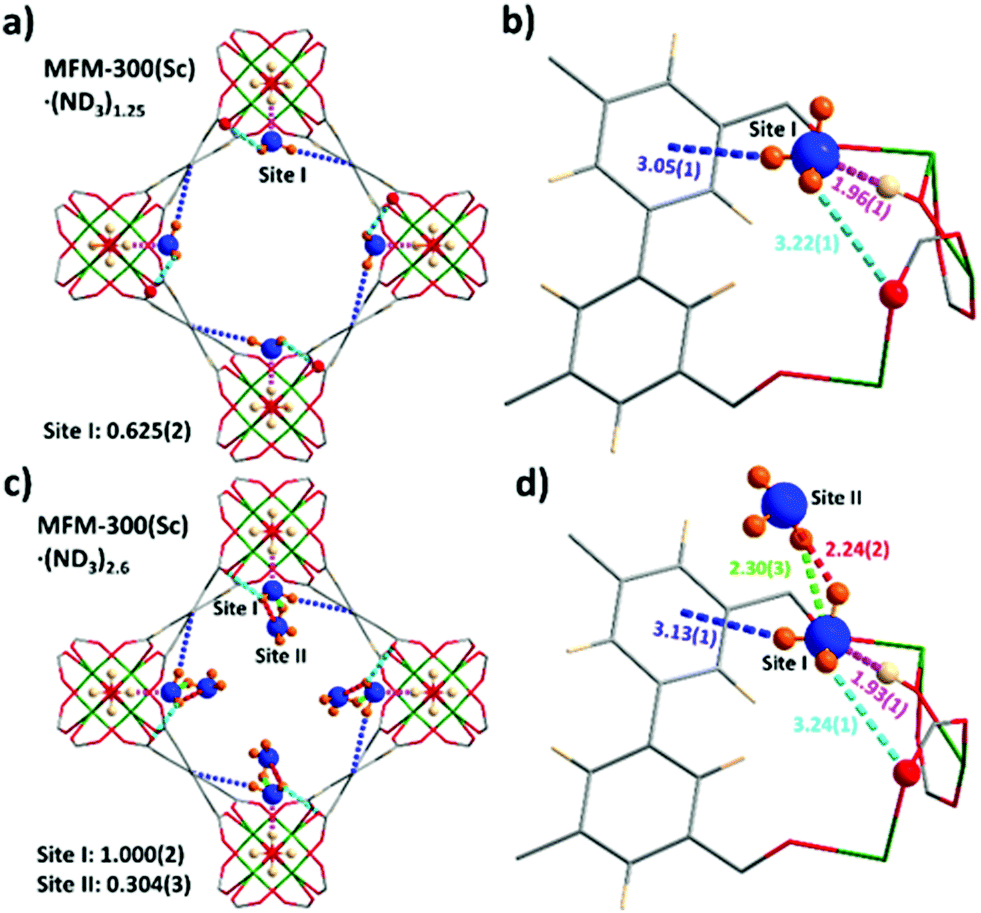 | ||
| Fig. 2 Views of binding sites for ND3 in MFM-300(Sc) determined by NPD at 10 K. The occupancy of each site has been converted into ND3/Sc for clarity. (a and c) Views along the c-axis showing packing of the guest molecules of ND3 in MFM-300(Sc)·(ND3)1.25 and MFM-300(Sc)·(ND3)2.6, respectively. (b and d) Detailed views of host–guest interactions between MFM-300(Sc) and adsorbed molecules of ND3. | ||
The analysis of the NPD data were entirely consistent with information from solid-state NMR spectroscopy. Upon loading MFM-300(Sc) with NH3, only slight structural modifications were observed, and the crystalline nature of the framework was retained. 45Sc magic angle spinning (MAS) NMR spectroscopy confirmed that the geometry around the Sc(III) centre was not notably distorted by interaction with NH3, with the μ2-OH groups (Fig. S11a, ESI†) and {1H-}13C CPMAS NMR spectra showing that the carboxyl resonance from the linker is unaffected upon NH3 loading (i.e. minimal metal site distortion). However, the resonances assigned to the aromatic carbons do shift, reflecting an interaction of the rings with the guest molecules (Fig. S11b, ESI†). The interaction of NH3 with the MOF was also investigated using 2D 1H-45Sc dipolar correlation (HETCOR) NMR spectroscopy (Fig. 3). The spectrum of pristine MFM-300(Sc) (Fig. 3a) shows clear cross peaks between aromatic protons (from the linker), as well as from hydroxyl protons (μ2-OH groups), with the Sc(III) site, demonstrating a close proximity between these atomic environments. The corresponding spectrum of NH3-loaded MFM-300(Sc) is substantially different. Whilst cross peaks with aromatic protons unchanged, cross peaks with μ2-OH groups have moved to higher chemical shifts, indicating the presence of hydrogen-bonding, and a new weak cross peak is observed and assigned to pore-confined NH3 protons. In situ synchrotron FTIR micro-spectra were recorded at 298 K (Fig. 3e and f). The characteristic O–H stretching mode of the μ2-OH group is observed at 3678 cm−1, which reduces in intensity and broadens upon loading of NH3. The band at 3404 cm−1 is assigned to the N–H stretching of NH3, and this exhibits a red shift to 3390 cm−1.6,14 The bands at 1614 and 1440 cm−1, assigned to νas(COO−) and νs(COO−), respectively,20 show small red shifts upon adsorption of NH3 (Δ = 4 − 7 cm−1), consistent with interaction between NH3 and carboxylate groups. The bands between 3800 and 1400 cm−1 for the local framework remain unchanged upon re-activation, confirming the high structural stability of MFM-300(Sc). Thus, the ssNMR and FTIR studies verify that NH3 is hydrogen-bonded to the μ2-OH groups via the lone pair of electrons on nitrogen, fully consistent with the NPD analysis.
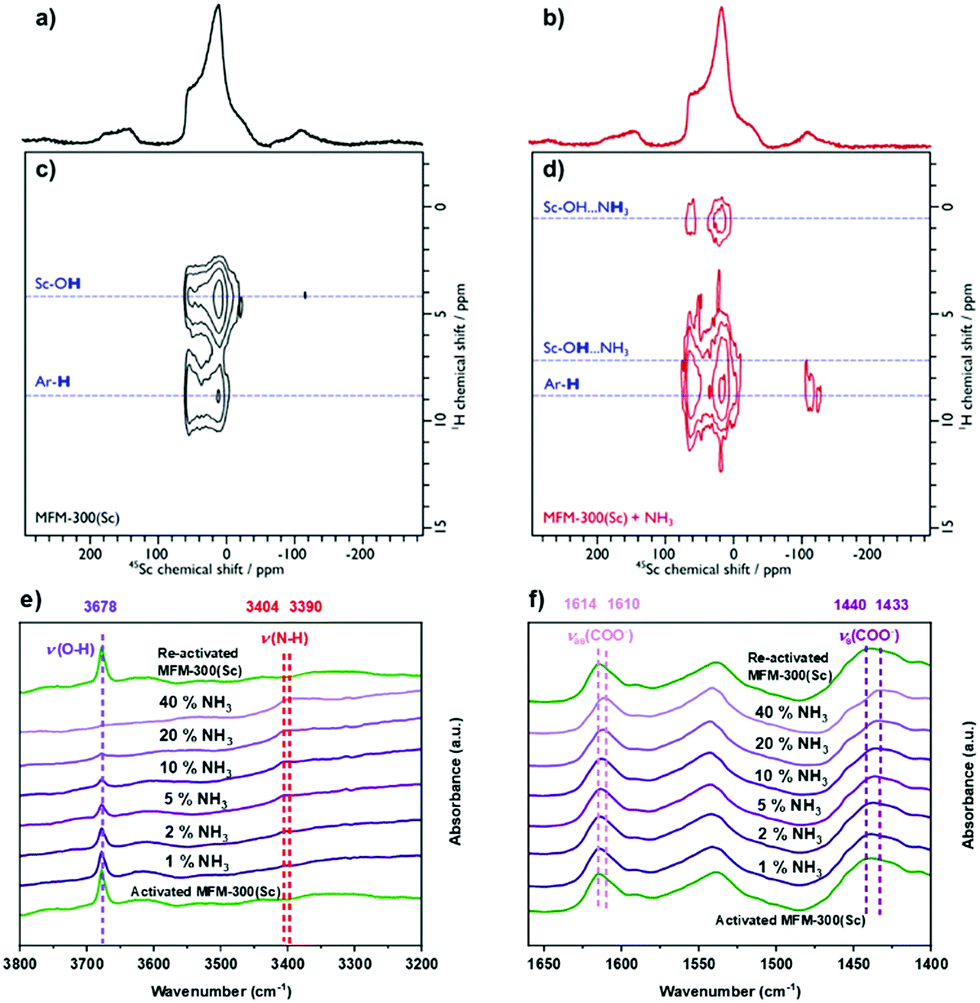 | ||
| Fig. 3 1H–45Sc Heteronuclear dipolar correlation spectroscopy (HETCOR) MAS NMR spectra of (a, c) pristine and (b, d) NH3-loaded MFM-300(Sc), with corresponding 45Sc MAS NMR spectra (top). The spectra were recorded at 9.4 T using a MAS frequency of 12 kHz. The dashed blue lines highlight correlations between the Sc(III) site and various proton environments. In situ FTIR spectra of MFM-300(Sc) as a function of NH3 loading (diluted in dry N2) and re-activated under a flow of dry N2 at 100 mL min−1 at 298 K for 2 h: (e) 3800–3200 cm−1, (f) 1650–1400 cm−1. | ||
INS spectra of bare and NH3-loaded MFM-300(Sc) were also collected (Fig. S12, ESI†) and simulated using DFT calculations based upon the structural models derived from NPD analyses (Fig. S13, ESI†). The difference spectra (Fig. 4a), which were obtained by subtracting the spectrum of the bare MOF from that of the NH3-loaded MOF, show the vibrational features of both the adsorbed NH3 molecules and the changes for the MOF host. The peaks in the low energy region (Fig. 4b) are primarily due to the vibrational modes of adsorbed NH3 molecules, with a small contribution due to changes in the lattice modes of the framework. The agreement between experimental and simulated spectra in terms of the overall profile allows unambiguous assignment of all major peaks. Specifically, the bands between 45 and 116 cm−1 are assigned to the translational motion of the NH3, which includes the vibration of NH3 molecules perpendicular to and along the molecular C3 axis and the hybrid of these modes. Peaks at 132 and 172 cm−1 are due to rotational motion of the NH3 around its C3 axis. Bands between 207 and 334 cm−1 are assigned to the rocking modes of the NH3. Compared to the spectrum of NH3 in the solid state, where each NH3 forms a 3D hydrogen bonding network with 6 adjacent NH3 molecules, bands in all regions for the adsorbed NH3 shift to lower energy and exhibit more broad features, indicating more dynamic environment for the adsorbed NH3. The features in the higher energy region mainly reflect the modes of the framework (Fig. 4c). Features I and III are due to the broadening of the peaks at 692 and 934 cm−1 for bare MFM-300(Sc), and are assigned to the C–H rocking out of the C6 plane, in-phase and anti-phase, respectively. Feature VI at higher frequency between 1090 and 1163 cm−1 shows reduced intensity upon adsorption of NH3, and is assigned to the Hring rocking within the C6 plane. Features II and IV originate from a significant blue shift of the peak at 754 cm−1 in the spectrum of bare MFM-300(Sc) to 987 cm−1 in the spectrum of NH3-loaded MFM-300(Sc). This is assigned to the rocking of μ2-OH within the Sc–O–Sc plane. Interestingly, the features involving the motions of Hring show only broadening and a decrease in intensities, while the features involving the motion of the μ2-OH experience changes in energy. This indicates a stronger interaction between NH3 and the μ2-OH than with Hring centres. Feature V in the difference spectrum is due to the umbrella motion of adsorbed NH3. Thus, the combined INS and DFT study has visualised directly the host–guest binding dynamics, consistent with the reversible and high adsorption of NH3 in MFM-300(Sc).
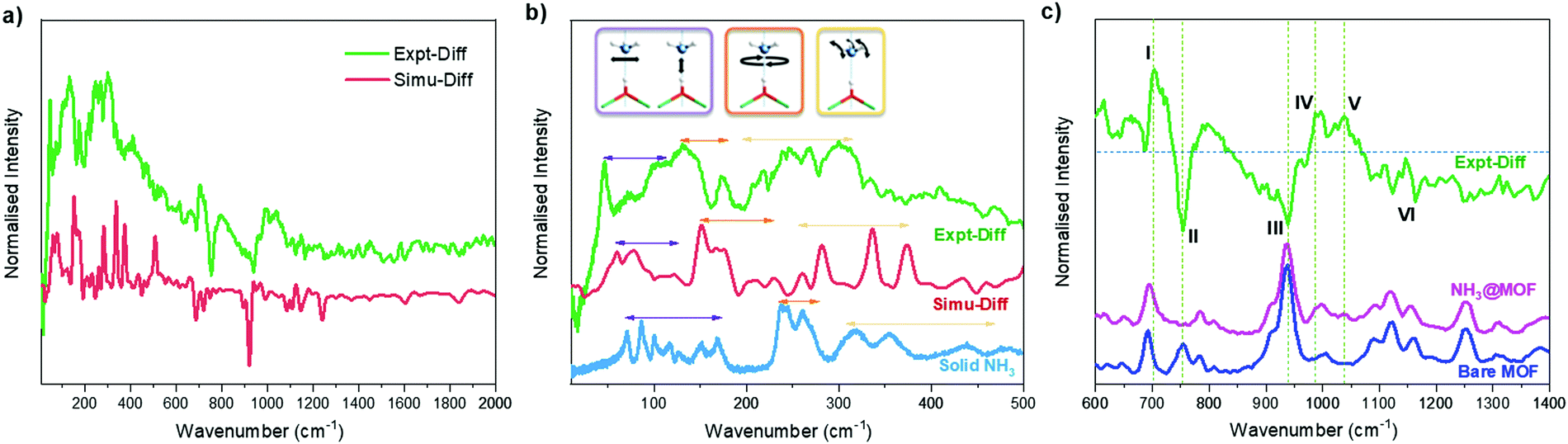 | ||
| Fig. 4 Experimental and simulated INS difference spectra of the adsorbed NH3 within MFM-300(Sc), denoted as Expt-Diff and Simu-Diff, respectively. (b) Comparison of the INS spectra of adsorbed NH3 with solid NH3. (c) Experimental INS spectra of bare MFM-300(Sc), NH3-loaded MFM-300(Sc) and the difference spectrum at the higher energy region. | ||
In summary, MFM-300(Sc) comprised of metal-oxide chains with bridging –OH groups shows exceptional adsorption capacity (19.5 mmol g−1 at 273 K and 1 bar) and regenerability for NH3. In situ NPD analysis, 45Sc ssNMR spectroscopy, synchrotron FTIR and INS/DFT studies have unambiguously visualised the binding interactions and dynamics of NH3 within the pores of MFM-300(Sc). This in-depth understanding of the structure–function relationship with these host-guest systems will enable the rational design of potential materials with desired properties.
We thank EPSRC (grant EP/I011870), The Royal Society, and University of Manchester for funding. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No 742401, NANOCHEM). We are grateful to STFC/ISIS facility for access to Beamlines TOSCA and WISH. We thank Diamond Light Source for access to Beamline B22. LG and YM thank the China Scholarship Council (CSC) for funding. The computing resources were made available through the VirtuES and the ICE-MAN projects, funded by Laboratory Directed Research and Development program and Compute and Data Environment for Science (CADES) at ORNL.
Conflicts of interest
The authors declare no competing interest.Notes and references
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. Ferrero Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- S. P. Mirajkar, B. S. Rao, M. J. Eapen and V. P. Shiralkar, J. Phys. Chem. B, 2001, 105, 4356–4367 CrossRef CAS.
- T. Zeng, H. Huang, N. Kobayashi and J. Li, Nat. Resour., 2017, 08, 611–631 CAS.
- C. J. Doonan, D. J. Tranchemontagne, T. G. Glover, J. R. Hunt and O. M. Yaghi, Nat. Chem., 2010, 2, 235–238 CrossRef CAS PubMed.
- H. Demir, K. S. Walton and D. S. Sholl, J. Phys. Chem. C, 2017, 121, 20396–20406 CrossRef CAS.
- X. Han, W. Lu, Y. Chen, I. da Silva, J. Li, L. Lin, W. Li, A. M. Sheveleva, H. G. W. Godfrey, Z. Lu, F. Tuna, E. J. L. McInnes, Y. Cheng, L. L. Daemen, L. J. M. McPherson, S. J. Teat, M. D. Frogley, S. Rudic, P. Manuel, A. J. Ramirez-Cuesta, S. Yang and M. Schröder, J. Am. Chem. Soc., 2021, 143, 3153–3161 CrossRef CAS PubMed.
- T. N. Nguyen, I. M. Harreschou, J. H. Lee, K. C. Stylianou and D. W. Stephan, Chem. Commun., 2020, 56, 9600–9603 RSC.
- C. Marsh, X. Han, J. Li, Z. Lu, S. P. Argent, I. da Silva, Y. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, S. P. Thompson, A. J. Blake, S. Yang and M. Schröder, J. Am. Chem. Soc., 2021, 143, 6586–6592 CrossRef CAS PubMed.
- S. Yang, J. Sun, A. J. Ramirez-Cuesta, S. K. Callear, W. I. David, D. P. Anderson, R. Newby, A. J. Blake, J. E. Parker, C. C. Tang and M. Schröder, Nat. Chem., 2012, 4, 887–894 CrossRef CAS PubMed.
- G. Smith, J. Eyley, X. Han, X. Zhang, J. Li, N. Jacques, H. Godfrey, S. Argent, L. McPherson, S. Teat, Y. Cheng, M. Frogley, G. Cinque, S. Day, C. Tang, T. Easun, S. Rudic, A. Cuesta, S. Yang and M. Schröder, Nat. Mater., 2019, 18, 1358–1365 CrossRef CAS PubMed.
- D. Saha and S. Deng, J. Colloid Interface Sci., 2010, 348, 615–620 CrossRef CAS PubMed.
- A. J. Rieth and M. Dincă, J. Am. Chem. Soc., 2018, 140, 3461–3466 CrossRef CAS PubMed.
- A. J. Rieth, Y. Tulchinsky and M. Dincă, J. Am. Chem. Soc., 2016, 138, 9401–9404 CrossRef CAS PubMed.
- H. G. W. Godfrey, I. da Silva, L. Briggs, J. H. Carter, C. G. Morris, M. Savage, T. L. Easun, P. Manuel, C. A. Murray, C. C. Tang, M. D. Frogley, G. Cinque, S. Yang and M. Schröder, Angew. Chem., Int. Ed., 2018, 57, 14778–14781 CrossRef CAS PubMed.
- Y. Chen, F. F. Zhang, Y. Wang, C. Y. Yang, J. F. Yang and J. P. Li, Microporous Mesoporous Mater., 2018, 258, 170–177 CrossRef CAS.
- D. W. Kim, D. W. Kang, M. Kang, J. H. Lee, J. H. Choe, Y. S. Chae, D. S. Choi, H. Yun and C. S. Hong, Angew. Chem., Int. Ed., 2020, 132, 22720–22725 CrossRef.
- P. Lyu, A. M. Wright, A. L. Olvera, P. G. M. Mileo, J. A. Zárate, E. M. Ahumada, V. Martis, D. R. Williams, M. Dincă, I. A. Ibarra and G. Maurin, Chem. Mater., 2021, 33, 6186–6192 CrossRef CAS.
- X. Zhang, I. da Silva, H. G. W. Godfrey, S. K. Callear, S. A. Sapchenko, Y. Cheng, I. Vitorica-Yrezabal, M. D. Frogley, G. Cinque, C. C. Tang, C. Giacobbe, C. Dejoie, S. Rudic, A. J. Ramirez-Cuesta, M. A. Denecke, S. Yang and M. Schröder, J. Am. Chem. Soc., 2017, 139, 16289–16296 CrossRef CAS PubMed.
- S. Yang, L. Liu, J. Sun, K. M. Thomas, A. J. Davies, M. W. George, A. J. Blake, A. H. Hill, A. N. Fitch, C. C. Tang and M. Schröder, J. Am. Chem. Soc., 2013, 135, 4954–4957 CrossRef CAS PubMed.
- K. I. Hadjiivanov, D. A. Panayotov, M. Y. Mihaylov, E. Z. Ivanova, K. K. Chakarova, S. M. Andonova and N. L. Drenchev, Chem. Rev., 2021, 121, 1286–1424 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2142629, 2142630 and 2142631. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc01197b |
| This journal is © The Royal Society of Chemistry 2022 |