
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyclohexanone ammoximation via in situ H2O2 production using TS-1 supported catalysts†
Richard J.
Lewis
 *a,
Kenji
Ueura
b,
Yukimasa
Fukuta
b,
Thomas E.
Davies
a,
David J.
Morgan
ac,
Charlie B.
Paris
d,
James
Singleton
a,
Jennifer. K.
Edwards
d,
Simon J.
Freakley
e,
Yasushi
Yamamoto
b and
Graham J.
Hutchings
*a
*a,
Kenji
Ueura
b,
Yukimasa
Fukuta
b,
Thomas E.
Davies
a,
David J.
Morgan
ac,
Charlie B.
Paris
d,
James
Singleton
a,
Jennifer. K.
Edwards
d,
Simon J.
Freakley
e,
Yasushi
Yamamoto
b and
Graham J.
Hutchings
*a
aMax Planck–Cardiff Centre on the Fundamentals of Heterogeneous Catalysis FUNCAT, Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: LewisR27@cardiff.ac.uk; Hutch@cardiff.ac.uk
bUBE Corporation, 1978-5, Kogushi, Ube, Yamaguchi 755-8633, Japan
cHarwellXPS, Research Complex at Harwell (RCaH) Didcot, OX11 0FA, UK
dCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK
eDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
First published on 9th September 2022
Abstract
The ammoximation of cyclohexanone to the corresponding oxime via in situ H2O2 formation offers an attractive alternative to the current industrial means of production, overcoming the significant economic and environmental concerns associated with the manufacture of a key reagent, H2O2. Herein we demonstrate the efficacy of a composite catalyst, consisting of precious metal nanoparticles supported on a commercial TS-1, towards the in situ synthesis of cyclohexanone oxime, bridging the wide condition gap that exists between the two distinct reaction pathways: H2O2 direct synthesis and cyclohexanone ammoximation. In particular, the alloying of Au with Pd and the introduction of low concentrations of Pt into AuPd nanoalloys are found to be key in promoting high catalytic performance. The improved catalytic activity of optimal catalysts is found to result from a combination of a disruption of contiguous Pd ensembles and the modification of Pd oxidation states, which in turn dictate catalytic activity towards the production and subsequent degradation of H2O2.
Introduction
The ammoximation of cyclohexanone to cyclohexanone oxime is a key process in the production of Nylon-6, with annual global production of the polyamide predicted to reach 8.9 million tons by 2024, a concurrent increase in the demand for cyclohexanone oxime is likewise expected.1 The application of the titanosilicate TS-1, first developed by EniChem,2 in conjunction with preformed H2O2 and NH3 overcame many of the drawbacks associated with the traditional route to oxime production, chiefly the formation of large quantities of the low-value fertilizer, ammonium sulphate. Key to the efficacy of the EniChem route is the high co-ordination ability of TiIV sites present within the TS-1 framework, which are considered responsible for the formation of the hydroxylamine intermediate from H2O2 and NH3, which subsequently reacts with cyclohexanone to produce the oxime with high selectivity.3Although the H2O2/TS-1 approach is able to achieve high selectivity towards cyclohexanone oxime, there are still significant improvements that can be achieved through decoupling the ammoximation process from the industrial means of H2O2 production, the anthraquinone oxidation process. Indeed, like all processes that utilise preformed H2O2 there are considerable economic and environmental costs associated with transport and storage, with the energy utilised in concentration of the oxidant effectively wasted upon dilution of the oxidant to appropriate levels prior to use.4 Additionally, the instability of H2O2 even at relatively mild temperatures requires the use of acidic stabilizing agents to prevent decomposition to H2O,5 with such promoters able to promote both decreased reactor and catalyst longevity as well as leading to the formation of complex product streams, adding significant cost to any industrial process.6
The direct synthesis of H2O2 from molecular H2 and O2 has been well reported in the literature, with AuPd-based catalysts in particular well studied,7–9 indeed there is growing interest in the use of such materials to generate H2O2in situ for the selective oxidation of a range of chemical feedstocks.10–16 However, such approaches have yet been unable to demonstrate a viable alternative to the existing industrial processes they aim to replace, often suffering from relatively low rates of conversion and selectivity based on H2.17 Additionally, the generation of considerable quantities of undesirable by-products, often driven by the presence of the H2 required to produce H2O2 is a cause for concern, with the utilisation of inhibitors to suppress competitive side reactions, which add further complexity to the process, typical.15,17
Recently we reported that through the in situ production of H2O2 from molecular H2 and O2 it is possible to catalyse the ammoximation of a range of ketones to the corresponding oxime, with product selectivities comparable to the current industrial route of production, while also achieving high utilisation of H2.18 This earlier investigation, focussed on a wet impregnation approach to catalyst synthesis, due in part to its simplicity and industrial applicability. However, such routes to catalyst preparation often fail to achieve high dispersion of active metals and in the case of bi-metallic systems can lead to incomplete alloy formation and inhomogeneity between nanoparticles.19,20 Furthermore, a significant focus of this earlier work was placed on the use of a physical mixture of TS-1 in conjunction with AuPd nanoparticles supported on a secondary carrier, however for industrial application a single-phase composite catalyst would clearly be preferential.
The solution phase formation of nanoalloys prior to their immobilisation onto a carrier has been widely demonstrated to offer enhanced control of particle size, shape and composition21–23 when compared to alternative routes to catalyst synthesis such as wet impregnation. Typically colloidal approaches involve the rapid chemical reduction of metal precursor species and the inhibition of particle agglomeration through the use of capping agents, with the application of such species, often reported to result in the modification of catalyst reactivity through steric effects and modification of metal oxidation states.23–25
With this in mind and in an attempt to better utilise precious metal sites and further improve on the rate of cyclohexanone ammoximation we have recently reported, within this current study we have evaluated the efficacy of TS-1 immobilised AuPd nanoparticles, synthesised by a facile sol-immobilisation technique,26 towards the in situ ammoximation of cyclohexanone to the corresponding oxime.
Experimental
Catalyst preparation
Mono-, bi- and tri-metallic 0.66%AuPdPt/TiO2 catalysts have been prepared (on a weight basis) by a sol-immobilisation procedure, based on a methodology previously reported in the literature,22 which has been shown to result in enhanced precious metal dispersion by limiting particle growth, while also allowing for improved interaction between active metal components.23,26 The procedure to produce the 0.275%Au–0.275%Pd–0.11%Pt/TS-1 catalyst (2 g) is outlined below with a similar methodology utilised for all catalysts.Aqueous solutions of HAuCl4·3H2O (0.449 mL, [Au] = 12.25 mg mL−1, Strem Chemicals), PdCl2 (0.917 mL, [Pd] = 6.0 mg mL−1, Merck) and H2PtCl6·3H2O (0.231 mL, [Pt] = 9.5 mg mL−1, Merck), were added to deionised water (800 mL) under vigorous stirring, at room temperature. The resulting solution was allowed to stir for 2 minutes prior to the addition of polyvinylalcohol (PVA) (1.584 mL, 1 wt.% MW = 9000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, 80% hydrolysed, Merck) such that the weight ratio of metal:PVA was 1:1.2, unless otherwise stated. The resulting solution was stirred for 2 minutes prior to the addition of a freshly prepared solution of NaBH4 (4.776 mL, 0.1 M, Merck), such that the molar ratio of NaBH4:(Au + Pd) was 5:1 and the molar ratio of NaBH4:Pt was 10:1. Upon the addition of NaBH4 the mixture turned dark brown and was stirred vigorously for an additional 30 minutes followed by the addition of TS-1 (1.987 g, HighChem). The solution was acidified to pH 1 via the addition of H2SO4 (>95%, Fischer Scientific) and allowed to stir for a further 1 h. The need for acidification of the catalyst synthesis solution can be related to the kinetics of nanoalloy immobilisation, through acidification it is possible to promote deposition of the PVA-encapsulated metal species and therefore good control over nanoparticle size. Following this, the suspension was filtered under vacuum, washed thoroughly with distilled water until the pH of the washings were neutral, then dried (110 °C, 16 h, static air). The resulting material was subsequently ground and calcined (400 °C, 3 h, 10 °C min−1, static air), unless otherwise stated.
000 g mol−1, 80% hydrolysed, Merck) such that the weight ratio of metal:PVA was 1:1.2, unless otherwise stated. The resulting solution was stirred for 2 minutes prior to the addition of a freshly prepared solution of NaBH4 (4.776 mL, 0.1 M, Merck), such that the molar ratio of NaBH4:(Au + Pd) was 5:1 and the molar ratio of NaBH4:Pt was 10:1. Upon the addition of NaBH4 the mixture turned dark brown and was stirred vigorously for an additional 30 minutes followed by the addition of TS-1 (1.987 g, HighChem). The solution was acidified to pH 1 via the addition of H2SO4 (>95%, Fischer Scientific) and allowed to stir for a further 1 h. The need for acidification of the catalyst synthesis solution can be related to the kinetics of nanoalloy immobilisation, through acidification it is possible to promote deposition of the PVA-encapsulated metal species and therefore good control over nanoparticle size. Following this, the suspension was filtered under vacuum, washed thoroughly with distilled water until the pH of the washings were neutral, then dried (110 °C, 16 h, static air). The resulting material was subsequently ground and calcined (400 °C, 3 h, 10 °C min−1, static air), unless otherwise stated.
The TS-1 utilised in this study was provided by HighChem and manufactured by Hengyi Chemical (Batch number 17052, manufacturer website: https://www.hengyichemical.com/product/36-en.html).
Catalyst testing
Note 1: Reaction conditions used within this study operate below the flammability limits of gaseous mixtures of H2 and O2.Note 2: The conditions used within this work for H2O2 synthesis and degradation have previously been investigated, with the presence of CO2 as a diluent for reactant gases and a methanol co-solvent identified as key to maintaining high catalytic efficacy towards H2O2 production.27
Note 3: In all cases reactions were run multiple times, over multiple batches of catalyst, with the data presented being an average of these experiments.
Direct synthesis of H2O2
Hydrogen peroxide synthesis was evaluated using a Parr Instruments stainless steel autoclave with a nominal volume of 100 mL, equipped with a PTFE liner and a maximum working pressure of 2000 psi. To test each catalyst for H2O2 synthesis, the autoclave was charged with catalyst (0.01 g), solvent (5.6 g MeOH and 2.9 g H2O, both HPLC grade, Fischer Scientific). The charged autoclave was then purged three times with 5%H2/CO2 (100 psi) before filling with 5% H2/CO2 (420 psi), followed by the addition of 25%O2/CO2 (160 psi) to give a H2:O2 ratio of 1:2. The temperature was then decreased to 2 °C (using a HAAKE K50 bath/circulator and an appropriate coolant) followed by stirring (1200 rpm) of the reaction mixture for 0.5 h. Gas pressures are given as gauge pressures, the reagents were not continuously introduced into the reactor. The above reaction parameters represent the optimum conditions we have previously used for the synthesis of H2O2.24 H2O2 productivity was determined by titrating aliquots of the final solution after reaction with acidified Ce(SO4)2 (0.01 M) in the presence of ferroin indicator. Catalyst productivities are reported as molH2O2 kgcat−1 h−1.
Degradation of H2O2
Catalytic activity towards H2O2 degradation (via hydrogenation and decomposition pathways) was determined in a manner similar to that used for measuring the H2O2 direct synthesis activity of a catalyst. The autoclave was charged with methanol (5.6 g, HPLC grade, Fisher Scientific), H2O2 (50 wt% 0.69 g, Merck), H2O (2.21 g, HPLC grade, Fisher Scientific) and catalyst (0.01 g), with the solvent composition equivalent to a 4 wt.% H2O2 solution. From the solution, two aliquots of 0.05 g were removed and titrated with acidified Ce(SO4)2 solution using ferroin as an indicator to determine an accurate concentration of H2O2 at the start of the reaction. The charged autoclave was then purged three times with 5%H2/CO2 (100 psi) before filling with 5%H2/CO2 (420 psi). Gas pressures are given as gauge pressures, the reagents were not continuously introduced into the reactor. The reaction mixture was cooled to 2 °C prior to the reaction commencing upon stirring (1200 rpm). The reaction was allowed to proceed for 0.5 h, after which, the catalyst was removed from the reaction solvents via filtration and as described previously, two aliquots of 0.05 g were titrated against the acidified Ce(SO4)2 solution using ferroin as an indicator. Catalyst degradation activity is reported as molH2O2 kgcat−1 h−1.Cyclohexanone ammoximation via the in situ synthesis of H2O2
Catalysts were evaluated for their activity towards ketone ammoximation with the procedure for cyclohexanone ammoximation outlined below using a stainless-steel autoclave (Parr Instruments) with a nominal volume of 100 mL, equipped with a PTFE liner and a maximum working pressure of 2000 psi.The autoclave was charged with the catalyst (0.075 g), solvent (H2O (7.5 g, HPLC grade, Fisher Scientific) and t-BuOH (5.9 g, Merck), cyclohexanone (0.196 g, 2.0 mmol, Merck) and ammonium bicarbonate (0.32 g, 4.0 mmol, Merck). With t-BuOH chosen as co-solvent due to enhanced solubility of H2, in comparison to H2O, and the ability of t-BuOH to aid in the maintenance of the –Ti(IV)–O–Si– moiety, considered to be responsible for the high activity of TS-1.25 The reactor was purged three times with 5% H2/N2 (100 psi) and then filled with 5% H2/N2 (420 psi) and 25% O2/N2 (160 psi) to give a H2:O2 ratio of 1:2. Reactant gas pressures are reported as gauge pressures, the reagent gases were not continuously introduced into the reactor. The reactor was stirred (100 rpm) while the reaction temperature was raised to 80 °C at which time stirring was increased to 800 rpm. The reaction was allowed to run for 3 h, unless otherwise stated, after which the reactor was cooled to 25 °C while stirring (100 rpm), using ice water. To the reaction solution ethanol (6 g, Fischer Scientific) and diethylene glycol monoethyl ether (0.15 g, Merck) were added, with the former used to ensure the complete homogeneity of the post-reaction solution, while the latter was chosen as an external standard. Following this the catalyst was removed by filtration and the resulting solution was analysed by gas chromatography using a Varian 3800 equipped with FID and a CP-Wax 52 CB column.
Ketone conversion and selectivity towards the oxime were calculated on the basis of starting amount of the ketone, according to eqn (1) and (2), respectively.
 | (1) |
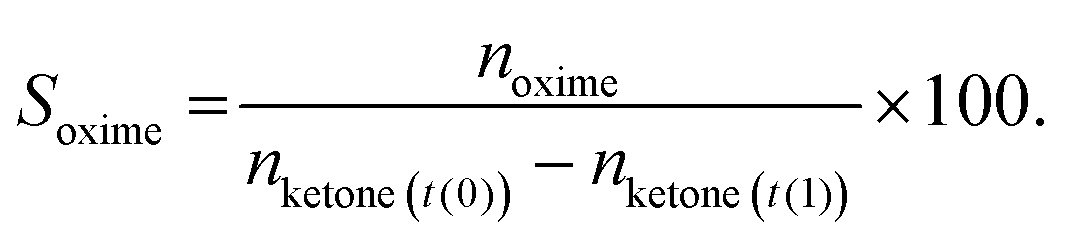 | (2) |
Catalyst characterisation
Investigation of the bulk structure of the crystalline materials was carried out using a (θ–θ) PANalytical X'pert Pro powder diffractometer using a Cu Kα radiation source, operating at 40 KeV and 40 mA. Standard analysis was carried out using a 40 min run with a back filled sample, between 2θ values of 10–80°. Phase identification was carried out using the International Centre for Diffraction Data (ICDD).X-ray diffractograms of the as-prepared 0.66%AuPd/TS-1 catalysts in Fig. S.1,† with no reflections associated with either Au or Pd observed, indicative of the low total loading and the high dispersion of the immobilised metals, which is typical of the route to catalyst synthesis.
N2 isotherms were collected on a Micromeritics 3Flex. Samples (ca. 0.020 g) were degassed (150 °C, 6 h) prior to analysis. Analyses were carried out at 77 K with P0 measured continuously. Free space was measured post-analysis with He. Pore size analysis was carried out using Micromeritics 3Flex software, N2-Cylindrical Pores-Oxide Surface DFT Model.
Note 4: Surface area analysis of the as received TS-1 and 0.33%Au–0.33%Pd/TS-1 (PVA:metal = 1.2) catalyst exposed to an oxidative heat treatment (static air, 400 °C, 3 h) is reported in Fig. S.2,† with a minor reduction in both surface area and pore volume observed with the immobilisation of precious metals onto the TS-1 support and subsequent exposure to an oxidative heat treatment.
X-ray photoelectron spectroscopy (XPS) analyses were made on a Kratos Axis Ultra DLD spectrometer. Samples were mounted using double-sided adhesive tape and binding energies were referenced to the C (1s) binding energy of adventitious carbon contamination that was taken to be 284.8 eV. Monochromatic AlKα radiation was used for all measurements; an analyser pass energy of 160 eV was used for survey scans while 40 eV was employed for detailed regional scans. The intensities of the Au(4f) and Pd(3d) features were used to derive the Au/Pd surface composition ratios. All transmission function corrected data was processed using CasaXPS v2.3.2 after removal of a Shirley background using modified Wagner sensitivity factors as supplied by the instrument manufacturer. Fitting of the spectra was performed using Voigt functions the parameters, which were derived from bulk materials.
Transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM) were performed on a JEOL JEM-2100 operating at 200 kV. Samples were prepared by dispersion in ethanol via sonication and deposited on 300 mesh copper grids coated with holey carbon film.
Thermal gravimetric analysis (TGA) was performed using a PerkinElmer TGA 4000. Catalytic samples (ca. 20–30 mg) were loaded into a ceramic crucible and then heated to 800 °C (5 °C min−1) under a flow of air (50 mL min−1).
Metal leaching was quantified using an Agilent 7900 ICP-MS equipped with an I-AS auto-sampler using a 5-point calibration using certified reference materials from PerkinElmer and certified internal standard from Agilent. All calibrants were matrix matched.
Results and discussion
Our initial studies established the efficacy of TS-1 supported precious metal catalysts towards the direct synthesis of H2O2 and its subsequent degradation, via hydrogenation and decomposition pathways, under conditions previously optimised for H2O2 synthesis. These include the presence of sub-ambient reaction temperatures, a methanol co-solvent and CO2 gaseous diluent, all of which have been shown to inhibit H2O2 degradation pathways and promote H2O2 stability (Table S.1†).27 In keeping with previous investigations,9,28–30 the incorporation of Au into supported Pd nanoparticles, so that Au and Pd content were equal on a weight basis, was observed to lead to a significant enhancement in catalytic activity towards H2O2 synthesis (104 molH2O2 kgcat−1 h−1) compared to the analogous monometallic Au- (9 molH2O2 kgcat−1 h−1) and Pd-only (12 molH2O2 kgcat−1 h−1) catalysts. The synergistic enhancement that results from the alloying Au and Pd has been well reported,31–33 with numerous studies identifying the ability of Au to promote the desorption of H2O2 as well as reaction intermediates (*OH and *OOH) from catalytic surfaces.33,34 Notably the activity of the 0.33%Au–0.33%Pd/TS-1 catalyst investigated within this study significantly exceeds that recently reported for the analogous catalyst formulation prepared via a wet co-impregnation procedure (44 molH2O2 kgcat−1 h−1) highlighting a key benefit of the sol-immobilisation route to catalyst synthesis.18Subsequent evaluation of catalytic performance towards cyclohexanone ammoximation, under conditions considered to be detrimental towards H2O2 production, namely the presence of ammonia and elevated temperatures,35 revealed the limited activity of both the monometallic Au and Pd catalysts towards cyclohexanone oxime production (3 and 13% oxime yield respectively). In comparison, the 0.33%Au–0.33%Pd/TS-1 catalyst (66% oxime yield) was observed to be far superior to the monometallic analogues (Fig. 1), which, alongside our studies into the direct synthesis of H2O2 (Table S.1†) highlights the key role of AuPd nanoalloy formation.
 | ||
| Fig. 1 Catalytic activity of 0.66%AuPd/TS-1 catalysts prepared via sol-immobilisation towards the ammoximation of cyclohexanone via in situ production of H2O2. Ammoximation reaction conditions: cyclohexanone (2 mmol), NH4HCO3 (4 mmol), 5%H2/N2 (420 psi), 25%O2/N2 (160 psi), catalyst (0.075 g), t-BuOH (5.9 g), H2O (7.5 g), 3 h, 80 °C 800 rpm. Key: cyclohexanone conversion (black bar), selectivity towards oxime (red bar), cyclohexanone oxime yield (blue bar), carbon balance (crosses). Note: all catalysts were exposed to an oxidative heat treatment prior to use (static air, 400 °C, 3 h). | ||
The disruption of contiguous Pd ensembles through the alloying with Au has been widely considered a major underlying cause for the synergy that often results from the combination of these two metals, although other factors, such as the control of Pd oxidation state and nanoparticle morphology clearly also play a role in altering catalytic performance.36–39 Pertinent to H2O2 production, Goodman and co-workers have reported the increased selectivity toward O–O bond maintenance (necessary for the formation of H2O2), which can be achieved through the formation of AuPd alloys, with the combination of electronic perturbation and isolation of Pd sites considered to make Pd more “atom-like”, and subsequently results in the weaker binding of reactants and products.40,41 In order to achieve optimal catalytic performance, there is a clear need to balance the dissociation of H2, favoured over Pd ensembles, with the stability of *O2 and *OOH intermediate surface species, favoured over AuPd alloy surfaces.42
We subsequently investigated the effect of the Au:Pd ratio on catalytic activity towards both the direct synthesis of H2O2 (Fig. S.3†) and cyclohexanone ammoximation (Fig. 2, with determination of apparent reaction rates reported in Table S.2†). These studies revealed the enhanced activity of the Au-rich 0.44%Au–0.22%Pd/TS-1 catalyst, which offered a H2O2 synthesis activity of 115 molH2O2 kgcat−1 h−1 and cyclohexanone oxime yield of 76%, with both metrics decreasing with the further introduction of Pd. Such a correlation perhaps unsurprisingly may be considered indicative of the direct relationship between the two separate processes (H2O2 synthesis and cyclohexanone ammoximation).
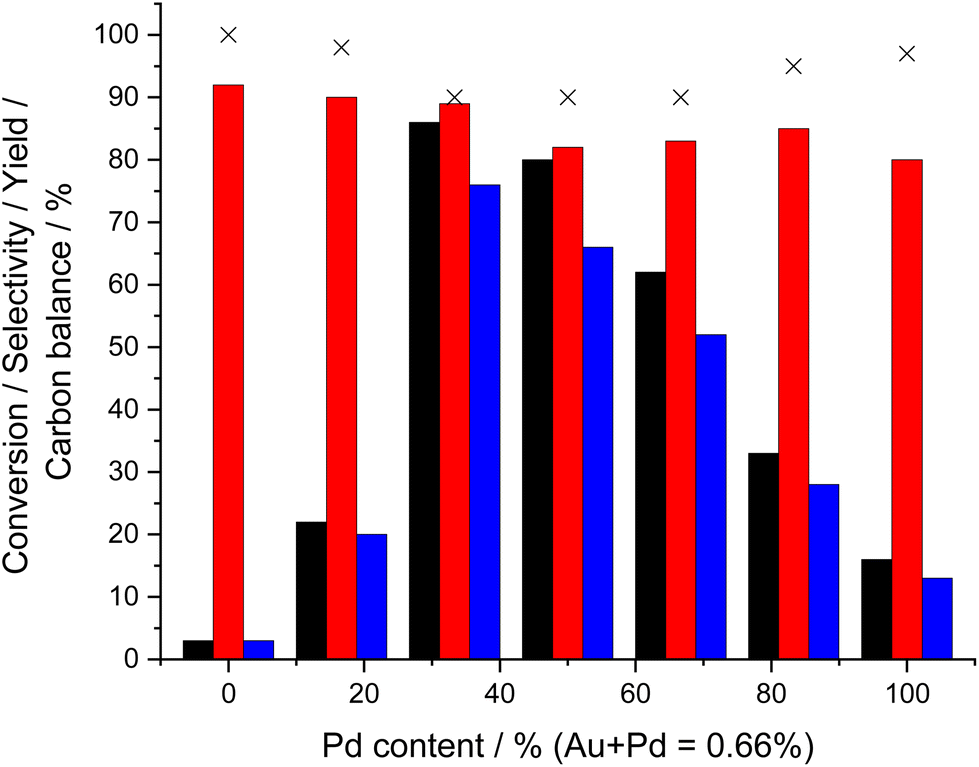 | ||
| Fig. 2 The effect of Au:Pd ratio on catalytic activity towards the ammoximation of cyclohexanone via in situ H2O2 production. Ammoximation reaction conditions: cyclohexanone (2 mmol), NH4HCO3 (4 mmol), 5%H2/N2 (420 psi), 25%O2/N2 (160 psi), catalyst (0.075 g), t-BuOH (5.9 g), H2O (7.5 g), 3 h, 80 °C 800 rpm. Key: cyclohexanone conversion (black bar), selectivity towards oxime (red bar), cyclohexanone oxime yield (blue bar), carbon balance (crosses). Note: all catalysts were exposed to an oxidative heat treatment prior to use (static air, 400 °C, 3 h). | ||
Determination of mean particle size via transmission electron microscopy (TEM) revealed that differences in catalytic performance cannot be attributed to variation in this metric, which is an often-key factor in determining catalytic selectivity towards H2O2.43–45 Indeed, good control of particle distribution was observed regardless of nominal catalyst formulation (mean particle size reported in Table 1, with corresponding micrographs reported in Fig. S.4.A and B†), which may have been expected given the chosen route to catalyst synthesis.23 Analysis of the as-prepared materials via X-ray photoelectron spectroscopy (XPS) (Table S.3†), revealed the presence of Pd0 in all catalysts, despite exposure to an oxidative heat treatment (static air, 400 °C, 3 h). Notably, the Au-rich materials (0.55%Au–0.11%Pd/TS-1 and 0.44%Au–0.22%Pd/TS-1), were found to consist entirely of Pd0, highlighting the ability of Au to influence Pd speciation. With the known ability of Pd0 to be highly active toward H2O2 synthesis we can attribute the modification of Pd oxidation state to be a key cause of the catalytic performance towards both H2O2 production and cyclohexanone ammoximation.
:Pd ratio on the mean particle size of 0.66%AuPd/TS-1 catalysts, prepared via sol-immobilisation (PVA:metal = 1.2)
| Catalyst | Mean particle size/nm (S.D) | Productivitya/molH2O2 kgcat−1 h−1 | Oxime yieldb/% |
|---|---|---|---|
| a H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 2 °C 1200 rpm. b Ammoximation reaction conditions: cyclohexanone (2 mmol), NH4HCO3 (4 mmol), 5%H2/N2 (420 psi), 25%O2/N2 (160 psi), catalyst (0.075 g), t-BuOH (5.9 g), H2O (7.5 g), 3 h, 80 °C 800 rpm. Note: all catalysts were exposed to an oxidative heat treatment prior to use (static air, 400 °C, 3 h). | |||
| 0.66%Au/TS-1 | 3.4 (0.93) | 9 | 3 |
| 0.55%Au–0.11%Pd/TS-1 | 3.5 (0.79) | 20 | 20 |
| 0.44%Au–0.22%Pd/TS-1 | 3.4 (0.82) | 115 | 76 |
| 0.33%Au–0.33%Pd/TS-1 | 3.4 (1.10) | 104 | 66 |
| 0.22%Au–0.44%Pd/TS-1 | 3.4 (0.80) | 86 | 52 |
| 0.11%Au–0.55%Pd/TS-1 | 2.7 (0.70) | 63 | 28 |
| 0.66%Pd/TS-1 | 3.5 (1.08) | 12 | 13 |
The role of the capping agent on the performance of the 0.33%Au–0.33%Pd/TS-1 catalyst towards both the direct synthesis of H2O2 and in situ ammoximation of cyclohexanone was next investigated (Table S.4† and Fig. 3 respectively), with numerous studies demonstrating the ability of such ligands to influence catalytic efficiency through control of the three-dimensional environment of the metal active site.46,47 With catalytic activity towards H2O2 synthesis widely reported to be dependent on particle size43,44,48 it is perhaps understandable that a decrease in this metric upon PVA incorporation during catalyst preparation (Table 2, with corresponding micrographs reported in Fig. S.5†), correlates with a considerable improvement in catalytic performance towards oxime production, with mean nanoparticle size decreasing from 6.7 nm for the catalyst prepared in the absence of PVA (44% oxime yield), to 2.9 nm after PVA introduction (PVA:metal = 0.6) (56% oxime yield). Interestingly, the further addition of PVA during colloid formation was found to result in a significant increase in catalytic activity towards both the direct synthesis of H2O2 and the in situ ammoximation of cyclohexanone, with the enhanced rate of cyclohexanone conversion particularly notable (100% cyclohexanone conversion and 89% oxime yield observed over the 0.66%AuPd/TS-1 (PVA:metal = 5) catalyst). However, this improvement in catalyst performance does not coincide with a further reduction in mean particle size, which may have been considered the cause for such promotion, with this metric remaining fairly stable (2.5–3.4 nm) for all catalysts prepared in the presence of PVA. In keeping with TEM analysis of this sub-set of materials, further investigation by XPS revealed a broadening of the Au 4f signal with the introduction of PVA, which is indicative of the formation of smaller nanoparticles.49,50 Additionally, and perhaps more importantly a greater proportion of Pd0 was also observed with the introduction of increasing amounts of PVA (Fig. S.6†). As such it is possible to attribute the enhanced H2O2 synthesis and ammoximation activity of the AuPd/TS-1 catalysts prepared in the presence of the PVA capping agent to result from both a reduction in mean nanoparticle size and, in a manner to our earlier studies into Au:Pd ratio, a shift in Pd oxidation state towards Pd0.
 | ||
| Fig. 3 Effect of PVA:metal ratio on catalytic activity of the 0.33%Au–0.33%Pd/TS-1 catalyst activity towards the ammoximation of cyclohexanone oxime via in situ H2O2 synthesis. Ammoximation reaction conditions: cyclohexanone (2 mmol), NH4HCO3 (4 mmol), 5%H2/N2 (420 psi), 25%O2/N2 (160 psi), catalyst (0.075 g), t-BuOH (5.9 g), H2O (7.5 g), 3 h, 80 °C 800 rpm. Key: cyclohexanone conversion (black bar), selectivity towards oxime (red bar), cyclohexanone oxime yield (blue bar), carbon balance (crosses). Note: all catalysts were exposed to an oxidative heat treatment prior to use (static air, 400 °C, 3 h). | ||
:metal ratio on the mean particle size of 0.33%Au–0.33%Pd/TS-1 catalysts, prepared via sol-immobilisation
| PVA:metal |
Mean particle size/nm (S.D) | Productivitya/molH2O2 kgcat−1 h−1 | Oxime yieldb/% |
|---|---|---|---|
| a H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5%H2/CO2 (420 psi), 25%O2/CO2 (160 psi), 0.5 h, 2 °C, 1200 rpm. b Ammoximation reaction conditions: cyclohexanone (2 mmol), NH4HCO3 (4 mmol), 5%H2/N2 (420 psi), 25%O2/N2 (160 psi), catalyst (0.075 g), t-BuOH (5.9 g), H2O (7.5 g), 3 h, 80 °C, 800 rpm. Note: all catalysts were exposed to an oxidative heat treatment prior to use (static air, 400 °C, 3 h). | |||
| 0 | 6.7 (2.51) | 56 | 44 |
| 0.6 | 2.9 (0.74) | 82 | 56 |
| 1.2 | 3.4 (1.10) | 104 | 66 |
| 2.4 | 2.5 (0.77) | 136 | 72 |
| 5.0 | 2.6 (0.72) | 165 | 89 |
The utilisation of organic capping agents to both direct nanoparticle morphology and prevent agglomeration is common for colloidal routes to catalyst synthesis. However, in some cases, the resulting organic layer which encapsulates the nanoalloy may inhibit catalyst performance by limiting active site accessibility.51,52 Notably, this has not been observed for the direct synthesis of H2O2, where sol-immobilisation prepared catalysts have been widely studied.22,47 For those reactions where the presence of the ligand can have deleterious effects, there are many elegant approaches utilised to ensure their removal.52 Thermal treatment is perhaps the most obvious and can be highly effective even at relatively low temperatures,53 although both the resulting carbonaceous residues54 and nanoparticle agglomeration can drastically reduce the performance of the resulting material, particularly in the case of monometallic Pd catalysts.55 Regardless, the application of thermal treatment is often necessitated to promote catalytic stability through the promotion of strong metal–support interactions (SMSI), which inhibits the leaching of active metals in liquid phase application.56 Notably in the case of oxide supports SMSI have been attributed to the development of an oxide overlayer, which partially covers metal nanoparticles.57
Subsequently, with a focus on the 0.33%Au–0.33%Pd/TS-1 (PVA:metal = 1.2) catalyst, the role of calcination temperature on catalytic performance was next investigated. In keeping with previous studies into AuPd nanoparticles immobilised on a range of supports58–60 the dried-only material (110 °C, 16 h, static air), offered particularly high activity towards both the direct synthesis (212 molH2O2 kgcat−1 h−1) and subsequent degradation (637 molH2O2 kgcat−1 h−1) of H2O2 (Table S.5†). A minimum calcination temperature of 400 °C was identified to impart catalyst stability, under H2O2 direct synthesis conditions (Table S.5†), with earlier studies also revealing the poor stability of AuPd/TS-1 materials calcined at lower temperatures.60 Perhaps as expected the application of increasing calcination temperatures was also observed to coincide with a reduction in H2O2 direct synthesis activity.
Subsequent analysis of the post-reaction solution revealed significant leaching of Pd after use in the H2O2 direct synthesis reaction, particularly in the case of the dried-only sample, with this metric decreasing substantially upon exposure to high-temperature calcination (>400 °C) (Table S.6†). By comparison, the extent of Au leaching observed was considerably less and decreased to zero upon exposure to relatively low heat treatment temperatures (>200 °C). Similar observations have been reported for AuPd-supported catalysts for a range of reactions, including H2O2 direct synthesis, with the improved stability which results from high-temperature calcination attributed to enhanced metal–support interactions,56,58 and aligns well with our observations within this study.
Evaluation of catalytic performance towards the in situ ammoximation of cyclohexanone (Fig. 4), revealed that despite offering high H2O2 synthesis rates the activity of the dried-only material toward the ammoximation reaction was limited (46% cyclohexanone conversion), while exposure to elevated calcination temperatures improved this metric considerably.
 | ||
| Fig. 4 Effect of calcination temperature on catalytic activity of the 0.33%Au–0.33%Pd/TS-1 catalyst activity towards the ammoximation of cyclohexanone oxime via in situ H2O2 synthesis. Ammoximation reaction conditions: cyclohexanone (2 mmol), NH4HCO3 (4 mmol), 5%H2/N2 (420 psi), 25%O2/N2 (160 psi), catalyst (0.075 g), t-BuOH (5.9 g), H2O (7.5 g), 3 h, 80 °C 800 rpm. Key: cyclohexanone conversion (black bar), selectivity towards oxime (red bar), cyclohexanone oxime yield (blue bar), carbon balance (crosses). Note: the dried-only material was exposed to low-temperature oxidative heat treatment (110 °C, 16 h, static air) prior to use. | ||
In an attempt to identify the underlying relationship between catalyst performance and calcination temperature the dried-only material was subsequently analysed by thermal gravimetric analysis (TGA) (Fig. S.7†), which revealed two distinct regions of interest (0–200 and 200–800 °C). The initial mass loss observed below 200 °C can be associated with the removal of physisorbed water,61 while that observed at temperatures exceeding 200 °C can be attributed to the thermal decomposition of the PVA capping agent. While such analysis may indicate that the relatively limited activity of the dried-only sample towards cyclohexanone ammoximation can be associated with the presence of the organic overlayer, possibly limiting reagent availability at metal active sites, it is important to note the high activity of the catalyst towards H2O2 production. As such it is considered that the presence of the ligand is not the underlying cause for the limited activity of the dried-only material.
We have recently reported that the formation of AuPd alloys is crucial in achieving both high catalytic performance and improved stability during application in the in situ ammoximation of cyclohexanone.18 Notably, in this previous study the AuPd/TS-1 catalyst, which was synthesised via a wet impregnation process, displayed a degree of nanoparticle inhomogeneity, typical of the route to catalyst preparation.10 These concerns around poor control of nanoparticle composition can be largely avoided through colloidal approaches to catalyst synthesis and it can be considered that AuPd alloys are the predominant species present within the 0.33%Au–0.33%Pd/TS-1 (PVA:metal = 1.2) catalyst, regardless of calcination temperature. However, one could also expect the presence of a minority of elementally segregated Au- and Pd-only species, although to a far lesser extent than that observed in the impregnation prepared analogue. As such, and informed by the observation of metal leaching during the H2O2 direct synthesis reaction (Table S.6†), we consider that it is possible to attribute the limited ammoximation activity of the dried-only catalyst to a combination of metal leaching and incomplete alloy formation. However, further study is still required to determine the extent to which each factor is responsible for the observed reactivity.
Evaluation of the catalytic series by TEM (Table 3, with corresponding micrographs reported in Fig. S.8†), revealed a relatively minor increase in mean particle size upon exposure to high-temperature calcination, which is indicative of the strong interaction between the nanoalloys and catalyst support, under non-reaction conditions, and is in keeping with our evaluation of AuPd/TiO2 catalysts, prepared by a similar synthesis route.59 However, it is important to note that due to limits of detection it was not possible to rule out the presence of atomic species or metal clusters and indeed the presence of such species may be expected given the high rates of H2O2 degradation observed, particularly over the dried material. Perhaps as expected given the oxidative heat treatment utilised, XPS analysis demonstrates a shift in Pd speciation from Pd0 in the as-prepared, dried catalyst towards a mixed Pd0–Pd2+ state in those materials exposed to calcination (Table S.7†). This coincides with a significant decrease in catalytic activity towards H2O2 degradation (Table S.5†), notably the H2O2 degradation rate of the dried only catalyst is exceptionally high under conditions optimised for H2O2 selectivity (637 molH2O2 kgcat−1 h−1), with this metric decreasing substantially upon calcination. As such it is possible to consider that while catalytic activity towards H2O2 production is a key metric in oxime production there is also a need to minimise competitive H2O2 degradation pathways which involve precious metal nanoalloys. This is likely to be particularly pertinent if the ability of the TS-1 component to utilise H2O2 in the formation of hydroxylamine limits overall process efficiency.
:metal = 1.2)
| Calcination temperature/°C | Mean particle size/nm (S.D) | Productivitya/molH2O2 kgcat−1 h−1 | Oxime yieldb/% |
|---|---|---|---|
| a H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5%H2/CO2 (420 psi), 25%O2/CO2 (160 psi), 0.5 h, 2 °C, 1200 rpm. b Ammoximation reaction conditions: cyclohexanone (2 mmol), NH4HCO3 (4 mmol), 5%H2/N2 (420 psi), 25%O2/N2 (160 psi), catalyst (0.075 g), t-BuOH (5.9 g), H2O (7.5 g), 3 h, 80 °C, 800 rpm. c Dried only sample exposed to 110 °C, 16 h, static air. | |||
| Dried onlyc | 2.5 (0.84) | 212 | 46 |
| 200 | 3.4 (1.09) | 176 | 76 |
| 300 | 3.5 (1.02) | 138 | 68 |
| 400 | 3.4 (1.10) | 104 | 66 |
| 500 | 3.5 (0.98) | 98 | 54 |
The inclusion of dopant concentrations of transition metals, in particular Pt, into supported Pd and AuPd catalysts has been reported to greatly enhance catalytic performance towards a range of chemical transformations,62,63 including the direct synthesis of H2O2.22,64–67 With these earlier works in mind, we subsequently investigated the effect of Pt incorporation on catalytic performance. A considerable increase in H2O2 synthesis rate was observed upon the inclusion of relatively low concentrations of Pt (0.11 wt%) into the 0.66%AuPd/TS-1 catalyst (135 molH2O2 kgcat−1 h−1), coinciding with a decrease in H2O2 degradation rate (172 molH2O2 kgcat−1 h−1) (Table S.8†). Correlating well with catalytic trends observed for H2O2 direct synthesis, the 0.275%Au–0.275%Pd–0.11%Pt/TS-1 catalyst was found to offer improved yields of cyclohexanone oxime (76%), with this metric decreasing substantially with further Pt incorporation (Fig. 5). Again, this highlights the close relationship between catalyst activity towards H2O2 production and the ammoximation of cyclohexanone. The enhanced activity of the optimal AuPdPt/TS-1 catalyst is further highlighted through comparison of apparent reaction rates (Table S.9†).
 | ||
| Fig. 5 Effect of Pt incorporation on the catalytic activity of 0.66%AuPd/TS-1 catalysts towards the ammoximation of cyclohexanone oxime via in situ H2O2 synthesis. Ammoximation reaction conditions: cyclohexanone (2 mmol), NH4HCO3 (4 mmol), 5%H2/N2 (420 psi), 25%O2/N2 (160 psi), catalyst (0.075 g), t-BuOH (5.9 g), H2O (7.5 g), 3 h, 80 °C 800 rpm. Key: cyclohexanone conversion (black bar), selectivity towards oxime (red bar), cyclohexanone oxime yield (blue bar), carbon balance (crosses). Note: all catalysts were exposed to oxidative heat treatment prior to use (static air, 400 °C, 3 h). | ||
Earlier works investigating the promotive effects of Pt incorporation into AuPd nanoalloys have typically focussed on a wet co-impregnation route to catalyst synthesis60 and as such it has been difficult to determine the key parameters responsible for the enhancement in activity upon the introduction of the dopant, with the modification of Pd oxidation states and changes in nanoparticle size proposed as possible causes for the observed improvement in catalytic performance. However, owing to the increased control offered by the sol-immobilisation route to catalyst synthesis, it is possible to rule out the latter as the cause for the observed catalytic trends, with negligible changes in mean particle size observed across the catalytic series (Table 4, with corresponding micrographs reported in Fig. S.9†).
:metal = 1.2)
| Catalyst | Mean particle size/nm (S.D) | Productivitya/molH2O2 kgcat−1 h−1 | Oxime yieldb/% |
|---|---|---|---|
| a H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5%H2/CO2 (420 psi), 25%O2/CO2 (160 psi), 0.5 h, 2 °C, 1200 rpm. b Ammoximation reaction conditions: cyclohexanone (2 mmol), NH4HCO3 (4 mmol), 5%H2/N2 (420 psi), 25%O2/N2 (160 psi), catalyst (0.075 g), t-BuOH (5.9 g), H2O (7.5 g), 3 h, 80 °C, 800 rpm. Note: all catalysts were exposed to an oxidative heat treatment prior to use (static air, 400 °C, 3 h). | |||
| 0.33%Au–0.33%Pd/TS-1 | 3.4 (1.10) | 104 | 66 |
| 0.275%Au–0.275%Pd–0.11%Pt/TS-1 | 2.7 (0.66) | 135 | 76 |
| 0.22%Au–0.22%Pd–0.22%Pt/TS-1 | 3.0 (0.98) | 106 | 60 |
| 0.11%Au–0.11%Pd–0.44%Pt/TS-1 | 3.1 (0.75) | 72 | 39 |
| 0.66%Pt/TS-1 | 3.1 (1.25) | 58 | 20 |
Analysis of the as-prepared, 0.66%AuPdPt/TS-1 catalysts by XPS is reported in Fig. 6. Upon the introduction of Pt the surface Pd:Au ratio was found to remain largely unchanged, however, perhaps more interestingly a considerable increase in Pd0 content was observed with the introduction of low concentrations of the dopant, which coincides with an enhancement in catalytic activity towards H2O2 (Table S.8†) and production of cyclohexanone oxime (Fig. 5), with Pd0 content continuing to increase upon further Pt incorporation. Again, in keeping with our earlier studies within this work, these observations clearly highlight the inherent relationship between H2O2 production and cyclohexanone ammoximation.
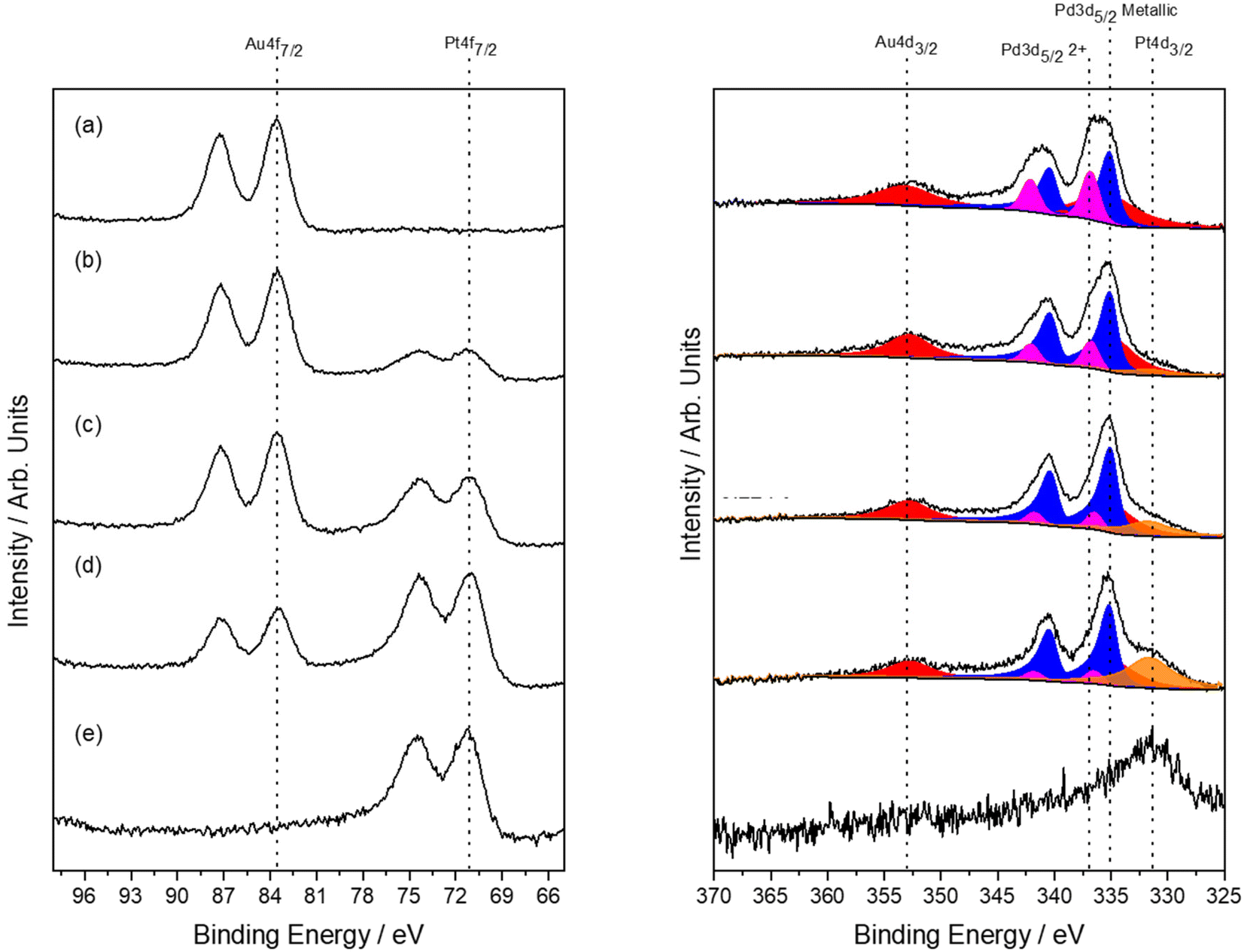 | ||
| Fig. 6 XPS Au(4f)/Pt(4f) and Pd(3d)/Au(4d) spectra for as-prepared 0.66%AuPdPt/TS-1 catalysts, prepared by sol-immobilisation (PVA:metal = 1.2). (a) 0.33%Au–0.33%Pd/TS-1, (b) 0.275%Au–0.275%Pd–0.11%Pt/TS-1, (c) 0.22%Au–0.22%Pd–0.22%Pt/TS-1 (d) 0.11%Au–0.11%Pd–0.44%Pt/TS-1 and (e) 0.66%Pt/TS-1. Note: all catalysts were exposed to an oxidative heat treatment prior to use (static air, 400 °C, 3 h). | ||
While the catalysts developed within this study have been demonstrated to offer high yields of cyclohexanone oxime, the need to improve catalytic performance, in particular selectivity towards the desired product is clear. With the comparatively high H2O2 degradation rates observed over the precious metal-loaded catalysts identified, under conditions considered optimal for H2O2 stability, the unselective catalytic formation of H2O from H2O2via hydrogenation and decomposition pathways must also be considered a key factor in overall ammoximation process efficiency. Indeed, in order to rival the current route to cyclohexanone oxime production the selective utilisation of H2 is a key parameter to consider and may be achieved through better balancing rates of H2O2 synthesis, over the active metal surfaces, and the subsequent utilisation of the synthesised H2O2 in the formation of the hydroxylamine intermediate. Returning to the 0.33%Au–0.33%Pd/TS-1 (PVA:metal = 1.2) catalyst, additional bare TS-1 (0.075 g) was used in conjunction with the metal-loaded catalyst, with a resulting improvement in overall efficacy (oxime yield = 93%) compared to that observed when using AuPd/TS-1 alone (oxime yield = 66%) (Fig. S.10†). We consider that this highlights the need for the iterative development of both catalytically active centres (i.e., the metal nanoparticles and TiIV within the TS-1 framework), and may direct attention towards the utilisation and development of titanosilicates with more readily accessible TiIV centres than those within TS-1.68–71
Finally, the relationship between catalytic performance towards the direct synthesis of H2O2 and in situ cyclohexanone ammoximation was compared, with a strong correlation between the two distinct reaction pathways observed (Fig. 7). Such observations may direct future attention towards the improvement of catalyst activity towards H2O2, potentially at the cost of selectivity. However, our studies have also revealed the need to minimise competitive H2O2 degradation pathways while also ensuring the efficacy of the overall ammoximation process does not become limited by the ability of the TS-1 component to synthesise hydroxylamine.
 | ||
| Fig. 7 The relationship between catalytic activity towards the direct synthesis of H2O2 and the in situ ammoximation of cyclohexanone. H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5%H2/CO2 (420 psi), 25%O2/CO2 (160 psi), 0.5 h, 2 °C, 1200 rpm. Ammoximation reaction conditions: cyclohexanone (2 mmol), NH4HCO3 (4 mmol), 5%H2/N2 (420 psi), 25%O2/N2 (160 psi), catalyst (0.075 g), t-BuOH (5.9 g), H2O (7.5 g), 3 h, 80 °C, 800 rpm. Key: 0.66%AuPd/TS-1 catalysts with varying Au:Pd ratio (black squares), varying the calcination temperature of the 0.33%Au–0.33%Pd/TS-1(PVA:metal = 1.2) catalyst (red circles), varying the PVA:metal ratio of the 0.33%Au–0.33%Pd/TS-1 catalysts (blue triangles), varying Pt content in 0.66%AuPdPt/TS-1 catalysts (green diamonds). | ||
Conclusion
The in situ synthesis of H2O2 from molecular H2 and O2 is demonstrated to offer an efficient route to the formation of cyclohexanone oxime, a major precursor to Nylon-6. With a focus on a composite TS-1 based catalyst, prepared via a sol-immobilisation protocol the need to improve both H2O2 synthesis rates through control of the precious metal component, in addition to the ability of the titanosilicate to readily utilise the synthesised H2O2 is identified. In particular, the role of alloying Pd with Au and the introduction of relatively small concentrations of Pt into AuPd nanoparticles are both revealed as measures which promote the synthesis of H2O2 and in situ production of the oxime.Author contributions
R.J.L, K.U and C.B.P conducted catalyst synthesis, testing, and data analysis. R.J.L, T.E.D, D.J.M and C.B.P conducted catalyst characterisation and corresponding data processing. R.J.L, K.U, Y.F, J.S, J.K.E, S.J.F, Y.Y, and G.J.H contributed to the design of the study and provided technical advice and result interpretation. R.J.L wrote the manuscript and supporting information, with all authors commenting on and amending both documents. All authors discussed and contributed to this work.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors wish to thank UBE Corporation for financial support. R. J. L and G. J. H acknowledge the Max Planck Centre for Fundamental Heterogeneous Catalysis (FUNCAT) for financial support. In addition, S. J. F acknowledges the award of a Prize Research Fellowship from the University of Bath. XPS data collection was performed at the EPSRC National Facility for XPS (‘Harwell XPS’). Operated by Cardiff University and UCL, under contract no. PR16195. The authors would like to thank the CCI-Electron Microscopy Facility which has been part-funded by the European Regional Development Fund through the Welsh Government and The Wolfson Foundation.References
- HDIN Research, Global Nylon 6 production capacity to reach 8.86 million tons in 2024 (2022) https://www.hdinresearch.com/news/56.
- P. Roffia, G. Leofanti, A. Cesana, M. Mantegazza, M. Padovan, G. Petrini, S. Tonti and P. Gervasutti, Stud. Surf. Sci. Catal., 1990, 55, 43–52 CrossRef CAS.
- A. Zecchina, S. Bordiga, C. Lamberti, G. Ricchiardi, C. Lamberti, G. Ricchiardi, D. Scarano, G. Petrini, G. Leofanti and M. Mantegazza, Catal. Today, 1996, 32, 97–106 CrossRef CAS.
- R. J. Lewis and G. J. Hutchings, ChemCatChem, 2019, 11, 298–308 CrossRef CAS.
- J. R. Scoville and I. A. Novicova (Cottrell Ltd.), US 5900256, 1996.
- G. Gao, Y. Tian, X. Gong, Z. Pan, K. Yong and B. Zong, Chin. J. Catal., 2020, 41, 1039–1047 CrossRef CAS.
- J. K. Edwards, B. Solsona, E. N. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef CAS PubMed.
- N. M. Wilson, P. Priyadarshini, S. Kunz and D. W. Flaherty, J. Catal., 2018, 357, 163–175 CrossRef.
- J. Brehm, R. J. Lewis, D. J. Morgan, T. E. Davies and G. J. Hutchings, Catal. Lett., 2021, 152, 254–262 CrossRef.
- C. M. Crombie, R. J. Lewis, D. Kovačič, D. J. Morgan, T. E. Davies, J. K. Edwards, M. S. Skjøth-Rasmussen and G. J. Hutchings, Catal. Lett., 2020, 151, 164–171 CrossRef.
- C. M. Crombie, R. J. Lewis, R. L. Taylor, D. J. Morgan, T. E. Davies, A. Folli, D. M. Murphy, J. K. Edwards, J. Qi, H. Jiang, C. J. Kiely, X. Liu, M. S. Skjøth-Rasmussen and G. J. Hutchings, ACS Catal., 2021, 11, 2701–2714 CrossRef CAS.
- Z. Jin, L. Wang, E. Zuidema, K. Mondal, M. Zhang, J. Zhang, C. Wang, X. Meng, H. Yang, C. Mesters and F. Xiao, Science, 2020, 367, 193–197 CrossRef CAS PubMed.
- R. J. Lewis, A. Bara-Estaun, N. Agarwal, S. J. Freakley, D. J. Morgan and G. J. Hutchings, Catal. Lett., 2019, 149, 3066–3075 CrossRef CAS.
- L. Torrente-Murciano, T. Villager and D. Chadwick, ChemCatChem, 2015, 7, 925–927 CrossRef CAS.
- Q. Chen and E. J. Beckman, Green Chem., 2008, 10, 934–938 RSC.
- G. Dodekatos, L. Abis, S. J. Freakley, H. Tüysüz and G. J. Hutchings, ChemCatChem, 2018, 10, 1351–1359 CrossRef CAS.
- G. Wang, W. Du, X. Duan, Y. Cao, Z. Zhang, J. Xu, W. Chen, G. Qian, W. Yuan, X. Zhou and D. Chen, Chem. Catal., 2021, 1, 885–895 CrossRef.
- R. J. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. E. Davies, D. J. Morgan, L. Chen, J. Qi, J. Singleton, J. K. Edwards, S. J. Freakley, C. J. Kiely, Y. Yamamoto and G. J. Hutchings, Science, 2022, 376, 615–620 CrossRef CAS PubMed.
- T. Richards, R. J. Lewis, D. J. Morgan and G. J. Hutchings, Catal. Lett., 2022 DOI:10.1007/s10562-022-03967-8.
- J. K. Edwards, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Faraday Discuss., 2008, 138, 225–239 RSC.
- L. M. Rossi, J. L. Fiorio, M. A. S. Garcia and C. P. Ferraz, Dalton Trans., 2018, 47, 5889–5915 RSC.
- X. Gong, R. J. Lewis, S. Zhou, D. J. Morgan, T. E. Davies, X. Liu, C. J. Kiely, B. Zong and G. J. Hutchings, Catal. Sci. Technol., 2020, 10, 4635–4644 RSC.
- A. Villa, D. Wang, G. M. Veith, F. Vindigni and L. Prati, Catal. Sci. Technol., 2013, 3, 3036–3041 RSC.
- G. M. Lari, B. Puértolas, M. Shahrokhi, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2017, 56, 1775–1779 CrossRef CAS.
- R. J. Lewis, M. Koy, M. Macino, M. Das, J. H. Carter, D. J. Morgan, T. E. Davies, J. B. Ernst, S. J. Freakley, F. Glorius and G. J. Hutchings, J. Am. Chem. Soc., 2022, 144(34), 15431–15436 CrossRef CAS.
- C. B. Paris, A. G. Howe, R. J. Lewis, D. Hewes, D. J. Morgan, Q. He and J. K. Edwards, ACS Catal., 2022, 12, 4440–4454 CrossRef CAS PubMed.
- A. Santos, R. J. Lewis, G. Malta, A. G. R. Howe, D. J. Morgan, E. Hampton, P. Gaskin and G. J. Hutchings, Ind. Eng. Chem. Res., 2019, 58, 12623–12631 CrossRef CAS.
- S. J. Freakley, R. J. Lewis, D. J. Morgan, J. K. Edwards and G. J. Hutchings, Catal. Today, 2015, 248, 10–17 CrossRef CAS.
- M. Chen, D. Kumar, C.-W. Yi and D. W. Goodman, Science, 2005, 310, 291–293 CrossRef CAS PubMed.
- J. K. Edwards, J. Pritchard, L. Lu, M. Piccinini, G. Shaw, A. F. Carley, D. J. Morgan, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2014, 53, 2381–2384 CrossRef CAS PubMed.
- N. M. Wilson, P. Priyadarshini, S. Kunz and D. W. Flaherty, J. Catal., 2018, 357, 163–175 CrossRef.
- T. Ricciardulli, S. Gorthy, J. S. Adams, C. Thompson, A. M. Karim, M. Neurock and D. W. Flaherty, J. Am. Chem. Soc., 2021, 143, 5445–5464 CrossRef CAS PubMed.
- J. Li, T. Ishihara and K. Yoshizawa, J. Phys. Chem. C, 2011, 115, 25359–25367 CrossRef CAS.
- T. Richards, J. H. Harrhy, R. J. Lewis, A. G. R. Howe, G. M. Suldecki, A. Folli, D. J. Morgan, T. E. Davies, E. J. Loveridge, D. A. Crole, J. K. Edwards, P. Gaskin, C. J. Kiely, Q. He, D. M. Murphy, J. Maillard, S. J. Freakley and G. J. Hutchings, Nat. Catal., 2021, 4, 575–585 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- C. J. Wrasman, A. Boubnov, A. R. Riscoe, A. S. Hoffman, S. R. Bare and M. Cargnello, J. Am. Chem. Soc., 2018, 140, 12930–12939 CrossRef CAS PubMed.
- S. Han and C. B. Mullins, ACS Catal., 2018, 8, 3641–3649 CrossRef CAS.
- S. Han and C. B. Mullins, Acc. Chem. Res., 2021, 54, 379–387 CrossRef CAS PubMed.
- Y. Han, Z. Zhong, K. Ramesh, F. Chen, L. Chen, T. White, Q. Tay, S. N. Yaakub and Z. Wang, J. Phys. Chem. C, 2007, 111, 8410–8413 CrossRef CAS.
- F. Gao, Y. Wang and D. W. Goodman, J. Am. Chem. Soc., 2009, 131, 5734–5735 CrossRef CAS PubMed.
- F. Gao and D. W. Goodman, Chem. Soc. Rev., 2012, 41, 8009–8020 RSC.
- J. Li, T. Ishihara and K. Yoshizawa, J. Phys. Chem. C, 2011, 115, 7392–7398 CrossRef CAS.
- P. Tian, D. Ding, Y. Sun, F. Xuan, X. Xu, J. Xu and Y. Han, J. Catal., 2019, 369, 95–104 CrossRef CAS.
- P. Tian, L. Ouyang, X. Xu, C. Ao, X. Xu, R. Si, X. Shen, M. Lin, J. Xu and Y. Han, J. Catal., 2017, 349, 30–40 CrossRef CAS.
- C. Williams, J. H. Carter, N. F. Dummer, Y. K. Chow, D. J. Morgan, S. Yacob, P. Serna, D. J. Willock, R. J. Meyer, S. H. Taylor and G. J. Hutchings, ACS Catal., 2018, 8, 2567–2576 CrossRef CAS.
- L. F. de L. e Freitas, B. Puértolas, J. Zhang, B. Wang, A. S. Hoffman, S. R. Bare, J. Pérez-Ramírez, J. W. Medlin and E. Nikolla, ACS Catal., 2020, 10, 5202–5207 CrossRef.
- S. J. Freakley, N. Agarwal, R. U. McVicker, S. Althahban, R. J. Lewis, D. J. Morgan, N. Dimitratos, C. J. Kiely and G. J. Hutchings, Catal. Sci. Technol., 2020, 10, 5935–5944 RSC.
- S. Abate, K. Barbera, G. Centi, G. Giorgianni and S. Perathoner, J. Energy Chem., 2016, 25, 297–305 CrossRef.
- R. J. Lewis, E. N. Ntainjua, D. J. Morgan, T. E. Davies, A. F. Carley, S. J. Freakley and G. J. Hutchings, Catal. Commun., 2021, 161, 106358 CrossRef CAS.
- G. K. Wertheim, S. B. DiCenzo and D. N. E. Buchanan, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 5384–5390 CrossRef CAS.
- D. R. Jones, S. Iqbal, P. J. Miedziak, D. J. Morgan, J. K. Edwards, Q. He and G. J. Hutchings, Top. Catal., 2018, 61, 833–843 CrossRef CAS.
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551–556 CrossRef CAS PubMed.
- Y. Borodko, H. S. Lee, S. H. Joo, Y. Zhang and G. Somorjai, J. Phys. Chem. C, 2010, 114, 1117–1126 CrossRef CAS.
- L. R. Baker, G. Kennedy, J. M. Krier, M. Van Spronsen, R. M. Onorato and G. A. Somorjai, Catal. Lett., 2012, 142, 1286–1294 CrossRef CAS.
- Y. Zhao, L. Jia, J. A. Medrano, J. R. H. Ross and L. Lefferts, ACS Catal., 2013, 3, 2341–2352 CrossRef CAS.
- S. Cattaneo, S. J. Freakley, D. J. Morgan, M. Sankar, N. Dimitratos and G. J. Hutchings, Catal. Sci. Technol., 2018, 8, 1677–1685 RSC.
- J. C. Matsubu, S. Zhang, L. DeRita, N. S. Marinkovic, J. G. Chen, G. W. Graham, X. Pan and P. Christopher, Nat. Chem., 2017, 9, 120–127 CrossRef CAS PubMed.
- J. K. Edwards, J. Pritchard, M. Piccinini, G. Shaw, Q. He, A. F. Carley, C. J. Kiely and G. J. Hutchings, J. Catal., 2012, 292, 227–238 CrossRef CAS.
- J. Pritchard, M. Piccinini, R. Tiruvalam, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, D. J. Morgan, A. F. Carley, J. K. Edwards, C. J. Kiely and G. J. Hutchings, Catal. Sci. Technol., 2013, 3, 308–317 RSC.
- R. J. Lewis, K. Ueura, Y. Fukuta, S. J. Freakley, L. Kang, R. Wang, Q. He, J. K. Edwards, D. J. Morgan, Y. Yamamoto and G. J. Hutchings, ChemCatChem, 2019, 11, 1673–1680 CrossRef CAS.
- Y. K. Chow, N. F. Dummer, J. H. Carter, R. J. Meyer, R. D. Armstrong, C. Williams, G. Shaw, S. Yacob, M. M. Bhasin, D. J. Willock, S. H. Taylor and G. J. Hutchings, ChemPhysChem, 2018, 19, 402–411 CrossRef CAS PubMed.
- S. A. Kondrat, P. J. Miedziak, M. Douthwaite, G. L. Brett, T. E. Davies, D. J. Morgan, J. K. Edwards, D. W. Knight, C. J. Kiely, S. H. Taylor and G. J. Hutchings, ChemSusChem, 2014, 7, 1326–1334 CrossRef CAS PubMed.
- J. W. M. Crawley, I. E. Gow, N. Lawes, I. Kowalec, L. Kabalan, C. R. A. Catlow, A. J. Logsdail, S. H. Taylor, N. F. Dummer and G. J. Hutchings, Chem. Rev., 2022, 122, 6795–6849 CrossRef CAS PubMed.
- G. Bernardotto, F. Menegazzo, F. Pinna, M. Signoretto, G. Cruciani and G. Strukul, Appl. Catal., A, 2009, 358, 129–135 CrossRef CAS.
- S. Quon, D. Y. Jo, G.-H. Han, S. S. Han, M. Seo and K. Lee, J. Catal., 2018, 368, 237–247 CrossRef CAS.
- T. Deguchi, H. Yamano, S. Takenouchi and M. Iwamoto, Catal. Sci. Technol., 2018, 8, 1002–1015 RSC.
- H. V. Nguyen, K. Y. Kim, H. Nam, S. Y. Lee, T. Yu and T. S. Seo, Lab Chip, 2020, 20, 3293–3301 RSC.
- J. Ding and P. Wu, Appl. Catal., A, 2014, 488, 86–95 CrossRef CAS.
- F. Song, Y. Liu, L. Wang, H. Zhang, M. He and P. Wu, Appl. Catal., A, 2007, 327, 22–31 CrossRef CAS.
- S. Zhao, W. Xie, J. Yang, Y. Liu, Y. Zhang, B. Xu, J. Jiang, M. He and P. Wu, Appl. Catal., A, 2011, 394, 1–8 CrossRef CAS.
- J. S. Reddy, S. Sivasanker and P. Ratnasamy, J. Mol. Catal., 1991, 69, 383–392 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc02689a |
| This journal is © The Royal Society of Chemistry 2022 |