
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poly(lactic-co-glycolic acid) encapsulated platinum nanoparticles for cancer treatment†
Aida
López Ruiz
 ,
Evaristo
Villaseco Arribas
and
Kathleen
McEnnis
*
,
Evaristo
Villaseco Arribas
and
Kathleen
McEnnis
*
Department of Chemical and Materials Engineering, New Jersey Institute of Technology, 161 Warren St, Newark, NJ 07102-1982, USA. E-mail: mcennis@njit.edu
First published on 16th February 2022
Abstract
Metal nanoparticles have been of interest to the field of nanomedicine due to their potential for cancer treatment, diagnosis, and antimicrobial activity. Particularly, platinum nanoparticles (PtNPs) have been reported to have remarkable anticancer effects and low toxicity in healthy cells. In this paper, we propose a delivery system for PtNPs as a substitute for common chemotherapy treatment. The anticancer effect of PtNPs was tested against triple negative breast cancer (TNBC) showing no cell viability for most of the concentrations used, while not being toxic against fibroblasts. To improve accumulation at the tumor site, a delivery system based on poly(lactic-co-glycolic acid) (PLGA) particles was used. Encapsulation was achieved by nanoemulsion, resulting in 59% PLGA particles containing PtNPs, and 12.5% PtNPs by weight. To improve the circulation time and specificity of the system, the PLGA particle surface was modified. Passive targeting with poly(ethylene glycol) (PEG) and active targeting with an antibody (anti-EGFR) were used, obtaining two different sets of particles, Pt–PLGA–PEG and Pt–PLGA–PEG–EGFR. To evaluate both sets of particles, cell studies were carried out against TNBC. Cell experiments demonstrated that Pt–PLGA–PEG–EGFR particles were more effective as a cancer treatment than Pt–PLGA–PEG particles, however, free PtNPs were more effective than the encapsulated PtNPs. This was likely due to the time needed for PtNPs to release from PLGA, however, the amount of Pt internalized in the cells was greater with the Pt–PLGA–PEG–EGFR particles. These results suggest that the Pt NPs encapsulated in PLGA modified with a combination of PEG and EGFR targeting will provide the most effective therapy for future in vivo experiments. In summary, this study lays the groundwork for future cancer treatments based on PtNPs delivered with targeted PLGA particles.
Introduction
Nanotechnology is a multidisciplinary field with numerous applications including cancer treatment and detection. Noble metal based nanoparticles, such as palladium, platinum, gold, and silver have been studied in the field of medicine and pharmaceuticals due to their therapeutic potential.1–7 In particular, platinum nanoparticles (PtNPs) have been reported with promising anticancer results.8–14 There is evidence of PtNPs’ capacity to enter the cell and damage DNA by releasing platinum ions.15,16 The toxicity of PtNPs needs to be further studied as there are conflicting reports on whether they are toxic for all cell types or just cancer cells.17–19 For instance, PtNPs altered the heart rate in mice but did not produce a deadly toxicity.20 Li et al. tested PtNPs with mice in vivo finding no toxicity by histological analysis on several organs, however, a large accumulation of PtNPs was observed in the liver, spleen and kidney.21 The Food and Drug Administration (FDA), however, has concluded that platinum in a zero oxidation state has the lowest toxicity compared to other oxidation states.22 The low toxicity of PtNPs compared to common chemotherapeutic drugs like cisplatin is an advantage that needs to be further studied.The toxicity of metallic nanoparticles when in contact with oxidizing agents from blood is a major limitation to their use in nanomedicine.19 A drug delivery system, however, can decrease toxicity for the patient by directly supplying the drug to the target, resulting in less drug needed overall.23 The usage of particles for drug delivery increases drug efficacy, specificity, tolerability, and therapeutic index.24,25 Poly(lactic-co-glycolic acid) (PLGA) based systems are frequently employed as delivery vehicles due to their biodegradability, biocompatibility, and minimal toxicity.26–31 There are reports where PLGA particles have been used with traditional drugs for cancer treatment, such as docetaxel and paclitaxel.25,26,32 Nonetheless, the toxicity of these drugs in off-target accumulation areas, such as the liver, typically prevents the application of these particles in clinical trials.33–35 Delivering PtNPs with targeted PLGA particles, however, is a novel, synergistic approach to minimize toxicity to healthy cells.
Nanoparticles that circulate in the bloodstream are recognized by proteins and these proteins attach to the surface forming a nanoparticle–protein complex (protein corona) that triggers the phagocytic system recognition and removal from the bloodstream.36,37 Surface modification of the particles with polymers such as polyethylene glycol (PEG) have been found to enhance the circulation life-time of the particles.38 Surface modifications with PEG are a commonly used procedure to enhance the properties of drug delivery systems through passive targeting. Additionally, ligands of specific biomarkers, monoclonal antibodies, peptides, or aptamers can be used to provide the most effective targeted therapy through active targeting.35,39–41 In the current project, the anticancer activity of PtNPs was tested against triple negative breast cancer (TNBC). Triple negative breast cancer is defined by the lack of overexpression of the three common receptors used to classify breast cancer.42 Therefore, there are very few available targets and no targeted therapies currently available.43–45 Epidermal growth factor (EGF) is a receptor that promotes cell proliferation migration and angiogenesis (development of new blood vessels)46 and is overexpressed in many tumors including breast cancer.47 Therefore, many strategies based on antibodies have been studied to block EGFR.46 The anti-EGFR monoclonal antibody, Cetuximab, has shown promising results for targeting metastatic TNBC.48,49
In the present study, we synthesized PtNPs to use as an anticancer treatment with no toxicity for healthy cells. The PtNPs were encapsulated with PLGA and surface modifications were performed to achieve increased cellular uptake (Fig. 1). This study adds further evidence of the effectiveness of PtNPs as a possible cancer treatment. Additionally, the encapsulation of PtNPs in a delivery system like PLGA and further surface modifications has not been investigated before. The cytotoxicity of PtNPs was tested with TNBC cells and fibroblasts showing that the PtNPs were highly effective against cancer cells with limited toxicity to the healthy fibroblast cells. Cisplatin, a common chemotherapeutic drug that contains a central platinum ion, was used as a control group that showed toxicity at all concentrations tested for both kinds of cells. PtNPs were encapsulated with PLGA by nanoemulsion, and then surface modifications were performed to enhance the effect of PtNPs on the targeted cells by adding PEG and an anti-EGFR ligand. Finally, cytotoxicity with TNBC of actively and passively targeted particles was compared, demonstrating increased cell death by particles conjugated with the antibody. This research lays the groundwork for an alternative cancer treatment for triple negative breast cancer with low toxicity to healthy cells.
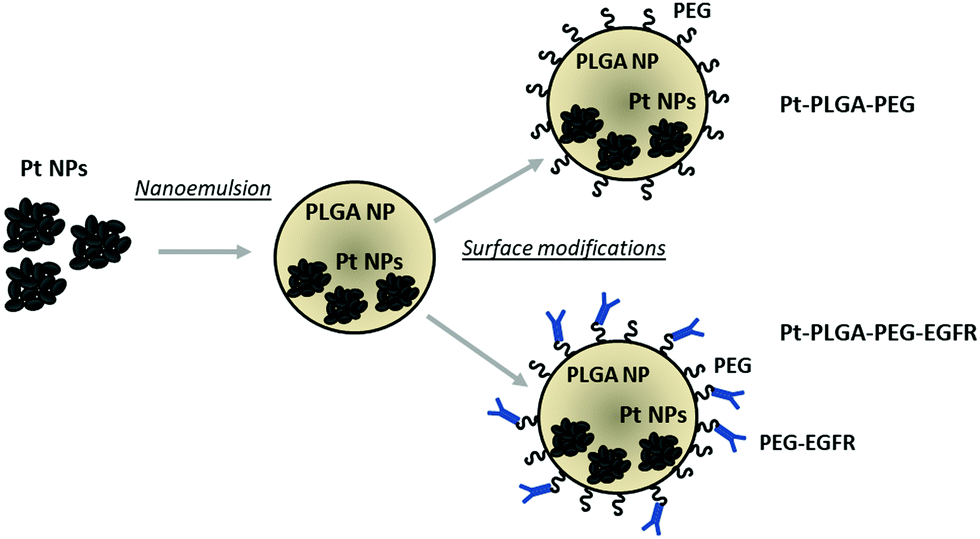 | ||
| Fig. 1 Schematic representation of PtNPs encapsulation with PLGA and surface modifications. | ||
Experimental
Materials
Hexadecyltrimethylammonium bromide (CTab), poly(ethylene glycol) 2-aminoethyl ether biotin (PEG–biotin) (Mn 3400), dichloromethane, N,N-dimethylformamide, TWEEN® 20, poly(vinyl alcohol) (PVA), chloroform, Brij® 58, L-ascorbic acid, potassium tetrachloroplatinate(II) and acetonitrile anhydrous 99.8% were purchased from Sigma Aldrich. 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide–HCl (EDC), sulfo-NHS and neutravidin biotin binding protein were purchased from Thermo Scientific. PLGA (Purasorb 5002A) with a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ratio of glycolic acid: lactic acid and 5000 g mol−1 molecular weight was provided by Corbin Purac. Amino-poly(ethylene glycol) (Mw 5000 Da) was purchased from Nanocs. Rabbit mAB Cetuximab Biotin and Goat anti-Rabbit IgG (H + L) secondary [Dylight 488] were purchased from Novus Biologicals. Cisplatin CRS was purchased from European Pharmacopeia Reference Standard. Dulbecco's modification of Eagle's medium (DMEM) was purchased from Corning. Fetal bovine serum (FBS), antibiotic–antimycotic, and phosphate buffered saline (PBS) were purchased from Gibco. Fibroblast Growth Basal Medium (FBM) was purchased from Lonza Corp. CellTiter 96® AQueous One Solution Cell Proliferation assays (MTS) were purchased from Promega. Live/dead cell imaging kit was purchased from Invitrogen. Two cell lines were used, triple negative breast cancer (TNBC) (MDA-MB-231) purchased from American Type Culture Collection (ATCC) and human ventricular cardiac fibroblasts (NHCF-V) from Lonza Corp.
:50 ratio of glycolic acid: lactic acid and 5000 g mol−1 molecular weight was provided by Corbin Purac. Amino-poly(ethylene glycol) (Mw 5000 Da) was purchased from Nanocs. Rabbit mAB Cetuximab Biotin and Goat anti-Rabbit IgG (H + L) secondary [Dylight 488] were purchased from Novus Biologicals. Cisplatin CRS was purchased from European Pharmacopeia Reference Standard. Dulbecco's modification of Eagle's medium (DMEM) was purchased from Corning. Fetal bovine serum (FBS), antibiotic–antimycotic, and phosphate buffered saline (PBS) were purchased from Gibco. Fibroblast Growth Basal Medium (FBM) was purchased from Lonza Corp. CellTiter 96® AQueous One Solution Cell Proliferation assays (MTS) were purchased from Promega. Live/dead cell imaging kit was purchased from Invitrogen. Two cell lines were used, triple negative breast cancer (TNBC) (MDA-MB-231) purchased from American Type Culture Collection (ATCC) and human ventricular cardiac fibroblasts (NHCF-V) from Lonza Corp.
Methods
PEGylation. The previously freeze-dried particles were redispersed in 1 mL of MES buffer solution containing 0.4 mg EDC. The solution was left reacting for 10 min at room temperature on a rotator. After the particles reacted with EDC, 20 μL of a solution of 1.1 mg sulfo-NHS with 40 μL MES was added to the particle solution, vortexed, and left reacting on the rotator for 10–15 min at room temperature. The particles were centrifuged at 22
136 RCF for 1 h, then the supernatant was removed and 1 mL of PBS was added and tip sonicated. Different amounts of PEG (5 kDa) and PEG–Biotin (3 kDa) were added to the solution and left reacting for 2 h on the rotator at room temperature. Finally, to remove the excess of PEG, unreacted particles were washed three times by centrifuging at 22136 RCF for 1 h and replacing the supernatant with PBS.
Antibody attachment. The biotin labeled antibody was attached to the particles through conjugation with neutravidin. A solution of 10 mg neutravidin in 0.5 mL 0.1% Tween PBS was added into a well on a 4-well plate with a stir bar. The particles previously tip sonicated were added dropwise into the neutravidin solution and left reacting for 3 h at room temperature. After the particles reacted, the solution was centrifuged at 22
136 RCF for 1 h and washed three times with PBS, to remove the unreacted neutravidin. The particles were then tip sonicated and 13.3 μL of biotin–antibody was added to the tube and left reacting for 3 h on a rotator at room temperature. Particles were then centrifuged at 22136 RCF for 1 h and washed three times in PBS, to remove the unreacted antibody.
CellTiter 96® AQueous One Solution Cell Proliferation assays (MTS) were performed for 1, 3, and 5 days. Cells were cultured for 24 hours at 37 °C (50000 cells mL−1) in a 96-well plate. After incubation, the media was replaced with different concentrations of treatment and then incubated for 1, 3, or 5 days without replacing the media. After incubating for 1, 3, or 5 days, cell viability was then determined by removing the media and substituting with the MTS solution, followed by incubation for 4 hours at 37 °C. Cell viability was quantified by measuring absorbance at 490 nm using a spectrophotometer (SpectraMax Paradigm, Molecular Devices). All experiments were performed in triplicate for each time point and on three different days.
000 cells mL−1) were cultured on a 6-well plate with glass slides. After a day of incubation, media was removed, and a solution of media and treatment was added to the cells. Cisplatin (50 μg mL−1), PtNPs (200 μg mL−1), Pt–PLGA–PEG (100 μg mL−1) or Pt–PLGA–PEG–EGFR (100 μg mL−1) was used. After the required days of treatment (1, 3, and 5 days), cells were washed with PBS. Then different fluorescent dyes (1.25 μL of each), calcein-AM, and ethidium homodimer-1 from the live/dead cell imaging kit were added to a PBS solution (1 mL). The plate was kept in dark conditions for 15 min. Finally, the glass slides were removed from the plate and placed on a microscope slide for imaging.
Samples (50 μL) were mixed with concentrated double-distilled nitric acid (DD HNO3; 200 μL) in metal-free polypropylene centrifuge tubes, sonicated for 60 min, then digested using a CEM microwave system (CEM, Matthews, NC) in 5 minute intervals for a total of 30 min at 300 W (75% power). Samples were diluted with MilliQ water to produce an acid concentration of 5% HNO3 for analysis via ICP-MS (dilution factor = 80×).
Pt concentrations in digested samples were quantified using a Nu AttoM Inductively Coupled Plasma Mass Spectrometer (ICP-MS), maintained in the Chemical Analysis Facility Core at the Environmental and Occupational Health Sciences Institute of Rutgers University, and operated at low resolution (300). The operating conditions were as follows: RF power of 1550 W, carrier gas flow of 1.00 L min−1 Ar, and nebulizer gas flow of ∼34 psi Ar. Three replicates at masses 194 Pt, 195 Pt, and 196 Pt were measured in deflector jump mode with 1.5 ms peak dwell time, 300 sweeps, and 10 cycles, and averaged (RSD <5%). Calibration standards were prepared daily with Pt concentrations ranging from 0.001– 1ppb, in 5% HNO3, with an instrument detection limit <0.005 ppb and a method detection limit of 0.4–0.8 ppb. Sample concentrations were determined using a linear regression through seven standards, with a correlation coefficient > 0.999 for all runs. Quality control standard (SPEX CertiPrep Multi-element Solution 3) was repeatedly measured after every sixth sample to account for instrument drift and reproduced with RSD < 4% (n = 6–11 dependent on batch size). Four (4) procedural blanks were prepared alongside samples to account for acid background contribution and/or contamination during sample preparation; all blanks measured below the detection limit (<0.005 ppb). Four (4) matrix matched standards were prepared by spiking Pt into control cell media (0.25 ppb) and measured to account for matrix effects (98–103% recovery). The percent uptake of Pt was determined by comparing the concentration of Pt found in the cells to the concentration of Pt in the original Pt solution incubated with the cells.
Statistical analysis
Data were analyzed by a one-way ANOVA, followed by Tukey's post-test. Data were considered statistically significant when the p-values were <0.05, and p-values >0.05 were considered not statistically significant.Results and discussion
Evaluation of platinum nanoparticles
In recent years platinum nanoparticles have shown promising results as an anticancer agent.52 Several studies have found that PtNPs of 50 nm have an anticancer effect, however other studies showed that PtNPs of 5 nm and 20 nm had no anticancer effect.10,21,50,53,54 Therefore, in the present study, platinum nanoparticles were synthesized by chemical reduction following the methodology of Shim et al., to achieve a size of 50 nm.50 The size, shape, and crystallinity of the PtNPs were determined by TEM and X-ray diffraction (XRD). By image analysis, a dendrimer shape was observed with a size distribution of 45 nm ± 10 nm, consistent with the size and shape previously reported10,21,50,55 (Fig. 2). Crystallinity was determined by using XRD (Fig. 2), showing peaks corresponding to the (111), (200), (220), (311), and (222) planes, in agreement with the data reported by Shim et al.50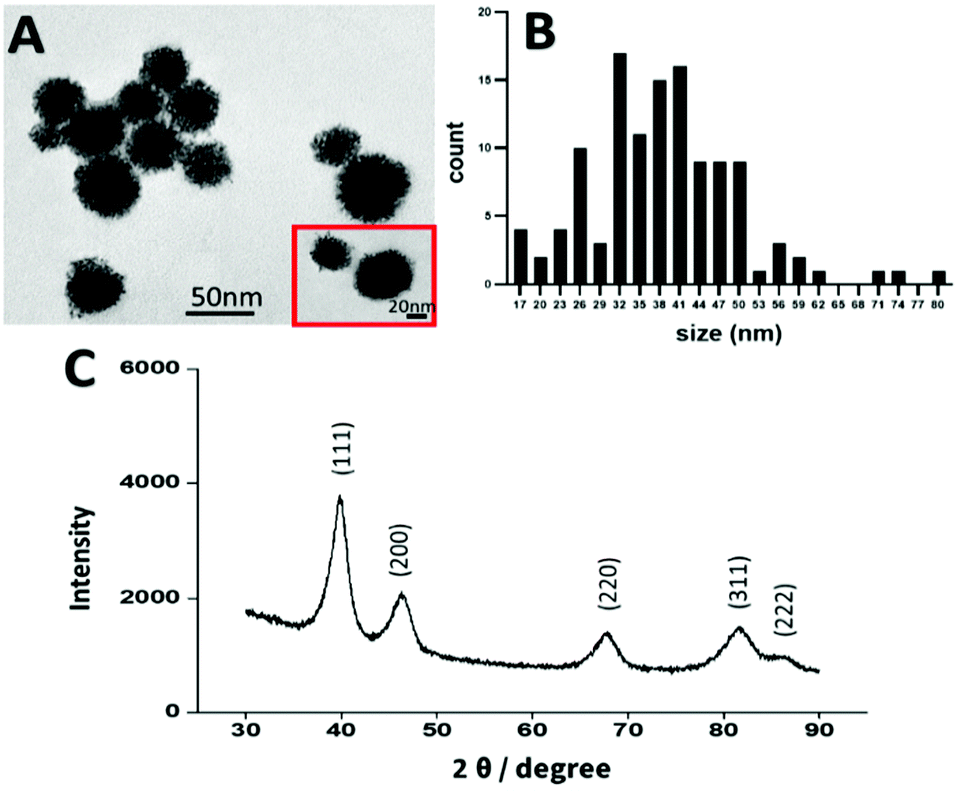 | ||
| Fig. 2 (A) TEM images of PtNPs, (B) Size distribution, (C) X-ray diffraction of PtNPs. | ||
To assess the effectiveness of PtNPs as an anticancer agent and the toxicity for healthy cells, two different cell lines were used: MDA-MB-231 (TNBC) and NHCF-V (fibroblasts). Cisplatin was used as a control representing a current FDA approved treatment for breast cancer. Cell viability studies were performed using an MTS assay with different concentrations of PtNPs and cisplatin for 1, 3, and 5 days for both cell lines. Through these experiments, the inhibitory concentration of treatment needed (IC50) was calculated.
PtNPs incubated with TNBC show a dose dependent anticancer effect. Cell viability declined visibly by using PtNPs for all concentrations tested and showed statistically significant differences compared to the untreated control cells. By increasing PtNP concentrations, cell viability decreased (Fig. 3A). At the lowest concentration tested (10 μg mL−1 PtNPs) cell viability was 80% whereas for the highest concentration (200 μg mL−1 PtNPs) cell viability decreased to 2% at 5 days incubation period. A time-dependent effect was also observed, with longer treatment with PtNPs resulting in less viability.
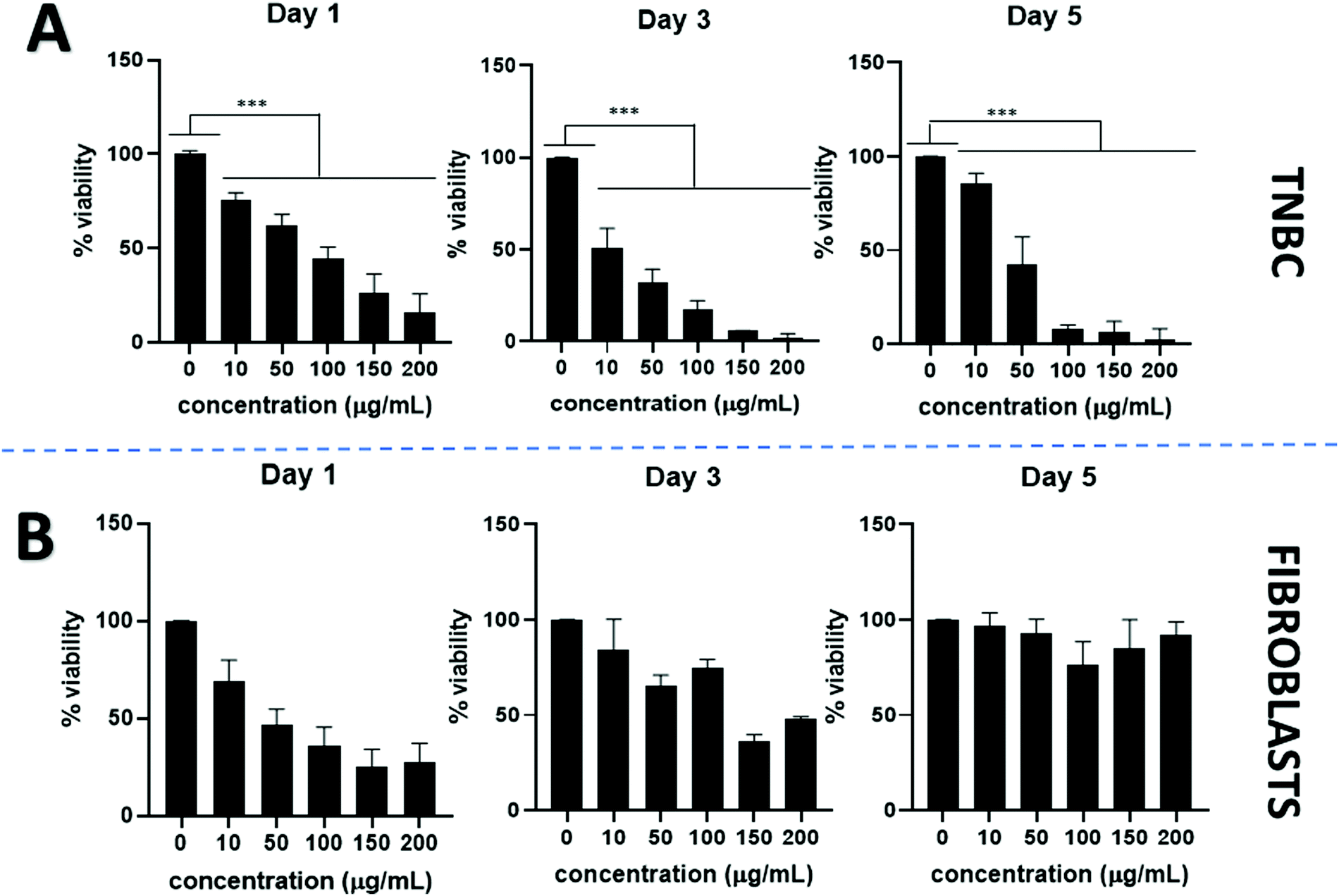 | ||
| Fig. 3 Cell viability for PtNPs using (A) TNBC and, (B) fibroblasts, for 1, 3, and 5 days with different concentrations of treatment. Results are expressed as mean + SD (***p < 0.001). Therefore, PtNPs are effective against TNBC but do not affect fibroblasts. | ||
Cytotoxicity with healthy cells (fibroblasts) was tested at the same concentrations used for TNBC treatments (Fig. 3B). An initial decrease in the cell viability was observed after 1 day of treatment, however, after a period of 3 and 5 days, the cell viability increased until there were no statistically significant differences in the cell viability between the untreated control and the use of PtNP treatment after 5 days at all concentrations.
Cisplatin was used as a comparison with a common chemotherapeutic drug. Although cisplatin is more effective than PtNPs at treating TNBC (Fig. 4A) its toxicity to healthy cells constitutes one of its main drawbacks. Lower concentrations of cisplatin were further tested against TNBC to assess a more accurate minimum inhibitory concentration (Fig. S1, ESI†). As shown in Fig. 4B, cisplatin did not display selectivity and showed high toxicity for both healthy and cancer cells.
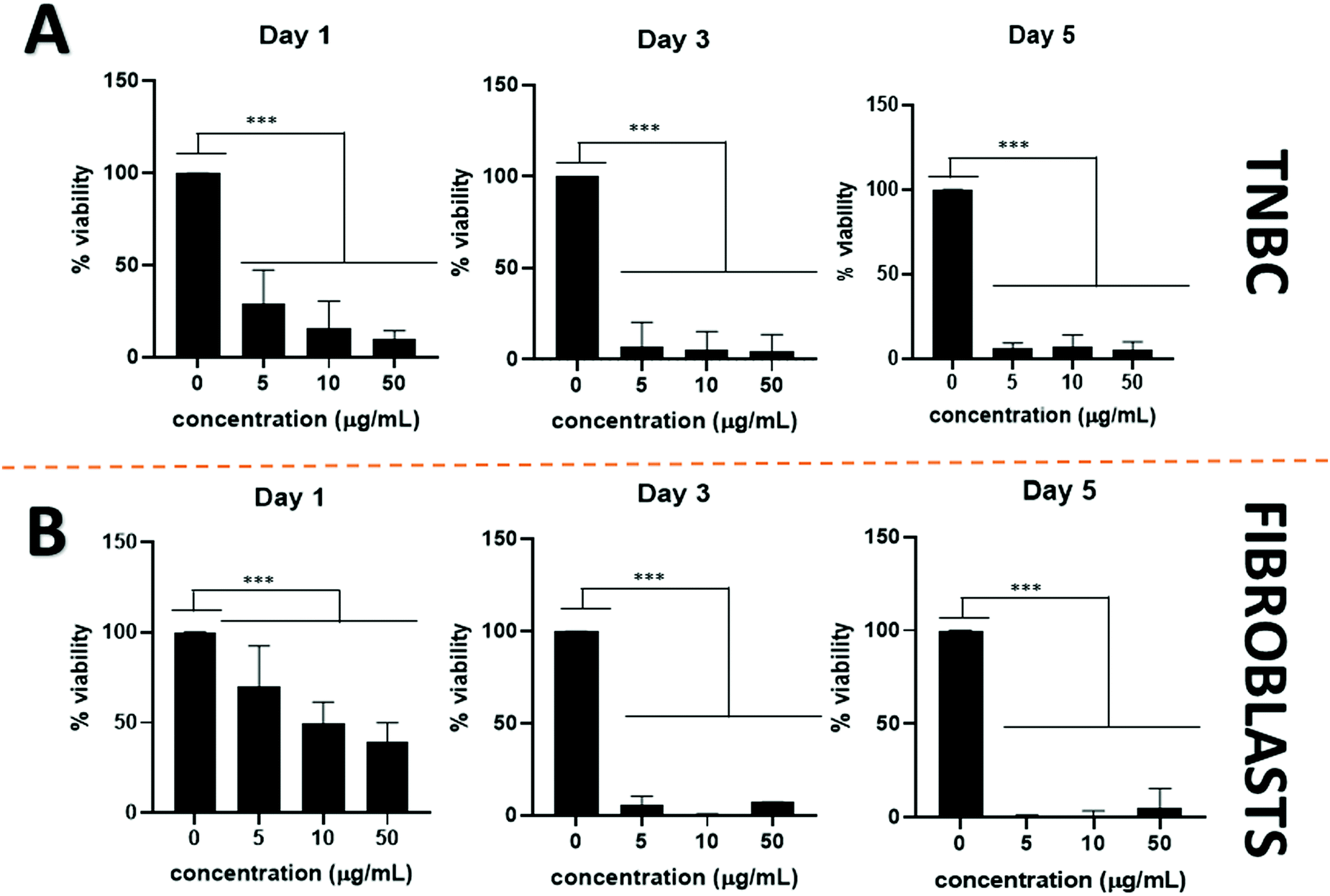 | ||
| Fig. 4 Cell viability for cisplatin using (A) TNBC and, (B) fibroblasts, for 1, 3, and 5 days with different concentrations of treatment. Results are expressed as mean + SD (***p < 0.001). Therefore, cisplatin was toxic for both cell lines. | ||
To assess the minimum concentration to decrease cell viability by half, IC50 was calculated (Fig. S2, ESI†). When using PtNPs against TNBC, the concentration needed decreased from 44 μg mL−1 at 1 day incubation period to 21 μg mL−1 at 5 days incubation period, confirming the time-dependent effect (Table 1). Although cisplatin is more effective than PtNPs at treating TNBC, achieving an IC50 of less than 1 μg mL−1 for any time point (Table 1), a similar IC50 was obtained for fibroblasts. However, for PtNPs it was not possible to calculate IC50 against fibroblasts since they showed no toxicity.
| IC50 μg mL−1 | ||||
|---|---|---|---|---|
| PtNPs–TNBC | PtNPs–fibroblasts | Cisplatin–TNBC | Cisplatin–fibroblasts | |
| 1 Day | 44 | NA | 0.4 | 18 |
| 3 Days | 14 | NA | 0.3 | 3 |
| 5 Days | 22 | NA | 0.2 | 3 |
MTS assays used for this cell viability data are a measure of cell metabolism. If the number of cells present is low, then the intensity of the signal will also be low, leading to the assumption of cell death. The initial decrease in cell viability using PtNPs followed by an increase is likely associated with an inhibition of cell growth caused by PtNPs, as opposed to cell death. To confirm this hypothesis, live/dead experiments were performed. These experiments represent a qualitative measure of cell viability. Cells will appear green if they are alive and red otherwise. To evaluate both cell viability and confluence, the highest concentrations of PtNPs (200 μg mL−1) and cisplatin (50 μg mL−1) were compared with the control group.
With this experiment, it was confirmed that the PtNPs were not toxic to fibroblasts, and the confluence of fibroblasts did increase after 3 days of culture (Fig. 5). The absence of red cells when using PtNPs, and the increasing number of cells by day 3 showed that cells are growing slower but not dying. By day 5 there was no difference between the use of PtNPs and the control group. The mechanism of action of PtNPs has been of interest for several years, however, there are conflicting reports suggesting the mechanism is related to ROS production or the release of platinum ions. It has been demonstrated that PtNPs increase the level of ROS produced inside the cell, damaging the DNA of the cells, stopping replication, and causing apoptosis.56 Furthermore, Bendale et al. tested PtNPs against red blood cells (PBMCs) finding no toxicity for healthy cells while causing toxicity for cancer cells (A375).13 They concluded that platinum enters the nucleus of the cell, causing fragmentation of the DNA. However, even though PtNPs are generally considered to have low toxicity, the harmless effect against cardiac fibroblast has never been found before. Moreover, the selectivity of PtNPs has not been studied. We hypothesize that the selectivity of PtNPs can be associated with the oxidative state of PtNPs, which may be altered by the more acidic pH of the cancer environment.57 Cisplatin, however, manifests high toxicity for fibroblasts corroborating the MTS results. Based on the obtained results, PtNPs were further investigated in combination with a PLGA particle delivery system.
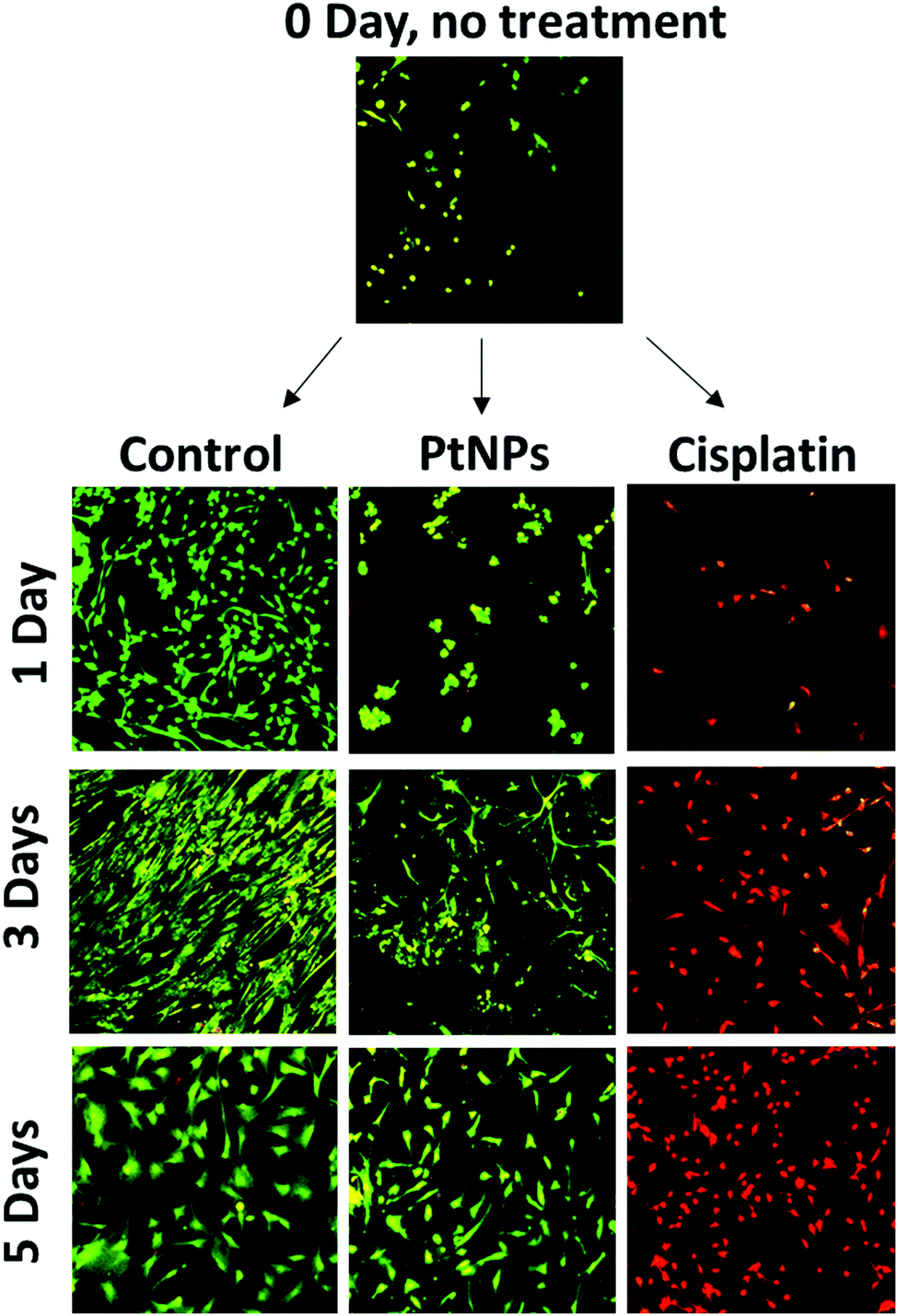 | ||
| Fig. 5 Live/dead assay with fibroblasts (NHCF-V), an initial image (day 0) before adding the treatment, and then using PtNPs (200 μg mL−1) and cisplatin (50 μg mL−1) after 1, 3, and 5 days of treatment. PtNPs appear to slow the growth of fibroblasts without killing them, but growth began to recover at 3 days incubation. | ||
Encapsulation
Systemic administration of drugs has many drawbacks for patients. Delivery systems can result in a larger dose of the drug to the target, increasing the pharmacokinetics of the material.58 Due to the drug dosage limitation of PtNPs and potential accumulation in the liver, spleen, and kidneys, the use of a delivery system was studied. PLGA is an FDA approved material for drug delivery with no toxicity (Fig. S3, ESI†).26,28,59 Due to its biocompatibility, PLGA was chosen as a preferred material for the preparation of a delivery system for PtNPs. Different techniques were used to encapsulate PtNPs including nanoprecipitation, and electrohydrodynamic (EHD) co-jetting (Fig. S4 and S5, ESI†). Nanoprecipitation was tested with two different organic solvents, acetone and acetonitrile, but the hydrophilic nature of PtNPs led to large aggregates when dispersed with an organic solvent (Fig. S4, ESI†). By EHD, different concentrations of CTab were tested to modulate PLGA sizes, however, results included large aggregates of PtNPs or PLGA particles that were too small (Fig. S5, ESI†). Therefore, the highest encapsulation rate was achieved by nanoemulsion.Emulsification has been widely studied for the synthesis of polymeric particles. Specifically, the water/oil/water (W/O/W) double emulsion has been used to encapsulate hydrophilic drugs.60,61 Specifically, nanoemulsion has been used to encapsulate cisplatin inside PLGA particles.62,63 For the first emulsion, a water phase containing the PtNPs was introduced into the organic phase with PLGA dissolved. Afterward, the mixture was emulsified by probe sonication. This step forms water droplets containing PtNPs inside the organic phase. Then, the first emulsion is introduced dropwise into a second water solution with surfactant and probe sonicated to form a second emulsion that will contain PtNPs encapsulated in PLGA particles (Fig. 6A). Three different formulations were tested with varying amounts of PtNPs. The first formulation used 0.01 mg μL−1 of PtNPs in aqueous solution, and TEM images showed that most of the PLGA particles were empty (Fig. 6B). Therefore, higher concentrations of PtNPs, 0.015 mg μL−1 and 0.02 mg μL−1, were tested in an attempt to increase the loading. For the greatest concentration used (0.02 mg μL−1), the PtNPs aggregated, decreasing encapsulation efficiency (Fig. 6C). However, with 0.015 mg μL−1 of PtNPs in aqueous solution, the encapsulation of PtNPs was successfully achieved with few empty PLGA particles. TEM analysis showed PLGA particles with many PtNPs encapsulated, as well as some single PtNPs with a thin PLGA coating (Fig. 6D).
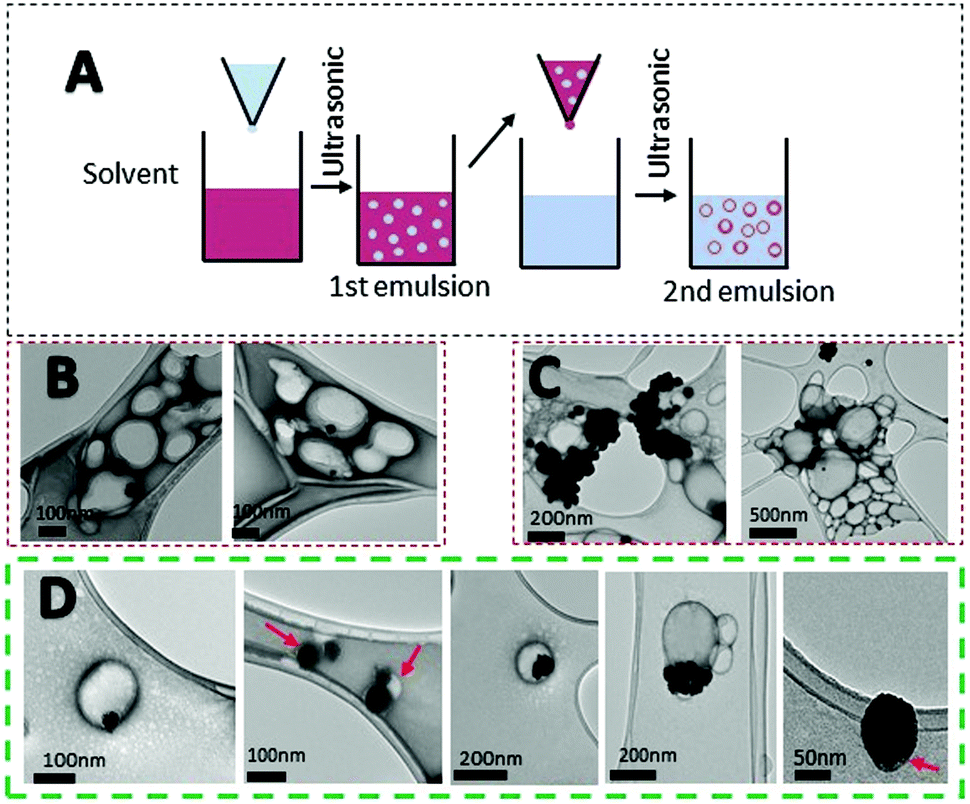 | ||
| Fig. 6 Nanoemulsion encapsulation of PtNPs with PLGA particles. (A) Schematic illustration of the double emulsion process to form particles. TEM images of double emulsion encapsulation with three different concentrations of PtNPs (B) 0.01 mg μL−1 (C) 0.02 mg μL−1 (D) 0.015 mg μL−1. Concentration tested on (D) 0.015 mg μL−1 resulted in the highest encapsulation rate of PtNPs, and therefore was used for further experiments. | ||
After confirming the encapsulation of PtNPs with PLGA particles, further characterization was performed for the 0.015 mg μL−1 PtNPs formulation. By image analysis of the TEM images (70 images) of 3 batches of this formulation, 59% of the PLGA particles (218 out of 436 particles) were found to contain PtNPs. These results are comparable to other formulations where cisplatin is encapsulated with PLGA, achieving an encapsulation efficiency of 60%.62 Furthermore, thermogravimetric analysis (TGA) of the three batches of the 0.015 mg μL−1 PtNPs formulation resulted in a 12.5 weight% loading of PtNPs in PLGA (Fig. 7A). Size analysis using nanoparticle tracking analysis (NTA) measurements reported a mode size of 140 nm ± 2.7 nm (Fig. 7B).
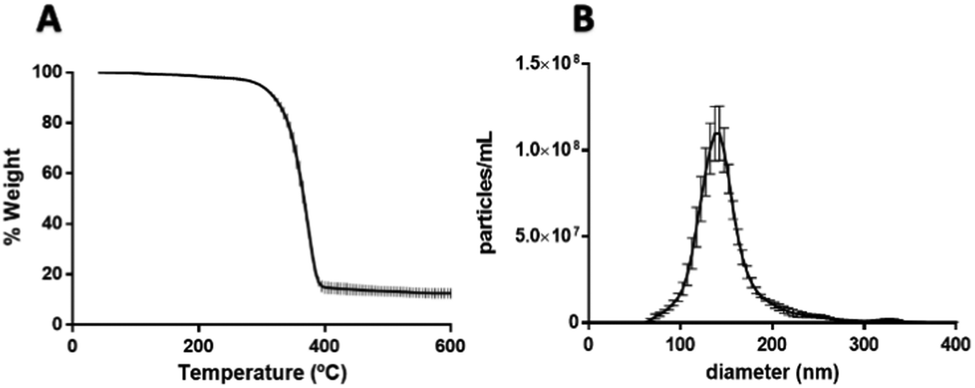 | ||
| Fig. 7 (A) Thermogravimetric analysis of 0.015 mg μL−1 PtNPs with PLGA particles by nanoemulsion shows 12.5% weight percent loading of PtNPs. Performed 3 times with 3 different batches. (B) Size distribution by nanoparticle tracking analysis of 0.015 mg μL−1 PtNPs with PLGA particles by nanoemulsion. Shows a mode size of 140 nm. | ||
Surface modifications
The addition of passive and active targeting ligands can provide a more effective treatment for TNBC, therefore two different surface modifications were performed (Fig. 8). PEGylation has been reported as an effective way to prevent protein adsorption onto the surface of the nanoparticles by creating a hydration layer,64 while the addition of an antibody gives specificity to the system to target a specific cancer cell type.46 The overexpression of EGFR on TNBC cells makes it a promising target for drug delivery systems.46,47 Therefore to compare the effect of a targeted delivery system with a passively targeted system, two groups of particles were synthesized, PEGylated particles (Pt–PLGA–PEG) and antibody attached particles (Pt–PLGA–PEG–EGFR). The PEGylated particles were decorated with 5 kDa PEG, while the antibody particles were decorated with 5 kDa PEG and 3 kDa PEG–biotin used to attach the EGFR by bioaffinity. Zeta potential and NMR were used to determine PEGylation of the nanoparticles (Fig. S6, ESI†). The attachment of PEG was confirmed by an increase of the zeta potential from −34 ± 1.04 mV for bare PLGA particles to −19 ± 1.06 mV for the decorated particles. The presence of the peak at 3.62 ppm associated with CH2 group in PEG also confirmed the presence of PEG on the particles. NTA was used to determine the size and the attachment of the EGFR to the particles (Fig. S6, ESI†). By using a secondary fluorescent antibody, the attachment of the EGFR was confirmed through visualization of the fluorescent signal. The mode size was 188.0 ± 6.3 nm for Pt–PLGA–PEG and 211.6 ± 15.2 nm for Pt–PLGA–PEG–EGFR (Fig. S6, ESI†).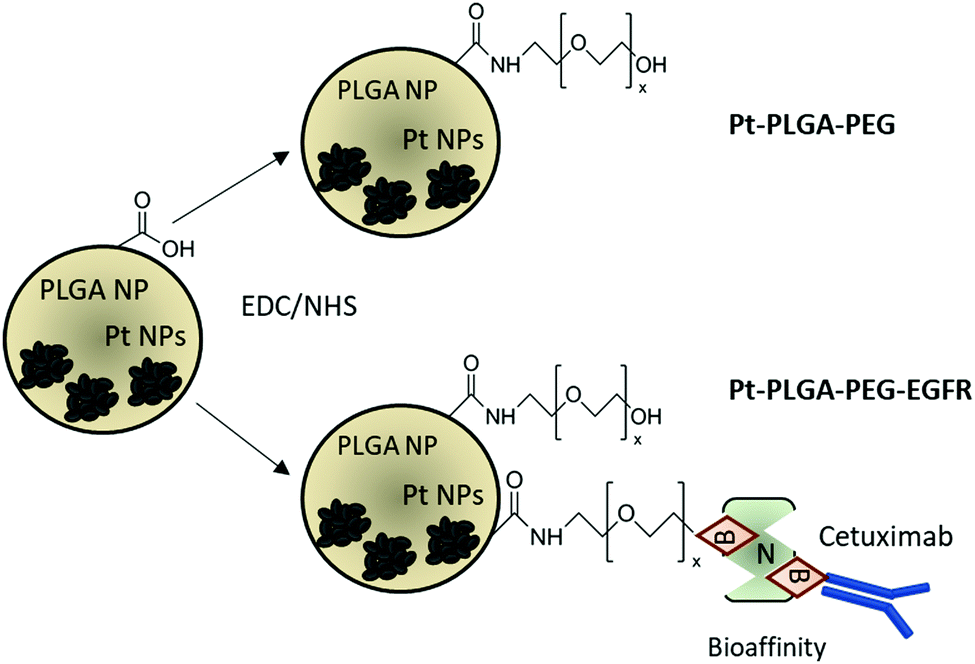 | ||
| Fig. 8 Schematic representation of surface modifications performed on PLGA particles. Pegylated particles (Pt–PLGA–PEG) and antibody modified particles (Pt–PLGA–PEG–EGFR). B stands for biotin, N for neutravidin and Cetuximab is the commercial name of the monoclonal anti-EGFR antibody. | ||
Cell viability experiments were performed with both sets of particles against TNBC cells. For these studies, 4 different concentrations of particles were tested, 10, 100, 200, and 400 μg mL−1, over 1, 3, and 5 days (Fig. 9). Both sets of particles showed statistically significant results between the control group and the highest concentrations of particles on day 1 and day 3. Therefore, both sets of particles showed a therapeutic effect against TNBC. However, TNBC cells were more affected by the particles delivered with the targeting antibody, showing lower cell viability for all the concentrations when compared with Pt–PLGA–PEG. To further prove that the targeting with EGFR was more effective, the IC50 (Fig. S7, ESI†) for both sets of particles was calculated. Table 2 shows that the minimum concentration to decrease cell viability by half was higher when using Pt–PLGA–PEG than for Pt–PLGA–PEG–EGFR. Therefore, the targeting of the particles with an antibody provides the most effective treatment against TNBC. However, if we compare IC50 values in Table 1 for PtNPs with TNBC and Table 2 for Pt–PLGA–PEG–EGFR with TNBC, the concentration of PtNPs needed to achieve the IC50 (14–44 μg mL−1) is lower for bare PtNPs than for Pt–PLGA–PEG–EGFR (109–250 μg mL−1). This phenomenon is likely associated with the release of PtNPs from the PLGA particles. The encapsulation of PtNPs increases the time to be released, increasing the concentration of particles needed to achieve the same effectivity as free PtNPs. Moreover, size of encapsulated PtNPs is significantly increased compared with free PtNPs. The optimum size for particle internalization is around 50 nm, and cellular uptake decreases with increasing particle size.65 Particles of 30–50 nm are internalized via receptor-mediated endocytosis, however, particles with a size smaller than 200 nm are internalized via caveolae-mediated endocytosis.66 Therefore, particle size is generally a key factor for cellular uptake. However, the cellular uptake data in this study (Fig. 11) suggests that the size increase does not affect internalization. Hence, release time of PtNPs is the most likely phenomenon associated to the need to increase concentration of encapsulated PtNPs compared to free PtNPs.
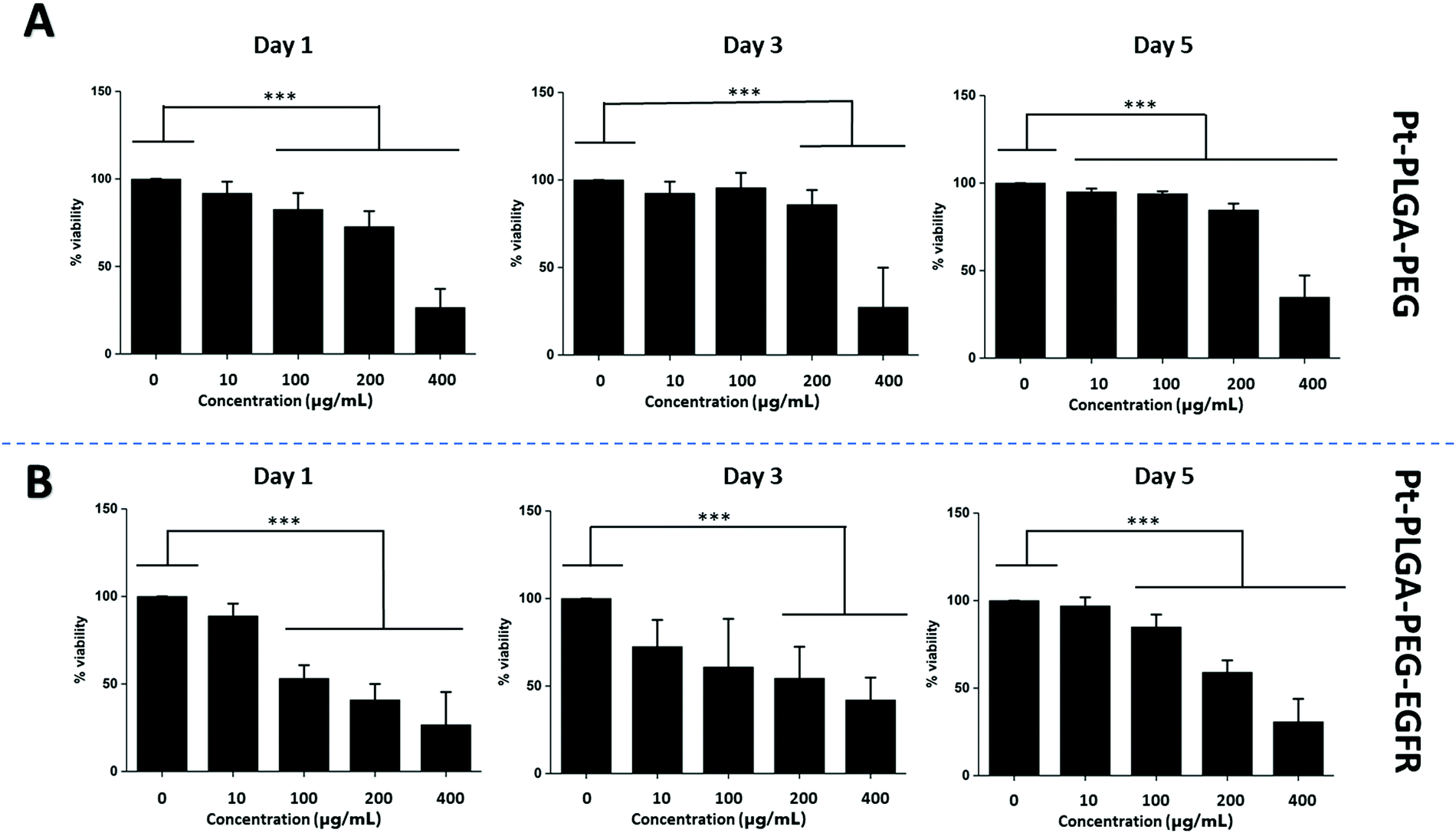 | ||
| Fig. 9 Cell viability with TNBC using (A) Pt–PLGA–PEG particles and, (B) Pt–PLGA–PEG–EGFR, for 1, 3, and 5 days with different concentrations of treatment. Results are expressed as mean + SD (***p < 0.001). The attachment of an antibody increases cancer cell death. | ||
| IC50 μg mL−1 | ||
|---|---|---|
| Pt–PLGA–PEG against TNBC | Pt–PLGA–PEG–EGFR against TNBC | |
| 1 Day | 261 | 109 |
| 3 Days | 298 | 239 |
| 5 Days | 328 | 250 |
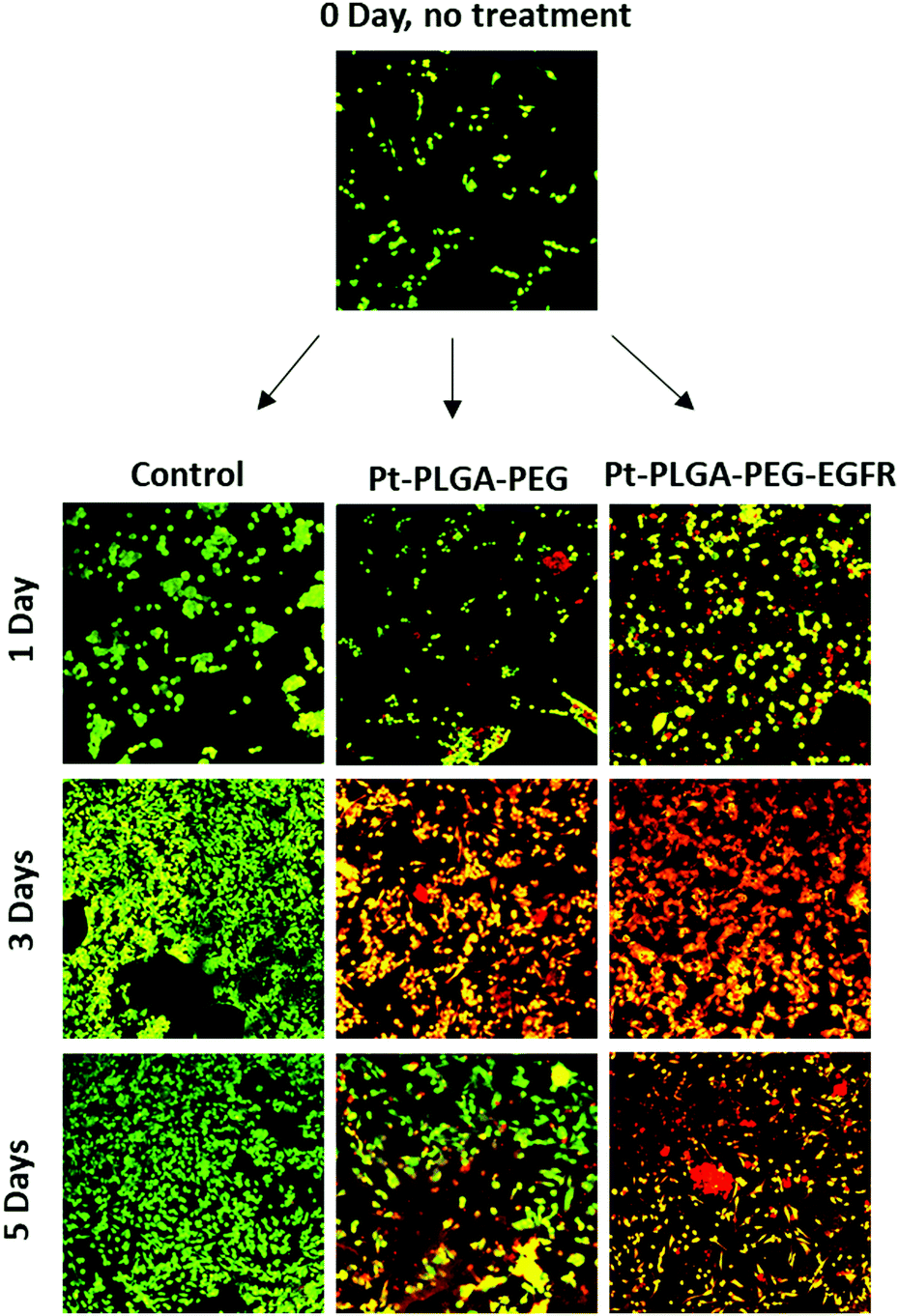 | ||
| Fig. 10 Live/dead assay with TNBC (MBA-MD-231), an initial image (day 0) before adding the treatment, and then incubation with Pt–PLGA–PEG (100 μg mL−1) and Pt–PLGA–PEG–EGFR (100 μg mL−1) after 1, 3, and 5 days of treatment. | ||
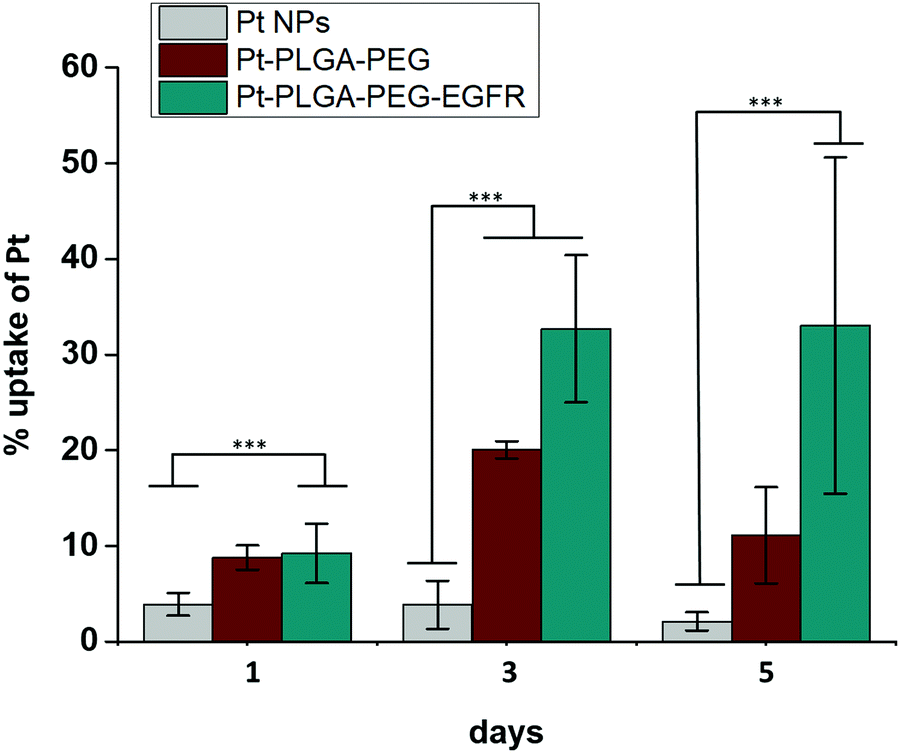 | ||
| Fig. 11 Cellular uptake of Pt with Pt NPs, Pt–PLGA–PEG and Pt–PLGA–PEG–EGFR against TNBC for 1, 3 and 5 days. Results are expressed as mean + SD (***p < 0.001). | ||
To further compare passively targeted and actively targeted particles, live/dead experiments with Pt–PLGA–PEG and Pt–PLGA–PEG–EGFR against TNBC were performed, with a concentration of 100 μg mL−1 over 1, 3, and 5 days (Fig. 10). In Fig. 10, as time increases the cells incubated with either type of particles show more cell death (red cells) compared to the control, which appears healthy (green cells). Moreover, the cells incubated with the actively targeted particles (Pt–PLGA–PEG–EGFR) have more cell death than the passively targeted particles (Pt–PLGA–PEG), agreeing with the IC50 values in Table 2. These experiments were performed with a concentration of 100 μg mL−1 for both types of particles, showing that even a lower concentration of particles can achieve a significant change in cell viability. In addition to providing information about cell viability, live/dead experiments can also indicate when the cells are affected by the treatment but not dead yet. In Fig. 10, when incubated with both sets of particles, the cells show a yellow color, indicating stress, in comparison to the control group composed of healthy cells (green). Longer incubation periods led to more cell death, observed by the shift in cell color (from yellow to red). This was a confirmation of the need for longer exposure times of the particles with the cells, to allow for the release of the particles and internalization by the cells.
Surface modifications on particles with active targeting should increase the internalization of the particles by the cells. Therefore, to test particle internalization, cellular uptake experiments were performed based on ICP-MS analysis. Fig. 11 compares the cellular uptake of bare PtNPs and the two combinations of surface modifications, Pt–PLGA–PEG and Pt–PLGA–PEG–EGFR with TNBC cells. Particles combined with the antibody (Pt–PLGA–PEG–EGFR) reported more cellular uptake with an increase in uptake over time compared to the other particles tested. Statistically significant differences were documented between Pt–PLGA–PEG–EGFR and PtNPs, demonstrating that the combination with an antibody increases cellular uptake of the particles. When comparing both sets of surface modifications, statistically significant differences were not found with cellular uptake, however, the cell viability and live/dead experiments indicate a stronger anticancer effect of Pt–PLGA–PEG–EGFR than Pt–PLGA–PEG.
Conclusions
In this study, synthesized PtNPs were found to have a potent anticancer effect against TNBC and no toxicity for healthy cells, an improvement when compared with common chemotherapeutics drugs like cisplatin, which was toxic for both cell lines. These results are consistent with other published studies that show an anti-cancer effect with Pt NPs1,20 and no toxicity in healthy cells.10,19 To increase PtNPs cellular uptake, a delivery system of targeted PLGA particles were used. By using nanoemulsion, encapsulation of the PtNPs was achieved with 59% of the PLGA particles containing PtNPs, and 12.5% of PtNP loading by weight. To achieve passive and active targeting of the particles, surface modifications were performed, obtaining two different sets of particles: Pt–PLGA–PEG (PEGylated) and Pt–PLGA–PEG–EGFR (conjugated with an EGFR antibody). Cell experiments were performed with both sets of particles against TNBC, showing more cell death with Pt–PLGA–PEG–EGFR than with Pt–PLGA–PEG, which confirmed the targeting of the cells by the antibody. When Pt–PLGA–PEG–EGFR particles were compared with free PtNPs a stronger anticancer effect was observed by free PtNPs, likely due to the time needed to release PtNPs from PLGA for the Pt–PLGA–PEG–EGFR particles. The internalization of Pt by the cells, however, was larger with the EGFR targeted particles, suggesting that with longer times, the anti-cancer effect will be more pronounced. This work demonstrates that PtNPs can be delivered through a targeted delivery system and have the potential to be effective cancer treatments. For future studies, in vivo experiments and release profiles should be performed to have a better understanding of the behavior of PtNPs.Author contributions
A. L. R. conducted the majority of the experiments and wrote the manuscript, E. V. A. conducted some experiments and helped writing the manuscript and K. M. provided advice and support. The manuscript was read and corrected by all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Sean Bannon, Ann Therese Ramirez, Suseendran Umakath, Ashish Kokkula, and Guangliang Liu for their help with cell culture, jetting nanoparticles, and NMR. We thank Dr Doherty, director of the inorganic analysis facility at Rutgers University for the ICP-MS analysis. This research has been funded by a grant from New Jersey Health Foundation (#PC 23-19).References
- B. Şahin, A. Aygün, H. Gündüz, K. Şahin, E. Demir, S. Akocak and F. Şen, Cytotoxic Effects of Platinum Nanoparticles Obtained from Pomegranate Extract by the Green Synthesis Method on the MCF-7 Cell Line, Colloids Surf., B, 2018, 163, 119–124 CrossRef PubMed
.
- A. K. Sahoo, U. Goswami, D. Dutta, S. Banerjee, A. Chattopadhyay and S. S. Ghosh, Silver Nanocluster Embedded Composite Nanoparticles for Targeted Prodrug Delivery in Cancer Theranostics, ACS Biomater. Sci. Eng., 2016, 2, 1395–1402 CrossRef CAS PubMed
- A. S. D. S. Indrasekara, B. J. Paladini, D. J. Naczynski, V. Starovoytov, P. V. Moghe and L. Fabris, Dimeric Gold Nanoparticle Assemblies as Tags for SERS-Based Cancer Detection, Adv. Healthcare Mater., 2013, 2, 1370–1376 CrossRef CAS PubMed
- M. Rai, A. P. Ingle, I. Gupta and A. Brandelli, Bioactivity of Noble Metal Nanoparticles Decorated with Biopolymers and Their Application in Drug Delivery, Int. J. Pharm., 2015, 496, 159–172 CrossRef CAS PubMed
- A. Mulgaonkar, S. Moeendarbari, W. Silvers, G. Hassan, X. Sun, Y. Hao and W. Mao, Hollow Gold Nanoparticles as Efficient in Vivo Radiosensitizing Agents for Radiation Therapy of Breast Cancer, J. Biomed. Nanotechnol., 2017, 13, 566–574 CrossRef CAS
- P. Jin, X. Niu, Z. Gao, X. Xue, F. Zhang, W. Cheng, C. Ren, H. Du, A. Manyande and H. Chen, Ultrafine Platinum Nanoparticles Supported on Covalent Organic Frameworks As Stable and Reusable Oxidase-Like Catalysts for Cellular Glutathione Detection, ACS Appl. Nano Mater., 2021, 4, 5834–5841 CrossRef CAS
- P. Patel, V. M. Nadar, D. Umapathy, S. Manivannan, R. Venkatesan, V. A. Joseph Arokiyam, S. Pappu, P. A. Prakash, M. S. Mohamed Jabir, B. Gulyás, P. Padmanabhan, S. T. Selvan and P. Kumar, Doxorubicin-Conjugated Platinum Theranostic Nanoparticles Induce Apoptosis via Inhibition of a Cell Survival (PI3K/AKT) Signaling Pathway in Human Breast Cancer Cells, ACS Appl. Nano Mater., 2021, 4, 198–210 CrossRef CAS
- H. Gehrke, J. Pelka, C. G. Hartinger, H. Blank, F. Bleimund, R. Schneider, D. Gerthsen, S. Bräse, M. Crone, M. Türk and D. Marko, Platinum Nanoparticles and Their Cellular Uptake and DNA Platination at Non-Cytotoxic Concentrations, Arch. Toxicol., 2011, 85, 799–812 CrossRef CAS PubMed
- S. F. Adil, M. E. Assal, M. Khan, A. Al-Warthan, M. R. H. Siddiqui and L. M. Liz-Marzán, Biogenic Synthesis of Metallic Nanoparticles and Prospects Toward Green Chemistry, Dalton Trans., 2015, 44, 9709–9717 RSC
- A. López Ruiz, C. Bartomeu García, S. Navarro Gallón and T. J. Webster, Novel Silver-Platinum Nanoparticles for Anticancer and Antimicrobial Applications, Int. J. Nanomed., 2020, 15, 169–179 CrossRef PubMed
- H. Mohammadi, A. Abedi, A. Akbarzadeh, M. J. Mokhtari, H. E. Shahmabadi, M. R. Mehrabi, S. Javadian and M. Chiani, Evaluation of Synthesized Platinum Nanoparticles on the MCF-7 and HepG-2 Cancer Cell Lines, Int. Nano Lett., 2013, 3, 28 CrossRef
- J. Rajput, J. R. Moss, A. T. Hutton, D. T. Hendricks, C. E. Arendse and C. Imrie, Synthesis, Characterization and Cytotoxicity of Some Palladium(II), Platinum(II), Rhodium(I) and Iridium(I) Complexes of Ferrocenylpyridine and Related Ligands. Crystal and Molecular Structure of Trans-Dichlorobis(3-Ferrocenylpyridine)Palladium(II), J. Organomet. Chem., 2004, 689, 1553–1568 CrossRef CAS
- Y. Bendale, V. Bendale, S. Paul and S. S. Bhattacharyya, Green Synthesis, Characterization and Anticancer Potential of Platinum Nanoparticles Bioplatin, Zhongxiyi Jiehe Xuebao, 2012, 10, 681–692 CAS
- Y. Bendale, V. Bendale and S. Paul, Evaluation of Cytotoxic Activity of Platinum Nanoparticles against Normal and Cancer Cells and Its Anticancer Potential through Induction of Apoptosis, Integr. Med. Res., 2017, 6, 141–148 CrossRef PubMed
- M. Yamada, M. Foote and T. W. Prow, Therapeutic Gold, Silver, and Platinum Nanoparticles, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 428–445 CAS
- J. Gao, G. Liang, B. Zhang, Y. Kuang, X. Zhang and B. Xu, FePt@CoS2 Yolk-Shell Nanocrystals as a Potent Agent to Kill HeLa Cells, J. Am. Chem. Soc., 2007, 129, 1428–1433 CrossRef CAS PubMed
- D. Pedone, M. Moglianetti, E. De Luca, G. Bardi and P. P. Pompa, Platinum Nanoparticles in Nanobiomedicine, Chem. Soc. Rev., 2017, 46, 4951–4975 RSC
- P. G. González-Larraza, T. M. López-Goerne, F. J. Padilla-Godínez, M. A. González-López, A. Hamdan-Partida and E. Gómez, IC50 Evaluation of Platinum Nanocatalysts for Cancer Treatment in Fibroblast, HeLa, and DU-145 Cell Lines, ACS Omega, 2020, 5, 25381–25389 CrossRef PubMed
- P. V. Asharani, Y. Lianwu, Z. Gong and S. Valiyaveettil, Comparison of the Toxicity of Silver, Gold and Platinum Nanoparticles in Developing Zebrafish Embryos, Nanotoxicology, 2011, 5, 43–54 CrossRef CAS PubMed
- C. X. Lin, J. L. Gu and J. M. Cao, The Acute Toxic Effects of Platinum Nanoparticles on Ion Channels, Transmembrane Potentials of Cardiomyocytes in Vitro and Heart Rhythm in Vivo in Mice, Int. J. Nanomed., 2019, 14, 5595–5609 CrossRef CAS PubMed
- Y. Li, K. H. Yun, H. Lee, S. H. Goh, Y. G. Suh and Y. Choi, Porous platinum nanoparticles high-Z and oxygen generating nanozyme for enhanced radiotherapy in vivo, Bio Mater., 2019, 197, 12–19 CrossRef CAS PubMed
- SUMMARY OF SAFETY AND EFFECTIVENESS DATA (SSED). https://www.accessdata.fda.gov/cdrh_docs/pdf4/P040046b.pdf, (accessed September 6, 2018).
- S. Dhar, F. X. Gu, R. Langer, O. C. Farokhzad and S. J. Lippard, Targeted Delivery of Cisplatin to Prostate Cancer Cells by Aptamer Functionalized Pt(IV) Prodrug-PLGA-PEG Nanoparticles, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17356–17361 CrossRef CAS PubMed
- A. Kumari, S. K. Yadav and S. C. Yadav, Biodegradable Polymeric Nanoparticles Based Drug Delivery Systems, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS PubMed
- T. Safra, F. Muggia, S. Jeffers, D. D. Tsao-Wei, S. Groshen, O. Lyass, R. Henderson, G. Berry and A. Gabizon, Pegylated Liposomal Doxorubicin (Doxil): Reduced Clinical Cardiotoxicity in Patients Reaching or Exceeding Cumulative Doses of 500 Mg/M2, Ann. Oncol., 2000, 11, 1029–1033 CrossRef CAS PubMed
- M. L. Hans and A. M. Lowman, Biodegradable Nanoparticles for Drug Delivery and Targeting, Curr. Opin. Solid State Mater. Sci., 2002, 6, 319–327 CrossRef CAS
- J. Keyvan Rad, Z. Alinejad, S. Khoei and A. R. Mahdavian, Controlled Release and Photothermal Behavior of Multipurpose Nanocomposite Particles Containing Encapsulated Gold-Decorated Magnetite and 5-FU in Poly(lactide-co-glycolide), ACS Biomater. Sci. Eng., 2019, 5, 4425–4434 CrossRef CAS PubMed
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Préat, PLGA-Based Nanoparticles: An Overview of Biomedical Applications, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS PubMed
- H. Lee, C.-H. Ahn and T. G. Park, Poly[lactic-co-(glycolic acid)]-Grafted Hyaluronic Acid Copolymer Micelle Nanoparticles for Target-Specific Delivery of Doxorubicin, Macromol. Biosci., 2009, 9, 336–342 CrossRef CAS PubMed
- M. M. Stevanović, S. D. Škapin, I. Bračko, M. Milenković, J. Petković, M. Filipič and D. P. Uskoković, Poly(lactide-co-glycolide)/Silver Nanoparticles: Synthesis, Characterization, Antimicrobial Activity, Cytotoxicity Assessment and ROS-Inducing Potential, Polymer, 2012, 53, 2818–2828 CrossRef
- J. C. Ramirez, S. E. Flores-Villaseñor, E. Vargas-Reyes, J. Herrera-Ordonez, S. Torres-Rincón and R. D. Peralta-Rodríguez, Preparation of PDLLA and PLGA Nanoparticles Stabilized with PVA and a PVA-SDS Mixture: Studies on Particle Size, Degradation and Drug Release, J. Drug Delivery Sci. Technol., 2020, 60, 101907 CrossRef CAS
- J. Plch, K. Venclikova, O. Janouskova, J. Hrabeta, T. Eckschlager, K. Kopeckova, Z. Hampejsova, Z. Bosakova, J. Sirc and R. Hobzova, Paclitaxel-Loaded Polylactide/Polyethylene Glycol Fibers with Long-Term Antitumor Activity as a Potential Drug Carrier for Local Chemotherapy, Macromol. Biosci., 2018, 18, 1800011 CrossRef PubMed
- J. Patel, J. Amrutiya, P. Bhatt, A. Javia, M. Jain and A. Misra, Targeted Delivery of Monoclonal Antibody Conjugated Docetaxel Loaded PLGA Nanoparticles into EGFR Overexpressed Lung Tumour Cells, J. Microencapsulation, 2018, 35, 204–217 CrossRef CAS PubMed
- A. Saneja, D. Arora, R. Kumar, R. D. Dubey, A. K. Panda and P. N. Gupta, CD44 Targeted PLGA Nanomedicines for Cancer Chemotherapy, Eur. J. Pharm. Sci., 2018, 121, 47–58 CrossRef CAS PubMed
- B. B. S. Cerqueira, A. Lasham, A. N. Shelling and R. Al-Kassas, Development of Biodegradable PLGA Nanoparticles Surface Engineered with Hyaluronic Acid for Targeted Delivery of Paclitaxel to Triple Negative Breast Cancer Cells, Mater. Sci. Eng., C, 2017, 76, 593–600 CrossRef CAS PubMed
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Rapid Formation of Plasma Protein Corona Critically Affects Nanoparticle Pathophysiology, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS PubMed
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, PEGylation as a Strategy for Improving Nanoparticle-Based Drug and Gene Delivery, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed
- R. Gref, Y. Minamitake, M. T. Peracchia, V. Trubetskoy, V. Torchilin and R. Langer, Biodegradable Long-Circulating Polymeric Nanospheres, Science, 1994, 263, 1600–1603 CrossRef CAS PubMed
- M. E. Davis, Z. Chen and D. M. Shin, Nanoparticles Therapeutics: An Emerging Treatment Modality for Cancer, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed
- P. J. Kennedy, F. Sousa, D. Ferreira, C. Pereira, M. Nestor, C. Oliveira, P. L. Granja and B. Sarmento, Fab-Conjugated PLGA Nanoparticles Effectively Target Cancer Cells Expressing Human CD44v6, Acta Biomater., 2018, 81, 208–218 CrossRef CAS PubMed
- B. Bahrami, M. Hojjat-Farsangi, H. Mohammadi, E. Anvari, G. Ghalamfarsa, M. Yousefi and F. Jadidi-Niaragh, Nanoparticles and Targeted Drug Delivery in Cancer Therapy, Immunol. Lett., 2017, 190, 64–83 CrossRef CAS PubMed
- S. Cleator, W. Heller and R. C. Coombes, Triple-Negative Breast Cancer: Therapeutic Options, Lancet Oncol., 2007, 8, 235–244 CrossRef PubMed
- J. Collignon, L. Lousberg, H. Schroeder and G. Jerusalem, Triple-Negative Breast Cancer: Treatment Challenges and Solutions, Breast Cancer: Targets Ther., 2016, 8, 93–107 CAS
- J. J. Arroyo-Crespo, A. Armiñán, D. Charbonnier, C. Deladriere, M. Palomino-Schätzlein, R. Lamas-Domingo, J. Forteza, A. Pineda-Lucena and M. J. Vicent, Characterization of Triple-Negative Breast Cancer Preclinical Models Provides Functional Evidence of Metastatic Progression, Int. J. Cancer, 2019, 145, 2267–2281 CrossRef CAS PubMed
- G. Bianchini, J. M. Balko, I. A. Mayer, M. E. Sanders and L. Gianni, Triple-negative Breast Cancer: Challenges and Oppotunities of a Heterogenous Disease, Nat. Rev. Clin. Oncol., 2016, 13, 674–690 CrossRef CAS PubMed
- F. Fay and C. J. Scott, Antibody-Targeted Nanoparticles for Cancer Therapy, Immunotherapy, 2011, 3, 381–394 CrossRef CAS PubMed
- J. Gu, L. Yin, J. Wu, N. Zhang, T. Huang, K. Ding, H. Cao, L. Xu and X. He, Cetuximab and Cisplatin Show Different Combination Effect in Nasopharyngeal Carcinoma Cells Lines via Inactivation of EGFR/AKT Signaling Pathway, Biochem. Res. Int., 2016, 2016, 7016907 Search PubMed
- R. V. Kutty and S. S. Feng, Cetuximab Conjugated Vitamin E TPGS Micelles for Targeted Delivery of Docetaxel for Treatment of Triple Negative Breast Cancers, Biomaterials, 2013, 34, 10160–10171 CrossRef CAS PubMed
- W. S. Liao, Y. Ho, Y. W. Lin, E. Naveen Raj, K. K. Liu, C. Chen, X. Z. Zhou, K. P. Lu and J. I. Chao, Targeting EGFR of Triple-Negative Breast Cancer Enhances the Therapeutic Efficacy of Paclitaxel- and Cetuximab-Conjugated Nanodiamond Nanocomposite, Acta Biomater., 2019, 86, 395–405 CrossRef CAS PubMed
- K. Shim, J. Kim, Y.-U. Heo, B. Jiang, C. Li, M. Shahabuddin, K. C.-W. Wu, M. S. A. Hossain, Y. Yamauchi and J. H. Kim, Synthesis and Cytotoxicity of Dendritic Platinum Nanoparticles with HEK-293 Cells, Chem. – Asian J., 2017, 12, 21–26 CrossRef CAS PubMed
- S. Fatima, Z. Iqbal, A. K. Panda, M. Samim, S. Talegaonkar and F. J. Ahmad, Polymeric Nanoparticles as a Platform for Permeability Enhancement of Class III Drug Amikacin, Colloids Surf., B, 2018, 169, 206–213 CrossRef CAS PubMed
- E. Porcel, S. Liehn, H. Remita, N. Usami, K. Kobayashi, Y. Furusawa, C. Le Sech and S. Lacombe, Platinum Nanoparticles: A Promising Material for Future Cancer Therapy?, Nanotechnology, 2010, 21, 085103 CrossRef PubMed
- A. Borowik, R. Banasiuk, N. Derewonko, M. Rychlowski, M. Krychowiak-Masnicka, D. Wyrzykowski, M. Ziabka, A. Woziwodzka, A. Krolicka and J. Piosik, Interactions of newly synthesized platinum nanoparticles with ICR-191 and their potential application, Sci. Rep., 2019, 9, 4987 CrossRef PubMed
- M. Moglianetti, E. De Luca, D. Pedone, R. Marotta, T. Catelani, B. Sartori, H. Amenitsch, S. F. Retta and P. P. Pompa, Platinnum nanozymes recover cellular ROS homeostasis in an oxidative stress-mediated disease model, Nanoscale, 2016, 8, 3739–3752 RSC
- X. Teng, X. Liang, S. Maksimuk and H. Yang, Synthesis of Porous Platinum Nanoparticles, Small, 2006, 2, 249–253 CrossRef CAS PubMed
- S. Gurunathan, M. Jeyaraj, M.-H. Kang and J.-H. Kim, Graphene Oxide-Platinum Nanoparticle Nanocomposites: A Suitable Biocompatible Therapeutic Agent for Prostate Cancer, Polymers, 2019, 11, 733 CrossRef CAS PubMed
- A. Sharma, A. K. Goyal and G. Rath, Recent Advances in Metal Nanoparticles in Cancer Therapy, J. Drug Targeting, 2018, 8, 617–632 CrossRef PubMed
- J. Zhuang, C.-H. Kuo, L.-Y. Chou, D.-Y. Liu, E. Weerapana and C.-K. Tsung, Optimized Metal-Organic-Framework Nanospheres for Drug Delivery: Evaluation of Small-Molecule Encapsulation, ACS Nano, 2014, 8, 2812–2819 CrossRef CAS PubMed
- S. Sharma, A. Parmar, S. Kori and R. Sandhir, PLGA-Based Nanoparticles: A New Paradigm in Biomedical Applications, TrAC, Trends Anal. Chem., 2016, 80, 30–40 CrossRef CAS
- G. Wang and H. Uludag, Recent Developments in Nanoparticle-Based Drug Delivery and Targeting Systems with Emphasis on Protein-Based Nanoparticles, Expert Opin. Drug Delivery, 2008, 5, 499–515 CrossRef CAS PubMed
- M. Iqbal, N. Zafar, H. Fessi and A. Elaissari, Double Emulsion Solvent Evaporation Techniques Used for Drug Encapsulation, Int. J. Pharm., 2015, 496, 173–190 CrossRef CAS PubMed
- P. Dana, S. Bunthot, K. Suktham, S. Surassmo, T. Yata, K. Namdee, W. Yingmema, T. Yimsoo, U. R. Ruktanonchai, S. Sathornsumetee and N. Saengkrit, Active targeting liposome-PLGA composite for cisplatin delivery against cervical cancer, Colloids Surf., B, 2020, 196, 111270 CrossRef CAS PubMed
- E. C. Gryparis, M. Hatziapostolou, E. Papadimitriou and K. Avgoustakis, Anticancer activity of cisplatin-loaded PLGA-mPEG nanoparticles on LNCaP prostate cancer cells, Eur. J. Pharm. Biopharm., 2007, 67, 1–8 CrossRef CAS PubMed
- N. Bertrand, P. Grenier, M. Mahmoudi, E. M. Lima, E. A. Appel, F. Dormont, J. M. Lim, R. Karnik, R. Langer and O. C. Farokhzad, Mechanistic Understanding of in Vivo Protein Corona Formation on Polymeric Nanoparticles and Impact on Pharmacokinetics, Nat. Commun., 2017, 8, 777 CrossRef PubMed
- L. Tang, X. Yang, Q. Yin, K. Cai, H. Wang, I. Chaudhury, C. Yao, Q. Zhou, M. Kwon, J. A. A. Hartman, I. T. Dobrucki, L. W. Dobrucki, L. B. Borst, S. Lezmi, W. G. Helferich, A. L. Ferguson, T. M. Fan and J. Cheng, Investigating the optimal size of anticancer nanomedicine, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15344–15349 CrossRef CAS PubMed
- P. Foroozandeh and A. A. Aziz, Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles, Nanoscale Res. Lett., 2018, 13, 339 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma01155c |
| This journal is © The Royal Society of Chemistry 2022 |