
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Analyte sensing with unselectively binding synthetic receptors: virtues of time-resolved supramolecular assays†
Amrutha
Prabodh
 ,
Stephan
Sinn
and
Frank
Biedermann
*
,
Stephan
Sinn
and
Frank
Biedermann
*
Karlsruhe Institute of Technology (KIT), Institute of Nanotechnology (INT), Hermann-von-Helmholtz-Platz 1, Eggenstein-Leopoldshafen 76344, Germany. E-mail: frank.biedermann@kit.edu
First published on 30th November 2022
Abstract
The cross-reactivity to many analytes is one major limitation of most synthetic receptors (SRs) known so far. Herein, we show that through time-resolved competitive binding assays, even unselectively binding SRs can be utilized for analyte distinction and quantification. Furthermore, our methodology has also been applied to analyte mixtures and can be used in a microplate format.
Chemosensors have great prospects for medicinal diagnostics, drug discovery and food safety monitoring, as they can be superior to biosensors in terms of chemical and thermal stability, equilibration time, price and scope for small molecule detection.1–5 Many synthetic receptor molecules, macrocyclic hosts, and host-dye assemblies have already been utilized as chemosensors in analyte sensing assays.5–16 In most designs, it was attempted to achieve complementarity of the chemosensor to the target analyte, e.g., in size, shape, and charge, such that they can engage in selectivity-ensuring lock-and-key-type non-covalent interactions with each other.17–19 Unfortunately, the rational design of chemosensors has not often yielded systems that possess sufficiently high binding strength besides the desired selectivity for detecting the target analyte at physiologically relevant concentrations.4,5
In contrast, macrocyclic hosts with a deep hydrophobic binding pocket, such as cucurbit[n]urils (CBn), deep cavitands, and pillar[n]arenes, are amongst the most strongly analyte-binding systems available to date.7,8,20,21 However, such systems suffer from a low binding selectivity due to their highly symmetric cavity interior that is diploid of selectivity-providing functional groups. For instance, CBn bind a wide range of analytes, including amino acids and their derivatives, peptides, proteins, drugs, dyes, and hydrocarbons, with a high affinity in aqueous media and often outperforming all other synthetic binders in terms of affinity.6,7,22–24 Consequently, many reports utilizing CBn and their chromophoric CBn⊃dye complexes for label-free analyte detection and reaction monitoring through absorbance, fluorescence, circular dichroism, and NMR have emerged.23,25–28 However, despite this, it remains challenging to apply CBn-type chemosensors for selective sensing applications, see Fig. 1a.29–31 For e.g., while CB8 has been used for capturing and solubilizing steroids, its immediate utility for steroid-sensing applications is limited because many hydrophobic compounds are bound with micromolar to nanomolar affinities by this macrocyclic host.6,32 For instance, the three structurally closely related steroids testosterone (Tes), progesterone (Prog) and nandrolone (Nan) possess very similar binding affinity values for CB8 (Fig. 1b), and were thus chosen as model analytes to develop a kinetics-based sensing assay as an alternative to common thermodynamics-based binding assays.
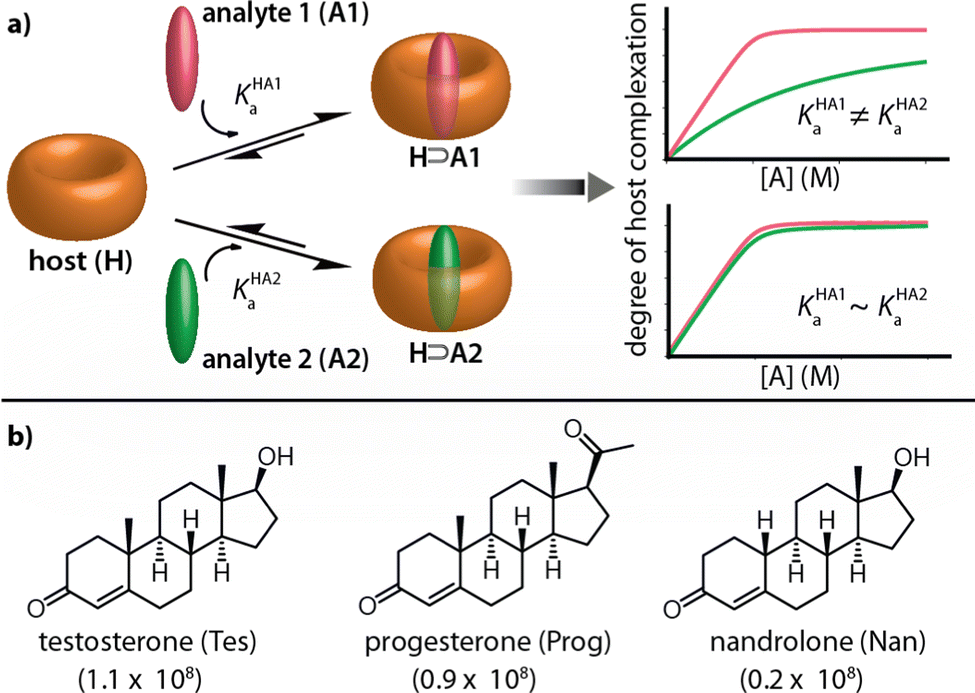 | ||
| Fig. 1 (a) Schematic representation of two scenarios for the complexation of two analyte molecules, analyte 1 (A1) and analyte 2 (A2), by a macrocyclic host (H). Scenario i: If the binding affinities of analyte 1 (KHA1a) and analyte 2 (KHA2a) for H are sufficiently distinct; differentiation of the two analytes by conventional binding assays is possible. Scenario ii: (A1) and (A2) show a similar affinity for H, preventing their distinction. (b) Chemical structures of the steroids investigated in this study. Their KHAa values with CB8 host in deionized water are given in parentheses. | ||
In this contribution, we demonstrate that access to the kinetic data dimension can – at least in parts – alleviate the cross-reactivity problems of CBn as representative synthetic receptors. Specifically, kinetic methods that enable analyte identification and quantification even in situations of poor thermodynamic selectivity are introduced herein.
Recently, the importance of elucidating both thermodynamic and kinetic parameters of evolving supramolecular systems has become more evident.33–36 We contributed to this field by the development of time-resolved competitive assays, i.e., the kinetic analyte-displacement assay (kinADA, Fig. 2a), the kinetic indicator-displacement assay (kinIDA, Fig. 2b), and the kinetic simultaneous-analyte-indicator binding assay (kinSBA).37,38 These assays allow assessing the kinetic parameters of host-analyte inclusion complexes with spectroscopically silent analytes and thereby address the physicochemical supramolecular community.37,38 In this work, we introduce a twist into the assay design to assess the potential of time-resolved competitive binding assays for sensing applications.
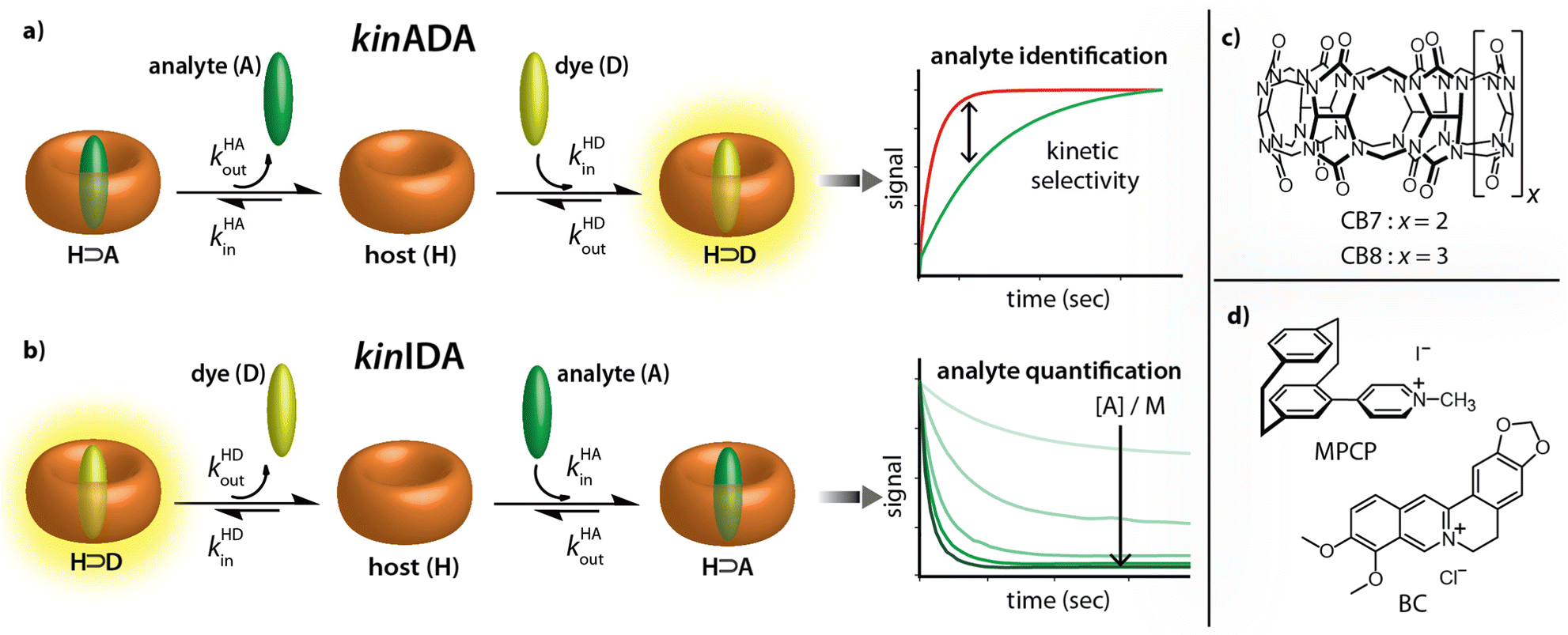 | ||
| Fig. 2 (a and b) Working principle of the kinetic analyte displacement assay (kinADA) and kinetic indicator displacement assay (kinIDA). The kinetic parameters kHAin and kHAout describe the rate constants for association and dissociation of the H⊃A complexes. Analogously, kHDin and kHDout refer to the kinetic parameters of the H⊃D complex. (c and d) Chemical structures of the macrocycles CB7 and CB8 and the environment-sensitive fluorophores MPCP and BC utilized in this study as host (H) and indicator dyes (D), respectively. | ||
While the assay concept may appear similar at first glance, the requirements for developing sensing assays differ from those for fundamental physiochemical binding studies. In the latter case, the accuracy and reproducibility of the measured and fitted parameters are crucial. The operator can thereby choose and control the concentrations of all molecular compounds and also has a free choice of the experimental methods and equipment. Conversely, in a practical sensing task, one typically encounters a situation where both the identity and the quantity of the target analyte in the sample is unknown and where additional cross-reactive compounds can be present.
Starting from these considerations, neither the kinIDA nor the kinSBA method appeared promising as a starting point for developing a sensing assay that can identify an analyte; Firstly, kinSBA shows a fast signal change directly after assay initiation. Thus, a sophisticated technical setup (e.g., rapid mixing) is needed for kinSBA, complicating its adaptation to the high-throughput microplate reader format. Secondly, for both kinIDA and kinSBA, the identity and concentration of the analyte influence the measurable kinetic traces. In our experience, such parameter coupling somewhat complicates the data analysis of kinSBA and kinIDA traces, as finding suitable parameter guesses for curve fitting typically involves several trial-and-error rounds.
Fortunately, the experimental setup and data analysis become facile through the pseudo-first-order kinetic analyte-displacement assay (kinADAPFO), a modified version of kinADA.
In kinADAPFO, the analyte-containing solution is first premixed and equilibrated with a solution of the host. Then a solution of a high-affinity indicator dye is introduced by a spiked addition, and the signal recording is started. In the course of the dynamic re-equilibration of the system, the analyte leaves the binding cavity of the host, which is then rapidly and nearly irreversibly re-occupied by the indicator dye. In essence, the supramolecular kinADAPFO setup produces a behaviour akin to SN1 reactions of a substrate R-X with a nucleophile Nu in organic chemistry. Thus, kinADAPFO kinetic curves are only dependent on the concentration and the dissociation-rate constant of the host⊃analyte complex but not on the type or concentration of the indicator dye. Importantly, kinADAPFO curves are also independent of analyte concentration if [host⊃analyte] → const. and [host]free → 0 at t = 0, i.e., if (nearly) all of the added host is bound to the analyte. This can readily be achieved by adding only a substoichiometric amount of host to the analyte-containing sample. Furthermore, as a result of the desirable parameter decoupling, the fitting of kinADAPFO traces is convenient and fast as the underlying coupled linear differential equations network for time-resolved kinetic assays simplifies to a single exponential equation (see Section 5.3 in the ESI†):
 | (1) |
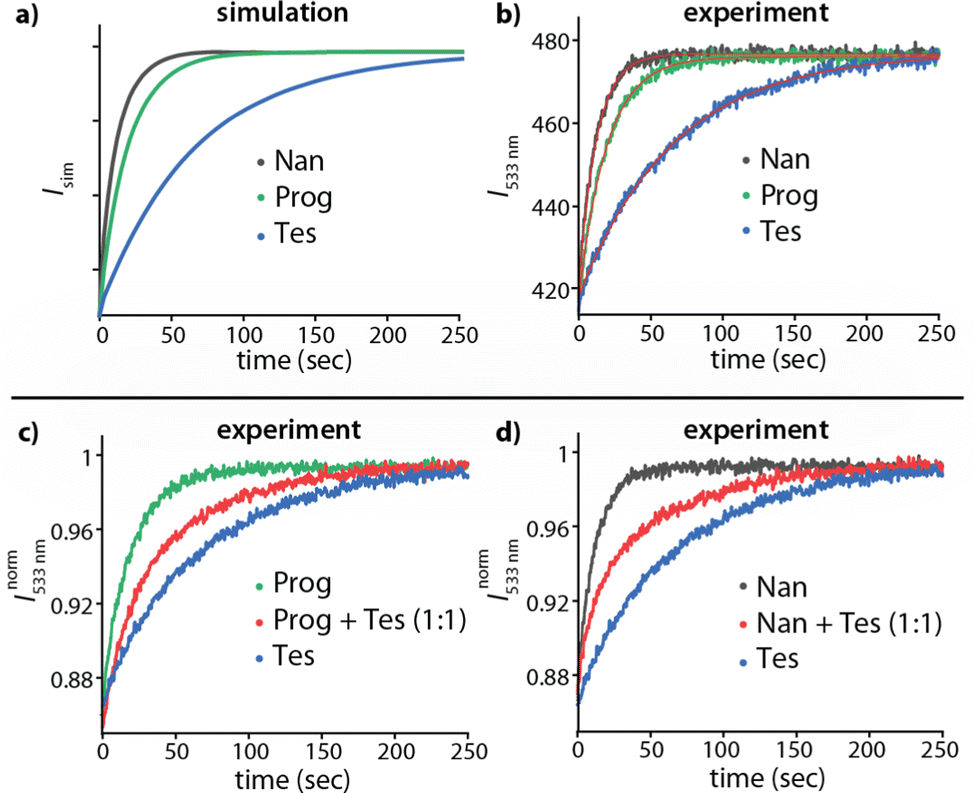
Based on these thoughts, we expected that the kinADAPFO can be utilized to identify the analyte in an analyte-concentration-independent fashion. A comparison of the simulated and measured kinetic traces presented in Fig. 3 supports this prediction. As a first showcase, we explored the behaviour of the steroid Nan as an analyte in the concentration range from 1.2 μM to 35.3 μM in water, to which each a substoichiometric amount (1.0 μM) of CB8 as the host was added. Subsequently, the ultra-high affinity indicator dye MPCP39 was introduced in a spiked addition to this host-analyte mixture, upon which the time-resolved sensing assay was initiated. Indeed, the simulated and experimentally recorded fluorescence-monitored kinetic traces (Fig. 3a and b) were completely independent of the analyte concentration. This is also evident from the slope obtained from a linear fitting of the initial linear part of the kinetic traces, which was independent of the analyte concentration (Fig. 3a and b insets).
 | ||
| Fig. 3 (a) Simulated kinADA and (b) experimental kinADAPFO traces monitored at 533 nm (λexc = 376 nm) upon varying analyte concentrations of Nan (1.2–35.3 μM) in the presence of 1 μM CB8 in water, followed by the spiked addition of MPCP indicator dye (50 μM). See Fig. S2 in ESI† for further details and parameters used for the simulations. The insets show the slope of the initial linear part of the kinetic traces for different analyte concentrations (c) Simulated kinIDA and (d) Experimental kinIDA traces monitored at 540 nm (λexc = 462 nm) upon varying analyte concentrations for Nan (1.2–35.3 μM) while keeping the concentration of the CB7⊃berberine complex at 1 μM. See Fig. S4 in ESI† for further details and parameters used for the simulations. (e and f) Plots of the initial slopes of the kinetic traces versus the analyte concentration. The calibration curves obtained from a linear fit are shown as a red line. | ||
Fitting of the kinADAPFO trace shown in Fig. 3b by eqn (1) yielded the kinetic parameter of 8.14 × 10−2 s−1 (Fig. S2a and Table S1 in ESI†), which is within error in agreement with the expected kHAout value for the analyte Nan, and succinctly different from the expectation for Tet and Prog (see Table S3 in ESI†). Future use of the herein introduced time-resolved supramolecular competitive assays for analyte identification will require setting up a reference database of kinetic parameters for combinations of analytes, hosts and indicator dyes at a defined media composition (e.g., temperature, pH, salt additives, co-solvents). In our opinion, the open-access repository SupraBank.org is ideally suited for this task.40
In addition to the possibility for analyte identification by kinADAPFO, also the quantity of the analyte can be determined through a kinetic sensing methodology. For instance, the kinetic traces strongly depend on the analyte concentration for a kinIDA setup, which can be utilized for quantitive analyte sensing. Fig. 3c and d shows a comparison of the simulated and measured kinetic traces for Nan (1.2 μM to 35.3 μM in water) to which a pre-complexed host⊃dye solution containing CB7 and BC (each at 1.0 μM) was introduced. Unlike for kinADAPFO, all concentration-dependent kinIDA traces are distinct, and the initial slope shows a linear dependence on the concentration of the analyte (Fig. 3e and f). Hence a linear calibration curve can be obtained from the measured slopes for a range of analyte concentrations.
In a practical sensing assay, one must also account for the interference from other cross-reactive analytes in the media. For instance, the steroids Tes, Prog, and Nan exhibit a lack of thermodynamic selectivity for the host CB8. A kinetic-based sensing assay is depicted in Fig. 4a and b, which shows the simulated and experimentally recorded fluorescence-monitored kinetic traces upon spiked addition of MPCP to the three solutions containing each one of the CB8⊃steroid complexes in water. The CB8⊃steroid indicative kHAout parameter obtained by fitting the traces to the exponential decay function (eqn (1)) is listed in Table S3 in the ESI.† The CB8 host exhibits kinetic selectivity for all three steroids studied. For instance, CB8 exhibit a thermodynamic selectivity (=KCB8Tesa/KCB8Proga) of only 1.2 for Tes over Prog, while the kinetics analysis displays a much higher kinetic selectivity (=kCB8Progout/kCB8Tesout) of 3.5. Hence, investigations into the binding kinetics aid in analyte differentiation and selective sensing even in the absence of thermodynamic selectivity.
 | ||
| Fig. 4 (a) Simulated kinADA and (b) Experimental kinADAPFO traces monitored at 533 nm (λexc = 376 nm) for CB8⊃Tes (1 μM, blue), CB8⊃Prog (1 μM, green) and CB8⊃Nan (1 μM, black) host⊃analyte complex upon spiked addition of MPCP dye (50 μM) in water. See Fig. S5 and S6 in the ESI† for further details and parameters used for the simulation. The red line in Fig. 4b depicts the fitted data according to the kinADAPFO binding model (eqn (1)). Experimental kinADAPFO traces monitored at 533 nm (λexc = 376 nm) for a mixture of two steroids as analytes (red curve) in case of (c) CB8 (1 μM), Tes (0.5 μM) and Prog (0.5 μM) and (d) CB8 (1 μM), Tes (0.5 μM) and Nan (0.5 μM) upon spiked addition of MPCP (50 μM) in water. The kinetic traces for individual steroids are shown for comparison. | ||
Finally, as the most complicated scenario, we investigated the occurrence of mixtures of analytes. Fig. 4c and d displays the kinetic traces for a solution containing CB8 and an equimolar mixture of two steroids as analytes, followed by a spiked addition of MPCP. The kinetic traces obtained were initially compared to the traces obtained for individual analytes along with a bi-exponential decay fit which gave a sufficient indication for the components of the mixture (Fig. S7 and S8 in ESI†). In addition, comparing the experimental data with the simulated kinADA curves for a mixture of two analytes (refer to Section 5.2 in ESI† for kinADA equations) enabled both the differentiation of the mixture's components and the determination of the concentration ratio of the analytes (Fig. S9 in ESI†). Indeed, under kinADAPFO conditions, our simulations revealed desirable independence of the absolute concentrations, while the relative analyte concentration ratio can be extracted from the data (Fig. S9(a) in ESI†).
Furthermore, we also adopted the time-resolved sensing method to a microplate reader format (Fig. S10 in ESI†).
In conclusion, time-resolved competitive assays that evaluate receptor-analyte binding and unbinding kinetics enable selective analyte sensing even with unselectively binding synthetic receptors. Both analyte identification and quantification are feasible by combining the kinADAPFO and kinIDA methods. Therefore, we believe our method can become a valuable addition to existing sensing concepts as it provides an alternative to the challenging development of selectively-binding synthetic receptors. A further increase in the data dimensions is envisioned when additional parameters that differentially affect the binding and unbinding kinetics of host-analyte complexes are tuned, e.g., temperature, viscosity, and presence of salts.
This work was financially supported through grants by the Emmy-Noether Programme of the DFG, and the DAAD.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC
.
- M. A. Beatty and F. Hof, Chem. Soc. Rev., 2021, 50, 4812–4832 RSC
- D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Chem. Soc. Rev., 2017, 46, 7105–7123 RSC
- T. W. Bell and N. M. Hext, Chem. Soc. Rev., 2004, 33, 589–598 CAS
- J. Krämer, R. Kang, L. M. Grimm, L. De Cola, P. Picchetti and F. Biedermann, Chem. Rev., 2022, 122, 3459–3636 CrossRef PubMed
- A. I. Lazar, F. Biedermann, K. R. Mustafina, K. I. Assaf, A. Hennig and W. M. Nau, J. Am. Chem. Soc., 2016, 138, 13022–13029 CrossRef CAS PubMed
- S. Sinn and F. Biedermann, Isr. J. Chem., 2018, 58, 357–412 CrossRef CAS
- W. Xue, P. Y. Zavalij and L. Isaacs, Angew. Chem., Int. Ed., 2020, 59, 13313–13319 CrossRef CAS PubMed
- H. Yao, H. Ke, X. Zhang, S.-J. Pan, M.-S. Li, L.-P. Yang, G. Schreckenbach and W. Jiang, J. Am. Chem. Soc., 2018, 140, 13466–13477 CrossRef CAS PubMed
- T. Schrader, G. Bitan and F.-G. Klärner, Chem. Commun., 2016, 52, 11318–11334 RSC
- S. A. Zebaze Ndendjio and L. Isaacs, Supramol. Chem., 2019, 31, 432–441 CrossRef CAS
- K. W. Bentley, Y. G. Nam, J. M. Murphy and C. Wolf, J. Am. Chem. Soc., 2013, 135, 18052–18055 CrossRef CAS PubMed
- M. A. Beatty, A. J. Selinger, Y. Li and F. Hof, J. Am. Chem. Soc., 2019, 141, 16763–16771 CrossRef CAS PubMed
- A. F. Sierra, D. Hernández-Alonso, M. A. Romero, J. A. González-Delgado, U. Pischel and P. Ballester, J. Am. Chem. Soc., 2020, 142, 4276–4284 CrossRef CAS PubMed
- W. Zhong and R. J. Hooley, Acc. Chem. Res., 2022, 55, 1035–1046 CrossRef CAS PubMed
- A. D. Gill, B. L. Hickey, W. Zhong and R. J. Hooley, Chem. Commun., 2020, 56, 4352–4355 RSC
- H.-J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 CrossRef CAS PubMed
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed
- R. Pinalli, A. Pedrini and E. Dalcanale, Chem. Soc. Rev., 2018, 47, 7006–7026 RSC
- J. Murray, K. Kim, T. Ogoshi, W. Yao and B. C. Gibb, Chem. Soc. Rev., 2017, 46, 2479–2496 RSC
- W.-I. K. Chio, S. Moorthy, J. Perumal, U. S. Dinish, I. P. Parkin, M. Olivo and T.-C. Lee, J. Mater. Chem. C, 2020, 8, 7051–7058 RSC
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed
- L. Cao, M. Šekutor, P. Y. Zavalij, K. Mlinarić-Majerski, R. Glaser and L. Isaacs, Angew. Chem., Int. Ed., 2014, 53, 988–993 CrossRef CAS PubMed
- J. Liu, H. Lambert, Y.-W. Zhang and T.-C. Lee, Anal. Chem., 2021, 93, 4223–4230 CrossRef CAS PubMed
- F. Biedermann and W. M. Nau, Angew. Chem., Int. Ed., 2014, 53, 5694–5699 CrossRef CAS PubMed
- A. Prabodh, Y. Wang, S. Sinn, P. Albertini, C. Spies, E. Spuling, L.-P. Yang, W. Jiang, S. Bräse and F. Biedermann, Chem. Sci., 2021, 12, 9420–9431 RSC
- A. Hennig, H. Bakirci and W. M. Nau, Nat. Methods, 2007, 4, 629–632 CrossRef CAS PubMed
- J. M. Chinai, A. B. Taylor, L. M. Ryno, N. D. Hargreaves, C. A. Morris, P. J. Hart and A. R. Urbach, J. Am. Chem. Soc., 2011, 133, 8810–8813 CrossRef CAS PubMed
- S. Lim, Y. Kuang and H. A. M. Ardoña, Front. Chem., 2021, 9 DOI:10.3389/fchem.2021.723111
- C. Zhong, C. Hu, R. Kumar, V. Trouillet, F. Biedermann and M. Hirtz, ACS Appl. Nano Mater., 2021, 4, 4676–4687 CrossRef CAS
- A. D. Gill, L. Perez, I. N. Q. Salinas, S. R. Byers, Y. Liu, B. L. Hickey, W. Zhong and R. J. Hooley, Chem. – Eur. J., 2019, 25, 1740–1745 CrossRef CAS PubMed
- A. Stahl, A. I. Lazar, V. N. Muchemu, W. M. Nau, M. S. Ullrich and A. Hennig, Anal. Bioanal. Chem., 2017, 409, 6485–6494 CrossRef CAS PubMed
- C. Bohne, Chem. Soc. Rev., 2014, 43, 4037–4050 RSC
- C. Márquez, R. R. Hudgins and W. M. Nau, J. Am. Chem. Soc., 2004, 126, 5806–5816 CrossRef PubMed
- E. Masson, M. Raeisi and K. Kotturi, Isr. J. Chem., 2018, 58, 413–434 CrossRef CAS
- L. Avram, A. D. Wishard, B. C. Gibb and A. Bar-Shir, Angew. Chem., Int. Ed., 2017, 56, 15314–15318 CrossRef CAS PubMed
- Z. Miskolczy, M. Megyesi, S. Sinn, F. Biedermann and L. Biczók, Chem. Commun., 2021, 57, 12663–12666 RSC
- A. Prabodh, S. Sinn, L. Grimm, Z. Miskolczy, M. Megyesi, L. Biczók, S. Bräse and F. Biedermann, Chem. Commun., 2020, 56, 12327–12330 RSC
- S. Sinn, E. Spuling, S. Bräse and F. Biedermann, Chem. Sci., 2019, 10, 6584–6593 RSC
- Suprabank, accessed 01/02/2022, https://suprabank.org/.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc04831k |
| This journal is © The Royal Society of Chemistry 2022 |