
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterisation of an integratively self-sorted [Fe4L6]8+ tetrahedron†
Lauren L. K.
Taylor
 ,
Rebecca
Andrews
,
April C. Y.
Sung
,
Iñigo J.
Vitorica-Yrezabal
and
Imogen A.
Riddell
*
,
Rebecca
Andrews
,
April C. Y.
Sung
,
Iñigo J.
Vitorica-Yrezabal
and
Imogen A.
Riddell
*
Department of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: imogen.riddell@manchester.ac.uk
First published on 18th October 2022
Abstract
Isolating metal–organic cage structures which incorporate more than one distinct ligand has been challenging due to competing pressures from narcissistic and social sorting phenomena. Here we report the first example of exclusive formation of a single tetrahedral product from a reaction mixture containing two different bidentate ligands. Exclusive formation of the tetrahedron, which incorporates one unique metal vertex, relies on a triamine to orientate the heteroditopic ligand. Inclusion of perchlorate counterions during the self-assembly process is also found to be a requirement if social sorting is to be avoided. The C3-symmetric structure is characterised by HR-MS, NMR spectroscopy and X-ray crystallography, and provides proof of principle for use of heteroditopic ligands in classical M4L6 supramolecular structures, opening exciting possibilities for their use in separation, storage and catalysis applications.
Within a subcomponent mixture, self-sorting principles can control the spontaneous self-assembly process and promote the formation of specific products.1,2 This phenomenon is prevalent throughout nature, governing processes such as base pairing in DNA,3 the self-fractioning of white blood cells in human blood4 and the highly-ordered, spontaneous grouping of alike neurons during foetal brain development.5 Recently there has been a growing interest in understanding and controlling complex self-sorting phenomenon in synthetic systems.6–12 In the field of metallo-supramolecular architectures the desire to controllably generate complex structures with unusual and useful functionality has led researchers to explore incorporation of reduced symmetry ligands13–17 as well as the formation of heteroleptic structures.18–24 Generation of discrete structures from these components requires that ligands are controllably orientated through sorting phenomena.
Self-sorting processes have been defined as either narcissistic or integrative,25,26 where narcissistic self-sorting describes the phenomenon of self-association2,27 and integrative self-sorting describes the combination of multiple different structural components within a single system.11,27 Commonly these terms are applied to the self-assembly of complexes from more than one homoditopic ligand, however, the self-assembly of reduced symmetry heteroditopic ligands can also be defined using these descriptors. In the latter case it is the coordination environment around the metal ion that is considered.7,11 For systems where neither narcissistic nor integrative sorting dominates, it is also possible to generate equilibrium mixtures containing several thermodynamic products of similar energies. This socially sorted mixture is termed a dynamic library.8,28 Gaining control over sorting to enable inclusion of more than one ligand within a metal–organic structure can however be challenging as narcissistic associations often dominate.27–30 For heteroditopic ligands orientation of the ligand with respect to the metal centre can be governed using steric and electronic arguments to bias either narcissistic or integrative sorting.11,14,31
Herein we detail self-assembly reactions incorporating two similar ligands (Scheme 1; L1 and L2) with a choice of amine and iron(II) salt. The system is shown to display integrative sorting generating C1 exclusively when TREN and iron perchlorate are employed in the self-assembly reaction. Alternatively, social sorting to form a mixture of two tetrahedral cages C1 and C2, and the mononuclear complex C3, is observed when iron triflimide is used in place of iron perchlorate.
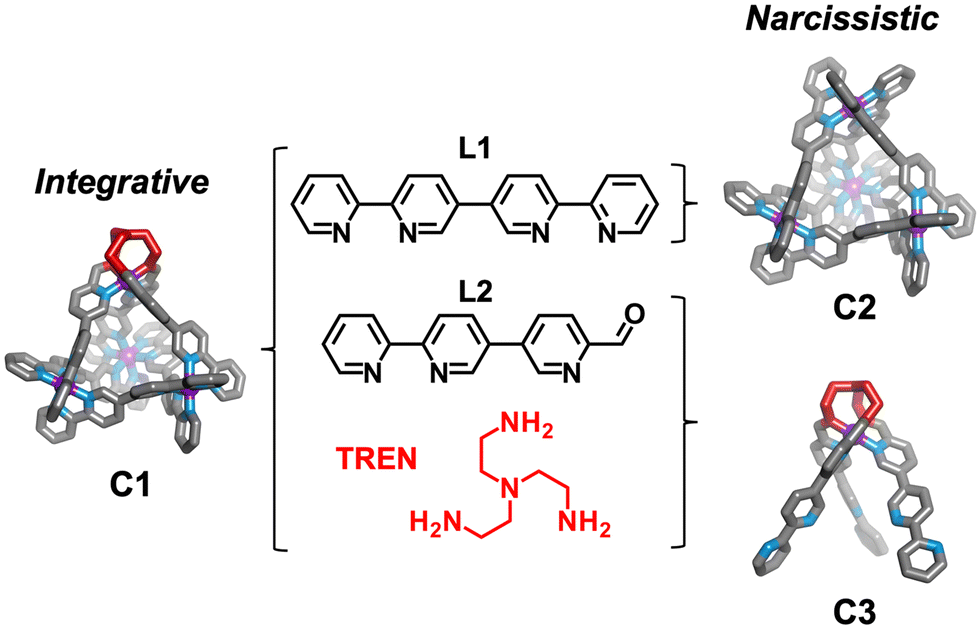 | ||
| Scheme 1 Left: Self-assembly of a heteroleptic tetrahedral cage C1 by integrative sorting. Right: Formation of homoleptic cage C2 and the mononuclear complex C3. Exclusive formation of C1 or social sorting is possible when using L1, L2, TREN and Fe(II) starting materials. | ||
The bisbidentate ligands L1 and L2 both bind octahedral metal ions through their bidentate N^N binding sites. Ligand 1 (L1) contains two bipyridine binding sites and was previously reported to form [Mn+4L16]4n+ tetrahedral architectures in the presence of octahedrally coordinating metal ions including Fe(II)32 and Co(III).33 Ligand 2 (L2) features a bidentate bipyridyl binding site at one end and a pyridine aldehyde moiety capable of reacting with a range of amines to generate a bidentate pyridylimine at the other.34 The relative orientation of the bidentate binding sites in these ligands is comparable, leading us to hypothesis the ligands could be interchanged within [M4L6]8+ tetrahedra.
To the best of our knowledge, the Fe4L6 tetrahedron (C1), which incorporates both L1 and L2, is the only example of an M4L6 tetrahedron formed through integrative sorting behaviour. Incorporation of L1 and L2 generates a structure which has overall C3-symmetry and incorporates one unique vertex which may allow for introduction of functionality not possible at the tris(bipyridine) sites,35 or affect host guest binding through alteration of guest access and egress.36
In initial studies L1, L2, Fe(ClO4)2 and the monoamine p-toluidine were combined in CD3CN to determine if the system displayed any self-sorting behaviour. Analysis of the 1H NMR from this reaction showed major peaks corresponding to the homoleptic cage, C2, alongside a broad baseline. Mass spectrometric analysis supported the formation of a library of M4L6 tetrahedra with between three and six L1 ligands incorporated (Fig. S5, ESI†). No evidence for formation of cages with greater than three equivalents of L2 was found suggesting that under the conditions of ionisation cages incorporating significant numbers of this component are less stable.
We next added one equivalent of TREN (T; tris(2-aminoethyl)amine) per four metal centres to the library generated with p-toluidine, hypothesising that the triamine would displace three equivalents of p-toluidine and orientate three pyridyl imines around one metal centre (Fig. 1). Analysis of the 1H NMR spectrum following addition of TREN (Fig S4, ESI†) indicated formation of a single set of resonances corresponding to C1, in addition to those of the liberated p-toluidine. Single crystals of C1 (Fig. 2) were grown through slow diffusion of diethyl ether into acetonitrile over the course of seven days. Measurement of the Fe(II) to N bond lengths in the crystal structure of C1 did not indicate any significant bond strain. All bond lengths were consistent with coordination of low-spin Fe(II) within a N6 coordination environment. Moreover, these values were comparable with those obtained from the single crystal structure of homoleptic tetrahedron C2 (Fig. S32, ESI†).
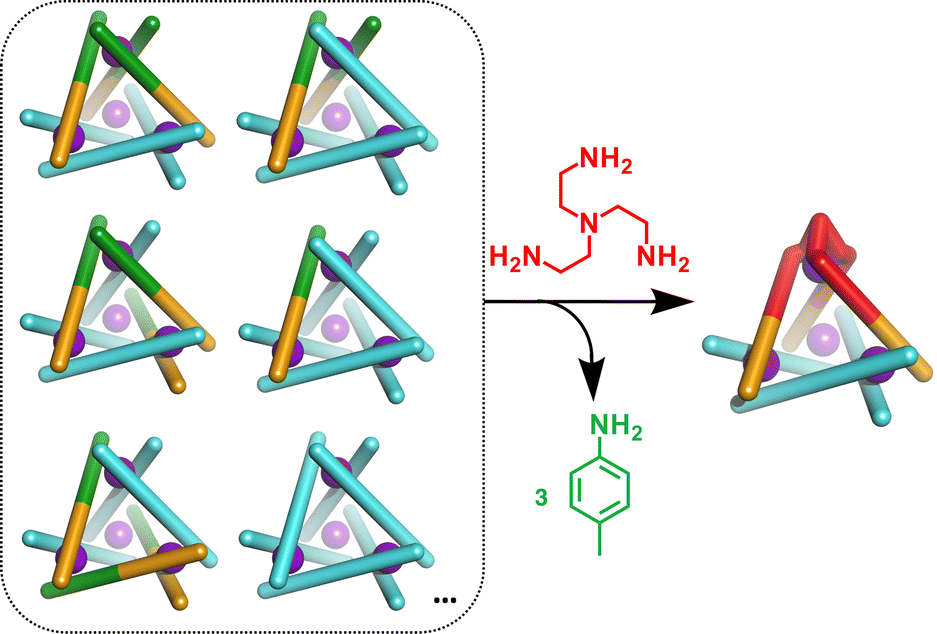 | ||
| Fig. 1 Displacement of p-toluidine (3 equiv.) by TREN(red) to generate C1 exclusively from a library of cages. The initial library includes a mixture of cages with varying numbers and orientations of L1 and L2. | ||
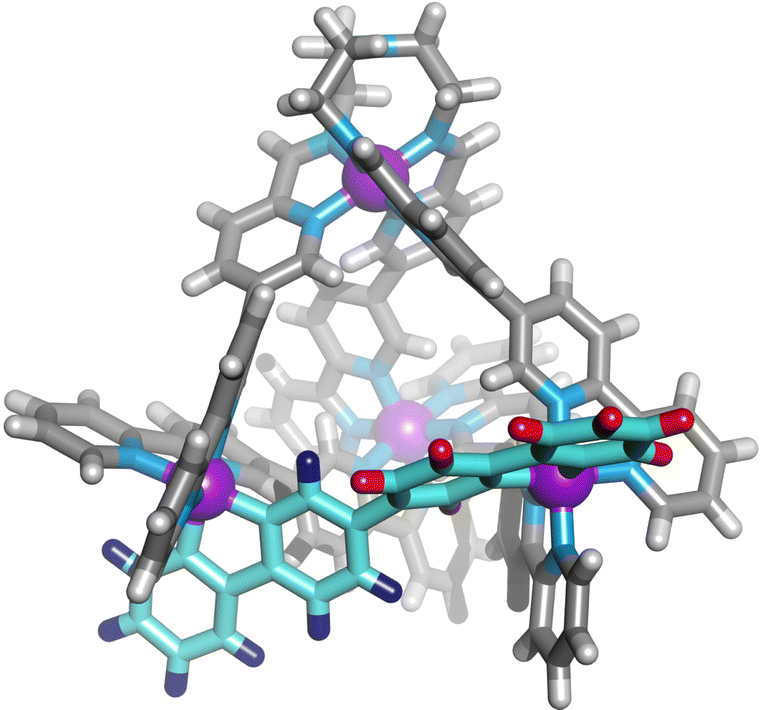 | ||
| Fig. 2 Single crystal X-ray structure of the cationic portion of C1. The two different bipyridine environments on L1 are coloured blue and red. Counterions and solvent removed for clarity. | ||
Assignment of the 1H NMR spectrum revealed that upon incorporation in C1, the two adjacent bipyridine units of L1 became inequivalent, consistent with formation of the C3-symmetric tetrahedral architecture (Fig. 2). Refinement of the single crystal X-ray data did not provide definitive evidence for encapsulation of the perchlorate counterion, however, structurally related M4L6 tetrahedra have been reported to bind perchlorate anions with high affinity.33,37
To try and better understand the factors governing the p-toluidine to TREN amine exchange process we next added three equivalents of 2-pyridinecarboxaldehyde and one equivalent of TREN per Fe(II) ion to a solution of purified C2 constituted from L1 and Fe(II). After heating the mixture at 65 °C for 24 hours the 1H NMR spectrum contained no evidence of C2 (Fig. S22, ESI†). Analysis of the reaction mixture confirmed exclusive formation of a mononuclear trispyridylimine complex, and the displaced L1 ligand precipitated from solution following decomplexation. We therefore propose that displacement of p-toluidine from complexes within the library is not the sole driving force for the structural rearrangement observed upon addition of TREN. Demetallation of the M4L6 structures, driven by the addition of TREN, also plays a significant role in the structural rearrangement of the mixture. This is consistent with previously reported mechanistic studies.30
Next we evaluated the effect the perchlorate counterion had on the 1H NMR profile of the self-assembly reaction mixture. When iron(II) triflimide (4 equiv.) was used in the self-assembly reaction alongside three equivalents of L1 and L2 and one equivalent of TREN a complex 1H NMR spectrum was recorded which could not be simplified through common purification techniques. Analysis of the spectrum identified two distinct sets of peaks corresponding to the heteroleptic and homoleptic tetrahedra (C1 and C2, Fig. S20, ESI†). Mass spectroscopic analysis of the reaction mixture also indicated the presence of the mononuclear species C3 (Fig. S21, ESI†). The large size of the triflimide anion37 ensures it cannot act as an internal template for either C1 or C2 and thus enabled the formation of a mixture of cages in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 C1:C2 ratio. Subsequent addition of potassium perchlorate to the crude mixture of C1, C2 and C3 synthesised with Fe(NTf2)2 revealed changes in the 1H NMR spectrum of the mixture that were consistent with encapsulation of this anion within both cages (C1 and C2). The observation that C1 and C2 are both capable of binding perchlorate anions is not unexpected given their structural similarities. Both complexes have comparable average Fe-Fe distances (9.36(0.007) Å and 9.44(0.050) Å, respectively) and enclose similarly shaped void cavities of comparable volume (ESI,† S3).
:2 C1:C2 ratio. Subsequent addition of potassium perchlorate to the crude mixture of C1, C2 and C3 synthesised with Fe(NTf2)2 revealed changes in the 1H NMR spectrum of the mixture that were consistent with encapsulation of this anion within both cages (C1 and C2). The observation that C1 and C2 are both capable of binding perchlorate anions is not unexpected given their structural similarities. Both complexes have comparable average Fe-Fe distances (9.36(0.007) Å and 9.44(0.050) Å, respectively) and enclose similarly shaped void cavities of comparable volume (ESI,† S3).
Inspection of the crystal lattice of C3 with each counterion indicated that the perchlorate ion sits in the C3-axis at the centre of the threefold symmetric metalloligand, while in the case of NTf2− this site is occupied by an acetonitrile molecule. The exclusive formation of C1 when synthesised from Fe(ClO4)2 starting materials is thus a kinetic effect, with the metal counterion identified as the discriminating factor in the self-assembly process. Additional studies with BF4−, OTf−, SbF6−, AsF6−, PF6− and ReO4− all generated socially sorted mixtures of products incorporating C1, C2 and C3 (ESI,† S1.3.6), indicating the preferential role of perchlorate in the formation of C1.
The formation and structural characterisation of an integratively sorted M4(L1)3(L2)3T metal–organic tetrahedral architecture is reported. M4L6 complexes are well documented to have desirable functional properties but, to date, examples of reduced-symmetry tetrahedra are rare. Moreover, formation of M4L6 tetrahedra which incorporate more than one ligand or metal ion have all previously been reported as components of statistical libraries. Here we show for the first-time exclusive formation of an integratively sorted M4L6 tetrahedron.
The heteroleptic tetrahedron is synthesized through addition of Fe(ClO4)2 into a system containing subcomponents L1, L2 and TREN. Replacing either iron(II) perchlorate or TREN generated mixtures of products. Understanding the parameters that govern the exclusive formation of C1 will enable the rational design of non-T-symmetric tetrahedra that incorporate dissimilar metal coordination sites. Such structures have increased functionality compared with their symmetric counterparts and present exciting possibilities for further development of metal–organic cages with applications ranging from separation to storage and catalysis.
This research was supported by a Royal Society University Research Fellowship (IAR), the Engineering and Physical Sciences Research Council (grants EP/K039547/1 and EP/R00482X/1), and the Diamond Light Source for the allocated time on the beamline I19 (Cy23480).
Data associated with this article, including experimental procedures, compound characterisation, X-ray crystallography and VOIDOO calculations are available in the ESI.†
I. A. R. conceived and supervised the project; L. L. K. T. designed and performed the experiments and characterised the compounds; R. A. performed experiments and characterised compounds; A. C. Y. S. synthesized novel ligand L2; I. J. Y. R. performed all crystallographic characterisation.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Frank, L. Krause, R. Herbst-Irmer, D. Stalke and G. H. Clever, Dalton Trans., 2014, 43, 4587–4592 RSC.
- M. Lal Saha and M. Schmittel, Org. Biomol. Chem., 2012, 10, 4651–4684 RSC.
- F. Crick and J. Watson, Nature, 1953, 171, 737–738 CrossRef PubMed.
- R. H. Carlson, C. V. Gabel, S. S. Chan, R. H. Austin, J. P. Brody and J. W. Winkelman, Phys. Rev. Lett., 1997, 79, 2149–2152 CrossRef CAS.
- S. Ackerman, Discovering the Brain, National Academies Press (US), Washington (DC), 1992 Search PubMed.
- M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
- Z. He, W. Jiang and C. A. Schalley, Chem. Soc. Rev., 2015, 44, 779–789 RSC.
- W. M. Bloch and G. H. Clever, Chem. Commun., 2017, 53, 8506–8516 RSC.
- S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC.
- M. Konopka, P. Cecot, J. M. Harrowfield and A. R. Stefankiewicz, J. Mater. Chem. C, 2021, 9, 7607–7614 RSC.
- M. M. J. Smulders, A. Jiménez and J. R. Nitschke, Angew. Chem., Int. Ed., 2012, 51, 6681–6685 CrossRef CAS PubMed.
- J. F. Ayme and J. M. Lehn, Chem. Sci., 2020, 11, 1114–1121 RSC.
- S. Cardona-Serra, E. Coronado, P. Gaviña, J. Ponce and S. Tatay, Chem. Commun., 2011, 47, 8235–8237 RSC.
- J. E. M. Lewis, A. Tarzia, A. J. P. White and K. E. Jelfs, Chem. Sci., 2020, 11, 677–683 RSC.
- A. D. Faulkner, R. A. Kaner, Q. M. A. Abdallah, G. Clarkson, D. J. Fox, P. Gurnani, S. E. Howson, R. M. Phillips, D. I. Roper, D. H. Simpson and P. Scott, Nat. Chem., 2014, 6, 797–803 CrossRef CAS PubMed.
- J. E. M. Lewis and J. D. Crowley, ChemPlusChem, 2020, 85, 815–827 CrossRef CAS PubMed.
- H. Ube, K. Endo, H. Sato and M. Shionoya, J. Am. Chem. Soc., 2019, 141, 10384–10389 CrossRef CAS PubMed.
- Y. Q. Zou, D. Zhang, T. K. Ronson, A. Tarzia, Z. Lu, K. E. Jelfs and J. R. Nitschke, J. Am. Chem. Soc., 2021, 143, 9009–9015 CrossRef CAS.
- J. Tessarolo, H. Lee, E. Sakuda, K. Umakoshi and G. H. Clever, J. Am. Chem. Soc., 2021, 143, 6339–6344 CrossRef CAS PubMed.
- A. W. Markwell-Heys, M. L. Schneider, J. M. L. Madridejos, G. F. Metha and W. M. Bloch, Chem. Commun., 2021, 57, 2915–2918 RSC.
- T. K. Ronson, D. A. Roberts, S. P. Black and J. R. Nitschke, J. Am. Chem. Soc., 2015, 137, 14502–14512 CrossRef CAS PubMed.
- M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC.
- A. J. Metherell and M. D. Ward, Chem. Sci., 2016, 7, 910–915 RSC.
- Q. F. Sun, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2014, 53, 13510–13513 CrossRef CAS.
- A. Wu and L. Isaacs, J. Am. Chem. Soc., 2003, 125, 4831–4835 CrossRef CAS PubMed.
- F. J. Rizzuto and J. R. Nitschke, J. Am. Chem. Soc., 2020, 142, 7749–7753 CrossRef CAS.
- L. R. Holloway, P. M. Bogie and R. J. Hooley, Dalton Trans., 2017, 46, 14719–14723 RSC.
- L. R. Holloway, Narcissistic Self-Sorting, Reactivity and Post-Assembly Modifications of Metal-Ligand Cage Complexes, UC Riverside, California, United States of America, 2018, https://escholarship.org/uc/item/6bw2b9gg.
- X. Zhao, H. Wang, B. Li, B. Zheng, D. Yang, W. Xu, X. Li, X. J. Yang and B. Wu, Chem. Commun., 2021, 57, 6078–6081 RSC.
- L. R. Holloway, M. C. Young, G. J. O. Beran and R. J. Hooley, Chem. Sci., 2015, 6, 4801–4806 RSC.
- K. Mahata, M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2010, 132, 15933–15935 CrossRef CAS PubMed.
- C. R. K. Glasson, J. K. Clegg, J. C. McMurtrie, G. V. Meehan, L. F. Lindoy, C. A. Motti, B. Moubaraki, K. S. Murray and J. D. Cashion, Chem. Sci., 2011, 2, 540–543 RSC.
- B. P. Burke, W. Grantham, M. J. Burke, G. S. Nichol, D. Roberts, I. Renard, R. Hargreaves, C. Cawthorne, S. J. Archibald and P. J. Lusby, J. Am. Chem. Soc., 2018, 140, 16877–16881 CrossRef CAS PubMed.
- M. L. Saha, S. Neogi and M. Schmittel, Dalton Trans., 2014, 43, 3815–3834 RSC.
- S. L. Han, J. Yang, D. Tripathy, X. Q. Guo, S. J. Hu, X. Z. Li, L. X. Cai, L. P. Zhou and Q. F. Sun, Inorg. Chem., 2020, 59, 14023–14030 CrossRef CAS PubMed.
- L. Zhang, Y. Jin, G. H. Tao, Y. Gong, Y. Hu, L. He and W. Zhang, Angew. Chem., Int. Ed., 2020, 59, 20846–20851 CrossRef CAS PubMed.
- I. A. Riddell, Y. R. Hristova, J. K. Clegg, C. S. Wood, B. Breiner and J. R. Nitschke, J. Am. Chem. Soc., 2013, 135, 2723–2733 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2112326, 2112327, 2112329 and 2112640. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc04624e |
| This journal is © The Royal Society of Chemistry 2022 |