
Recent advances in high-crystalline conjugated organic polymeric materials for photocatalytic CO2 conversion
Fengyi
Yang
a,
Jiafu
Qu
a,
Yang
Zheng
c,
Yahui
Cai
 b,
Xiaogang
Yang
a,
Chang Ming
Li
a and
Jundie
Hu
*a
b,
Xiaogang
Yang
a,
Chang Ming
Li
a and
Jundie
Hu
*a
aSchool of Materials Science and Engineering, Suzhou University of Science and Technology, Suzhou 215009, China. E-mail: hjd@usts.edu.cn
bCollege of Materials Science and Engineering, Nanjing Forestry University, Nanjing 210037, China
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
First published on 26th September 2022
Abstract
The photocatalytic conversion of carbon dioxide (CO2) to high-value-added fuels is a meaningful strategy to achieve carbon neutrality and alleviate the energy crisis. However, the low efficiency, poor selectivity, and insufficient product variety greatly limit its practical applications. In this regard, conjugated organic polymeric materials including carbon nitride (g-C3N4), covalent organic frameworks (COFs), and covalent triazine frameworks (CTFs) exhibit enormous potential owing to their structural diversity and functional tunability. Nevertheless, their catalytic activities are largely suppressed by the traditional amorphous or weakly crystalline structures. Therefore, constructing relevant high-crystalline materials to ameliorate their inherent drawbacks is an efficient strategy to enhance the photocatalytic performance of conjugated organic polymeric materials. In this review, the advantages of high-crystalline organic polymeric materials including reducing the concentration of defects, enhancing the built-in electric field, reducing the interlayer hydrogen bonding, and crystal plane regulation are highlighted. Furthermore, the strategies for their synthesis such as molten-salt, solid salt template, and microwave-assisted methods are comprehensively summarized, while the modification strategies including defect engineering, element doping, surface loading, and heterojunction construction are elaborated for enhancing their photocatalytic activities. Ultimately, the challenges and opportunities of high-crystalline conjugated organic polymeric materials in photocatalytic CO2 conversion are prospected to give some inspiration and guidance for researchers.
 Fengyi Yang | Fengyi Yang is currently a Postgraduate at Suzhou University of Science and Technology. Her current research direction is on photoenzymatic catalytic carbon dioxide conversion reactions. |
 Chang Ming Li | Chang Ming Li is a Professor and the Director of School of Materials Science and Engineering in Suzhou University of Science and Technology, China. His main research interests involve cross-field sciences including functional nanomaterials and green energies. He has published more than 700 peer-reviewed journal papers with more than 30 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 citations and H-index of 85 as well as 240 patents. He is a Fellow of the American Institute of Medicine and Biological Engineering, Foreign Member of the Russian Academy of Engineering and Fellow of the Royal Society of Chemistry, serving as an Editorial Board Member of RSC Advances, member of Advisory Board for Nanoscales, RSC and Joule, Cell as well as the Chief Editor of Materials on Energy.
000 citations and H-index of 85 as well as 240 patents. He is a Fellow of the American Institute of Medicine and Biological Engineering, Foreign Member of the Russian Academy of Engineering and Fellow of the Royal Society of Chemistry, serving as an Editorial Board Member of RSC Advances, member of Advisory Board for Nanoscales, RSC and Joule, Cell as well as the Chief Editor of Materials on Energy. Jundie Hu | Jundie Hu received her PhD at the College of Chemistry, Chemical Engineering and Materials Science from Soochow University in 2019 under the supervision of Prof. Jianmei Lu. Subsequently, she joined Suzhou University of Science and Technology as an Associate Professor. Her current research interests mainly focous on engineering nanoporous materials including 2D materials, heterojunctions, conjugated organic polymeric materials, and their applications in photocatalysis, such as water splitting, carbon dioxide conversion, and environmental remediation. |
1. Introduction
The excessive emission of CO2 has caused irreversible harm to the environment, such as the greenhouse effect, climate change, rising sea levels, and marine ecological destruction.1 According to the World Meteorological Organization, the CO2 concentration has exceeded 400 ppm, and many global extreme climate records are being broken. Simultaneously, energy shortage is another crisis accompanying the rapid consumption of fossil fuels. In this regard, low-carbon sustainable development strategies, including energy saving and emission reduction, renewable energy, and carbon capture and storage, have been launched worldwide.2,3 Therefore, how to effectively reduce the CO2 content while developing a sustainable carbon-neutral economy is a valuable approach. In this case, directly converting CO2 into high-value-added hydrocarbon fuels or chemicals (e.g., CO, CH4, CH3OH, HCOOH, HCHO, C2H5OH, and C2H4) has proven to be an effective way, where many technologies can be employed including organic catalysis,4 electrocatalysis,5,6 photocatalysis,7,8 biocatalysis,9 thermocatalysis,10 and photoenzymatic catalysis.11 Among these, artificial photosynthesis to achieve photocatalytic CO2 reduction is an extremely promising route due to its advantages of clean, eco-friendly, green and mild conditions, etc.12–14 As we all know, the energy provided by the sun in one hour far exceeds the energy consumption of all human beings in one year. Therefore, efficient photocatalytic CO2 conversion can simultaneously alleviate the two global challenges of environment and energy.15Photocatalysts are the critical factor to achieve efficient CO2 conversion, which involves a series of complex processes including light-harvesting, separation and transfer of photogenerated charge carriers, adsorption and activation of CO2, and redox reactions on the surface of catalysts. However, it is difficult to improve the reactivity and selectivity of photocatalytic CO2 conversion due to the thermodynamic stability of the CO2 molecule (dissociation energy of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond: ∼750 kJ mol−1) and complex intermediates and products (Fig. 1). Therefore, an ideal photocatalyst is one that can satisfy both the thermodynamic and kinetic conditions of CO2 conversion. At present, many inorganic semiconductor materials including metal oxides,16–18 sulfides,19,20 nitrides,21,22 and perovskite halides23 have been shown to be effective catalysts for CO2 conversion. Nevertheless, compared with organic polymeric materials, inorganic materials are difficult for adjusting band structures (including band gap, conduction band, and valence band), narrow visible light absorption range, and rapid recombination of photogenerated carriers, which greatly limit their catalytic efficiency and product selectivity.24 Although their band structure can be changed by element doping,25 this process is very cumbersome. In contrast, organic semiconductor materials exhibit numerous advantages.26
O bond: ∼750 kJ mol−1) and complex intermediates and products (Fig. 1). Therefore, an ideal photocatalyst is one that can satisfy both the thermodynamic and kinetic conditions of CO2 conversion. At present, many inorganic semiconductor materials including metal oxides,16–18 sulfides,19,20 nitrides,21,22 and perovskite halides23 have been shown to be effective catalysts for CO2 conversion. Nevertheless, compared with organic polymeric materials, inorganic materials are difficult for adjusting band structures (including band gap, conduction band, and valence band), narrow visible light absorption range, and rapid recombination of photogenerated carriers, which greatly limit their catalytic efficiency and product selectivity.24 Although their band structure can be changed by element doping,25 this process is very cumbersome. In contrast, organic semiconductor materials exhibit numerous advantages.26
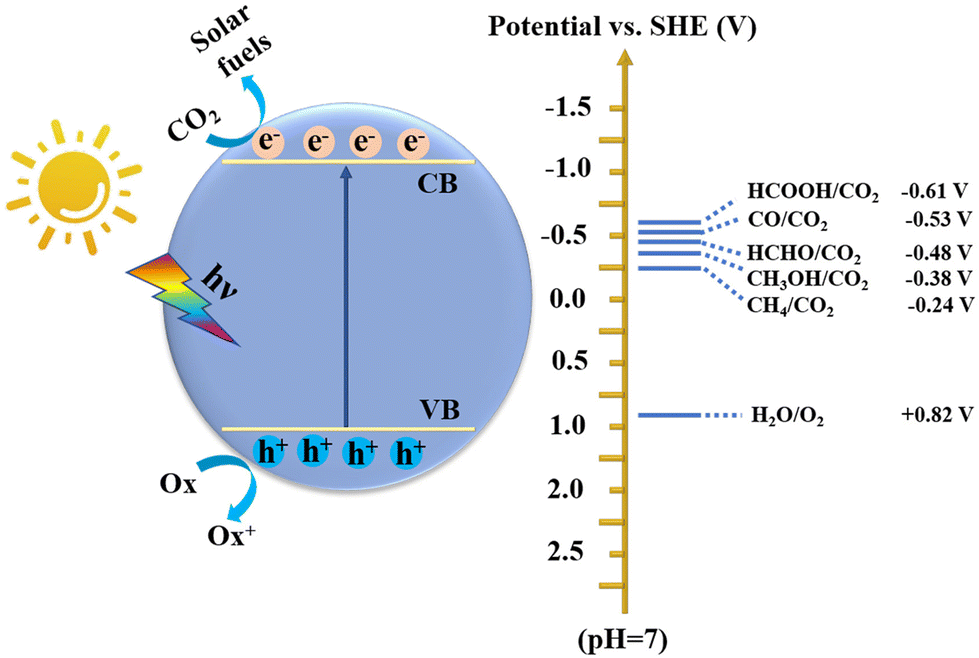 | ||
| Fig. 1 Schematic diagram of photocatalytic CO2 reduction over a semiconductor. | ||
Conjugated organic polymeric materials such as graphite carbon nitride (g-C3N4),27,28 covalent organic frameworks (COFs),29 covalent triazine frameworks (CTFs),30 and conjugated microporous polymers (CMPs)31 are arousing interests from researchers because of their tunable molecular or electronic structure,32 adjustable band gap by controlling different monomers,33,34 excellent visible light response, and unique properties of photocarrier separation and transport enabled by their π conjugated stacking structures. For example, g-C3N4 is a graphene-like semiconductor, which shows good photochemical stability due to the strong covalent bond between carbon and nitrogen.35 COFs have a periodic arrangement, uniform apertures and precise adjustable structures, and their physical and chemical properties can be accurately regulated by combining specific groups or molecules.36 CTFs are a new type of porous metal-free material with an ordered structure, large specific surface area, diverse structure and stable physical and chemical properties.37 These conjugated organic polymeric materials all show great potential for photocatalytic CO2 conversion, and thus are widely reported in the literature. However, most of the reported organic polymer photocatalysts are amorphous or possess low crystallinity, which greatly affects the separation and transport of charge carriers, limiting their photocatalytic activities.38 Crystallinity is an important factor affecting the photoreactivity of conjugated organic polymeric materials because high crystallinity can effectively improve their photoelectric kinetics by eliminating defects, enhancing the built-in electric field, reducing the interlayer hydrogen bonding and regulating the crystal planes.39–42 Thus, the construction of high-crystalline conjugated organic polymeric materials is a promising strategy to achieve enhanced photocatalytic CO2 conversion.
Herein, we review the recent advances in high-crystalline conjugated organic polymeric materials for photocatalytic CO2 conversion. Firstly, we comprehensively review the synthesis methods of high-crystalline conjugated organic polymeric materials (mainly focusing on crystalline g-C3N4, COFs and CTFs) for convenient reference (Fig. 2). Then, the advantages of high-crystalline conjugated organic polymeric materials are elaborated, which will provide theoretical guidance for the design of efficient photocatalysts. Some modification strategies and applications of high-crystalline conjugated organic polymeric materials for improved CO2 conversion are summarized, especially the relationship between crystallinity and CO2 conversion performance is emphasized. Finally, the current problems and challenges in this field are summarized, and the future development prospects of high-crystalline conjugated organic polymeric materials in photocatalytic CO2 conversion are discussed.
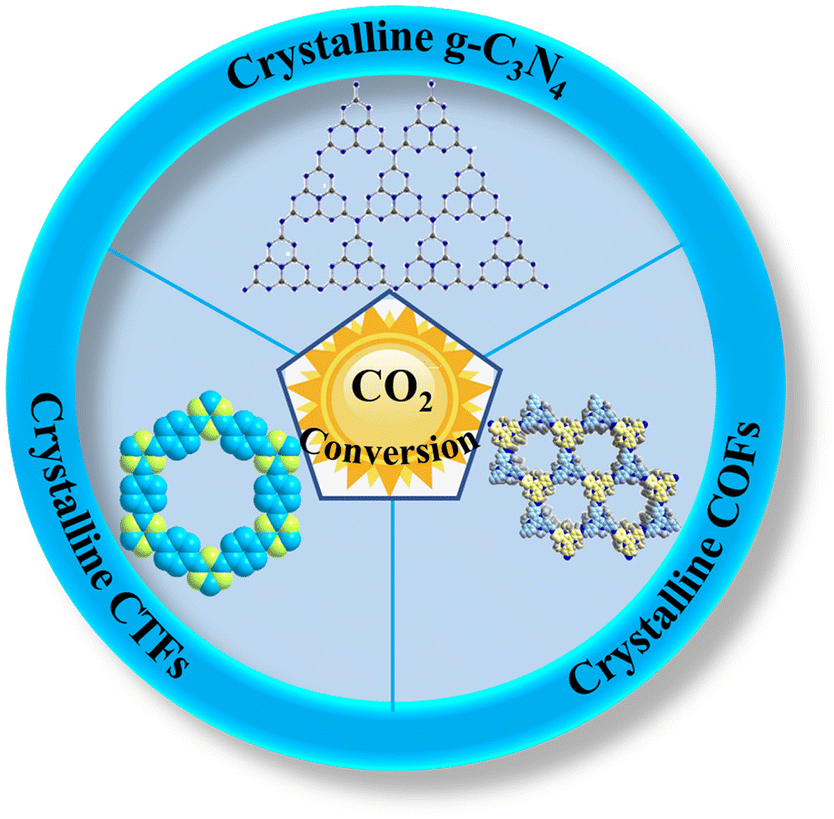 | ||
| Fig. 2 Overview of high-crystalline conjugated organic polymeric materials for the photocatalytic conversion of CO2. | ||
2. Synthesis of high-crystalline conjugated organic polymeric materials
At present, high-crystalline conjugated organic polymeric materials are arousing interest from researchers, and thus many methods have been developed to synthesize these high-crystalline materials. More importantly, different synthetic methods will affect the structure, size and morphology of the catalysts, which in turn affect the photocatalytic performance of the photocatalysts. In the case of the common g-C3N4 materials, they are mainly prepared via high-temperature pyrolysis, solvothermal method and physicochemical vapor deposition, and their precursors are usually small nitrogen-containing molecules. Other organic polymers including COFs, CTFs, and CMPs are synthesized by coupling two or more different monomers, generally by coupling reactions and condensation reactions.43,44 Now, most COFs are synthesized in solvothermal systems under harsh conditions. However, most of them possess low crystallinity or are amorphous. In this section, the main synthetic methods for high-crystalline conjugated organic polymeric materials are summarized according to the classification of different materials (such as g-C3N4, COFs and CTFs).2.1 Synthesis of crystalline g-C3N4
In the early stages, the g-C3N4 first applied for photocatalytic water splitting and hydrogen (H2) evolution was amorphous,45 and its H2 yield was very low even with the use of Pt as a cocatalyst. The main reason for this is that g-C3N4 with low crystallinity exhibits poor light absorption, slow electron transport, and rapid recombination of photogenerated carriers. Thus, to solve this problem, various common methods to synthesize highly crystalline g-C3N4 have been developed, such as molten salt method, microwave-assisted method and solid salt template method.Different precursors have an effect on the formation of the crystalline phase. For example, many highly crystalline g-C3N4 possessing triazinyl groups have been synthesized from dicyandiamide,46 and heptazine highly crystalline g-C3N4 can usually be prepared using melamine as the precursor system.47
The different methods for the synthesis of highly crystalline g-C3N4 have advantages and disadvantages. For example, the molten salt method, which is one of the most widely used methods to synthesize high-crystalline g-C3N4, is easy to perform, simple, environmentally friendly, etc.; however, this method requires a large amount of molten salt, water-sensitive salt LiCl, and its yield is relatively low. Different from the widely used molten salt method employing multiple salts, with the use of a single salt as a template, its limited space can guide the growth of g-C3N4 and yield g-C3N4 samples with high crystallinity and obvious long-range periodicity and large grain size. Also, the modification temperature is lower than the melting point of the salt, and thus this method is milder.48
On the one hand, in the two-step method, nitrogen-rich precursors (e.g., dicyandiamide, melamine, and urea) are thermally polymerized to form large blocks of g-C3N4, and the bulk g-C3N4 is mixed with molten salts for secondary pyrolysis to obtain crystalline g-C3N4.52 The current crystalline g-C3N4 mainly includes two structures of poly(triazine imides) (PTI) and poly(heptazine imides) (PHI). Bhunia et al. prepared highly crystalline PTI for improved photocatalytic hydrogen generation using melamine and 2,4,6-triaminopyrimidine as precursors to obtain g-C3N4, and then LiCl and KCl as molten salts to get PTI.53 Studies also showed that the stability of triazine-based g-C3N4 is less than heptazine-based g-C3N454,55 because heptazine has a larger π conjugated system, which is beneficial to widen the light absorption range, promote the charge migration rate, and enhance the photocatalytic activity. With the assistance of hydrochloric acid, Li et al. obtained bulk g-C3N4 by thermal polymerization with melamine, and then synthesized heptazine structure g-C3N4 (HCCN) with KCl and LiCl as molten salts and found that the amount of hydrochloric acid can change the crystallinity and surface defects of g-C3N4 by removing the K+ ions from the terminal amino. Furthermore, the high crystallinity of g-C3N4 was confirmed in its TEM images (Fig. 3a),52 where the clear lattice fringes indicate the successful regulation of high crystallization in g-C3N4.
 | ||
| Fig. 3 (a) HRTEM images of CCN. Reproduced from ref. 52 with permission from the Chinese Academy of Sciences, Copyright 2020. (b and c) SEM images of CAN. Reproduced from ref. 57 with permission from Elsevier, Copyright 2022. (d) HRTEM images of CNA (inset is the enlarged part). Reproduced from ref. 57 with permission from Elsevier, Copyright 2022. (e) Proposed salt melt synthetic processes of PTI/Li+Cl− catalyst using different binary alkali metal chlorides with different melting points. Reproduced from ref. 51 with permission from Wiley-VCH, Copyright 2021. | ||
On the other hand, the one-step molten-salt method involves mixing the precursors for the preparation of g-C3N4 with molten salts for calcination. For example, in our recent work, crystalline g-C3N4 was synthesized via a one-step molten salt method. In detail, electron-deficient 5-aminotetrazolium was used as the precursor and NaCl/KCl as the molten salt, and a well-ordered CNA nanoarray was obtained by high-temperature polymerization. These results show that the highly crystalline g-C3N4 synthesized by the molten salt method also has a certain effect on the morphology of g-C3N4, where g-C3N4 is transformed from the general bulk or nanosheets to nanorods (Fig. 3b–d). It is speculated that the reason for the formation of this rod-like shape is the accumulation in the molten salt state. The adhesion between NaCl/KCl and heptazine is relatively low, and thus the nanorod structure was formed in the space of the salt. In addition, as shown in Fig. 3e, Liu et al. used an LiCl/NaCl binary salt as the molten salt to synthesize crystalline g-C3N4, provided milder reaction conditions because its melting point is very close to the polymerization temperature.51 Dontsova et al. synthesized potassium-doped PHI using triazole derivatives as the precursors via the melt salt method, which showed improved visible light absorption and changed electronic structure.56
 | ||
| Fig. 4 (a) Illustration of the synthesis process of PCNNs-IHO by on-surface polymerization. Reproduced from ref. 62 with permission from Wiley-VCH, Copyright 2021. (b) Schematic illustration of constructing crystalline g-C3N4 by salt template method. Reproduced from ref. 63 with permission from Elsevier, Copyright 2022. (c) Schematical illustration of microwave thermolysis of urea pellets. Reproduced from ref. 42 with permission from Elsevier, Copyright 2021. | ||
Some materials with tunable nanostructures are often prepared by using some templates such as the inexpensive and versatile porous anodic alumina (AAO). Li et al. used AAO templates impregnated with a mixture of cyanamide and distilled water to limit the synthesis of g-C3N4 nanorods (CNRs) through thermal condensation within a single nanochannel, thereby improving the crystallinity of g-C3N4.67
2.2. Synthesis of crystalline COFs
Since the first report on COFs by Cote et al. in 2005,68 COFs have been widely used in photocatalysis due to their high porosity, large specific surface area, wide visible light absorption range and strong charge separation ability. Compared with some amorphous conjugated organic polymeric materials, the highly ordered π-electron channels in COFs enable better carrier transport. Therefore, crystalline COFs have attracted significant attention because of their unique structures and properties. However, the construction of crystalline COFs is a great challenge due to their poor reversibility. At present, crystalline COFs are mainly synthesized via the solvothermal method, microwave-assisted method, sonochemical method, vapor-assisted conversion, ionothermal, interfacial polymerization, etc.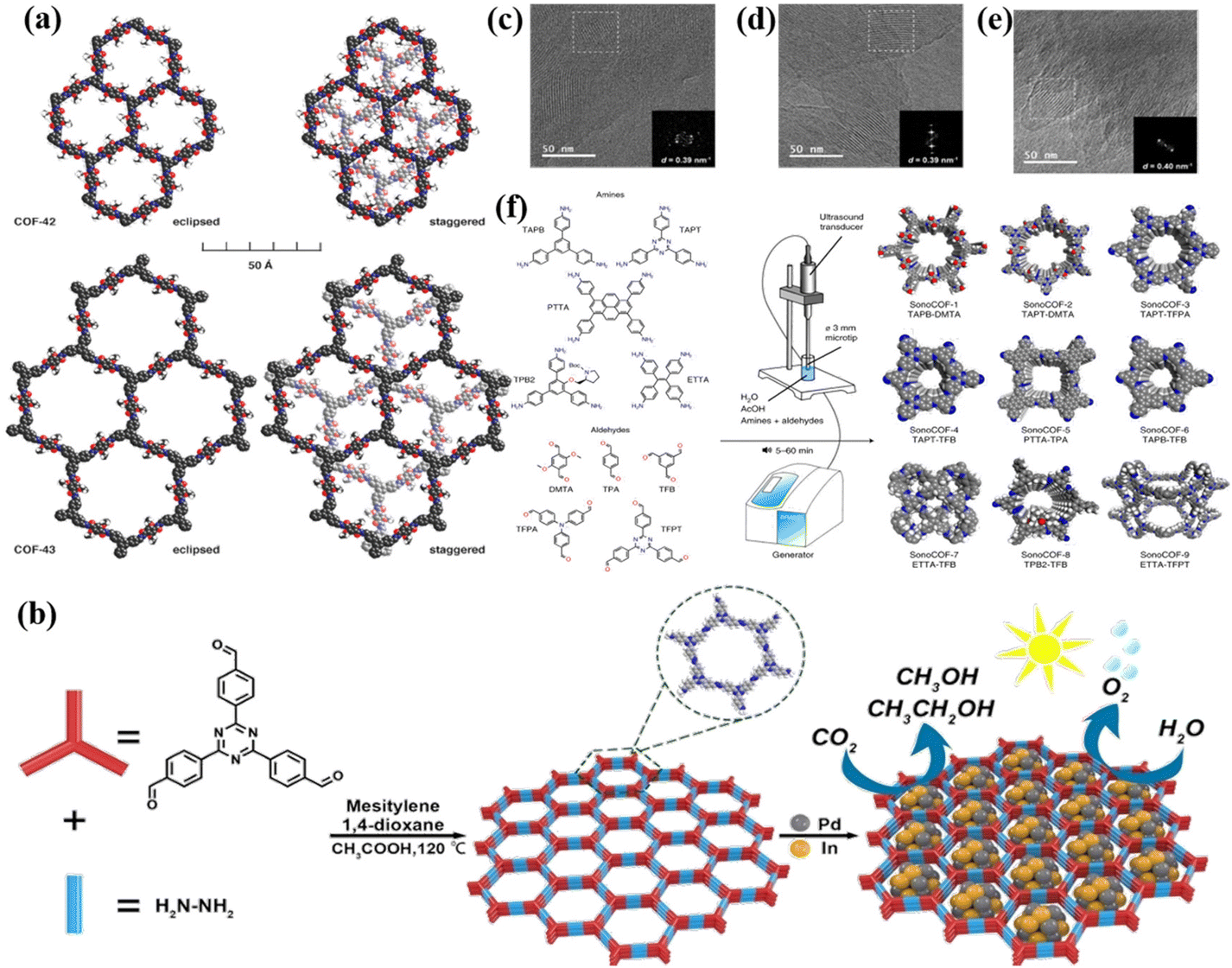 | ||
| Fig. 5 (a) Space-filling models of COF-42 and COF-43. Reproduced from ref. 70 with permission from the American Chemical Society, Copyright 2011. (b) Synthesis of PdIn@N3-COF. Reproduced from ref. 71 with permission from Elsevier, Copyright 2021. (c–e) TEM images of high-crystalline hydrazide COFs. Reproduced from ref. 74 with permission from the American Chemical Society, Copyright 2020. (f) Apparatus and conditions used for sonochemical synthesis, the COFs studied and the monomers used to synthesize them. Reproduced from ref. 83 with permission from Springer, Copyright 2022. | ||
2.3 Synthesis of crystalline CTFs
Covalent triazine frameworks are promising catalysts due to their unique triazine structure, abundant porosity, high conjugation, and excellent chemical and thermal stability, exhibiting great potential in the field of photocatalysis. In 2008, Thomas et al. synthesized a series of polymers with triazine bonds by condensation reaction, and first proposed the concept of CTFs.84 However, similar to the above-mentioned g-C3N4 and COFs, the CTFs synthesized at present are mostly low-crystalline or amorphous, while the methods for synthesizing high-crystalline CTFs are limited, which are discussed in this section.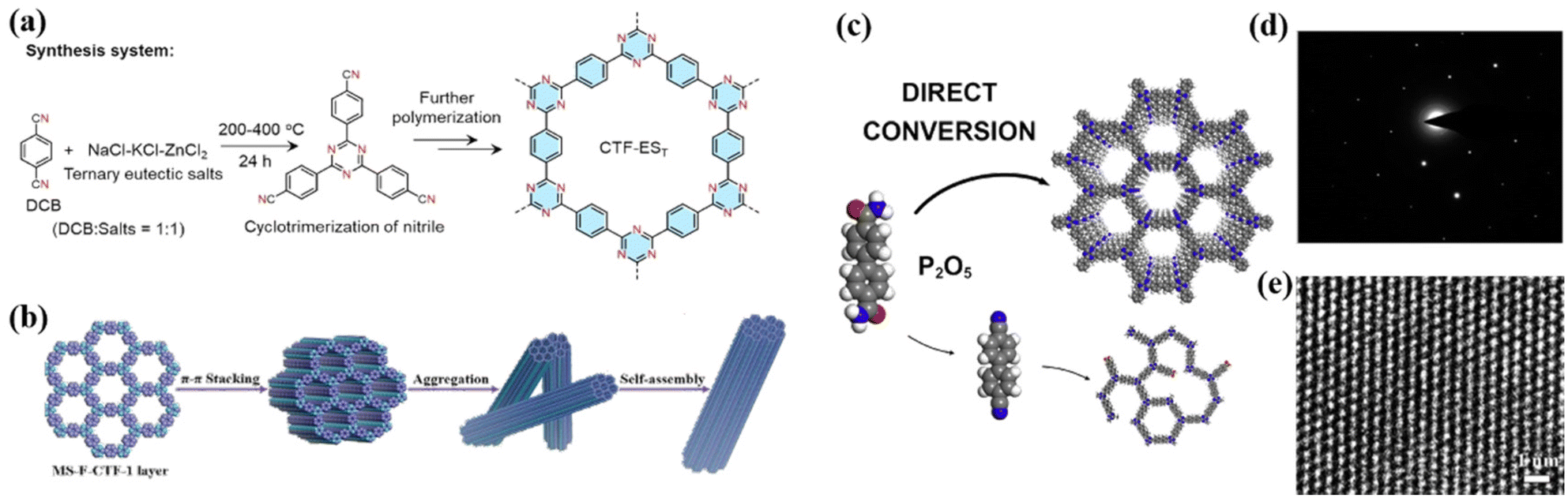 | ||
| Fig. 6 (a) Synthesis of CTF-EST in ternary NaCl-KCl-ZnCl2 eutectic salt system. Reproduced from ref. 86 with permission from Wiley-VCH, Copyright 2022. (b) Suggested mechanism for the formation of fiber for MS-F-CTF-1. Reproduced from ref. 91 with permission from Wiley-VCH, Copyright 2022. (c) Strategy of P2O5-assisted synthesis of CTFs. Reproduced from ref. 93 with permission from Elsevier, Copyright 2021. (d) Higher-magnification HR-TEM image and (e) SAED pattern of 2DP. Reproduced from ref. 96 with permission from the American Chemical Society, Copyright 2017. | ||
O)–NH2] into an s-triazine ring under P2O5 catalysis.94 The resulting CTFs also exhibited good crystallinity.
2.4 Other crystalline polymeric materials
Besides the above-mentioned three high-crystalline g-C3N4, COFs and CTFs, there are some other highly crystalline organic polymeric catalysts. For instance, Chu et al. developed a crystalline polyimide photocatalyst by polymerizing melamine (MA) with pyromellitic dianhydride (PMDA), which exhibited excellent photocatalytic activity under visible light.95 Liu et al. synthesized a two-dimensional triazine crystal polymer (2DP) (Fig. 6d and e) via the trimerization reaction of carbonitrile catalyzed by trifluoromethanesulfonic acid.963. Advantages of high-crystalline conjugated organic polymeric materials
Conjugated organic polymeric materials are good visible light-responsive photocatalysts, which exhibit structural diversity and functional tenability; however, their intrinsic stability and photocatalytic activity are largely limited by their traditional amorphous or weakly crystalline structures,81,97 together with abundant structural defects or trapping centers for the recombination of photogenerated electron–hole (e−–h+) pairs.98 Therefore, the crystallinity of organic photocatalysts is critical for their improved stability and catalytic activity, which can be used to effectively regulate their electronic structures in various ways, and further used to measure their degree of ordered arrangement in the solid state. Some of the advantages of high crystallinity in conjugated organic polymeric materials are summarized below.3.1 Reduces the concentration of defects
Plenty of defects will be introduced during the thermal polymerization process of precursors to prepare organic polymeric photocatalysts,99,100 which is the pivotal factor leading to the random migration and rapid recombination of carriers,101 resulting in an unsatisfactory photocatalytic effect. According to the literature, most conjugated organic polymeric materials (e.g., g-C3N4, COFs and CTFs) are low-crystalline or amorphous with many defects, which act as charge trap sites or charge recombination centers, leading to inhibited photocatalytic activities.38 In this regard, constructing the relevant high-crystalline materials to eliminate the defects of charge trapping sites is an important approach to improve the charge transport and inhibit photogenerated charge recombination,38 which are critical factors to enhance the photocatalytic performance, especially for the photoreduction of CO2.As shown in Fig. 7, a series of characterization methods was used to verify the presence of defects or vacancies in materials, such as X-ray absorption fine structure (XAFS) (Fig. 7a),102 X-ray photoelectron spectroscopy (XPS) (Fig. 7b),103 Raman spectroscopy (Fig. 7c),104 scanning transmission electron microscopy (STEM) (Fig. 7d),105 electron paramagnetic resonance spectroscopy (EPR) (Fig. 7e),57 and positron annihilation lifetime spectroscopy (PALS) (Fig. 7f).106 Specifically, XAFS can be employed to determine the defect levels by observing the migration distances between adjacent coordinating atoms and their peak intensities.107 However, defects in materials can change their bonding energy, which can be observed in shifted peaks or emerging peaks. Therefore, XPS is also an effective method to detect the defect sites in materials.108 Similarly, different chemical bonds exhibit different vibration modes, and a variation in the molecular vibration level causes a Raman shift. The presence of defects in materials can affect the vibration mode, leading to Raman shifts or new peaks.109 STEM can be employed to observe the atomic number in the crystal structure and the way each atom is arranged and find defects in materials.110 PALS is based on the positron lifetime to determine the defect type and the relative concentration of defects.111 Among them, EPR technology is a relatively convenient and fast detection way for the detection of defects, which can be judged according to the signal of the g factor in the Lorentzian line (at about 2.00) of unpaired electrons on the surface of materials.112 Many reports on the defects of highly crystalline materials have employed EPR.47,62 For example, in the recent work by our group, EPR was used to test the defects of the prepared catalysts. As shown in Fig. 7e through the EPR test, it can be concluded that the EPR signal intensity of the prepared highly crystalline carbon nitride (CNA) is low because the regulation of high crystallization eliminates the deep trapping sites of g-C3N4, and this low defect density also makes the catalyst have better ability to inhibit photogenerated electron–hole pair recombination and promote electron transfer.57 In addition to the improvement in crystallinity in g-C3N4, the improvement in crystallinity in other organic polymeric materials can also improve the defect situation. For instance, Khan et al. developed a “liquid–gas phase-switching method” to synthesize COF films, and the resulting COF films had high crystallinity and a dense surface without defects. Compared with the initial COF films, the surface defects also disappeared after the crystallinity increased.113 All these results indicate the merits of high-crystalline materials to eliminate defects.
 | ||
| Fig. 7 Results from the methods used to characterize defects or vacancies: (a) XAFS. Reproduced from ref. 102 with permission from The Royal Society of Chemistry, Copyright 2021. (b) XPS. Reproduced from ref. 103 with permission from the American Chemical Society, Copyright 2020. (c) Raman spectra. Reproduced from ref. 104 with permission from The Royal Society of Chemistry, Copyright 2019. (d) STEM. Reproduced from ref. 105 with permission from the American Chemical Society, Copyright 2019. (e) ERP. Reproduced from ref. 57 with permission from Elsevier, Copyright 2022. (f) PALS. Reproduced from ref. 106 with permission from Wiley-VCH, Copyright 2022. | ||
3.2 Enhances the built-in electric field
A built-in-electric field in photocatalysts can drive the separation and migration of photogenerated e−–h+ pairs to the surface of photocatalysts.114,115 Achieving a strong built-in electric field depends on two properties, as follows: (1) a high degree of crystallinity is necessary to obtained highly ordered structures and (2) the molecular dipole guarantees the ability of photogenerated charge separation. Therefore, the photocatalytic performance is closely related to the strength of the built-in-electric field. For instance, Zhang et al.116 synthesized a high-crystalline perylene imide polymer photocatalyst (urea-PDI). The advantages of high crystallinity and large dipole structure are conducive to the formation of a strong built-in electric field, making it possible for urea-PDI to have good charge separation and transport ability. Compared with other synthesized polymers (e.g., hydrazine-PDI and ethylenediamine-PDI), urea-PDI exhibited superior crystallinity, which ensures the stable existence of an internal dipole-induced electric field in polymers. Fig. 8a shows the process of charge transfer under the action of an internal electric field in Li's report, according to the built-in electric field characterization,117 where the surface charge density and zeta potential of urea-PDI were measured by atomic force microscopy with a Kelvin probe (KPFM), which can be used to index the built-in electric field. The results in Fig. 8b and c show that the surface potential and zeta potential of urea-PDI were much higher than that of other relevant catalysts, indicated its stronger built-in electric field with significantly enhanced charge separation and transportation, and it was further inferred that the high crystallinity allows the catalyst molecules to be arranged in an oriented space to provide channels for the rapid transfer of photogenerated charges. This conclusion was also confirmed in the report by Zhang et al.,40 where the highly crystalline triazine-PDI achieved a remarkable photocatalytic performance due to the enhanced built-in electric field. Recently, Guo et al.118 reported that a carbon nitride homogeneous junction (triple-crystallinity) with different crystallinity induced a high-efficiency photocatalytic performance due to the electric field enhancement at the interface. The results showed that the increase in crystallinity led to a decrease in the Fermi level and band position, resulting in the band bending of space charge region and interface, which in turn created a built-in electric field. As shown in Fig. 8d and e, the difference in electron density was used to analyze the built-in electric field of tri-crystallinity carbon nitride (CN) after photoexcitation, where more photogenerated holes were accumulated at high and medium crystallinity CN at the interface of high/medium and medium/low crystallinity CN, respectively. These charge accumulation characteristics also confirmed the existence and direction of the built-in-electric field. The built-in electric field improved the charge separation and transfer efficiency, and thus its photocatalytic efficiency is much higher than that of the general g-C3N4. The advantages of high crystallinity to achieve an internal electric field are also reflected in other conjugated organic polymeric materials. For example, the amorphous g-C3N4 with nitrogen vacancy (g-C3N4(NH)) synthesized by Wang et al.,119 and the crystalline COF (TP-TTA COF), which was easily deposited on g-C3N4(NH), formed a heterojunction through π–π interaction, leading to the formation of a strong built-in electric field. This is beneficial for the separation of charge carriers in the g-C3N4(NH)/COF photocatalytic reaction, thus improving the catalytic performance. | ||
| Fig. 8 (a) Charge transfer process at the interface. Reproduced from ref. 117 with permission from Springer Nature, Copyright 2016. (b) Surface potential of different polymers detected with KPFM. Reproduced from ref. 116 with permission from Wiley-VCH, Copyright 2020. (c) Zeta potential of different polymers. Reproduced from ref. 116 with permission from Wiley-VCH, Copyright 2020. (d) High/medium-crystallinity interface and (e) medium/low-crystallinity interface. Reproduced from ref. 118 with permission from Elsevier, Copyright 2022. | ||
3.3 Reduces interlayer hydrogen bonds
Usually, there are abundant interlayer hydrogen bonds in conjugated organic polymeric materials; however, interlayer hydrogen bonds act as channels for charge transport and recombination in photocatalytic reactions, and excess interlayer hydrogen bonds will greatly limit the specific surface area and active sites, thereby inhibiting the catalytic efficiency. Iqbal et al. found that the interlayer hydrogen bonds can be broken by preparing high-crystalline g-C3N4via a two-step heat treatment process. Compared with the usual bulk g-C3N4, highly crystalline g-C3N4 possess significantly decreased interlayer hydrogen bonds and ordered π conjugate system, which is conducive to realizing a high efficiency separation and suppressed recombination of photogenerated e−–h+ pairs, leading to a drastic improvement in photocatalytic activity.41 Therefore, high-crystalline materials can effectively reduce the interlayer hydrogen bonds, thereby promoting the photocatalytic activity.In addition, Kandambeth et al. found that construction of appropriate intramolecular hydrogen bonds also realized the transition of COFs from amorphous to crystalline state, and promoted a transition from disordered to ordered skeleton.72 Hydrogen bonds can also lock the molecular structure of COF materials and improve their planarity, thus further increasing their crystallinity.
3.4 Crystal plane regulation
It is well known that the crystal plane of semiconductors greatly affects their catalytic activities, where different crystal planes exhibit different electronic structures and surface reaction performances. Thus, crystal plane regulation is crucial for improving the photocatalytic performance. However, the general conjugated organic polymeric materials prepared via the conventional thermal polymerization methods possess low crystallinity and instability, which impede the understanding of the atomic structure and the identification of the role of the reaction surface role in promoting photocatalytic reactions on materials.The construction of high-crystalline materials is an effective strategy to regulate their crystal planes. For example, Lin et al. prepared(PTIs)/Li+Cl− high-crystalline polytriazine imides with different crystal faces and different aspect ratios via an ion-thermal method for photocatalytic overall water splitting. As shown in Fig. 9a, all the crystals form prismatic crystals surrounded by two hexagonal {0001} planes as base planes and six rectangular prismatic planes {10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0}. The aberration-corrected integrated differential phase contrast (AC-iDPC) image (Fig. 9b–d) showed the highly ordered atomic structure of PTI/Li+Cl−. Some in situ experimental characterizations and theoretical calculations showed that the {100} plane is beneficial for electron transfer and catalytic reaction, where its overall water splitting performance is linearly related to the relative surface area of the {100} and {0001} planes.39 Therefore, the construction of high-crystalline conjugated organic polymeric materials and screening the optimal crystal planes are efficient strategies for enhanced photocatalytic performance.
0}. The aberration-corrected integrated differential phase contrast (AC-iDPC) image (Fig. 9b–d) showed the highly ordered atomic structure of PTI/Li+Cl−. Some in situ experimental characterizations and theoretical calculations showed that the {100} plane is beneficial for electron transfer and catalytic reaction, where its overall water splitting performance is linearly related to the relative surface area of the {100} and {0001} planes.39 Therefore, the construction of high-crystalline conjugated organic polymeric materials and screening the optimal crystal planes are efficient strategies for enhanced photocatalytic performance.
 | ||
| Fig. 9 (a) Left, schematic illustration of the PTI/Li+Cl− crystal. Right, crystal structure of PTI/Li+Cl−. (b) AC-iDPC images of a typical PTI/Li+Cl− crystal. The inset shows the corresponding diffraction pattern. (c) Expanded view of the red box in (b). (d) AC-iDPC images of a typical PTI/Li+Cl− crystal aligned along the {2110} direction. Reproduced from ref. 39 with permission from Springer Nature, Copyright 2020. | ||
4. Modification of high-crystalline conjugated organic polymeric materials
High-crystalline conjugated organic polymeric materials are candidates for solar energy conversion due to their various advantages; however, their photocatalytic performance is still limited due to their limited visible light response and electron transfer efficiency. Thus, to further improve their photocatalytic efficiency, various modification strategies have been developed, such as defect engineering, element doping, surface loading, and construction of heterojunctions.4.1 Defect engineering
Improving the crystallinity and introducing defects are two effective ways to ameliorate the photocatalytic activities of conjugated organic polymeric materials, but these two methods are inherently contradictory. As mentioned above herein, high crystallinity may lead to a decrease in surface defects in catalysts; nevertheless, the introduction of defects can lead to a decrease in the crystallinity of materials. Therefore, it is necessary to balance the crystallinity and defects in organic catalysts. Photocatalysts can only possess excellent catalytic activity when they have an appropriate amount of defects, while maintaining high crystallinity.Some works have been devoted to this research. For instance, Ren et al. introduced some additional groups to promote the dissociation of excitons and inhibit the recombination of photogenic e−–h+ pairs. Cyanogen defects were introduced in the polymer backbone without affecting the crystallinity (Fig. 10a and b), which enhanced the light absorption capacity, accelerated the charge separation, and enhanced the photocatalytic performance.120–122
 | ||
| Fig. 10 (a) Illustration of the defect engineering strategy. Reproduced from ref. 120 with permission from Wiley-VCH, Copyright 2019. (b) TEM images of D-CCN. Reproduced from ref. 120 with permission from Wiley-VCH, Copyright 2019. (c) Proposed structure of CN-NVs-CTe (nitrogen vacancies are marked by yellow circles). Reproduced from ref. 123 with permission from The Royal Society of Chemistry, Copyright 2020. (d) HRTEM image of CN-NVs-CTe. Reproduced from ref. 123 with permission from The Royal Society of Chemistry, Copyright 2020. | ||
The reasonable introduction of vacancies can serve as additional active sites to facilitate photocatalytic reactions, and excitons can accelerate dissociation near the defect sites. As shown in Fig. 10c, Liu et al. reported a facile alkali-assisted molten salt method to prepare nitrogen-vacancy-rich crystalline g-C3N4, in which LiCl/KCl was used to obtain crystalline CN to enhance the crystallinity, and potassium hydroxide was used to remove some nitrogen atoms in situ to generate nitrogen vacancies.123 The HRTEM results, as shown in Fig. 10d, indicated that the polymer still retained high crystallinity after introducing nitrogen defects.
4.2 Element doping
Heteroatom doping is a well-known and effective way to improve the photocatalytic performance of materials, and it is a simple and valid method to adjust the light absorption, electronic structure and band gap of catalysts. This includes metal doping (e.g., Ni, Cu, and Co),124,125 alkaline earth metal doping (e.g., K and Na),61 and non-metal doping (e.g., C, B, F, and Cl).126 For instance, Zhang et al. synthesized a series of highly crystalline g-C3N4 with K intercalation by a molten salt method using melamine as the precursor. The intercalation of K element regulated the surface active sites on the g-C3N4 framework and greatly improved the photocatalytic activity.127 Li et al. inserted Cl element into the intermediate layer of CTF-1 through a covalent bridge by ball-milling stripping-assisted acidification (Fig. 11a), resulting in a unique electronic structured COF(Cl-ECF). In this Cl-ECF, a covalent interlayer channel of Cl–C and Cl–N covalent bond was formed, which promoted the electron delocalization and separation and migration of carriers. In addition, the insertion of Cl element can narrow the band gap of Cl-ECF, which can effectively activate more photoinduced charge carriers, thereby improving the photocatalytic performance.128 Zhu et al. prepared a series of P-, S-, Se-doped CTF-1 by high-temperature annealing (Fig. 11b), in which a built-in electric field was generated by the spin polarization, resulting in the promoted separation and transfer of e−–h+ pairs, which greatly improved the photocatalytic efficiency.129 Chen et al. prepared K+-, Ni- and N-doped crystalline g-C3N4 by Ni2+ anchoring and the supramolecular thermal polymerization of encapsulated melamine-cyanic acid, and then recalcination in a mixture of KCl/LiCl. The obtained strip-like K+, Ni and C co-doped crystalline g-C3N4 displayed excellent charge separation efficiency, light absorption capability, and photoreduction ability.130 It can be seen that element doping is an effective method to improve the photocatalytic performance of high-crystalline g-C3N4. | ||
| Fig. 11 (a) Schematic diagram of the preparation for Cl-ECF photocatalysts. Reproduced from ref. 128 with permission from Elsevier, Copyright 2020. (b) Schematic illustration of the synthetic process of HA-CTF-1. Reproduced from ref. 129 with permission from the American Chemical Society, Copyright 2022. Proposed molecular structure for (c) CN and (d) KCCN. Reproduced from ref. 132 with permission from the American Chemical Society, Copyright 2021. (e) HRTEM image of KCCN. Reproduced from ref. 132 with permission from the American Chemical Society, Copyright 2021. | ||
More interestingly, element doping and defect engineering can synergistically regulate the electronic structure of catalysts, which can more effectively enhance their visible light absorption, promote charge separation, and capture more useful electrons to participate in surface reactions.131 For example, Zhang et al. realized the construction of K-doped together with cyanide-defected crystalline g-C3N4 using KCl as a template. In this process, K+ ions were doped gradiently into heptazine-based CCN (KCCN) by controlling their diffusion rates from surface to body (Fig. 11c and d), resulting in gradient changes in the electronic structure, band position and band gap in the same crystalline g-C3N4.132 The high crystallinity of this bifunctional g-C3N4 can be demonstrated by its clear lattice fringes in the HRTEM image (Fig. 11e). More importantly, the K+ concentration gradient and newly formed cyanide defects can induce a built-in electric field, which greatly facilitates exciton dissociation, charge transport and separation. Wu et al.133 also obtained a bifunctional crystalline g-C3N4 catalyst (ACNN) with both alkali metal doping and N vacancies via a molten salt method. This bifunctional ACNN successfully expanded the visible light absorption range, suppressed charge recombination, and reduced the band gap from 2.85 to 2.63 eV. Thus, this synergistic effect of element doping and defect engineering has good prospects in improving the utilization of photogenerated charges and promoting photocatalytic reactions.
4.3. Surface loading
Conjugated organic polymeric materials are also good carriers with abundant tunable sites and specific geometric and electronic structures, which are suitable for the loading of a cocatalyst or grafting of organic molecules to enhance their catalytic activities. In recent years, surface-modified catalysts with single atoms have developed rapidly and attracted attention from many researchers because a single metal can maximize the utilization of catalytic sites and form strong interactions with carriers to promote the catalytic efficiency. Li et al. synthesized a CTF-PDDA-TPDH catalyst by stabilizing Pt single atoms (SA) in the N3 sites of ultrathin CTF nanosheets. DFT calculation showed that the single Pt atom was anchored by the –N3 site (Fig. 12a), and the Pt–N3 coordination structure also became the photocatalytic active site. The wavelet transform extended X-ray absorption fine structure (WT-EXAFS) diagram (Fig. 12b) confirmed that Pt existed in the catalyst as a single atom. The photogenerated electrons in the conduction band of CTF could be quickly transferred to Pt single atoms, and further facilitated the separation and transfer of charges.134 Huang et al. used the special geometric and electronic structure of CTF-1 to prepare a Co-SA/CTF (Co-SA/CTF) catalyst through the space constraint effect and coordination interaction for photocatalytic CO2 reduction. The introduction of Co single atoms not only enhanced the adsorption of CO2, but also accelerated the separation and transfer of photocarriers, thereby promoting the photocatalytic performance.135 Shen and co-workers reported a one-step method for the synthesis of single-atom molten Co salt coordinated with a crystalline g-C3N4/PTI heterojunction based on the built-in electric field in a homogeneous junction, which facilitates charge separation and single-atom loading for accelerating surface catalytic kinetics. The WT-EXAFS (Fig. 12c) analysis confirmed the existence of single-atom Co. Therefore, while improving the crystallinity, the charge transfer was promoted by adjusting the electronic structure of the Co single atom to achieve high-efficiency photocatalytic performances.124 | ||
| Fig. 12 (a) DFT-optimized structure of the Pt-SACs/CTF catalyst. Reproduced from ref. 134 with permission from the American Chemical Society, Copyright 2020. (b) WT-EXAFS of Pt-SACs/CTF. Reproduced from ref. 134 with permission from the American Chemical Society, Copyright 2020. (c) Topographical maps of WT-EXAFS of Co-CCN/PTI-4 and Co-CCN/PTI-40. Reproduced from ref. 124 with permission from Science China Press, Copyright 2021. (d) Schematic illustration of PDI/CNA catalyst fabrication. Reproduced from ref. 57 with permission from Elsevier, Copyright 2022. | ||
Another surface-loading strategy is to graft organic molecules with excellent photocatalytic properties on the surface of g-C3N4 to construct an electron donor–acceptor (D–A) structure. Our team reported the grafting of a high-crystalline g-C3N4 nanoarray (CNA) of the organic small molecule perylene tetracarboxylic dianhydride (PTCDA) to build a D–A structure to improve the visible light response of the catalyst (Fig. 12d). Consequently, this accelerated the charge transport and promoted the separation of redox centers, namely electronic PTCDA molecular absorption and pyridine N atoms can be used as the reduction center and seven oxazine units as the oxidation site.57 Thus, high crystallization eliminates defects in the material and prevents rapid charge reorganization, while the D–A structure enables the separation of redox centers to facilitate the photocatalytic hydrogen peroxide (H2O2) synthesis process. In addition, while ensuring crystallinity, many small molecular materials and functional groups have been used to modify conjugated organic polymers to improve their catalytic performance, such as cyclized quaternary ammonium bipyridine molecules,136 polyethylene glycol,137 and acetylene (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–) and diacetylene (–CC–CC–) moieties.138 This strategy of surface loading is one of the effective methods to improve the properties of conjugated organic polymeric materials.
C–) and diacetylene (–CC–CC–) moieties.138 This strategy of surface loading is one of the effective methods to improve the properties of conjugated organic polymeric materials.
4.4 Construction of heterojunctions
The construction of semiconductor heterojunctions is also one of the most common and effective methods to improve the catalyst efficiency, and thus is widely used in the design of catalysts.15,139 The photocatalytic mechanisms of heterojunctions generally include type-I, type-II, Z-scheme, and S-scheme, which can improve the catalytic activity from different angles. The main purpose of constructing heterojunctions is to regulate the redox potential, enhance the visible light response, accelerate the separation of carriers, and promote the transfer and migration of photogenerated charges through the formed built-in electric field at the hetero-interface and effectively suppresses their recombination, thereby facilitating the photocatalytic effect.The charge transfer mode, advantages and disadvantages of different heterostructures are also different. In the type-I heterojunction, both the VB and CB edges of one semiconductor are aligned to the band gap of the other half of the conductor, forming a straddle alignment. Although efficient photocatalytic activity exists in the type-I heterojunction, the photogenerated electrons and holes of the semiconductor may be transferred to the other half of the conductor, leading to electron hole pair recombination. In type-II semiconductor heterojunctions, the CB and VB are higher than the other half conductor and the chemical potential difference between them makes the band bending result in the formation of a built-in electric field. This promotes the movement of the photoproduced electron–hole pairs in opposite directions, leading to different heterojunction side electron–hole space separation.140 Considering the shortcomings of type-II charge transfer from a thermodynamic point of view, improving the charge separation efficiency is achieved by sacrificing the redox ability, and from a kinetic point of view, the existence of an electrostatic effect will also make the original e−–h+ in the photocatalyst inhibit the transfer of electron holes in other catalysts, which is not conducive to the photocatalytic reaction.141 The advantage of the Z-scheme heterojunction is that it has one photocatalyst with strong reducing electrons and another photocatalyst with strong oxidizing holes. However, the traditional Z-scheme heterojunction is limited to reacting only in an aqueous environment. In the new Z-scheme heterojunction system, including all-solid Z-scheme heterojunction and direct Z-scheme heterojunction, this problem is alleviated, but the direction of electron hole transfer is still controversial.142 S-scheme heterojunctions can realize the spatial separation of photogenerated e− and h+ through the built-in electric field, band bending and electrostatic effect.143 Therefore, the construction of suitable heterojunction catalysts is still a promising means to improve the photocatalytic reactivity.
Our group obtained highly crystalline g-C3N4 (CCN) and ReS2via a template-mediated method and solvent stripping method, respectively. The ReS2 and CCN heterojunction was obtained via self-assembly. As shown in Fig. 13a, the photogenerated electrons on the CB of CCN would be transferred to ReS2, and the accumulated electrons would be used for photocatalytic reduction reaction. High crystallinity and heterogeneous interfaces contribute to photogenerated carrier migration, and thus enhance the catalytic activity.63 Lin et al. achieved a good photocatalytic performance using a CCN/LaOCl heterojunction via the in situ growth of highly crystalline g-C3N4 on the surface of LaOCl. The high crystallinity of g-C3N4 is favorable for the transport of photoexcited charges, and the built-in electric field formed at the hetero-interface can promote the separation of excited electron-holes, thereby inhibiting their recombination, and finally achieving a good photocatalytic performance.146 In addition, the heterojunctions with different crystal phases of the same material can remarkably eliminate the interfacial defects.144,147,148 The built-in electric field formed by this close contact heterogeneous interface (Fig. 13b) and improved crystallinity make the catalyst much better. He et al. constructed a metal-free CN/CTF heterojunction by combining CTF and g-C3N4 through a simple self-assembly method. The addition of highly crystalline CTF greatly promoted the photocatalytic activity of g-C3N4 due to the stable structure of the CTF crystal.145 This metal-free CN/CTF heterojunction exhibited excellent photocatalytic CO2 reduction activity due to the hetero-interface effect (Fig. 13c). Furthermore, Xu et al. synthesized a covalent organic framework-based (P-COF-1)/CTF heterojunction with a 2D-2D heterojunction structure through intermolecular π–π interaction. The construction of COF and CTF heterojunctions increases the visible light absorption and the separation efficiency of photogenerated carriers.149 Cheng et al. prepared a heterojunction photocatalyst for the directional charge movement of an S-scheme heterojunction by anchoring single-atom Co to CeO2 and by introducing it in highly crystalline g-C3N4 (named CeCo-PTI). The efficient and selective photocatalytic reduction of CO2 to methane was achieved through the site regulation of the directed charge transfer of this special S-scheme heterojunction.150
 | ||
| Fig. 13 (a) 2D/2D ReS2/CCN heterojunction. Reproduced from ref. 63 with permission from Elsevier, Copyright 2022. (b) Built-in electric field formed by the heterojunction. Reproduced from ref. 144 with permission from Elsevier, Copyright 2020. (c) Schematic mechanism of CN/CTF heterostructure for photocatalytic CO2 reduction. Reproduced from ref. 145 with permission from the Chinese Academy of Sciences, Copyright 2022. | ||
5. Application in photocatalytic CO2 reduction
Carbon dioxide conversion is an effective way to convert this waste carbon source into renewable energy via low-carbon sustainable development strategies, including thermochemical catalysis,151 electrochemical reduction,152 biological catalysis,153 photoelectric chemical catalysis154 and photocatalytic reduction.155 Among them, photocatalytic CO2 reduction is an extremely promising route. It is well known that a typical semiconductor photocatalytic CO2 reduction process involves the five steps, as follows: light absorption, charge separation, CO2 adsorption, surface redox reaction, and product desorption, which are all critical for efficient CO2 conversion. In this regard, high-crystalline conjugated organic polymeric materials (such as g-C3N4, COFs, and CTFs) exhibit remarkable photocatalytic CO2 conversion effects, and thus are attracting attention from researchers because of their advantages of tunable molecular or electronic structure, excellent visible light response, and unique properties of photocarrier separation and transport enabled by their π conjugated stacking structures. In this section, we separately introduce the application of different high-crystalline conjugated organic polymeric materials in photocatalytic CO2 reduction and their photocatalytic activities are summarized in Table 1.| Photocatalyst | Light source | Products | Selectivity | Yield (μmol g−1 h−1) | Ref. | |
|---|---|---|---|---|---|---|
| Crystalline g-C3N4 based photocatalysts | CN-ATZ-NaK | 350 W Xe lamp (AM1.5 filter) | CO, CH4, CH3OH | CO | CO (14 μmol g−1 h−1) | 156 |
| CNNA/rGO | 350 W Xe lamp (AM1.5 filter) | CH4, CO, CH3OH, C2H5OH | CO (64.6%) | CO (3.67 μmol g−1 h−1) | 157 | |
| Cu-CCN | 300 W Xe lamp | CO | CO (100%) | CO (3.086 μmol g−1 h−1) | 158 | |
| CeCo-PTI | 350 W Xe lamp | CO, CH4 | CH4 (88.3%) | CH4 (45.4 μmol g−1 h−1) | 150 | |
| Crystalline COFs based photocatalysts | TTCOF-Zn | 300 W Xe lamp | CO | CO (100%) | CO (12.33 μmol after 60 h) | 159 |
| sp2C-COF | 300 W Xe lamp (λ ≥ 420 nm) | CO | CO (84%) | CO (1040 μmol g−1 h−1 over 17.5 h illumination) | 160 | |
| HBC-TFPN | 300 W Xe lamp (λ ≥ 420 nm) | CO, H2 | CO (92%) | CO (7.7 μmol g−1 h−1) | 161 | |
| TRITER-2 | 20 W white LED irradiation | — | CH3OH | — | 162 | |
| PdxIny@N3-COF | A 300 W Xe lamp (with a 400 nm filter) | CH3OH, CH3CH2OH | CH3OH (74%) | CH3OH (590.52 μmol g−1 in 24 h) | 71 | |
| MCOF-Ti6Cu3 | 300 W Xe lamp (AM1.5 filter) | — | HCOOH | HCOOH (77.3 μmol g−1 h−1) | 163 | |
| Crystalline CTFs based photocatalysts | PT-SA/CTF-1 | 300 W Xe lamp (with a 420 nm filter) | CO, CH4 | CO | CO (4.5 μmol g−1 h−1) | 164 |
| CN/CTF | 300 W Xe lamp | CO | CO (99.9%) | CO (151.1 μmol g−1 h−1) | 145 | |
5.1 Crystalline g-C3N4 for photocatalytic CO2 reduction
The g-C3N4 catalyst is a typical conjugated organic polymeric material and one of the most commonly used catalysts for the photocatalytic reduction of CO2.165 It is widely used in the field of photocatalysis due to its suitable CB position, metal-free inherent property, and good catalytic activity. However, the photocatalytic performance of general low-crystalline or amorphous g-C3N4 is limited by its disordered electronic structure, low charge utilization, rapid recombination of carriers, and limited light absorption. More importantly, it is difficult to control the product selectivity of photocatalytic CO2 reduction with the general amorphous g-C3N4, often producing some complex mixtures, which makes the separation of products in practical applications challenging. In this case, crystalline g-C3N4-based photocatalysts exhibit excellent efficiency and high product selectivity in CO2 conversion due to their multiple advantages.For example, Zhang et al. prepared crystalline g-C3N4 by the molten salt method, which exhibited a significantly enhanced visible light response compared to the general amorphous g-C3N4. It exhibited excellent photocatalytic activity without the use of metal cocatalysts and sacrificial agents during CO2 conversion, the product was dominated by CO and the production rate is as high as about 14 μmol/g h−1, which is much higher than the currently reported g-C3N4-based materials.156 The high-crystalline g-C3N4 obtained by the molten salt method exhibited an excellent photocatalytic performance due to the enhanced light absorption and the improved π–π* electronic transitions.
Compared with the research on the photocatalytic CO2 reduction with pristine crystalline g-C3N4, more research is devoted to further improving the photocatalytic activity of crystalline g-C3N4 by some modification measures. While maintaining good crystallinity, introducing defects (such as cyanide group and vacancy) and ions125,166,167 and constructing heterojunctions are efficient ways to promote the adsorption and activation of the CO2 molecule, and improve photoelectron transfer ability and utilization rate, thereby improving the CO2 reduction ability. As is shown in Fig. 14a, Xia et al. constructed a unique 2D/1D graphene/polyheptazinimide heterojunction (denoted as CNNA/rGO). Fig. 14b shows the ordered 1D crystalline carbon nitride nanorod crystal structure, which provides a pathway for light harvesting to achieve a fast reduction process and a direct path for photoelectron transfer to the graphene surface, improving the light absorption and interfacial charge transfer. The total CO2 conversion achieved was 12.63 μmol g−1 h−1 without the addition of a cocatalyst and sacrificial agent.157
 | ||
| Fig. 14 (a) 1D/2D CNNA/rGO. Reproduced from ref. 157 with permission from Elsevier, Copyright 2019. (b) HRTEM image of the inner structure of a CN nanorod in CNNA/r-GO. Reproduced from ref. 157 with permission from Elsevier, Copyright 2019. (c) HAADF–STEM image of Cu-CCN samples. Reproduced from ref. 158 with permission from the American Chemical Society, Copyright 2020. In situ Fourier transform infrared spectra (ISFTIR) of (d) Cu-CCN samples and (e) CCN samples. (f) Reaction pathways for photocatalytic CO2 reduction and the corresponding chemical molecular structure on Cu-CCN sample. Reproduced from ref. 158 with permission from the American Chemical Society, Copyright 2020. (g) Photocatalytic activity of samples for CO2 reduction. Reproduced from ref. 158 with permission from the American Chemical Society, Copyright 2020. (h) Time-resolved transient PL decay of PTI, Ce-PTI, and CeCo-PTI. (i) Femtosecond TA spectra of CeCo-PTI at different probe delays under 350 nm excitation. The inset shows interfacial electronic modification in CeCo-PTI. Reproduced from ref. 150 with permission from John Wiley-VCH, Copyright 2022. (j) In situ Raman spectra of CoTPP/g-C3N4 in CO2-saturated aqueous solution. Reproduced from ref. 168 with permission from The Royal Society of Chemistry, Copyright 2017. | ||
The main product is CO, but an interesting phenomenon is that the increase in crystallinity makes the content of C2H5OH in the product increase, which may be because the introduction of rGO helps to accumulate photogenerated charges and improve the adsorption of CO2. C2H5OH is produced through the coupling of the intermediate methoxyl (˙OCH3) and methyl (˙CH3) radicals. The obtained composite catalyst with crystalline microstructure significantly improved the light absorption, exciton splitting, charge transport, and selective CO2 binding, resulting in the efficient photoreduction of CO2 to chemical fuel.157 In addition, as shown in Fig. 14c, Li et al. supported a Cu single atom on crystalline g-C3N4 (Cu-CCN) based on the molten salt method and reflow method to improve the photocatalytic CO2 reduction performance. The presence of excessive defects in amorphous g-C3N4 limits the enhancement in catalytic activity with the introduction of single atoms. Thus, it is a good strategy to replace amorphous g-C3N4 with crystalline g-C3N4. Here, Cu atoms can provide sites for CO2 activation. In situ Fourier transform infrared spectroscopy (ISFTIR) (Fig. 14d and e) was used to accurately detect the CO2 reduction process and to determine the reaction mechanism by capturing the intermediates. The results showed that the CO2 molecule was absorbed on the surface of Cu-CCN samples in the form of HCO3−, and the main intermediate in this CO2 reduction process was HCOO−. Furthermore, DFT theoretical simulation calculations (Fig. 14f) showed that the CO2 adsorbed on the Cu-CCN samples initially generated *HCOO, which was then converted to *CO and *CHO. In this process, the transition from *HCOO to *CO is more thermally advantageous, and thus it is easier for this process to reduce CO2 to CO. Combined with the ISFTIR and DFT results, it was found that CO2 is more favorable for the conversion to CO than CH4 due to the introduction of Cu single atoms. However, the photocatalytic activity of highly crystalline g-C3N4 is more likely to be enhanced after the introduction of single atoms due to the elimination of its defects. Thus, the Cu-CCN samples can achieve nearly 100% photocatalytic conversion of CO2 to CO (Fig. 14g).158 Therefore, high-crystalline g-C3N4-based photocatalysts exhibit excellent effects and prospects in CO2 conversion. Differently, Cheng and her team proposed a new method to improve the photocatalytic CO2 reduction performance of highly crystalline g-C3N4. They constructed sigmoid heterojunctions with specific carrier migration by using single-atom Co anchored in a CeO2 cocatalyst instead of a carbon nitride support. The specific unsaturated structure of bimetallic CeCo accelerated the movement of electrons from the PTI donor to the specific active site Co to achieve the directed electron transfer in specific interface structures, thus enabling the efficient photocatalytic conversion of CO2 to CH4. Fig. 14(h and i) show the time-resolved transient PL decay and femtosecond transient absorption (fs-TA) spectra of CeCo-PTI, reflecting the decay kinetics of the dynamic charge separation in CeCo-PTI, which reduces the charge recombination ability, and thus improves the catalytic performance.150 Liu et al. designed and synthesized a compound of g-C3N4 and cobalt porphyrin (CoTPP/g-C3N4) based on π–π supramolecular interaction, and studied the formation of chemical bonds during CO2 reduction by in situ Raman testing in saturated CO2 solution (Fig. 14j). It was confirmed that the peaks at 423 cm−1 and 496 cm−1 were due to the insertion of active CO2 in the Co–C bond, and the peaks at 638 cm−1 and 753cm−1 were due to the production of –COO− and –COOH intermediates, respectively. The results showed that CO2 could be reduced to formic acid by the catalyst.168
5.2 Crystalline COFs for photocatalytic CO2 reduction
Crystalline COF materials are promising porous crystal materials for artificial photosynthesis due to their tunable structures and chemical stability. Their large specific surface area provides more sites for CO2 adsorption and activation, and their ordered structure can effectively promote the efficient separation and transport of charge carriers.169 For instance, Lu et al. designed a series of crystalline porphyrintetrathiafulvalene covalent organic frameworks (TTCOF-M), in which the electron-deficient metalloporphyrin complexes (TAPP) possessed good visible light absorption ability and potential CO2 reduction ability, while the electron-rich tetrathiafulvalene (TTF) possessed rapid electron transfer ability. The effective coupling between TAPP and TTF in the COFs enables photogenerated electrons to be effectively separated and transferred from TTF to TAPP, thereby achieving redox center separation. TTCOF-Zn or TTCOF-Cu has a good photocatalytic redox potential for CO2 reduction because its conduction band minimum (CBM) is more negative than that of the standard reduction potential of CO/CO2. The fast Fourier transformation image of the HRTEM image of TTCOF-Zn (Fig. 15a) showed its hexagonal void structure arranged along the axis of the 001 crystal, demonstrating its AA stacking pattern. This mode facilitated the transport of photogenerated electrons and holes and inhibited their recombination. The results showed that TTCOF-Zn exhibited excellent CO2 reduction capability in the absence of a photosensitizer, sacrificial agent and cocatalyst. Furthermore, TTCOF-Zn could achieve nearly 100% CO2 photoreduction to CO with only H2O as the electron donor, with a maximum yield of 12.33 μmol after 60 h illumination (Fig. 15b).159 Due to the covalent coupling between the TAPP and TTF molecules in TTCOFs, visible light induced the effective separation and transfer of e−–h+ pairs, which promoted the photocatalytic reaction. Moreover, due to the affinity between the metal and CO2 molecules, TTCOF-Zn had strong adsorption capacity for CO2. | ||
| Fig. 15 (a) HRTEM image of TTCOF-Zn, inset: the pores are highlighted. Reproduced from ref. 159 with permission from Wiley-VCH, Copyright 2019. (b) CO2RR performances of TTCOF-M and COF366-Zn. Reproduced from ref. 159 with permission from Wiley-VCH, Copyright 2019. (c) Transient absorption spectra of Re-Bpy-sp2c-COF. Reproduced from ref. 160 with permission from The Royal Society of Chemistry, Copyright 2020. (d) Complete transient absorption surface probed Re-Bpy-sp2c-COF. Reproduced from ref. 160 with permission from The Royal Society of Chemistry, Copyright 2020. (e) Photocatalytic formation of methanol from CO2. Reproduced from ref. 162 with permission from Elsevier, Copyright 2022. (f and g) Reaction energy for photocatalytic reduction of CO2. Reproduced from ref. 162 with permission from Elsevier, Copyright 2022. (h) Photocatalytic performance for CO2 reduction of PdxIny@N3-COF. Reproduced from ref. 71 with permission from Elsevier, Copyright 2021. (i and j) In situ XPS spectra of c Ti 2p and d Cu 2p for the simulated solar-driven CO2 reduction process over MCOF-Ti6Cu3. Reproduced from ref. 163 with permission from Springer Nature, Copyright 2022. (k) In situ DRIFTS spectra for the simulated solar-driven CO2 reduction process over MCOF-Ti6Cu3. Reproduced from ref. 163 with permission from Springer Nature, Copyright 2022. | ||
However, in many cases, additional sacrificial agents, photosensitizers, and cocatalysts are required to enhance the photocatalytic CO2 conversion ability of crystalline COFs. Fu et al. synthesized a bipyridine-containing crystalline sp2c-COF, which was modified by a rhenium complex to improve its photocatalytic CO2 reduction performance. In this process, dye-sensitization played a critical role, enhancing the CO2 conversion capacity by 84% with a CO productivity of 1040 μmol g−1 h−1 over 17.5 h illumination. The transient absorption spectra (TA) (Fig. 15c and d) combined with the lifetime increase in the ground state bleach showed the formation of a long-lived charge separation state in Re-sp2c-COF. This also proved the high photocatalytic activity of the catalyst. Compared with the highly crystalline Re-sp2c-COF, the CO2 reduction activity of the analogous amorphous material was significantly reduced, which further confirmed the huge advantage of highly crystallized materials.160 Yu et al. used the twisted binding of hexabenzocoronene (HBC) to form a dioxin-linked super stable two-dimensional high-crystalline COF (denoted as HBC-TFPN) with a wavy lattice. Increasing the crystallinity of covalent organic frame photocatalysts is beneficial to improve their photocatalytic activity. The results showed that in the presence of a photosensitizer and electron donor, the CO yield reached 7.7 μmol g−1 h−1 and the selectivity was as high as 92%.161 Chakrabortty and colleagues synthesized recyclable crystalline ordered COFs based on triazine (TAPT) via the combined copolymerization of 4,4′-biphenyldicarbaldehyde and a triamine 1,3,5-tris-(4-aminophenyl) triazine. This highly crystalline organic polymer with excellent specific surface area and active sites could efficiently convert CO2 to methanol under the action of triethylamine electron donor and NiO cocatalyst (Fig. 15e). According to the energy calculation in the CO2 reduction process (Fig. 15f and g), the stable intermediate HCOO* is formed by the hydrogenation of CO2. The highest reaction energy of formic acid hydrogenation is the rate-determining step, and then the dissociated intermediate (H2COOH*) is converted into hydroxyl (*OH) and formaldehyde (formaldehyde). Then, by further intermediate addition (H*), this process occurs until methanol is produced.162 In the study of Huang et al., they prepared highly crystalline COFs (PdxIny@N3-COF), which limited the growth of PdIn nanoclusters. The highly conjugated structure of the highly crystalline COFs promotes carrier separation and the bimetallic synergism stabilizes the C1 intermediate, which is more conducive to the formation of C2+ products. Therefore, the final products of photocatalytic CO2 conversion were mainly multi-carbon alcohols (Fig. 15h).71 Zhou et al. prepared a crystalline skeleton material of heterometallic REDOX clusters based on a covalent organic framework, MCOF-Ti6Cu3, which effectively coupled photocatalytic CO2 reduction and water oxidation. The electron transfer process during the photocatalytic reaction was confirmed by in situ XPS test. As shown in Fig. 15i and j, upon exposure to illumination, the binding energy of Ti 2p increases, while that of Cu 2p decreases. More importantly, the binding energy changes to its initial value after CO2 is introduced, indicating that some of the electrons flow to the Cu cluster under photoexcitation and can undergo CO2 reduction reaction on the Cu cluster. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTs) (Fig. 15k) further elucidated the reason for its good photocatalytic performance and formic acid conversion selectivity. With an increase in the light exposure time, the *HCOOH peak gradually increased, which is an important intermediate in the formation of HCOOH. This photocatalyst could convert CO2 to HCOOH with high selectivity.163
5.3 Crystalline CTFs for photocatalytic CO2 reduction
Nitrogen-rich crystalline CTFs are promising photocatalytic CO2 conversion catalysts due to their high specific surface area, adjustable pore size and designable surface active sites. For instance, Huang et al. reported the preparation of a new catalyst via the photo-deposition of Pt on the surface of ethylene glycol (EG)-modified CTFs (PT-SA/CTF-1) (Fig. 16a). WT-EXAFS (Fig. 16b) further revealed the local coordination environment of the Pt-SA/CTF-1 catalyst, and the Pt–N structure provided the active site for photocatalytic CO2 reduction. CTF-1 is a crystalline material with a large specific surface area, which provides a scaffold for CO2 capture and activation. The modification of Pt single atoms not only improved the adsorption and activation of CO2, but also accelerated the separation and transfer of photogenerated carriers in CTF-1, which enabled efficient photocatalytic CO2 methanation. Consequently, the yield of methane, as the main product, was about 4.5 μmol g−1 h−1 (Fig. 16c).164 Moreover, He et al. reported a metal-free 2D/2D CN/CTF heterojunction with a long-range polymer framework for the highly selective conversion of CO2 to CO, reaching 151.1 μmol g−1 h−1 with a 30 h stabilization time (Fig. 16d). The enhanced photocatalytic activity is mainly attributed to the interaction of the two catalysts to generate a large number of photogenerated electrons, which also reduces the probability of charge recombination.145 | ||
| Fig. 16 (a) Schematic illustration of the photocatalytic CO2 reduction over Pt-SA/CTF-1 under visible light. Reproduced from ref. 164 with permission from Elsevier, Copyright 2021. (b) 3D contour WTEXAFS plots of Pt L3-edge EXAFS signals in Pt-SA/CTF-1 sample, PtO2 and Pt foil. Reproduced from ref. 164 with permission from Elsevier, Copyright 2021. (c) CO2 reduction performance under various conditions of Pt-SA/CTF-1. Reproduced from ref. 164 with permission from Elsevier, Copyright 2021. (d) Comparison of CO yield for CO2 reduction under 200 mW cm−2 visible light within 3 h with blank, CTF, CN, CN/CTF in acetonitrile/Co(bpy)3Cl2 TEOA system. Reproduced from ref. 145 with permission from the Chinese Academy of Sciences, Copyright 2022. | ||
5.4 Other crystalline organic polymeric materials for photocatalytic CO2 reduction
In addition to the above-mentioned high-crystalline g-C3N4, COFs and CIFs, other high-crystalline organic polymeric materials have been reported for the photocatalytic conversion of CO2. Verma et al. synthesized crystalline polymeric gels for photocatalytic CO2 reduction via amide bond binding of tetrathiafulvalene (TTF) and terpyridine (TPY) derivatives.170 The ordered crystal structure led to an increase in π–π stacking between the TTF and TPY molecules, which enhanced the visible light absorption range and promoted the photocatalytic CO2 reduction.6. Conclusions and outlook
Photocatalytic CO2 conversion is increasingly becoming an effective way to convert waste carbon into high value-added clean energy and fuels, as well as a valid way to achieve carbon neutrality. In this process, compared with traditional inorganic semiconductor materials, high-crystalline conjugated organic polymeric materials (e.g., g-C3N4, COFs, and CTFs) exhibit various advantages of tunable molecular or electronic structure, adjustable band gap by controlling different monomers, excellent visible light response, and unique properties of photocarrier separation and transport enabled by their π-conjugated stacking structures. Recently, an increasing number of high-crystalline organic polymeric materials are being reported for photocatalytic reactions, such as water splitting for H2-evolution and CO2 reduction. Compared with H2-evolution, photocatalytic CO2 conversion is still in its infancy, and thus relatively less reported, but is attracting interest from a large number of researchers. In this review, we summarized the recent advances of high-crystalline conjugated organic polymeric materials in the conversion of CO2 into high value-added chemical fuels via photocatalytic technology. Especially, the advantages of high-crystalline conjugated organic polymeric materials including absence of defects, enhanced built-in electric field, reduced interlayer hydrogen bonding, and crystal plane regulation were highlighted. The methods for the synthesis of crystalline g-C3N4, COFs, and CTFs were elaborated, such as molten-salt method, solid salt template method, microwave-assisted method, superacid-catalyzed strategy, and in situ oxidation. Subsequently, a series of methods for the modification of these materials was summarized to enhance their photocatalytic activities, which can give readers some reference and guidance. Finally, the applications of high-crystalline materials in photocatalytic CO2 conversion were presented in detail.Although some progress has been made in the preparation and application of highly crystalline conjugated organic polymeric materials, the related development in this field is still in its infancy, and the reported photocatalytic efficiencies are far from practical requirements. Some challenges and problems are faced in this research direction, which also shows great potential and prospects in many aspects:
(1) The current research is still focused on improving the yields of CO2 conversion products, while there is a lack of in-depth research on product selectivity, conversion efficiency (such as turnover number (TON) and turnover frequency (TOF)), and solar energy utilization. In the case of the diversity of products (e.g., CO, CH4, CH3OH, HCOOH, HCHO, C2H5OH, and C2H4), the rational design of photocatalysts is critical to improve their selectivity. Furthermore, the photoenzyme synergistic catalysis reaction that mimics plant photosynthesis is also a new approach for high-selectivity CO2 conversion, in which the specificity and high activity of the enzyme can be combined.
(2) At present, the products of photocatalytic CO2 conversion are still mainly C1 derivatives, the most common ones are CO, CH4 and HCOOH, mainly because these reaction processes are easier to realize. However, there are very few reports on higher value-added C2 (e.g., C2H5OH, C2H4, and C2H6) and multi-carbon (e.g., polycarbon alcohols and polycarbon hydrocarbons) products, which have been achieved by methods such as electrocatalysis and thermocatalysis. More excitedly, some carbon and nitrogen compounds (such as urea and amino acids), which are the basic substances of life metabolism, are also expected to be realized through complex processes such as CO2 conversion, C–C bond coupling, and C–N bond coupling. Therefore, various valuable carbon-containing products will be expected to be achieved by photocatalytic CO2 conversion.
(3) To realize the practical application of the photocatalytic conversion of CO2 to high-value products and overcome the limitation of sunlight by alternating night-and-day, the combination of photocatalytic technology with solar photovoltaic, electrocatalysis and other technologies may be a meaningful way. Moreover, in this process, it is also very important for practical application to get rid of the dependence on cocatalysts, sacrificial agents, photosensitizers and organic solvents.
(4) Compared with crystalline g-C3N4, the species of crystalline COFs and CTFs are very few, and thus there is an urgent need to develop and prepare efficient crystalline COF and CTF photocatalysts for CO2 reduction, especially organic catalysts with spatially separated redox centers. In addition, the stability of high-crystalline conjugated organic polymeric materials need to be further improved.
(5) To deeply explore the molecular structure of high-crystalline conjugated organic polymeric catalysts and real-time tracking of their surface species changes during the photocatalytic CO2 conversion process to reveal the reaction mechanism, many advanced electronic and spectroscopic tools (e.g., synchrotron radiation-based XAFS spectroscopy, aberration-corrected scanning transmission electron microscopy, femtosecond transient absorption spectrometry (fs-TAS), and time-resolved fluorescence spectroscopy), in situ characterization techniques (e.g., in situ Raman, FTIR, and XPS) and DFT theoretical calculation are necessary.
In conclusion, high-crystalline conjugated organic polymeric materials have been proven to be excellent catalysts for photocatalytic CO2 conversion. Highly crystalline organic polymers will also be attractive for practical applications, which can provide some guidance to alleviate the environmental and energy issues. Combined with the in-depth understanding of the structure, properties and modification methods of high-crystalline organic polymers through more theoretical and experimental studies, more efficient high-crystalline organic polymers will be developed for photocatalytic CO2 reduction in the future.
Author contributions
F. Y. wrote the draft of the manuscript, organized the figures, and selected the appropriate references. J. Q. and Y. Z. conceived the outline of the study. Y. C., X. Y. and C. M. Li revised the manuscript. J. H. conceived the theme and the outline of this manuscript, directed the research program, and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support provided by National Natural Science Foundation of China (22008163), Postgraduate Research & Practice Innovation Program of Jiangsu Province, Natural Science Research Project of Higher Education Institutions in Jiangsu Province (20KJB150042 and 21KJB150038), Natural Science Foundation of Jiangsu Province (BK20210867), Doctor Project of Mass Entrepreneurship and Innovation in Jiangsu Province.References
- M. Pera-Titus, Chem. Rev., 2014, 114, 1413–1492 CrossRef CAS
.
- I. Sullivan, A. Goryachev, I. A. Digdaya, X. Li, H. A. Atwater, D. A. Vermaas and C. Xiang, Nat. Catal., 2021, 4, 952–958 CrossRef CAS
- S. Sgouridis, M. Carbajales-Dale, D. Csala, M. Chiesa and U. Bardi, Nat. Energy, 2019, 4, 456–465 CrossRef CAS
- H. Zhou, Y. Zhang, W. Chen, W. Zhang and X. Lu, Asian J. Org. Chem., 2022, 11, 202100270 Search PubMed
- P. Li, J. Bi, J. Liu, Q. Zhu, C. Chen, X. Sun, J. Zhang and B. Han, Nat. Commun., 2022, 13, 1–9 Search PubMed
- K. M. Lee, J. H. Jang, M. Balamurugan, J. E. Kim, Y. I. Jo and K. T. Nam, Nat. Energy, 2021, 6, 733–741 CrossRef CAS
- E. Nikoloudakis, I. Lopez-Duarte, G. Charalambidis, K. Ladomenou, M. Ince and A. G. Coutsolelos, Chem. Soc. Rev., 2022, 51, 6965–7045 RSC
- J. Li, H. Huang, W. Xue, K. Sun, X. Song, C. Wu, L. Nie, Y. Li, C. Liu, Y. Pan, H. Jiang, D. Mei and C. Zhong, Nat. Catal., 2021, 4, 719–729 CrossRef CAS
- J. Hou, G. Dong, B. Xiao, C. Malassigne and V. Chen, J. Mater. Chem. A, 2015, 3, 3332–3342 RSC
- Z. Li, Y. Qu, J. Wang, H. Liu, M. Li, S. Miao and C. Li, Joule, 2019, 3, 570–583 CrossRef CAS
- P. Zhang, J. Hu, Y. Shen, X. Yang, J. Qu, F. Du, W. Sun and C. Li, ACS Appl. Mater. Interfaces, 2021, 13, 46650–46658 CrossRef CAS PubMed
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed
- J. Hu, C. Chen, Y. Zheng, G. Zhang, C. Guo and C. Li, Small, 2020, 16, 2002988 CrossRef CAS
- Y. Shao, J. Hu, T. Yang, X. Yang, J. Qu, Q. Xu and C. Li, Carbon, 2022, 190, 337–347 CrossRef CAS
- C. Chen, J. Hu, X. Yang, T. Yang, J. Qu, C. Guo and C. Li, ACS Appl. Mater. Interfaces, 2021, 13, 20162–20173 CrossRef CAS
- H. Zhang, Y. Li, J. Wang, N. Wu, H. Sheng, C. Chen and J. Zhao, Appl. Catal., B, 2021, 284, 119692 CrossRef CAS
- Y. Feng, C. Wang, P. Cui, C. Li, B. Zhang, L. Gan, S. Zhang, X. Zhang, X. Zhou, Z. Sun, K. Wang, Y. Duan, H. Li, K. Zhou, H. Huang, A. Li, C. Zhuang, L. Wang, Z. Zhang and X. Han, Adv. Mater., 2022, 34, 2109074 CrossRef CAS
- Y. Pan, Y. You, S. Xin, Y. Li, G. Fu, Z. Cui, Y. Men, F. Cao, S. Yu and J. B. Goodenough, J. Am. Chem. Soc., 2017, 139, 4123–4129 CrossRef CAS PubMed
- S. Yin, X. Zhao, E. Jiang, Y. Yan, P. Zhou and P. Huo, Energy Environ. Sci., 2022, 15, 1556–1562 RSC
- A. Sabbah, I. Shown, M. Qorbani, F. Fu, T. Lin, H. Wu, P. Chung, C. Wu, S. R. M. Santiago, J. Shen, K. Chen and L. Chen, Nano Energy, 2022, 93, 106809 CrossRef CAS
- Y. Cao, R. Zhang, T. Zhou, S. Jin, J. Huang, L. Ye, Z. Huang, F. Wang and Y. Zhou, ACS Appl. Mater. Interfaces, 2020, 12, 9935–9943 CrossRef CAS PubMed
- R. Shankar, M. Sachs, L. Francàs, D. Lubert-Perquel, G. Kerherve, A. Regoutz and C. Petit, J. Mater. Chem. A, 2019, 7, 23931–23940 RSC
- X. Shi, X. A. Dong, Y. He, P. Yan and F. Dong, Sci. Bull., 2022, 67, 1137–1144 CrossRef CAS
- X. Chen and A. Selloni, Chem. Rev., 2014, 114, 9281–9282 CrossRef CAS PubMed
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef PubMed
- D. Zhao, Y. Wang, C.-L. Dong, Y.-C. Huang, J. Chen, F. Xue, S. Shen and L. Guo, Nat. Energy, 2021, 6, 388–397 CrossRef CAS
- J. Hu, P. Zhang, T. Yang, Y. Cai, J. Qu and X. Yang, Appl. Surf. Sci., 2022, 576, 151841 CrossRef CAS
- W. Zhang, L. Chen, S. Dai, C. Zhao, C. Ma, L. Wei, M. Zhu, S. Chong, H. Yang, L. Liu, Y. Bai, M. Yu, Y. Xu, X. Zhu, Q. Zhu, S. An, R. S. Sprick, M. A. Little, X. Wu, S. Jiang, Y. Wu, Y. Zhang, H. Tian, W. Zhu and A. I. Cooper, Nature, 2022, 604, 72–79 CrossRef CAS PubMed
- C. Wu, Z. Teng, C. Yang, F. Chen, H. B. Yang, L. Wang, H. Xu, B. Liu, G. Zheng and Q. Han, Adv. Mater., 2022, 34, 2110266 CrossRef CAS
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., Int. Ed., 2013, 52, 1432–1436 CrossRef CAS
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS
- L. Li, Z. Cai, Q. Wu, W.-Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef CAS
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS
- F. He, Z. Wang, Y. Li, S. Peng and B. Liu, Appl. Catal., B, 2020, 269, 118828 CrossRef CAS
- Z. Xie, B. Wang, Z. Yang, X. Yang, X. Yu, G. Xing, Y. Zhang and L. Chen, Angew. Chem., Int. Ed., 2019, 58, 15742–15746 CrossRef CAS PubMed
- P. Puthiaraj, Y.-R. Lee, S. Zhang and W.-S. Ahn, J. Mater. Chem. A, 2016, 4, 16288–16311 RSC
- Z. Wang, Y. Luo, T. Hisatomi, J. J. M. Vequizo, S. Suzuki, S. Chen, M. Nakabayashi, L. Lin, Z. Pan, N. Kariya, A. Yamakata, N. Shibata, T. Takata, K. Teshima and K. Domen, Nat. Commun., 2021, 12, 1005 CrossRef CAS PubMed
- L. Lin, Z. Lin, J. Zhang, X. Cai, W. Lin, Z. Yu and X. Wang, Nat. Catal., 2020, 3, 649–655 CrossRef CAS
- H. Zhang, X. Chen, Z. Zhang, K. Yu, W. Zhu and Y. Zhu, Appl. Catal., B, 2021, 287, 119957 CrossRef CAS
- W. Iqbal, B. Qiu, Q. Zhu, M. Xing and J. Zhang, Appl. Catal., B, 2018, 232, 306–313 CrossRef CAS
- X. Lin, H. Du, D. Jiang, P. Zhang, Z. Yu, H. Bi and Y. Yuan, J. Energy Chem., 2022, 65, 541–547 CrossRef CAS
- J. Xiao, X. Liu, L. Pan, C. Shi, X. Zhang and J.-J. Zou, ACS Catal., 2020, 10, 12256–12283 CrossRef CAS
- M. Mansha, T. Ahmad, N. Ullah, S. A. Khan, M. Ashraf, S. Ali, B. Tan and I. Khan, Chem. Rec., 2022, 22, e202100336 CAS
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS
- E. Wirnhier, M. Doblinger, D. Gunzelmann, J. Senker, B. V. Lotsch and W. Schnick, Chemistry, 2011, 17, 3213–3221 CrossRef CAS
- L. Lin, H. Ou, Y. Zhang and X. Wang, ACS Catal., 2016, 6, 3921–3931 CrossRef CAS
- Y. Xu, X. He, H. Zhong, D. J. Singh, L. Zhang and R. Wang, Appl. Catal., B, 2019, 246, 349–355 CrossRef CAS
- M. J. Bojdys, J.-O. Muller, M. Antonietti and A. Thomas, Chem. – Eur. J., 2008, 14, 8177–8182 CrossRef CAS PubMed
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 17030 CrossRef CAS
- M. Liu, C. Wei, H. Zhuzhang, J. Zhou, Z. Pan, W. Lin, Z. Yu, G. Zhang and X. Wang, Angew. Chem., Int. Ed., 2022, 61, 202113389 Search PubMed
- Y. Li, D. Zhang, X. Feng and Q. Xiang, Chin. J. Catal., 2020, 41, 21–30 CrossRef CAS
- M. K. Bhunia, K. Yamauchi and K. Takanabe, Angew. Chem., Int. Ed., 2014, 53, 11001–11005 CrossRef CAS
- E. Kroke, M. Schwarz, E. Horath-Bordon, P. Kroll, B. Noll and A. D. Norman, New J. Chem., 2002, 26, 508–512 RSC
- J. Gracia and P. Kroll, J. Mater. Chem., 2009, 19, 3013 RSC
- D. Dontsova, S. Pronkin, M. Wehle, Z. Chen, C. Fettkenhauer, G. Clavel and M. Antonietti, Chem. Mater., 2015, 27, 5170–5179 CrossRef CAS
- J. Hu, C. Chen, H. Yang, F. Yang, J. Qu, X. Yang, W. Sun, L. Dai and C. Li, Appl. Catal., B, 2022, 317, 121723 CrossRef CAS
- C. Qiu, Y. Xu, X. Fan, D. Xu, R. Tandiana, X. Ling, Y. Jiang, C. Liu, L. Yu, W. Chen and C. Su, Adv. Sci., 2019, 6, 1801403 CrossRef PubMed
- J. Yuan, Y. Tang, X. Yi, C. Liu, C. Li, Y. Zeng and S. Luo, Appl. Catal., B, 2019, 251, 206–212 CrossRef CAS
- G. Zhang, J. Zhu, Y. Xu, C. Yang, C. He, P. Zhang, Y. Li, X. Ren and H. Mi, ACS Catal., 2022, 12, 4648–4658 CrossRef CAS
- T. Huo, Q. Deng, F. Yu, G. Wang, Y. Xia, H. Li and W. Hou, ACS Appl. Mater. Interfaces, 2022, 14, 13419–13430 CrossRef CAS PubMed
- F. Guo, B. Hu, C. Yang, J. Zhang, Y. Hou and X. Wang, Adv. Mater., 2021, 33, 2101466 CrossRef CAS PubMed
- T. Yang, Y. Shao, J. Hu, J. Qu, X. Yang, F. Yang and C. Li, Chem. Eng. J., 2022, 448, 137613 CrossRef CAS
- Y. Yuan, L. Yin, S. Cao, L. Gu, G. Xu, P. Du, H. Chai, Y. Liao and C. Xue, Green Chem., 2014, 16, 4663–4668 RSC
- L. Lin, P. Ye, C. Cao, Q. Jin, G. Xu, Y. Shen and Y. Yuan, J. Mater. Chem. A, 2015, 3, 10205–10208 RSC
- Q. Liu, X. Wang, Q. Yang, Z. Zhang and X. Fang, Appl. Catal., B, 2018, 225, 22–29 CrossRef CAS
- X. Li, J. Zhang, X. Chen, A. Fischer, A. Thomas, M. Antonietti and X. Wang, Chem. Mater., 2011, 23, 4344–4348 CrossRef CAS
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed
- Q. Yang, M. Luo, K. Liu, H. Cao and H. Yan, Appl. Catal., B, 2020, 276, 119174 CrossRef CAS
- F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478–11481 CrossRef CAS PubMed
- Y. Huang, P. Du, W.-X. Shi, Y. Wang, S. Yao, Z.-M. Zhang, T.-B. Lu and X. Lu, Appl. Catal., B, 2021, 288, 120001 CrossRef CAS
- S. Kandambeth, D. B. Shinde, M. K. Panda, B. Lukose, T. Heine and R. Banerjee, Angew. Chem., Int. Ed., 2013, 52, 13052–13056 CrossRef CAS PubMed
- X. Chen, M. Addicoat, E. Jin, L. Zhai, H. Xu, N. Huang, Z. Guo, L. Liu, S. Irle and D. Jiang, J. Am. Chem. Soc., 2015, 137, 3241–3247 CrossRef CAS PubMed
- X. Li, J. Qiao, S. Chee, H. Xu, X. Zhao, H. S. Choi, W. Yu, S. K. Quek, U. Mirsaidov and K. P. Loh, J. Am. Chem. Soc., 2020, 142, 4932–4943 CrossRef CAS PubMed
- C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef CAS PubMed
- N. L. Campbell, R. Clowes, L. K. Ritchie and A. I. Cooper, Chem. Mater., 2009, 21, 204–206 CrossRef CAS
- H. Wei, S. Chai, N. Hu, Z. Yang, L. Wei and L. Wang, Chem. Commun., 2015, 51, 12178–12181 RSC
- R. Matsuoka, R. Sakamoto, K. Hoshiko, S. Sasaki, H. Masunaga, K. Nagashio and H. Nishihara, J. Am. Chem. Soc., 2017, 139, 3145–3152 CrossRef CAS PubMed
- K. Dey, M. Pal, K. C. Rout, H. S. Kunjattu, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 13083–13091 CrossRef CAS PubMed
- D. D. Medina, J. M. Rotter, Y. Hu, M. Dogru, V. Werner, F. Auras, J. T. Markiewicz, P. Knochel and T. Bein, J. Am. Chem. Soc., 2015, 137, 1016–1019 CrossRef CAS PubMed
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 5328–5331 CrossRef CAS PubMed
- G. Li, J. Ye, Q. Fang and F. Liu, Chem. Eng. J., 2019, 370, 822–830 CrossRef CAS
- W. Zhao, P. Yan, H. Yang, M. Bahri, A. M. James, H. Chen, L. Liu, B. Li, Z. Pang, R. Clowes, N. D. Browning, J. W. Ward, Y. Wu and A. I. Cooper, Nat. Synth., 2022, 1, 87–95 CrossRef
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed
- M. J. Bojdys, J. Jeromenok, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 2202–2205 CrossRef CAS PubMed
- Z.-A. Lan, M. Wu, Z. Fang, Y. Zhang, X. Chen, G. Zhang and X. Wang, Angew. Chem., Int. Ed., 2022, 61, 202201482 Search PubMed
- Z. Yang, H. Chen, S. Wang, W. Guo, T. Wang, X. Suo, D. E. Jiang, X. Zhu, I. Popovs and S. Dai, J. Am. Chem. Soc., 2020, 142, 6856–6860 CrossRef CAS PubMed
- J. Liu, P. Lyu, Y. Zhang, P. Nachtigall and Y. Xu, Adv. Mater., 2018, 30, 1705401 CrossRef PubMed
- K. Wang, L. Yang, X. Wang, L. Guo, G. Cheng, C. Zhang, S. Jin, B. Tan and A. Cooper, Angew. Chem., Int. Ed., 2017, 56, 14149–14153 CrossRef CAS PubMed
- V. Nguyen and M. Grunwald, J. Am. Chem. Soc., 2018, 140, 3306–3311 CrossRef CAS PubMed
- J. Liu, M. Liu, X. Wang, X. Wang and B. Tan, Small, 2022, 18, 2200984 CrossRef CAS PubMed
- M. Liu, Q. Huang, S. Wang, Z. Li, B. Li, S. Jin and B. Tan, Angew. Chem., Int. Ed., 2018, 57, 11968–11972 CrossRef CAS PubMed
- S.-Y. Yu, J. C. Kim, H.-J. Noh, Y.-K. Im, J. Mahmood, I.-Y. Jeon, S. K. Kwak and J.-B. Baek, Cell Rep. Phys. Sci., 2021, 2, 100653 CrossRef CAS
- S.-Y. Yu, J. Mahmood, H.-J. Noh, J.-M. Seo, S.-M. Jung, S.-H. Shin, Y.-K. Im, I. I.-Y. Jeon and J.-B. Baek, Angew. Chem., Int. Ed., 2018, 57, 8438–8442 CrossRef CAS PubMed
- S. Chu, Y. Wang, C. Wang, J. Yang and Z. Zou, Int. J. Hydrogen Energy, 2013, 38, 10768–10772 CrossRef CAS
- J. Liu, W. Zan, K. Li, Y. Yang, F. Bu and Y. Xu, J. Am. Chem. Soc., 2017, 139, 11666–11669 CrossRef CAS PubMed
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef
- J. Wang, Z. Yang, W. Yao, X. Gao and D. Tao, Appl. Catal., B, 2018, 238, 629–637 CrossRef CAS
- X. Bu, Y. Bu, S. Yang, F. Sun, L. Tian, Z. Peng, P. He, J. Sun, T. Huang, X. Wang, G. Ding, J. Yang and X. Xie, RSC Adv., 2016, 6, 112210–112214 RSC
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC
- U. Diebold, Nat. Chem., 2011, 3, 271–272 CrossRef CAS PubMed
- J. Ge, J. Zheng, J. Zhang, S. Jiang, L. Zhang, H. Wan, L. Wang, W. Ma, Z. Zhou and R. Ma, J. Mater. Chem. A, 2021, 9, 14432–14443 RSC
- F. Jiang, S. Wang, B. Liu, J. Liu, L. Wang, Y. Xiao, Y. Xu and X. Liu, ACS Catal., 2020, 10, 11493–11509 CrossRef CAS
- M. Dimitrievska, F. Oliva, M. Guc, S. Giraldo, E. Saucedo, A. Pérez-Rodríguez and V. Izquierdo-Roca, J. Mater. Chem. A, 2019, 7, 13293–13304 RSC
- D. Edelberg, D. Rhodes, A. Kerelsky, B. Kim, J. Wang, A. Zangiabadi, C. Kim, A. Abhinandan, J. Ardelean, S. Scully, D. Scullion, L. Embon, R. Zu, E. J. G. Santos, L. Balicas, C. Marianetti, K. Barmak, X. Zhu, J. Hone and A. N. Pasupathy, Nano Lett., 2019, 19, 4371–4379 CrossRef CAS PubMed
- Y. Liu, C. Chen, Y. He, Z. Zhang, M. Li, C. Li, X.-B. Chen and Z. Shi, Small, 2022, 2201556, DOI:10.1002/smll.202201556
- M. Denecke, Coord. Chem. Rev., 2006, 250, 730–754 CrossRef CAS
- C. S. Fadley, J. Electron Spectrosc. Relat. Phenom., 2010, 178–179, 2–32 CrossRef CAS
- M. Kalbac, Y. P. Hsieh, H. Farhat, L. Kavan, M. Hofmann, J. Kong and M. S. Dresselhaus, Nano Lett., 2010, 10, 4619–4626 CrossRef CAS PubMed
- D. A. Muller, Nat. Mater., 2009, 8, 263–270 CrossRef CAS PubMed
- F. Tuomisto and I. Makkonen, Rev. Mod. Phys., 2013, 85, 1583–1631 CrossRef CAS
- G. Gabrielse, D. Hanneke, T. Kinoshita, M. Nio and B. Odom, Phys. Rev. Lett., 2006, 97, 030802 CrossRef CAS PubMed
- N. A. Khan, R. Zhang, X. Wang, L. Cao, C. S. Azad, C. Fan, J. Yuan, M. Long, H. Wu, M. A. Olson and Z. Jiang, Nat. Commun., 2022, 13, 1–8 Search PubMed
- J. Li, L. Cai, J. Shang, Y. Yu and L. Zhang, Adv. Mater., 2016, 28, 4059–4064 CrossRef CAS PubMed
- Y. Guo, W. Shi and Y. Zhu, EcoMat, 2019, 1, 12007 CrossRef
- Z. Zhang, X. Chen, H. Zhang, W. Liu, W. Zhu and Y. Zhu, Adv. Mater., 2020, 32, 1907746 CrossRef CAS PubMed
- J. Li, G. Zhan, Y. Yu and L. Zhang, Nat. Commun., 2016, 7, 11480 CrossRef CAS PubMed
- Y. Guo, S. Huang, Y. Guo, Z. Ye, J. Nan, Q. Zhou and Y. Zhu, Appl. Catal., B, 2022, 312, 121388 CrossRef CAS
- J. Wang, Y. Yu, J. Cui, X. Li, Y. Zhang, C. Wang, X. Yu and J. Ye, Appl. Catal., B, 2022, 301, 120814 CrossRef CAS
- W. Ren, J. Cheng, H. Ou, C. Huang, M. M. Titirici and X. Wang, ChemSusChem, 2019, 12, 3257–3262 CrossRef CAS PubMed
- Y. Wang, M. Sun, W. Tang, Q. Li, Z. Ren, Y. Liu, Y. Zhang, C. Zeng, Z. Wang, Y. Wu, J. Hao, X. Wu and R. Yang, Mater. Today Phys., 2022, 23, 100634 CrossRef CAS
- B. Zhao, D. Gao, Y. Liu, J. Fan and H. Yu, J. Colloid Interface Sci., 2022, 608, 1268–1277 CrossRef CAS PubMed
- X. Liu, B. Jing, G. Lun, Y. Wang, X. Wang, C. Fang, Z. Ao and C. Li, Chem. Commun., 2020, 56, 3179–3182 RSC
- S. Shen, J. Chen, Y. Wang, C. Dong, F. Meng, Q. Zhang, Y. Huangfu, Z. Lin, Y. Huang, Y. Li, M. Li and L. Gu, Sci. Bull., 2022, 67, 520–528 CrossRef CAS
- L. Cheng, P. Zhang, Q. Wen, J. Fan and Q. Xiang, Chin. J. Catal., 2022, 43, 451–460 CrossRef CAS
- S. Cao, H. Li, T. Tong, H.-C. Chen, A. Yu, J. Yu and H. Chen, Adv. Funct. Mater., 2018, 28, 1802169 CrossRef
- J. Zhang, C. Yu, J. Lang, Y. Zhou, B. Zhou, Y. Hu and M. Long, Appl. Catal., B, 2020, 277, 119225 CrossRef CAS
- S. Li, M. Wu, T. Guo, L. Zheng, D. Wang, Y. Mu, Q. Xing and J. Zou, Appl. Catal., B, 2020, 272, 118989 CrossRef CAS
- C. Zhu, Q. Fang, R. Liu, W. Dong, S. Song and Y. Shen, Environ. Sci. Technol., 2022, 56, 6699–6709 CrossRef CAS PubMed
- Y. Chen, X. Yan, J. Xu and L. Wang, J. Mater. Chem. A, 2021, 9, 24056–24063 RSC
- Z. Luo, C. Li, S. Liu, T. Wang and J. Gong, Chem. Sci., 2017, 8, 91–100 RSC
- G. Zhang, Y. Xu, D. Yan, C. He, Y. Li, X. Ren, P. Zhang and H. Mi, ACS Catal., 2021, 11, 6995–7005 CrossRef CAS
- S. Wu, H. Yu, S. Chen and X. Quan, ACS Catal., 2020, 10, 14380–14389 CrossRef CAS
- J. Li, P. Liu, Y. Tang, H. Huang, H. Cui, D. Mei and C. Zhong, ACS Catal., 2020, 10, 2431–2442 CrossRef CAS
- G. Huang, G. Lin, Q. Niu, J. Bi and L. Wu, J. Mater. Sci. Technol., 2022, 116, 41–49 CrossRef
- Z. Mi, T. Zhou, W. Weng, J. Unruangsri, K. Hu, W. Yang, C. Wang, K. A. I. Zhang and J. Guo, Angew. Chem., Int. Ed., 2021, 60, 9642–9649 CrossRef CAS PubMed
- T. Zhou, L. Wang, X. Huang, J. Unruangsri, H. Zhang, R. Wang, Q. Song, Q. Yang, W. Li, C. Wang, K. Takahashi, H. Xu and J. Guo, Nat. Commun., 2021, 12, 3934 CrossRef CAS PubMed
- L. Chen, L. Wang, Y. Wan, Y. Zhang, Z. Qi, X. Wu and H. Xu, Adv. Mater., 2020, 32, 1904433 CrossRef CAS PubMed
- X. Li, J. Hu, T. Yang, X. Yang, J. Qu and L. C, Nano Energy, 2022, 92, 106714 CrossRef CAS
- R. Marschall, Adv. Funct. Mater., 2014, 24, 2421–2440 CrossRef CAS
- L. Zhang, J. Zhang, H. Yu and J. Yu, Adv. Mater., 2022, 34, 2107668 CrossRef CAS PubMed
- H. Li, Y. Zhou, W. Tu, J. Ye and Z. Zou, Adv. Funct. Mater., 2015, 25, 998–1013 CrossRef CAS
- Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6, 1543–1559 CAS
- Y. Li, F. Gong, Q. Zhou, X. Feng, J. Fan and Q. Xiang, Appl. Catal., B, 2020, 268, 118381 CrossRef CAS
- J. He, C. Wang, X. Jin, S. Liu, Q. Zhao and M. Zhu, Chin. J. Catal., 2022, 43, 1306–1315 CrossRef CAS
- Y. Lin, X. Wang, X. Fu and W. Su, J. Mater. Chem. A, 2022, 10, 8252–8257 RSC
- Z. Zeng, H. Yu, X. Quan, S. Chen and S. Zhang, Appl. Catal., B, 2018, 227, 153–160 CrossRef CAS
- Z. Sun, H. Dong, Q. Yuan, Y. Tan, W. Wang, Y. Jiang, J. Wan, J. Wen, J. Yang, J. He, T. Cheng and L. Huang, Chem. Eng. J., 2022, 435, 134865 CrossRef CAS
- N. Xu, Y. Liu, W. Yang, J. Tang, B. Cai, Q. Li, J. Sun, K. Wang, B. Xu, Q. Zhang and Y. Fan, ACS Appl. Energy Mater., 2020, 3, 11939–11946 CrossRef CAS
- L. Cheng, X. Yue, J. Fan and Q. Xiang, Adv. Mater., 2022, 34, 2200929 CrossRef CAS PubMed
- S. Mehla, A. E. Kandjani, R. Babarao, A. F. Lee, S. Periasamy, K. Wilson, S. Ramakrishna and S. K. Bhargava, Energy Environ. Sci., 2021, 14, 320–352 RSC
- Z. Zhang, G. Wen, D. Luo, B. Ren, Y. Zhu, R. Gao, H. Dou, G. Sun, M. Feng, Z. Bai, A. Yu and Z. Chen, J. Am. Chem. Soc., 2021, 143, 6855–6864 CrossRef CAS PubMed
- V. D. Luca, D. Vullo, A. Scozzafava, V. Carginale, M. Rossi, C. T. Supuran and C. Capasso, Bioorg. Med. Chem., 2013, 21, 1465–1469 CrossRef CAS PubMed
- Z. Wen, S. Xu, Y. Zhu, G. Liu, H. Gao, L. Sun and F. Li, Angew. Chem., Int. Ed., 2022, 61, e202201086 CAS
- Y. Wang, Y. Qu, B. Qu, L. Bai, Y. Liu, Z. Yang, W. Zhang, L. Jing and H. Fu, Adv. Mater., 2021, 33, 2105482 CrossRef CAS PubMed
- G. Zhang, G. Li, T. Heil, S. Zafeiratos, F. Lai, A. Savateev, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 3433–3437 CrossRef CAS PubMed
- Y. Xia, Z. Tian, T. Heil, A. Meng, B. Cheng, S. Cao, J. Yu and M. Antonietti, Joule, 2019, 3, 2792–2805 CrossRef CAS
- Y. Li, B. Li, D. Zhang, L. Cheng and Q. Xiang, ACS Nano, 2020, 14, 10552–10561 CrossRef CAS PubMed
- M. Lu, J. Liu, Q. Li, M. Zhang, M. Liu, J. Wang, D. Yuan and Y. Lan, Angew. Chem., Int. Ed., 2019, 58, 12392–12397 CrossRef CAS PubMed
- Z. Fu, X. Wang, A. M. Gardner, X. Wang, S. Y. Chong, G. Neri, A. J. Cowan, L. Liu, X. Li, A. Vogel, R. Clowes, M. Bilton, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 543–550 RSC
- H.-Y. Yu, J.-S. Wang, F.-Y. Xie, Q. Yang, Y. Chen, L. Zhao, Y. Li and W.-J. Ruan, Chem. Eng. J., 2022, 445, 136713 CrossRef CAS
- P. Chakrabortty, S. Ghosh, A. Das, A. Khan and S. M. Islam, Catal. Sci. Technol., 2022, 12, 3484–3497 RSC
- J. Zhou, J. Li, L. Kan, L. Zhang, Q. Huang, Y. Yan, Y. Chen, J. Liu, S. L. Li and Y. Q. Lan, Nat. Commun., 2022, 13, 4681 CrossRef CAS PubMed
- G. Huang, Q. Niu, J. Zhang, H. Huang, Q. Chen, J. Bi and L. Wu, Chem. Eng. J., 2022, 427, 131018 CrossRef CAS
- J. Hu, T. Yang, X. Yang, J. Qu, Y. Cai and C. Li, Small, 2022, 18, 2105376 CrossRef CAS PubMed
- H. Li, B. Zhu, S. Cao and J. Yu, Chem. Commun., 2020, 56, 5641–5644 RSC
- P. Xia, M. Antonietti, B. Zhu, T. Heil, J. Yu and S. Cao, Adv. Funct. Mater., 2019, 29, 1900093 CrossRef
- J. Liu, H. Shi, Q. Shen, C. Guo and G. Zhao, Green Chem., 2017, 19, 5900–5910 RSC
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS PubMed
- P. Verma, A. Singh, F. A. Rahimi, P. Sarkar, S. Nath, S. K. Pati and T. K. Maji, Nat. Commun., 2021, 12, 1–14 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2022 |