
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly dispersed silica-supported iridium and iridium–aluminium catalysts for methane activation prepared via surface organometallic chemistry†
Léon
Escomel
 a,
Daniel F.
Abbott
b,
Victor
Mougel
b,
Laurent
Veyre
a,
Chloé
Thieuleux
a and
Clément
Camp
*a
a,
Daniel F.
Abbott
b,
Victor
Mougel
b,
Laurent
Veyre
a,
Chloé
Thieuleux
a and
Clément
Camp
*a
aUniversité de Lyon, Institut de Chimie de Lyon, Laboratory of Catalysis, Polymerization, Processes & Materials, CP2M UMR 5128 CNRS-UCB Lyon 1-CPE Lyon, CPE Lyon 43 Bd du 11 Novembre 1918, F-69616 Villeurbanne, France. E-mail: clement.camp@univ-lyon1.fr
bLaboratorium für Anorganische Chemie, ETH Zürich, Vladimir-Prelog-Weg 1, CH-8093 Zürich, Switzerland
First published on 24th June 2022
Abstract
The grafting of an iridium–aluminium precursor onto silica followed by thermal treatment under H2 yields small (<2 nm), narrowly distributed nanoparticles used as catalysts for methane H/D exchange. This Ir–Al/SiO2 catalyst demonstrated enhanced catalytic performances in comparison with the monometallic Ir/SiO2 analogue (TOFs of 339 h−1versus 117 h−1 respectively), highlighting the promoting effect of aluminium. TON up to 900 is obtained after 9 hours, without evidence of catalyst deactivation, and identical performances are achieved after air exposure, underlining the good robustness of both Ir–Al/SiO2 and Ir/SiO2 catalytic materials.
Promoters are ubiquitous in heterogeneous catalysis, and a large number of precious metal catalysts are modified by the addition of a second metal component to enhance the catalysts’ activity, selectivity or robustness as compared to monometallic analogues. Methane hydrogen/deuterium (H/D) exchange is the simplest methane transformation, yet of high fundamental importance for understanding the activation of relatively inert C–H bonds, and of potential interest for labelling studies. To date, a range of transition metal films were found capable of catalytic CH4 H/D exchange, with different efficiencies and selectivities.1 Dehydrated aluminas are also proficient in methane deuteration2via a mechanism involving heterolytic C–H bond splitting across Al–O moieties.3 Combining Lewis acidic Al sites with transition metals thus appears attractive for potentially promoting this transformation, yet this strategy has not been described to the best of our knowledge. Precise synthesis of systems with a homogeneous distribution of two metallic components is challenging, yet critical for a rational understanding of structure/activity relationships. Surface OrganoMetallic Chemistry (SOMC) is a powerful approach to address this challenge, that was used with success to generate well-defined surface organometallic fragments and highly dispersed nanoparticles (Nps) homogeneously distributed at the surface of solid supports.4,5 Accordingly, in recent years, attractive catalysts prepared by SOMC and containing two metal components emerged in the literature as a way to enhance activity and selectivity performances for instance in CO2 reduction6 or alkane dehydrogenation.7 Our contribution to this field was the discovery of original silica-supported Ta/Ir catalysts highly active in H/D exchange reactions of arenes, and prepared from molecular heterobimetallic complexes.8
We recently described the synthesis of an original iridium–aluminium complex, [(Cp*IrH3)3Al], 1 (Scheme 1), and have demonstrated the lability of the [Cp*IrH3]− iridate moieties surrounding the Al(III) cation, notably in presence of protic species.9 We hypothesized that such protonolysis reactivity could be used with profit for the grafting of 1 onto silica.
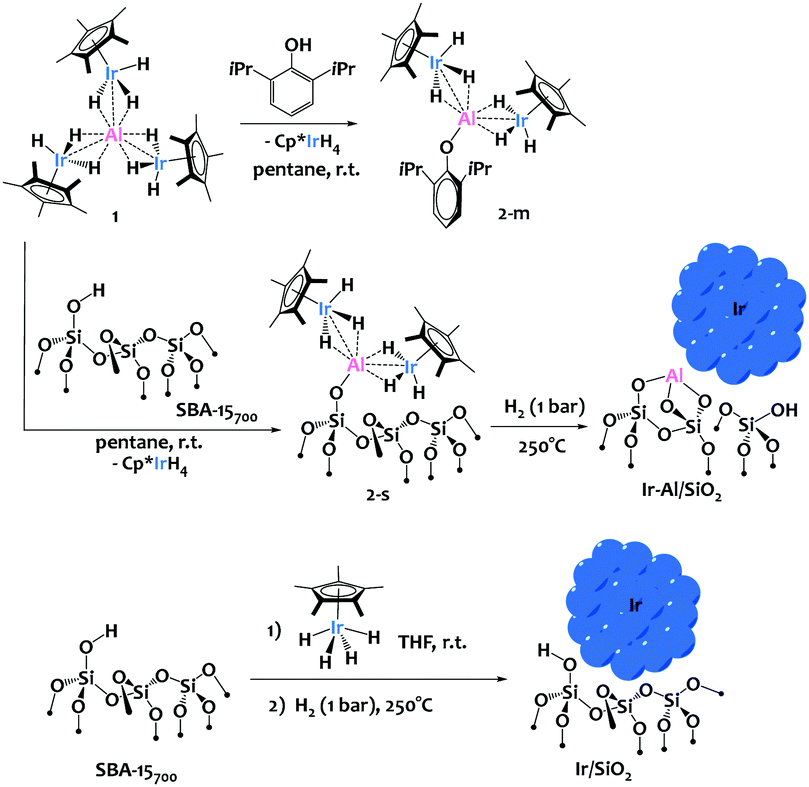 | ||
| Scheme 1 SOMC approach for the preparation of Ir and Ir–Al surface species and Nps. | ||
To anticipate the reactivity of 1 with silica surface silanols, we first investigated the reaction of 1 with 1 equiv. of 2,6-diisopropylphenol. This yielded selectively the mono-substituted [Al(OAr)(Cp*IrH3)2] (Ar = 2,6-(iPr)C6H3) complex, 2-m, along with one equivalent of IrCp*H4 as coproduct of reaction (Scheme 1). Complex 2-m was fully characterized (see ESI†), and displayed a structure typical of Ir–Al species as previously described by our group where the iridate centers are held around the Al3+ cation through bridging hydrides and polarized metal–metal interactions.9 This preliminary investigation validated the proposed ligand exchange reactivity, which is facile, selective and results in the formation of a robust Al–O bond.
We then treated mesoporous silica, namely SBA-15, dehydroxylated at 700 °C (SBA-15700)4,8 with a pentane solution of 1 at room temperature, leading to material 2-s (Scheme 1). The IR spectra show the consumption of the surface isolated silanols (ν(OH) = 3748 cm−1, see Fig. 1a and b) and the appearance of new signals attributed to metal-hydride stretches at 2131 and 2000 cm−1 and ν(C–H) from the Cp* ligands at 2913–2989 cm−1 (Fig. 1). This IR signature is similar to the IR spectrum of 2-m (see Fig. S8, ESI†), which testifies to the relevance of 2-m as molecular model for the surface species 2-s. During the reaction, 1 equiv. of IrCp*H4 is formed per grafted Al, which is removed from the material by successive pentane washings, and quantified by 1H-NMR spectroscopy (see ESI†). These analyses are in agreement with the chemical grafting of the precursor onto silica via protonolysis of one [Cp*IrH3]− group by a surface silanol, as seen in the preparation of 2-m, yielding the monopodal surface species (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)Al(Cp*IrH3)2, 2-s. Solid-state 1H and 13C NMR spectroscopy analyses (see ESI†) support the proposed structure for 2-s, with notably a broad Ir–H resonance averaged at δ(1H) = −17.14 ppm (vs. −16.55 in 2-m). The elemental analysis data for this solid (expected wt%: C 10.62, H 1.60, Ir 17.00, Al 1.19; found: C 10.87, H 1.69, Ir 16.40, Al 1.21; expected ratios: C/Al = 20.0, Ir/Al = 2.0; found: C/Al = 20.1, Ir/Al = 1.9) are also in excellent agreement with the proposed formula for 2-s and correspond to 0.53 surface organometallic sites per nm2 of silica, as expected.8
SiO)Al(Cp*IrH3)2, 2-s. Solid-state 1H and 13C NMR spectroscopy analyses (see ESI†) support the proposed structure for 2-s, with notably a broad Ir–H resonance averaged at δ(1H) = −17.14 ppm (vs. −16.55 in 2-m). The elemental analysis data for this solid (expected wt%: C 10.62, H 1.60, Ir 17.00, Al 1.19; found: C 10.87, H 1.69, Ir 16.40, Al 1.21; expected ratios: C/Al = 20.0, Ir/Al = 2.0; found: C/Al = 20.1, Ir/Al = 1.9) are also in excellent agreement with the proposed formula for 2-s and correspond to 0.53 surface organometallic sites per nm2 of silica, as expected.8
 | ||
| Fig. 1 Diffuse reflectance infrared Fourier transform spectra of SBA-15700 (a), SiOAl(Cp*IrH3)22-s (b), Ir-Al/SiO2 (c), Cp*IrH4 physisorbed on SBA-15700 (d) and Ir/SiO2 (e) recorded on powder materials under argon. | ||
Next, thermal treatment of 2-s under H2 (1 bar, 250 °C) produced material Ir-Al/SiO2 as a deep brown powder. The IR spectrum of Ir-Al/SiO2 (Fig. 1c) shows drastic changes in the Ir–H bands, with the disappearance of the signal at 2131cm−1, and a slight shift of the signal at 2000 cm−1 to 2011 cm−1. Furthermore, the C–H stretches vanished compared to those in 2-s, and some isolated silanols are restored (νOH = 3748 cm−1). These spectral data suggest decomposition of surface species and removal of the organic Cp* ligands. This is confirmed by elemental analysis, with low C and H weight percent (%C = 0.69 and %H = 0.24, C/Al ratio = 1.2). The Ir/Al atomic ratio of 1.9 remains unchanged, indicating that only the organic ligands are affected by the thermal treatment under H2. HAADF-STEM images of Ir-Al/SiO2 show the formation of small, narrowly distributed and well dispersed Nps at the surface of the SBA-15 support, with a mean size of 1.6 ± 0.4 nm (see Fig. 2A and ESI†).
 | ||
| Fig. 2 STEM-HAADF micrographs and Nps size distribution of Ir-Al/SiO2 (A) and Ir/SiO2 (B) materials (bar scales: 20 nm). | ||
EDS analyses show that Ir and Al are homogeneously distributed on the silica support. Unfortunately the metal Nps size is too small to provide a definite composition by electron microscopy or X-ray diffraction, yet reasonable hypotheses can be drawn. Al3+ sites are hardly reduced and are highly oxophilic, typically forming robust bonds with silica supports.10 This was confirmed by X-ray photoelectron (XPS) analysis, which shows that the Al 2p peak position remains fixed at 74.6 eV both before and after the thermal treatment under H2 and is consistent with Al3+ (see SI).11 In contrast, iridium can be easily reduced under hydrogen atmosphere to form Ir(0) Nps.12,13 In fact, the reduction of Ir in 2-s can easily be followed in the Ir 4f7/2 XPS spectra. Initially, 2-s displays an Ir 4f7/2 binding energy (B.E.) of 61.7 eV, which is in line with an Ir3+ complex containing electron-rich ligands such as Cp*.14 After H2 treatment, the formation of Ir(0) is confirmed by the appearance of a second pair of peaks with a lower Ir 4f7/2 B.E. of 60.6 eV. We thus propose that the Ir-Al/SiO2 material features small Ir(0) Nps surrounded by a high density of Al3+ sites located at the Nps/support interface. Similar situation was described by Copéret and coworkers, who generated Cu Nps on silica containing Zr(IV)15 or Ti(IV)16 isolated sites. Still, the partial incorporation of Al atoms into the metal Nps cannot be ruled out.
In parallel, we prepared a monometallic iridium analogous material to serve as benchmark for catalysis. The sample was prepared using incipient wetness impregnation (IWI), by the addition of an appropriate volume (340 μL) of a 1.24M THF solution of Cp*IrH4 onto 295 mg of SBA-15700 to reach a similar iridium loading than that obtained in the Ir-Al/SiO2 material, and the solvent was further evacuated under vacuum at room temperature. We reported before that Cp*IrH4 does not react with silica surface silanols,8 and is physisorbed at the surface of the support, which is notably confirmed by the ν(Ir–H) signature in this material (Fig. 1d) which is identical to that of the IrCp*H4 precursor17 (σ = 2150 cm−1). We then reduced this material under the same conditions (1 bar H2, 250 °C) used for the aluminium–iridium catalyst, to yield material Ir/SiO2 as a dark brown powder (Scheme 1, bottom). The elimination of the Cp* ligands during such treatment is confirmed by the disappearance of the ν(C–H) signals around 2950 cm−1 (Fig. 1e). A strong shift of the ν(Ir–H) band from 2150 to 2024 cm−1 is observed on the IR spectrum of Ir/SiO2 This signature is very similar to that of Ir-Al/SiO2 (Fig. 1c). This suggests that the Nps resulting from the decomposition of physisorbed Cp*IrH4 have similar hydride sites than those resulting from the decomposition of species 2-s, which is in favour of the formation of monometallic Ir(0) Nps in both cases. STEM analyses of Ir/SiO2 reveal the presence of small Ir Nps at the surface of the SBA-15700 support (see Fig. 2B and ESI†). The Nps size distribution of Ir/SiO2 is the same to that of Ir-Al/SiO2 with a mean size of 1.6 ± 0.4 nm. A few Ir aggregates (3–15 nm) were also noticed in some silica grains by STEM (see ESI†). The Ir 4f7/2 XPS spectra (see ESI†) clearly show the decrease of the Ir3+ peak at 61.6 eV and the appearance of the Ir(0) peak at 60.1 eV after the reduction step. Although the B.E. is low for metallic Ir, this is quite consistent with the reduced average coordination number of surface atoms found in Nps.18 A small contribution of an Ir3+ peak is still present after H2 treatment for both Ir/SiO2 and Ir-Al/SiO2, corresponding to a Ir(0)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ir3+ ratio of ca. 3:1 (see Fig. 3 and ESI†). This is either due to incomplete reduction or to the presence of Ir(III) hydride sites at the surface of the Nps.
:Ir3+ ratio of ca. 3:1 (see Fig. 3 and ESI†). This is either due to incomplete reduction or to the presence of Ir(III) hydride sites at the surface of the Nps.
 | ||
| Fig. 3 XPS Ir 4f spectra of materials 2-s (top) and Ir-Al/SiO2 (bottom). | ||
Importantly, H2 chemisorption studies on Ir-Al/SiO2 and Ir/SiO2 showed similar Ir dispersions of 67% and 56% respectively. This corresponds to a Np average diameter of 1.4 nm and 1.7 nm respectively using a truncated cubic octahedron geometry (see ESI†), which is in good agreement with the STEM analyses, and indicates a comparable amount of accessible surface metal atoms for both catalysts.
To evaluate the potential of these new materials in catalysis we studied the hydrogen/deuterium isotope exchange (HIE) reaction between CH4 and D2 (Scheme 2). The reaction was carried out in a batch reactor at 250 °C. The Ir-Al/SiO2 (1.0 mol% ±0.1 mol% Ir compared to methane) and Ir/SiO2 (1.2 mol% ± 0.1 mol%) catalysts were exposed to a mixture of dry CH4 (42 mbars) and dry D2 (980 mbars) such as the D/H atomic ratio in the gas phase is ca. 12. The catalyst loading of Ir/SiO2 is slightly higher than that of Ir-Al/SiO2 to compensate the small dispersion difference, and achieve a similar Ir surface sites quantity for both catalysts (0.67 mol% ± 0.03 mol%).
 | ||
| Scheme 2 – Deuteration of methane (42 mbars) into CH4−xDx isotopomers at T = 250 °C using D2 gas (980 mbars) as a deuterium source. | ||
In the course of the reaction, methane isotopomers – noted CH4−xDx (x being from 0 to 4) – are produced and quantified by GC-MS. The precise amount of each isotopomer was determined as a function of methane conversion (Fig. 4) according to the fragmentation models given by Dibeler and Mohler.19 To consolidate these results, the isotopic distribution was also determined by a complementary mathematical approach developed by Schoofs and his team.20 From these data, it is possible to calculate the deuteration rate of methane – noted τ thereafter – by using the following relation: τ = 0.25CH3D + 0.5CH2D2 + 0.75CHD3 + CD4. Note that since D/H ≈ 12 in our case, the thermodynamic equilibrium – noted τeq – of the reaction is reached for a deuteration rate of 92%.
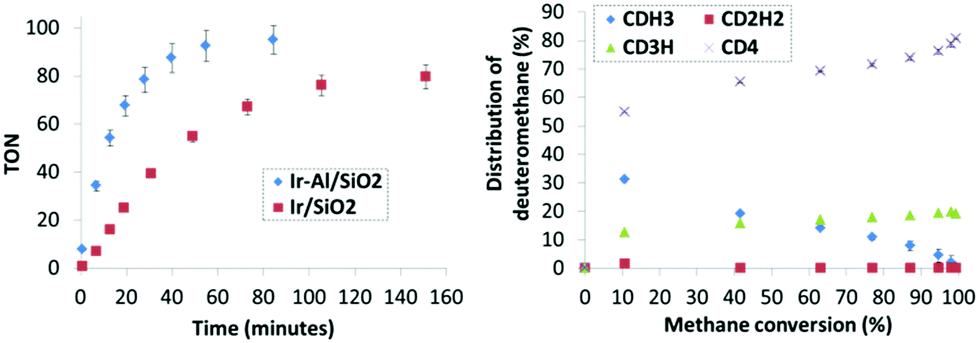 | ||
| Fig. 4 Monitoring catalytic H/D exchange of methane. Left: Turnover number as a function of reaction time for catalysts Ir-Al/SiO2 and Ir/SiO2 at 250 °C and 0.67 mol% Ir surface sites. Right: Distribution of deuteromethanes as a function of CH4 conversion for Ir-Al/SiO2. | ||
Comparative TONs for both catalysts are plotted on Fig. 4 left. The Ir-Al/SiO2 catalyst is more active and operates the H/D exchange of methane at a maximum turnover frequency (TOF) of 339 h−1versus 117 h−1 for Ir/SiO2. Note that similar trends are obtained when turnover numbers are corrected from the Ir dispersion extracted from the H2 chemisorption studies (Fig. S25, ESI†). Furthermore, 50% deuterium incorporation into methane is observed after 12 minutes for Ir-Al/SiO2versus 34 minutes for Ir/SiO2 and the thermodynamic equilibrium is reached within 65 minutes for Ir-Al/SiO2versus 145 minutes for Ir/SiO2. These kinetic data testify to the higher catalytic activity of the aluminium–iridium catalyst with respect of its monometallic iridium counterpart by a factor of about three, which is not negligible. Note that this kinetic behaviour was reproduced on independently synthesized samples. Importantly, the spent Ir-Al/SiO2 and Ir/SiO2 catalysts were exposed to air and then re-used in catalysis, and identical performances were obtained in this second catalytic run (Fig. S26 and S27, ESI†), highlighting the good robustness of these materials. STEM analyses of the spent catalyst show no modification of the particles size distribution. Decreasing the catalyst loading to 0.1 mol% resulted in an increased TON of 900 after 9 hours, without evidence of catalyst deactivation. 2-s was also tested in catalysis, and was active only after a prolonged induction period of several hours, corresponding to the removal of the Cp* ligands and the in situ formation of Nps (Fig. S23, ESI†).
To get insights into the mechanisms in place here between the gas phase (CH4−xDx and D2) and the surface of the catalysts, we paid attention to the distribution of deuteromethane isotopomers as a function of methane conversion rate (Fig. 4, right). For both catalysts, similar selectivity is observed (see ESI†). CD4 is the major isotopomer at low CH4 conversion (10 to 20%), but a subsequent amount of CH3D is also observed. Then, the relative amount of CD4 increases while the CH3D proportion drops as CH4 is consumed. Interestingly, the amount of CH2D2 stays negligible in the reactional medium (<2%) whatever the CH4 conversion. Two main mechanisms are known for methane H/D exchange: (i) stepwise exchange, in which CDH3 is the major product at low conversion with negligible amount of CD4, and (ii) multiple exchange, in which the C–H activations subsequent to adsorption are faster than the desorption step, which gives CD4 as major product at low CH4 conversion (see ESI† for details).1 In the present case, the data suggest an important contribution of a multiple exchange mechanism, in which the rate determining step is the CH4 dissociative adsorption step, which may be facilitated by the presence of the Al3+ sites at the direct proximity of the Ir particles.
In summary, a SOMC synthetic methodology is used to generate the surface species (SiO)Al(Cp*IrH3)2, 2-s, ensuring a good dispersion of both metallic components (Al & Ir) on the silica support. Thermal hydrogenolysis of 2-s leads to the formation of small (<2 nm), narrowly distributed Ir Nps surrounded by a high density of Al3+ sites located at the Nps/silica support interface. This Ir-Al/SiO2 catalyst demonstrated enhanced catalytic performances in the H/D exchange of methane in comparison with the monometallic Ir/SiO2 analogue, highlighting the promoting effect of aluminum. Although H/D exchange in methane has been studied with a large number of transition metal catalysts,1 the promoting effect of a Lewis acidic metal has not been described to the best of our knowledge. Both Ir-Al/SiO2 and Ir/SiO2 catalysts are air-stable and do not deactivate (at least down to 0.1 mol%), which is a clear advantage compared to silica-supported molecular metal hydrides, which are highly active in methane deuteration, but extremely air sensitive and deactivate easily.21
This research was funded by the French National Research Agency (ANR) (Grant number ANR-21-CE07-0009-01 (SHICC)).
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Kemball, Proc. R. Soc. London, Ser. A, 1953, 217, 376–389 Search PubMed; A. Frennet, Catal. Rev., 1974, 10, 37–68 CrossRef CAS; A. Sattler, ACS Catal., 2018, 8, 2296–2312 CrossRef.
- P. J. Robertson, M. S. Scurrell and C. Kemball, J. Chem. Soc., Faraday Trans. 1, 1975, 71, 903–912 RSC.
- J. Joubert, A. Salameh, V. Krakoviack, F. Delbecq, P. Sautet, C. Copéret and J. M. Basset, J. Phys. Chem. B, 2006, 110, 23944–23950 CrossRef CAS PubMed.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed.
- C. Copéret, Acc. Chem. Res., 2019, 52, 1697–1708 CrossRef PubMed.
- S. R. Docherty, N. Phongprueksathat, E. Lam, G. Noh, O. V. Safonova, A. Urakawa and C. Copéret, JACS Au, 2021, 1(4), 450–458 CrossRef CAS PubMed; E. Lam, G. Noh, K. W. Chan, K. Larmier, D. Lebedev, K. Searles, P. Wolf, O. V. Safonova and C. Copéret, Chem. Sci., 2020, 11, 7593–7598 RSC; E. Lam, G. Noh, K. Larmier, O. V. Safonova and C. Copéret, J. Catal., 2021, 394, 266–272 CrossRef.
- J. Camacho-Bunquin, M. S. Ferrandon, H. Sohn, A. J. Kropf, C. Yang, J. Wen, R. A. Hackler, C. Liu, G. Celik, C. L. Marshall, P. C. Stair and M. Delferro, ACS Catal., 2018, 8, 10058–10063 CrossRef CAS; K. Searles, K. W. Chan, J. A. Mendes Burak, D. Zemlyanov, O. Safonova and C. Copéret, J. Am. Chem. Soc., 2018, 140, 11674–11679 CrossRef PubMed.
- S. Lassalle, R. Jabbour, P. Schiltz, P. Berruyer, T. K. Todorova, L. Veyre, D. Gajan, A. Lesage, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2019, 141, 19321–19335 CrossRef CAS PubMed; S. Lassalle, R. Jabbour, I. Del Rosal, L. Maron, E. Fonda, L. Veyre, D. Gajan, A. Lesage, C. Thieuleux and C. Camp, J. Catal., 2020, 392, 287–301 CrossRef; I. Del Rosal, S. Lassalle, C. Dinoi, C. Thieuleux, L. Maron and C. Camp, Dalton Trans, 2021, 50, 504–510 RSC.
- L. Escomel, N. Soulé, E. Robin, I. Del Rosal, L. Maron, E. Jeanneau, C. Thieuleux and C. Camp, Inorg. Chem., 2022, 61(15), 5715–5730 CrossRef CAS PubMed; L. Escomel, I. Del Rosal, L. Maron, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2021, 143, 4844–4856 CrossRef PubMed.
- I. B. Moroz, P. Florian, J. Viger-Gravel, C. P. Gordon, A. Lesage and C. Copéret, Angew. Chem., Int. Ed., 2020, 59(37), 16167–16172 CrossRef CAS PubMed.
- J. Moulder and J. Chastain, Handbook of X-ray photoelectron spectroscopy: a reference book of standard spectra for identification and interpretation of XPS data, 1992.
- F. Héroguel, G. Siddiqi, M. D. Detwiler, D. Y. Zemlyanov, O. V. Safonova and C. Copéret, J. Catal., 2015, 321, 81–89 CrossRef.
- F. Héroguel, D. Gebert, M. D. Detwiler, D. Y. Zemlyanov, D. Baudouin and C. Copéret, J. Catal., 2014, 316, 260–269 CrossRef.
- B. Van Dijk, G. M. Rodriguez, L. Wu, J. P. Hofmann, A. MacChioni and D. G. H. Hetterscheid, ACS Catal., 2020, 10, 4398–4410 CrossRef CAS PubMed.
- E. Lam, K. Larmier, P. Wolf, S. Tada, O. V. Safonova and C. Copéret, J. Am. Chem. Soc., 2018, 140, 10530–10535 CrossRef CAS PubMed.
- G. Noh, E. Lam, J. L. Alfke, K. Larmier, K. Searles, P. Wolf and C. Copéret, ChemSusChem, 2019, 12, 968–972 CrossRef CAS PubMed.
- T. M. Gilbert, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1985, 107, 3508–3516 CrossRef CAS.
- J. Radnik, C. Mohr and P. Claus, Phys. Chem. Chem. Phys., 2003, 5, 172–177 RSC.
- F. L. Mohler, V. H. Dibeler and E. Quinn, J. Res. Natl. Bur. Stand., 1934, 1958(61), 171 Search PubMed.
- B. Schoofs, J. A. Martens, P. A. Jacobs and R. A. Schoonheydt, J. Catal., 1999, 183, 355–367 CrossRef CAS.
- G. L. Casty, M. G. Matturro, G. R. Myers, R. P. Reynolds and R. B. Hall, Organometallics, 2001, 20, 2246–2249 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, NMR, IR, XRD, STEM, EDS, XPS, chemisorption, catalysis data. CCDC 2149676. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc02139k |
| This journal is © The Royal Society of Chemistry 2022 |