
Photoactive perylenediimide metal–organic framework for boosting iodoperfluoroalkylation of alkenes and oxidative coupling of amines†
Zhengfen
Liu
,
Chao
Li
,
Jian
Chen
,
Xiaobo
Li
,
Fumang
Luo
,
Feixiang
Cheng
 * and
Jian-Jun
Liu
*
* and
Jian-Jun
Liu
*
College of Chemistry and Environmental Science, Qujing Normal University, Qujing 655011, China. E-mail: chengfx2019@163.com; jjliu302@163.com
First published on 5th November 2021
Abstract
The incorporation of photoactive units into metal–organic frameworks (MOFs) is of particular interest due to their ability to endow the MOFs with additional properties such as photomagnetism, photomodulated electrical conductivity, and photocatalytic properties. Perylenediimides (PDIs), as a class of organic dye molecules with strong visible-light absorption ability and high photochemical activity, have attracted considerable interest because of their consecutive photoinduced electron transfer processes for the generation of energetic PDI radical anions. In this context, a novel photoactive MOF based on a perylenediimide derivative, Zn2(diPyPI-Cl4)(NDC)2·3DMF (MOF 1) (diPyPI-Cl4 = N,N′-bis(pyridyl)-1,6,7,12-tetrachloroperylene-3,4,9,10-tetracarboxy bisimide, H2NDC = 2,6-naphthalenedicarboxylic acid), has been successfully constructed, which exhibits a porous three-dimensional framework with a pcu topology. MOF 1 that acts as an efficient and recyclable electron-transfer photocatalyst is applied for boosting the iodoperfluoroalkylation of alkenes and oxidation of amines to imines under mild conditions with high yields and excellent substrate application ranges. This photocatalyst has the advantages of high efficiency, low catalytic loading and easy availability, which is highly feasible from a synthetic point of view.
Introduction
Perfluoroalkylation reactions have received significant interest in recent years because the introduction of fluorine-containing groups can prominently change the physicochemical features of organic molecules.1–4 Therefore, perfluoroalkyl compounds are important organic molecules in agrochemistry, medicinal chemistry, and materials science.5–9 Accordingly, an extensive amount of effort has pursued practical and efficient fluorination and perfluoroalkylation. Iodoperfluoroalkylation of alkenes is an ideal strategy for synthesizing this class of compounds because they could be easily transformed into high-value products and more complex molecules by employing iodine atoms due to their ability to act as superb leaving groups.10–14 Over the past decades, some reported synthetic methods for the iodoperfluoroalkylation of alkenes include the atom transfer radical addition (ATRA) reaction between perfluoroalkyl iodides and alkenes using both photochemical and thermal activation.15–19 However, homolytic cleavage of the C–I bond in perfluoroalkyl iodides as being responsible for the perfluoroalkyl radical formation requires high temperatures and/or potentially explosive radical initiators and/or UV-light irradiation, which cause energy waste and environmental pollution. Although some relatively mild photocatalysts such as Ir/Ru/Cu noble-metal complexes have been employed in iodoperfluoroalkylation,20–23 issues such as high cost, often in a relatively high loading and under UV irradiation, and use of a significant excess of the perfluoroalkyl iodide reagent severely impede their extensive application in photocatalytic organic synthesis.In the past years, some organic photocatalysts for the iodoperfluoroalkylation of alkenes have been proposed to develop green catalytic processes to resolve the above problems.24–27 These organic photocatalysts have to be inexpensive, efficient, less toxic and readily available and should have an extremely low loading. To this end, perylene diimides (PDIs), a class of fluorescent dye molecules with outstanding optical, electronic, and light fastness properties, are promising photoactive materials suitable for the exploitation of novel organic green transformations.28–33 Compared to traditional organic dyes, PDIs with redox-active and strong visible-light absorption ability can undergo consecutive photoinduced electron transfer processes for the generation of energetic PDI radical anions.34–36 These energetic PDI radical anions overcome the energy limitation of visible light and enable the strong chemical bonds to undergo photocatalytic conversion in organic synthesis. König's group reported a significant finding that PDIs as homogeneous photocatalysts can catalyze the reduction of aryl halides under visible light irradiation through consecutive photoinduced electron transfer processes.37 The high planarity and big π-electron-deficient conjugated system result in a strong tendency to aggregate and poor solubility of PDIs, which severely hinder the development of consecutive photoinduced electron transfer processes in other photocatalytic transformations.38,39
In contrast to homogeneous photocatalysts, heterogeneous photocatalysts can be easily separated and recycled in practical applications.40 As a unique and relatively new class of porous hybrid materials, metal–organic frameworks (MOFs) have received considerable interest in heterogeneous photoconversion owing to their high crystallinity and porosity.41–46 Incorporating PDI derivatives into MOFs represents a promising approach for heterogenizing this photocatalysis especially concerning the consecutive photoinduced electron transfer processes and for overcoming the above limitations of PDIs as homogeneous photocatalysts.47,48 Hence, we report the successful incorporation of a PDI derivative ligand within a porous MOF, namely, Zn2(diPyPI-Cl4)(NDC)2·3DMF (MOF 1) (diPyPI-Cl4 = N,N′-bis(pyridyl)-1,6,7,12-tetrachloroperylene-3,4,9,10-tetracarboxy bisimide, H2NDC = 2,6-naphthalenedicarboxylic acid) for enhancing the photocatalytic efficiency. MOF 1 acts as an efficient visible light driven photocatalyst for promoting the iodoperfluoroalkylation of alkenes and oxidative coupling of amines with high yields, excellent substrate application ranges and extremely low catalytic loadings.
Experimental
Materials and measurements
Elemental analysis of CHN was performed on an Elementar Vario EL Cube elemental analyzer. Powder XRD measurements were performed at room temperature on a Bruker D8 ADVANCE X-Ray diffractometer using CuKα radiation in the range of 5–50°. Thermogravimetric analysis (TGA) of the sample was performed on a Mettler Toledo TGA2 thermogravimetric system under a nitrogen atmosphere with a heating rate of 10 °C min−1. The nuclear magnetic resonance (NMR) spectrum was recorded on a Bruker Avance III 400 NMR spectrometer. The Fourier transform infrared (FTIR) spectrum of the sample was taken on a Nicolet iS50 spectrometer.Synthesis of N,N′-bis(pyridyl)-1,6,7,12-tetrachloroperylene-3,4,9,10-tetracarboxy bisimide (diPyPI-Cl4)
4-Aminopyridine (0.534 g, 5.6 mmol) was added to a mixture of 1,6,7,12-tetrachloroperylene-3,4,9,10-tetracarboxylic dianhydride (1.0 g, 1.88 mmol) and pyridine (50 mL) under an argon atmosphere, and the reaction mixture was stirred and heated to reflux overnight. After being cooled to room temperature, the resultant precipitate was filtered and washed with a large amount of water, acetone, and hexanes and allowed to dry in air. The crude product was purified by silica gel column chromatography with dichloromethane/methanol (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as an eluent to yield the diPyPI-Cl4 product as a red solid. 1H NMR (400 MHz, TFA-d): δ 9.05 (d, 4H), 8.80 (s, 4H), 8.30 (d, 4H).
:1) as an eluent to yield the diPyPI-Cl4 product as a red solid. 1H NMR (400 MHz, TFA-d): δ 9.05 (d, 4H), 8.80 (s, 4H), 8.30 (d, 4H).
Synthesis of Zn2(diPyPI-Cl4)(NDC)2·3DMF (1)
A mixture of Zn(NO3)2·6H2O (30 mg, 0.1 mmol), diPyPI-Cl4 (6 mg, 0.008 mmol) and 2,6-naphthalenedicarboxylic acid (H2NDC, 21.6 mg, 0.1 mmol) were dissolved in 6 mL of N,N-dimethylformamide (DMF) solution, and sonicated to create a fine suspension. The resulting reaction mixture was sealed in a scintillation vial and heated to 95 °C. A dark red crystalline material formed within two days. The crystalline solid was removed from the scintillator, isolated by filtration, washed with DMF, and allowed to dry in air. The yield of 1 is ca. 52%. Anal. Calcd for C76H66Cl4N10O18Zn2: C 54.88, H 3.22, N 6.71%. Found: C 55.12, H 3.03, N 6.63%. IR (KBr, cm−1): 3065(w), 2985(w), 2886(w), 1728(s), 1685(s), 1643(s), 1454(m), 1326(s), 1253(s), 1165(m), 786(m), 628(m). Crystal data for MOF 1. C67H45Cl4N7O15Zn2, M = 1460.68, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 13.0525(4) Å, b = 13.1212(5) Å, c = 26.5491(8) Å, α = 95.769(3)°, β = 95.504(3)°, γ = 97.774(3)°, V = 4454.6(3) Å3, Z = 2, Dc = 0.925 g cm−3, F000 = 1248, T = 153(2) K, 2θmax = 60.5°, 31498 reflections collected, 14644 unique (Rint = 0.0300). GoF = 1.126, R1 = 0.0672, wR2 = 0.2050, R indices based on 11282 reflections with I > 2θ.
, a = 13.0525(4) Å, b = 13.1212(5) Å, c = 26.5491(8) Å, α = 95.769(3)°, β = 95.504(3)°, γ = 97.774(3)°, V = 4454.6(3) Å3, Z = 2, Dc = 0.925 g cm−3, F000 = 1248, T = 153(2) K, 2θmax = 60.5°, 31498 reflections collected, 14644 unique (Rint = 0.0300). GoF = 1.126, R1 = 0.0672, wR2 = 0.2050, R indices based on 11282 reflections with I > 2θ.
General procedure for the iodoperfluoroalkylation of alkenes
An oven-dried 10 mL reaction tube equipped with a stir bar was charged with alkenes (0.2 mmol), perfluoroalkyl iodides (0.4 mmol) and MOF 1 (1.24 mg, 0.001 mmol, 0.5 mol%) under an argon atmosphere in a glove box. A solution of sodium ascorbate (19.81 mg, 0.1 mmol) in 1.9 mL of dry CH3CN was added at room temperature. The reaction tube was sealed with a cap and filled with 100 μL of H2O from the cap using a micro-injector. The reaction mixture was stirred at room temperature under a blue light-emitting diode (LED, λ = 450 nm) for 4 hours in total. The reaction opened to air and then extracted with ethyl acetate (3 × 20 mL). The organic phases were combined and washed with brine (10 mL) and then dried over sodium sulfate. The solvent was removed under reduced pressure and the crude material was purified by flash column chromatography to give the corresponding iodoperfluoroalkylation product.General procedure for the oxidative coupling of amines
An oven-dried 10 mL reaction vial equipped with a stir bar was charged with alkenes (0.2 mmol) and MOF 1 (1.24 mg, 0.001 mmol, 0.5 mol%) under an oxygen atmosphere. Anhydrous DMF (2.0 mL) was added to the reaction vial. Then, the reaction mixture was stirred for 6 hours under a 300 W Xe lamp with a cutoff below 420 nm at room temperature. The reaction mixture was opened in air, and the resulting biphasic solution was extracted with ethyl acetate (10 mL × 3). The combined organic layer was washed with brine, dried over Na2SO4, and the combined solutions were concentrated in vacuo. The crude material was loaded onto a deactivated silica gel column and purified by flash chromatography (deactivated silica gel was prepared by the addition of 15 mL of Et3N to 1 L of silica gel).Results and discussion
Due to the strong π–π interactions, perylenediimide is almost insoluble in common organic solvents. For increasing the solubility of the perylenediimide ligand, four chloride atoms were introduced into the bay positions, leading to a noncoplanar conjugate structure, which will increase its solubility for synthesizing MOFs. Similar to the reported procedure, the reaction of diPyPI-Cl4, Zn(NO3)2·6H2O, and 2,6-naphthalenedicarboxylic acid (H2NDC) in DMF solvent afforded MOF 1. The purity of the bulk MOF 1 has been investigated by the PXRD technique. As shown in Fig. S5,† the major peak positions of the synthesized bulk MOF 1 match well with those of the simulated pattern, suggesting the presence of mainly one crystalline phase. Single-crystal X-ray analysis reveals that MOF 1 crystallizes in the triclinic space group P with a three-dimensional framework. The asymmetric unit consists of two Zn(II) cations, one diPyPI-Cl4 ligand, and two deprotonated NDC2− ligands (Fig. 1a). Each of the zinc cations has a slightly distorted square pyramidal coordination environment, with the site being occupied by four carboxylate oxygen atoms from four NDC2− ligands and one nitrogen atom from the diPyPI-Cl4 ligand (dZn–O = 2.017–2.063 Å, dZn–N = 2.033 Å). In the structure of MOF 1, the diPyPI-Cl4 ligand with the twist angles between the two naphthalene units of 39° is found, as a result of the bulky chlorine substituents avoiding steric interaction with one another. Each deprotonated NDC2− ligand acts as a μ4-linker and connects two paddle wheel [Zn2(COO)4] units, while each paddle wheel [Zn2(COO)4] unit is surrounded by four carboxylate groups from four NDC2− ligands with a Zn⋯Zn distance of 2.932 Å. These [Zn2(COO)4] units are bridged by naphthalene of NDC2− ligands, forming a two-dimensional layer with a square grid (Fig. 1b), which are further pillared by diPyPI-Cl4 ligands to generate a porous three-dimensional framework with a pcu topology (Fig. 1c). The single framework leaves enough voids that are filled by mutual interpenetration of another independent equivalent framework in a two-fold interpenetrating structure (Fig. 1d). In spite of the structure feature a two-fold interpenetration with a substantial solvent accessible void volume remained 54% as calculated by the SQUEEZE routine in the PLATON software.49 Face-to-face π⋯π interactions are observed between the NDC2− ligand and phenyl rings of diPyPI-Cl4 ligands (dπ⋯π = 3.519–3.752 Å). This successive π⋯π interaction facilitates the electron-transfer from the electron-rich substrate to the electron deficient diPyPI-Cl4 ligands for photocatalytic reactions.
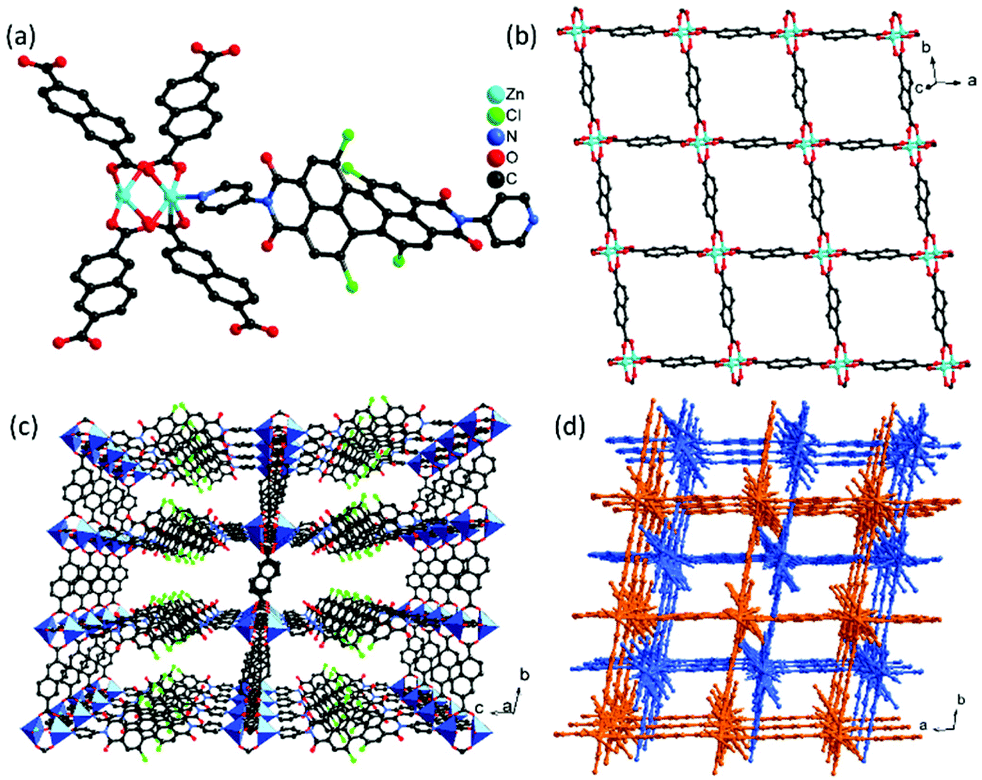 | ||
| Fig. 1 (a) Coordination environment of zinc(II) cations in MOF 1; (b) two-dimensional square grid of the [Zn2(NDC)2]n layer; (c) the pillared-layer three-dimensional framework in MOF 1; (d) view of the two-fold interpenetration in MOF 1 along the c axis. | ||
To examine the thermal stability of MOF 1, TGA was performed under a N2 flow from room temperature to 800 °C with a heating rate of 10 °C min−1. As shown in Fig. S1,† the TGA curve of MOF 1 shows a weight loss of 15.1% at 150 °C, corresponding to the release of free solvent molecules (calculated 14.7%, about 3 DMF molecules). Then almost no weight loss is observed until about 350 °C. After this, the framework and other components also begin to decompose (Fig. S1, ESI†). N2 adsorption–desorption isotherms were measured at 77 K to determine the surface areas (Fig. S4, ESI†), and the result shows an inferior BET surface area of 183 m2 g−1. The UV-Vis diffuse reflectance spectrum of MOF 1 shows a board absorption band at around 350–600 nm, which should be attributed to the π–π* transition of the diPyPI-Cl4 ligands (Fig. S3, ESI†). The band gap energies of MOF 1 is estimated to be 2.06 eV according to the Kubelka–Munk function, indicating that MOF 1 can be photoexcited by visible light irradiation (Fig. S2, ESI†).
As is known, PDIs have many special features such as high stability, excellent optical and redox properties, and so on, so they are utilized as color pigments and fluorescent dyes, and in optoelectronic applications including optical sensors, fluorescent solar light collectors, and optical power limiters.50–53 In spite of their interesting photochemical properties, PDIs have found very few applications in organic photocatalysis. Currently, PDIs have been limited to the photooxidation of benzyl alcohols and the reduction of aryl halides.34,37,47 To explore the photocatalytic activity of MOF 1 for other organic photocatalytic reactions, the iodoperfluoroalkylation of alkenes and oxidative coupling of amines were selected as test models.
In order to explore the photocatalytic performance of MOF 1 as a heterogeneous catalyst in the iodoperfluoroalkylation of alkenes, we first studied the ATRA reaction between alkene 1a and perfluoroalkyl iodide 2a using 0.5 mol% MOF 1 as a catalyst and sodium ascorbate as a reducing agent (0.5 equiv.) that were added to a CH3CN/H2O (95:5) solution at room temperature for 4 h (Table 1). Interestingly, the desired ATRA product 3a was obtained in 98% assay yield (AY, determined by 1H NMR integration against an internal standard, Table 1, entry 1). Subsequently, we conducted a series of controlled experiments to clarify the unique features of this method (entries 2–5, Table 1). First, the reaction cannot occur in the absence of light or the presence of MOF 1 (entries 2–3, Table 1), which indicates that MOF 1 has significant photochemical activity. Next, in the absence of sodium ascorbate and TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl, 1 equiv.), the reaction was completely inhibited. This may be because the excited MOF 1 could not be reduced and the reaction followed a radical mechanism (entries 4–5, Table 1). Finally, the reaction will not react when exposed to air (entry 6, Table 1).
|
|
||
|---|---|---|
| Entry | Deviation from the standard conditions | Yielda |
| a Reactions conducted on a 0.1 mmol scale. Assay yield determined by 1H NMR spectroscopy of the crude reaction mixtures using C2H2Cl4 as an internal standard. b Isolated yield after chromatographic purification. | ||
| 1 | None | >99% (98%)b |
| 2 | In the dark | 0% |
| 3 | No MOF 1 | 0% |
| 4 | No sodium ascorbate | 0% |
| 5 | TEMPO (1 equiv.) | 0% |
| 6 | In air | 0% |

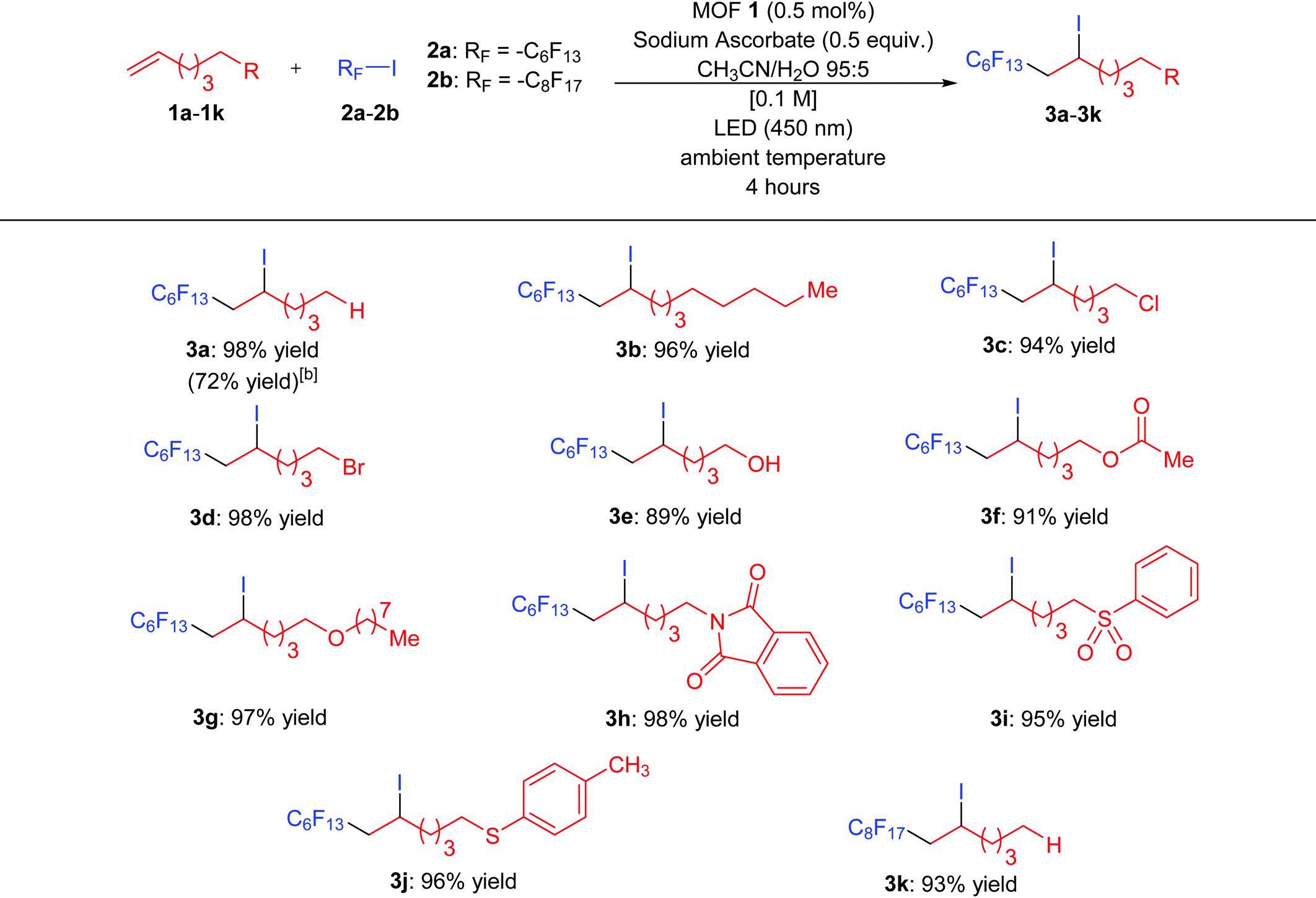
In an effort to explore the scope of this transformation, we first examined alkenes under our optimal conditions (Table 1, entry 1). In general, we found that a wide variety of substrates with alkyl chains and halogens, alcohol, ester, ether, imide and sulfone moieties underwent the iodoperfluoroalkylation process (89–98% isolated yields). The scope of the iodoperfluoroalkylation of alkenes is listed in Table 2. For alkenes bearing alkyl chain (1a) and (1b) substituents, the yields were obtained in 98 and 96%, respectively. The halide substituted alkenes were well tolerated, leading to products 3c and 3d in 94 and 98%. Substrates bearing an alcohol (1e), ester (1f) and ether (1g) furnished products 3e to 3g in 89–98% yields. Interestingly, terminal olefinic compounds containing an aryl group were also good substrates under the iodoperfluoroalkylation conditions, affording products 3h, 3i, and 3j with imide, sulfone, and thioether moieties in 98, 95 and 96% yields, respectively. In addition to the perfluorohexyl chain (2a), a longer perfluorinated substituent (2b) was installed in excellent yields to afford 3k (93% yield). It is not difficult to find that the yield of compound 3e is slightly lower than that of other compounds. The main reason is that a small amount of compound 3e continues to be oxidized into an aldehyde compound. Therefore, our newly developed MOF 1 may have catalytic oxidation performance and will be further explored in the future. Finally, to study the scalability of this method, we performed the transformation of alkene 1a with perfluoroalkyl iodide 2a on a 1 mmol scale under the optimal conditions, the desired product 3a was isolated in 72% yield.
Encouraged by the results with iodoperfluoroalkylation of alkenes, we further explored the oxidative coupling of amines catalyzed by MOF 1 and selected benzylamine as the experimental model substrate. We performed a control experiment to prove that the prerequisite for the oxidative coupling to occur smoothly is that catalyst 1, oxygen and light must exist at the same time (entries 1–3, Table 3). Then, when the reaction was placed at 60 °C under no light conditions, only a trace product was obtained, which indicated that the coupling reaction was a photocatalytic reaction rather than a thermocatalytic reaction (entry 4, Table 3). Surprisingly, in the presence of a heterogeneous photocatalyst MOF 1 and oxygen, using DMF as a solvent, after 6 h of visible light irradiation, the yield of N-benzylidenebenzylamine was as high as 100% assay yield (Table 1, entry 5), and the isolated yield after chromatographic purification was 97%. This result is much higher than the yield (61%) of its equivalent ligand (Table 1, entry 6) which means that MOF 1 not only greatly improves the yield and can be recycled and reused, thereby reducing the reaction cost.
|
|
||
|---|---|---|
| Entry | Conditions | Yielda |
| a Reactions conducted on a 0.1 mmol scale. Assay yield determined by 1H NMR spectroscopy of the crude reaction mixtures using C2H2Cl4 as an internal standard. b Isolated yield after chromatographic purification. | ||
| 1 | No catalyst | 1% |
| 2 | No oxygen | 4% |
| 3 | No light | 0% |
| 4 | No light at 60 °C | 1% |
| 5 | MOF 1 | 100% (97%)b |
| 6 | Equivalent ligand of MOF 1 | 61% |

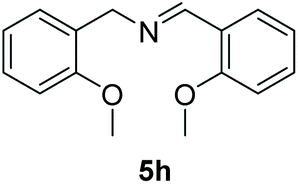
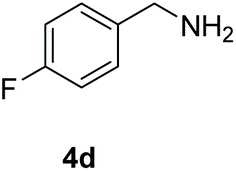
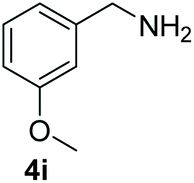
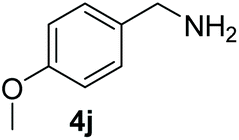
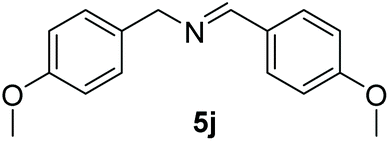
With the optimal conditions in hand (Table 1, entry 5), we studied the oxidative coupling of amines bearing various substituents (Table 4). First of all, for the substrates with electron withdrawing groups (–F, –Cl) in the ortho, meta or para positions, excellent yields (90–98%) of coupling products (5b–5g) were obtained. Next, we tested the amines with electron-donating groups (–OCH3) at the ortho, meta and para positions and obtained yields of 82–91% (5g–5i). To demonstrate the scalability and utility of the oxidative coupling process on a 1 mmol scale under the optimal conditions, the desired product 5a was isolated in 95% yield. Finally, we also explored alkylamine substrates, but unfortunately only traces of oxidative coupling products were obtained. The reason for this result may be that the saturated alkylamine radical cation intermediate generated during the reaction cannot exist stably, and the subsequent process cannot be carried out.






Recyclability, especially of metal–organic framework materials, is critical for achieving the widespread use of photocatalysts. Therefore, the recyclability of MOF 1 as a heterogeneous photocatalyst has also been explored. The used photocatalyst can be easily regenerated by centrifugation, drastically washed with acetonitrile, and used in consecutive runs for three cycles. The results indicated the photocatalytic activity of MOF 1 without significant loss after three photocatalytic cycles (Fig. S6, ESI†). The PXRD patterns demonstrated that MOF 1 maintained well its structure after recycling (Fig. S5, ESI†). These results suggest the good photostability and recyclability of MOF 1 in the photocatalytic system. Although the iodoperfluoroalkylation of alkenes and oxidative coupling of amines have already been reported to be promoted by several expensive Ir/Ru/Cu metal complexes,5 no one exhibited such bifunctional heterogeneous photocatalysis. Considering the possible applications of the perfluoroalkyl compounds and imine derivatives in pharmaceuticals, the photocatalyst MOF 1 is anticipated to open up a synthetic method for such intermediates.
We tried to clarify the photochemical conversion of MOF 1 in these two types of reactions (Scheme 1). MOF 1 was excited to form [MOF 1]* under the radiation of light and then undergoes a single electron transfer process with Asc− and benzylamine (4a) to generate [MOF 1]˙−. On the one hand, [MOF 1]˙− and perfluoroalkyl iodide 2a again underwent a single electron transfer process to produce C6F13˙ and regenerate MOF 1 to complete a photocatalytic cycle. Immediately afterwards, C6F13˙ and alkene 1a underwent an ATRA reaction mechanism, and the expected product 3a was obtained. On the other hand, [MOF 1]˙− also underwent a single electron transfer process with O2 to obtain O2˙−, and regenerate MOF 1 to achieve another photocatalytic cycle. Then, O2˙− interacted with the benzylamine radical cation generated in the previous step to obtain H2O2 and phenylmethanimine. Finally, a molecule of NH3 is removed during the reaction between phenylmethanimine and benzylamine, thereby obtaining the target product 5a. In summary, the heterogeneous MOF 1 plays a very important role in the current photochemical transformation.
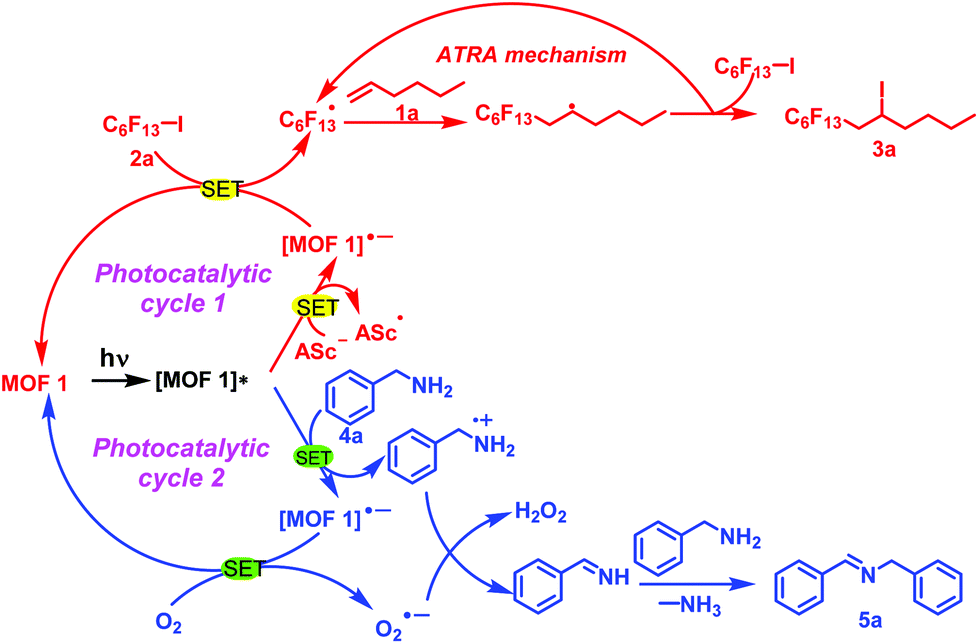 | ||
| Scheme 1 Proposed photochemical conversion mechanism of MOF 1. | ||
Conclusions
In conclusion, a novel photoactive MOF based on a perylenediimide derivative has been successfully synthesized, which possesses a porous three-dimensional framework and pcu topology. MOF 1 acts as an efficient electron-transfer photocatalyst for boosting the iodoperfluoroalkylation of alkenes and photooxidation of amines to imines under mild conditions with high yields and excellent substrate application ranges. The heterogeneous photocatalyst was easily separated and reused in the reaction process, and was recycled without a significant loss of photocatalytic activity. The current photocatalytic system has the advantages of high efficiency, low catalytic loading and easy availability, providing a promising strategy to overcome the obstacles that iodoperfluoroalkylation of alkenes and coupling of amines are currently facing.Author contributions
Zhengfen Liu and Jian-Jun Liu conceived the experiments and studies; Chao Li, Jian Chen, Xiaobo Li and Fumang Luo synthesized the compounds and analyzed the experimental data; Feixiang Cheng contributed to the materials/reagents/analysis tools; Zhengfen Liu and Jian-Jun Liu wrote the original draft; Feixiang Cheng reviewed and supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21961030), the Application Basis Research Project of Yunnan Province Science and Technology Department (202101AU070004), the Yunnan Province Thousand Youth Talents Plan, the Program of Innovative Research Team (in science and technology) in University of Yunnan Province and the Scientific and Technological Innovation Team for Green Catalysis and Energy Material in Yunnan Institutions of Higher Learning.Notes and references
- J. Wang, M. Sànchez-Rosellò, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade, Chem. Rev., 2014, 114, 2432 CrossRef CAS
.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. AceÇa, V. A. Soloshonok, K. Izawa and H. Liu, Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Organic fluorine compounds: a great opportunity for enhanced materials properties, Chem. Soc. Rev., 2011, 40, 3496 RSC
- T. Liang, C. N. Neumann and T. Ritter, Introduction of Fluorine and Fluorine-Containing Functional Groups, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS
- K. Muller, C. Faeh and F. Diederich, Fluorine in Pharmaceuticals: Looking Beyond Intuition, Science, 2007, 317, 1881 CrossRef PubMed
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, Applications of Fluorine in Medicinal Chemistry, J. Med. Chem., 2015, 58, 8315 CrossRef CAS
- T. Fujiwara and D. O'Hagan, Successful fluorine-containing herbicide agrochemicals, J. Fluorine Chem., 2014, 167, 16 CrossRef CAS
- A. C. Stuart, J. R. Tumbleston, H. Zhou, W. Li, S. Liu, H. Ade and W. You, Fluorine Substituents Reduce Charge Recombination and Drive Structure and Morphology Development in Polymer Solar Cells, J. Am. Chem. Soc., 2013, 135, 1806 CrossRef CAS PubMed
- S. Barata-Vallejo, M. V. Cooke and A. Postigo, Fluorine Substituents Reduce Charge Recombination and Drive Structure and Morphology Development in Polymer Solar Cells, ACS Catal., 2018, 8, 7287 CrossRef CAS
- Z.-L. Li, X.-H. Li, N. Wang, N.-Y. Yang and X.-Y. Liu, Radical-Mediated 1,2-Formyl/Carbonyl Functionalization of Alkenes and Application to the Construction of Medium-Sized Rings, Angew. Chem., Int. Ed., 2016, 55, 15100 CrossRef CAS PubMed
- W. R. Dolbier, Structure, Reactivity, and Chemistry of Fluoroalkyl Radicals, Chem. Rev., 1996, 96, 1557 CrossRef CAS PubMed
- Q. Wang, B. Wang, H. Deng, Y. Shangguang, Y. Lin, Y. Zhang, Z. Zhang, Y. Xiao, H. Guo and C. Zhang, Silver-Catalyzed Three-Component Difunctionalization of Alkenes via Radical Pathways: Access to CF3-Functionalized Alkyl-Substituted 1,4-Naphthoquinone Derivatives, J. Org. Chem., 2019, 84, 1006 CrossRef CAS
- D. Zheng and A. Studer, Photoinitiated Three-Component α-Perfluoroalkyl-β-heteroarylation of Unactivated Alkenes via Electron Catalysis, Org. Lett., 2019, 21, 325 CrossRef CAS PubMed
- S. Zhang, L. Li, J. Zhang, J. Zhang, M. Xue and K. Xu, Electrochemical fluoromethylation triggered lactonizations of alkenes under semi-aqueous conditions, Chem. Sci., 2019, 10, 3181 RSC
-
T. Pintauer and K. Matyjaszewski, Encyclopedia of Radicals, Wiley, Hoboken, 2012, vol. 4, pp. 1851–1894 Search PubMed
- J. Banus, H. J. Emeleus and R. N. Haszeldine, Reactions of the perfluoroalkyl radicals, J. Chem. Soc., 1950, 3041 RSC
- A. Bravo, H.-R. Bjørsvik, F. Fontana, L. Liguori, A. Mele and F. Minisci, New Methods of Free-Radical Perfluoroalkylation of Aromatics and Alkenes. Absolute Rate Constants and Partial Rate Factors for the Homolytic Aromatic Substitution by n-Perfluorobutyl Radical, J. Org. Chem., 1997, 62, 7128 CrossRef CAS
- N. O. Brace, Syntheses with perfluoroalkyl radicals from perfluoroalkyl iodides. A rapid survey of synthetic possibilities with emphasis on practical
applications. Part one: alkenes, alkynes and allylic compounds, J. Fluorine Chem., 1999, 93, 1 CrossRef CAS
- S. Barata-Vallejo and A. Postigo, Metal-mediated radical perfluoroalkylation of organic compounds, Coord. Chem. Rev., 2013, 257, 3051 CrossRef CAS
- C. J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, Visible Light-Mediated Atom Transfer Radical Addition via Oxidative and Reductive Quenching of Photocatalysts, J. Am. Chem. Soc., 2012, 134, 8875 CrossRef CAS
- J. D. Nguyen, J. W. Tucker, M. D. Konieczynska and C. R. J. Stephenson, Intermolecular Atom Transfer Radical Addition to Olefins Mediated by Oxidative Quenching of Photoredox Catalysts, J. Am. Chem. Soc., 2011, 133, 4160 CrossRef CAS
- T. Rawner, E. Lutsker, C. A. Kaiser and O. Reiser, The Different Faces of Photoredox Catalysts: Visible-Light-Mediated Atom Transfer Radical Addition (ATRA) Reactions of Perfluoroalkyl Iodides with Styrenes and Phenylacetylenes, ACS Catal., 2018, 8, 3950 CrossRef CAS
- R. Beniazza, L. Remisse, D. Jardel, D. Lastécouères and J.-M. Vincent, Light-mediated iodoperfluoroalkylation of alkenes/alkynes catalyzed by chloride ions: role of halogen bonding, Chem. Commun., 2018, 54, 7451 RSC
- E. Arceo, E. Montroni and P. Melchiorre, Photo-Organocatalysis of Atom-Transfer Radical Additions to Alkenes, Angew. Chem., Int. Ed., 2014, 53, 12064 CrossRef CAS PubMed
- R. Beniazza, R. Atkinson, C. Absalon, F. Castet, S. A. Denisov, N. D. McClenaghan, D. Lastécouères and J.-M. Vincent, Benzophenone vs. Copper/Benzophenone in Light-Promoted Atom Transfer Radical Additions (ATRAs): Highly Effective Iodoperfluoroalkylation of Alkenes/Alkynes and Mechanistic Studies, Adv. Synth. Catal., 2016, 358, 2949 CrossRef CAS
- T. Zhang, P. Wang, Z. Gao, Y. An, C. He and C. Duan, Pyrene-based metal–organic framework NU-1000 photocatalysed atom-transfer radical addition for iodoperfluoroalkylation and (Z)-selective perfluoroalkylation of olefins by visible-light irradiation, RSC Adv., 2018, 8, 32610 RSC
- T. Yajima and M. Ikegami, Metal-Free Visible-Light Radical Iodoperfluoroalkylation of Terminal Alkenes and Alkynes, Eur. J. Org. Chem., 2017, 2126 CrossRef CAS
- F. Würthner, C. R. Saha-Moller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Perylene Bisimide Dye Assemblies as Archetype Functional Supramolecular Materials, Chem. Rev., 2016, 116, 962 CrossRef PubMed
- D. Liu, J. Wang, X. Bai, R. Zong and Y. Zhu, Self-Assembled PDINH Supramolecular System for Photocatalysis under Visible Light, Adv. Mater., 2016, 28, 7284 CrossRef CAS PubMed
- D. M. Schultz and T. P. Yoon, Solar Synthesis: Prospects in Visible Light Photocatalysis, Science, 2014, 343, 1239176 CrossRef
- A. Stergiou and N. Tagmatarchis, Fluorene-Perylene Diimide Arrays onto Graphene Sheets for Photocatalysis, ACS Appl. Mater. Interfaces, 2016, 8, 21576 CrossRef CAS
- W. Wei, D. Liu, Z. Wei and Y. Zhu, Short-Range π–π Stacking Assembly on P25 TiO2 Nanoparticles for Enhanced Visible-Light Photocatalysis, ACS Catal., 2017, 7, 652 CrossRef CAS
- P. Hao, H. Zhu, Y. Pang, J. Shen and Y. Fu, The impact of positional isomerism on electronic and photochromic properties of 1D zinc-based naphthalene diimide coordination polymers, CrystEngComm, 2020, 22, 3371–3377 RSC
- H.-X. Gong, Z. Cao, M.-H. Li, S.-H. Liao and M.-J. Lin, Photoexcited perylene diimide radical anions for the reduction of aryl halides: a bay-substituent effect, Org. Chem. Front., 2018, 5, 2296 RSC
- X. Bai, C. Wang, X. Wang, T. Jia, B. Sun, S. Yang, D. Li, J. Li and H. Li, Strong electron affinity PDI supramolecules form anion radicals for the degradation of organic pollutants via direct electrophilic attack, Catal. Sci. Technol., 2021, 11, 1899 RSC
- Y. Xu, J. Zheng, J. O. Lindner, X. Wen, N. Jiang, Z. Hu, L. Liu, F. Huang, F. Würthner and Z. Xie, Consecutive Charging of a Perylene Bisimide Dye by Multistep Low-Energy Solar-Light-Induced Electron Transfer Towards H2 Evolution, Angew. Chem., Int. Ed., 2020, 59, 10363 CrossRef CAS PubMed
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Reduction of aryl halides by consecutive visible light-induced electron transfer processes, Science, 2014, 346, 725 CrossRef CAS PubMed
- J. J. Han, A. D. Shaller, W. Wang and A. D. Q. Li, Architecturally Diverse Nanostructured Foldamers Reveal Insightful Photoinduced Single-Molecule Dynamics, J. Am. Chem. Soc., 2008, 130, 6974 CrossRef CAS PubMed
- A. D. Shaller, W. Wang, A. Li, G. Moyna, J. J. Han, G. L. Helms and A. D. Q. Li, Sequence-Controlled Oligomers Fold into Nanosolenoids and Impart Unusual Optical Properties, Chem. – Eur. J., 2011, 17, 8350 CrossRef CAS
- P. Hao, Y. Xu, X. Li, J. Shen and Y. Fu, Photochromism and photocatalysis of organic–inorganic hybrid iodoargentates modulated by argentophilic interactions, Inorg. Chem. Front., 2020, 7, 3184–3194 RSC
- X. Feng, Y. Pi, Y. Song, Z. Xu, Z. Li and W. Lin, Integration of Earth-Abundant Photosensitizers and Catalysts in Metal–Organic Frameworks Enhances Photocatalytic Aerobic Oxidation, ACS Catal., 2021, 11, 1024 CrossRef CAS
- S. Li, H.-M. Mei, S.-L. Yao, Z.-Y. Chen, Y.-L. Lu, L. Zhang and C.-Y. Su, Well-distributed Pt-nanoparticles within confined coordination interspaces of self-sensitized porphyrin metal–organic frameworks: synergistic effect boosting highly efficient photocatalytic hydrogen evolution reaction, Chem. Sci., 2019, 10, 10577 RSC
- P. Hao, W. Wang, L. Zhang, J. Shen and Y. Fu, Metal-dependent electronic and photochromic behaviors of dimethylbenzotriazolium iodometallate hybrids, Inorg. Chem. Front., 2019, 6, 287–292 RSC
- J. Cai, L. Zhao, C. He, Y. Li and C. Duan, A host–guest semibiological photosynthesis system coupling artificial and natural enzymes for solar alcohol splitting, Nat. Commun., 2021, 12, 5092 CrossRef CAS PubMed
- A. M. Rice, C. R. Martin, V. A. Galitskiy, A. A. Berseneva, G. A. Leith and N. B. Shustova, Photophysics Modulation in Photoswitchable Metal–Organic Frameworks, Chem. Rev., 2020, 120, 8790 CrossRef CAS
- P. Hao, C. Guo, J. Shen and Y. Fu, A novel photochromic hybrid containing trinuclear [Cd3Cl12]6− clusters and protonated tripyridyl-triazines, Dalton Trans., 2019, 48, 16497–16501 RSC
- L. Zeng, T. Liu, C. He, D. Shi, F. Zhang and C. Duan, Organized Aggregation Makes Insoluble Perylene Diimide Efficient for the Reduction of Aryl Halides via Consecutive Visible Light-Induced Electron-Transfer Processes, J. Am. Chem. Soc., 2016, 138, 3958 CrossRef CAS
- B. Lü, Y. Chen, P. Li, B. Wang, K. Müllen and M. Yin, Stable radical anions generated from a porous perylenediimide metal-organic framework for boosting near-infrared photothermal conversion, Nat. Commun., 2019, 10, 767 CrossRef
- L. Spek, PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef PubMed
- K. D. Belfield, M. V. Bondar, F. E. Hernandez and O. V. Przhonska, Photophysical Characterization, Two-Photon Absorption and Optical Power Limiting of Two Fluorenylperylene Diimides, J. Phys. Chem. C, 2008, 112, 5618 CrossRef CAS
- M. G. Debije and P. P. C. Verbunt, Thirty Years of Luminescent Solar Concentrator Research: Solar Energy for the Built Environment, Adv. Energy Mater., 2012, 2, 12 CrossRef CAS
- M. Xu, J.-M. Han, Y. Zhang, X. Yang and L. Zang, A selective fluorescence turn-on sensor for trace vapor detection of hydrogen peroxide, Chem. Commun., 2013, 49, 11779 RSC
- X. Feng, Y. An, Z. Yao, C. Li and G. Shi, A Turn-on Fluorescent Sensor for Pyrophosphate Based on the Disassembly of Cu2+-Mediated Perylene Diimide Aggregates, ACS Appl. Mater. Interfaces, 2012, 4, 614 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures, PXRD, TGA, NMR, X-ray crystallographic information, and other figures. CCDC 2108376. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qi01206a |
| This journal is © the Partner Organisations 2022 |