
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Risk for the release of an enormous amount of nanoplastics and microplastics from partially biodegradable polymer blends†
Xin-Feng
Wei
 *ab,
Mikael S.
Hedenqvist
a,
Luyao
Zhao
a,
Andreas
Barth
b and
Haiyan
Yin
*c
*ab,
Mikael S.
Hedenqvist
a,
Luyao
Zhao
a,
Andreas
Barth
b and
Haiyan
Yin
*c
aDepartment of Fibre and Polymer Technology, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden. E-mail: xinfengbio@gmail.com
bDepartment of Biochemistry and Biophysics (DBB), Stockholm University, SE-106 91, Stockholm, Sweden
cRISE Research Institutes of Sweden, Division Bioeconomy and Health, Material and Surface Design, Box 5607, SE-11486 Stockholm, Sweden. E-mail: haiyan.yin@ri.se
First published on 28th October 2022
Abstract
Nanoplastics and microplastics (NMPs) in natural environments are an emerging global concern and understanding their formation processes from macro-plastic items during degradation/weathering is critical for predicting their quantities and impacts in different ecological systems. Here, we show the risk of enormous emissions of NMPs from polymer blends, a source that has not been specifically studied, by taking immiscible (most common case) partially biodegradable polymer blends as an example. The blends have the common “sea-island” morphology, where the minor non-biodegradable polymer phase (polyethylene and polypropylene) is dispersed as NMP particles in the major continuous biodegradable matrix (poly(ε-caprolactone)). The dispersed NMP particles with spherical and rod-like shapes are gradually liberated and released to the surrounding aquatic environment during the biodegradation of the matrix polymer. Strikingly, the number of released NMPs from the blend is very high. The blend film surface erosion process, induced by enzymatic hydrolysis of the matrix, involving fragmentation, hole formation, and hole wall detachment, was systematically investigated to reveal the NMP release process. Our findings present direct evidence and detailed insights into the high risk of emissions of NMPs from partially biodegradable immiscible polymer blends with a widespread “sea-island” morphology. Efforts from authorities, developers, manufacturers, and the public are needed to avoid the use of non-biodegradable polymers in blends with biodegradable polymers.
Introduction
Nanoplastics (NPs) and microplastics (MPs), defined as plastic particles with a size <1 μm (ref. 1) and of 1 μm–5 mm,2 respectively, have become an emerging global concern because of their persistence, widespread presence in freshwater and seawater, sediments, biota, soils, and ambient air, and because of their potential adverse effects on biota and ecosystems.3–7 They originate from primary sources such as personal care products and industrial cleaners containing nano/micro-beads, as well as from secondary sources, i.e. from the fragmentation of plastic items and wastes upon weathering/environmental degradation.1,8–11 The number of nano/micro-plastic particles (NMPs) accumulated in nature is enormous. For instance, 14 million tons of MPs are estimated to reside on the ocean floor,12 and 24.4 trillion pieces (82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–578000 tons) of MPs exist in regions closer to the surface in the world's oceans.13
000–578000 tons) of MPs exist in regions closer to the surface in the world's oceans.13
Many countries, such as the USA, China, and Sweden, have banned microbead-added products to reduce the primary MP emissions.14,15 However, the emissions of secondary NMPs, the main origin of the MPs in marine environments,8 continue to grow rapidly because of continuously growing plastic production and largely uncontrolled waste.16,17 Besides, more than 5000 million tons of plastic waste are accumulated either in landfills or natural environments (mostly in the oceans)18 and they are continuously emitting numerous NMPs because of mechanical stress, heat, UV radiation, and biological degradation.9,16,19 In addition, an increasing number of studies show that the highly degraded secondary NMPs contain a greater content of hazardous substances than pristine NMPs due to the presence of degradation products such as monomers, oligomers and oxidative products.20–23 Therefore, it is particularly crucial to understand the degradation-induced formation process of NMPs in various environmental conditions for better prediction of NMP emissions.16,24–28 Most studies focus on pure plastics,24–27 and overlook an important source for NMPs: the degradation of polymer blends.
Polymer blends, mixes of at least two polymers, account for more than 20% of the total engineering polymer consumption, and the global market is expected to exceed 50 million tons per year.29 Polymers are generally immiscible and their blends form generally a sea-island morphology, i.e., a minor phase dispersed as droplets in a major continuous phase.30,31 The dispersed phase shows shapes of spheres, cylinders, ribbons, and ellipsoids with sizes ranging from hundreds of nanometers to tens of micrometers, depending on blend ratio, interfacial adhesion, and processing conditions.30 We have pointed out that these dispersed particles in polymer blends should be regarded as NMPs, which could be liberated and released into the environment during degradation.32 However, the release of the dispersed NMP particles in a polymer blend has not been investigated despite the great potential threat of NMP emissions.
Concerns over sustainability and plastic pollution from traditional polymers have driven the rapid development of biodegradable polymers.11,33–37 In contrast to traditional plastics, biodegradable plastics can break down into low molecular weight compounds such as water, methane, and carbon dioxide by bacteria, fungi, and algae. They are often blended with other polymers, including non-biodegradable polymers, to tailor their processability and mechanical properties for particular applications.38,39 For instance, non-biodegradable rubbery polymers such as ethylene-co-vinyl acetate,40 thermoplastic polyurethane,41 and olefin block copolymers42 are often added into polylactic acid (PLA) to improve the toughness or/and ductility of PLA.38 For a summary of the different types of existing partially biodegradable polymer blends and their properties, please refer to ref. 43. In addition, a large amount of partially biodegradable polymer blends may be inadvertently produced as a consequence of the recycling of biodegradable polymers together with conventional non-biodegradable polymers. The reason for this is the difficulty to separate biodegradable polymers from non-biodegradable polymers in wastes. This may result in the release of non-biodegradable polymer particles from the blend into the environment during biodegradation.
In this study, we demonstrated the enormous emission of NMPs from polymer blends and elucidated the corresponding release process for the first time. In particular, two immiscible polymer blends of poly(ε-caprolactone) (PCL, a commonly used biodegradable polymer) and polyethylene (PE) or polypropylene (PP) (these two are by far the most commonly used polymers) were selected as the model blend, and its enzymatic hydrolysis in an aquatic environment was modeled by a microbial hydrolytic enzyme (Amano Lipase PS). Enzymatic hydrolysis was studied because it is the dominant process of biodegradation of biodegradable polyesters into small molecules, catalyzed by esterase released from surrounding microorganisms. The prepared blends were aged in phosphate buffered saline solution containing lipase at 40 °C for up to 6 days. The mass loss and thickness reduction of the blend films were examined to reveal the hydrolysis rate. The NMPs released from the blend films during the hydrolysis were determined, and the film surface morphological changes during the degradation, were carefully evaluated to reveal the NMP release process.
Results and discussion
Morphology of the blend
The prepared immiscible PCL/PE 90/10 blend had a typical sea-island morphology with the minor PE phase dispersed in the major continuous PCL matrix (Fig. 1a). The PE phase existed primarily in the form of spherical/elliptical particles, except in the surface region up to a depth of ca. 80 μm (16% of film thickness), where oblate spheroidal/rod-shaped particles dominated (Fig. 1b). Orientated structures such as rods and fibrils are often observed for the dispersed domains in immiscible polymer blends due to the existence of extension and shear forces during the conventional processing processes such as extrusion and injection molding.44–46 Here, when the melted polymer blend was pressed to fill the entire mold during the compression process, the melt experienced the greatest shear force in the region close to the mold surface, leading to the formation of rod-shape particles and their orientation parallel to the surface in the surface region of the samples. The diameter of the spherical PE particles was 0.1–10 μm and the rod-shaped particles were much larger, >10 μm long and ca. 3 μm wide (Fig. 1a and d). The particles analyzed had an average size of ca. 2.8 μm and an average volume of 35.3 μm3. The particles were regarded as cylinders when their length was greater than 2 times their width, otherwise, they were treated as spheres to simplify the calculation of particle volume.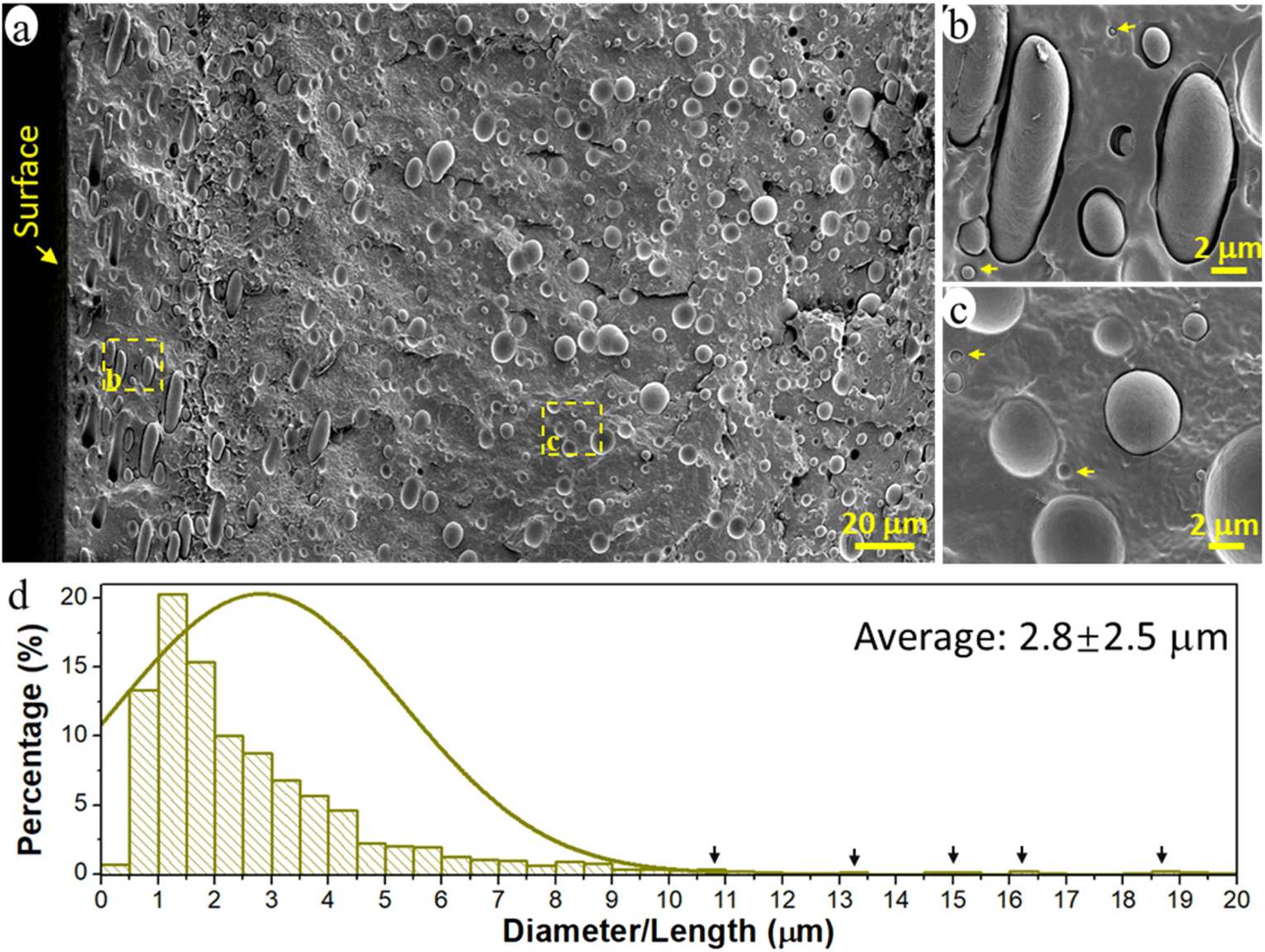 | ||
| Fig. 1 (a–c) Scanning Electron Microscope (SEM) images of the cryo-fractured cross-section of the PCL/PE 90/10 blend film and (d) the size distribution of the dispersed PE particles in (a). The arrows in (b) and (c) highlight particles with a size smaller than 1 μm. The total number of particles accounted for in (d) was ca. 1800. Note that only the size of the uncovered/visible part of the particles in the cross-section was measured, the actual size of the particles could be slightly different, especially for the rod-shaped particles. | ||
Nanoplastic and microplastic emission
The blend film started to fragmentate after ca. 4 days of ageing and it broke into many small pieces after 6 days of enzymatic hydrolysis ageing (Fig. 2a) in an aquatic environment in the presence of a microbial hydrolytic enzyme (Amano Lipase PS). The weight of the blend film decreased significantly to 32.6%, 4.8%, and 1.6% of its original weight after 2, 4, and 6 days of ageing, respectively, and the thickness decreased correspondingly (Fig. 2d). This was due to a very rapid enzymatic hydrolysis of the PCL matrix under the actual temperature and pH conditions with Amano Lipase PS as the enzyme. Despite the large reduction in thickness and weight, the overall shape of the sample remained unchanged until the start of the fragmentation (Fig. 2a). This is because the erosion takes place at the surface as enzymes are not able to diffuse into polymers and can only catalyze hydrolysis from the surface.47 The hydrolysis-induced surface erosion also changed the surface morphology and roughness of the film during the ageing (Fig. 2a).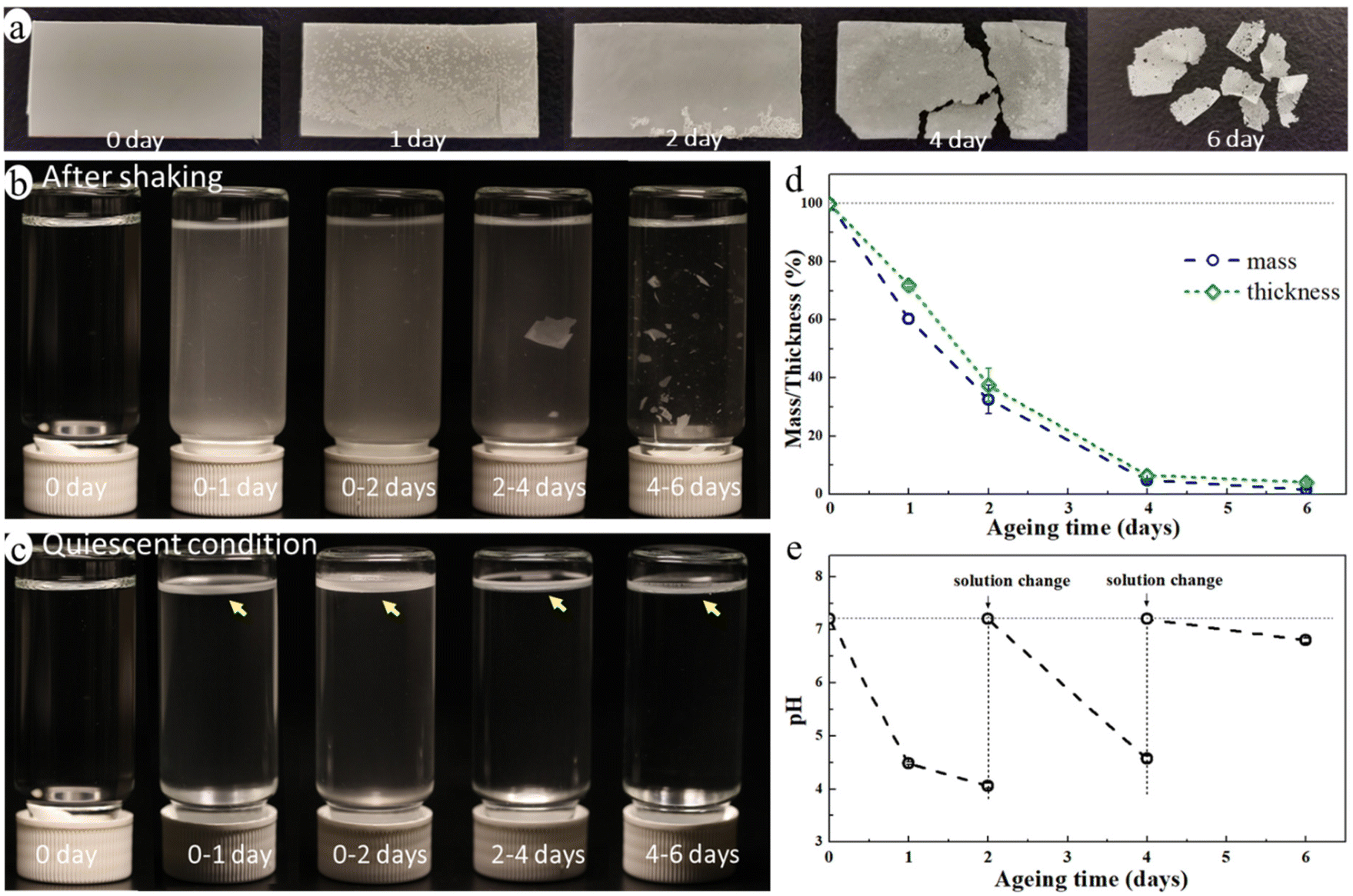 | ||
| Fig. 2 Images of the samples after different ageing times: blend films (a), buffer solution (b) after shaking by hand and (c) after standing still for one day; (d) the changes in mass and thickness of the film and (e) changes in the pH of the buffer solution during the ageing. The arrows in c point to the NMP layer floating on the solution surface. | ||
The buffer solution became turbid in all aged samples due to the suspension of many small particles (Fig. 2b), indicating the release of MP particles from the blend during ageing. Noting that the buffered enzyme solution was changed every second day of ageing to maintain enzyme activity, Fig. 2b and c represent only the MPs formed during the indicated ageing periods. The solution after 4–6 days of ageing was less turbid than that of other aged samples, but it contained many large film fragments. When letting the solution stand still for one day, the large film fragments sank to the bottom, while the small MP particles floated on the liquid surface forming a white floating layer (Fig. 2c). This finding indicated that the released MP particles were essentially from the PE phase of the blend as PE's density is lower than that of water, whereas the PCL's density is higher. The pH of the buffer solution decreased from 7.2 to 4.1 after the first two days of ageing, and it also decreased upon further ageing after each buffer change (Fig. 2e). The decrease in pH was caused by the release of acidic monomers and oligomers from PCL during the hydrolysis.48
The released particles were clearly observed in an optical microscope when suspended in the buffer solution (Fig. 3), and in SEM when collected on a filter membrane (Fig. 4a–e). Particles with sizes of a couple of hundred nanometers, e.g., those in the insert of Fig. 4d and e, were frequently observed under high magnification in SEM, which indicated a formation of NPs together with MPs. In addition, many nanoparticles were attached to the surface of micron-sized PE particles in Fig. 4d and e. Both optical microscopy and SEM images showed that the NMP particles had spherical and rod-like shapes, the same as the PE particles observed in Fig. 1, and infrared (IR) bands of PE (CH2 absorption at 2913, 2850, 1463, and 720 cm−1)49 dominated the IR spectrum of the formed NMPs (Fig. 4f). Besides, the IR spectra of individual particles, obtained by using an IR microscope, showed the IR bands of PE but not the characteristic bands of PCL (for instance the dominating carbonyl peak at 1736 cm−1, Fig. S1†). These findings confirmed that the observed microplastics were exactly the non-biodegradable PE particles that were liberated and released during the hydrolysis of the PCL matrix. The rod-shaped particles were observed mainly within the first two days of ageing because they were primarily located in the surface region, which was the first to be eroded through enzymatic hydrolysis. Besides, the released PE particles showed a fibrous surface texture (the inserts in Fig. 4d and e). This nano-structured surface morphology was also observed for the PE particles in the unaged blend sample (not shown here) and was due to protruding PE crystal lamellae in the interface of PCL and PE.
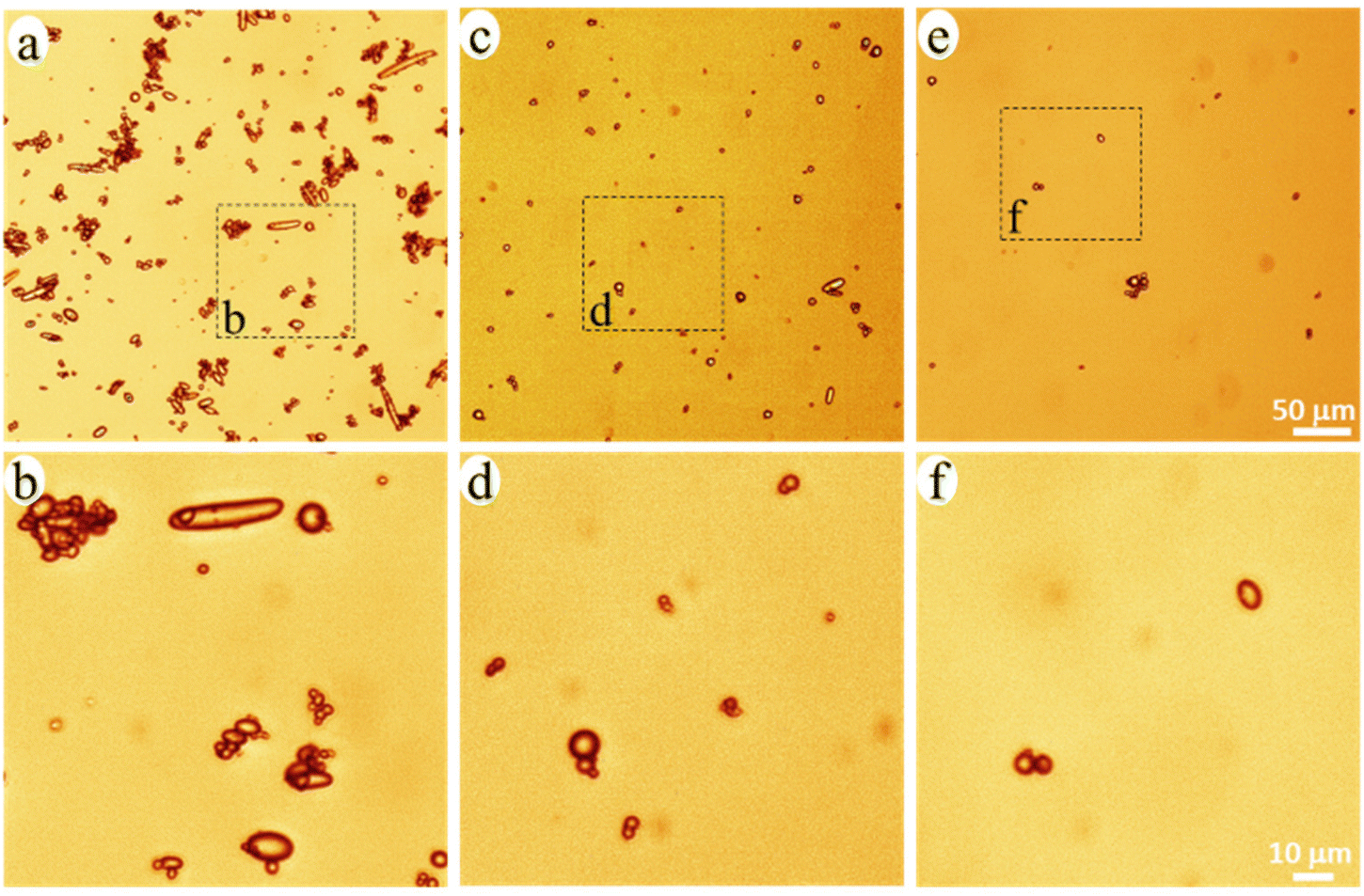 | ||
| Fig. 3 Optical microscopy images of the NMP particles in the buffer solution with 0–2 (a and b), 2–4 (c and d), and 4–6 (e and f) days of ageing. | ||
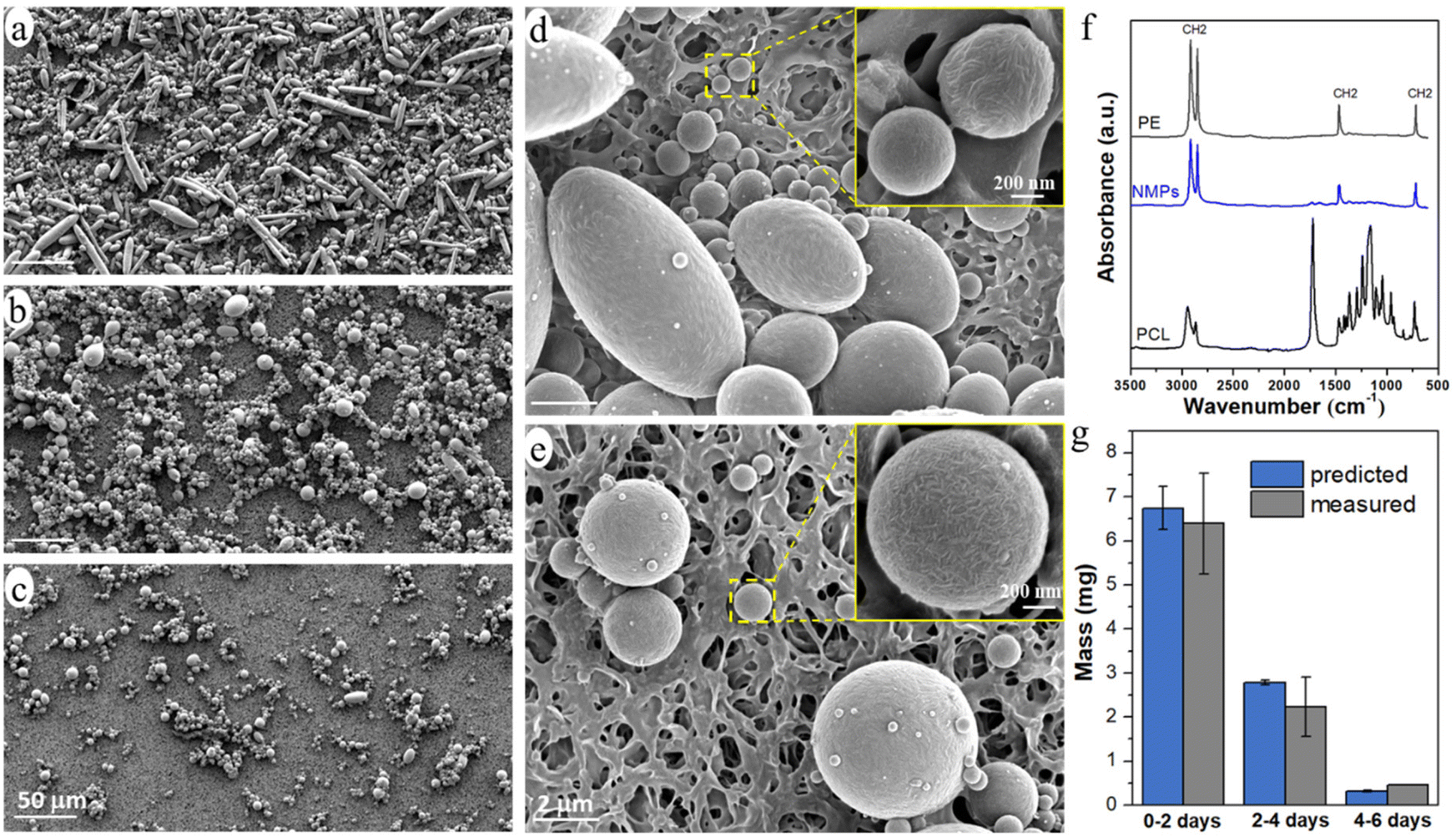 | ||
| Fig. 4 SEM images of the NMP particles on the membrane, released during 0–2 (a and d), 2–4 (b), and 4–6 (c and e) days of ageing; (f) IR spectra of neat PE, NMPs formed during 0–2 days of ageing, and neat PCL and (g) predicted and measured mass of the NMPs formed during different ageing periods. The inset in d and e shows typical NPs released during ageing. | ||
PE occurred either as individual particles or in particle aggregates in the solution (Fig. 3). Some of the aggregated particles were small fragments of the blend film, where the PE particles were held together by the heavily eroded PCL matrix between them (Fig. S2†). The bridging PCL matrix underwent further hydrolysis, ultimately liberating all the PE particles in the fragments. Therefore, these detached fragments (matrix MPs) were just an intermediate product of the enzymatic hydrolysis of the blend. From the SEM images, it was observed that most of the particles showed no PCL matrix connection with adjacent particles (Fig. 4d, and Fig. S3†), indicating that they were fully liberated particles. Besides, the EDS spectra of the naked particles did not show any oxygen, expected for PCL, which further confirms that the particles consisted of PE (Fig. S2†).
The number of particles was greatest during 0–2 days of ageing and least during 4–6 days of ageing (Fig. 3 and 4), which agreed well with the extent of the mass loss of the film during every two-day period of ageing (Fig. 2d) and the pH drop (Fig. 2e). Striking was that the NMP particles formed in the 30 ml solution during the first two days of ageing were so many that they fully covered the membrane with a diameter of 35 mm (Fig. 4a). Fig. 4g shows that the measured mass of the formed NMPs agreed well with that predicted by multiplying the mass loss of the film during each period with the PE content in the blend (10%). The number of the formed PE NMPs was calculated according to:
| n = m/(ρV0) |
Release process
The evolution of the surface morphology of the blend films during ageing was carefully investigated to understand the release process of NMPs from the PCL/PE blend under enzymatic hydrolysis (Fig. 5). Many holes were formed on the film surface after one day of ageing and the walls around the hole formed a continuous network on top of the bulk film (Fig. 5a and e). The walls consisted of partially uncovered PE particles and heavily eroded PCL matrix (Fig. 5e). This “hole-wall” morphology is also observed for neat PCL films in the initial stage of hydrolysis under the same ageing condition, which is attributed to the uneven hydrolysis/erosion rate across the polymer surface.48 The walls were ca. 80 μm deep with a very thin part (less than 5 μm thick) connecting to the bulk film (Fig. 5i and j). Subsequently, the thin connecting part was easily eroded, leading to the detachment of the protruding walls from the film and the formation of the observed small fragment particles in the solution (Fig. S2†). | ||
| Fig. 5 SEM images of the surface of the film samples aged for 1 (a and e), 2 (b and f), 4 (c and g), and 6 (d and h) days. (e) shows the “hole-wall” structure formed on the surface after one day of ageing. (i), (k), and (l) show the cryofractured cross-section of the films aged for 1, 2, and 4 days of ageing, respectively. (j) and (m) show the enlarged hole wall structure. (n) illustrates the evolution of the surface morphology of the PCL/PE blend film during the enzymatic hydrolysis process. (o) illustrates the release process of a single PE particle from the matrix during ageing. The degrading enzymes have access only to the top surface of the shown box, whereas its other sides are in contact with bulk material. The arrows indicate the direction of the enzymatic attack. | ||
As shown in Fig. 5b, most of the protruding hole walls had detached from the bulk film after 2 days of ageing, leaving a relatively flat surface (Fig. 5k). The “hole-wall” morphology was formed again in the sample after 4 days of ageing (Fig. 5c and l), and the thickness of the residual bulk film was reduced to less than 50 μm (Fig. 5l). Under further ageing, many through-holes were observed in the thinned film (Fig. 5d and h), indicating that the film was penetrated by the erosion from the surface and down, which eventually led to the massive fragmentation of the bulk film and the formation of large film fragments (Fig. 1a). Fig. 5n illustrates the observed surface morphology during the enzymatic attack, involving hole formation, wall detachment, new hole formation, film fragmentation, and the associated NMP release.
Apart from the above morphology evolution of the blend surface, all eroded surfaces consisted of a large amount of partially uncovered PE particles (Fig. 5a–m). Based on the observed structures of the PE particles with different degrees of uncovering, the release process of a single particle from the blend during the ageing is summarized in Fig. 5o. At first, the hydrolysis gradually eroded the PCL matrix from the surface (i.e., from top to bottom in Fig. 5o), uncovering the PE particle accordingly. Once the surface erosion reached the “equator” of the PE particle, further erosion of the PCL matrix beneath the PE particle was hindered by the presence of the PE particle and had to occur from the side rather than from above. Hence, this region was eroded at a slower rate than those at the same depth but with no particles above. As a result, the PE particle was still bonded to the bulk film by the uneroded PCL matrix below it, when most of the surrounding PCL matrix was eroded (Fig. 5h, illustrated in Fig. 5o). Under further ageing, the PCL connecting part was eroded from the side, releasing the particle into the solution.
PCL/PP blend
To reveal that the demonstrated NMPs emission from the PCL/PE blend was representative for partially biodegradable polymer blends, PCL was also blended with another commonly used non-biodegradable polymer, i.e., PP at a greater mass ratio (80/20), and its NMP release behavior under the same ageing conditions was revealed. The prepared PCL/PP blend also showed a sea-island morphology and the dispersed PP particles were similarly released as NMPs into the buffer solution during the hydrolysis (Fig. S4†). The number of the NMP particles released was also large, giving the solution a milky white color (Fig. S4a†). Apart from the blends presented here, in an ongoing parallel investigation we have observed the release of a large amount of NMP particles from immiscible blends where the non-biodegradable particles consist of PE, PP or polystyrene and the biodegradable matrix consists of poly (vinyl alcohol), the latter having quite different chemistry and properties than PCL. These findings confirm the remarkable potential NMP emissions of partially biodegradable polymer blends with a sea-island morphology, regardless of the type of non-biodegradable polymer in the dispersed phase and the biodegradable matrix. Despite that only one aquatic environment (buffer solution) was used here, the release of the non-biodegradable dispersed particles would also occur in other environments where the matrix polymer is biodegradable, such as soil, industrial composting sites, freshwater, and seawater (as illustrated in Fig. 6). | ||
| Fig. 6 Illustration of the release of NMP particles from a plastic bag that is made of partially biodegradable polymer blends upon the biodegradation of the matrix polymers under different environmental conditions. | ||
Conclusions
This study demonstrated the possibility of a huge NMP emission from partially biodegradable polymer blends by taking PCL/PE and PCL/PP blends as examples (PE and PP being the most used plastics). The choice was made to represent the most common case of polymer blends, i.e., where the two polymers are immiscible and formed the commonly observed “sea-island” morphology. A “hole-wall” structure was formed on the surface during the hydrolysis of the PCL matrix, and the subsequent detachment of protruded walls during further surface erosion led to the release of small fragments as intermediate products. The fragmentation of the bulk sample finally occurred in the strongly thinned film after prolonged degradation, leading to the further release of fragments. At the same time, the dispersed non-biodegradable particles were gradually liberated and released during the erosion of the surrounding matrix polymer. The number of released NMPs was very large.The special sea-island morphology leads to that all dispersed polymer phases exist as numerous NMPs particles in polymer blends. As long as the NMP particles remain dispersed in the polymer matrix, they do not pose any problem, but the risk of NMP emission is particularly high if the surrounding polymer matrix degrades faster than the dispersed phase during environmental degradation. Examples for such polymer blends are (i) non-biodegradable polymers in a matrix of a biodegradable polymer (as revealed in this study), (ii) blends of biodegradable polymers of which the matrix polymer degrades faster, and (iii) blends of biodegradable polymers where the dispersed polymer is selectively not biodegradable in its habitat environment (for instance, PLA is biodegradable in industrial composting sites, but not in aquatic environments). Hence, the degree of difference in the degradability of the matrix polymer and dispersion polymer under different environments is a key factor in determining the emission of the dispersed NMPs from polymer blends. In addition, the NMP emission from partially biodegradable polymer blends could pose problems in the recycling of biodegradable polymers, because it is difficult to fully separate biodegradable polymers from other conventional non-biodegradable polymer wastes. Hence, a large amount of partially biodegradable polymer blends with potentially high NMP emissions are inadvertently incorporated into recycled polymer products. Our work strongly suggests that adding non-biodegradable polymers into biodegradable polymers should be avoided, as they not only reduce the degree of biodegradation, but also cause a high risk of substantial emission of NMPs.
Experimental section
Materials and sample preparation
PCL pellets with a trademark of Capa™ FB100 and a density of 1130 kg m−3 were purchased from Perstorp Holding, Sweden. Low-density PE having a trademark of FA6224 and a density of 922 kg m−3 and isotactic PP having a trademark of BB125MO and a density of 905 kg m−3 were supplied from Borealis, Sweden. Amano Lipase PS, from Burkholderia (≥30000 U g−1, optimum pH and temperature, pH 7.0 and 50 °C) was purchased from Merck, Sweden. Gibco™ phosphate-buffered saline (PBS) with a pH of 7.2 was purchased from ThermoFisher Scientific, Sweden. The formulation of PBS was 9000 mg L−1 of NaCl, 726 mg L−1 of Na2HPO4–7H2O, and 210 mg L−1 KH2PO4 according to the supplier.
PCL and PE were blended with a mass ratio of 90/10 in a twin-screw mini extruder (Xplore instruments, the Netherlands) with a recirculating channel at 160 °C with a screw rotation rate of 100 rpm for 5 min. PCL pellets were dried overnight in a ventilated oven at 55 °C before the melt blending. The extruded PCL/PE 90/10 blend filaments were chopped and pressed into 0.5 ± 0.05 mm thick films in a hot-press (Fontijne TP-400, the Netherland) with a force of 200 kN at 160 °C for 5 min. Film samples, weighing 0.10 ± 0.01 g with a rectangular shape (20 × 10 mm2), were cut from the pressed films using a scalpel. The prepared film samples were stored in a desiccator containing silica gel before any tests. PCL/PP blend film samples with a mass ratio of 80/20 were also prepared using the same methods but a different temperature of 180 °C was used for the extrusion and compression. The 90/10 and 80/20 mass ratios studied are commonly used in polymer blends.
Biodegradation test/enzymatic hydrolysis
The same biodegradation test used for neat PCL in previous work48 was also employed here to hydrolyze the PCL/PE and PCL/PP blend. In brief, one rectangular film sample was placed in a 35 ml capped glass vial containing 30 mL buffer-lipase enzymatic solution (lipase concentration: 0.5 mg mL−1). The glass vials were then placed/aged in an orbital incubator shaker (Thermo Scientific MaxQ™ 6000) at 40 °C and 200 rpm for up to 6 days. The aged film sample was taken out and placed in a new glass vial containing a new buffer-enzymatic solution after every second day of ageing to maintain the enzymatic activity. The solution pH was monitored with a pH meter (Mettler Toledo SevenCompact Duo S213) during the ageing. Glass vial samples with three replicates were collected after 1, 2, 4, and 6 days of ageing. The aged films were carefully removed from the solution, rinsed with Milli-Q water, and dried in a desiccator containing silica gel. The weight and thickness of the dried film samples were measured with a Mettler AE100 balance and Mitutoyo thickness gauge, respectively. The remaining glass vials containing the formed NMPs and buffer solution were collected and stored in a refrigerator at 4 °C before further tests.Characterizations
Ca. 100 μL buffer solution containing the formed NMPs was dropped on a glass slide and observed under a light microscope (Inverted Laboratory Microscope Leica DM IL LED). The 30 mL buffer NMP suspension was filtered using a polyvinylidene fluoride membrane with a pore size of 0.1 μm, followed by three washings with 30 mL Milli-Q water to remove the remaining enzyme on the membrane. The obtained membrane was first dried under ambient conditions and then in a silica gel desiccator. The weight of the membrane before and after the filtration and drying was recorded to calculate the mass of the collected NMPs. The NMPs collected on the membrane and the surface morphology of the aged films were observed in a field-emission scanning electron microscope (FE-SEM, Hitachi S-4800). Energy dispersive spectroscopy (EDS) data were collected from an 80 mm2 X-Max Large Area Silicon Drift Detector sensor (Oxford Instruments Nanotechnology) and were evaluated using AZtec INCA software. Before SEM analysis, the film samples were cryo-fractured in liquid nitrogen for cross-section observation and all samples were coated with palladium using an Agar high-resolution sputter coater, model 208RH. The Fourier Transform Infrared (FTIR) spectra of the collected NMPs and neat PE and PCL were recorded (32 scans, 4 cm−1 resolution) on a PerkinElmer Spotlight 400 (USA) equipped with a single attenuated total reflection accessory (Golden Gate, Graseby Specac, UK). A PerkinElmer Spotlight 400 FTIR imaging system equipped with a liquid-nitrogen-cooled mercury-cadmium-telluride detector was employed to measure the IR spectra of the individual particles (a more detailed description of the experiment is presented in the caption of Fig. S1†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Formas research council [grant no. 2016-01362 to HY and 2019-00388 to AB] and Vinnova [grant no. 2017-02712 to HY] is gratefully acknowledged.References
- N. B. Hartmann, T. Hüffer, R. C. Thompson, M. Hassellöv, A. Verschoor, A. E. Daugaard, S. Rist, T. Karlsson, N. Brennholt, M. Cole, M. P. Herrling, M. C. Hess, N. P. Ivleva, A. L. Lusher and M. Wagner, Environ. Sci. Technol., 2019, 53, 1039–1047 CrossRef CAS PubMed
.
- M. C. Rillig and A. Lehmann, Science, 2020, 368, 1430–1431 CrossRef CAS PubMed
- Y.-Q. Zhang, M. Lykaki, M. T. Alrajoula, M. Markiewicz, C. Kraas, S. Kolbe, K. Klinkhammer, M. Rabe, R. Klauer, E. Bendt and S. Stolte, Green Chem., 2021, 23, 5247–5271 RSC
- X. Z. Lim, Nature, 2021, 593, 22–25 CrossRef CAS PubMed
- C. D. Rummel, A. Jahnke, E. Gorokhova, D. Kühnel and M. Schmitt-Jansen, Environ. Sci. Technol. Lett., 2017, 4, 258–267 CrossRef CAS
- X.-F. Wei, T. Rindzevicius, K. Wu, M. Bohlen, M. Hedenqvist, A. Boisen and A. Hakonen, Chem. Eng. J., 2022, 442, 136117 CrossRef CAS
- I. V. Kirstein, F. Hensel, A. Gomiero, L. Iordachescu, A. Vianello, H. B. Wittgren and J. Vollertsen, Water Res., 2021, 188, 116519 CrossRef CAS PubMed
- D. Eerkes-Medrano, R. C. Thompson and D. C. Aldridge, Water Res., 2015, 75, 63–82 CrossRef CAS PubMed
- K. Min, J. D. Cuiffi and R. T. Mathers, Nat. Commun., 2020, 11, 1–11 CrossRef PubMed
- S. T. Sait, L. Sørensen, S. Kubowicz, K. Vike-Jonas, S. V. Gonzalez, A. G. Asimakopoulos and A. M. Booth, Environ. Pollut., 2021, 268, 115745 CrossRef CAS PubMed
- S. Ju, G. Shin, M. Lee, J. M. Koo, H. Jeon, Y. S. Ok, D. S. Hwang, S. Y. Hwang, D. X. Oh and J. Park, Green Chem., 2021, 23, 6953–6965 RSC
- J. Barrett, Z. Chase, J. Zhang, M. M. B. Holl, K. Willis, A. Williams, B. D. Hardesty and C. Wilcox, Front. Mar. Sci., 2020, 7, 576170 CrossRef
- A. Isobe, T. Azuma, M. R. Cordova, A. Cózar, F. Galgani, R. Hagita, L. D. Kanhai, K. Imai, S. Iwasaki, S. i. Kako, N. Kozlovskii, A. L. Lusher, S. A. Mason, Y. Michida, T. Mituhasi, Y. Morii, T. Mukai, A. Popova, K. Shimizu, T. Tokai, K. Uchida, M. Yagi and W. Zhang, Micropl. Nanopl., 2021, 1, 16 CrossRef
- L. Anagnosti, A. Varvaresou, P. Pavlou, E. Protopapa and V. Carayanni, Mar. Pollut. Bull., 2021, 162, 111883 CrossRef CAS PubMed
- C. F. Hunt, W. H. Lin and N. Voulvoulis, Nat. Sustain., 2021, 4, 366–372 CrossRef
- K. Zhang, A. H. Hamidian, A. Tubić, Y. Zhang, J. K. Fang, C. Wu and P. K. Lam, Environ. Pollut., 2021, 274, 116554 CrossRef CAS PubMed
- L. Lebreton, J. Van Der Zwet, J.-W. Damsteeg, B. Slat, A. Andrady and J. Reisser, Nat. Commun., 2017, 8, 1–10 CrossRef PubMed
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed
- O. S. Alimi, D. Claveau-Mallet, R. S. Kurusu, M. Lapointe, S. Bayen and N. Tufenkji, J. Hazard. Mater., 2022, 423, 126955 CrossRef CAS PubMed
- C. D. Rummel, H. Schäfer, A. Jahnke, H. P. H. Arp and M. Schmitt-Jansen, Anal. Bioanal. Chem., 2022, 414, 1469–1479 CrossRef CAS PubMed
- T. Kim, K. Park and J. Hong, J. Hazard. Mater., 2022, 424, 127630 CrossRef CAS PubMed
- G. Biale, J. La Nasa, M. Mattonai, A. Corti, V. Vinciguerra, V. Castelvetro and F. Modugno, Polymers, 2021, 13, 1997 CrossRef CAS PubMed
- F. Zha, M. Shang, Z. Ouyang and X. Guo, Gondwana Res., 2021, 108, 60–71 CrossRef
- A. K. Sarkar, A. E. Rubin and I. Zucker, Environ. Sci. Technol., 2021, 55, 10491–10501 CrossRef CAS PubMed
- M. Huber, V.-M. Archodoulaki, E. Pomakhina, B. Pukánszky, E. Zinöcker and M. Gahleitner, Polym. Degrad. Stab., 2022, 195, 109794 CrossRef CAS
- X.-F. Wei, M. Bohlén, C. Lindblad, M. Hedenqvist and A. Hakonen, Water Res., 2021, 198, 117123 CrossRef CAS PubMed
- T. Wang, Y. Ma and R. Ji, Bull. Environ. Contam. Toxicol., 2021, 107, 736–740 CrossRef CAS PubMed
- K. V. Thomas, Nat. Sustain., 2021, 1–3 Search PubMed
-
N. F. A. Zainal and C. H. Chan, in Compatibilization of Polymer Blends, ed. A. R. Ajitha and S. Thomas, Elsevier, 2020, pp. 391–433, DOI:10.1016/B978-0-12-816006-0.00014-1
-
A. Ajitha and S. Thomas, Compatibilization of polymer blends, Elsevier, 2020, pp. 1–29 Search PubMed
- X.-Q. Liu, Z.-Y. Sun, R.-Y. Bao, W. Yang, B.-H. Xie and M.-B. Yang, RSC Adv., 2014, 4, 41059–41068 RSC
- X.-F. Wei, F. Nilsson, H. Yin and M. S. Hedenqvist, Environ. Sci. Technol., 2021, 55, 4190–4193 CrossRef CAS PubMed
- C. DelRe, Y. Jiang, P. Kang, J. Kwon, A. Hall, I. Jayapurna, Z. Ruan, L. Ma, K. Zolkin, T. Li, C. D. Scown, R. O. Ritchie, T. P. Russell and T. Xu, Nature, 2021, 592, 558–563 CrossRef CAS PubMed
- K. L. Law and R. Narayan, Nat. Rev. Mater., 2022, 7, 104–116 CrossRef CAS
- S. Agarwal, Macromol. Chem. Phys., 2020, 221, 2000017 CrossRef CAS
- S. Kubowicz and A. M. Booth, Environ. Sci. Technol., 2017, 51, 12058–12060 CrossRef CAS PubMed
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310–6322 RSC
- V. Nagarajan, A. K. Mohanty and M. Misra, ACS Sustainable Chem. Eng., 2016, 4, 2899–2916 CrossRef CAS
-
L. Rajeshkumar, Biodegradable Polymers, Blends and Composites, Elsevier, 2022, pp. 527–549 Search PubMed
- S. Moradi and J. K. Yeganeh, Polym. Test., 2020, 91, 106735 CrossRef CAS
- H.-C. Zhang, B.-h. Kang, L.-S. Chen and X. Lu, Polym. Test., 2020, 87, 106521 CrossRef CAS
- M. Wu, Z. Wu, K. Wang, Q. Zhang and Q. Fu, Polymer, 2014, 55, 6409–6417 CrossRef CAS
- K. Hamad, M. Kaseem, Y. G. Ko and F. Deri, Polym. Sci., Ser. A, 2014, 56, 812–829 CrossRef CAS
- N. Mechbal and M. Bousmina, Rheol. Acta, 2004, 43, 119–126 CrossRef CAS
- X.-C. Xia, Q.-P. Zhang, L. Wang, J.-M. Feng and M.-B. Yang, Macromol. Chem. Phys., 2014, 215, 1146–1151 CrossRef CAS
- S. Bärwinkel, A. Seidel, S. Hobeika, R. Hufen, M. Mörl and V. Altstädt, Materials, 2016, 9, 659 CrossRef PubMed
- B. Laycock, M. Nikolić, J. M. Colwell, E. Gauthier, P. Halley, S. Bottle and G. George, Prog. Polym. Sci., 2017, 71, 144–189 CrossRef CAS
- X. F. Wei, A. J. Capezza, Y. Cui, L. Li, A. Hakonen, B. Liu and M. S. Hedenqvist, Water Res., 2022, 211, 118068 CrossRef CAS PubMed
- B. Smith, Spectroscopy, 2021, 36, 17–22 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc02388a |
| This journal is © The Royal Society of Chemistry 2022 |