
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An unusual self-assembling columnar mesogen prepared by tethering a planar naphthalenediimide acceptor to bent phenothiazine donors†
Kim D.
Thériault
a,
Chase L.
Radford
 b,
Gowdaianapallya P.
Nagabhushana
a,
David T.
Hogan
a,
Vance E.
Williams
c,
Timothy L.
Kelly
*b and
Todd C.
Sutherland
*a
b,
Gowdaianapallya P.
Nagabhushana
a,
David T.
Hogan
a,
Vance E.
Williams
c,
Timothy L.
Kelly
*b and
Todd C.
Sutherland
*a
aDepartment of Chemistry, University of Calgary, 2500 University Drive NW, Calgary, Alberta T2N 1N4, Canada. E-mail: todd.sutherland@ucalgary.ca
bDepartment of Chemistry, University of Saskatchewan, 110 Science Place, Saskatoon, Saskatchewan S7N 5C9, Canada. E-mail: tim.kelly@usask.ca
cDepartment of Chemistry, Simon Fraser University, 8888 University Drive, Burnaby, British Columbia V5A 1S6, Canada
First published on 4th November 2021
Abstract
We describe a synthetic approach to achieving columnar stacked mesogens of aromatic electron donors (D) and acceptors (A) as an orthogonal strategy to segregate charge carrying channels in films. An unusual columnar liquid crystalline mesogen was designed, such that a planar naphthalenediimide acceptor flanked by two bent-shaped phenothiazine donor moieties self-assembled into a liquid crystalline phase. The use of different shapes in the D–A–D arrangement could allow segregated charge transport columns, thus limiting both intercolumnar and intramolecular D–A interactions. In solution, the mesogen showed independent optical and electronic properties of its constituent D and A components and annealed films of the columnar liquid crystalline material showed a near doubling of photocurrent generation compared to amorphousfilms.
Introduction
Organic semiconductors have made a significant impact in the technology industry with the development of ultrathin and lightweight displays that have been applied to flexible substrates. While organic light emitting diodes (OLEDs) have seen commercial success, organic field-effect transistors and photovoltaics lag, in part owing to challenges in controlling the nanostructure within the organic material. Self-assembly of films into organised channels that carry charges and limit recombination, as illustrated in Fig. 1a with columnar arrays, are attractive structural motifs that could enable hole and electron transport. Ideally, molecular scale separation between Donor (D) and Acceptor (A) units that are organized could offer efficient hole and electron transport channels, or ambipolar charge transport, and have been suggested as candidate film architectures.1–6 | ||
| Fig. 1 (a) Illustration of columnar phases demonstrating that overlapping tethered donor and acceptor units could lead to migration of electrons and holes once assembled. (b) Structure of triad 1 and pi-functional units 2 and 3. | ||
Columnar liquid crystalline phases, as illustrated in Fig. 1a, possess many of the organizational features necessary to accomplish efficient charge transport. However, there are examples of columnar liquid crystalline constructs that possess both D and A units, and most molecular structures consist of planar π-conjugated units.6–14 Flexible films that possess efficient n- and p-conduction channels would enable devices to take advantage of organic attributes and lead to next generation organic solar cells, field effect transistors or light emitting diodes. There is an urgent need to develop diversity in the molecular scaffolds that make up columnar, or discotic, assemblies.
An approach to producing single component materials that conduct both holes and electrons is to link D and A units via covalent bonds resulting in improved interfacial contact between D and A units.15 A few architectures have been developed that tether D and A building blocks, such as fullerene–oligomer dyads,16,17 di-block copolymers,18–24 and D–A small molecules.25–28 Recent examples of columnar mesophases that exhibit ambipolar charge transport include the work of Mu and co-workers29 that explore fullerene–triphenylene hybrid dendrimers, and Bala and co-workers’30 use of triphenylene–pentaalkynylbenzene dyads to yield blue OLED emitters.
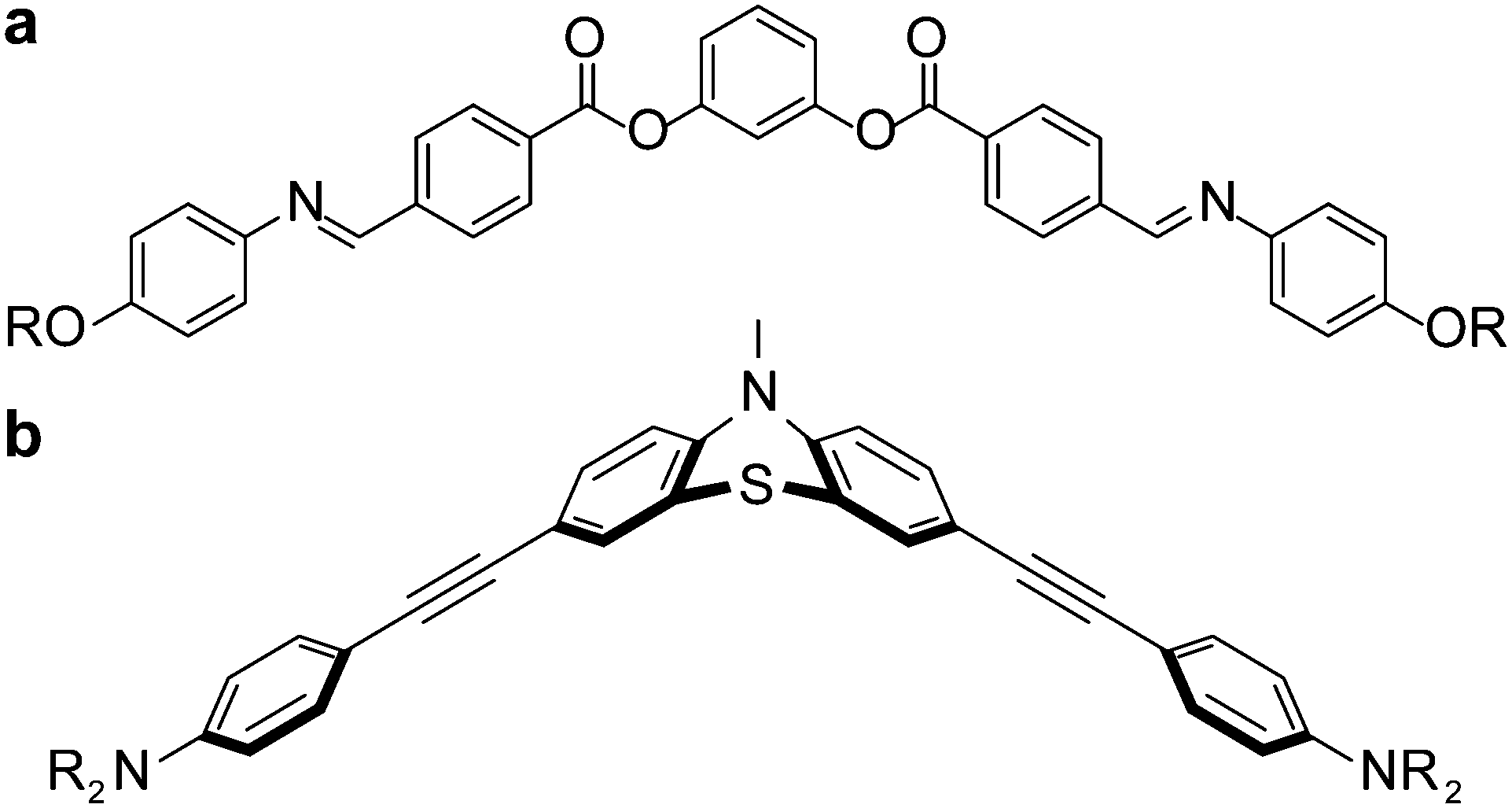
Despite these strategies, the morphology of the D–A films has been difficult to control as many blended films show favorable intermolecular interactions between D- and A-units, resulting in alternating D–A stacks, which are ideal arrangements for charge recombination, not propagation.31–34 Consequently, some researchers have relied upon lipophilic/hydrophilic amphiphiles,7,35–37 H-bonding38–41 or disc-rod incompatibilities42 to favor self-assembly into isolated D–A channels. A recent review by Lehmann and co-workers43 describes progress in liquid crystal design using shape persistent mesogens, such as macrocycles, gear wheels, stars, bowls and tubes, in the context of ‘free space’ to guide assemblies. Herein, we demonstrate an alternate strategy to control the nanostructure based on tethering mismatched molecular shapes.44–49 Bent-core, or banana-shaped, mesogens, represented by a generic structure shown in Fig. 2a, were first characterised in 1996 by Niori and co-workers50 and have been explored extensively. Bent-core mesogens reported are commonly based on a central planar, rigid, aromatic ring and the bend arises from the meta-substitution pattern of the central ring, as depicted in Fig. 2a, to form a 2-D structure. Here, we report a bent-core that is the result of a central non-planar phenothiazine that adopts a 3-D structure, as highlighted in Fig. 2b. Clearly, the shape of molecules in Fig. 2 are similar, yet there are no reports of liquid crystalline materials derived from structures based on a scaffold shown in Fig. 2b. There are two reports of smectic51 and nematic52 liquid crystal phases based on heavily-substituted anthraquinone cores that adopt a similar bent shape as shown in Fig. 2b by virtue of steric crowding of the central carbonyls with neighbouring alkoxy or alkoxyaryl groups.
 | ||
| Fig. 2 (a) Common structural motif of bent-core, or banana, mesogens. (b) Bent-core, or butterfly, shape motif used in this work. | ||
Structurally more intriguing complexity arises by using covalently tethered bent-core structures and assessing the resulting shape mismatched partners. To date, columnar liquid crystals that exhibit molecular-scale segregation between D and A components are formed from planar, rigid cores that are surrounded by saturated alkyl chains. The concept of using shape to induce molecular scale segregation was pioneered by Tschierske in non-conventional liquid crystals.1,2,53,54 Exploiting molecular shape using D and A building blocks to organize D–A structures has been reported using linked planar conjugated systems, such as triphenylene and PDI7–9 or spherical shapes such as, fullerene-disc shape-amphiphiles,6,10 fullerene-oligo(phenylenevinylene)11 or fullerene-dendritic structures.12 To the best of our knowledge, there are no reports of a D–A conjugate that contains bent-core structures (as defined in Fig. 2b) and exhibits columnar liquid crystalline phases, as a design strategy for columnar mesogens.
In pursuit of developing novel mesogenic structures that result in phase separation of D and A units, we present triad 1 comprised of bent phenothiazine (PTZ, 2) donors tethered via C2 alkyl chains to a planar naphthalenediimide (NDI, 3) acceptor (Fig. 1b). D–A bridged molecules that are isolated electronically exhibit independent optical and electrochemical properties in solution, similar to the individual D and A components. In addition, the films of 1 exhibited self-assembly into columnar stacks providing favourable long-range ordering to carry charges in the solid state. The photoresponse of the material was also assessed to demonstrate the potential of this type of molecular architecture in organic electronics. The combination of exploiting shape and D–A moieties to induce functional columnar assemblies has not been explored.
Experimental
All chemicals were purchased from Aldrich and used as received without further purification, aside from the following: 1,4,5,8-naphthalenetetracarboxylic dianhydride was purchased from TCI and AlCl3 was purchased from Fluka. Solvents were used as received or dried using an MBraun solvent purification system. THF and DMF were stored over sieves before use and triethylamine was distilled in the presence of CaH2 and stored over sieves. N,N-didecyl-4-ethynylaniline was prepared in our lab following methods described previously.55 Column chromatography was performed on a SiliCycle SilicaFlash P60 silica gel (230–400 mesh). Thin-layer chromatography was carried out on Merck silica gel F254 aluminium-backed TLC plates.NMR spectra were recorded on a Bruker 400 MHz spectrometer and referenced to residual CHCl3 in CDCl3 (1H 7.26 ppm, 13C 77.00) or residual CD3COCD2H in (CD3)2CO (1H 2.05 ppm). ESI and APCI mass spectra were recorded with an Agilent 6520 LC Q-TOF spectrometer run in either ESI or APCI mode by direct infusion with the column bypassed. All samples were dissolved in DCM and run with an eluent flow rate of 0.2 mL min−1 MeOH, source temperature of 200 °C, fragmenter voltage of 120 V, Nebulizer pressure of 12 psi and a drying gas flow rate of 7 L min−1. MALDI mass spectra were recorded with a Bruker Daltonics Autoflex MALDI-TOF spectrometer. FTIR spectra were recorded in diffuse reflectance mode with drop-cast films on ground KBr on a Varian FTS-7000 spectrometer. UV/Vis spectra were recorded using a Cary 5000 spectrophotometer in dual beam mode. Emission spectra were recorded using a Photon Technology International (PTI) Quanta Master spectrofluorimeter operated in CW mode, with samples in a quartz Suprasil cell (1 cm width and path length, Hellma, Canada). Differential pulse voltammetry (DPV) and cyclic voltammetry (CV) experiments were carried out on an Autolab PGSTAT302 potentiostat that was controlled by a PC running Autolabs's GPES v 4.9 software in a temperature-controlled, three-electrode cell (15 mL). The working electrode was a Pt button, the reference electrode was a Ag wire, and the counter electrode was a Pt wire. All potentials were referenced to an internal ferrocene/ferrocenium redox couple. Each DPV experiment consisted of approximately 7 mM redox active product dissolved in 0.1 M [NBu4]PF6 in degassed dichloromethane, maintaining an argon blanket during the entire experiment. Spectroelectrochemical spectra were recorded with the Cary 5000 linked to a CHI 650 potentiostat using a thin layer cell (OTTLE) with a Pt mesh working electrode in the beam path, a Pt wire counter electrode, and a Ag wire reference electrode; electrolyte and solvent were used as above. Differential scanning calorimetry was collected with a Setaram Instrumentation microSC and thermal gravimetric analysis was carried out with a TA-Q50 TGA under a N2 atmosphere. Cross-polarized optical microscopy was carried out on an Olympus BX-41 microscope equipped with a heating stage (Linkam T95). Electrochemical impedance data were collected using an Zahner IM6 impedance analyzer from 0.1 Hz to 100 kHz under open circuit and applied potentials and the results were fit to equivalent circuits using Thales software package, with an ITO substrate as the working electrode, Pt wire as counter electrode, as well as a Ag wire reference electrode; supporting electrolyte was 0.1 M [NBu4]PF6 in CH3CN. The flat cell used in impedance measurements is available through ALS. Electrochemical photo-response data were collected using a Zahner CIMPS-3 system comprising of IM6 potentiostat, XPot light source potentiostat, optical bench sensor, LDA photosense-amplifier-illuminator supply unit loaded with KBL01 LED (455 nm). Experiments were run using the Thales software package, with an ITO substrate as the working electrode loaded in an PECC-1 electrochemical cell (3.1 cm2 geometric area) comprised of a Pt wire as counter electrode, as well as a Ag wire reference electrode; supporting electrolyte was 0.1 M [NBu4]PF6 in CH3CN.
GIWAXS experiments were performed at the Canadian Light Source (CLS) using the Hard X-ray MicroAnalysis (HXMA) beamline. A photon energy of 12.69 keV was selected using a Si(111) monochromator. The beam size was defined by slits having a 0.2 mm vertical gap and a 0.3 mm horizontal gap, and the angle of incidence was set to 0.06°. The sample was deposited on a 〈100〉 silicon wafer by drop-casting. GIWAXS patterns were collected with a Rayonix SX165 CCD detector (80 μm pixel size; 16.3 cm diameter), which was placed 209 mm from the sample center. The GIWAXS data were calibrated against a silver behenate standard and analyzed using the GIXSGUI software package.56 Both polarization and solid-angle corrections were applied.
5: 10-cyanomethyl-10H-phenothiazine
Chloroacetonitrile (7.0 mL, 110 mmol) was added to an olive solution of phenothiazine (6.9127 g, 34.7 mmol) in dry DMF (14 mL). The mixture was equally divided into 7 microwave vials and sequentially irradiated up to 80 °C for 20 minutes at 100 W while continuously cooling the sample with compressed N2 to maximize the microwave power. The solution becomes darker and more viscous. After rinsing the vials, the solvent was removed in vacuo and the resulting mixture was fused to silica prior to purification via silica gel chromatography (hexanes![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DCM (1:1)) to afford 5 as a beige powder (1.8370 g, 22%). 1H NMR (400 MHz, acetone-d6) δ 7.30 (ddd, J = 8.2, 7.3, 1.6 Hz, 2H), 7.22 (dd, J = 7.7, 1.5 Hz, 2H), 7.17 (dd, J = 8.2, 1.2 Hz, 2H), 7.06 (td, J = 7.5, 1.2 Hz, 2H), 4.99 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 143.02, 127.76, 127.51, 124.20, 123.96, 115.63, 114.21, 37.37. MS (ESI HRMS) m/z calcd for C14H10N2S + H: 239.06375; found 239.06352.
:DCM (1:1)) to afford 5 as a beige powder (1.8370 g, 22%). 1H NMR (400 MHz, acetone-d6) δ 7.30 (ddd, J = 8.2, 7.3, 1.6 Hz, 2H), 7.22 (dd, J = 7.7, 1.5 Hz, 2H), 7.17 (dd, J = 8.2, 1.2 Hz, 2H), 7.06 (td, J = 7.5, 1.2 Hz, 2H), 4.99 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 143.02, 127.76, 127.51, 124.20, 123.96, 115.63, 114.21, 37.37. MS (ESI HRMS) m/z calcd for C14H10N2S + H: 239.06375; found 239.06352.
6: 3,7-dibromo-10-cyanomethyl-10H-phenothiazine
An olive solution of 5 (0.9458 g, 4.0 mmol) in dry THF (100 mL) was purged with N2, cooled in an ice bath and shielded from direct light. NBS (2.2604 g, 12.7 mmol) was added in three equal portions one hour between each addition. One hour after the last addition of NBS, the solvent was removed in vacuo. The resulting brown oil was purified with silica gel chromatography (DCM). Further purification by crystallizing the product from DCM and layering with pentane afforded 6 as fine white needles (0.9291 g, 59%). 1H NMR (400 MHz, CDCl3) δ 7.36 (dd, J = 8.6, 2.2 Hz, 2H), 7.31 (d, J = 2.2 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 4.56 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 141.94, 130.72, 130.00, 125.88, 116.86, 115.50, 114.97, 37.51. MS (ESI HRMS) m/z calcd for C14H8Br2N2S + Cl: 430.84485; found 430.84285.7: 3,7-dibromo-10-(2-aminoethyl)-10H-phenothiazine
A solution of AlCl3 (0.6759 g, 5.1 mmol) in dry Et2O (50 mL) was cooled in an ice bath and purged with N2, after which LiAlH4 (0.1913 g, 5.0 mmol) was added and left to disproportionate for 90 minutes. A suspension of 6 (0.8542 g, 2.2 mmol) in Et2O (150 mL) was then added to the reaction mixture and left to stir under N2 overnight and allowing the ice bath to reach room temperature. The reaction mixture was quenched with MeOH, filtered over Celite and the solvent was removed in vacuo to afford a beige oil. The crude product was purified by silica gel chromatography (NH4OH:MeOH:DCM (1:9:90)) to afford 7 as a yellow oil (0.6682 g, 77%). 1H NMR (400 MHz, CDCl3) δ 7.24–7.20 (m, 4H), 6.69 (dd, J = 8.0, 1.0 Hz, 2H), 3.86 (t, J = 5.9 Hz, 2H), 2.99 (t, J = 5.9 Hz, 2H), 1.37 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 143.91, 130.14, 129.80, 127.11, 116.93, 115.11, 50.57, 38.51. MS (APCI HRMS) m/z calcd for C14H12Br2N2S + Cl: 434.87615; found 434.87491.
8: 4,4′-(10-(2-aminoethyl)-10H-phenothiazine-3,7-diyl)bis(ethyne-2,1-diyl)bis(N,N-didecylaniline)
A solution of 7 (1.0056 g, 2.5 mmol) in dry NEt3 (180 mL) was purged with N2 prior to adding bis(triphenylphosphine)palladium(II) dichloride (0.1776 g, 0.25 mmol), triphenyl phosphine (0.2156 g, 0.82 mmol) and copper(I) iodide (0.0737 g, 0.39 mmol). The yellow mixture was heated to 80 °C under N2 for 30 min before adding a purged solution of N,N-didecyl-4-ethynylaniline (2.9897 g, 7.5 mmol) in dry NEt3 (24 mL) via syringe at a rate of 1 mL min−1. The brown mixture was stirred under N2 at 80 °C for 3 hours and the solvent was removed in vacuo. The crude product was purified by alumina chromatography (DCM → NH4OH:MeOH:DCM (1:9:190)) to afford 8 as a yellow oil (1.9209 g, 74%). 1H NMR (400 MHz, CDCl3) δ 7.32 (d, J = 8.9 Hz, 4H), 7.27 (dd, J = 5.5, 1.9 Hz, 4H), 6.81 (d, J = 9.0 Hz, 2H), 6.55 (d, J = 8.9 Hz, 4H), 3.97 (t, J = 6.0 Hz, 2H), 3.33 – 3.20 (m, 8H), 3.07 (t, J = 5.9 Hz, 2H), 1.63–1.54 (m, 8H), 1.35–1.22 (m, 58H), 0.91–0.86 (m, 12H). 13C NMR (101 MHz, CDCl3) δ 147.88, 143.95, 132.73, 130.39, 130.05, 125.12, 118.92, 115.45, 111.23, 108.76, 90.87, 86.15, 50.97, 50.68, 38.87, 31.89, 29.65, 29.56, 29.52, 29.31, 27.23, 27.14, 22.68, 14.10. MS (MALDI HRMS, HCCA) m/z calcd for C70H104N4S+: 1032.7982; found 1032.7934.
1: N,N′-bis(4,4′-(10-(ethyl)-10H-phenothiazine-3,7-diyl)bis(ethyne-2,1-diyl)bis(N,N-didecylaniline))naphthalene-1,4,5,8-tetracarboxylic diimide
To a dark yellow solution of 8 (0.3141 g, 0.30 mmol) in dry DMF (30 mL) was added 1,4,5,8-naphthalenetetracarboxylic dianhydride (0.0393 g, 0.15 mmol) and the mixture was heated in an oil bath to 130 °C overnight. The solvent was removed in vacuo and the residue was reconstituted in ether, washed with H2O and the aqueous layer was extracted with chloroform. The organic layer was dried with MgSO4 and the organic solvents were removed in vacuo. The resulting dark yellow solid was purified by silica gel chromatography (hexane:chloroform (1:9)) to yield 1 as a green solid (0.2127 g, 61%). 1H NMR (400 MHz, CDCl3) δ 8.59 (s, 4H), 7.24 (d, J = 8.3 Hz, 8H), 7.15 (q, J = 8.0, 7.1 Hz, 8H), 6.85 (s, 4H), 6.51 (d, J = 8.5 Hz, 8H), 4.29 (s, 4H), 3.90 (s, 4H), 3.29–3.12 (m, 16H), 1.62–1.46 (m, 16H), 1.31 (d, J = 6.2 Hz, 112H), 0.91 (t, J = 6.7 Hz, 24H). 13C NMR (101 MHz, CDCl3) δ 162.85, 147.75, 142.01, 132.66, 130.72, 128.84, 126.60, 126.09, 121.44, 118.39, 114.19, 111.07, 108.65, 90.98, 86.09, 50.86, 31.90, 29.71, 29.59, 29.52, 29.34, 27.20, 27.13, 22.69, 14.12. MS (MALDI HRMS, HCCA) m/z calcd for C154H208N8O4S2+: 2297.5760; found 2297.5689.
Results and discussion
The synthesis of triad 1 is shown in Scheme 1. Bent phenothiazine, 4, was N-alkylated with chloroacetonitrile using microwave conditions to give the cyano derivative in 22% yield. Conventional heating methods were also employed and resulted in in poor yields (8–14%), whereas, under microwave heating conditions, the yields were improved with the benefits of shorter reaction times and unreacted PTZ could be recovered and recycled. Phenothiazine derivate 5 was then electrophilically dibrominated with NBS in the 2,7-positions to give 6 in 59% yield. Dibrominated nitrile 6 was then reduced with LiAlH4/AlCl3 to give amine 7 in 77% yield. Altering the order of bromination and reduction was attempted but resulted in challenging purifications and was abandoned. The penultimate step involved a Sonogashira coupling of the ethynylaniline with dibrominated 7 to give amine 8 in 74% yield after alumina column chromatography. The final step to producing triad 1, involved the condensation of two equivalents of amine 8 with naphthalenetetracarboxylic dianhydride to give diimide 1 in 61% yield. | ||
| Scheme 1 Synthesis of triad 1. Reaction conditions: (i) chloroacetonitrile, microwave, 22% yield (ii) NBS, 59% yield (iii) LiAlH4, AlCl3, 77% yield (iv) N,N-didecyl-4-ethynylaniline, 74% yield (v) 1,4,5,8-naphthalenetetracarboxylic dianhydride, 61% yield. | ||
Because triad 1 comprises both electron-rich PTZs and an electron-poor NDI component, it is critical to investigate whether the triad exhibits intramolecular charge transfer absorption features, intermolecular associations, or aggregates, in solution. The chromophoric properties of triad 1 and its independent building blocks PTZ 2 and NDI 3 were assessed by absorption and fluorescence spectroscopy as shown in Fig. 3. The absorption profile of 1 (squares) overlays well with the linear combination of the NDI and PTZ building blocks 2 and 3 (dashed grey line), suggesting each chromophoric unit of triad 1 behaves independently in solution. The peak shapes of both absorption and emission spectra for chromophores 1–3 exhibit shifts in different solvents (Fig. S24–S29, ESI†), which are consistent with other reports.55,57,58 A sharp peak at 375 nm, attributed to the π–π* transition of NDI is evident in polar solvents DCM and DMF, whereas the sharp peak is absent in the solid state and in less polar solvents like toluene and n-hexane. The absorption profile of a film of 1 lacks a sharp absorption feature, which is consistent with the lower dielectric medium in the solid-state.
 | ||
| Fig. 3 Molar absorption coefficients in DCM of 1 (squares, 10−5 M) and its fluorescence spectrum (excitation 381 nm, circles) compared to building blocks PTZ 2 (red fill) and NDI 3 (blue fill) in DCM. The grey dashed line represents the addition of 2 and 3 in a 2:1 summative ratio and the black line (grey fill) represents the normalized solid-state film absorption profile of 1. | ||
The fluorescence spectrum of triad 1 matches the fluorescence peak profile of PTZ 2. The low quantum yield of 1 (Φf = 0.002, DCM) is interesting since the fluorescence quantum yield of PTZ 2 is near 0.3, which indicates a significant quenching of the excited state of 1, through energy or electron transfer mechanisms. Importantly, the excitation spectrum of 1 (λexc = 458 nm) matches the absorption profile of PTZ 2, suggesting that the weak emission of 1 is dominated by the PTZ portion of the triad (Fig. S26, ESI†). As expected, NDI 3 is weakly emissive in DCM and hexanes solutions, but shows emissive ground-state charge-transfer complexes with toluene (Fig. S27, ESI†), as demonstrated by others.59 The absorption spectra of a spin-cast film of 1, shown in Fig. 3, before and after annealing (Fig. S29, ESI†) shows a similar profile to the solution spectra, save a sharp transition at 375 nm, which is mirrored in non-polar solvents hexane and toluene (Fig. S28, ESI†). The film absorption profile of 1 has a lower energy onset compared to solution spectra, however the film does not show new low energy electronic transitions, suggesting intermolecular donor–acceptor interactions do not play a role in the solid-state. TD-DFT (B3LYP/6-31G+(d) including CH2Cl2 PCM solvent model) computations (Fig. S36, ESI†) of 1 add further support of independent chromophores, since the calculated absorption spectrum shows only PTZ- and NDI-localized optical transitions. Indeed, Chen and co-workers60 explored a PTZ donor tethered to a triazole acceptor core that demonstrated non-interacting building blocks. Similarly, a bent-shaped PTZ cyclophane bearing an acceptor core showed minimal interaction between donor and acceptor units was also reported by Hempe and co-workers.61
Electrochemically, triad 1 shows several oxidation and reduction peaks during cyclic voltammetry (CV), shown in Fig. S30 (ESI†). Triad 1 has a low irreversible oxidation potential of 0.15 V (vs. Fc/Fc+), characteristic of the phenothiazine group and corresponds to a HOMO energy level estimate of −5.0 eV versus vacuum level. Triad 1 also displays two quasireversible reduction waves at −1.1 V (LUMO estimate = −3.7 eV) and −1.5 V (vs. Fc/Fc+) that are nearly identical to N-alkylated-NDI.62 Clearly, the optical gap – assessed from the intersection of the normalized absorption and emission spectra – at 2.9 eV and the electrochemical HOMO–LUMO estimate at 1.3 eV do not agree, again supporting the notion that each building block of triad 1 is electronically isolated.
Spectroelectrochemical investigations of triad 1 confirms the non-interacting building blocks. Under oxidizing potentials, spectroscopic signatures identified as the radical cation of PTZ with broad absorption features spanning from 400 nm to 800 nm emerge from the baseline, while the absorption features of NDI are unaffected.57 Likewise, upon applying reducing potentials, characteristic features consistent with the first and second electron reduction of NDI appear (Fig. S31, ESI†).62 Upon electrochemical reduction at the first reduction potential, the NDI band at 381 nm decreases commensurate with several new peaks increasing in intensity that are consistent with other NDI radical anion reports.63–66 Decreasing the reduction potential to the second peak in CV results in two sharp peaks at 399 nm and 420 nm and two broad transitions at 555 nm and 602 nm, consistent with reports of the NDI dianion.66,67 In all electrochemically reduced spectra, only the NDI absorption features change and the PTZ absorption features remain unchanged, indicating that the NDI moiety is reduced independently of the PTZ moieties. The reduction chemistry in the spectroelectrochemical experiments were reversible and the neutral triad 1 spectrum was regenerated upon applying positive potentials.
The solid-state properties of triad 1 were first assessed thermally with data shown in Fig. S22 and S23 (ESI†). Triad 1 showed reasonable thermal stability with decomposition starting 350 °C by thermal gravimetric analysis (TGA) and a differential scanning calorimetry (DSC) thermogram indicated a clearing transition at 140 °C (transition enthalpy of 2.4 J g−1). Upon cooling, the DSC thermogram showed the same liquid to mesophase transition with a hysteresis of 6 °C. The DSC results indicate the presence of a single mesophase. The magnitude of hysteresis rules out nematic phases, and the transition enthalpy is consistent with a columnar phase.
A drop-cast film of 1 on a microscope slide is shown in Fig. 4a as cross-polarized optical micrographs (POM) to assess the textures as a function of temperature. Fig. 4a is a drop-cast film that was held beyond its clearing temperature (150 °C) then cooled and held at 133 °C. Thin films of triad 1 were also formed successfully by spin-casting techniques and a POM image is included in Fig. S32 (ESI†).
 | ||
| Fig. 4 Top: Cross-polarized optical microscope images of drop cast films of 1 on glass cooled from its isotropic phase temperature and held at 133 °C (100× magnification). Note this image was taken with a quarter-wave plate to improve viewing contrast. Bottom: 2D GIWAXS data – log scale. | ||
Pseudo-focal conic textures emerge in Fig. 4a, consistent with hexagonal columnar phases.68–70 To further probe the structure of the sample, Grazing-Incidence Wide-Angle X-ray Scattering (GIWAXS) patterns were collected for films of triad 1 on a Si(100) surface, Fig. 4b. Azimuthally-integrated linecuts from χ = −85° to 0° are included in the Fig. S34 (ESI†). The diffraction peaks are few, but salient features are noteworthy for structural analysis.70–72 First, there is a single strong feature at q = 0.18 Å−1 that corresponds to a d-spacing of 3.5 nm. The d-spacing of this peak is consistent with intercolumnar spacing. No higher-order reflections were observed, making it impossible to definitively index the peak. But assuming that the packing is hexagonal columnar, the d-spacing of 3.5 nm would correspond to a lattice parameter, a, of 4.0 nm, which is equal to the molecular diameter (Fig. 5). In addition, a weak diffuse ring at q = 1.48 Å−1 is attributed to a weakly ordered intermolecular stacking distance of 4.3 Å, which is a feature commonly termed an ‘alkyl-halo’ region.71,72 The diffuse scattering observed in Fig. 4b indicates the structural distribution is isotropic, implying there is no preferred orientation with respect to the Si(100) surface, and that further isotropic evidence is provided by the orientation analysis in the Fig. S35 (ESI†). Despite the lack of peaks in GIWAXS and the small DSC peaks, there remains the possibility that the films could consist of a weakly crystalline phase. Fig. 5b shows the side view of DFT-energy minimized triad 1 to highlight the bent versus planar regions of the molecule and alternate views are shown in Fig S21 (ESI†). Fig. 5c proposes a packing model consistent with the structural, optical and X-ray data of a hexagonal columnar liquid crystalline phase highlighting the separated donor and acceptor portions of triad 1 in the packing of the bent PTZs and flat NDIs. The grey circles in Fig. 5c represent a randomly organized alkyl region, which implies that the bent-core PTZ moieties do not necessarily exhibit orbital overlap with subsequent columnar layers, leading to less-than-ideal charge transport pathways.
 | ||
| Fig. 5 (a) Overlay of 1 denoting the planar, bent and alkyl regions. (b) Side view of DFT-optimized 1 highlighting the bent versus planar cores. (c) 2-D schematic of packing arrangement into a hexagonal columnar phase showing phase segregation by shape. | ||
The self-assembly of 1 into columnar phases led us to assess the functionality of the material using chopped light voltammetry to quantify photocurrent generation. A cross-polarized optical microscope image of annealed spin-cast films of 1 (Fig. S32, ESI†) shows micro-size domains on ITO surfaces that are not homeotropically aligned. Note that these films were thinner spin-cast films compared to drop-cast films on glass shown in Fig. 4a, thus have smaller domains on this conductive substrate. Experiments that assess charge transfer through columns should result in enhanced photoresponses when ordered, since donor and acceptor regions could be separate. Fig. 6 shows the photocurrent generated from films of 1 being exposed to 472 nm light (100 mW cm−2) at different applied potentials under chopped light conditions. The applied potential was ramped from 0 V to 400 mV (vs. open circuit potentials) over 40 seconds. Beyond 400 mV, the films became unstable as assessed by defects that were visible under cross-polarized optical microscopy. Note, negative potential ranges were explored and resulted in film instability. Films of 1 showed photoresponses upon application of applied positive potentials that were enhanced when the amorphous spin-cast films were annealed The films were annealed by heating the film to above the clearing temperature, allowing the film to cool to at a rate of 1 °C min,1 and held at 150 °C for 30 min before removing the film from the heat source. The photocurrent enhancement of the annealed films compared to the amorphous films of 1 is shown by the integrated currents (photocharge) at 40 s, or 400 mV, and reveals approximately 2-fold enhancement (1.73 μC annealed vs. 0.87 μC amorphous). The enhanced photocurrent of the annealed films is speculated to result from two factors. First, the organization in the annealed films causes improved overlap between donor regions and acceptor regions, thus perhaps providing molecular-scale conduits to carry charges. Note that annealed films are not homeotropically aligned to the substrate, yet show enhanced photocurrents relative to amorphous films, which could suggest aligned microdomains within the film are responsible for the enhancement. Second, the annealed films also show enhanced absorption (1.7 times larger absorption) at the 472 nm excitation wavelength, leading to more photoexcited states being generated and thus potentially more charge carriers (Fig. S29, ESI†).
 | ||
| Fig. 6 Photocurrent generated from a film of 1 under a potential ramp from 0 V to 400 mV over 40 seconds with light chopped at 2 Hz at room temperature (24 °C). Lines show the photocurrents of ITO blank (black), unannealed film of 1 (red) and annealed film of 1 (blue). The right vertical axis shows the photocharge accumulated by the difference between the integrated currents of the on and off light source for the ITO blank (squares), unannealed film of 1 (circles) and annealed film of 1 (triangles). | ||
Given the thickness of the film estimated at 100 nm, as assessed by its capacitance from impedance spectroscopy, the magnitude of the photocurrent produced is quantifiable (three film samples, measure 10 times each). A phase separated comparator system that couples an oligothiophene donor with a perylenediimide using the hydrophobic/hydrophilic approach resulted in a photocurrent of 4 nA,36 compared to 400 nA here. However, note the photocurrents measured by Li and co-workers36 were carried out with an all-solid-state method and thus magnitudes are not comparable, especially considering the large area (3.1 cm2) electrode used in this work. A more recent report by Hecht and co-workers13 has shown a highly oriented donor–acceptor liquid crystalline film that exhibits a photocurrent of 45 nA using a two-contact device. Additionally, a columnar liquid crystalline film of a sub-phthalocyanine sandwiched between two ITO electrodes by Zhang and coworkers73 has demonstrated impressive light on–off current ratios of 103 and a windmill-shaped molecule, truxene, that form columnar liquid crystals disclosed by Liu and co-workers has demonstrated ambipolar charge transport, as assessed by transient photocurrents.74 Nevertheless, reasonable photocurrent magnitudes are achieved with films of 1, but a low Ion/Ioff ratio of 2.8 at 400 mV. High speed chronoamperometry of the chopped-light experiments was carried out and included in Fig. S33 (ESI†). The high temporal resolution data shows that the peak photocurrent is achieved within 1 ms of light exposure.
Conclusions
In summary, triad 1 represents a new class of mesogen that self-assembles into a columnar liquid crystalline phase demonstrating a novel approach to using bent molecular shape to achieve D–A segregation with a charge-carrying function. Furthermore, the self-assembled structure shows enhanced photocurrent generation over its amorphous phase, supporting the functional electronic aspect of triad 1. This D–A segregation approach may be generalizable and represent an orthogonal strategy to achieve ambipolar self-assembled functional materials. The unusual-shaped mesogen reported herein opens opportunities to exploit molecular shape whilst maintaining a columnar assembly for functional materials. However, much effort is needed to find suitable conditions that address the following issues: (1) homeotropic alignment of columnar stacks; (2) design of bent-shaped molecules that stack with effective overlap of pi-systems to tune charge transport properties; (3) detailed understanding of the balance of intermolecular forces that leads to effective columnar arrays between the alkyl dispersion, the planar aromatic interactions and bent structures. With more precise structural parameters of the columnar arrangement, an illuminated path forward to diverse applications could be possible, such as single component OLEDs or OPVs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. C. S. thanks NSERC of Canada Discovery Grants program (RGPIN-2019-04851) for funding. K. D. T. and D. T. H. thank NSERC PGS-D program and Alberta Innovates for scholarship support. T. L. K. thanks NSERC (RGPIN-2017-03732) and the University of Saskatchewan for financial support. T. L. K. is a Canada Research Chair in Photovoltaics. The research was undertaken, in part, thanks to funding from the Canada Research Chair program. C. L. R. thanks NSERC for scholarship support. The Canadian Light Source (CLS) is supported by CFI, NSERC, the University of Saskatchewan, the Government of Saskatchewan, Western Economic Diversification Canada, the National Research Council Canada, and the Canadian Institutes of Health Research. Technical support from HXMA beamline scientist Dr Chang-Yong Kim is gratefully acknowledged.Notes and references
- A. Pegenau, T. Hegmann, C. Tschierske and S. Diele, Chem. – Eur. J., 1999, 5, 1643–1660 CrossRef CAS.
- C. Tschierske, J. Mater. Chem., 1998, 8, 1485–1508 RSC.
- B. R. Kaafarani, Chem. Mater., 2011, 23, 378–396 CrossRef CAS.
- T. Wöhrle, I. Wurzbach, J. Kirres, A. Kostidou, N. Kapernaum, J. Litterscheidt, J. C. Haenle, P. Staffeld, A. Baro, F. Giesselmann and S. Laschat, Chem. Rev., 2016, 116, 1139–1241 CrossRef PubMed.
- W.-S. Li, Y. Yamamoto, T. Fukushima, A. Saeki, S. Seki, S. Tagawa, H. Masunaga, S. Sasaki, M. Takata and T. Aida, J. Am. Chem. Soc., 2008, 130, 8886–8887 CrossRef CAS PubMed.
- H. Hayashi, W. Nihashi, T. Umeyama, Y. Matano, S. Seki, Y. Shimizu and H. Imahori, J. Am. Chem. Soc., 2011, 133, 10736–10739 CrossRef CAS PubMed.
- T. Sakurai, Y. Tsutsui, K. Kato, M. Takata and S. Seki, J. Mater. Chem. C, 2016, 4, 1490–1496 RSC.
- Y. Xiao, X. Su, L. Sosa-Vargas, E. Lacaze, B. Heinrich, B. Donnio, D. Kreher, F. Mathevet and A.-J. Attias, CrystEngComm, 2016, 18, 4787–4798 RSC.
- K. Zhao, L. An, X. Zhang, W. Yu, P. Hu, B. Wang, J. Xu, Q. Zeng, H. Monobe, Y. Shimizu, B. Heinrich and B. Donnio, Chem. – Eur. J., 2015, 21, 10379–10390 CrossRef CAS PubMed.
- A. Watarai, K. Ohta and M. Yasutake, J. Porphyrins Phthalocyanines, 2016, 20, 1444–1456 CrossRef CAS.
- M. Lehmann and M. Hügel, Angew. Chem., Int. Ed., 2015, 54, 4110–4114 CrossRef CAS PubMed.
- D. Felder-Flesch, D. Guillon and B. Donnio, Fullerene-Containing Liquid Crystals in Handbook of Liquid Crystals, 2013 Search PubMed.
- M. Hecht, T. Schlossarek, S. Ghosh, Y. Tsutsui, A. Schmiedel, M. Holzapfel, M. Stolte, C. Lambert, S. Seki, M. Lehmann and F. Würthner, ACS Appl. Nano Mater., 2020, 3, 10234–10245 CrossRef CAS.
- M. Dechant, M. Lehmann, G. Uzurano, A. Fujii and M. Ozaki, J. Mater. Chem. C, 2021, 9, 5689–5698 RSC.
- J. Roncali, Adv. Energy Mater., 2011, 1, 147–160 CrossRef CAS.
- S. Izawa, K. Hashimoto and K. Tajima, Phys. Chem. Chem. Phys., 2012, 14, 16138–16142 RSC.
- T. Nishizawa, K. Tajima and K. Hashimoto, J. Mater. Chem., 2007, 17, 2440–2445 RSC.
- C. Guo, Y.-H. Lin, M. D. Witman, K. A. Smith, C. Wang, A. Hexemer, J. Strzalka, E. D. Gomez and R. Verduzco, Nano Lett., 2013, 13, 2957–2963 CrossRef CAS PubMed.
- S. M. Lindner, S. Hüttner, A. Chiche, M. Thelakkat and G. Krausch, Angew. Chem., Int. Ed., 2006, 45, 3364–3368 CrossRef CAS PubMed.
- M. Sommer, S. Hüttner, U. Steiner and M. Thelakkat, Appl. Phys. Lett., 2009, 95, 183308 CrossRef.
- M. Sommer, A. S. Lang and M. Thelakkat, Angew. Chem., Int. Ed., 2008, 47, 7901–7904 CrossRef CAS PubMed.
- M. Sommer, S. M. Lindner and M. Thelakkat, Adv. Funct. Mater., 2007, 17, 1493–1500 CrossRef CAS.
- S.-S. Sun, C. Zhang, A. Ledbetter, S. Choi, K. Seo, C. E. Bonner, M. Drees and N. S. Sariciftci, Appl. Phys. Lett., 2007, 90, 043117 CrossRef.
- Q. Zhang, A. Cirpan, T. P. Russell and T. Emrick, Macromolecules, 2009, 42, 1079–1082 CrossRef CAS.
- A. Cravino, P. Leriche, O. Alévêque, S. Roquet and J. Roncali, Adv. Mater., 2006, 18, 3033–3037 CrossRef CAS.
- O. Hagemann, M. Jørgensen and F. C. Krebs, J. Org. Chem., 2006, 71, 5546–5559 CrossRef CAS PubMed.
- C. He, Q. He, Y. He, Y. Li, F. Bai, C. Yang, Y. Ding, L. Wang and J. Ye, Sol. Energy Mater. Sol. Cells, 2006, 90(7), 1815–1827 CrossRef CAS.
- X. Sun, Y. Liu, X. Xu, C. Yang, G. Yu, S. Chen, Z. Zhao, W. Qiu, Y. Li and D. Zhu, J. Phys. Chem. B, 2005, 109, 10786–10792 CrossRef CAS PubMed.
- B. Mu, J. Chen, K. Chen, C. Zhang and D. Chen, J. Mater. Chem. C, 2021, 9, 8029–8036 RSC.
- I. Bala, W.-Y. Yang, S. P. Gupta, J. De, R. A. K. Yadav, D. P. Singh, D. K. Dubey, J.-H. Jou, R. Douali and S. K. Pal, J. Mater. Chem. C, 2019, 7, 5724–5738 RSC.
- A. G. Bé, C. Tran, R. Sechrist and J. J. Reczek, Org. Lett., 2015, 17, 4834–4837 CrossRef.
- A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2038–2054 CrossRef CAS PubMed.
- P. H. J. Kouwer, W. F. Jager, W. J. Mijs and S. J. Picken, Macromolecules, 2001, 34, 7582–7584 CrossRef CAS.
- J. Yamada, H. Akutsu, H. Nishikawa and K. Kikuchi, Chem. Rev., 2004, 104, 5057–5084 CrossRef CAS PubMed.
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS PubMed.
- W.-S. Li, A. Saeki, Y. Yamamoto, T. Fukushima, S. Seki, N. Ishii, K. Kato, M. Takata and T. Aida, Chem. – Asian J., 2010, 5, 1566–1572 CrossRef CAS PubMed.
- U. Scherf, A. Gutacker and N. Koenen, Acc. Chem. Res., 2008, 41, 1086–1097 CrossRef CAS PubMed.
- I. Bala, J. De, S. P. Gupta, U. K. Pandey and S. K. Pal, J. Mater. Chem. C, 2021, 9, 8552–8561 RSC.
- F. Wurthner, S. Yao, B. Heise and C. Tschierske, Chem. Commun., 2001, 2260–2261 RSC.
- D. Goldmann, D. Janietz, C. Schmidt and J. H. Wendorff, J. Mater. Chem., 2004, 14, 1521–1525 RSC.
- B. Feringán, P. Romero, J. L. Serrano, C. L. Folcia, J. Etxebarria, J. Ortega, R. Termine, A. Golemme, R. Giménez and T. Sierra, J. Am. Chem. Soc., 2016, 138, 12511–12518 CrossRef PubMed.
- H. K. Bisoyi, V. A. Raghunathan and S. Kumar, Chem. Commun., 2009, 7003–7005 RSC.
- M. Lehmann, M. Dechant, M. Lambov and T. Ghosh, Acc. Chem. Res., 2019, 52, 1653–1664 CrossRef CAS PubMed.
- X. Zeng, G. Ungar, Y. Liu, V. Percec, A. E. Dulcey and J. K. Hobbs, Nature, 2004, 428, 157 CrossRef CAS PubMed.
- C.-L. Wang, W.-B. Zhang, X. Yu, K. Yue, H.-J. Sun, C.-H. Hsu, C.-S. Hsu, J. Joseph, D. A. Modarelli and S. Z. D. Cheng, Chem. – Asian J., 2013, 8, 947–955 CrossRef CAS PubMed.
- S. R. Varanasi, O. A. Guskova, A. John and J. U. Sommer, J. Chem. Phys., 2015, 142, 224308 CrossRef CAS PubMed.
- F.-A. Teng, Y. Cao, Y.-J. Qi, M. Huang, Z.-W. Han, S. Z. D. Cheng, W.-B. Zhang and H. Li, Chem. – Asian J., 2013, 8, 1223–1231 CrossRef CAS PubMed.
- H.-J. Sun, Y. Tu, C.-L. Wang, R. M. Van Horn, C.-C. Tsai, M. J. Graham, B. Sun, B. Lotz, W.-B. Zhang and S. Z. D. Cheng, J. Mater. Chem., 2011, 21, 14240–14247 RSC.
- R. W. Date and D. W. Bruce, J. Am. Chem. Soc., 2003, 125, 9012–9013 CrossRef CAS PubMed.
- T. Niori, T. Sekine, J. Watanabe, T. Furukawa and H. Takezoe, J. Mater. Chem., 1996, 6, 1231–1233 RSC.
- S. Norvez, F.-G. Tournilhac, P. Bassoul and P. Herson, Chem. Mater., 2001, 13, 2552–2561 CrossRef CAS.
- M. Lehmann, S. Maisch, N. Scheuring, J. Carvalho, C. Cruz, P. J. Sebastião and R. Y. Dong, Soft Matter, 2019, 15, 8496–8511 RSC.
- C. Tschierske, Microsegregation in Liquid Crystalline Systems: Basic Concepts in Handbook of Liquid Crystals, 2014 Search PubMed.
- A. Pegenau, X. Hong Cheng, C. Tschierske, P. Goring and S. Diele, New J. Chem., 1999, 23, 465–467 RSC.
- K. D. Theriault, C. Radford, M. Parvez, B. Heyne and T. C. Sutherland, Phys. Chem. Chem. Phys., 2015, 17, 20903–20911 RSC.
- Z. Jiang, J. Appl. Crystallogr., 2015, 48, 917–926 CrossRef CAS.
- K. D. Theriault and T. C. Sutherland, Phys. Chem. Chem. Phys., 2014, 16, 12266–12274 RSC.
- T. C. Barros, S. Brochsztain, V. G. Toscano, P. B. Filho and M. J. Politi, J. Photochem. Photobiol., A, 1997, 111, 97–104 CrossRef CAS.
- C. Kulkarni, G. Periyasamy, S. Balasubramanian and S. J. George, Phys. Chem. Chem. Phys., 2014, 16, 14661–14664 RSC.
- D.-G. Chen, T.-C. Lin, Y.-A. Chen, Y.-H. Chen, T.-C. Lin, Y.-T. Chen and P.-T. Chou, J. Phys. Chem. C, 2018, 122, 12215–12221 CrossRef CAS.
- M. Hempe, A. K. Harrison, J. S. Ward, A. S. Batsanov, M. A. Fox, F. B. Dias and M. R. Bryce, J. Org. Chem., 2021, 86, 429–445 CrossRef CAS PubMed.
- S. V. Bhosale, C. H. Jani and S. J. Langford, Chem. Soc. Rev., 2008, 37, 331–342 RSC.
- S. M. Mackinnon and Z. Y. Wang, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3467–3475 CrossRef CAS.
- L. Wang, G. W. Goodloe, B. J. Stallman and V. Cammarata, Chem. Mater., 1996, 8, 1175–1181 CrossRef CAS.
- V. V. Roznyatovskiy, D. M. Gardner, S. W. Eaton and M. R. Wasielewski, Org. Lett., 2014, 16, 696–699 CrossRef CAS PubMed.
- A. J. Taylor, E. S. Davies, J. A. Weinstein, I. V. Sazanovich, O. V. Bouganov, S. A. Tikhomirov, M. Towrie, J. McMaster and C. D. Garner, Inorg. Chem., 2012, 51, 13181–13194 CrossRef CAS PubMed.
- I. V. Sazanovich, M. A. H. Alamiry, J. Best, R. D. Bennett, O. V. Bouganov, E. S. Davies, V. P. Grivin, A. J. H. M. Meijer, V. F. Plyusnin, K. L. Ronayne, A. H. Shelton, S. A. Tikhomirov, M. Towrie and J. A. Weinstein, Inorg. Chem., 2008, 47, 10432–10445 CrossRef CAS PubMed.
- S. Leng, L. H. Chan, J. Jing, J. Hu, R. M. Moustafa, R. M. V. Horn, M. J. Graham, B. Sun, M. Zhu, K.-U. Jeong, B. R. Kaafarani, W. Zhang, F. W. Harris and S. Z. D. Cheng, Soft Matter, 2009, 6, 100–112 RSC.
- H. Bock and W. Helfrich, Liq. Cryst., 1992, 12, 697–703 CrossRef CAS.
- S.-J. Yoon, J. H. Kim, K. S. Kim, J. W. Chung, B. Heinrich, F. Mathevet, P. Kim, B. Donnio, A.-J. Attias, D. Kim and S. Y. Park, Adv. Funct. Mater., 2012, 22, 61–69 CrossRef CAS.
- E. J. Foster, R. B. Jones, C. Lavigueur and V. E. Williams, J. Am. Chem. Soc., 2006, 128, 8569–8574 CrossRef CAS PubMed.
- P. Herwig, C. W. Kayser, K. Müllen and H. W. Spiess, Adv. Mater., 1996, 8, 510–513 CrossRef CAS.
- C. Zhang, K. Nakano, M. Nakamura, F. Araoka, K. Tajima and D. Miyajima, J. Am. Chem. Soc., 2020, 142, 3326–3330 CrossRef CAS PubMed.
- X. Liu, T. Usui and J. Hanna, Chem. Mater., 2014, 26, 5437–5440 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental; synthesis; 1H- and 13C-NMR spectra; FTIR; absorption spectra; emission spectra; DFT computations; DSC, TGA; P-XRD; GIWAXS; transient photocurrents. See DOI: 10.1039/d1ma00452b |
| This journal is © The Royal Society of Chemistry 2022 |