
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLipase-catalyzed acylation of levoglucosan in continuous flow: antibacterial and biosurfactant studies†
Marcelo A. do Nascimentoae,
Juan P. C. Vargasb,
José G. A. Rodriguesc,
Raquel A. C. Leãoa,
Patricia H. B. de Mourad,
Ivana C. R. Leald,
Jonathan Bassut e,
Rodrigo O. M. A. de Souzaa,
Robert Wojcieszake and
Ivaldo Itabaiana Jr*ef
e,
Rodrigo O. M. A. de Souzaa,
Robert Wojcieszake and
Ivaldo Itabaiana Jr*ef
aBiocatalysis and Organic Synthesis Group, Chemistry Institute, Federal University of Rio de Janeiro, CEP 21941-909, Brazil
bNanotechnology Engineering Program, COPPE, Universidade Federal do Rio de Janeiro, UFRJ, Rio de Janeiro, Brazil
cInstitute of Chemistry, Federal University of Rio de Janeiro, University City, 21941-909, Rio de Janeiro, Brazil
dLaboratory of Natural Products and Biological Assays, Department of Natural Products and Food, Pharmacy Faculty, Federal University of Rio de Janeiro, 21941-902, Rio de Janeiro, Brazil
eUniv. Lille, CNRS, Centrale Lille, Univ. Artois, UMR 8181 – UCCS – Unité de Catalyse et Chimiedu Solide, F-59000, Lille, France
fLaboratory of Technological Biochemistry and Biocatalysis, Department of Biochemical Engineering, School of Chemistry, Federal University of Rio de Janeiro, 21941-909, Rio de Janeiro, Brazil. E-mail: ivaldo@eq.ufrj.br
First published on 21st January 2022
Abstract
Studies involving the transformation of lignocellulosic biomass into high value-added chemical products have been intensively conducted in recent years. Its matrix is mainly composed of cellulose, hemicellulose and lignin, being, therefore, an abundant and renewable source for obtaining several platform molecules, with levoglucosan (LG) standing out. This anhydrous carbohydrate can be acylated to obtain carbohydrate fatty acid esters (CFAEs). Here, these compounds were obtained via enzymatic acylation of LG, commercially obtained (Start BioScience®), with different acyl donors in continuous flow. Through the experimental design using a model reaction, it was possible to optimize the reaction conditions, temperature and residence time, obtaining a maximum conversion at 61 °C and 77 min. In addition, there was a productivity gain of up to 100 times in all comparisons made with the batch system. Finally, CFAEs were applied in tests of interfacial tension and biological activity. For a mixture of 4- and 2-O-lauryl-1,6-anhydroglucopyranose (MONLAU), the minimum interfacial tension (IFTmin) obtained was 96 mN m−1 and the critical micelle concentration (CMC) was 50 mM. Similar values were obtained for a mixture of 4- and 2-O-palmitoyl-1,6-anhydroglucopyranose (MONPAL), not yet reported in the literature, of 88 mN m−1 in 50 mM. For a mixture of 4- and 2-O-estearyl-1,6-anhydroglucopyranose (MONEST) and 4- and 2-O-oleoyl-1,6-anhydroglucopyranose (MONOLE), CMC was higher than 60 mM and IFTmin of 141 mN m−1 and 102 mN m−1, respectively. Promising data were obtained for minimal inhibitory concentration (MIC) and minimal bactericidal concentration (MBC) of MONLAU against Staphylococcus aureus strains at 0.25 mM.
Introduction
Carbohydrates, also called sugars, are the most abundant organic substances in the biological world that make up more than 50% of the dry weight of the earth's biomass, being essential for life.1,2 These compounds can be partially acylated via chemo or enzymatic reactions in batch or continuous flow conditions with different acyl donors to access a variety of mono- or oligosaccharide esters which may have valuable properties with wide applications in the food, pharmaceutical, detergent, agricultural, fine chemical and personal care industries.3,4In this scenario, levoglucosan (1,6-anhydro-β-D-glucopyranose) (LG) might be a promising chemical platform for the synthesis of carbohydrate fatty acid esters, denoted as CFAEs. LG is a major product of cellulose pyrolysis and it can be converted to different high added-value chemicals such as glucose, 5-hydroxymethylfurfural, furfural, sorbitol, levoglucosenone and, as highlighted in this work, CFAEs (Fig. 1).5
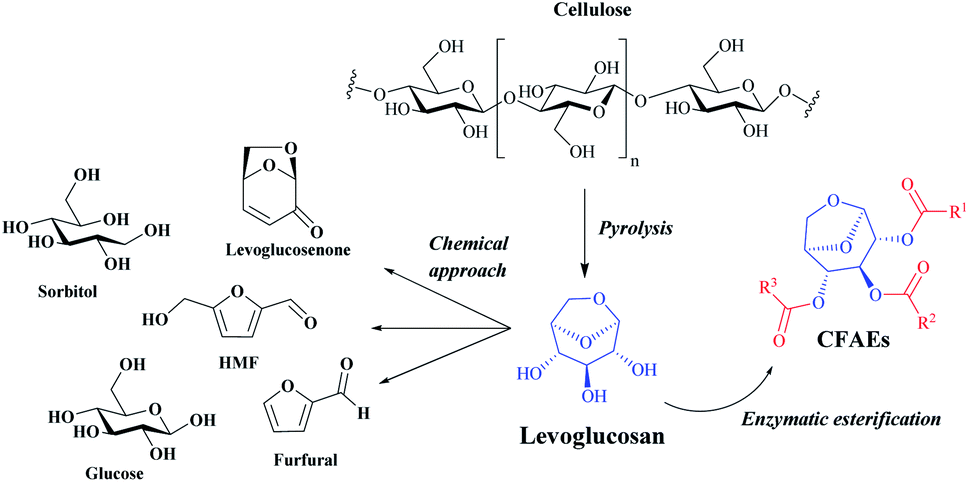 | ||
| Fig. 1 General representation for highly valued products obtained by chemical and biochemical transformation of LG. | ||
The structure–activity relationship of CFAEs depends on the degree of substitution of the hydroxyl groups, in addition to the size and amount of unsaturation in the alkyl chains,3,6 which can generate compounds with a hydrophobic–lipophilic balance (HLB) of great industrial interest as emulsifying agents, stabilizers in conventional systems and also promising biological activities as already mentioned.7–9 It is important to highlight that these compounds are also biodegradable and non-toxic.10
A challenging task in the chemical synthesis of CFAEs is the selective acylation of a single secondary hydroxyl group due to the presence of multiple functional groups.11 Thus, several protection and deprotection steps are necessary to achieve the selective incorporation of a single ester function, consuming a large amount of energy and/or reagents contrary to the principles defined by “Green Chemistry” that requires the development of sustainable technologies with decreased environmentally impact and lesser possibility of side products.12
Accordingly, an alternative is enzymatic processes, where lipases (triacylglycerol hydrolases, EC 3.1.1.3) are the most suitable class of enzymes for esterification and transesterification reactions. In this context, Pulido and Gotor (1994)13 observed the regioselective monoacylation of the secondary hydroxyl, 4-O-, of carbohydrates with ester oximes in 1,4-dioxane. Galletti et al. (2007)14 proposed the acetylation of levoglucosan in the presence of several acyl donors using acetonitrile and ionic liquids as solvent. The esterification occurred mainly in the 4-O-hydroxyl. The same was observed by Nascimento et al. (2019)11 for long-chain acyl donor. Holmstrøm and Pedersen (2020)15 obtained high degrees of regioselectivity in the acetylation of functionalized glycosides.
As shown in several studies,11,13–17 high yields and regioselectivity can be obtained under milder operating conditions alongside high purity of the final product.18 However, a recurrent barrier persist,19 i.e., instability of the enzyme at high temperatures, inhibition by solvent and/or reagents and products, or under turbulent flow conditions, making application difficult for application on an industrial scale. Nevertheless, studies on enzymatic immobilization have been widely disseminated by several research group that report better robustness and stability of these biocatalysts, what are suitable for these applications.12,19,20
Here, we used Lipase B from Candida antarctica (CALB) immobilized in epoxy resin,21 denominated CalB_Epoxy, in the synthesis of CFAEs via LG esterification/transesterification reaction with different acyl donors. We have also shown that CFAEs which were synthetized in batch11 can be readily scalable to a continuous flow system where temperature and residence time conditions have been optimized with the aid of the design of experiments (DoE) statistical approach.
Finally, the obtained CFAEs for a mixture of 4 and 2: O-lauryl-1,6-anhydroglucopyranose (MONLAU), O-palmitoyl-1,6-anhydroglucopyranose (MONPAL), O-estearyl-1,6-anhydroglucopyranose (MONEST) and O-oleoyl-1,6-anhydroglucopyranose (MONOLE), respectively, were applied in biological and surface activity tests, where promising results were obtained.
Results and discussion
One of the biggest obstacles in continuous flow chemistry is the handling of solids that can form during the chemical reaction or in the piping that precedes the entry of reagents into the reactor, usually leading to clogging of the channels. Furthermore, the low solubility of the starting materials can make flow-through synthesis a challenging task.22,23 Thus, and in order to obtain the best reaction conditions, the reagents were prepared at a concentration of 40 and 100 mM, respectively, and submitted to solubility tests at different temperatures (see ESI – 1.4 – Table S3†).According to the results obtained, it is possible to observe the insolubility of levoglucosan in acetonitrile at temperatures less than or equal to 50 °C. For the acyl donors: lauric acid, vinyl palmitate and vinyl oleate were soluble in all conditions analyzed, whereas laurate and vinyl stearate were soluble at temperatures greater than or equal to 50 °C. In addition, for the molar concentrations analyzed, the solubility profile remained constant, varying only with the temperature change. Therefore, due to the low solubility of levoglucosan at temperature below 50 °C and for the experimental conditions available in the laboratory, it was necessary to heat the starting material before pumping it into the fixed bed reactor.
Optimization using DoE in a model reaction
The model reaction used was the esterification of levoglucosan with lauric acid (Scheme 1). In the application of the experimental planning, the input variables used were temperature (X1) and residence time (X2). And as response variable (Y) the conversion (%). In the CCD, 11 experiments were performed at random and the matrix of experiments with the obtained conversion (%) responses are shown in Table S1 (see ESI – 1.2†). After the execution of all experiments, a second-order equation was obtained:| Y = b0 + b1T + b2t + b1T2 + b1t2 + b1Tt | (1) |
| b = (XtX)−1(XtY) | (2) |
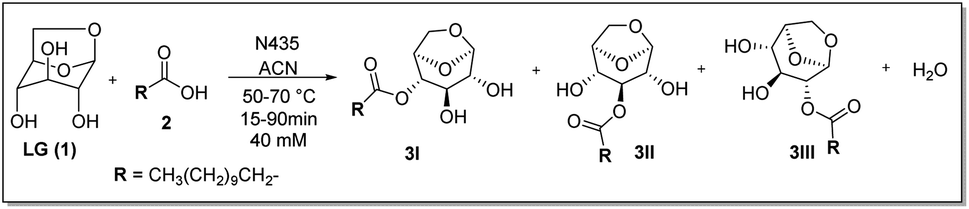 | ||
| Scheme 1 Esterification reaction of levoglucosan with lauric acid. * Reactor volume packed with N435: 7.854 mL. | ||
From the generated model it was possible to build the response surface and the level curve (Fig. 2a and b).
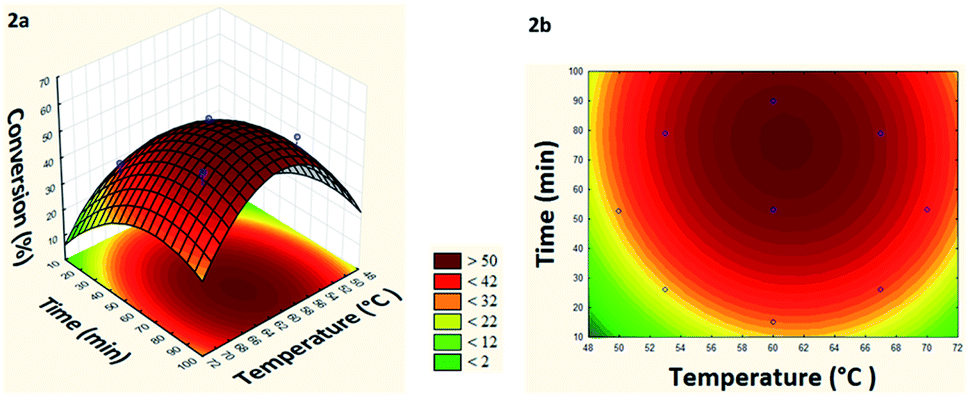 | ||
| Fig. 2 (a) Response surface and (b) level curve of the conversion in the model reaction of esterification of levoglucosan with lauric acid relating temperature and time. | ||
Based on response surface and levels curve analysis (Fig. 2a and b) can verify that at temperatures below 54 °C and short residence times (10–25 min) the conversion remained below 20%, increasing linearly with the increase of these two parameters, until reaching a maximum conversion (55%) at 61 °C and 77 min. It was also observed that for temperatures above 69 °C and residence times less than 30 min, the conversion tends to remain below 22%, indicating that there is dependence between these two factors. Besides, there is an ideal working range between 60–62 °C and 70–80 min for maximizing conversion.
In the esterification reaction using carboxylic acid, one of the products formed is water, a strong nucleophile in deacylation of the acyl enzyme, shifting the balance in the opposite direction, i.e., hydrolysis of the product (this is a thermodynamically controlled synthesis)27,28 and, consequently, decreasing the reaction yield. To shift the balance towards the products and increase yield, it is common to use in excess of one of the reagents or remove one of the products, usually water.29
Alternatively, it is possible to obtain the products desired by changing the leaving group of the acylating agent, avoiding, for example, formation of water in the reaction medium and, furthermore, improve the solubility of the starting materials. Thus, after optimizing the temperature and residence time, the best conditions obtained were applied in the model reaction by varying the acyl donor output group (Fig. 3).
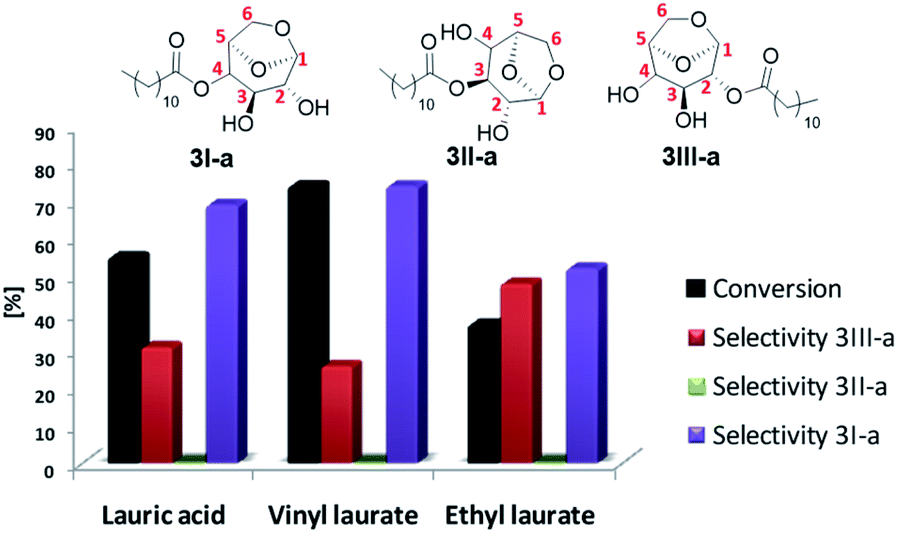 | ||
| Fig. 3 Acylation of levoglucosan with different acyl donors in 61 °C and residence time 77 min using N435 as biocatalyst. Selectivity 3I-a: 4-O-lauryl-1,6-anhydroglucopyranose; selectivity 3II-a: 3-O-lauryl-1,6-anhydroglucopyranose and selectivity 3III-a: 2-O-lauryl-1,6-anhydroglucopyranose. Analyses were performed in a gas chromatograph equipped with a mass spectrometry detector (GC-MS). | ||
Better results were obtained using vinyl laurate as acylating agent, with conversion 74%, selectivity 3I-a of 75% and 3III-a of 25%. The formation of 3-O-lauryl-1,6-anhydro-glucopyranose (selectivity 3II-a) was not observed. According to Boissièere-Junot et al.30 the orientation of the hydroxyls (axial or equatorial) or neighboring substituent governs the regioselectivity of acetylation catalyzed by lipase. When the C3-OH is axial a high regioselectivity is observed for the adjacent axial C4- or C2-OH.30 Moreover, studies carried out by Uriarte et al.31 suggest that in chair conformation, interactions by intramolecular hydrogen bonds that occur between C3-OH and oxygen in C6, both in the axial position, stabilize the molecule, decreasing its reactivity as a nucleophile. As a result a preference for 3I-a or 3III-a must be observed.30
The low conversion and loss of selectivity obtained using ethyl laurate 40% may be related to its partial solubility under applied experimental conditions. Contrary to that, the reaction in the presence of completely soluble lauric acid showed a conversion 55%, 19% lower compared to the vinyl donor, however, better than the values reported by Galletti et al.14 40% conversion using lauric acid for the batch reaction for about five days.
Another point to be highlighted is the vinyl alcohol formed that tautomerizes to acetaldehyde irreversibly, a more stable compound that also contributes to shifting the chemical balance in the direct direction of the reaction, thereby increasing the yield of the desired ester.32
The results suggest the great potential of vinyl acyl donors for the synthesis of levoglucosan esters in continuous flow, not only for their solubility, but also for the best conversions and selectivities obtained.
Synthesis of long-chain CFAEs
Finally, after screening, the promising results obtained in the acylation of levoglucosan with vinyl donor motivated us to evaluate a complementary in flow approach regarding the regioselectivity presented by lipase Candida antarctica B immobilized in epoxy support by our research group, called CalB_Epoxy, applied to obtain CFAEs of levoglucosan in the batch condition.11The transesterification reaction was carried out in the presence of a series of vinyl esters containing alkyl chains of variable length (C12–C18), as well as in the presence of biocatalyst N435 used as reference. The results are shown in Table 1.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Immob. biocat. | Acyl donor | Regioselectivity (%) | |||
| 3I | 3II | 3III | By-products | |||
| a N435 = Novozym 435 (Candida antarctica lipase B), immobilized on macroporous acrylic type ion exchange resin; CalB_Epoxy = Candida antarctica lipase B immobilized on epoxy support by our research group 2; equivalents, a = vinyl laurate, b = vinyl palmitate, c = vinyl stearate and d = vinyl oleate. | ||||||
| 1 | N435 | a | 75 | — | 25 | — |
| 2 | b | 76 | — | 24 | — | |
| 3 | c | 73 | — | 27 | — | |
| 4 | d | 75 | — | 25 | — | |
| 5 | CalB_Epoxy | a | 33 | — | 10 | 57 |
| 6 | b | 50 | — | 15 | 35 | |
| 7 | c | 43 | — | 17 | 40 | |
| 8 | d | 45 | — | 14 | 41 | |
As a result, the formation of by-products using N435 was not observed. In contrast, there was the formation of by-products in the presence of CalB_Epoxy, respectively (Table 1).
Regarding regioselectivity, N435 showed similar values between 23% for 3IIIa–d, and 77% for 3Ia–d for all acyl donors used, i.e., there was a preference for the hydroxyl linked to C4 for the acylation of levoglucosan. However, there was the formation of by-products using CalB_Epoxy, reducing the selectivity for the desired products (Table 1 – entries 5–8). In contrast to observed by Do Nascimento et al.11 for the batch reaction, with N435 being more selective to produce lauric esters (99% at 50 °C) while CalB_Epoxy was more selective to produce oleic esters (83% at 55 °C) of LG, respectively.
Compared to the batch system,11 the productivity values obtained for the continuous flow reaction were higher in all samples, even with the loss of selectivity for CFAEs using CalB_Epoxy (Table 2). Calculations obtained for specific activity showed that CalB_Epoxy presented 41.33 U mg−1 of protein, a value approximately 2.5 times higher than that obtained for N435 of 16.53 U mg−1 of protein. These results suggest that the high specific activity for CalB_Epoxy may have contributed to significant gains for productivity, although the selectivity values for CFAEs are lower than for N435.
| Entry | Immob. enz.a | Acyl donorb | Conversion (%) | Productivityc | |
|---|---|---|---|---|---|
| Batch | Continuous flow | ||||
| a N435 = Novozym 435 (Candida antarctica lipase B), immobilized on macroporous acrylic type ion exchange resin; CaLB_Epoxy = Candida antarctica lipase B immobilized on epoxy support by our research group.b 2 equivalent, a: vinyl laurate, b: vinyl palmitate, c: vinyl stearate and d: vinyl oleate.c Productivity = mg of product. h−1 SA−1 of biocatalyst. | |||||
| 1 | N435 | a | 74 | 0.030 | 3.660 |
| 2 | b | 76 | 0.034 | 4.370 | |
| 3 | c | 57 | 0.034 | 3.511 | |
| 4 | d | 85 | 0.035 | 5.211 | |
| 5 | CalB_Epoxy | a | 59 | 0.003 | 0.511 |
| 6 | b | 99 | 0.010 | 1.500 | |
| 7 | c | 99 | 0.009 | 1.481 | |
| 8 | d | 97 | 0.014 | 1.420 | |
For a better understanding of the results, it is important to highlight the differences between the N435 and CalB_Epoxy biocatalysts. According to Ortiz et al.33 N435 is a CALB immobilized by hydrophobic adsorption via interfacial activation on a Lewatit VP OC 1600 resin used as a support. The same protocol was applied by Do Nascimento et al.21 in the immobilization of CALB, but in epoxy acrylate resin ECR 8205 (Purolite®), denominated CalB_Epoxy. Therefore, the difference between the two biocatalysts lies in the support used for their immobilization and, consequently, in the way these enzymes attach themselves to the solid material, which reflects on their specific chemical, biochemical, mechanical and kinetic properties.33
As mentioned, in both biocatalysts the lipase was adsorbed on the hydrophobic surface of the support via an interfacial activation protocol.21 This method of immobilization has some advantages, for example, they can almost completely retain their activity.34 Moreover, it is less sensitive to variation in experimental conditions because there is no conformational equilibrium to be changed.35,36 However, as it is a reversible immobilization and based on hydrophobic interactions, enzymes can detach from the support and go to the reaction medium, resulting in a loss of enzymatic charge with a subsequent decrease in its activity.
For biocatalysts N435 and CalB_Epoxy, leaching of the enzyme was not observed after the passage of pure solvent through the fixed bed reactor. However, the reagents, as well as the products formed during the reaction can lead to the desorption of the lipase molecules into to reaction medium as well as the polymeric components of the support, contaminating the product, as reported by Hirata et al.37 where free fatty acids and di or monoglycerides, which have recognized detergent properties, favored enzyme desorption. Furthermore, bimolecular aggregates may have been formed in the immobilization and/or leaching during the reaction with subsequent interaction between two open forms of the lipase.38 When this occurs, the effect can be quite negative because these dimers will be immobilized along with the monomeric enzyme molecules. This phenomenon may have occurred with CalB_Epoxy, changing its properties.38,39
Evaluation of interfacial tension between oil/water in different concentrations of CFAEs
Table 3 shows the theoretical values of HLB calculated for CFAs using the Becher equation (eqn (1)).40 These values between 4.42 and 5.91 and the low solubility in water, especially for compounds from 2-4, suggest an application as a wetting agent or as oil-soluble hydrophobic (w/o) emulsifiers.41| Entry | Compounda | Oil solubility (mg mL−1) | HLB value | IFTmin (mN m−1) | CMC (mM) |
|---|---|---|---|---|---|
| a Mixture of 4- and 2-: O-lauryl-1,6-anhydroglucopyranose (MONLAU), O-palmitoyl-1,6-anhydroglucopyranose (MONPAL), O-estearyl-1,6-anhydroglucopyranose (MONEST) and O-oleoyl-1,6-anhydroglucopyranose (MONOLE). | |||||
| 1 | MONLAU | >100 | 5.91 | 92 | 53 |
| 2 | MONEST | >100 | 4.80 | 88 | 50 |
| 3 | MONPAL | >100 | 4.42 | 137 | 64 |
| 4 | MONOLE | >100 | 4.61 | 105 | 62 |
In this context, behavioral trend values of CFAEs were measured through experimental studies by the pendant drop method,42 of the minimum interfacial tension (IFTmin) in function of the concentration of CFAEs in oil-water (Fig. 4).
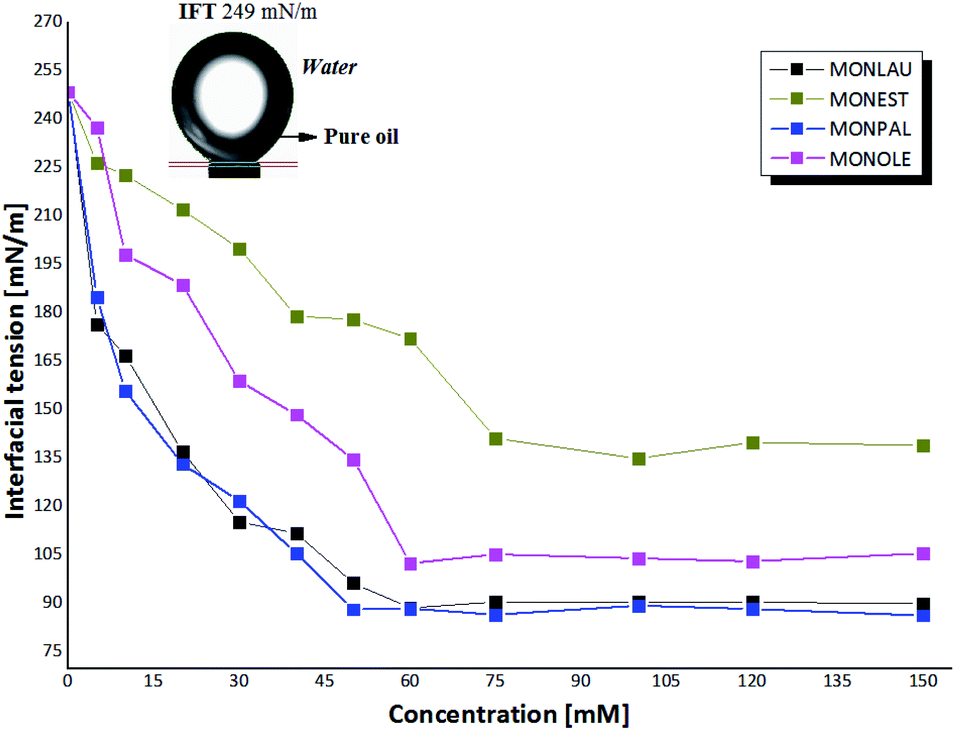 | ||
| Fig. 4 Relationship between the concentration of CFAs and interfacial tension. | ||
The IFTmin is one of the key parameters that govern the compatibility between the components of the blend.43 It is results from the difference in energy between molecules at a fluid interface when compared to their bulk counterparts.44 Compounds with properties such as surfactants tend to distribute themselves at interfaces between phases with different degrees of polarity, forming a molecular film that reduces interfacial and surface tension.
In this context, a drop in the voltage value at the water–oil interface was observed with the increase of the molar concentration of CFAEs in the oil and it remained practically constant after 50 mM, thus revealing a state in which the surfaces of the tested samples are saturated with the CFAEs molecules. From this specific concentration, called critical micelle concentration (CMC), thermodynamically stable molecular aggregates begin to form. The structure of the surfactant and the experimental condition define how these molecules are submerged within the fluid.45
Also, it is possible to note that compounds 1-2 (MONLAU and MONPAL) showed a similar behavior, with a considerable decrease in IFTmin below CMC, in the range of 248 mN m−1 and 86 mN m−1, and with non-significant variations for high concentrations of CFAEs, above the CMC. Thus, it is suggested a greater affinity of these molecules with the aqueous medium, compared to compounds 3 and 4, so that these compounds were able to spread, and also reduce the tension at the interface with the aqueous medium more effective, at 50 mM. In contrast, MONEST and MONOLE the CMC was greater than 60 mM and IFTmin in the range of 137-105 mN m−1.
Preliminary evaluation of the active properties of the MONLAU product were reported by Galletti et al.,14 which obtained an HLB value of 5.9 and 45 mN m−1 at 0.019 ng mm2. Furthermore, Kootery et al.46 suggest that monoacylated levoglucosan esters can be considered as representative renewable surfactants.
These results corroborate those obtained in this work of the 92 mN m−1 at 53 mM for MONLAU. Moreover, similar results were obtained for MONPAL e MONOLE, without description in the literature, with 88 mN m−1 at 53 mM and 102.48 mN m−1 at 60 mM, which indicates lipophilic water in oil emulsifier.
Antibacterial assays
The results referring the minimal inhibitory concentration and minimal bactericidal concentration, of esterified compounds against S. aureus strains, are shown in Table 4. Among the tested substances MONEST and levoglocusan were not able to inhibit the growth of the selected strains at concentration ≤ 1 mM. Concerning the esterified products, MONLAU and MONOLE showed higher activity, highlighting MONLAU which presented MIC values lower than that of the lauric acid substrate. MONLAU was able to inhibit the growth of all MSSA and MRSA strains at 0.25 mM. Since, MRSA presents a multidrug-resistant pattern for variable antimicrobial classes, such macrolides, fluoroquinolones, aminoglycosides, tetracyclines, and lincosamides,47 this is a promising result, once the World Health Organization (WHO) considers methicillin-resistant and vancomycin intermediate, and resistant, S. aureus to be a high priority pathogen.| Compound | ATCC 25923 MSSA | ATCC 29213 MSSA | ATCC 33591 MRSA | 517 MRSA |
|---|---|---|---|---|
| a Minimal bactericidal concentration (MBC). MSSA: methicillin-sensitive Staphylococcus aureus. MRSA: methicillin-resistant Staphylococcus aureus. | ||||
| MONLAU | 0.25 mM | 0.25 mM | 0.25 mM | 0.25 mM |
| 0.25 mMa | 0.25 mMa | 0.25 mMa | 0.25 mMa | |
| MONEST | >1.0 mM | >1.0 mM | >1.0 mM | >1.0 mM |
| MONPAL | >1.0 mM | >1.0 mM | 1.0 mM | >1.0 mM |
| MONOLE | 1.0 mM | 0.5 mM | 0.25 mM | 1.0 mM |
| Lauric acid | 0.5 mM | 0.5 mM | 0.5 mM | 0.5 mM |
| Levoglucosan | >1.0 mM | >1.0 mM | >1.0 mM | >1.0 mM |
| Methicillin | 0.5 μg mL−1 | 0.25 μg mL−1 | 4.0 μg mL−1 | 4.0 μg mL−1 |
Due to this promising result, bactericidal and bacteriostatic modes of action of MONLAU were evaluated, and the MBC assay indicated the bactericidal effect of MONLAU against all strains (MIC = MBC), including for methicillin-resistant Staphylococcus aureus. This was the first report on the antibacterial activity of levoglucosan fatty acid esters. MONLAU, against S. aureus, showed higher activities compared to other CFAEs reported in literature, such sucrose monolaurate (MIC = MBC = 0.4 mM),48 methyl α-D-glucopyranoside monolaurate (MIC = 188 μg mL−1)49 and sucrose monocaprate (MIC > 400 μg.mL−1).50
Experimental
Materials
N435 (immobilized CALB on Lewatit VP OC 1600 resin), Lipase B from Candida antarctica (CALB, soluble form) was purchased from Novozymes®, 1,6-anhydroglucopyranose (levoglucosan) was purchased from Start BioScience®. Epoxy resin Purolite® ECR8205F was purchased from Purolite International Limited. Aliphatic carboxylic acids, vinyl esters, NO-bis (trimethylsilyl) trifluoroacetamide (BSTFA) were purchased from Sigma-Aldrich. All other reagents used were of analytical grade.Lipase-catalyzed acylation of levoglucosan in continuous flow
In a flask, the solution containing the starting materials was prepared and immersed in a shaking bath. Syrris® syringe pumps were used to transport the solution containing the reagents into the fixed bed reactor at different flow rates, according to the previously defined residence time. Solubility tests were performed (see ESI – 1.4 – Table S3†).
Design of experiments
In order to find the optimal experimental conditions of the variables (temperature and residence time) in the synthesis of levoglucosan esters in continuous flow, the experimental design approach was used. The DoE main objective was to increase the conversion in the esterification reaction of levoglucosan with lauric acid, according to the Scheme 1.It is important to point out that the significant experimental variables that interfere in the system were selected according to the screening performed for the model reaction in a batch system.11 After the selection of the significant variables, the optimization step was performed using the Central Composite Design (CCD). At this stage, the previously selected variables were studied with five levels, which allowed establishing the best working conditions in the synthesis of levoglucosan esters in continuous flow.6
The CCD makes it possible to carry out the response surface methodology because the variables used are worked on at five levels. These levels (−α, −1, 0, +1, +α), are coded by convention and correspond to the five levels studied for the two variables (X1 and X2), where (−α is the lowest level, +α is the top level and 0 is the center point). Table 5 shows the coded levels and the experimental values studied for the two variables temperature and residence time in the CCD.
| Variable | Level | |||||
|---|---|---|---|---|---|---|
| Code | (−1414) | (−) | 0 | (+) | (+1414) | |
| (X1) Temperature (°C) | T | 50 | 53 | 60 | 67 | 70 |
| (X2) Residence time (min) | tr | 15 | 26 | 52.5 | 79 | 90 |
These levels have been defined to ensure that all areas of the parameter space can be explored, regardless of the range of factors (see ESI – 1.2 – Table S1†).
A stock solution containing levoglucosan and lauric acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 40 mM) in acetonitrile was prepared and added to a 50 mL flask and kept under agitation at 300 rpm at 55 °C. Then, was pumped into the fixed bed reactor packed with N435, 2.0 g, to a total volume of 7.854 mL. Flow rates of 87–524 μL min−1 was used and, in addition, different temperatures were evaluated, ranging from 50 to 70 °C. It is known that in a continuous flow, the reaction time is determined by the ratio between the reactor volume and the flow used in the system. A triplicate was performed for the best experimental condition found.
:1, 40 mM) in acetonitrile was prepared and added to a 50 mL flask and kept under agitation at 300 rpm at 55 °C. Then, was pumped into the fixed bed reactor packed with N435, 2.0 g, to a total volume of 7.854 mL. Flow rates of 87–524 μL min−1 was used and, in addition, different temperatures were evaluated, ranging from 50 to 70 °C. It is known that in a continuous flow, the reaction time is determined by the ratio between the reactor volume and the flow used in the system. A triplicate was performed for the best experimental condition found.
After optimizing the temperature and residence time, the influence of the acyl donor on the LG esterification or transesterification reaction was analyzed.
Transesterification of levoglucosan
After the screening step where the influence of temperature, residence time and acyl donor were investigated in the levoglucosan acetylation reaction, the best results were applied for the synthesis of levoglucosan esters containing alkyl chains of variable length, with up to 18 saturated carbons and unsaturated, biocatalyzed by N435 and CalB_Epoxy, CALB immobilized in epoxy resin by our research group,21 according to the Scheme 2.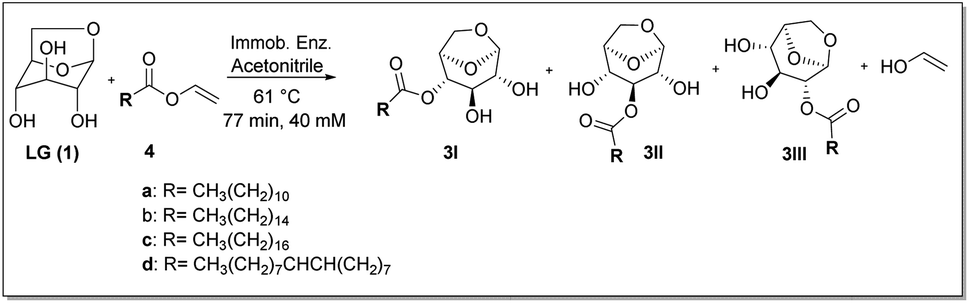 | ||
| Scheme 2 Esterification reaction of levoglucosan with different vinyl esters. * Reactor volume: 7.854 mL (2.0 g of immobilized enzyme). (a) vinyl laurate, (b) vinyl palmitate, (c) vinyl stearate and (d) vinyl oleate. | ||
The reaction progress was monitored by TLC. After stopping the reaction, CFAEs were separated by flash chromatography (eluent:ethyl acetate/hexane 1:1—Developer: sulfuric acid/ethanol). To determine the selectivity of the lipases in the position of the hydroxyl groups, the final product, obtained as a mixture of 4-O′ and 2-O′ levoglucosan monoesters (MONLAU, MONPAL, MONEST and MONOLE) were silylated with BSTFA and analyzed by gas chromatography with a mass spectrometry detector (GC-MS) (see ESI – 2.1 – Fig. S3 and S4†).
The elucidation of CFAEs has been extensively carried out by NMR:1H, 13C, HMBC, HSQC, 1H, 1H-COSY (“Correlation Spectroscopy”) and NOEdiff (NOE “difference spectroscopy”) spectra, FTIR spectrum ν (cm−1, KBr), HRMS (High-Resolution Mass Spectrometry) by our research group and the results have been published by Do Nascimento et al.11 for further details, see ESI 2.4 – Fig. S5–S8 and S10–S13.†
Determination of productivity
In batch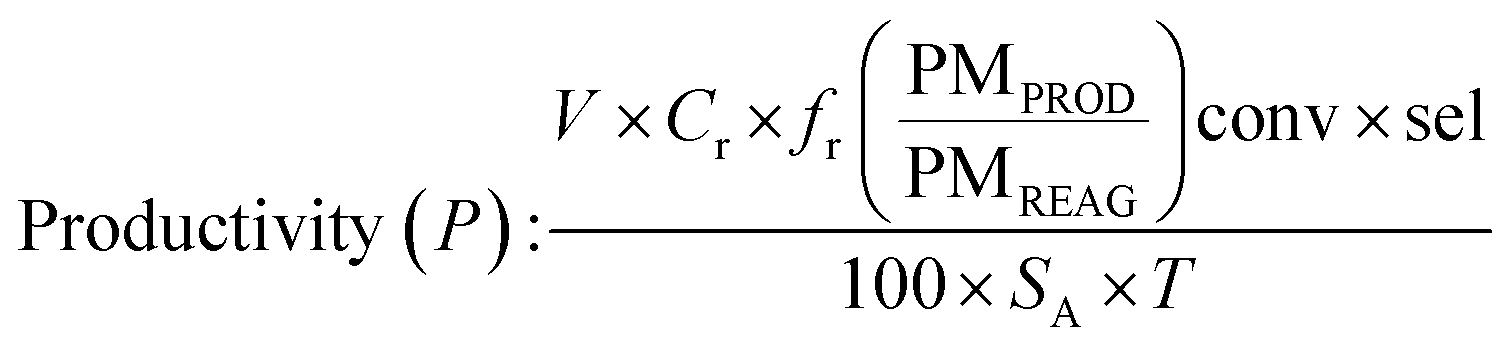
In flow
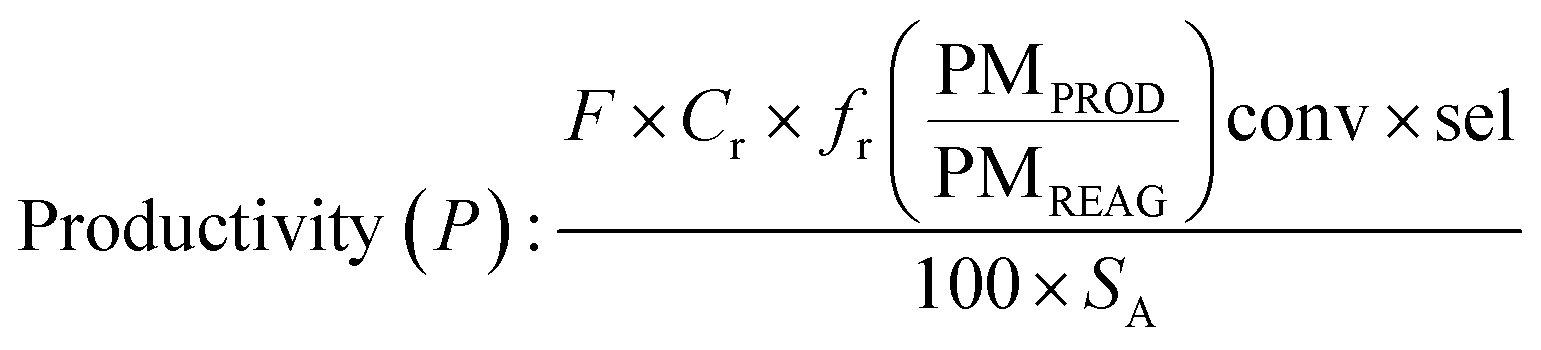
Determination of surface activity
The HLB value has been estimated by the 1H NMR spectrum through the integration of the proton signals from the lipophilic and hydrophilic parts of the molecule. These values represent an empirical numerical correlation of the emulsifying and solubilizing properties of different surface-active agents.41 The term Igph denotes the integration amplitude of the hydrophilic groups and Itot refers to the total integration amplitude of the protons in the molecule. The ratio H describes the relative hydrophilic part of the molecule and can be used to estimate HLB value applying the equation described of Rabaron et al.51 (eqn (3)).| H = Igph/Itot |
| HLB = 60H/(H + 2) | (3) |
The HLB scale ranges from 0 to 20. In the range of 3.5 to 6.0, surfactants are more suitable for use in W/O emulsions. Surfactants with HLB values in the 8 to 18 range are most commonly used in O/W emulsions (Becher, 1988).40
For the interface tension (IFT) measurements, a two-step process was used. First, compounds were dissolved in olive oil and pendant drop of organic solutions were formed in aqueous environments. To achieve this, the CFAs solutions were loaded into a syringe connected to a U-shape needle. Then, the needle was placed at the bottom of a quartz cell containing distilled water (dispersion medium), and by moving the plunger downward, a drop of volume 15 μL was formed at the tip of the needle. The so-formed drop is illuminated by means of a light source, and a high-resolution CCD camera was used to capture the drop profile (see ESI – 2.2 - Tables S4–S8†). The determination of the interfacial tension, was calculated according to the Young–Laplace equation defined for the pendant drop (PD) method. A Goniometer OCA25 (DataPhysics Instruments, Germany) was used at room temperature and 1 atm pressure, and the droplet profiles were captured every 1 s for a total period of 20 s. CMC values were determined by plotting the interfacial tension vs. the concentration (in mM).
Antibacterial assays
For the antibacterial activity assessment Staphylococcus aureus species were used, one clinical and three strains from American Type Culture Collection (ATCC). All information about the bacterial species, correspondent codes, isolation source, and susceptibility profile is described in Table S9 (see ESI – 2.3†). The clinical strain was isolated from patients of Clementino Fraga Filho University Hospital (HUCFF), UFRJ, RJ, Brazil.52Minimal inhibitory concentration (MIC)
The MIC of the compounds (MONLAU, MONPAL, MONOLE, MONEST, lauric acid and levoglucosan) was determined by the broth microdilution technique in 96-microtiter plates,53 using a bacterial solution equivalent to a 0.5 McFarland scale (108 CFU mL−1) subsequently diluted 1:10 (107 CFU mL−1). The samples were prepared at final concentrations of 1.0, 0.5, 0.250, 0.125, 0.0625 and 0.03125 mM. The bacterial growth was evaluated using resazurin. After 24 h, 20 μL of aqueous resazurin solution (0.85%) was added in each well, and then the microplate was incubated again for 4 h at 37 °C.
Minimal bactericidal concentration (MBC)
The MBC assay of MONLAU was determined according to Isenberg.54 To perform this assay three concentrations of active samples were used at concentrations 0.250 mM (MIC), 0.5 (2×-MIC), 1.0 mM (4×-MIC).Conclusions
The high potential of using immobilized enzymes in continuous processes is widely recognized. In this work, it was possible to maximize the synthesis of CFAEs in continuous flow through the DoE where a significant gain in productivity and regioselectivity for C4-OH compared to the batch process was observed. In addition, CFAEs were applied in surface activity tests through the analysis of minimum interfacial tension and in biological tests through the evaluation of antibacterial activity in species of Staphylococcus aureus, the clinical strain was isolated from patients of Clementino Fraga Filho University Hospital (UFRJ-RJ-Brazil) and three strains of American Type Culture Collection (ATCC).A preliminary analysis of MONLAU reported by Galletti et al. (2007)14 suggests good properties as a surfactant at low concentrations of the ester. Similar results were found for MONPAL, not yet reported in the literature, at different concentrations of CFAEs. Finally, analyses of the biological activity of CFAEs showed that MONLAU showed promising results in tests of antibacterial activity compared to other CFAEs reported in the literature.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro for Jovem Cientista do Nosso Estado Grant Number: E-203.267/2017, Conselho Nacional de Desenvolvimento Científico e Tecnológico (BR) for Universal Grant Number 429974/2018-3, Centrale Lille, the CNRS, and Lille University as well as the Centrale Initiative Foundation, for their financial contribution. LIA CNRS France-Brazil “Energy & Environment” and Métropole Européen de Lille (MEL) for “CatBioInnov” project are also acknowledged. Authors also thank Professor Dr Kátia Regina Netto dos Santos, coordinator of the Hospital Infection Laboratory at the UFRJ, Department of Medical Microbiology, Institute of Microbiology and Surfaces and Thin Films and Protective Coatings Laboratory-Metalmat/UFRJ.References
- C. C. Wang, J. C. Lee, S. Y. Luo, S. S. Kulkarni, Y. W. Huang, C. C. Lee, K. L. Chang and S. C. Hung, Nature, 2007, 446, 896–899 CrossRef CAS PubMed
.
- R. V. Stick, Carbohydrates: The Sweet Molecules of Life, 2001 Search PubMed
- N. Park and M. K. Walsh, LWT, 2021, 147, 111664 CrossRef CAS
- M. C. P. Gonçalves, J. P. Romanelli, J. R. Guimarães, A. C. Vieira, B. P. de Azevedo and P. W. Tardioli, Crit. Rev. Biotechnol., 2021, 41, 865–878 CrossRef PubMed
- I. Itabaiana Junior, M. Avelar Do Nascimento, R. O. M. A. De Souza, A. Dufour and R. Wojcieszak, Green Chem., 2020, 22, 5859–5880 RSC
- H. M. El-laithy, O. Shoukry and L. G. Mahran, Eur. J. Pharm. Biopharm., 2011, 77, 43–55 CrossRef CAS PubMed
- S. W. Chang and J. F. Shaw, Nat. Biotechnol., 2009, 26, 109–116 CAS
- N. Petkova, Biointerface Res. Appl. Chem., 2021, 11, 12055–12067 CAS
- J. Andrés, M. Vargas, J. Orduña, M. Bernardo, G. Metzker, E. Gomes, M. Boscolo, S. Paulo, E. Sciences and S. Jose, Carbohydr. Res., 2020, 489, 107957 CrossRef PubMed
- Y. Zheng, M. Zheng, Z. Ma, B. Xin, R. Guo and X. Xu, Sugar Fatty Acid Esters, AOCS Press, 2012 Search PubMed
- M. Avelar do Nascimento, L. Ester Gotardo, E. Miguez Bastos, F. C. L. Almeida, R. A. C. Leão, R. O. M. A. de Souza, R. Wojcieszak and I. Itabaiana, Sustainability, 2019, 11, 6044 CrossRef
- V. S. Gamayurova, M. E. Zinov'eva, K. L. Shnaider and G. A. Davletshina, Catal. Ind., 2021, 13, 58–72 CrossRef
- R. Pulido and V. Gotor, Carbohydr. Res., 1994, 252, 55–68 CAS
- P. Galletti, F. Moretti, C. Samorì and E. Tagliavini, Green Chem., 2007, 9, 987–991 RSC
- T. Holmstrøm and C. M. Pedersen, Eur. J. Org. Chem., 2020, 2020, 4612–4615 CrossRef
- V. Gotor, Org. Process Res. Dev., 2002, 6, 420–426 CrossRef CAS
- E. Busto, V. Gotor-Fernández and V. Gotor, Chem. Soc. Rev., 2010, 39, 4504–4523 RSC
- R. T. Otto, U. T. Bornscheuer, C. Syldatk and R. D. Schmid, J. Biotechnol., 1998, 64, 231–237 CrossRef CAS
- J. Chapman, A. Ismail and C. Dinu, Catalysts, 2018, 8, 238 CrossRef
- J. J. Bassi, L. M. Todero, F. A. P. Lage, G. I. Khedy, J. D. Ducas, A. P. Custódio, M. A. Pinto and A. A. Mendes, Int. J. Biol. Macromol., 2016, 92, 900–909 CrossRef CAS PubMed
- M. A. Do Nascimento, L. E. Gotardo, R. A. C. Leão, A. M. De Castro, R. O. M. A. De Souza and I. Itabaiana, ACS Omega, 2019, 4, 860–869 CrossRef CAS
- V. R. L. J. Bloemendal, M. A. C. H. Janssen, J. C. M. Van Hest and F. P. J. T. Rutjes, React. Chem. Eng., 2020, 5, 1186–1197 RSC
- M. Guidi, P. H. Seeberger and K. Gilmore, Chem. Soc. Rev., 2020, 49, 8910–8932 RSC
- B. Barros Neto, I. S. Scarminio and R. E. Bruns, Como Fazer Experimentos – Pesquisa e Desenvolvimento na Ciência e na Indústria, 4th edn, 2010 Search PubMed
- T. R. Rosa, J. G. A. Rodrigues and R. Q. de Ferreira, Quim. Nova, 2016, 39, 8–14 Search PubMed
- F. C. Vicentini, L. C. S. Figueiredo-Filho, B. C. Janegitz, A. Santiago, E. R. Pereira-Filho and O. Fatibello-Filho, Quim. Nova, 2011, 34, 825–830 CAS
- W. Chulalaksananukul, J. S. Condoret, P. Delorme and R. M. Willemot, FEBS Lett., 1990, 276, 181–184 CrossRef CAS PubMed
- J. S. Rhee, S. J. Kwon and J. J. Han, in Enzymes in Nonaqueous Solvents, Humana Press, New Jersey, 2003, vol. 15, pp. 135–150 Search PubMed
- U. T. Bornscheuer and R. J. Kazlauskas, in Hydrolases in Organic Synthesis, 2006, pp. 141–183 Search PubMed
- N. Boissièere-Junot, C. Tellier and C. Rabiller, J. Carbohydr. Chem., 1998, 17, 99–115 CrossRef
- I. Uriarte, P. Écija, R. Lozada-Garcia, P. Çarçabal and E. J. Cocinero, ChemPhysChem, 2018, 19, 766–773 CrossRef CAS
- G. D. Yadav and A. H. Trivedi, Enzyme Microb. Technol., 2003, 32, 783–789 CrossRef CAS
- C. Ortiz, M. L. Ferreira, O. Barbosa, J. C. S. dos Santos, R. C. Rodrigues, Á. Berenguer-Murcia, L. E. Briand and R. Fernandez-Lafuente, Catal. Sci. Technol., 2019, 9, 2380–2420 RSC
- E. A. Manoel, J. C. S. dos Santos, D. M. G. Freire, N. Rueda and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2015, 71, 53–57 CrossRef CAS
- A. Bastida, P. Sabuquillo, P. Armisen, R. Fernández-Lafuente, J. Huguet and J. M. Guisán, Biotechnol. Bioeng., 1998, 58, 486–493 CrossRef CAS
- R. Fernandez-Lafuente, P. Armisén, P. Sabuquillo, G. Fernández-Lorente and J. M. Guisán, Chem. Phys. Lipids, 1998, 93, 185–197 CrossRef CAS
- D. B. Hirata, T. L. Albuquerque, N. Rueda, J. J. Virgen-Ortíz, V. G. Tacias-Pascacio and R. Fernandez-Lafuente, J. Mol. Catal. B: Enzym., 2016, 133, 117–123 CrossRef CAS
- J. M. Palomo, M. Fuentes, G. Fernández-Lorente, C. Mateo, J. M. Guisan and R. Fernández-Lafuente, Biomacromolecules, 2003, 4, 1–6 CrossRef CAS PubMed
- J. M. Palomo, C. Ortiz, M. Fuentes, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, J. Chromatogr. A, 2004, 1038, 267–273 CrossRef CAS PubMed
- P. Becher, Encyclopedia of Emulsion Technology: Basic Theory, Measurement, Applications, New York, Marcel Dek., 1988, vol. 3 Search PubMed
- J. T. Davies, Proc. Int. Congr. Surf. Act., 2nd, 1957, 426–438 CAS
- E. Y. Arashiro and N. R. Demarquette, Mater. Res., 1999, 2, 23–32 CrossRef CAS
- S. Wu, Polym. Eng. Sci., 1987, 27, 335–343 CrossRef CAS
- J. D. Berry, M. J. Neeson, R. R. Dagastine, D. Y. C. Chan and R. F. Tabor, J. Colloid Interface Sci., 2015, 454, 226–237 CrossRef CAS PubMed
- G. Könnecker, J. Regelmann, S. Belanger, K. Gamon and R. Sedlak, Ecotoxicol. Environ. Saf., 2011, 74, 1445–1460 CrossRef
- K. Parambath Kootery, H. Jiang, S. Kolusheva, T. P. Vinod, M. Ritenberg, L. Zeiri, R. Volinsky, D. Malferrari, P. Galletti, E. Tagliavini and R. Jelinek, ACS Appl. Mater. Interfaces, 2014, 6, 8613–8620 CrossRef CAS PubMed
- A. M. Algammal, H. F. Hetta, A. Elkelish, D. H. H. Alkhalifah, W. N. Hozzein, G. E. S. Batiha, N. El Nahhas and M. A. Mabrok, Infect. Drug Resist., 2020, 13, 3255–3265 CrossRef CAS
- K. M. Park, S. J. Lee, H. Yu, J. Y. Park, H. S. Jung, K. Kim, C. J. Lee and P. S. Chang, Food Sci. Biotechnol., 2018, 27, 401–409 CrossRef CAS PubMed
- X. Zhang, W. Wei, X. Cao and F. Feng, J. Sci. Food Agric., 2015, 95, 1631–1637 CrossRef CAS PubMed
- J. D. Monk, A. K. Hathcox and L. R. Beuchat, Food Microbiol., 1996, 13, 213–225 CrossRef
- A. Rabaron, G. Cavé, F. Puisieux and M. Seiller, Int. J. Pharm., 1993, 99, 29–36 CrossRef CAS
- I. C. R. Leal, K. R. N. Dos Santos, I. I. Júnior, O. A. C. Antunes, A. Porzel, L. Wessjohann and R. M. H. Kuster, Planta Med., 2010, 76, 47–52 CrossRef CAS PubMed
- CLSI, Performance Standards for Antimicrobial Susceptibility Testing, Twenty-Second Informational Supplement Clinical and Laboratory Standards Institute, 2015, vol. 32 Search PubMed
- H. D. Isenberg, J. Antimicrob. Chemother., 1988, 22, 73–86 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08111j |
| This journal is © The Royal Society of Chemistry 2022 |