
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA review: research progress on the formation mechanism of porous anodic oxides
Chengyuan
Li
a,
Yilin
Ni
a,
Jingjing
Gong
b,
Ye
Song
 *a,
Tianle
Gong
a and
Xufei
Zhu
*a
*a,
Tianle
Gong
a and
Xufei
Zhu
*a
aKey Laboratory of Soft Chemistry and Functional Materials of Education Ministry, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: soong_ye@163.com; zhuxufei.njust@vip.163.com
bSchool of Design Art and Media, Nanjing University of Science and Technology, Nanjing 210094, China
First published on 20th November 2021
Abstract
Owing to the great development potential of porous anodic oxides (PAO) in many fields, research on their formation mechanisms, fabrication processes and applications has a history of more than ten years. Although compared with research on the fabrication processes and applications of PAO, research on their formation mechanisms started late, several mainstream theories have been formed in the academic community, including the field-assisted dissolution (FAD) theory, the field-assisted ejection (FAE) theory, the self-organization theory, the ionic and electronic current theory and the oxygen bubble mould effect. This review will focus on summarizing the core views of the mainstream mechanisms mentioned above and comparing the explanations for some of their classical experimental phenomena.
1. Introduction
In recent years, nanoporous materials have attracted wide scientific and technological interest owing to their superior performance in many fields, such as catalysts, biomaterials, supercapacitors, energy storage materials, gas sensors, and hydrogen generation.1–9 As nanotubes are being widely used, increasing numbers of nanotube preparation methods have been developed, such as hydrothermal, sol–gel, glancing angle deposition, multi-step anodization, and the pulse/stop anodization and template methods.5,10–15 Compared to other preparation methods, which have their own drawbacks, the anodization method can be convenient to prepare nanostructures with high regularity, and it has been widely used in research.5 Although porous anodic oxides of valve metals (Al, Ti, Sn, etc.) has been researched for quite a long time as a topic of active interest, its formation mechanism is still not very clear.15–20 Owing to the lack of research on the formation mechanism, a large number of experiments should be conducted to summarize the experiences when developing and improving the preparation technology of nanotubes; therefore, it is significant to clarify the formation mechanism of nanotubes in order to accurately control their morphology. After years of research and discussion, several mainstream theories have been formed in the academic community. The research on the formation mechanism of nanotubes began with the discussion of the growth process of porous anodic oxides (PAO). Initially, O'Sullivan et al.21 proposed the field-assisted dissolution (FAD) theory to explain the formation of porous anodic aluminum (PAA). Due to the similarities in the formation of PAO, FAD theory has also been applied by many scholars to explain the formation mechanism of other valve metal anodic oxides.10,22–25 With further research, the field-assisted ejection (FAE) theory derived from the FAD theory was proposed to improve the explanation of PAO.26–28 In 2006, Skeldon et al.29,30 used tracer atom technology to explore the growth mode of oxides in PAA, and they finally found that it was completely opposite to the growth mode expected by FAD theory. At this point, a new growth model, the plastic flow model, was proposed, which challenged the status of FAD theory.28–34 Unfortunately, although the experiment showed a growth model that was completely opposite to the traditional mechanism hypothesis, the growth kinetics of PAO is still not clear. In response to the unclear driving force of plastic flow, many scholars have conducted a series of studies and have proposed explanations such as self-ordering, electric field drive models, volume expansion models, ionic and electronic current theory, and oxygen bubble mould effects.11,28,35–49 In addition to the above mechanisms, the self-organization mechanism of PAO is supported by some scholars.23,28,36,42,50 In 2014, Schmuki et al.23 summarized the factors which may influence the self-organization during the growth of ATNTs in their paper; however, they still did not give a convincing explanation for the dynamics of self-organization. Insufficient analysis of the causes of the self-organization process is also a defect of self-organization theory. In view of the above situation, the main goal of this article is to review the mainstream theories about the formation mechanism of PAO, which has many details that are worthy of being discussed and studied. The FAD and FAE theories, the oxygen bubble mould effect, the ionic and electronic current theory based on the plastic flow model, and the self-organization theory are the focuses of this review.2. Discussion of the formation mechanism of PAO
According to the different anodizing conditions (mainly voltage, current and electrolyte), anodic oxides usually form three structures: compact layers, nanotubes and nano-porous sponge structures.5,23,28,42,50–52 Schmuki et al.42 mentioned that the distinction between the porous and tubular morphology is mechanistically not really justified. Therefore, it is unnecessary to distinguish porous oxides as nanotubes and nanopores in terms of mechanism research, and the anodic oxides with different morphologies can be divided into two types, compact anodic oxides (or compact layers) and PAO.When referring to the formation mechanism of PAO, the importance of PAA can never be ignored. The research on the mechanism of anodic oxides originated from the exploration of the phenomenon by which two structures of a compact layer and PAA are formed by anodization of aluminum under different conditions. To date, many scholars have cited the formation mechanism of PAA to try to explain the growth process of ATNTs and other PAO, which had a profound impact on the later development of the theory.28,53–55
2.1 The methods of anodization
There are many anodization methods, such as constant voltage anodization, constant current anodization, and pulse-stop voltage anodization. As shown in Fig. 1a, these methods usually use graphite as the cathode and the metal as the anode, then apply voltage or current between the two electrodes to achieve anodization of the anode metal. As shown in Fig. 1b, a PAO array is usually formed on the surface of the anode metal after anodizing.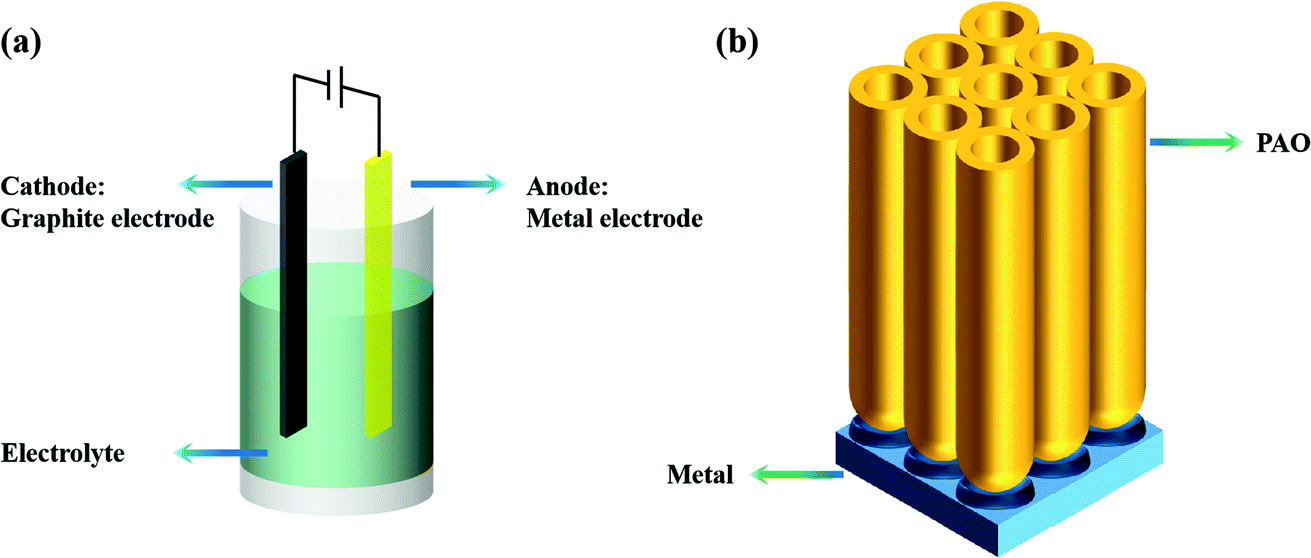 | ||
| Fig. 1 (a) Anodizing device; (b) PAO array. | ||
2.2 The field-assisted dissolution (FAD) theory and the field-assisted ejection (FAE) theory
This theory proposes that the FAD reaction plays a major role in the formation of the nanotubes.
In the growth progress of PAA, the FAD reaction is as follows:25,53,58
| Al2O3 + 6H+ → 2Al3+ + 3H2O | (1) |
Due to the participation of fluoride ion, the FAD reaction in the growth progress of ATNTs in fluorine-containing electrolyte is as follows:9,10,42,59
| TiO2 + 6F− + 4H+ → [TiF6]2− + 2H2O | (2) |
Moreover, the reaction in fluorine-free electrolyte can be described as follows:9,42,60
| M → MZ+ + ze− | (3) |
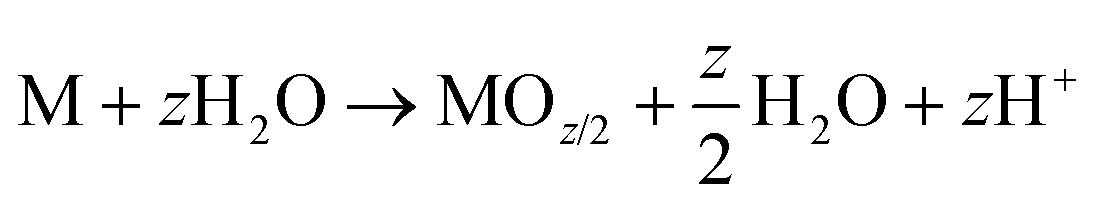 | (4) |
Fig. 2 shows the anodizing process described by FAD theory, among which Fig. 2a–c show the 3-D models of the various stages and Fig. 2d–f show the detailed diagrams. According to FAD theory, the growth of PAO can be divided into three stages: initial stage, channel deepening stage and growth termination stage.
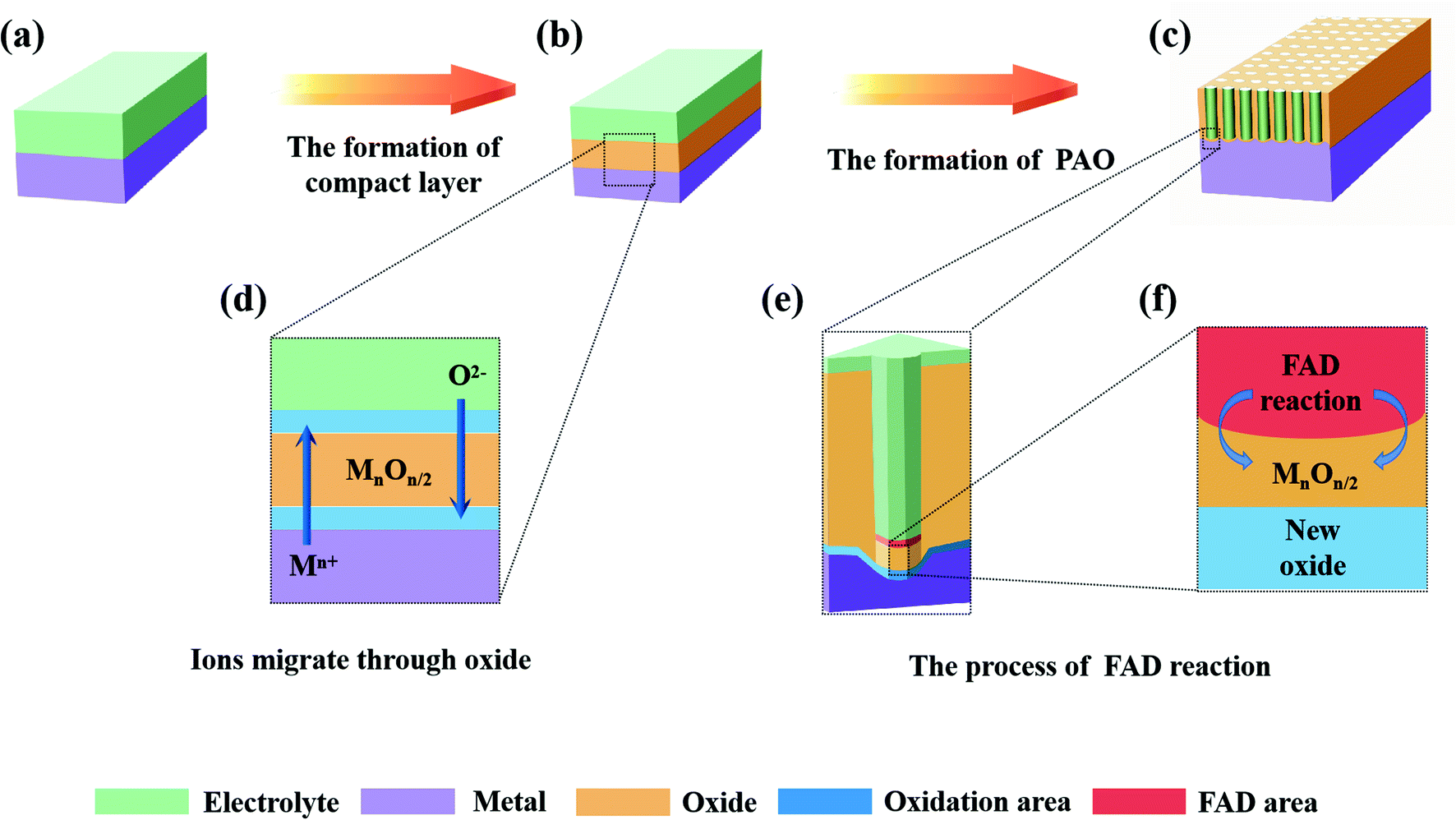 | ||
| Fig. 2 Schematic of the mechanism of FAD theory. | ||
(1) Initial stage
At the beginning of the reaction, a compact layer is formed between the metal and electrolyte (Fig. 2b), and then the metal ions (Mn+) migrate to the surface of the metal substrate and form a layer of oxides with the oxygen (O2− or other oxygen-containing groups) in the electrolyte (Fig. 2d). This process has nothing to do with whether the electrolyte contains fluorine ions.60 At this time, two interfaces of electrolyte/metal oxides (E/O) and metal oxides/metal matrix (O/M) are formed on the surface of the metal.28,42 In addition, there are early reports that the growth of oxides only occurred at the O/M interface.61,62
(2) Pore initiating stage
In this stage, oxides have been formed on the surface of the metal substrate, and fluoride ions (or other anions) begin to migrate to the E/O interface under the action of the electric field, where they participate in the FAD reaction (Fig. 2e). When the FAD reaction occurs, the formed oxides begins to dissolve; no new oxides are formed at the E/O interface (only the FAD reaction occurs at the E/O interface), and the metal oxides only form at the O/M interface (Fig. 2f).21 The pores start to form in this stage (Fig. 2c).63
(3) Channel deepening stage
FAD theory proposes that during the anodizing process, there is a competition between the formation of oxides and the dissolution of oxides on the anode surface. In this steady stage, due to the balance of two reactions, the thickness of the barrier layer remains constant and the pores of the PAO are stably deepened.21,22,24,35,59,63,64 It is important to note that the FAD reaction is always dominant during this process. In fact, the rate of the FAD reaction must be higher than the rate of oxide formation to ensure deepening of the channels. Since the constant voltage in the anodizing process, applied electric field and electrolyte will not change, the FAD reaction should continue to proceed. In other words, the FAD reaction will continue as long as the electric field conditions are established. From this, it is easy to draw the conclusion that nanotubes can grow indefinitely, which is contrary to the experimental facts.65 There is no explanation for this reaction stage in early FAD theory.
In summary, it can be seen that FAD theory constructs a growth model similar to ‘digging from top to bottom’, and it gives reasonable qualitative explanations for some common phenomena, such as the influences of the current, voltage, and fluoride ion content on PAO.
2.3 The self-organization theory
In addition to the FAD and FAE theories mentioned in Section 2.2, the self-organization theory as an explanation of the formation mechanism of PAO is also recognized by some scholars. Compared with the traditional theory, which tends to explain the mechanism, the explanation of the mechanism in the self-organization theory is more inclined toward the preparation.9,23,28,36,42,50,72–74 Self-organization theory inherits some of the assumptions of FAD theory (such as [TiF6]2− and the FAD reaction) and gives the boundary conditions for preparing PAO with different morphologies.23,72,74The self-organization theory attributes the formation of PAO to several key factors (voltage, solubility, stress, field effects, etc). These factors determine whether the metal can form PAO after anodization.23,28,53,74–76 In 2014, Schmuki et al. reviewed the self-organization theory. This review mainly adopts the viewpoints of Schmuki et al. in self-organization theory.23
Firstly, the voltage is a precondition for oxide formation. The anodizing process is a high-field ion formation/transport process in which the ion migration is controlled by the field across the oxide layer.23,35,58,74,77,78 As shown in Fig. 3a and b, a more compact layer will be formed on the surface of the metal substrate at the initial stage of anodization. Under the action of an electric field, Mn+ in the metal matrix passes through the oxides and reacts with O2− to form MOn/2 at the E/O interface, while O2− in the electrolyte passes through the oxides to form MOn/2 at the O/M interface with Mn+. The current intensity in this process can be expressed by the formula I = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(BF) = Aexp(BΔU/d), where I is the current; ΔU is the voltage across the oxide layer thickness d, defining the electric field (F = ΔU/d); and A and B are experimental constants.23 In addition, the formation of a self-organized nanotube array requires an appropriate voltage.79
exp(BF) = Aexp(BΔU/d), where I is the current; ΔU is the voltage across the oxide layer thickness d, defining the electric field (F = ΔU/d); and A and B are experimental constants.23 In addition, the formation of a self-organized nanotube array requires an appropriate voltage.79
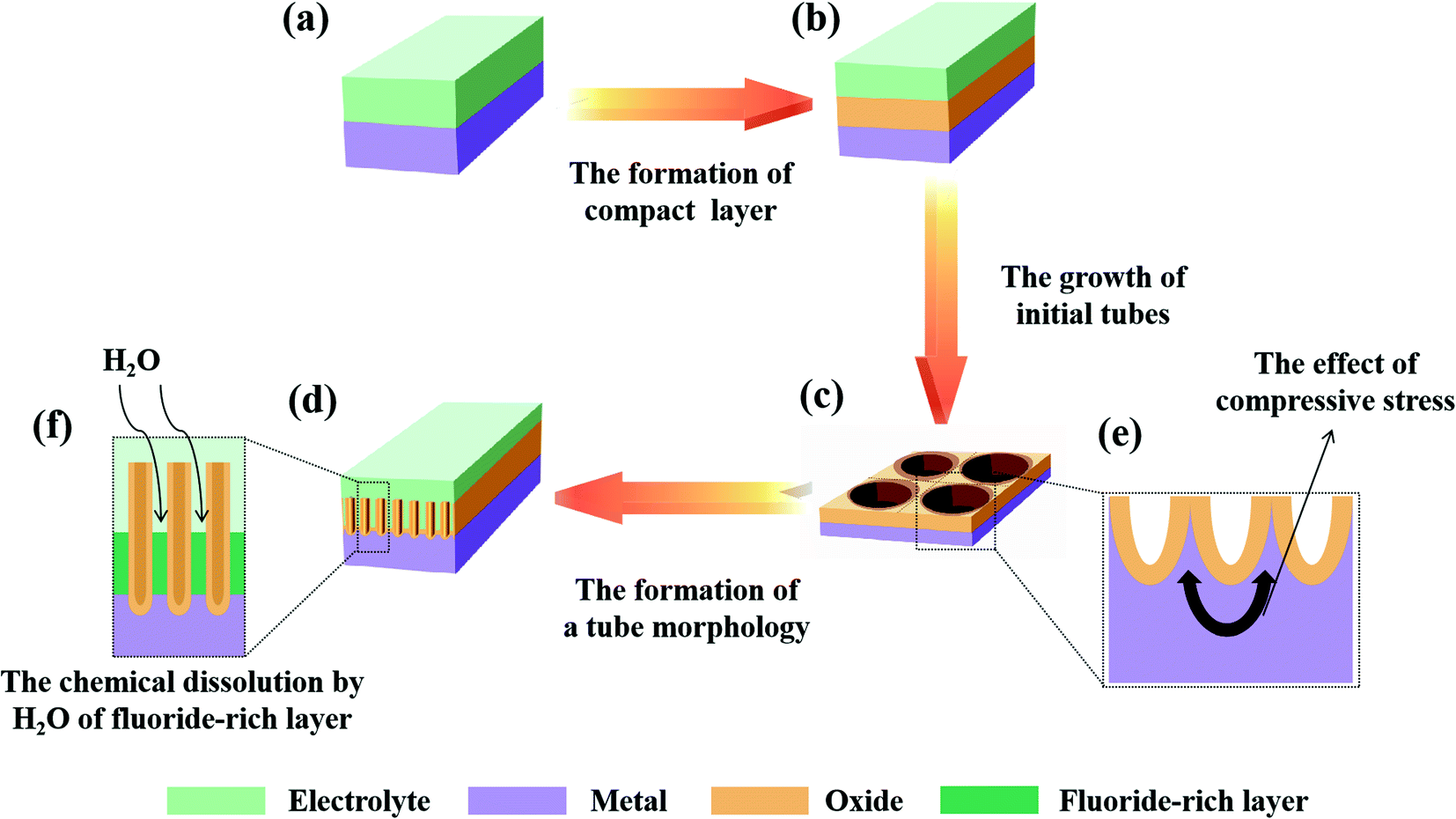 | ||
| Fig. 3 Schematic of the mechanism of self-organization theory. | ||
Secondly, the buildup of compressive stress during the early phase of anodic oxides is considered to be a reason for the formation of the hemispherical bottom of PAO, and Fig. 3c and e show how stress works in the formation of PAO. Moreover, the stress maybe enhanced by voltage-induced electrostrictive forces.23,80,81 It is worth mentioning that this process occurs in the early stage of the anodization reaction. Moreover, this view is contrary to the model of ‘dig from top to bottom’ based on the traditional FAD theory.
Thirdly, a proper solubility of the electrolyte is a necessary condition for oxide formation. The self-organization theory proposes that the formation of PAO is closely related to the solubility of metal oxides in the electrolyte.23,74,82 When the electrolyte has low solubility for the oxides, compact oxides will be formed on the metal surface. When the electrolyte has high solubility for the oxides, an irregular oxide film (sponge oxides) will be formed on the metal surface. When the electrolyte has optimized solubility for the oxides, self-organized oxides will be formed on the metal surface. Fig. 3d and f show the formation process of PAO in an electrolyte with suitable solubility. Here, we will take the anodizing process of Ti as an example to analyze the formation mechanism of PAO.
(1) The formation of compact layers (low solubility)
For a given voltage and an insoluble oxide, the anodizing process is self-limiting.23,74 During the anodizing process, the oxides grows thicker, and the electric field drops. When the electric field drops to the point where it becomes too weak to assist the migration of ions through the oxides, the compact layer reaches the final thickness d.
(2) The formation of self-organized oxides (optimized solubility)
In this case, the TiO2 at the E/O interface will be corroded by F− (or other anions) because the electrolyte has a dissolving effect on the formed oxides. Lately, a steady state will be established between oxide formation at the inner interface and dissolution at the outer interface (due to dissolution/complexation of Ti4+ as [TiF6]2−). Additionally, F− will rapidly migrate through the oxides under an electric field, which leads to the accumulation of fluoride at the O/M interface. Then, the initial tubes start to grow under the mutual effect of steady-state oxide formation/dissolution and compressive stress, which shapes the oxides and fluoride-rich layer into a hemisphere. The tube morphology is formed by chemical dissolution by H2O of the fluoride-rich layer between the hexagonal cells.23
(3) The formation of electropolishing (high solubility)
If the solubility of the oxides is further enhanced, in other words, the dissolution rate becomes too high, no steady-state oxide layer will be formed, and complete oxide dissolution occurs, which is similar to the electropolishing of Ti.9
Fourthly, once the interface curvature and oxide dissolution have started, distinct spots will show enhanced localized dissolution due to field concentration effects, which are also known as field effects.23
In addition to the above factors, other factors have a certain degree of influence on the final structure of PAO, such as the electrolyte composition (F− content, pH, conductivity, H2O content) and current efficiency. These factors together determine the minimum and maximum critical conditions for the formation of self-organizing oxides. If the condition cannot meet the minimum critical standard, a compact layer will be formed eventually. If the condition exceeds the maximum critical limit, the surface of Ti will be electropolished.9,23 The morphology of nanotubes can be achieved only in the right range. In 2007, Albu et al.83 proposed that the F− content and applied voltage can determine the ideal self-organization conditions in which the most efficient self-organized growth of nanotubes will be achieved.
2.4 Ionic and electronic current theory and the oxygen bubble mould effect
The plastic flow model originated from the description of the phenomenon in which the oxides at the bottom of the tube migrate to the wall of the tube, as observed in 18O-tracer studies.9,23,84 Moreover, the model shows a PAO growth model that is completely opposite to the traditional FAD theory. Besides, the reliability of the flow model has also been confirmed by many other researchers, even researchers who support FAD theory.29,30,33 However, it may be limited by the research conditions at the time; this model only summarizes the phenomenon, and it does not give an explanation of the internal mechanism. It only makes inferences about the growth of oxides related to ionic and electronic currents.85 But it still provides important enlightenment for the later ionic and electronic current theory and oxygen bubble mould effect theory.45,46In 1969, Diggle et al.86 pointed out in a classic review article that in the early stage of aluminum anodization, under the action of a high electric field, the conductance in the oxides is dominated by ion conductance. When the oxides grow to a certain thickness, the electric field is reduced to a certain extent, and the conductance in the oxides will be transformed into electronic conductance.15,42,46–49 This provided a theoretical basis for the formation of the electronic current and ionic current theory. In 1981, the classic Wood model of PAA reported the existence of anion-contaminated alumina (ACA) at the interface near the electrolyte in the porous layer.87 As mentioned in Section 2.3, the amount of oxygen released has an effect on the morphology of nanotubes. Unfortunately, these typical phenomena were overlooked in previous theories, such FAD theory and FAE theory. Based on previous research, Zhu et al.46 pointed out the oxygen bubble mould effect, which connected the changes in the current with the formation process of PAO and explained the dynamic basis of the plastic flow phenomenon in 2009. The mechanism explanation based on the ionic and electronic current theory and the oxygen bubble mould effect has been recognized by increasing numbers of scholars in recent years.28,53,69,88
The ionic current and electronic current theory and the oxygen bubble model effect explain the current curve and morphological changes during the growth of PAO as follows:
The current density during the anodizing process affects the rate of oxide growth, but it should be combined with the current efficiency (that is, how much current is ultimately involved in the growth of the oxides) for analysis.23,36–38,89 According to the electronic current and ionic current theory, the total current in the anodizing process is composed of ionic current and electronic current.46,69,74,85,88,90–97 Among them, the ionic current dominates the growth of oxides (main reaction), and the electronic current dominates the precipitation of oxygen (side reaction). In 2015, Chong et al.47 studied the transient current under constant anodizing voltage based on the ionic current and electronic current theories and the research results of Yang et al.,98 and they gave the following theoretical expressions of the time-dependent ionic current and electronic current:
| j = jion + je | (5) |
 | (6) |
 | (7) |
In 2018, Zhao et al.93 put forward a new decomposition method of electronic current and ionic current based on the above equations, which is in good agreement with actual experimental results.
(1) The current drop stage (the growth of the compact layer)
In this stage, Jtotal = Jion, that is, the current efficiency is close to 100%, which is recognized by mainstream theories.23,29,38 At this time, almost all the current comes from the migration of ions, and the oxides rapidly grow to form a compact layer. As the thickness of the oxides increases, it becomes more difficult for ions to cross the oxides; therefore, the migration rate of the ions gradually decreases, which causes continuous declination of the ionic current (the macroscopic manifestation is a decrease in the total current density). It is important to note here that not all ions entering the oxides can successfully cross the oxides (that is, the number of ions that pass through the oxides is not equal to the number of ions that enter the oxides), which means there is an ion-rich area in the oxides (this is consistent with reports that an anion-rich area was observed on the surface of PAO). The anion-rich area is called the anion contaminated layer (ACL) in the oxygen bubble mould effect theory.11,45,87,90 The above process is shown in Fig. 4a and b. Compared with other theories referred to in Section 2.2, we can find that FAD theory and oxygen bubble mould effect theory have similar descriptions of the ion movement in stage I of the anodization, that is, the oxides are formed at the E/O interface and O/M interface at the same time.
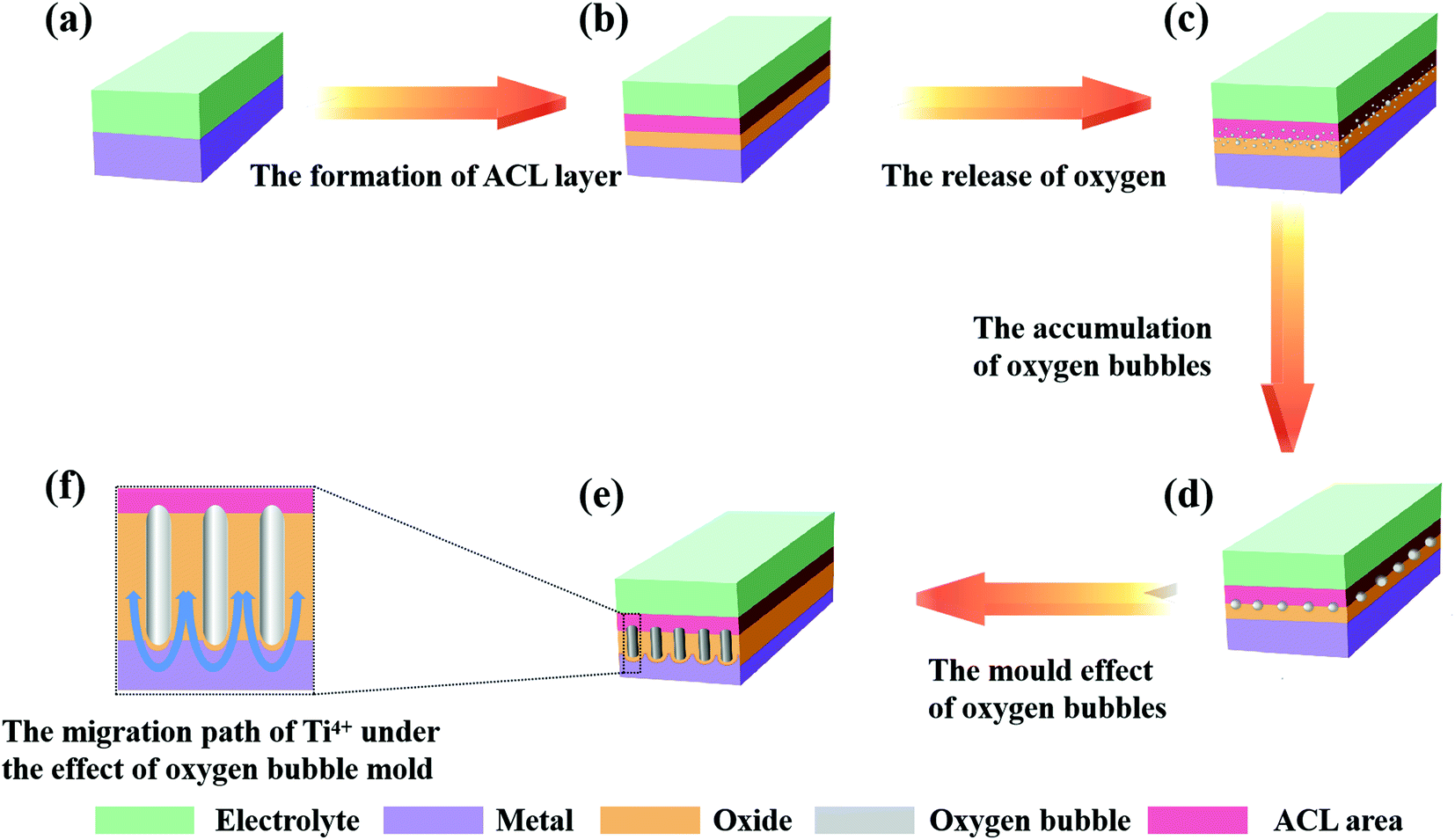 | ||
| Fig. 4 Schematic of the mechanism of the oxygen bubble mould effect theory. | ||
(2) The current rise stage (the release of oxygen)
As shown in Fig. 4b, the compact layer close to the electrolyte is transformed into an ACL. As the oxides thicken, the number of anions accumulated in the ACL continues to increase. When the compact layer reaches a certain thickness, the anion aggregated in the ACL has also reached a considerable number, which can cause a collision discharge of anions (2O2− → O2 + 4e−).46 Once the electronic current is formed, it will increase exponentially (refer to eqn (7)), which is called the avalanche electronic current.44,46,49,85 As shown in Fig. 4c, the electronic current increases rapidly with the release of oxygen, and it causes the total current to rise in this process (ionic current is still maintaining the growth of oxides). However, oxygen is released at the interface between the ACL and the compact layer, which means that oxygen bubbles cannot enter the electrolyte directly under the influence of atmospheric pressure, electrolyte pressure and ACL pressure; instead, the bubbles stay inside the oxides and gradually expand (Fig. 4d). Finally, under the influence of the pressure of the oxygen bubbles and the volume expansion stress of the oxides, the oxides at the bottom bend and deform to form a hemispherical bottom. It is worth mentioning that due to the existence of oxygen bubbles, ions cannot migrate through the inside of the oxygen bubbles; they can only grow upwards along both sides of the oxygen bubbles (Fig. 4e and f). In this stage, oxygen bubbles guide the formation of PAO like a mould.
(3) The current holding stage (the perforation of the channel)
As the side reaction (2O2− → O2 + 4e−) continues, the continuously released oxygen causes the accumulation of pressure inside the oxygen bubble. Once the pressure of the oxygen bubble reaches the critical point, the oxygen bubble will break through the ACL and enter the electrolyte, and the diameter of the channels will also be determined accordingly. At this time, Je and Jion no longer change, the release rate of oxygen and the growth rate of oxides reach equilibrium (macroscopically, the total current remains stable), and the oxides begin to grow steadily. In addition to these, a new pollution layer will form at the bottom of the PAO after the oxygen bubbles enter the electrolyte. Moreover, the oxygen bubbles evolved continuously from the bottom will maintain the tubular shape of the holes.46 In addition, it is reported that the effect of gas release on the formation of nanostructures cannot be ignored.74,99–104
3. The explanations of experimental phenomena based on different theories
Due to differences in the formation mechanism of PAO, some common experimental phenomena also have different explanations. However, the previous reviews on the formation mechanism of PAO rarely compared the explanations of these phenomena based on different theories; therefore, in this part, we will conduct a detailed analysis from this perspective.3.1 The influence of anodizing voltage
(1) FAD theoryWhen the anodizing voltage increases, the fluoride ions in the electrolyte will move faster under the action of the voltage, and the rate of the FAD reaction will also increase. The increase in the ‘digging’ rate of the tunnel is ultimately manifested as the increase in the growth rate of the PAO. However, many documents supporting the FAD theory have adopted the assumption that the movement speed of F− under the action of an electric field is twice that of O2−.70,71 According to the formula F = q × E, a = F/M, where F is the force received by the ion in the electric field, q is the amount of charge carried by the ion, E is the electric field strength at the position of the ion, M is the mass of the ion, and a is the acceleration of the ion. The relative atomic mass of F− is 18.998, and it has a negative charge. The relative atomic mass of O2− is 15.999, and it has two negative charges. The force of O2− in the electric field is twice that of F−, and its weight is about 84.21% that of fluoride ion; therefore, the movement speed of oxygen ion should be greater than that of fluoride ion, which is completely opposite to the speed assumption adopted in FAD theory. This means that the existence of [TiF6]2− cannot be directly inferred from the FAD theory, and the existence of [TiF6]2− remains to be demonstrated, as no one has directly detected it to date.
(2) Self-organization theory
Self-organization theory defines an anodic growth factor f (determined by the type of oxides, such as f(TiO2) = 2.5 nm V−1) and the applied voltage ΔU (the effective voltage needs to be considered for determination in different electrolytes), which can be used to estimate the radius of the hemispherical bottom of nanotubes based on the formula r = f × ΔU. To a large extent, this simple formula is able to explain the linear dependencies of pore/tube diameters on the anodizing voltage observed in the experiment. If too high a voltage is applied, this will trigger (dielectric) breakdown events in the oxides.23 The occurrence of the breakdown event is still in line with the expectation of the formula——the radius of the nanotubes is so large that the surface topography is continuously destroyed. Although this formula can be used to predict the size of the radius, it is only an empirical formula and does not analyze how the voltage affects the radius of the PAO from a theoretical level. Therefore, the mechanism of the influence of voltage on the radius of the PAO is not clear.
(3) Ionic and electronic current theory and oxygen bubble mould effect
According to a series of previous studies by scholars who support this theory, as the anodizing voltage increases, the ionic current and electronic current during the reaction increase correspondingly. In other words, the influence of voltage on the PAO morphology is realized by changing the current. Therefore, here, we will not go into detail; please refer to Chapter 3.3.
3.2. The influence of the electrolyte composition
(1) FAD theoryIn FAD theory, the content of F− is an important factor affecting the FAD reaction. The higher the content of F−, the faster the FAD reaction proceeds, and vice versa.74,105 According to experimental facts, when the content of F− in the electrolyte increases, the growth rate of PAO increases, and the tube walls of the nanotubes will also become thinner. From this perspective, the expectations of the FAD theory are in good agreement with the experimental results. However, some groups have successfully prepared PAO in fluorine-free electrolytes.15,24,106,107 FAD theory attributes the formation of nanotubes to the corrosion of oxides by F− under the action of the electric field. When the voltage is constant, the concentration of F− determines the speed of corrosion. From this, we can make the following two inferences: one is that the F− content determines the speed of the FAD reaction, and the other is that the electrolyte contains H2O (the moisture content is not important), which can cause the FAD reaction. This means that if the electrolyte has higher F− content, nanotubes with smoother walls will be obtained. However, the experimental fact is that ribs can be observed on the surface of the nanotubes in electrolytes with high F− content.36 However, the rib phenomenon of nanotubes is unlikely to appear during the corrosion process of the FAD reaction on the oxides from top to bottom. Moreover, Chong et al.108 believe that the ribs are formed by the reaction between the electrolyte entering the gap between the tube walls and the titanium substrate. Besides, experiments have proved that PAO cannot be formed if the H2O content of the electrolyte is below a certain critical point.99,103
(2) Self-organization theory
Self-organization theory proposes that appropriate solubility is the precondition for the formation of PAO. Moreover, the main factors affecting the solubility are the F− content and the H2O content.23,57,60,72,74,79 As the solubility of the electrolyte increases, the morphology will also change from pores and nanotubes to electropolishing oxides and sponge-oxides. This explanation is appropriate from a macro perspective. From a microscopic point of view, the erosion of oxides by fluoride ions only occurs in the gaps, and it does not contribute to the generation of nanopores in self-organization theory. In other words, the content of F− only changes the size of the gap between the tubes and cannot affect the diameter of the pores, which conflicts with experimental facts.
(3) Ionic and electronic current theory and the oxygen bubble mould effect
In 2020, Zhang et al.38 reported that the total volume of the nanotube array remained about the same for a given current, and the volume expansion factor of the nanotubes was around 2.0 in electrolytes with different NH4F contents, which is contrary to what would be expected on the basis of the FAD and FAE theories. They also found that upon increasing the NH4F concentration, the inner diameter of the nanotubes remained unchanged. They attributed these phenomena to oxygen bubbles, as the inner diameter of the nanotubes depends on the oxygen bubble mould effect.
3.3 The explanation of the current curve
As shown in Fig. 5, the preparation of PAO by constant voltage anodization can usually record a typical current–time curve, and different theories explain this process as follows: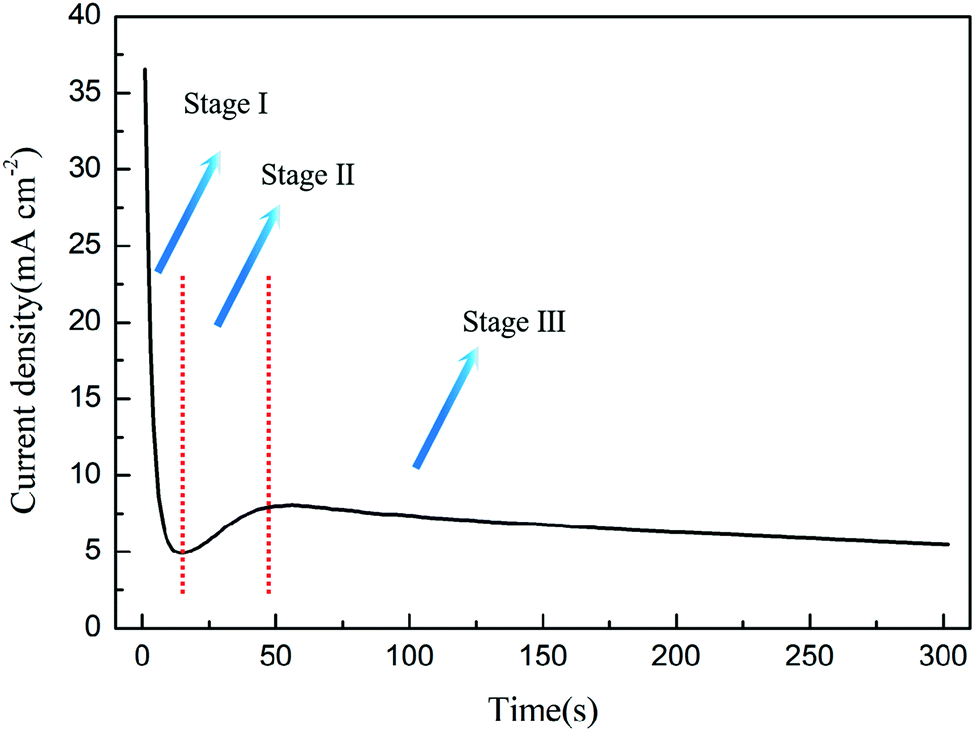 | ||
| Fig. 5 The current–time curve during anodization. | ||
(1) FAD theory
In the constant voltage anodization process, the current curve is usually used to record the PAO formation process. The current curve during the formation process of PAO is usually divided into three stages: falling, rising and steady. The FAD theory explains the change of the three-stage current as follows: (1) the first stage of the current falling is due to the formation of the compact layer at the initial stage of the reaction, which causes the resistance to increase. (2) The second stage of the current rising is due to the FAD reaction that reduces the oxide layer resistance. (3) The third stage of the steady current is due to the PAO reaching growth and dissolution equilibrium, and the resistance no longer changes.54,59,64 This is exactly in line with the three reaction stages proposed in the FAD theory above. The above explanation actually treats the entire anodization system as a black box for analysis. The FAD theory regards the electrolyte and the anode and cathode as a resistance, and it macroscopically analyzes the effect of resistance changes on the current under constant voltage conditions; however, it does not explain the microscopic changes, such as the contribution of FAD reactions to the current.
(2) Self-organization theory
Self-organization theory gives no explanation for the changes of currents, but it describes the growth of nanotubes at each current stage. In stage I, the fast formation process of the compact layer leads to an increase of the electrical resistance because the compact layer with an insulator nature becomes thicker with time, and this causes exponential decay of the current density. In stage II, the nucleation of the initial pores occurs with the increase of the current density, and it continues until a local maximum is reached. This process is accompanied by the development of an incipient disordered porous structure. When a steady state is reached as the density of pores is reduced, the growth of PAO comes to stage III. In this stage, the disordered pores start to merge, and pores parallel to each other and perpendicular to the surface start to grow. When the final steady state is achieved, the current density tends to stabilize.35,52 In addition to the change of the current density, self-organization theory takes the factor of current efficiency into account, which is more advanced than the previous FAD and FAE theories.23 The phenomenon that the current efficiency affects the morphology of PAO has been reported many times. Moreover, self-organization theory considers the contribution of different reactions to the current. It points out that not only the main reaction of oxide growth but side reactions that lead to the precipitation of oxygen occur during the anodizing process. In the electrolytes with high water content, excessive oxygen precipitation can lead to the formation of sponge-like oxides.23 At this point, the self-organization theory is in good agreement with experimental phenomena. In fact, the release of oxygen in the formation process of PAO is universal (only the amount is different), and the self-organization theory lacks quantitative analysis of the relationship between oxygen release and morphology. Thus, if the current efficiency is high enough to form PAO, how does oxygen affect the morphology of PAO? How does oxygen leave the PAO? These questions cannot be answered in the theory of self-organization.
(3) Ionic and electronic current theory and the oxygen bubble mould effect
The theory of ionic and electronic current proposes that the total current is composed of ionic current and electronic current. The ionic current dominates the growth of oxides, and the electronic current is related to the evolution of oxygen. In stage I, oxides form rapidly at the E/O interface and O/M interface, and the ionic current accounts for almost 100% of the total current.46,47 However, it becomes increasingly difficult for ions to pass through the oxides with the formation of the compacter layer, which leads to a rapid drop in the total current. In stage II, the anions between the ACL and the barrier layer release electrons due to the collision, and they form an avalanche electronic current, which is accompanied by the release of oxygen.11,108 Although the ionic current is declining, the continuously rising electronic current makes up for this part of the loss, and finally causes the total current to rise. In stage III, the electronic current reaches a steady state with the oxygen bubbles entering the electrolyte, and the thickness of the barrier layer hardly changes at this time; that is, the ionic current also reaches a steady state, so the total current does not change.95
3.4. The formation of the hemispherical bottom of PAO and the dynamics of plastic flow
(1) FAD theoryThe phenomena observed by Skeldon and his co-workers61 in the 18O-tracer study are also significantly different from the expectations of the FAD theory.
Fig. 6a shows a schematic of the tracer atom experimental results expected by the FAD theory, and Fig. 6b shows an experimental diagram of the actual experimental results. Comparing Fig. 6a and b, we can see that according to the FAD theory, the tracer atomic layer on the growth path of nanopores should be dissolved instead of being ‘squeezed’ onto the tube wall. Within the framework of FAD theory, it is impossible to find a reasonable explanation for this phenomenon.
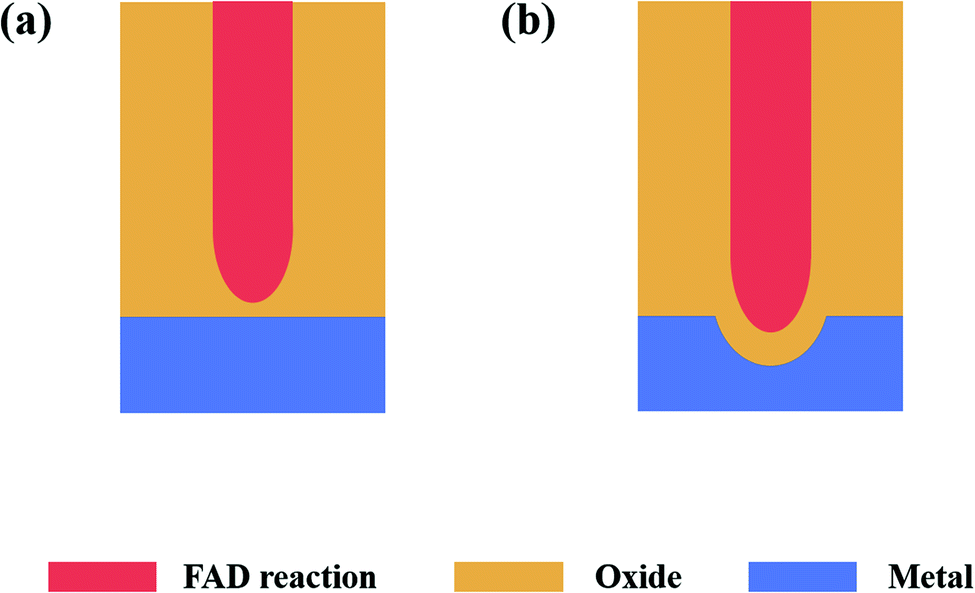 | ||
| Fig. 6 (a) Schematic of FAD theoretical expectations and (b) schematic of the experimental phenomena. | ||
(2) Self-organization theory
Because a highly ordered hexagonal close-packed TiO2 structure can be observed clearly in the vicinity of the tube bottom, the supporters of self-organization theory consider that this structure is self-organized in the early growth stage and so is PAA.
In the early growth stage, the buildup of compressive stress is a key element to the formation of the hemispherical bottom. Moreover, the compressive stress is created by plain volume expansion when converting metal to oxides (the Pilling–Bedworth ratio), which may be enhanced by voltage-induced electrostrictive forces.80 Especially, the view that the existence of stress contributes to the transfer of oxides from the bottom of the tube to the wall is recognized by many scholars.13,23 This explanation of the formation of the hemisphere is more advanced than the traditional FAD theory and FAE theory, because the model of ‘dig from top to bottom’ and the model of ‘ejection’ have no dynamic factors that reasonably promote the formation of a regular bottom surface. Although self-organization theory involves further analysis of the cause of the hemispherical bottom than previous theories (FAD theory and FAE theory), it still has the following shortcomings:
(a) The view that the volume expansion when the metal is transformed into oxides is the main reason for the compressive stress lacks support of experimental data. Moreover, if volume expansion is the main source of compressive stress, then how does the applied voltage influence the radius of the hemispherical bottom? This remains to be discussed.
(b) When the length of the nanotubes increases, more force is needed to promote the upward flow of oxides; therefore, the effect of stress (caused by the volume expansion at the O/M surface) which pushes the oxides from the bottom to the wall will be relatively weakened. Therefore, there must be other factors in the anodizing process to ensure that the nanotubes can grow vertically. The self-organization theory only mentions that voltage-induced electrostrictive forces may enhance the transfer of oxides. Therefore, this theory is incomplete in this area.23,80
(3) Ionic and electronic current theory and the oxygen bubble mould effect
According this theory, the formation of the hemispherical bottom of PAO and the plastic flow of oxides during the anodizing process are due to the oxygen bubble mould effect. As mentioned in Section 2.4, the oxygen bubbles released in the second stage of the anodizing process will stay between the ACL and the compact layer, which changes the migration path of ions in the oxides. Due to the existence of oxygen bubbles, the ions that migrate under the action of the electric field can only move around the oxygen bubbles, and they finally appear as plastic flow.14,97 Moreover, because of the external pressure, the oxygen bubbles cannot enter the electrolyte immediately after being released; they will form compressive stress on the oxides and the substrate, which reasonably explains the source of the compressive stress in the oxides and the formation of the hemispherical bottom.
4. Summary and outlook
The research on PAO initially focused on PAA and ATNTs, and with the development of preparation technology, research interest is also rising in the anodization of other valve metal oxides, such as Zr, Sn, Ni, Ta, V, Nb, W and Cu.23,28,54,69,88,109–113 These metals can be prepared by anodization to form films with regular nanotube arrays, which have shown wide application potential in a wide variety of fields, such as sensors, supercapacitors, and photocatalysis.111,114–126 Because of its simple and efficient preparation process and low cost, it also has good prospects in future industrial production. Therefore, further research on the mechanism is of great significance for the preparation of nanoporous materials with precise structures.This paper reviews the basic research on the formation mechanism of PAO, and it introduces four mainstream mechanism explanations. By comparing the explanations of these mechanisms for some common experimental phenomena, we attempt to analyze which theoretical model is more convincing. FAD theory and FAE theory are theoretical models that were proposed early and used to explain the formation mechanism of PAO; they have the advantages of simplicity, intuitiveness, and easy understanding. They have a guiding significance that cannot be ignored for the study of the PAO mechanism. However, with the development of characterization methods, they also showed increasing limitations, the two most critical points of which are the plastic flow of oxides and the relationship between the current and the reaction. The experimental phenomenon of plastic flow is completely contrary to the expectations of FAD theory and FAE theory. Moreover, the FAD and FAE reaction is a chemical reaction, and it lacks physical connection with the current in the anodizing process. Self-organization theory further explored the formation mechanism of PAO on the basis of FAD theory and FAE theory, which were combined with physical factors such as volume expansion and stress and strain to explain the formation mechanism of PAO, and it put forward some empirical formulas for predicting morphology based on experiments. However, its explanation of the changes in the current curve during the anodizing process still has defects. The ionic current and electronic theory combines the current change with the formation of oxides from the perspective of physics. Moreover, the oxygen bubble theory analyzes the dynamics of the plastic flow and the source of the compressive stress at the bottom of the PAO. It is worth mentioning that this theory also coincides with the influence of oxygen bubble evolution on the morphology mentioned in the self-organization theory.
Certainly, in addition to the mainstream mechanisms which were mentioned above, there are many other interesting studies. In 2008, Su et al.116 established the equifield strength model to explain the formation of the pore morphology. In 2020, Suliali et al.19 connected the anodic current measured during TiO2 growth and the kinetics of the redox reactions of water with a mathematical model of anodization reaction kinetics. In 2019, Byvaltsev13 summarized seven models, including the model of mechanical stress and the model of point defects.
In summary, PAO is an important future development direction in the field of materials; therefore, the formation mechanism of PAO still needs to be studied in order to enable its wide use in the field of materials.
Author contributions
Chengyuan Li: writing – original draft, methodology, data curation. Yilin Ni: review & editing, investigation. Jingjing Gong: software, Visualization. Ye Song: supervision. Tianle Gong: review & editing. Xufei Zhu: conceptualization, project administration.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 51777097, 51577093) and the National Undergraduate Training Program for Innovation and Entrepreneurship (202010288034Z). Thanks for Z. Y.References
- J. F. Liao, Z. Y. Ling, Y. Li and X. Hu, Electrochem. Commun., 2015, 58, 37–40 CrossRef CAS.
- H. Zhang, Z. J. Chen, Y. Song, M. Yin, D. Li, X. F. Zhu, X. Y. Chen, P. C. Chang and L. F. Lu, Electrochem. Commun., 2016, 68, 23–27 CrossRef CAS.
- R. Hang, Y. Zhao, L. Bai, Y. Liu, A. Gao, X. Zhang, X. Huang, B. Tang and P. K. Chu, Electrochem. Commun., 2017, 76, 10–14 CrossRef CAS.
- Y. L. Zhang, W. L. Cheng, F. Du, S. Y. Zhang, W. H. Ma, D. D. Li, Y. Song and X. F. Zhu, Electrochim. Acta, 2015, 180, 147–154 CrossRef CAS.
- K. L. Wang, G. H. Liu, N. Hoivik, E. Johannessen and H. Jakobsen, Chem. Soc. Rev., 2014, 43, 1476–1500 RSC.
- H. Tsuchiya and P. Schmuki, Nanoscale, 2020, 12, 8119–8132 RSC.
- V. Galstyan, E. Comini, G. Faglia and G. Sberveglieri, Sensors, 2013, 13, 14813–14838 CrossRef PubMed.
- R. Zazpe, J. Prikryl, V. Gartnerova, K. Nechvilova, L. Benes, L. Strizik, A. Jager, M. Bosund, H. Sopha and J. M. Macak, Langmuir, 2017, 33, 3208–3216 CrossRef CAS PubMed.
- F. Riboni, N. T. Nguyen, S. Soa and P. Schmuki, Nanoscale Horiz., 2016, 1, 445–466 RSC.
- N. Pishkar, M. Ghoranneviss, Z. Ghorannevis and H. Akbari, Results Phys., 2018, 9, 1246–1249 CrossRef.
- S. Qin, L. Z. Wu, J. H. Zhang, Y. Han, H. X. Zheng, J. Ma and W. H. Ma, Mater. Res. Bull., 2018, 106, 220–227 CrossRef CAS.
- H. Masuda, A. Abe, M. Nakao, A. Yokoo, T. Tamamura and K. Nishio, Adv. Mater., 2003, 15, 161–164 CrossRef CAS.
- S. V. Byvaltsev, Mater. Today: Proc., 2019, 19, 1949–1952 CAS.
- X. F. Zhu, Y. Song, D. L. Yu, C. S. Zhang and W. Yao, Electrochem. Commun., 2013, 29, 71–74 CrossRef CAS.
- A. Gao, R. Q. Hang, L. Bai, B. Tang and P. K. Chu, Electrochim. Acta, 2018, 271, 699–718 CrossRef CAS.
- Z. X. Su and W. Z. Zhou, J. Mater. Chem., 2011, 21, 8955–8970 RSC.
- R. Jin, H. W. Fan, X. Y. Yin, Q. Chen, J. Ma and W. H. Ma, Electrochim. Acta, 2016, 222, 983–989 CrossRef CAS.
- P. Y. Li, D. M. Niu, M. F. He, H. Huang, Z. R Ying, H. Q. Xu, J. W. Zhu and X. F. Zhu, J. Phys. Chem. C, 2020, 124, 3050–3058 CrossRef CAS.
- N. J. Suliali, C. M. Mbulanga, W. E. Goosen, R. Betz and J. R. Botha, Electrochim. Acta, 2020, 337, 135791 CrossRef CAS.
- Q. Dou, P. Shrotriya, W. F. Li and K. R. Hebert, Electrochim. Acta, 2019, 295, 418–426 CrossRef CAS.
- J. P. O'Sullivan and G. C. Wood, Proc. R. Soc. London, Ser. A, 1970, 317, 511–543 Search PubMed.
- V. Zwilling, M. Aucouturier and E. Darque-Ceretti, Electrochim. Acta, 1999, 45, 921–929 CrossRef CAS.
- X. M. Zhou, N. T. Nguyen, S. Özkan and P. Schmuki, Electrochem. Commun., 2014, 46, 157–162 CrossRef CAS.
- R. Hahn, J. M. Macak and P. Schmuki, Electrochem. Commun., 2007, 9, 947–952 CrossRef CAS.
- E. Palibroda, Electrochim. Acta, 1995, 40, 1051–1055 CrossRef CAS.
- S. Berger, J. Kunze, P. Schmuki, A. T. Valota, D. J. LeClere, P. Skeldon and G. E. Thompson, J. Electrochem. Soc., 2010, 157, C18–C23 CrossRef CAS.
- G. E. Thompson, Thin Solid Films, 1997, 297, 192–201 CrossRef CAS.
- W. Lee and S. J. Park, Chem. Rev., 2014, 114, 7487–7556 CrossRef CAS.
- P. Skeldon, G. E. Thompson, S. J. Garcia-Vergara, L. Iglesias-Rubianes and C. E. Blanco-Pinzon, Electrochem. Solid-State Lett., 2006, 9, B47–B51 CrossRef CAS.
- S. J. Garcia-Vergara, P. Skeldon, G. E. Thompson and H. Habazaki, Electrochim. Acta, 2006, 52, 681–687 CrossRef CAS.
- S. J. Garcia-Vergara, H. Habazaki, P. Skeldon and G. E. Thompson, Electrochim. Acta, 2010, 55, 3175–3184 CrossRef CAS.
- K. R. Hebert, S. Albu, I. Paramasivam and P. Schmuki, Nat. Mater., 2012, 11, 162–166 CrossRef CAS PubMed.
- J. E. Houser and K. R. Hebert, Nat. Mater., 2009, 8, 415–420 CrossRef CAS PubMed.
- J. E. Houser and K. R. Hebert, J. Electrochem. Soc., 2006, 153, B566–B573 CrossRef CAS.
- J. Li, Z. Y. Zhang, Y. X. Li, Y. J. Ma, L. Chen, Z. Y. Zhang and R. G. Sun, Electrochim. Acta, 2016, 213, 14–20 CrossRef CAS.
- D. Regonini, C. R. Bowen, A. Jaroenworaluck and R. Stevens, Mater. Sci. Eng. R Rep., 2013, 74, 377–406 CrossRef.
- M. S. Yu, Y. Chen, C. Li, S. Yan, H. M. Cui, X. F. Zhu and J. S. Kong, Electrochem. Commun., 2018, 87, 76–80 CrossRef CAS.
- Z. Y. Zhang, Q. Wang, H. Q. Xu, W. C. Zhang, Q. Y. Zhou, H. P. Zeng, J. L. Yang, J. W. Zhu and X. F. Zhu, Electrochem. Commun., 2020, 114, 106717 CrossRef CAS.
- K. Nielsch, J. Choi, K. Schwirn, R. B. Wehrspohn and U. Gösele, Nano Lett., 2002, 2, 677–680 CrossRef CAS.
- T. T. Kao and Y. C. Chang, Appl. Surf. Sci., 2014, 288, 654–659 CrossRef CAS.
- G. Knörnschild, A. A. Poznyak, A. G. Karoza and A. Mozalev, Surf. Coat. Technol., 2015, 275, 17–25 CrossRef.
- P. Roy, S. Berger and P. Schmuki, Angew. Chem., Int. Ed., 2011, 50, 2904–2939 CrossRef CAS PubMed.
- J. C. Wu, Y. Li, Z. X. Li, S. Z. Li, L. Shen, X. Hu and Z. Y. Ling, Electrochem. Commun., 2019, 109, 106602 CrossRef CAS.
- J. M. Albella, I. Montero and J. M. Martinez-Duart, Electrochim. Acta, 1987, 32, 255–258 CrossRef CAS.
- X. F. Zhu, L. Liu, Y. Song, H. B. Jia, H. D. Yu, X. M. Xiao and X. L. Yang, Monatsh. Chem., 2008, 139, 999–1003 CrossRef CAS.
- X. F. Zhu, Y. Song, L. Liu, C. Y. Wang, J. Zheng, H. Jia and X. Wang, Nanotechnology, 2009, 20, 475303 CrossRef PubMed.
- B. Chong, D. Yu, R. Jin, Y. Wang, D. Li, Y. Song, M. Gao and X. Zhu, Nanotechnology, 2015, 26, 145603 CrossRef PubMed.
- W. Q. Huang, H. Q. Xu, Z. R. Ying, Y. X. Dan, Q. Zhou, J. Zhang and X. F. Zhu, Electrochem. Commun., 2019, 106, 106532 CrossRef CAS.
- M. S. Yu, C. Li, Y. B. Yang, S. K. Xu, K. Zhang, H. M. Cui and X. F. Zhu, Electrochem. Commun., 2018, 90, 34–38 CrossRef CAS.
- R. Q. Hang, F. L. Zhao, X. H. Ya, B. Tang and P. K. Chu, Appl. Surf. Sci., 2020, 517, 146118 CrossRef CAS.
- W. Wei, S. Berger, C. Hauser, K. Meyer, M. Yang and P. Schmuki, Electrochem. Commun., 2010, 12, 1184–1186 CrossRef CAS.
- S. Singh, M. Festin, W. R. T. Barden, L. Xi, J. T. Francis and P. Kruse, ACS Nano, 2008, 2, 2363–2373 CrossRef CAS PubMed.
- A. Ruiz-Clavijo, O. Caballero-Calero and M. Martín-González, Nanoscale, 2021, 13, 2227–2265 RSC.
- Y. D. Xue and Y. T. Wang, Nanoscale, 2020, 12, 10912–10932 RSC.
- J. T. Domagalski, E. Xifre-Perez, A. Santos, J. Ferre-Borrull and L. F. Marsal, Microporous Mesoporous Mater., 2020, 303, 110264 CrossRef CAS.
- R. Sanz, M. A. Buccheri, M. Zimbone, V. Scuderi, G. Amiard, G. Impellizzeri, L. Romano and V. Privitera, Appl. Surf. Sci., 2017, 399, 451–462 CrossRef CAS.
- M. Martín-Gonzaleza, R. Martinez-Moro, M. H. Aguirre, E. Flores and O. Caballero-Calero, Electrochim. Acta, 2020, 330, 135241 CrossRef.
- J. H. Oh and C. V. Thompson, Electrochim. Acta, 2011, 56, 4044–4051 CrossRef CAS.
- D. S. Guan and Y. Wang, Nanoscale, 2012, 4, 2968–2977 RSC.
- J. M. Macak, H. Tsuchiya, A. Ghicov, K. Yasuda, R. Hahn, S. Bauer and P. Schmuki, Curr. Opin. Solid State Mater. Sci., 2007, 11, 3–18 CrossRef CAS.
- C. Cherki and J. Siejka, J. Electrochem. Soc., 1973, 120, 784–791 CrossRef CAS.
- C. Cheng and A. H. W. Ngan, Electrochim. Acta, 2011, 56, 9998–10008 CrossRef CAS.
- M. P. Kobylański, P. Mazierski, A. Malankowska, M. Kozak, M. Diak, M. J. Winiarski, T. Klimczuk, W. Lisowski, G. Nowaczyk and A. Zaleska-Medynska, Surf. Interfaces, 2018, 12, 179–189 CrossRef.
- L. Mohana, C. Dennis, N. Padmapriya, C. Anandan and N. Rajendran, Mater. Today Commun., 2020, 23, 101103 CrossRef.
- M. S. Yu, H. M. Cui, F. P. Ai, L. F. Jiang, J. S. Kong and X. F. Zhu, Electrochem. Commun., 2018, 86, 80–84 CrossRef CAS.
- H. Takahashi and M. Nagayama, Corros. Sci., 1978, 18, 911–917 CrossRef CAS.
- H. Habazaki, K. Shimizu, S. Nagata, P. Skeldon, G. E. Thompson and G. C. Wood, Corros. Sci., 2002, 44, 1047–1055 CrossRef CAS.
- H. Habazaki, P. Skeldon, K. Shimizu, G. E. Thompson and G. C. Wood, Corros. Sci., 1995, 37, 1497–1509 CrossRef CAS.
- S. X. Liu, J. L. Tian and W. Zhang, Nanotechnology, 2021, 32, 222001 CrossRef CAS PubMed.
- D. J. LeClere, A. Velota, P. Skeldon, G. E. Thompson, S. Berger, J. Kunze, P. Schmuki, H. Habazaki and S. Nagata, J. Electrochem. Soc., 2008, 155, C487–C494 CrossRef CAS.
- H. Habazaki, K. Fushimi, K. Shimizu, P. Skeldon and G. E. Thompson, Electrochem. Commun., 2007, 9, 1222–1227 CrossRef CAS.
- H. Tsuchiya and P. Schmuki, Electrochem. Commun., 2004, 6, 1131–1134 CrossRef CAS.
- H. Sopha, A. Jäger, P. Knotek, K. Tesař, M. Jarosova and J. M. Macak, Electrochim. Acta, 2016, 190, 744–752 CrossRef CAS.
- D. Kowalski, D. Kim and P. Schmuki, Nano Today, 2013, 8, 235–264 CrossRef CAS.
- A. Mazzarolo, K. Lee, A. Vicenzo and P. Schmuki, Electrochem. Commun., 2012, 22, 162–165 CrossRef CAS.
- H. Masuda and K. Fukuda, Science, 1995, 268, 1466–1468 CrossRef CAS PubMed.
- S. Berger, J. Kunze, P. Schmuki, D. LeClere, A. T. Valota, P. Skeldon and G. E. Thompson, Electrochim. Acta, 2009, 54, 5942–5948 CrossRef CAS.
- A. Jaroenworaluck, D. Regonini, C. R. Bowen, R. Stevens and D. Allsopp, J. Mater. Sci., 2007, 42, 6729–6734 CrossRef CAS.
- O. Jessensky, F. Müller and U. Gösele, Appl. Phys. Lett., 1998, 72, 1173–1175 CrossRef CAS.
- V. Zwilling, E. Darque-Ceretti, A. Boutry-Forveille, D. David, M. Y. Perrin and M. Aucouturier, Surf. Interface Anal., 1999, 27, 629–637 CrossRef CAS.
- J.-F. Vanhumbeeck and J. Proost, Electrochim. Acta, 2008, 53, 6165–6172 CrossRef CAS.
- W. Z. Cao, K. F. Chen and D. F. Xue, Materials, 2021, 14, 510 CrossRef CAS PubMed.
- S. P. Albu, A. Ghicov, J. M. Macak and P. Schmuki, Phys. Status Solidi RRL, 2007, 1, R65–R67 CrossRef CAS.
- J. Siejka and C. Ortega, J. Electrochem. Soc., 1977, 124, 883–891 CrossRef CAS.
- M. Propp, L. Young, D. L. Pulfrey and G. Olive, J. Electrochem. Soc., 1977, 124, 891–897 CrossRef CAS.
- J. W. Diggle, T. C. Downie and C. W. Goulding, Chem. Rev., 1969, 69, 365–405 CrossRef CAS.
- G. E. Thompson and G. C. Wood, Nature, 1981, 290, 230–232 CrossRef CAS.
- Y. Fu and A. Mo, Nanoscale Res. Lett., 2018, 13, 187–207 CrossRef PubMed.
- Q. V. Overmeere, D. Mercier, R. Santoro and J. Proost, Electrochem. Solid-State Lett., 2012, 15, C1–C4 CrossRef.
- M. S. Yu, H. M. Cui, H. Li, S. Y. Zhang, J. S. Kong, S. W. Zhao, F. P. Ai, Y. Song and X. F. Zhu, J. Phys. Chem. C, 2018, 122, 549–556 CrossRef CAS.
- F. Yang, X. J. Fenga, F. Ge, T. C. Zhang, J. R. Qi, D. Z. Li and X. F. Zhu, Electrochem. Commun., 2019, 103, 17–21 CrossRef CAS.
- S. W. Zhao, L. Z. Wu, C. Li, C. Y. Li, M. S. Yu, H. M. Cui and X. F. Zhu, Electrochem. Commun., 2018, 93, 25–30 CrossRef CAS.
- S. W. Zhao, C. Li, T. F. Wei, C. Y. Li, M. S. Yu, H. M. Cui and X. F. Zhu, Electrochem. Commun., 2018, 91, 60–65 CrossRef CAS.
- S. Y. Zhang, S. K. Xu, D. Y. Hu, C. Zhang, J. F. Che and Y. Song, Mater. Res. Bull., 2018, 103, 205–210 CrossRef CAS.
- J. J. Zhang, W. Q. Huang, K. Zhang, D. Z. Li, H. Q. Xu and X. F. Zhu, Electrochem. Commun., 2019, 100, 48–51 CrossRef CAS.
- K. Zhang, S. K. Cao, C. Li, J. R. Qi, L. F. Jiang, J. J. Zhang and X. F. Zhu, Electrochem. Commun., 2019, 103, 88–93 CrossRef CAS.
- Q. Y. Zhou, D. M. Niu, X. J. Feng, A. C. Wang, Z. R. Ying, J. P. Zhang, N. Lu, J. W. Zhu and X. F. Zhu, Electrochem. Commun., 2020, 119, 106815 CrossRef CAS.
- R. Q. Yang, L. F. Jiang, X. F. Zhu, Y. Song, D. L. Yu and A. J. Han, RSC Adv., 2012, 2, 12474–12481 RSC.
- K. S. Raja, T. Gandhi and M. Misra, Electrochem. Commun., 2007, 9, 1069–1076 CrossRef CAS.
- Z. D. Zhu, W. B. Tu, Y. L. Cheng and Y. L. Cheng, Surf. Coat. Technol., 2019, 361, 176–187 CrossRef CAS.
- A. Suleiman, T. Hashimoto, P. Skeldon, G. E. Thompson, F. Echeverria, M. J. Graham, G. I. Sproule, S. Moisa, H. Habazaki, P. Bailey and T. C. Q. Noakes, Corros. Sci., 2008, 50, 1353–1359 CrossRef CAS.
- D. L. Yu, Y. Song, X. F. Zhu, R. Q. Yang and A. J. Han, Appl. Surf. Sci., 2013, 276, 711–716 CrossRef CAS.
- Y. Konno, E. Tsuji, P. Skeldon, G. E. Thompson and H. Habazaki, J. Solid State Electrochem., 2012, 16, 3887–3896 CrossRef CAS.
- J. W. Cao, Z. Q. Gao, C. Wang, H. M. Muzammal, W. Q. Wang, Q. D. Gu, C. Dong, H. T. Ma and Y. P. Wang, Surf. Coat. Technol., 2020, 388, 125592 CrossRef CAS.
- S. P. Albu, P. Roy, S. Virtanen and P. Schmuki, Isr. J. Chem., 2010, 50, 453–467 CrossRef CAS.
- C. L. Chang, X. B. Huang, Y. P. Liu, L. Bai, X. N. Yang, R. Q. Hang, B. Tang and P. K. Chu, Electrochim. Acta, 2015, 173, 345–353 CrossRef CAS.
- J. Park and J. Choi, Appl. Surf. Sci., 2018, 448, 212–218 CrossRef CAS.
- B. Chong, D. L. Yu, M. Q. Gao, H. W. Fan, C. Y. Yang, W. H. Ma, S. Y. Zhang and X. F. Zhu, J. Electrochem. Soc., 2015, 162, H244–H250 CrossRef CAS.
- H. A. El-Sayed, C. A. Horwood, A. D. Abhayawardhana and V. I. Birss, Nanoscale, 2013, 5, 1494–1498 RSC.
- Z. Y. Zhang, C. P. Yu, J. H. Chen, Q. Y. Zhou, Y. J. Chen, J. Y. Xu, M. C. Xian, J. X. Wang, G. X. Yang, X. F. Zhu and W. C. Zhang, Chem. Eng. J., 2021, 425, 130676 CrossRef CAS.
- T. G. Novak, J. Kim, P. A. DeSario and S. Jeon, Nanoscale Adv., 2021, 3, 5166–5182 RSC.
- X. Li, C. Y. Li, T. L. Gong, J. H. Su, W. C. Zhang, Y. Song and X. F. Zhu, Ceram. Int., 2021, 47, 23332–23337 CrossRef CAS.
- T. L. Gong, C. Y. Li, X. Li, H. Y. Yue, X. F. Zhu, Z. Y. Zhao, R. Q. Lv and J. W. Zhu, Nanoscale Adv., 2021, 3, 4659–4668 RSC.
- J. J. Gong, Y. K. Lai and C. J. Lin, Electrochim. Acta, 2010, 55, 4776–4782 CrossRef CAS.
- Y. Y. Jiang, H. T. Zhao, J. Liang, L. C. Yue, T. S. Li, Y. L. Luo, Q. Liu, S. Y. Lu, A. M. Asiri, Z. J. Gong and X. P. Sun, Electrochem. Commun., 2021, 123, 106912 CrossRef CAS.
- Z. X. Su and W. Z. Zhou, Adv. Mater., 2008, 20, 3663–3667 CrossRef CAS.
- C. Chen, X. B. Wang, R. D. Xu, Y. Zhang, S. Y. Feng, A. Ju and W. H. Jiang, RSC Adv., 2021, 11, 6146–6158 RSC.
- C. C. Raj and L. Neelakantan, J. Electrochem. Soc., 2018, 165, E521–E526 CrossRef CAS.
- T. Yanagishita, R. Moriyasu, T. Ishii and H. Masuda, RSC Adv., 2021, 11, 3777–3782 RSC.
- M. S. Yu, W. Q. Huang, P. Y. Li, H. Huang, K. Zhang and X. F. Zhu, Electrochem. Commun., 2019, 98, 28–32 CrossRef CAS.
- S. K. Cao, W. Q. Huang, S. Y. Zhang, L. Z. Wu and Y. Song, J. Alloys Compd., 2019, 772, 173–177 CrossRef CAS.
- D. D. Li, L. Zhao, C. H. Jiang and J. G. Lu, Nano Lett., 2010, 10, 2766–2771 CrossRef CAS PubMed.
- D. M. Niu, Q. Y. Zhou, X. F. Zhu, X. J. Feng, S. Chen, A. C. Wang and Y. Song, Chem. Phys. Lett., 2020, 759, 137950 CrossRef CAS.
- J. W. Cao, C. Wang, Z. Q. Gao, S. Y. Shang, Q. D. Gu, N. Gao, Y. P. Wang and H. T. Ma, ECS J. Solid State Sci. Technol., 2020, 9, 104010 CrossRef CAS.
- X. Z. Lang, X. D. Wang, J. Ma and T. Qiu, Nanoscale Adv., 2021, 3, 2918–2923 RSC.
- X. H. Xia, Z. Y. Zeng, X. L. Li, Y. Q. Zhang, J. P. Tu, N. C. Fan, H. Zhang and H. J. Fan, Nanoscale, 2013, 5, 6040–6047 RSC.
| This journal is © The Royal Society of Chemistry 2022 |