
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanocubes of Mo6S8 Chevrel phase as active electrode material for aqueous lithium-ion batteries†
Amr
Elgendy
abc,
Athanasios A.
Papaderakis
 ab,
Rongsheng
Cai
d,
Kacper
Polus
e,
Sarah J.
Haigh
bdf,
Alex S.
Walton
ae,
David J.
Lewis
*d and
Robert A. W.
Dryfe
*ab
ab,
Rongsheng
Cai
d,
Kacper
Polus
e,
Sarah J.
Haigh
bdf,
Alex S.
Walton
ae,
David J.
Lewis
*d and
Robert A. W.
Dryfe
*ab
aDepartment of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: robert.dryfe@manchester.ac.uk; Tel: +44 (0) 161-306-4522
bHenry Royce Institute, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cEgyptian Petroleum Research Institute, 11727, Cairo, Egypt
dDepartment of Materials, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: david.lewis-4@manchester.ac.uk; Tel: +44 (0) 161-306-3561
ePhoton Science Institute, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
fNational Graphene Institute, University of Manchester, Oxford Road, Manchester M13 9PL, UK
First published on 23rd June 2022
Abstract
The development of intrinsically safe and environmentally sustainable energy storage devices is a significant challenge. Recent advances in aqueous rechargeable lithium-ion batteries (ARLIBs) have made considerable steps in this direction. In parallel to the ongoing progress in the design of aqueous electrolytes that expand the electrochemically stable potential window, the design of negative electrode materials exhibiting large capacity and low intercalation potential attracts great research interest. Herein, we report the synthesis of high purity nanoscale Chevrel Phase (CP) Mo6S8via a simple, efficient and controllable molecular precursor approach with significantly decreased energy consumption compared to the conventional approaches. Physical characterization of the obtained product confirms the successful formation of CP-Mo6S8 and reveals that it is crystalline nanostructured in nature. Due to their unique structural characteristics, the Mo6S8 nanocubes exhibit fast kinetics in a 21 m lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) electrolyte as a result of the shorter Li+ ion diffusion distance. Full battery cells comprised of Mo6S8 and LiMn2O4 as negative and positive electrode materials, respectively, operate at 2.23 V delivering a high energy density of 85 W h kg−1 (calculated on the total mass of active materials) under 0.2 C-rate. At 4 C, the coulombic efficiency (CE) is determined to be 99% increasing to near 100% at certain cycles. Post-mortem physical characterization demonstrates that the Mo6S8 anode maintained its crystallinity, thereby exhibiting outstanding cycling stability. The cell outperforms the commonly used vanadium-based (VO2 (B), V2O5) or (NASICON)-type LiTi2(PO4)3 anodes, highlighting the promising character of the nanoscale CP-Mo6S8 as a highly efficient anode material. In summary, the proposed synthetic strategy is expected to stimulate novel research towards the widespread application of CP-based materials in various aqueous and non-aqueous energy storage systems.
Introduction
The global “energy transition” that is currently underway requires the development of efficient and environmentally-friendly strategies for energy conversion and storage. Extensive research is being conducted towards the efficient exploitation of renewable energy sources. However, their intermittent nature in combination with a relatively low power output make the integration of renewable energy into the main electricity grid a challenging task.1 Development of a large-scale rechargeable energy storage system (ESS) is necessary to mitigate these issues. Among various energy storage systems, lithium-ion batteries have long been regarded as the primary power storage solution for portable electronic devices. Their long-term application in transportation and stationary systems is limited by safety issues associated with flammability and thermal instability of the organic electrolytes used.2,3 The latter under certain conditions of improper usage could potentially lead to fire or explosion.4,5 Aqueous Li-ion batteries (ARLIBs) are a highly promising alternative to conventional designs since they can potentially offer prolonged stability while maintaining high efficiency and complying with the necessary safety requirements.6 For these reasons, research on ARLIBs has gradually attracted attention since their introduction by Dahn et al.7 However, the main drawback of ARLIBs which prevents their wide application is the significantly narrow potential window of electrolyte stability (1.23 V) resulting from solvent degradation (water splitting).8 This means that the vast majority of the electrochemical couples used in conventional Li-ion batteries cannot be used for ARLIBs, since the anode suffers severe degradation even when a minimal amount of hydrogen gas is present at the anode side during cycling.8 These inherent limitations that arise from water electrolysis pose a major barrier to the development of high voltage ARLIBs (>2.0 V) and high energy density devices (>50 W h kg−1).9As a way of circumventing the limitations posed by the decomposition of water, Suo et al. have reported a new class of electrolytes, namely “water-in-salt” (WIS) electrolytes offering the possibility to form a solid electrolyte-interphase (SEI) in an aqueous electrolyte. In such systems, the potential window of electrolyte stability is significantly increased to 3.0 V from 1.23 V.10 The expanded potential window is due to the smaller water-to-salt ratio in the electrolyte, which results in the decrease of the number of free water molecules that can participate in the overall water oxidation process. Grimaud et al. studied the fate of water molecules in WIS electrolytes and demonstrated that the hydroxides produced during the hydrogen evolution reaction (HER), react chemically with the bis(trifluoromethanesulfonyl)imide TFSI− anions and catalyse the formation of the SEI that prevents further water reduction.11 An alternative mechanism for the SEI formation is the preferential decomposition of TFSI anions at the anode side, which contributes to the expanded potential window.12
The wide voltage window permits a wider choice of electrode materials than would otherwise be possible in conventional aqueous batteries.13 The proper selection of active electrode material is one of the key parameters that influences battery performance. So far, compounds such as LiCoO2, LiMn2O4, and LiFePO4 have been used as cathode materials in ARLIBs, which show a flat dis/charge plateau.14,15 However, few active materials demonstrate stable cycling performance when it comes to choosing an anode side with a suitable redox potential. Compared to the cathode, the proper selection of anode material is a more challenging task.16
Transition metal chalcogenides are promising anode materials, due to their unique structure and physical properties.17 In particular, Chevrel phase (CP) compounds with the formula MxMo6X8 (M = metal, X = S, Se, or Te),18 have been attracting great interest, because of their characteristic crystal structure, involving six Mo atoms embedded on the faces of a slightly distorted cube, formed by eight chalcogen atoms occupying its corners.19 In between the closely packed clusters, relatively open cavities are formed by three-dimensional (3D) channels, which allow the incorporation of various cationic species. Because of the unique structural characteristics, CPs have generated interest in electrocatalysis,20–22 superconductivity,23 and secondary ion batteries.24–26 Following their application to prototype Mg ion batteries,27 CPs have been also utilized in various metal-ion batteries, including mono and multivalent cations (Li+, Na+, Mg2+, Zn2+, Al3+).24 However, these devices suffer from a decreased cycling stability and low specific capacity, both arising from the sluggish kinetics of the prepared micrometre-sized Chevrel phases.24 In this respect, it has been suggested that electrode kinetics can be accelerated by switching to nanomaterials, due to the decrease in ion diffusion length.19 Such nanostructures also have the advantage of accommodating strains occurring during the ion insertion and removal processes.4 Developing synthetic routes to obtain controlled nanosized CPs is inherently attractive and directly related to the electrochemical performance of energy storage systems. So far, little progress has been made towards this goal. Jun Liu et al. reported a method for synthesizing CP nanocubes and showed that the material exhibits increased reversible capacity and improved kinetics, compared to larger particles.26 The preparation procedure was carried out at 1000 °C for 7 hours under the flow of forming gas (4% H2 and 96% Ar, 100 sccm). However, the use of hydrogen gas as a reducing agent at high temperatures requires rigorous safety protocols making the method less industrially attractive.25 Several alternative methods have been reported in the literature, including solid-state-reaction synthetic routes,18 molten salt approaches,28 high-energy mechanical milling strategies,25 homogeneous chemical methods with appropriate soluble precursors,29 a self-propagating high-temperature technique,30 and microwave-assisted methods.22 Most of these processes consume high amounts of energy, and the final product exhibits a low degree of purity with uncontrolled particle size most often in the micrometre range.
In recent years, the molecular precursor route was successfully used by one of the current authors to synthesize a series of binary, ternary, and quaternary metal chalcogenides, where in all cases the resultant materials exhibit structural features in the nanoscale range.31 Following a similar approach, we have recently reported the successful preparation of a nanoscale Mo6S8 which offers improved electrocatalytic performance towards HER in acidic media (0.5 M H2SO4).20 Inspired by the successful preparation of these nanostructured materials and their ability to host different cations, we investigate herein the performance of such nanoscale CP Mo6S8 electrodes as an anode material for ARLIBs. Since, the insertion potential of Mo6S8 is close to the stable potential limit of WIS electrolyte, the full cell can offer an average discharge voltage of 1.8 V when paired with an appropriate cathode material, such as LiMn2O4. Compared to the micro-structured Mo6S8 reported before,10,32,33 the prepared nanosized Mo6S8 anode shows excellent electrochemical performance and increased stability, with the main advantages being the simplicity of the preparation route and the improved cycle life of the electrode materials.
Experimental section
Synthesis of Mo6S8 Chevrel phases
Tetrakis(diethyldithiocarbamato) molybdenum(IV) [Mo(S2C(NEt2)4], abbreviated Mo(DTC)4 was synthesized following the method described by Lewis et al.31 In brief, 3.0 g molybdenum hexacarbonyl (1.0 eq., 7.57 mmol, Sigma Aldrich, 98%), and 6.75 g tetraethylthiuram disulfide (2.0 eq.,15.15 mmol, Sigma Aldrich, ≥97%) were refluxed in 50 mL acetone with a temperature set at 80 °C for 2 h. After cooling, the mixture was stored in the freezer overnight to facilitate complete crystallisation. Using vacuum filtration, dark brown crystals were collected and rinsed with n-hexane to collect Mo(DTC)4 (6.721 g, 85.84%). Fourier-transform infrared spectroscopy (FTIR) (solid) νmax/cm−1: 2974(w), 2947 (w), 2894 (w), 1518 (m), 1418 (sh), 1372 (sh), 1350 (m), 1316 (sh), 1288 (m), 1255 (sh), 1223 (m), 1166 (m), 1113 (sh), 1066 (m), 1018 (sh), 995 (m), 966 (sh), 932 (m), 865 (m), 805 (sh), 783 (m), 660 (m), 610 (m), 589 (m). Anal. Calc. for C20H40N4S8Mo: C, 34.86; H, 5.85; N, 8.13; S 37.23; Mo, 13.93%; Found: C, 35.04; H, 5.80; N, 8.08; S 37.38; Mo, 13.79%. The resultant FTIR spectrum was consistent with previous literature.34 For the preparation of CuxMo6S8, briefly, stoichiometric amounts of Mo(DTC)4 (1.4 g, 2 mmol), Mo (0.2 g, 2 mmol), and copper(II) dibutyldithiocarbamate (Cu(BDTC)2), (0.5 mg, 1.3 mmol)) powders were ground perfectly using a pestle and mortar. After that, the powdered mixture was compressed into pellets using an 11 mm diameter mold and a 20 Ton Hydraulic Press to ensure homogeneity. The pellet was placed in a ceramic boat inside a tube furnace, which was gradually heated to 800 °C at 3 °C min−1 and kept under Ar atmosphere for 7 hours at the peak temperature. Following this, Cu ions were chemically leached from the molecular precursor derived Chevrel phase by immersing the sample into a 6 M HCl (Fischer Scientific, Inc.) solution with stirring for 10 h under constant O2 bubbles in order to accelerate the Cu leaching process. Finally, the reaction solution was centrifuged, with the resulting Mo6S8 being washed with de-ionized water several times, followed by drying at 80 °C overnight to obtain the leached Mo6S8.Material characterisation
Powder X-ray diffraction (P-XRD) patterns were recorded from 10° to 80° (2θ) using Cu Kα radiation source operating at 40 kV and 40 mA using the XRD5-PANalytical X'Pert Pro diffractometer. The surface morphologies combined with the energy-dispersive X-ray (EDX) elemental analysis were performed with an FEI (Quanta 650) field emission gun scanning electron microscope (SEM) using an accelerating voltage of 15 kV. A substrate of conductive carbon tape was used to hold the samples during imaging to reduce the effect of charging. High-resolution scanning transmission electron microscopy (HR-STEM) was performed using a Thermo Fisher Titan STEM (G2 80–200) equipped with a Cs probe corrector (CEOS), a high-angle annular dark-field (HAADF) detector, detector and 0.7 srad solid angle Super-X EDX detector, operating at 200 kV. The particle diameter distributions of the CP were directly measured from HAADF-STEM images. The elemental composition of the sample was characterized by EDX in STEM mode. The STEM samples were prepared by dispersing the CPs powders in methanol, with subsequent sonication of the mixture for 5 min and drop-casting onto a copper grid coated with an amorphous holey carbon film. The Raman spectra measurements were performed using a Renishaw inVia Confocal Raman microscope with a 523 nm excitation laser and a 20× objective lens. X-ray photoelectron spectroscopy (XPS) spectra were recorded using a SPECS ultra-high vacuum (UHV) surface analysis system (base pressure 5 × 10−10 mbar) with a micro-focused Al-Kα source (1486.6 eV). The spectra were analysed using CasaXPS software, and the peak fitting was performed using Voigt-type functions and Shirley backgrounds. All binding energies were charge calibrated to the adventitious C 1s peak at 284.8 eV. For post-mortem analysis (ex situ), such as XRD measurements, the negative electrode (Chevrel phase) is recovered from the coin cells after cycling, washed with water to remove the electrolyte, and then dried before further measurement.Electrochemical characterisation
The electrochemical properties of the prepared Mo6S8 based ARLIBs were studied in a three-electrode configuration and CR2032-type coin cell. In three-electrode measurements, the working electrode was prepared by mixing the active material with 10% w/w of carbon black Super P (Alfa Aesar, 99+% metals basis) in a total quantity of 10 mg active material/carbon. Then, the mixture was dispersed in 1 ml of a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/ethanol mixture, to which 50 μL of Nafion® solution was added. To obtain a homogeneous slurry, the suspension was sonicated for 2 h. Using a micropipette, the desired amount of slurry was withdrawn from the suspension, and was then drop cast onto a polished glassy carbon (GC) substrate with a geometric area of 0.07 cm2. The final active material electrode loading was about 1.5 mg cm−2. For the half-cell configuration, Pt mesh was used as the counter and Ag/AgCl as the reference electrode. The aqueous electrolytes are 21 m solution (where m is moles solute per kg of solvent) of lithium bis-trifluoromethane sulfonyl-imide, (LiTFSI, Fluorochem, 99%). The electrolyte was made by dissolving the required amount of salt in ultra-pure water (Millipore Milli-Q), which was previously purged with N2 gas for 30 min. Cyclic voltammetry (CV) measurements were performed using Metrohm PGSTAT302N potentiostat, equipped with the FRA32 module and controlled via Nova 1.11 software. For full-cell assembly, either Mo6S8 for the anode or LiMn2O4 (MTI Corporation) for the cathode are mixed with 10 wt% carbon black and 10 wt% poly (vinylidene difluoride, PVDF) binders (60 wt% in water, Aldrich), to form a homogeneous free-standing slurry with the help of ethanol. Afterward, the blended slurries were pressed onto a 316 stainless steel grid for use as the negative electrode and a titanium substrate current collector for the positive electrode, followed by overnight drying at 80 °C. Full cell assembly was performed in a CR2032-type coin cell using a hydraulic crimping machine (MSK-160D), with Mo6S8 anode (4–6 mg cm−2), glass fibre (Whatman) as a separator, and a LiMn2O4 cathode (5–8 mg cm−2). The cells were galvanostatically charged and discharged on a Battery Test System (BaSyTec GmbH, Germany) under ambient atmospheric conditions.
:1 water/ethanol mixture, to which 50 μL of Nafion® solution was added. To obtain a homogeneous slurry, the suspension was sonicated for 2 h. Using a micropipette, the desired amount of slurry was withdrawn from the suspension, and was then drop cast onto a polished glassy carbon (GC) substrate with a geometric area of 0.07 cm2. The final active material electrode loading was about 1.5 mg cm−2. For the half-cell configuration, Pt mesh was used as the counter and Ag/AgCl as the reference electrode. The aqueous electrolytes are 21 m solution (where m is moles solute per kg of solvent) of lithium bis-trifluoromethane sulfonyl-imide, (LiTFSI, Fluorochem, 99%). The electrolyte was made by dissolving the required amount of salt in ultra-pure water (Millipore Milli-Q), which was previously purged with N2 gas for 30 min. Cyclic voltammetry (CV) measurements were performed using Metrohm PGSTAT302N potentiostat, equipped with the FRA32 module and controlled via Nova 1.11 software. For full-cell assembly, either Mo6S8 for the anode or LiMn2O4 (MTI Corporation) for the cathode are mixed with 10 wt% carbon black and 10 wt% poly (vinylidene difluoride, PVDF) binders (60 wt% in water, Aldrich), to form a homogeneous free-standing slurry with the help of ethanol. Afterward, the blended slurries were pressed onto a 316 stainless steel grid for use as the negative electrode and a titanium substrate current collector for the positive electrode, followed by overnight drying at 80 °C. Full cell assembly was performed in a CR2032-type coin cell using a hydraulic crimping machine (MSK-160D), with Mo6S8 anode (4–6 mg cm−2), glass fibre (Whatman) as a separator, and a LiMn2O4 cathode (5–8 mg cm−2). The cells were galvanostatically charged and discharged on a Battery Test System (BaSyTec GmbH, Germany) under ambient atmospheric conditions.
Results and discussion
In the conventional processing route for the preparation of Cu2Mo6S8, CuS, MoS2, and elemental Mo are mixed in a stoichiometric ratio and subsequently heated at 1000 °C for 24 h.19 Here, we employed a different molecular approach to prepare Cu2Mo6S8, where, Mo(DTC)2 acts as the MoS2 source, and Cu(BDTC)2 supplies the CuS upon its decomposition (see Experimental section). These precursors decompose concurrently in the presence of Mo metal to form the target CP as reported in our previous work.20 The P-XRD, Raman spectroscopy and XPS were used to characterise the materials structurally and to identify the chemical composition of the as-prepared materials. The XRD patterns of the synthesised Cu2Mo6S8, and Mo6S8 are shown in Fig. 1(a). The Cu2Mo6S8 provided a diffraction pattern that is in excellent agreement with the standard pattern of Cu2Mo6S8 (ICSD#158985). Moreover, the presence of a strong peak at 13.8° and the minimal appearance of the peak at 14.4° being indexed to MoS2, confirms the high purity of the synthesised Cu2Mo6S8 compound.30 Pure Mo6S8 was obtained following chemical leaching of Cu from Cu2Mo6S8 in 6 M HCl (see Experimental section). In this study, the obtained powder pattern in Fig. 1(a) can be indexed to rhombohedral Mo6S8 (ICSD#252376), which exhibits the intense peak of the (101) plane characteristic of CPs.35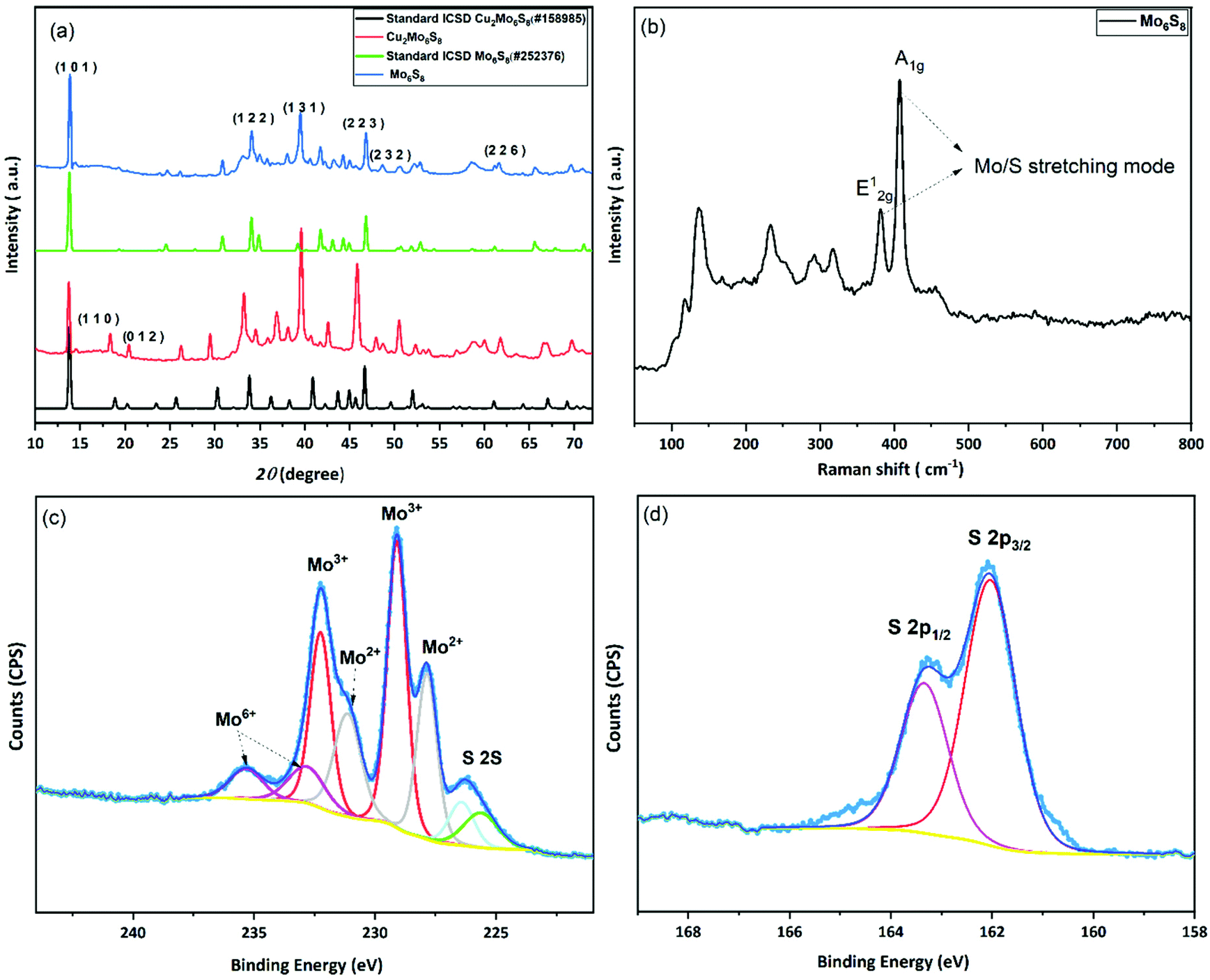 | ||
| Fig. 1 Characterization of the prepared materials: (a) P-XRD pattern of the as-synthesized Cu2Mo6S8 and Mo6S8 along with their corresponding standard patterns, (b) Raman spectrum of Mo6S8, (c) high resolution XPS spectra of the Mo 3d/S 2s region and (d) S 2p region. | ||
The Raman spectrum (Fig. 1(b)) of the material shows distinctive peaks at 136, 233, 316, 376 and 404 cm−1 corresponding to the various vibrational phonons of Mo/S in Mo6S8.32,36 The Raman spectra from aged and fresh leached Mo6S8 are also similar (Fig. S1†) suggesting that the Mo6S8 material is stable over a time period of 6 months. The surface states and chemical composition of Mo6S8 were examined by XPS and the survey spectrum is shown in Fig. S2.† The narrow window in the Mo 3d binding energy range (Fig. 1(c)), shows the characteristic doublet peaks centred at 228 and 231.1 eV attributed to Mo2+ and those at 229.1, 232.2 eV corresponding to the Mo3+ oxidation state in Mo6S8.19,20 Doublet peaks located at 232.7 and 235.3 eV are consistent with the presence of an oxide surface film due to exposure of the samples in ambient air prior to the measurements.37 The S 2p high resolution spectrum (Fig. 1(d)) can be fitted with two components related to S2p3/2 at 162.0 eV and S2p1/2 at 163.2 eV attributed to the sulfur with an oxidation state of S2− coordinated to Mo atoms.19 Notably, the peak profiles and their corresponding binding energy positions are commensurate with the previous literature reports on the Mo6S8 CP.9,38,39 For a detailed study of the surface state and chemical composition of the prepared Mo6S8 samples, we refer the reader to our previous work in which XPS photon depth profiling with tunable synchrotron radiation was presented.20
The surface morphology of the synthesised materials was examined by SEM. Fig. 2(a–d) shows the SEM images of Cu2Mo6S8 and Mo6S8. As we can see in Fig. 2, Chevrel phases with agglomerated nanocubes are revealed by the SEM. It is worth noting that the surface morphology is not affected by the acid leaching process and well-defined nanocubes similar to those in the as-prepared material are clearly seen in Fig. 2(c and d). The complete removal of Cu following the leaching process was confirmed by the disappearance of the Cu peak in the EDX spectrum of Mo6S8 (Fig. S3(a and b)†).
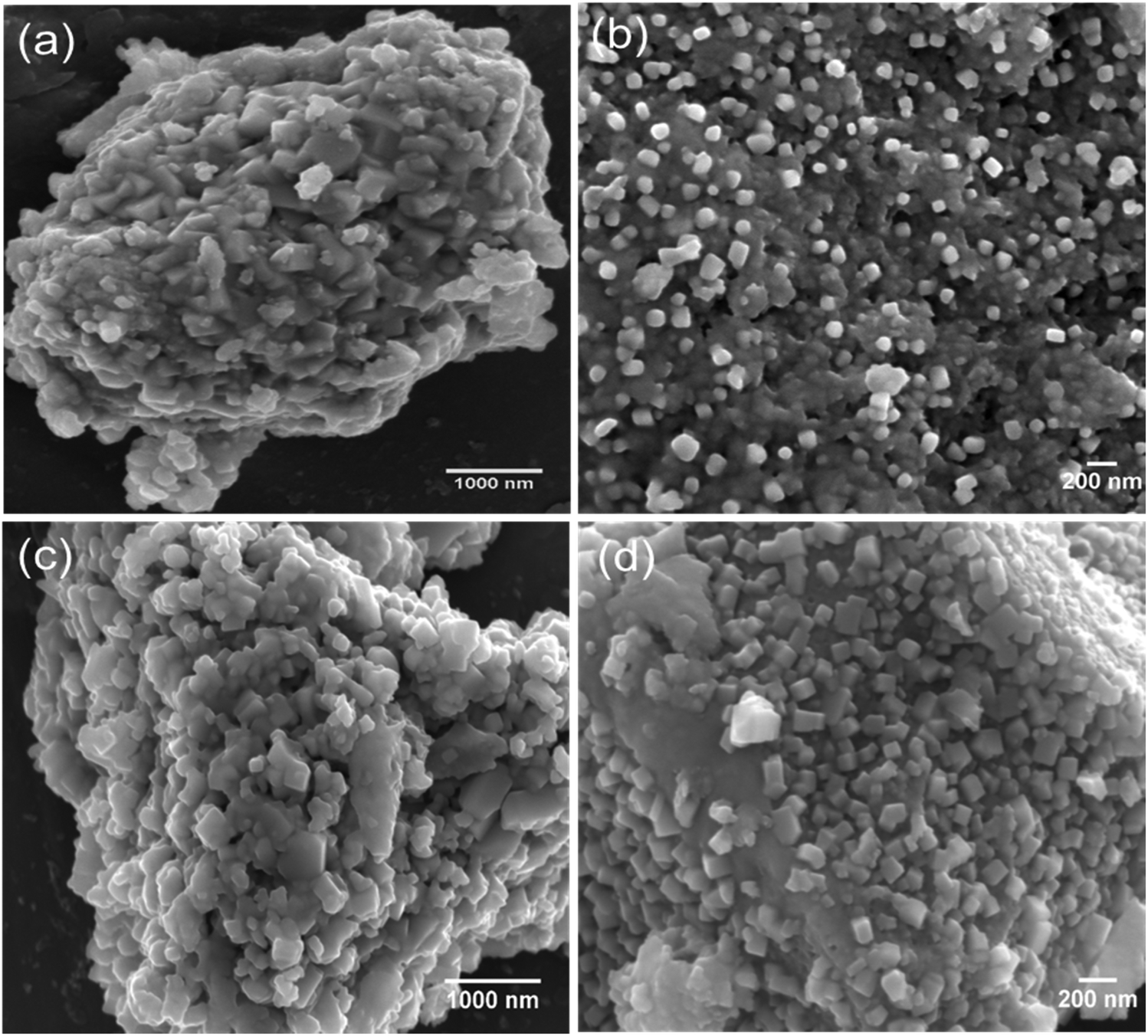 | ||
| Fig. 2 SEM images of the as-prepared (a and b) Cu2Mo6S8 and (c and d) Mo6S8 at different magnifications. | ||
To provide further insights into the surface morphology and crystal structure of the prepared material, we performed HAADF-STEM measurements (Fig. 3). The Mo6S8 CP crystal structure can be clearly observed from the atomic resolution HAADF-STEM images and is distinct from the layered structure of MoS2.26 Mo6S8 particles in the nanoscale range are observed (Fig. 3(a)) consistent with the SEM data. The interplanar spacing of the two sets of crystal planes marked by yellow lines in Fig. 3(b) are measured to be 0.614 nm and 0.629 nm, which correspond respectively to the (101) and (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 11) planes of the Mo6S8 CP. Fig. 3(d) shows the corresponding fast Fourier transform (FFT) pattern, which can be indexed by [12] zone-axis of Mo6S8 (P63/mmc space group). The overlay in Fig. 3(c) shows the model crystal structure of Mo6S8 viewed along the [12] direction, which matches our results, and indicates the single crystalline structure of the prepared Mo6S8 nanocubes.19 Furthermore, the chemical composition and elemental distributions were probed by STEM EDX mapping (Fig. S4†) where the uniform distribution of the elemental constituents in both the as-prepared and leached materials is highlighted.
11) planes of the Mo6S8 CP. Fig. 3(d) shows the corresponding fast Fourier transform (FFT) pattern, which can be indexed by [12] zone-axis of Mo6S8 (P63/mmc space group). The overlay in Fig. 3(c) shows the model crystal structure of Mo6S8 viewed along the [12] direction, which matches our results, and indicates the single crystalline structure of the prepared Mo6S8 nanocubes.19 Furthermore, the chemical composition and elemental distributions were probed by STEM EDX mapping (Fig. S4†) where the uniform distribution of the elemental constituents in both the as-prepared and leached materials is highlighted.
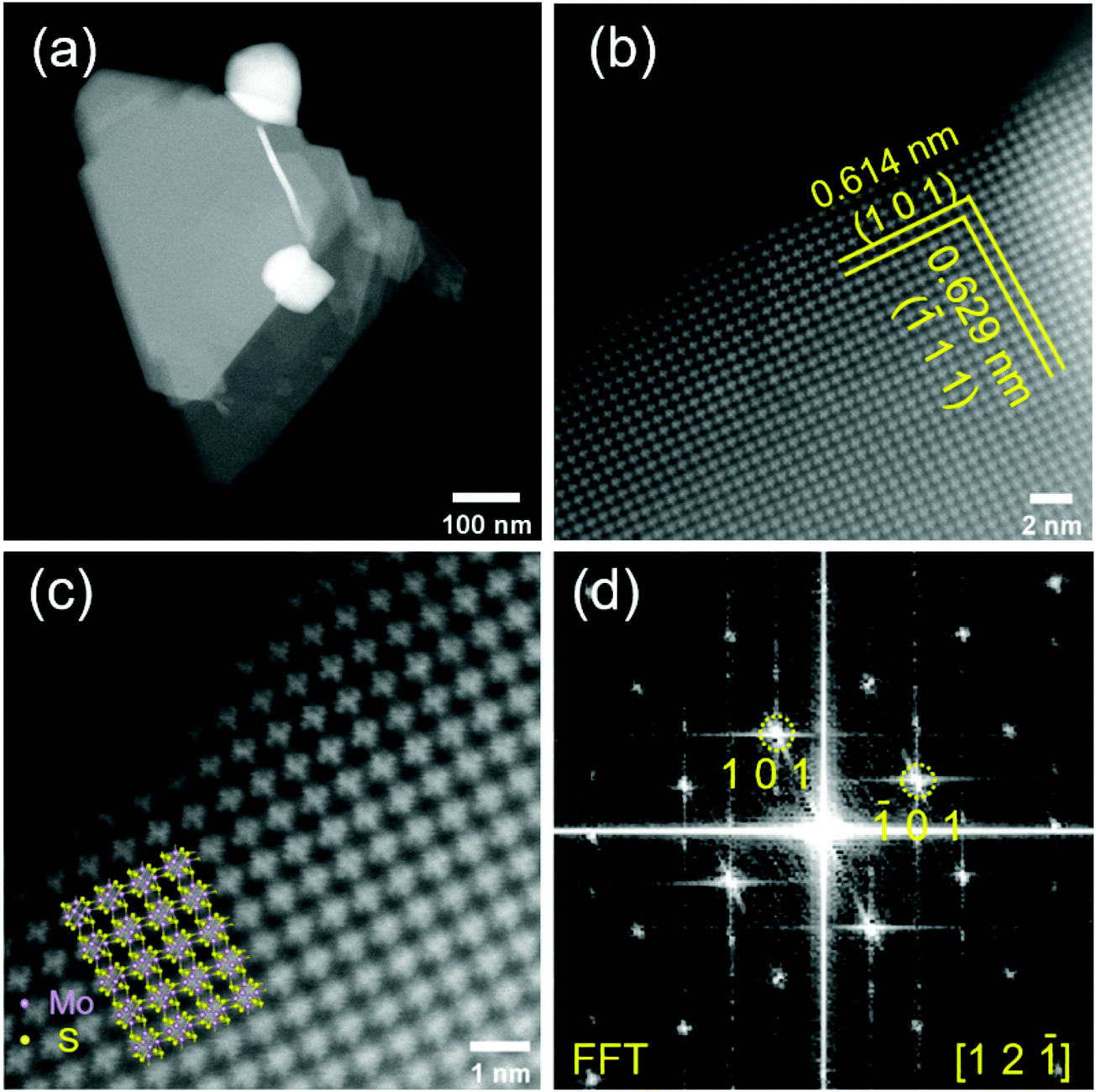 | ||
| Fig. 3 HAADF-STEM images of Mo6S8 particle at low (a), medium (b) and high magnifications (c). The atomic resolution image in (c) reveals the expected crystal structure of Mo6S8 viewed along [12] with an atomic model overlaid bottom left. (d) Fast Fourier Transform (FFT) obtained from (b) which can be indexed to the [12] zone-axis of Mo6S8. | ||
Energy storage performance of Chevrel phase Mo6S8
The prepared CP materials were used as anodes in ARLIBs and their performance was evaluated via a series of electrochemical and spectroscopic techniques. Initially, the electrochemical Li+ ion intercalation/deintercalation in both as-prepared and Cu-leached nanosized CPs was studied by means of cyclic voltammetry in a 21 m LiTFSI electrolyte, using a three-electrode configuration. As seen in Fig. 4(a), the non-leached Cu2Mo6S8 electrode exhibits a pair of redox peaks located at −1.0 V and −0.9 V vs. Ag/AgCl, that can be assigned to the insertion and removal of Li+, respectively. On the contrary, in the case of the pure Mo6S8 electrode (Fig. 4(b)), two distinct pairs of redox peaks appear, where the two cathodic peaks observed at −0.59 V and −1.01 V vs. Ag/AgCl are related to the insertion of Li+ ions into the LixMo6S8 (where, 0 < x < 4), with their anodic counterparts at −0.48 V and −0.81 V vs. Ag/AgCl corresponding to the removal of Li+ ions from the CP of LixMo6S8. These findings are in line with what has been previously reported for pure Mo6S8 electrodes in similar highly concentrated electrolytes.10 The appearance of two pairs of peaks in the Cu-leached electrode can be attributed to the widening of the pseudo-cubic cavity 1 in CP's crystal structure upon Cu chemical dissolution, which removes any energetic/steric barriers for Li+ ion insertion and thus results in a higher degree of Li+ ion accommodation within the structure of the material.40 Furthermore, it is noteworthy that Cu2Mo6S8 anodes show a much smaller Li+ ion insertion current response than Mo6S8, further indicating that the presence of Cu in the CP crystal structure partially hinders the insertion process.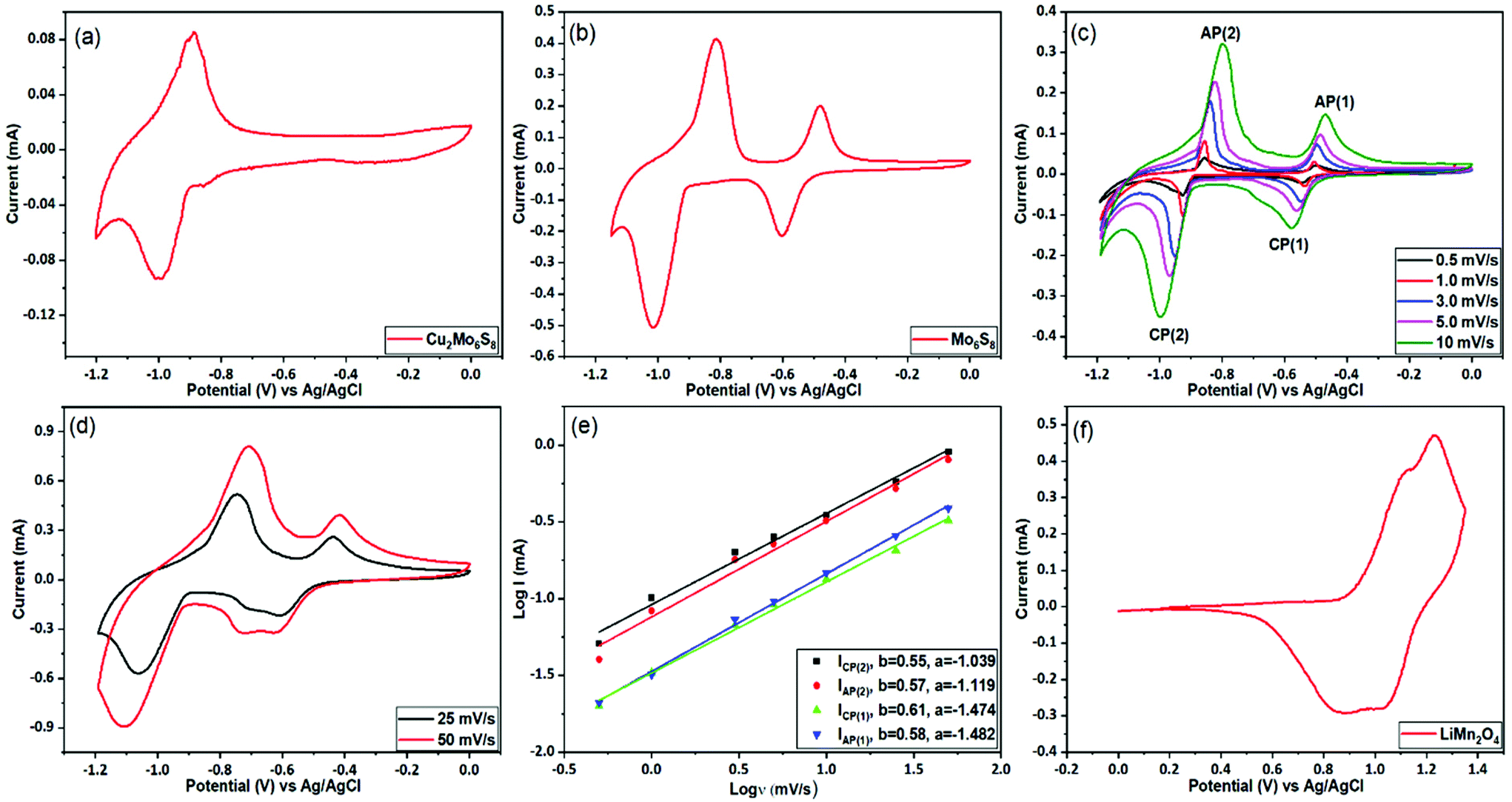 | ||
| Fig. 4 Electrochemical performance of active materials. Cyclic voltammetry (CV) of the Cu2Mo6S8 (a) and Mo6S8 (b), as measured at a scan rate of 5 mV s−1. CV of the Mo6S8 electrode at various scan rates (c and d). Power law dependence of peak currents for CP(1), CP(2) AP(1), AP(2) (peaks labelled in (c)) on scan rate (e). Exponents, b, of 0.5 and 1 indicate diffusion-limited and surface-controlled processes, respectively. CV traces of positive (LiMn2O4) materials measured at a scan rate of 0.5 mV s−1 (f). All data were collected in a three-electrode system including Mo6S8 and LiMn2O4 deposited on glassy carbon as the working electrode, Ag/AgCl and Pt mesh as the reference and counter electrode, respectively. | ||
The observed overpotential for Li+ ions intercalation/deintercalation in the prepared Mo6S8 is lower compared to that reported for conventional anode materials such as vanadium-based (VO2 (B), V2O5) or (NASICON)-type LiTi2(PO4)3 electrodes, while the small separation between the anodic and cathodic peaks in the cyclic voltammograms (CVs), demonstrates the faster Li+ intercalation kinetics. The latter can be attributed to the unique structural morphology in the nanoscale range of the prepared materials that decreases the diffusion length of Li+ ions [17]. The effect of potential scan rate on the intercalation/deintercalation process was also investigated and the results are displayed in Fig. 4(c and d). It can be seen that an increase in the potential scan rate leads to a larger peak separation between the anodic and cathodic peaks in the CVs of Fig. 4(c) (up to 200 mV for 10 mV s−1), which suggests the diffusion limitation of the ions at higher scan rates.41 By further increasing the potential scan rate at 25 and 50 mV s−1 (Fig. 4(d)), the anodic/cathodic peak separation increases to ca. 320 and 400 mV respectively, where the ions’ diffusion limitation phenomenon becomes more evident, in line with what is reported in the literature for various battery materials.42 To consider the charge storage mechanism of the prepared Mo6S8 during the charging process in more detail, the relationship between the peak current (i) and potential scan rate (v) was studied, based on the power-law formula i = aνb, where the exponent b (the slope of log (i) vs. log (v) plot) reveals the charge storage mechanism. For processes dependent on interactions at the surface, the current response is proportional to the scan rate (b = 1), whereas for a diffusion-controlled mechanism, i is proportional to the square root of v (b = 0.5). As seen in Fig. 4(e), the fitting results show that the calculated b values are ca. 0.6, indicating that the charge storage mechanism of nanosized Mo6S8 anodes is mainly controlled by the lithiation/delithiation during charge and discharge processes.
Since it has been reported that the CP can reversibly intercalate different cations from organic electrolytes, we investigated whether Na+, and Mg2+ could intercalate from aqueous electrolytes. Towards this end, we studied the performance of the prepared CPs in 2 M Na2SO4 and the WIS electrolyte mixture of 40 m CsOAc (OAc = acetate, Alfa Aesar, 99.9%) +10 m NaOAc (Sigma-Aldrich, 99%). As can be seen in Fig. S5(a),† no electrochemical features related to intercalation were observed in the 2 M Na2SO4 electrolyte, due to the restricted stable potential window (ESPW). On the contrary, when using the WIS-based acetate electrolyte mixture (Fig. S4(b)),† two clear de-intercalation peaks appear at −1.5 V and −1.2 V vs. Ag/AgCl, with an ill-defined cathodic shoulder at −1.55 V (within the HER potential range) that might be attributed to the reverse process. However, since the intercalation process was found to occur at potentials very close to the onset of HER, no reliable and reproducible charge/discharge data could be recorded. Similar studies were also conducted for the intercalation of Mg2+ ions in the synthesized Mo6S8 anodes using 1 M MgSO4. From the data presented in Fig. S6† it can be inferred that the process occurs at potentials located at the immediate vicinity of the HER potential range and thus Mg2+ cannot be reversibly intercalated. The same outcome has been recently reported using highly concentrated 4 m Mg(TFSI)2, where the cathodic limit was found to be ca. −0.9 V vs. Ag/AgCl.43 Despite these unsuccessful preliminary attempts, we believe that Mo6S8 will play a dominant role as anode material for aqueous Na/Mg-ion batteries with appropriately engineered electrolytes able to offer a wider potential window.
Having characterized the electrochemical processes at play in the Mo6S8 anode, we turn to investigate the cathode side. For these measurements, the spinel LiMn2O4 was selected as the cathode material. Similar to the studies involving the anode, a three-electrode configuration was used to evaluate the electrochemical performance of the LiMn2O4. Fig. 4(f) shows the typical CV plot of LiMn2O4 electrode at a scan rate of 0.5 mV s−1 in a potential range between 0 and 1.35 V vs. Ag/AgCl. Two pairs of redox peaks can be resolved from the relevant CVs, suggesting that Li+ ions are inserted into/extracted from the LiMn2O4 by a two-step process. It is known that these two pairs of current peaks originate from the different oxidation states of Mn3+/Mn4+ upon delithiation and lithiation processes.10
In the next stage, Mo6S8 and LiMn2O4 were assembled as coin cells and were tested for their rate capability and cycling stability. As shown in Fig. 5(a), the CV of the full cell recorded at a scan rate of 1 mV s−1, shows two distinct reversible redox couple peaks of lithiation/delithiation reactions at 1.57 V and 2.0 V for the anodic scan with their cathodic counterparts recorded at 1.42 V and 1.75 V, respectively. It is emphasized that no obvious water decomposition signature is seen within the applied potential range. Moreover, the CV profile remains stable, apart from the 1st cycle where some irreversible lithium consumption is caused by SEI formation.9 The galvanostatic charge/discharge profiles for the 1st, 3rd, 5th, and 10th cycles at 0.2 C show two sloping discharge plateaus at 1.6 V and 1.95 V. These observations, together with the differential capacity (dQ/dV) profile calculated from the charge/discharge profile (Fig. 5(b)), are in good agreement with the results of the CVs. Also, we emphasize the fact that the coulombic efficiency increased to 99% within the first five cycles, (Fig. 5(b)), revealing the formation of a stable and protective SEI over as few as five cycles. The latter is also consistent with the CV profile. To examine the rate capability of the fabricated Li-ion battery, the cell was cycled at different charging/discharging rates and the results are depicted in Fig. 5(d). At 4 C rate the full cell shows a capacity of ca. 18 mA h g−1 corresponding to 42% of the capacity obtained at 0.2 C (42 mA h g−1). This decrease in capacity at high rates (4 C) is mainly caused by the slow rate of internal Li+ ion diffusion, and not due to the electrode degradation (see Fig. 7 and the relevant discussion therein). Notably, when the cycling rate returns to 0.2 C, the recorded capacity is ca. 38 mA h g−1 which corresponds to a capacity retention of ca. 91% compared to the first cycle. This observation demonstrates the excellent high-rate performance of the cell. An approximate estimation of the energy density from the two average discharge voltages and their corresponding capacities leads to a value of ca. 85 W h kg−1 at a 0.2 C rate and based on the mass of active materials.
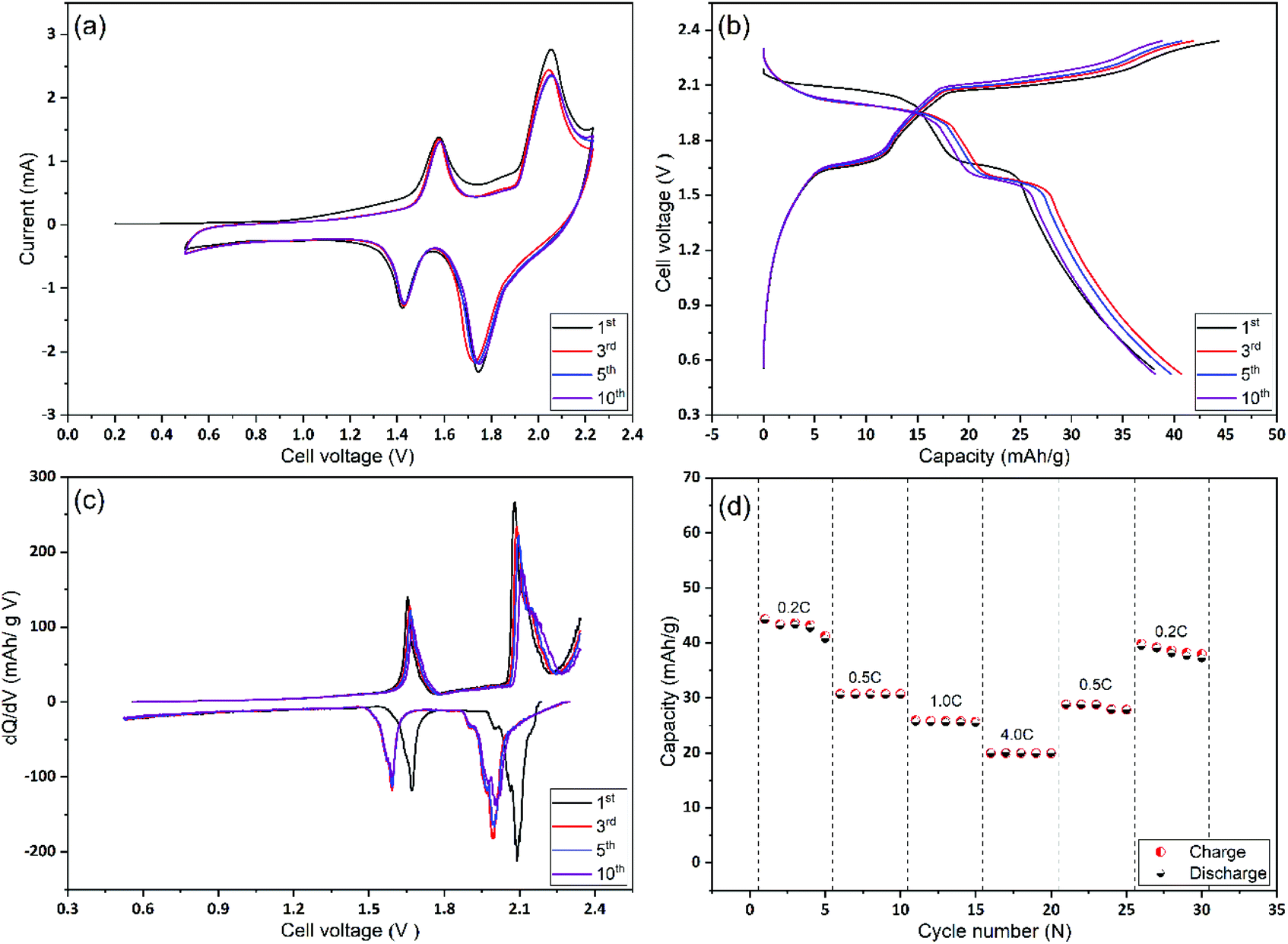 | ||
| Fig. 5 Electrochemical performance of full Li ion based Mo6S8//LiMn2O4 cell (a) CV of Mo6S8/LiMn2O4 in aqueous 21 m LiTFSI at a scan rate of 1 mV s−1. (b) The charge–discharge profiles of the electrochemical couple of LiMn2O4/Mo6S8 nanocubes between 0.5–2.2 V at the 1st, 3rd, 5th, and 10th cycles. (c) Differential capacity (dQ/dV) profile calculated from the 1st, 3rd, 5th, and 10th charge/discharge cycles of LiMn2O4/Mo6S8 nanocubes in ARLIBs. (d) Rate performance of LiMn2O4/Mo6S8 nanocubes with different currents. | ||
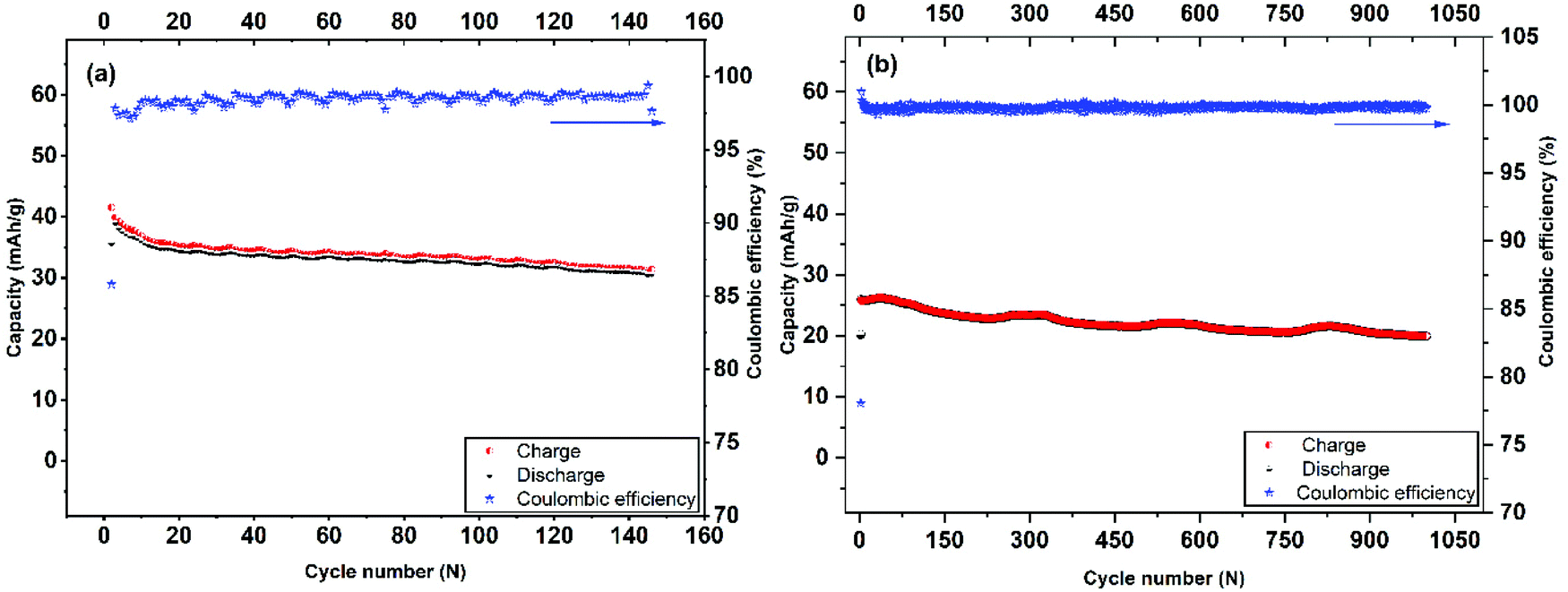 | ||
| Fig. 6 Long-term cycling performance and coulombic efficiency of Mo6S8 nanocubes at 0.2 C-rate (a), and 4 C-rate (b). | ||
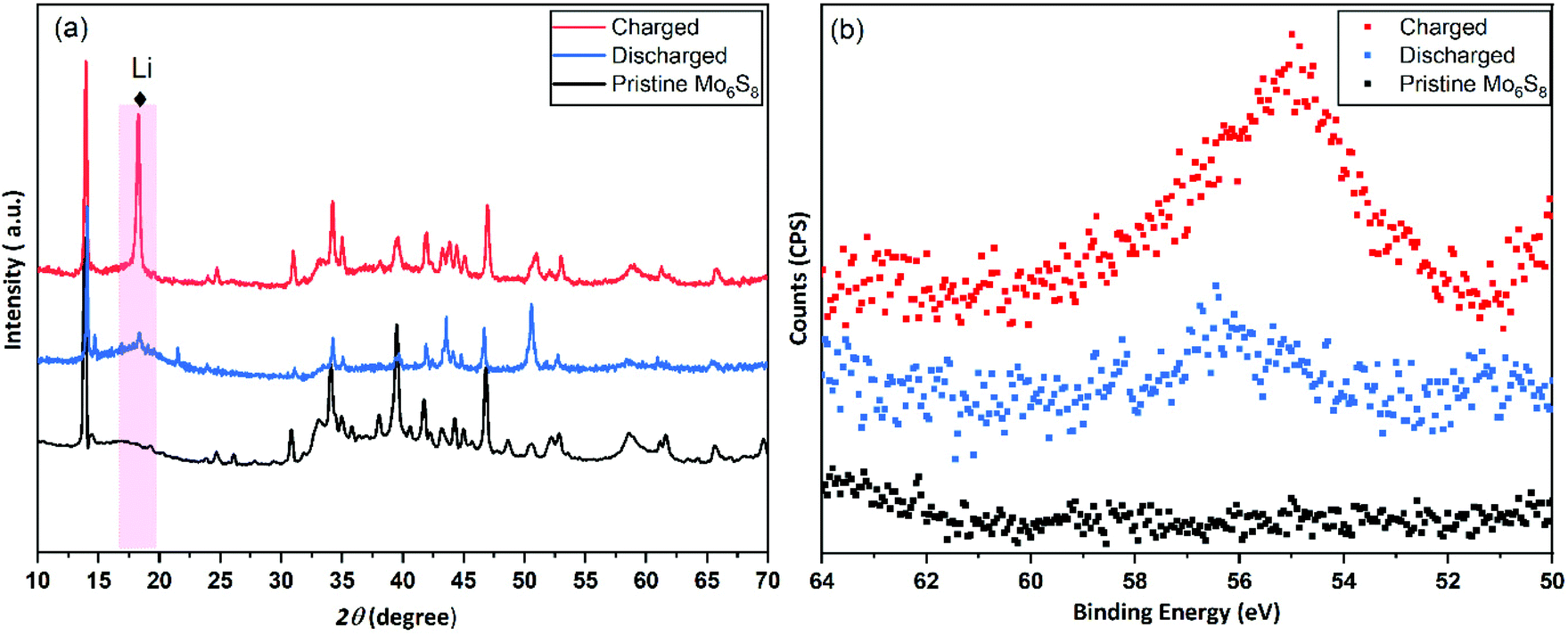 | ||
| Fig. 7 Post-mortem characterization on cycled Mo6S8 electrodes. (a) Post-mortem P-XRD characterization was conducted on the pristine, fully charged to 2.2 V, and fully discharged to 0.5 V Mo6S8 electrode. (b) High resolution XPS spectra of the Li 1s for the pristine, charged, and discharged Mo6S8 electrode. | ||
The cycling stability and capacity retention of the Mo6S8/LiMn2O4 cell was tested at 0.2 C and 4 C-rates for different cycles as shown in Fig. 6(a and b). The specific capacity with respect to the total mass was 40 mA h g−1 at 0.2 C and 25 mA h g−1 at 4 C, for the first cycle. Excellent cycling stability and high capacity retention of 31 mA h g−1 within 145 cycles, corresponding to a capacity retention of 77% under the harsh conditions imposed by the low (0.2 C) rate. Moreover, at high rates (4 C) the capacity retention is determined to be 80% after 1000 cycles. In both cases, the coulombic efficiency was ca. 100%. The excellent cycling capability can be interpreted in terms of the nanostructure crystalline features of the prepared CP material that (i) form three-dimensional Li+ ion diffusion paths and (ii) shortens the diffusion path of Li+ ions and ions, thereby accelerating ion transportation during the charge/discharge process. This is also supported by the electrochemical impedance spectra (EIS) measurements recorded at different charging/discharging cycles (Fig. S7†). The qualitative features of the Nyquist plots include: (i) the high-frequency intercept related to the electronic conductivity of the cell, (ii) a depressed semicircle in the intermediate frequency region where its diameter roughly corresponds to the charge transfer resistance associated with Li+ (de)intercalation processes at the electrode/electrolyte interface and (iii) an inclined line of higher than 45° angle slope relative to the real axis in the low-frequency range (Warburg resistance, showing the diffusion rate of Li+ in the active material).44 It can be inferred from the Nyquist plots that the interfacial resistance remains stable at around 30 Ω during the first 10 cycles, followed by a slight increase to 40 Ω after the 100th cycle, before reaching the final value of 54 Ω. The observed increase in the charge transfer value (as qualitatively estimated by the diameter of the depressed semicircle observed at the intermediate frequency range) may be attributed to the capacity fade of the positive electrode (LiMn2O4) during cycling, as has been recently reported for systems using 21 m LiTFSI electrolytes. This phenomenon is ascribed to the low conductivity of LiMn2O4 and the Jahn–Teller distortion arising from the thermodynamic instability of the spinel lattice.9,45,46
In order to investigate the intercalation of Li+ ions into Mo6S8 nanocubes, ex situ post-mortem XRD and XPS measurements were performed on cycled electrodes. The full cell was charged and discharged at a 0.2 C rate and the negative electrode was recovered from the cell and washed thoroughly with water before the measurements. As shown in the XRD pattern of the charged Mo6S8 (Fig. 7(a)), there is a phase transition from the Mo6S8 to LixMo6S8 (where, 0 < x < 4), which is identified as R-3 structure rhombohedral LixMo6S8 (PDF #01-081-0859). However, in the fully discharged state, the sharp crystalline peak at 18° due to the Li+ insertion and the Mo6S8 structure was recovered, indicating the reversible phase transition. This can be further supported by the XPS data obtained for Mo6S8 at different states of charge, where a high intensity peak for the Li 1s is observed in the fully charged Mo6S8 (Fig. 7(a)). On the other hand, this peak was barely seen when the cell was discharged to 0.5 V, demonstrating the reversible insertion/deinsertion process during the charge/discharge process. Furthermore, the stability of Mo6S8 nanocubes in the 21 m LiTFSI aqueous electrolyte upon cycling, was evaluated in a three-electrode cell system and in the full cell configuration. In the first case, Mo6S8 was scanned in the working electrolyte for more than 50 cycles at a scan rate of 1 mV s−1 in the potential range used for the data in Fig. 4(b). The electrolyte was then subjected to ICP-MS analysis to determine the molybdenum concentration (for the Experimental process please see the supplementary data). The results show that molybdenum concentration (−9 ppb) in the solution is less than the detection limit of the instrument, i.e., 5 ppb. For the full cell configuration, a CR2032-type coin cell comprised of Mo6S8 as anode and LiMn2O4 as cathode was assembled based on the procedure described in the Experimental section. The cell was then cycled for 15 cycles at low rate of 0.2 C. Following completion of the charge/discharge cycling procedure, the cell was disassembled, and the glass membrane separator soaked in the electrolyte was carefully removed. The latter was subsequently digested in nitric acid and the contents of the solution were analyzed by means of ICP-MS. A pristine membrane was also analyzed following the above procedure (blank sample). The analysis results revealed a molybdenum content of 297 ppb for the membrane removed from the coin cell and 18 ppb for the blank sample. The trace amounts of molybdenum detected after digestion of the membrane soaked in the working electrolyte are attributed to the presence of Mo6S8 residuals on the surface of the membrane upon cell disassembly as well as the intrinsic molybdenum content in the separator as evidenced by the analysis of the blank sample. The findings from both the three-electrode and coin cells strongly demonstrate the high chemical and electrochemical stability of Mo6S8 in this electrolyte during cycling.
Conclusions
An alternative strategy for the preparation of high purity Chevrel phase Mo6S8 is proposed, which is based on a facile, efficient and controllable molecular precursor approach with a significantly decreased energy input compared to the conventional approaches. The resultant Mo6S8 material exhibits unique structural characteristics in the nanoscale range, as confirmed by a series of physicochemical characterization techniques. Application of the nanostructured Mo6S8 as the anode in rechargeable aqueous Li-ion batteries using a concentrated electrolyte of 21 m LiTFSI, delivers a high electrochemical capacity of 45 mA h g−1 at 0.2 C with a discharge voltage as high as ca. 2 V. The cycling performance of the full cells using a LiMn2O4 cathode, is found to be highly reproducible as evidenced by the capacity retention of 77% for 145 cycles at 0.2 C and 80% at 4 C for 1000 cycles. In both cases the coulombic efficiency was determined to be close to 100% further confirming the increased stability of the system. These results are interpreted in terms of the three-dimensional Li+ ion diffusion network expanding in the nanometre range and the decreased Li+ ion diffusion path, compared to the conventional microstructured Mo6S8, that accelerates ion transportation during consecutive charge/discharge cycles. Both characteristics arise from the nanostructured morphology of the material, as a result of the synthetic approach followed. Post-mortem powder XRD of the fully charged/discharged anodes, demonstrate the stability of the material, as proven by the fact that its crystal structure remains unaffected, despite undergoing a phase transition during Li+ ion insertion/removal. In summary, we envisage that the overall strategy introduced in this work will contribute to the stimulation of novel research towards the wide application of CP Mo6S8 based materials in various electrochemical energy storage systems using both aqueous and non-aqueous electrolytes.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work presented in this paper.Acknowledgements
Amr Elgendy (Amr Ahmed Sadek) would like to thank the Newton–Mosharafa Fund (a partnership between the British Council and the Ministry of Higher Education – Missions Sector, Egypt) for supporting him in the form of a full PhD scholarship. Further support from EPSRC is gratefully acknowledged (EP/T01816X/1). The authors acknowledge the use of the Department of Materials X-ray Diffraction Suite at the University of Manchester and the technical support, advice, and assistance provided by Mr Gary Harrison. TEM access was supported by the Henry Royce Institute for Advanced Materials, funded through EPSRC grants EP/R00661X/1, EP/S019367/1, EP/P025021/1 and EP/P025498/1. S. J. H. acknowledges the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant ERC-2016-STG-EvoluTEM-715502). R. C and S. J. H. would further like to thank the UK Catalysis Hub for resources and support provided via the membership of the UK Catalysis Hub Consortium and funded by EPSRC grant: P/R027129/1.References
- F. Wang, Y. Lin, L. Suo, X. Fan, T. Gao, C. Yang, F. Han, Y. Qi, K. Xu and C. Wang, Energy Environ. Sci., 2016, 9, 3666–3673 RSC.
- J. Huang, J. Liu, J. He, M. Wu, S. Qi, H. Wang, F. Li and J. Ma, Angew. Chem., Int. Ed., 2021, 60, 20717–20722 CrossRef CAS PubMed.
- F. Li, J. Liu, J. He, Y. Hou, H. Wang, D. Wu, J. Huang and J. Ma, Angew. Chem., Int. Ed., 2022, e202205091 CAS.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- L. Xue, Q. Zhang, X. Zhu, L. Gu, J. Yue, Q. Xia, T. Xing, T. Chen, Y. Yao and H. Xia, Nano Energy, 2019, 56, 463–472 CrossRef CAS.
- W. Li, J. R. Dahn and D. S. Wainwright, Science, 1994, 264, 1115–1118 CrossRef CAS PubMed.
- H. Kim, J. Hong, K.-Y. Park, H. Kim, S.-W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS PubMed.
- L. Suo, F. Han, X. Fan, H. Liu, K. Xu and C. Wang, J. Mater. Chem. A, 2016, 4, 6639–6644 RSC.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- N. Dubouis, P. Lemaire, B. Mirvaux, E. Salager, M. Deschamps and A. Grimaud, Energy Environ. Sci., 2018, 11, 3491–3499 RSC.
- L. Suo, D. Oh, Y. Lin, Z. Zhuo, O. Borodin, T. Gao, F. Wang, A. Kushima, Z. Wang and H.-C. Kim, J. Am. Chem. Soc., 2017, 139, 18670–18680 CrossRef CAS PubMed.
- M. R. Lukatskaya, J. I. Feldblyum, D. G. Mackanic, F. Lissel, D. L. Michels, Y. Cui and Z. Bao, Energy Environ. Sci., 2018, 11, 2876–2883 RSC.
- N. Alias and A. A. Mohamad, J. Power Sources, 2015, 274, 237–251 CrossRef CAS.
- D. Bin, Y. Wen, Y. Wang and Y. Xia, J. Energy Chem., 2018, 27, 1521–1535 CrossRef.
- Y. Wang, J. Yi and Y. Xia, Adv. Energy Mater., 2012, 2, 830–840 CrossRef CAS.
- L. Zhang, H. B. Wu, Y. Yan, X. Wang and X. W. D. Lou, Energy Environ. Sci., 2014, 7, 3302–3306 RSC.
- R. Chevrel, M. Sergent and J. Prigent, J. Solid State Chem., 1971, 3, 515–519 CrossRef CAS.
- M. Mao, Z. Lin, Y. Tong, J. Yue, C. Zhao, J. Lu, Q. Zhang, L. Gu, L. Suo and Y.-S. Hu, ACS Nano, 2019, 14, 1102–1110 CrossRef PubMed.
- A. Elgendy, A. A. Papaderakis, C. Byrne, Z. Sun, J. V. Lauritsen, E. P. Higgins, A. Ejigu, R. Cernik, A. S. Walton and D. J. Lewis, ACS Appl. Energy Mater., 2021, 4, 13015–13026 CrossRef CAS.
- J. Strachan, A. F. Masters and T. Maschmeyer, Mater. Res. Bull., 2021, 139, 111286 CrossRef CAS.
- J. C. Ortiz-Rodríguez, N. R. Singstock, J. T. Perryman, F. P. Hyler, S. J. Jones, A. M. Holder, C. B. Musgrave and J. M. Velázquez, ACS Appl. Mater. Interfaces, 2020, 12, 35995–36003 CrossRef PubMed.
- R. Chevrel, M. Hirrien and M. Sergent, Polyhedron, 1986, 5, 87–94 CrossRef CAS.
- L. Mei, J. Xu, Z. Wei, H. Liu, Y. Li, J. Ma and S. Dou, Small, 2017, 13, 1701441 CrossRef PubMed.
- P. Saha, P. H. Jampani, M. K. Datta, D. Hong, B. Gattu, P. Patel, K. S. Kadakia, A. Manivannan and P. N. Kumta, Nano Res., 2017, 10, 4415–4435 CrossRef CAS.
- Y. Cheng, L. R. Parent, Y. Shao, C. Wang, V. L. Sprenkle, G. Li and J. Liu, Chem. Mater., 2014, 26, 4904–4907 CrossRef CAS.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- E. Lancry, E. Levi, A. Mitelman, S. Malovany and D. Aurbach, J. Solid State Chem., 2006, 179, 1879–1882 CrossRef CAS.
- P. Saha, P. H. Jampani, M. K. Datta, C. U. Okoli, A. Manivannan and P. N. Kumta, J. Electrochem. Soc., 2014, 161, A593 CrossRef CAS.
- G. Gershinsky, O. Haik, G. Salitra, J. Grinblat, E. Levi, G. D. Nessim, E. Zinigrad and D. Aurbach, J. Solid State Chem., 2012, 188, 50–58 CrossRef CAS.
- G. Murtaza, S. Alderhami, Y. T. Alharbi, U. Zulfiqar, M. Hossin, A. M. Alanazi, L. Almanqur, E. U. Onche, S. P. Venkateswaran and D. J. Lewis, ACS Appl. Energy Mater., 2020, 3, 1952–1961 CrossRef CAS PubMed.
- H. Chu, J. Pan, S. Bai, Y. Ma, Y. Feng, Y. Wen, Y. Yang, R. Luo and A. Chen, Electrochim. Acta, 2017, 258, 236–240 CrossRef CAS.
- Y. Huang, W. Sun, K. Xu, J. Zhang, H. Zhang, J. Li, L. He, L. Cai, F. Fu and J. Qin, Energy Storage Mater., 2022, 46, 577–582 CrossRef.
- E. P. Higgins, A. A. Papaderakis, C. Byrne, R. Cai, A. Elgendy, S. J. Haigh, A. S. Walton, D. J. Lewis and R. A. Dryfe, J. Phys. Chem. C, 2021, 125, 20940–20951 CrossRef CAS.
- M. S. Chae, J. W. Heo, S.-C. Lim and S.-T. Hong, Inorg. Chem., 2016, 55, 3294–3301 CrossRef CAS PubMed.
- D. Holmgren, R. Demers, M. Klein and D. Ginsberg, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 36, 1952 CrossRef CAS PubMed.
- J. Richard, A. Benayad, J.-F. Colin and S. Martinet, J. Phys. Chem. C, 2017, 121, 17096–17103 CrossRef CAS.
- K. Lu, F. Xia, B. Li, Y. Liu, I. B. Abdul Razak, S. Gao, J. Kaelin, D. E. Brown and Y. Cheng, ACS Nano, 2021, 15, 16887–16895 CrossRef CAS PubMed.
- F. Xia, B. Li, Y. Liu, Y. Liu, S. Gao, K. Lu, J. Kaelin, R. Wang, T. J. Marks and Y. Cheng, Adv. Funct. Mater., 2021, 31, 2104716 CrossRef CAS.
- E. Levi, G. Gershinsky, D. Aurbach and O. Isnard, Inorg. Chem., 2009, 48, 8751–8758 CrossRef CAS PubMed.
- J. Yang, M. A. Bissett and R. A. W. Dryfe, ChemSusChem, 2021, 14, 1700–1709 CrossRef CAS PubMed.
- L. Wang, J. Zou, S. Chen, G. Zhou, J. Bai, P. Gao, Y. Wang, X. Yu, J. Li and Y.-S. Hu, Energy Storage Mater., 2018, 12, 216–222 CrossRef.
- F. Wang, X. Fan, T. Gao, W. Sun, Z. Ma, C. Yang, F. Han, K. Xu and C. Wang, ACS Cent. Sci., 2017, 3, 1121–1128 CrossRef CAS PubMed.
- T. Quan, Y. Xu, M. Tovar, N. Goubard-Bretesché, Z. Li, Z. Kochovski, H. Kirmse, K. Skrodczky, S. Mei and H. Yu, Batteries Supercaps, 2020, 3, 747–756 CrossRef CAS.
- Y. Zhang, H. Xie, H. Jin, X. Li, Q. Zhang, Y. Li, K. Li, F. Luo, W. Li and C. Li, ACS Omega, 2021, 6, 21304–21315 CrossRef CAS PubMed.
- Y. Wen, C. Ma, H. Chen, H. Zhang, M. Li, P. Zhao, J. Qiu, H. Ming, G. Cao and G. Tang, Electrochim. Acta, 2020, 362, 137079 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr02014a |
| This journal is © The Royal Society of Chemistry 2022 |