
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrthogonally deconstructable and depolymerizable polysilylethers via entropy-driven ring-opening metathesis polymerization†
Alayna M.
Johnson
 ,
Keith E. L.
Husted
,
Landon J.
Kilgallon
and
Jeremiah A.
Johnson
*
,
Keith E. L.
Husted
,
Landon J.
Kilgallon
and
Jeremiah A.
Johnson
*
Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: jaj2109@mit.edu
First published on 12th July 2022
Abstract
The synthesis of novel polysilylethers via entropy-driven ring-opening metathesis polymerization (ED-ROMP) of cyclic bifunctional silyl ether-based monomers is reported. These polymers display good thermal stability and ultra-low Tg (−88 °C). Moreover, they are rapidly deconstructable via the cleavage of the silicon-oxygen linkages with acid or fluoride triggers, and they were partially depolymerizable by the addition of exogenous metathesis catalyst. Analysis of the deconstructed polymer products provided insight into the polymer microstructure, showing that the ED-ROMP process was regiorandom. Altogether, this work offers a new class of deconstructable polymers with a range of potential applications.
Ring-opening metathesis polymerization (ROMP) is a popular method for polymer synthesis due to its versatility, robust reactivity, and functional group tolerance, all of which were accelerated by the development of ruthenium alkylidene metathesis catalysts by Grubbs and coworkers.1–3 The driving force behind ROMP is typically release of ring strain, which compensates for the entropic costs associated with polymerization.4 When rings are large or equipped with especially flexible bonds, however, atoms can adopt near-ideal bond angles, so the enthalpic gain of polymerization becomes negligible and the reaction will exhibit a distribution of monomers, oligomers, and polymer chains at equilibrium.5 Such reactions can be driven towards polymerization if the number of rotational and vibrational microstates is larger in the polymer repeat unit than in the monomer—a phenomenon known as entropy-driven ring-opening metathesis polymerization (ED-ROMP).6 Grubbs and coworkers reported the first studies of modern ED-ROMP with crown ethers in 1997.7 More recent examples include ED-ROMP of cyclic olefins equipped with ester and disulfide functional groups.8,9
ED-ROMP is particularly useful for the synthesis of polymers with complex backbone compositions.10 Especially relevant is the introduction of degradable and/or bioderived moieties into otherwise recalcitrant hydrocarbon polymers. For example, Hillmyer and coworkers recently reported the synthesis of bio-based high-molecular weight poly(ricinoleic acid) via ED-ROMP.11
The exploration of chemically labile bonds and their applications in polymer sustainability is of continued interest. Silyl ethers are an attractive option for this purpose because they boast both usually high Si–O bond strengths (∼108 kcal mol−1) under typical conditions and facile degradability with a variety of triggers.12,13
Our group previously reported that 8- and 7-member cyclic bifunctional silyl ether olefins could be efficiently prepared and statistically copolymerized with a variety of ROMP-active norbornene derivatives with potential applications in drug delivery and recyclable thermosets.12,14,15 At the concentrations at which these copolymerizations were previously conducted (50 mM), however, these monomers were not found to homopolymerize.12 Stemming from this observation, we sought to identify conditions under which these monomers could be homopolymerized as we anticipated their respective polymers would possess desirable properties including facile deconstructability and thermal properties akin to polydimethylsiloxane (PDMS).16
To begin this investigation, seven- and eight-membered bifunctional silyl ether olefins (iPrSi7 and iPrSi8, respectively) were treated with 0.002 equivalents (with a target number-average degree of polymerization (DP) of 500) of third-generation Grubbs bispyridyl complex (G3) for two hours (Scheme 1). Initial screening was performed at [M]0 = 500 mM and the reaction was quenched after 2 h by adding excess ethyl vinyl ether. iPrSi7 was not observed to homopolymerize under these conditions, as no high molecular weight species were observed by SEC (Fig. S3, ESI†). Instead, a peak corresponding to ∼0.4 kDa was found, consistent with formation of iPrSi7 cyclic dimer. 1H NMR analysis of the crude product revealed a clean mixture of unreacted iPrSi7 (63%), the cyclic vinyl silyl ether analog of iPrSi7 (iPrSi7-vin, 32%), and traces of the postulated cyclic iPrSi7 dimer (iPrSi7-dim, 5%) (Fig. S4, ESI†). The cyclic silyl vinyl ether, an olefin isomerization product, was separately synthesized and isolated to confirm its structure (Fig. S5 and S6, ESI†).
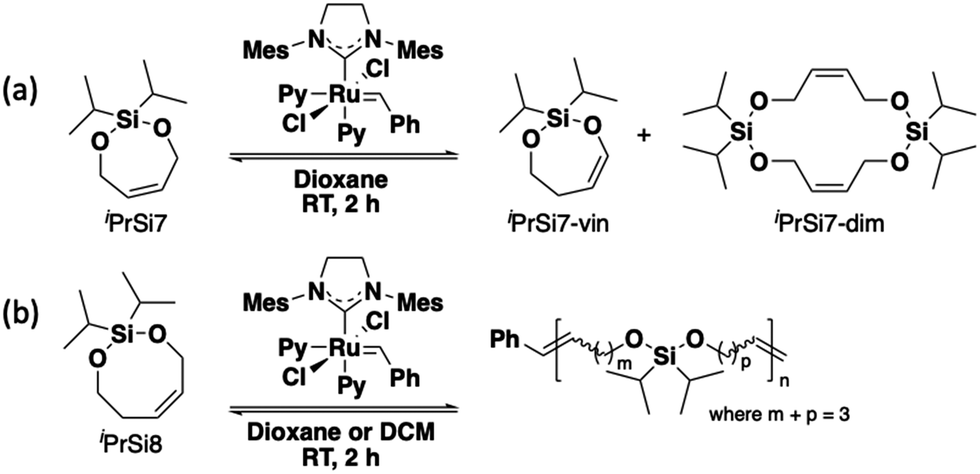 | ||
| Scheme 1 Attempts at homopolymerization of bifunctional silyl ether monomers via ROMP. (A) Seven-membered iPrSi7 forms isomerized cyclic vinyl silyl ether and cyclic dimer. (B) Eight-membered iPrSi8 forms high molecular weight polysilylethers. | ||
On the other hand, SEC analysis of the iPrSi8 homopolymerization reaction mixture revealed high molecular weight species, consistent with successful ROMP. Therefore, we screened a variety of temperatures to optimize the reaction conditions (Scheme 1 and Table 1, entries 1–5). Initial screening covered a range of low temperatures (–78 °C to 3 °C) because performing conventional enthalpy-controlled ROMP at low temperatures can mitigate the entropic penalty of polymerization and drive the reaction toward high-molecular weight polymer;5 however, we observed higher conversion and number-average molar mass (51.2% conversion, Mn = 47.7 kDa at 3 °C) at higher temperatures, indicative of ED-ROMP.17 Attempts to polymerize iPrSi8 at higher temperatures (35 °C to 60 °C) led to the formation of a side product we assign as the isomerized silyl vinyl ether (Fig. S11, ESI†).
| Entry | T (°C) | Time (min) | Conv.b (%) | M n,theo (kDa) | M n,SEC (kDa) | Đ |
|---|---|---|---|---|---|---|
a ROMP was performed in DCM (low T) or dioxane (high T) under N2 and quenched with ethyl vinyl ether. A feed ratio of 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 [M]0:[G3] and a monomer concentration of [iPrSi8]0 = 500 mM were used for all reactions.
b Monomer conversion was determined 1H NMR spectroscopy.
c Theoretical number-average molar mass Mn,theo = DPmonomer × MWmonomer where DP = feed ratio × conversion.
d Molar mass and dispersity were determined by size-exclusion chromatography in CHCl3 relative to low dispersity polystyrene standards. :1 [M]0:[G3] and a monomer concentration of [iPrSi8]0 = 500 mM were used for all reactions.
b Monomer conversion was determined 1H NMR spectroscopy.
c Theoretical number-average molar mass Mn,theo = DPmonomer × MWmonomer where DP = feed ratio × conversion.
d Molar mass and dispersity were determined by size-exclusion chromatography in CHCl3 relative to low dispersity polystyrene standards.
|
||||||
| 1 | −78 | 120 | 28.8 | 31.0 | 36.5 | 1.94 |
| 2 | −18 | 120 | 46.7 | 50.1 | 41.9 | 1.50 |
| 3 | −10 | 120 | 47.8 | 51.2 | 43.8 | 1.97 |
| 4 | 0 | 120 | 50.7 | 54.3 | 46.8 | 1.63 |
| 5 | 3 | 120 | 51.2 | 55.0 | 47.7 | 1.61 |
| 6 | RT | 0.25 | 7.5 | 16.1 | 3.5 | 1.10 |
| 7 | RT | 0.5 | 14.4 | 30.9 | 6.0 | 1.12 |
| 8 | RT | 1 | 23.3 | 50.2 | 9.1 | 1.18 |
| 9 | RT | 2 | 38.3 | 82.1 | 15.1 | 1.26 |
| 10 | RT | 5 | 63.6 | 136 | 22.8 | 1.53 |
| 11 | RT | 60 | 95.4 | 204 | 136 | 1.70 |
| 12 | RT | 120 | 96.1 | 206 | 159 | 1.76 |
We further investigated the polymerization of iPrSi8 by plotting ln([M]e) against 1/T (where [M]e is the equilibrium monomer concentration) and extracting thermodynamic parameters from the slope and y-intercept (Fig. S12, ESI†). From this analysis, the change in enthalpy (ΔHp) and entropy (ΔSp) upon polymerization were found to be 2.1 kJ mol−1 and 7.57 J mol−1 K−1, respectively. A small enthalpic component and a favorable entropic component are consistent with the thermodynamic parameters reported for polymerization of related 8-membered siloxanes such as octamethylcyclotetrasiloxane (which is used to generate PDMS under anionic conditions), as the highly flexible polysiloxane chain has more rotational and vibrational microstates available than the cyclic monomer.18,19
To investigate the nature of the iPrSi8 homopolymerization, simultaneous reactions were set up at room temperature and quenched at different times (Fig. 1A–C and Table 1, entries 6–10). The polymer number-average molar mass as measured by size-exclusion chromatography (Mn,SEC) increased linearly as a function of monomer conversion while maintaining moderate dispersities (1.10–1.53), suggesting a controlled polymerization (Fig. 1A). A plot of ln([M]/[M]0) as a function of time revealed a linear relationship to 5 min, consistent with pseudo-first order kinetics (Fig. S13 and S14, ESI†). Allowing the polymerization to proceed for 1 h produced high Mn,SEC species (136 kDa) and at 2 h, negligible changes to conversion and Mn were observed, suggesting that the reaction had reached equilibrium (Fig. 1B and Table 1, entries 11, 12).
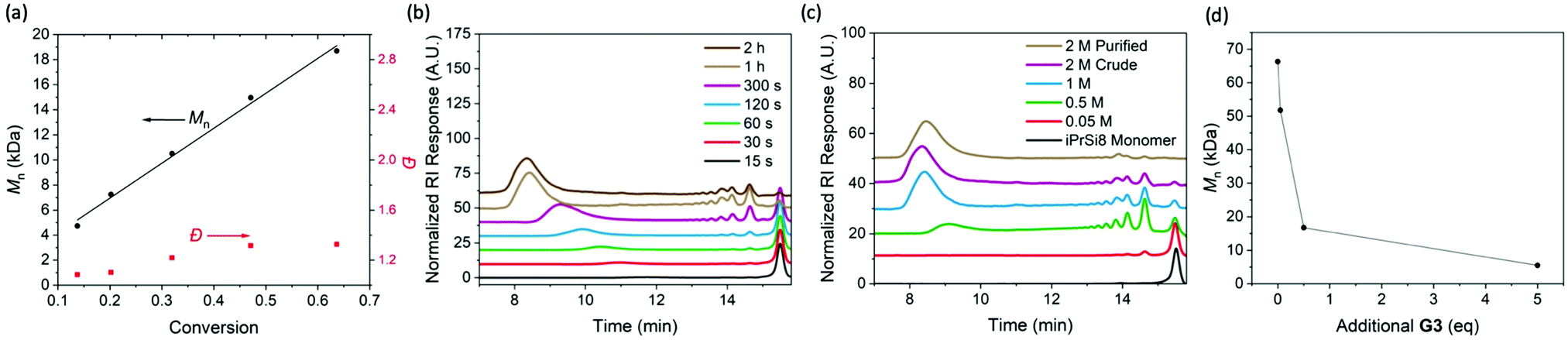 | ||
| Fig. 1 Characterization of the polymerization and depolymerization of iPrSi8. (A). Plot of Mn and Đ vs monomer conversion, obtained by a combination of SEC and 1H NMR analysis. (B) SEC traces (normalized RI) of poly(iPrSi8) quenched at different reaction times (correlated to Table 1, entries 6–12). (C) SEC traces of poly(iPrSi8) prepared at increasing monomer concentration in dioxane (correlated to Table 2, entries 1–5). (D) Polymer size (obtained by SEC) of homopolymer prepared from 500 mM iPrSi8 (Table 2, entry 2) as it is exposed to increasing amounts of additional G3. | ||
Low molecular weight features were present in the SEC traces of the unpurified polymers (Fig. 1B), which suggests the formation of cyclic oligomers by backbiting, a common side reaction in ED-ROMP.11 This behavior is further reflected in a significant difference between the theoretical number-average molar mass (Mn,theo) and Mn,SEC (Table 1). We hypothesized that oligomer formation could be suppressed by increasing the monomer concentration.11 Thus, polymerizations were carried out at increasing concentration for several hours to ensure sufficient time to reach equilibrium (Fig. 1C and Table 2, entries 1–5). The relative concentration of cyclic oligomer was estimated by SEC using an RI detector, assuming an equivalent molar RI response. Consistent with expectation, higher concentrations afforded higher molar masses and lower oligomer content; however, a modest increase in dispersity was observed. Precipitation of the quenched reaction into methanol yielded high molecular weight homopolymer (Mn,SEC = 87 kDa, Đ = 1.78) and reduced the cyclic oligomer content below 6%.
| Entry | [M]0/[G3]b | Conc.c (M) | M n,SEC (kDa) | Đ | Cyclic oligomer contente (%) |
|---|---|---|---|---|---|
| a ROMP was performed in dioxane under N2 for 2 h at RT and quenched with ethyl vinyl ether. b Feed ratio of monomer to G3. c Initial monomer concentration. d M n,SEC and Đ were determined by SEC in CHCl3 relative to low Đ polystyrene standards. e Calculated from the area percentage of RI chromatographs of SEC. | |||||
| 1 | 500 | 0.05 | 0.42 | 1.02 | 100 |
| 2 | 500 | 0.50 | 57.7 | 1.42 | 68 |
| 3 | 500 | 1.0 | 135 | 1.76 | 27 |
| 4 | 500 | 2.0 | 156 | 1.86 | 21 |
| 5 | 500 | 2.0e | 87 | 1.78 | 5.6 |
| 6 | 50 | 2.0 | 22.4 | 1.52 | — |
| 7 | 100 | 2.0 | 44.1 | 1.49 | — |
| 8 | 500 | 2.0 | 138 | 1.53 | — |
| 9 | 1000 | 2.0 | 183 | 1.68 | — |
Since ED-ROMP reactions lack a significant enthalpic driving force, the polymerization exists in an equilibrium of rings and chains.20 We sought to capitalize on this ring-chain equilibrium for depolymerization. Depolymerization was probed by preparing a stock solution of poly(iPrSi8) (Table 2, entry 2), collecting aliquots, and exposing each to different amounts of G3. SEC traces of each depolymerization reaction compared to a reference sample that was not treated with additional G3 demonstrate that the size of the polymer chain can be reduced by an order of magnitude (from 58 kDa to 5.5 kDa) under these conditions (Fig. 1D). 1H NMR spectra of the G3-treated mixture indicate that the depolymerization mechanism leads to the reformation of iPrSi8 monomer in ∼28% yield (Fig. S15, ESI†). A similar result was observed for larger homopolymers; treatment of DP 1000 poly(iPrSi8) (Table 2, entry 9) with 5 eq. G3 reduced its molar mass from 183 to 16.8 kDa (Fig. S16, ESI†).
As noted above, silyl ethers represent robust yet selectively cleavable functional groups that can be exploited for polymer backbone deconstruction;12 thus, we hypothesized that poly(iPrSi8) could be chemically deconstructed orthogonally to depolymerization. Samples of poly(iPrSi8) of various DP were synthesized and subjected to deconstruction conditions (Table 2, entries 6–9). Given the well-established precedent of cleaving Si–O bonds with Brønsted acids and fluoride ions, each polymer was separately exposed to 0.5 M HCl in dioxane or 0.2 M tetrabutylammonium fluoride (TBAF, Fig. 2A, step i) in THF at room temperature for fifteen minutes (Fig. S17 and S19, ESI†).12,14 Under both conditions, no high molecular weight species were detected by SEC (Fig. 2B), suggesting complete backbone deconstruction. We anticipate that by modifying the steric bulk and polarity of the silyl substituents, different backbone deconstruction rates could be achieved.12,21
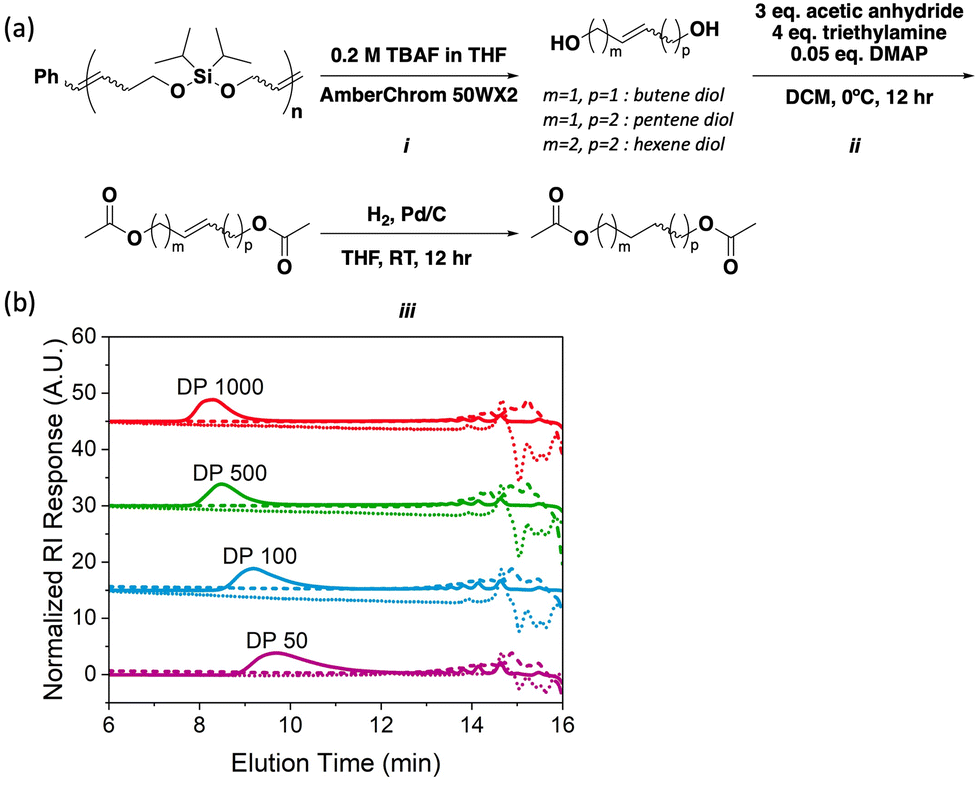 | ||
| Fig. 2 Poly(iPrSi8) deconstruction and regiochemical analysis. (A) Quantitative fluoride-triggered deconstruction (i), acetylation (ii), and hydrogenation (iii) of poly(iPrSi8) to yield a mixture of protected saturated diols. (B) SEC traces (normalized RI) of poly(iPrSi8) to different target DP (solid line, correlated to Table 2, entries 6–9) and subsequent deconstruction with 0.2 M TBAF in THF (dotted line) and 0.5 M HCl in dioxane (dashed line). | ||
ED-ROMP reactions are known to involve intra- and inter-molecular mechanisms that can alter the regio- and stereo-chemistry of the growing polymer chain.11 While it is possible to use methods like 1H NMR to characterize homopolymer regio- and stereo-regularity, as was done by Hillmyer and coworkers to determine that the ED-ROMP of ricinoleic acid forms a regiorandom polymer, new methods for the characterization of the microstructure of backbone degradable polymers are of interest. We noticed that the fluoride-mediated deconstruction of poly(iPrSi8) yields a mixture of isolable diols, the composition of which should encode information about the polymer microstructure (Fig. 2A). Thus, we designed a gas-chromatography mass-spectrometry (GC-MS) assay to characterize the deconstructed product mixture. Initial analysis of the fluoride-deconstructed mixture via GC-MS showed six distinct features: cis and trans isomers of butene, pentene, and hexene diols, which correspond to head-to-head (HH), head-to-tail or tail-to-head (HT/TH), and tail-to-tail (TT) polymer segments, respectively (Fig. S18, ESI†).
Unfortunately, due to the chemically similar nature of the cis and trans diol derivatives, complete resolution of this mixture was not possible. Initial attempts to hydrogenate the unprotected diol mixture and simplify analysis were unsuccessful; hydrogenation of small unsaturated diols is known to form several side products, which would bias the product ratio.22 This obstacle was circumvented by acetylating the unsaturated diol mixture (Fig. 2A, step ii, Fig. S20 and S21, ESI†) and then hydrogenating to yield a mixture of 1,4-diacetoxybutane, 1,5-diacetoxypentane and 1,6-diacetoxyhexane (Fig. 2A, step iii, Fig. S22 and S23, ESI†). Aliphatic acetylated calibration standards were prepared and measured separately (Fig. S24 and S25, ESI†). Using this method, the ratio of HH: HT/TH: TT ratio was found to be 1:2:1 (Fig. S26 and Table S1, ESI†). This observation strongly suggests that the homopolymerization is regiorandom, as a regioregular polymerization would lead to preferential formation of 1,5-diacetoxypentane under these conditions.
Finally, the thermal properties of poly(iPrSi8) were investigated. The purified polymers appeared as light pink viscous oils at room temperature, the latter of which is consistent with a low glass transition temperature (Tg) amorphous material. Thermogravimetric analysis (TGA) revealed high thermal stability of the precipitated poly(iPrSi8) under nitrogen. At a heating rate of 10 °C min−1, the fastest rate of mass loss did not occur until 486 °C as might be expected for siloxane- and olefin-containing polymers (Fig. S28, ESI†). Of note, TGA conducted on the crude polymer before precipitation showed multi-stage mass loss, consistent with the non-degradative loss of volatile low-molecular weight species such as the cyclic oligomers observed in the SEC traces of the crude mixture.
No major thermal transitions, including Tg, were detectable above −75 °C via differential scanning calorimetry (DSC) (Fig. S29, ESI†). This result is consistent with other siloxane-containing polymers such as PDMS, which have glass transition temperatures as low as −123 °C.23 Additionally, poly(iPrSi8) contains both cis and trans olefins in its backbone and a mixture of 4-, 5-, and 6-carbon linkages between silyl ethers; this combination of stereo- and regio-irregularity is likely to render crystallization difficult. Subsequently, ultra-low temperature DSC conditions were pursued to identify a Tg for these materials. A heating rate of 20 °C min−1 revealed that a discernible glass transition occurs at −88 °C (Fig. S30, ESI†). Therefore, poly(iPrSi8) represents an ultra-low-Tg polymer that is also highly resistant to crystallization, potentially facilitating applications in low-temperature environments.
In summary, previously unreported polysilylether homopolymers24,25 were synthesized via ED-ROMP, characterized, and deconstructed. ED-ROMP of an 8-membered cyclic silyl ether olefin yielded high molecular weight polymer with an ultra-low Tg (–88 °C), while the analogous 7-membered cyclic monomer did not homopolymerize under the same conditions. We demonstrate that these materials can undergo rapid backbone degradation upon both acid and fluoride treatment and partial depolymerization with G3. Further studies on the copolymerization of silyl ether monomers with ROMP-active olefins to afford deconstructable hydrocarbon-based polymers are underway.
We thank the National Science Foundation (Graduate Research Fellowship) for supporting this work.
Conflicts of interest
There are no conflicts to declare.References
- D. M. Lynn, S. Kanaoka and R. H. Grubbs, J. Am. Chem. Soc., 1996, 118, 784–789 CrossRef CAS.
- P. Schwab, M. B. France, J. W. Ziller and R. H. Grubbs, Angew. Chem., Int. Ed. Engl., 1995, 34, 2039–2041 CrossRef CAS.
- J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 41, 4035–4037 CrossRef CAS PubMed.
- A. R. Hlil, J. Balogh, S. Moncho, H. L. Su, R. Tuba, E. N. Brothers, M. Al-Hashimi and H. S. Bazzi, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3137–3145 CrossRef CAS.
- Z. Chen, J. P. Claverie, R. H. Grubbs and J. A. Komfield, Macromolecules, 1995, 28, 2147–2154 CrossRef CAS.
- P. Hodge and H. M. Colquhoun, Polym. Adv. Technol., 2005, 16, 84–94 CrossRef CAS.
- M. J. Marsella, H. D. Maynard and R. H. Grubbs, Angew. Chem., Int. Ed. Engl., 1997, 36, 1101–1103 CrossRef CAS.
- J. A. Nowalk, C. Fang, A. L. Short, R. M. Weiss, J. H. Swisher, P. Liu and T. Y. Meyer, J. Am. Chem. Soc., 2019, 141, 5741–5752 CrossRef CAS PubMed.
- F. N. Behrendt, A. Hess, M. Lehmann, B. Schmidt and H. Schlaad, Polym. Chem., 2019, 10, 1636–1641 RSC.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- R. Ogawa and M. A. Hillmyer, Polym. Chem., 2021, 12, 2253–2257 RSC.
- P. Shieh, H. V. T. Nguyen and J. A. Johnson, Nat. Chem., 2019, 11, 1124–1132 CrossRef CAS PubMed.
- P. Shieh, M. R. Hill, W. Zhang, S. L. Kristufek and J. A. Johnson, Chem. Rev., 2021, 121(12), 7059–7121 CrossRef CAS PubMed.
- P. Shieh, W. Zhang, K. E. L. Husted, S. L. Kristufek, B. Xiong, D. J. Lundberg, J. Lem, D. Veysset, Y. Sun, K. A. Nelson, D. L. Plata and J. A. Johnson, Nature, 2020, 583, 542–547 CrossRef CAS PubMed.
- K. E. L. Husted, P. Shieh, D. J. Lundberg, S. L. Kristufek and J. A. Johnson, ACS Macro Lett., 2021, 10, 805–810 CrossRef CAS PubMed.
- S. J. Clarson, K. Dodgson and J. A. Semlyen, Polymer, 1985, 26, 930–934 CrossRef CAS.
- A. K. Pearce, J. C. Foster and R. K. O’Reilly, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1621–1634 CrossRef CAS.
- C. L. Lee and O. K. Johannson, J. Polym. Sci., Part A: Polym. Chem., 1966, 4, 3013–3026 CrossRef CAS.
- A. Duda and A. Kowalski, Thermodynamics and Kinetics of Ring - Opening Polymerization, Wiley-VCH, 2009 Search PubMed.
- S. Strandman, J. E. Gautrot and X. X. Zhu, Polym. Chem., 2011, 2, 791–799 RSC.
- M. C. Parrott, J. C. Luft, J. D. Byrne, J. H. Fain, M. E. Napier and J. M. Desimone, J. Am. Chem. Soc., 2010, 132, 17928–17932 CrossRef CAS PubMed.
- M. G. Musolino, C. M. S. Cutrupi, A. Donato, D. Pietropaolo and R. Pietropaolo, J. Mol. Catal. A: Chem., 2003, 195, 147–157 CrossRef CAS.
- I. Bahar, I. Zuniga, R. Dodge and W. L. Mattice, Macromolecules, 1991, 24, 2986–2992 CrossRef CAS.
- C. Cheng, A. Watts, M. A. Hillmyer and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 11872–11876 CrossRef CAS PubMed.
- H. Fouilloux, M. N. Rager, P. Ríos, S. Conejero and C. M. Thomas, Angew. Chem., Int. Ed., 2022, 61, e202113443 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc02718f |
| This journal is © The Royal Society of Chemistry 2022 |