
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUmpolung strategies for the functionalization of peptides and proteins
Andrew M.
White†
 ab,
Isabella R.
Palombi†
ab and
Lara R.
Malins
*ab
ab,
Isabella R.
Palombi†
ab and
Lara R.
Malins
*ab
aResearch School of Chemistry, Australian National University, Canberra, ACT 2601, Australia. E-mail: lara.malins@anu.edu.au
bAustralian Research Council Centre of Excellence for Innovations in Peptide and Protein Science, Australian National University, Canberra, ACT 2601, Australia
First published on 2nd February 2022
Abstract
Umpolung strategies, defined as synthetic approaches which reverse commonly accepted reactivity patterns, are broadly recognized as enabling tools for small molecule synthesis and catalysis. However, methods which exploit this logic for peptide and protein functionalizations are comparatively rare, with the overwhelming majority of existing bioconjugation approaches relying on the well-established reactivity profiles of a handful of amino acids. This perspective serves to highlight a small but growing body of recent work that masterfully capitalizes on the concept of polarity reversal for the selective modification of proteinogenic functionalities. Current applications of umpolung chemistry in organic synthesis and chemical biology as well as the vast potential for further innovations in peptide and protein modification will be discussed.
 Andrew M. White | Andrew White is a Postdoctoral Research Fellow at the Research School of Chemistry at the Australian National University, Canberra, Australia. He received his PhD in 2020 from the University of Queensland under the supervision of Dr. Thomas Durek and Prof. David Craik, where he worked on the synthesis of cyclic peptides for applications in medicinal chemistry and chemical biology. His current research in the lab of Assoc. Prof. Malins focuses on the chemical synthesis of ribosomally-synthesized and post-translationally modified peptides. |
 Isabella R. Palombi | Isabella Palombi received her BSc (Hons) in Chemistry from the Australian National University (ANU) in 2020. She is currently in the first year of her PhD in Organic Chemistry within the group of Associate Professor Lara Malins at the Research School of Chemistry, ANU. Her research interests include working at the interface of synthetic chemistry and biology to design and test new peptide therapeutic compounds. |
 Lara R. Malins | Lara Malins is an Associate Professor at the Research School of Chemistry at the Australian National University (ANU). She completed her PhD at The University of Sydney with Professor Richard Payne, focusing on the development of new peptide ligation strategies. In 2015, Lara joined the laboratory of Professor Phil Baran at The Scripps Research Institute as a National Institutes of Health postdoctoral research fellow. She returned to Australia in November 2017 to begin her independent career at the ANU, where her group focuses on the development of new synthetic methods for drug discovery, natural product synthesis, and chemical biology. |
Introduction
With growing appreciation for the value of peptide and protein-based therapies,1 synthetic strategies for the precise modification of these complex biomolecules are in exceedingly high demand. Although modern synthetic and biosynthetic approaches are routinely applied for the assembly of the peptide or protein backbone, the need to fine-tune both structure and function through targeted modifications of side chain functionalities has continued to push the boundaries of synthetic chemistry. The precision of nature's enzymatic post-translational modification machinery, characterized by exquisite chemo- and regioselectivity, serves as powerful inspiration. Over the past several years, chemists seeking to extend the scope of peptide and protein modifications to include the site-specific, on-demand incorporation of both native and designer functionality have successfully mined the rich interface of peptide science and cutting-edge organic chemistry. Advances in transition-metal catalysis,2 photochemistry,3 and electrochemistry4 are actively reshaping the toolbox of bioconjugation strategies.Despite the advances offered by such technological improvements, modern bioconjugation strategies still rely disproportionately on the conventional reactivity profiles of just a small selection of amino acid side chains (Fig. 1). Cysteine (Cys), lysine (Lys) and tyrosine (Tyr) are common protagonists owing to their nucleophilicity under physiological conditions.5 As peptide chemists, we are highly cognizant of the innate reactivity attributable to various side chain functionalities and have overwhelmingly designed and pursued strategies which exploit these properties. For example, nucleophilic Cys residues have been widely targeted for alkylation and arylation with an increasing variety of valuable new electrophiles.6 By comparison, strategies which pair electrophilic Cys derivatives with external nucleophiles in a reversal of the conventional reactivity profile—termed reactivity umpolung—are exceedingly rare. Nevertheless, given the scarcity of innately electrophilic proteinogenic side chain functionalities, electrophilic Cys equivalents have obvious advantages for the development of new chemo- and regioselective transformations.
This perspective aims to highlight the emerging value and vast untapped potential of umpolung approaches to peptide and protein modification. Following a brief historical overview of the terminology and the rise of umpolung strategies as a tool for retrosynthetic planning in small molecule synthesis, we will highlight the growing body of literature which serves to extend this concept to the synthesis and modification of biomolecules, organized broadly by functional group beginning with the amide functionality and progressing to amino acid side chain motifs. In the spirit of broadening our repertoire of bioconjugation reactions and our capacity for strategic planning, we will also briefly highlight strategies which explore underutilized or overlooked modes of innate reactivity. Our hope is that emerging methods will increasingly leverage not only well-established and intrinsic side chain reactivity but will also powerfully exploit as yet untapped reactivity (innate or umpolung).
Defining umpolung in the context of peptide and protein modifications
Before exploring polarity reversal in the context of peptide and protein functionalization, it is first prudent to examine the terminology and provide a brief historical perspective. Reactivity umpolung is perhaps best exemplified in the context of carbonyl chemistry—the very framework in which the terminology was first coined. In the late 1960s, Corey referred to the temporary reversal of the reactivity of a specific atom in a functional group as “symmetrization of reactivity”,7 highlighting emerging chemistry for the conversion of the electrophilic carbonyl carbon of an aldehyde (e.g.1) into a nucleophilic center through preparation of the corresponding lithiated 1,3-dithiane 2, an acyl anion equivalent (Fig. 1A).8 Evans alternatively referred to the inversion of conventional reactivity patterns as “charge affinity inversion” in discussing the versatile chemistry of allylic sulfoxides.9 Ultimately Seebach's reference to reactivity umpolung10 etched this term into the common lexicon, aided by his authoritative review in 1979 which summarized the concepts underlying polarity reversal and formalized the nomenclature.11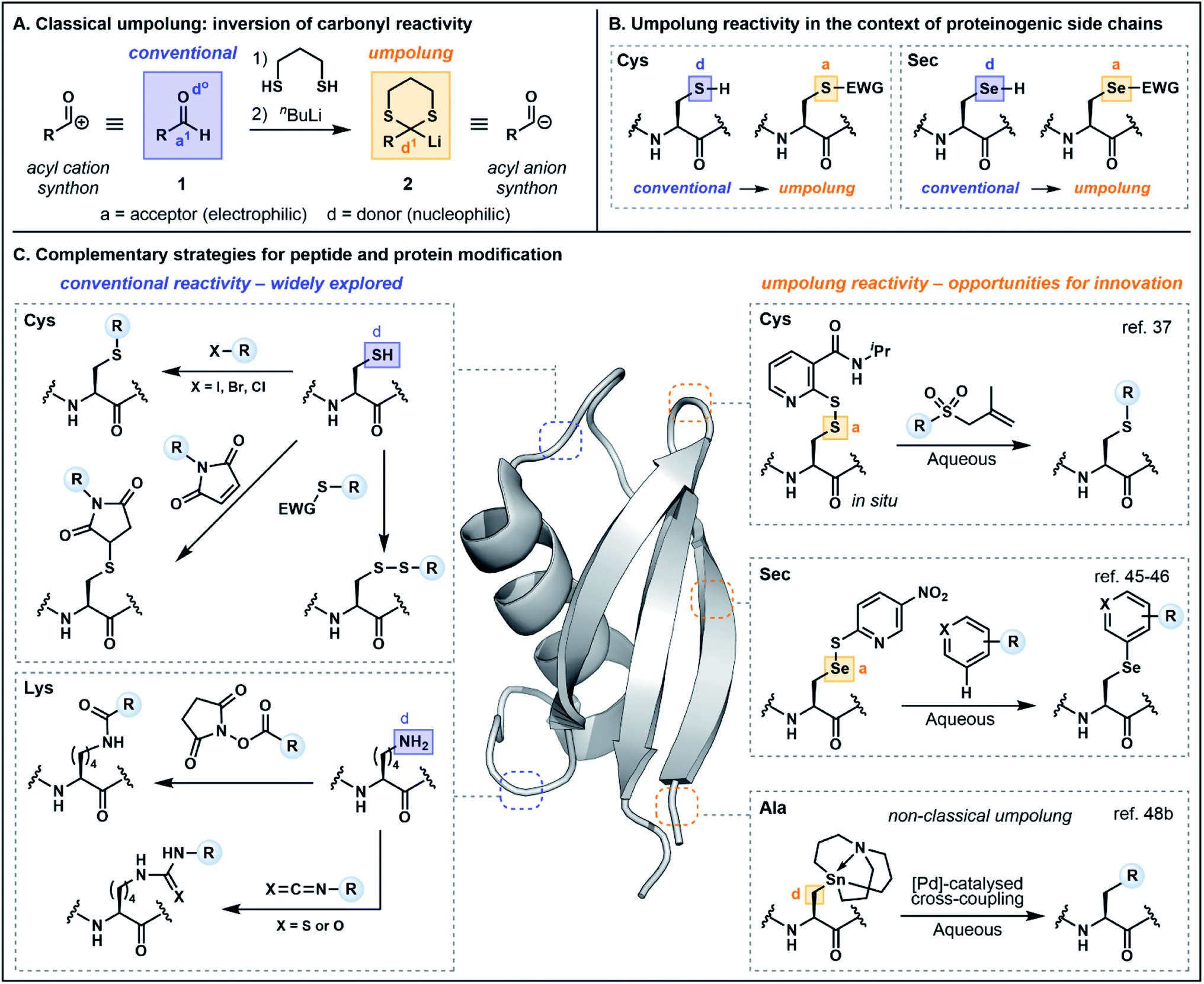 | ||
| Fig. 1 (A) Umpolung of carbonyl reactivity; (B) inversion of the innate reactivity of cysteine (Cys) and selenocysteine (Sec); (C) an overview of modern strategies highlighting conventional and umpolung approaches to bioconjugation. | ||
In Seebach's early exposition,11 conventional reactivity patterns are defined by alternating nucleophilic or donor (d) and electrophilic or acceptor (a) sites, with heteroatoms constituting donor centers (e.g. d0 describes the oxygen in aldehyde 1, Fig. 1A). Hence the electrophilic carbonyl carbon is an acceptor (a1, with superscript 1 referring to the distance from the heteroatom center). Accordingly, a reversal of the “a” and “d” centers constitutes reactivity umpolung, which is reflected in the reactivity of dithiane 2. The IUPAC definition of umpolung now broadly encompasses the inversion of any commonly accepted reactivity pattern;12 the present perspective will apply this more inclusive definition and highlight both “classical” as well as “non-classical” umpolung. The latter term is used in instances where a more liberal interpretation of the concept has been employed. For additional clarity, in figures and schemes herein, purple will be used to signify conventional reactivity while orange will signify reactivity umpolung. Atoms labeled as “a” and “d” will denote electron acceptors or donors, respectively, and for simplicity, the numerical superscripts have been omitted.
In the context of amino acids, simple heteroatom containing side chains are generally nucleophilic at the heteroatom center (e.g. the Cys sulfur and the selenocysteine (Sec) selenium, Fig. 1B). Exploiting the heteroatom as an acceptor constitutes reactivity umpolung. In the context of side chains with more nuanced reactivity patterns, umpolung is perhaps harder to distinguish from underexplored modes of innate reactivity. For example, at physiological pH, nitrogens in the histidine (His) imidazole side chain are nucleophilic, rendering C2 an acceptor atom based on the alternating donor/acceptor pattern proposed by Seebach.11 However, the electrophilicity of C2 has rarely been explored in bioconjugation chemistry (vide infra). Moreover, reactivity at C4/C5 is difficult to define based simply on alternating reactivity patterns. It should likewise be noted that while simple changes in pH may affect the protonation state—and accordingly the reactivity—of side chain functionality, protonation alone does not alter innate reactivity patterns. Deprotonation of Cys (pKa ∼ 8) is an illustrative example, in which the corresponding thiolate exhibits enhanced nucleophilicity. Nevertheless, changes in protonation state may serve as a valuable tool for exploiting underutilized modes of innate reactivity and for achieving enhanced chemoselectivity. The following sections will therefore provide a nuanced discussion of the multifaceted reactivity conventionally attributed to proteinogenic functionality, beginning with backbone amides and proceeding to side chain motifs, so as to more fully illuminate the key advantages afforded by umpolung approaches.
Amide umpolung strategies
Amide bonds are ubiquitous in nature, comprising the core component of peptide and protein backbones. Indeed, the amide functional group is present in 40% of bioactive compounds13 and is an important moiety in many agrochemicals and polymers.14 The chemical synthesis of amide bonds has almost universally been achieved via the condensation of carboxylic acids and amines, mimicking the protein synthesis process seen in nature.15 Typical strategies focus on enhancing the inherent electrophilicity of the carbonyl carbon associated with the carboxylic acid component through transformation into a more reactive acyl donor, such as an ester, acyl chloride or anhydride (Fig. 2A, acceptor), followed by nucleophilic substitution with an amine (donor). The in situ formation of activated esters with phosphonium or uronium salts (e.g. PyBOP, HATU, COMU) is a commonly employed method which generally proceeds in high yields. However, these reagents can be toxic and/or explosive,16 and the associated synthetic pathways typically require stoichiometric quantities of coupling reagents, producing large amounts of waste which may prove costly on industrial scales. Furthermore, sterically demanding nucleophiles (e.g. disubstituted amines) or poor nucleophiles (e.g. aniline derivatives) are challenging substrates, often resulting in low conversion and/or epimerization of the α-carbon of chiral carboxylic acids.16,17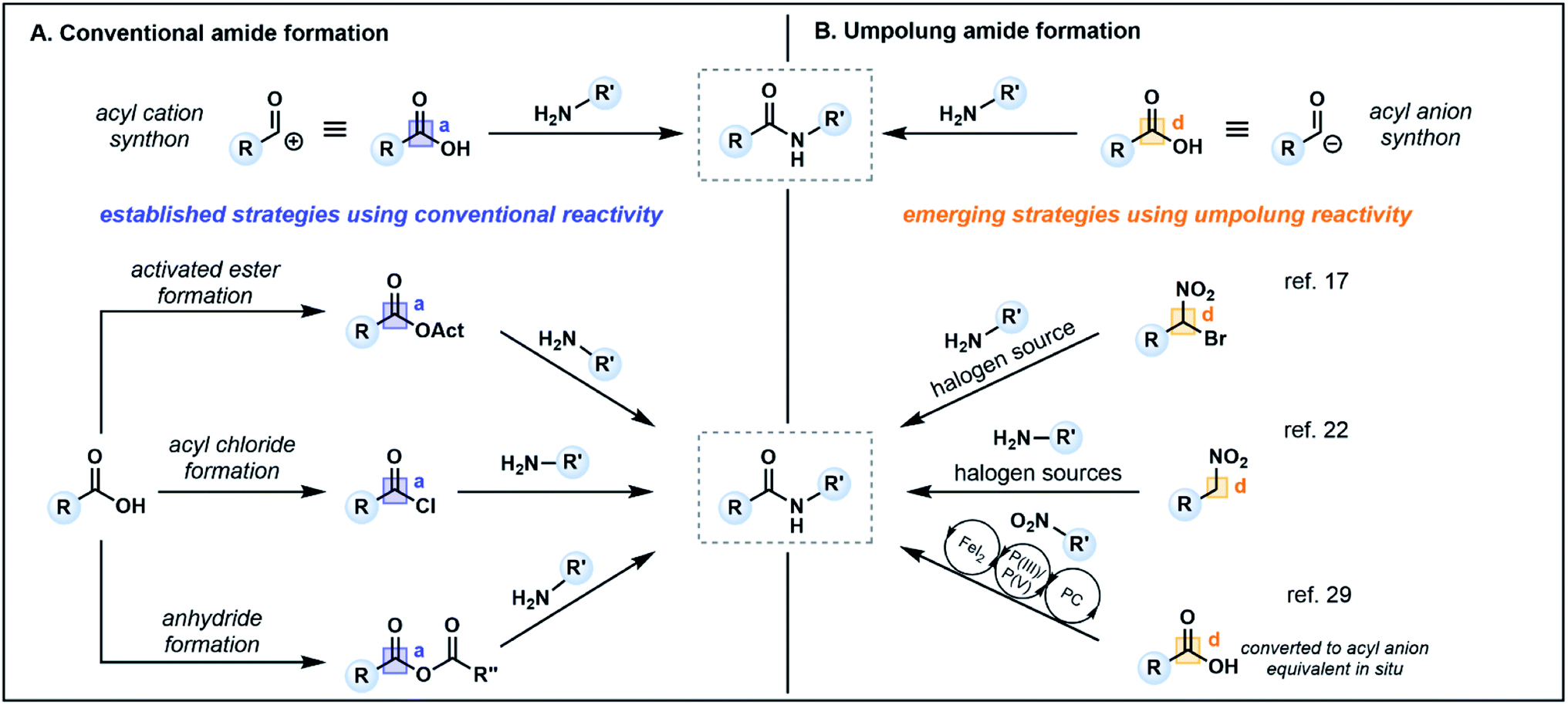 | ||
| Fig. 2 Conventional methods of amide synthesis vs. emerging strategies which utilize polarity reversed starting materials. PC = photocatalyst; Act = activating group. | ||
Recent alternatives to traditional amide synthesis methods have aimed to address these challenges.18 Amide synthesis involving polarity inversion of the carboxylic acid and amine reactants is an emerging strategy. So-called umpolung amide synthesis (UmAS), first coined by Johnston and co-workers,19 employs a nucleophilic acyl carbon (donor) and electrophilic nitrogen (acceptor) for carbon–nitrogen bond formation (Fig. 2B), in contrast to the polarization seen in conventional amide synthesis (Fig. 2A).17,19 Seminal work by the Johnston group utilized nitronates derived from α-bromo nitroalkanes (3) as nucleophilic acyl anion equivalents (cf.2, Fig. 1A) paired with an amine component (4) as a novel approach to amide bonds (Fig. 3A).17 The amine is converted into an electrophile in situ using N-iodosuccinimide (NIS) as a halonium source, with the resulting amide products (5) formed under mild reaction conditions in the presence of K2CO3. The methodology tolerates a variety of bulky substituents and functional groups on the nitroalkane and amine components, although aryl amines such as aniline were not tolerated. Tertiary amides and amides formed from lactonization-prone substrates, which are often hard to access using activated carboxylic acid approaches,17 were also compatible with this method (e.g.6). Moreover, UmAS was shown to be suitable in peptide synthesis (e.g.7, 23 peptide/amino acid examples), with no epimerization observed for chiral substrates.
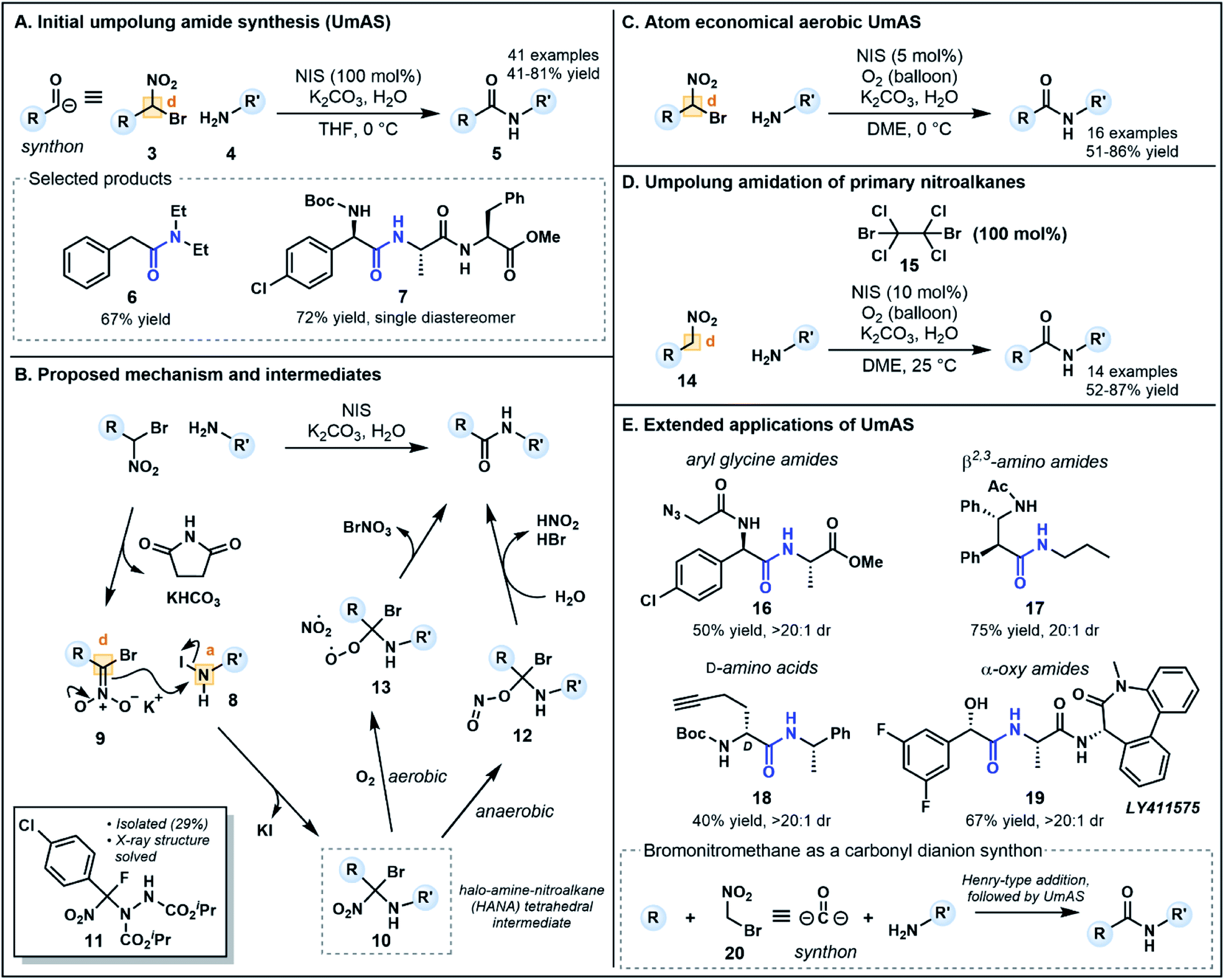 | ||
| Fig. 3 Iterations of umpolung amide synthesis (UmAS) developed by Johnston and co-workers and the proposed mechanism. Yields and diastereomeric ratios (dr) provided are for the UmAS step (amide bond formation highlighted in blue). | ||
Probing the mechanism has been pivotal in the refinement of UmAS. In their initial investigations, Johnston and co-workers hypothesized that the amine and halonium source afforded an electrophilic N-halo amine (8, acceptor), activating the nitrogen to nucleophilic attack from nitronate 9 (donor) under basic conditions (Fig. 3B). Following nucleophilic substitution, a 1,1,1 halo-amino-nitroalkane (HANA) tetrahedral intermediate (10) is formed. To date, this highly reactive intermediate has escaped observation, however, the structure of an intermediate (11) formed from a similar transformation between an α-fluoro nitroalkane and an azodicarboxylate electrophile has recently been elucidated (Fig. 3B).20 It was initially postulated that the tetrahedral intermediate is converted to the amide product by hydrolysis, however, various 18O-labeling experiments used to identify the origin of the amide oxygen highlighted the possibility of both anaerobic and aerobic pathways to the amide.
The anaerobic pathway is thought to involve isomerization of the nitro group to form a nitrite, followed by collapse of intermediate 12 to an amide by elimination of HBr and nitrous acid (Fig. 3B). Possible intermediates and the energy landscape of this pathway were probed using density functional theory calculations.20,21 The aerobic pathway instead involves competitive formation of a peroxide intermediate (13) via addition of O2 after fragmentation of the C–NO2 bond (Fig. 3B). Intermediate 13 can collapse to an amide upon elimination of bromonium nitrate.19 Interestingly, this pathway generates an alternative halonium source in situ, a finding that revealed substoichiometric equivalents of NIS (5–10 mol%) can be used in an aerobic setting without compromising yield (Fig. 3C).22 The one equivalent of electrophilic bromine (BrNO3) released in this pathway can provide an alternative halonium source for N-halo amine (c.f.8) formation. This development provides an advantageous atom economical approach to amidation that minimizes cost and waste generation compared to traditional methods which use large excesses of coupling reagents.
To increase the substrate scope available for the carbonyl feedstock, a one-pot amidation was later developed by Johnston and co-workers starting from primary nitroalkanes (14, Fig. 3D).23 In this second generation approach, the α-bromo nitroalkane required for UmAS is formed in situ in the presence of a brominating reagent. Screening of a range of electrophilic bromine reagents and bases revealed that dibromotetrachloroethane 15 (100 mol%) and K2CO3 were the best combination for this transformation, along with NIS (10 mol%) under an oxygen atmosphere for aerobic UmAS. Concurrently, Hayashi and co-workers developed an oxidative amidation starting from primary nitroalkanes as a means of improving substrate scope.24 Primary nitroalkanes reacted with amines in the presence of I2 and K2CO3 under an atmosphere of O2, providing efficient access to dipeptides (13 examples). Although the approach is ostensibly similar to UmAS, the group's detailed mechanistic insights suggest that the reaction exploits conventional reactant polarities, whereby the nitroalkane is converted to an electrophilic activated ester and the amine functions as a nucleophile.25
UmAS has been further extended to a considerable collection of synthetic applications (Fig. 3E) including the construction of aryl glycine derivatives (e.g.7, 16), β2,3-amino amides (e.g.17) and L- and D-amino acids (e.g.18) using bis(amidine) catalysts.26 Moreover, α-oxy amides can be synthesized using enantioenriched α-bromo nitroalkane precursors readily forged from chiral copper(II) complexes,27 for example γ-secretase inhibitor LY411575 19, the synthesis of which has previously incorporated the α-oxy motif as a racemic mixture, requiring chromatographic separation following the amidation step.28 Avoiding epimerization during the C–N bond formation, a common problem with existing methods, is a powerful advantage to the approach particularly in the formation of aryl glycine derivatives.17 Notably, in each application except the construction of β2,3-amino amides, bromo nitromethane (20) acts as a carbonyl dianion synthon (Fig. 3E) in which base-mediated formation of a nitronate first enables installation of the “R” group through a Henry-type addition. Subsequent umpolung amidation provides a doubly umpolung approach to the synthesis of amides that are not readily accessible via traditional methods.
Recently, Xie and co-workers developed an alternative umpolung amidation strategy involving a carboxylic acid (21) and modified amine reagent, in contrast to the technology introduced by Johnston.29 The approach employs a P(III)/P(V) catalytic cycle to generate a nucleophilic acyl anion equivalent in situ that can react with a nitroarene or nitroalkane (22) to form an amide (Fig. 4).30 Reactions proceed in acetonitrile with K2HPO4 and a catalytic system comprised of FeI2, an organophosphine precatalyst 23 and an iridium photocatalyst in the presence of PhSiH3 as the reductant. Working in synergy, the catalysts initially generate a phosphine radical cation (24) from 23, which can recombine with the carboxylate and undergo β-scission to form acyl radical 25 (donor). Concurrently, the nitro group is converted into a nitroso group (26) in the presence of FeI2 and silane reductant. Nucleophilic addition of the acyl radical into 26 (acceptor) forms an N-hydroxy amide (27) which is reduced to the corresponding amide 28 (Fig. 4). The methodology tolerates a variety of functional groups on the carboxylic acid and nitro components. Pleasingly, aryl amides can be readily accessed using nitroarenes, however, tertiary amides cannot be formed due to constraints imposed by the nitro group on the amine equivalent. Finally, the amidation of carboxylic acids within peptides and amino acids is possible (six examples), indicating a future avenue for this method as a late-stage peptide modification strategy—a prospect aided by the advantage of employing unmodified carboxylic acid starting materials.
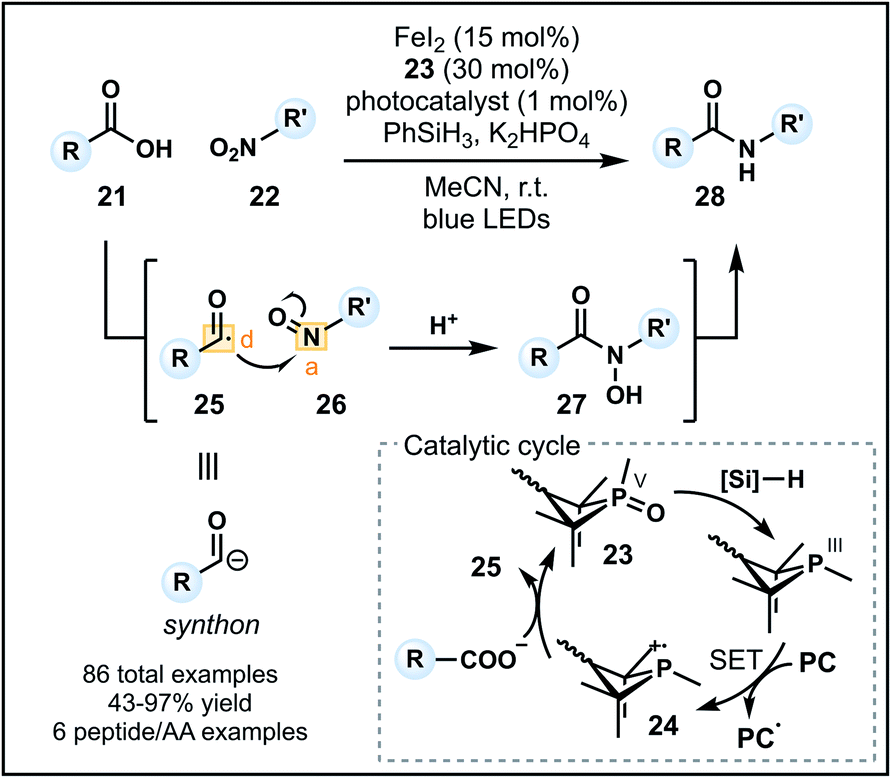 | ||
| Fig. 4 Umpolung amidation utilizing the P(III)/P(V) catalytic cycle. Key intermediates that are important in umpolung C–N bond formation are shown. PC = photocatalyst; SET = single electron transfer; AA = amino acid. | ||
Perspective
Alternative methods for amide bond formation, particularly those which overcome the limitations of classical approaches, continue to provide valuable new strategies for the chemist's toolbox. To this end, the umpolung amidation strategies discussed above have begun to address conventional limitations in reaction scope, exemplified by the successful formation of tertiary amides and aryl amides as well as compatibility with epimerization prone substrates. Moreover, such strategies may provide additional layers of orthogonality relative to traditional amidation chemistry by broadening the number of viable retrosynthetic strategies for the total syntheses of amide-rich natural products and providing new pathways for late-stage peptide modification. These initiatives are aided by the ability to form amide bonds in the presence of competing nucleophiles and sensitive functional groups, whilst minimizing protecting group manipulations. However, the routine adoption of such amidation strategies will likely require approaches that continue to exploit readily available carboxylic acid and amine feedstocks as starting materials, a challenge for the development of umpolung amidation platforms. Alternative methods have been developed with this limitation in mind, including strategies involving amine activation or catalytic amidation.31 The wide availability of amino acids and the compatibility of common coupling reagents with solid phase peptide synthesis (SPPS) are no doubt compelling reasons for the broad popularity of existing amidation strategies. Nevertheless, the valuable conceptual and mechanistic breakthroughs afforded by the pursuit of umpolung amidation strategies have challenged conventional thinking and will continue to inspire the development of innovative strategies for diverse applications in synthetic chemistry.24,31a,b,32Umpolung strategies for the functionalization of amino acid side chains
Side chain functionalities are frequently targeted for chemoselective peptide and protein modifications by taking advantage of the diversity and inherent reactivity of the canonical amino acids. A selection of recent strategies has exploited polarity inversion as an alternative means of achieving residue-specific modifications, including for valuable applications in total synthesis and medicinal chemistry. The following sections, organized by amino acid side chain functionality, will detail these advances.Cysteine (Cys) umpolung strategies
As noted in the introduction, the Cys thiol is well known for its nucleophilic character and is widely utilized in peptide and protein chemistry. Many such transformations, including oxidations, nucleophilic substitutions and Michael additions (see Fig. 1C for representative examples) have demonstrated applications in chemical biology, medicinal chemistry and organic synthesis, making Cys arguably the most versatile residue for the construction of functionalized biomolecules.5b,33 However, the Cys thiol can also be readily modified with electron withdrawing functionality to afford electrophilic Cys equivalents (Fig. 5A) in which the sulfur atom serves as an acceptor. The inverted polarity of these Cys derivatives primes them for nucleophilic attack, and this umpolung strategy has been exploited for the generation of novel approaches to challenging natural products and for the development of new late-stage functionalization methods.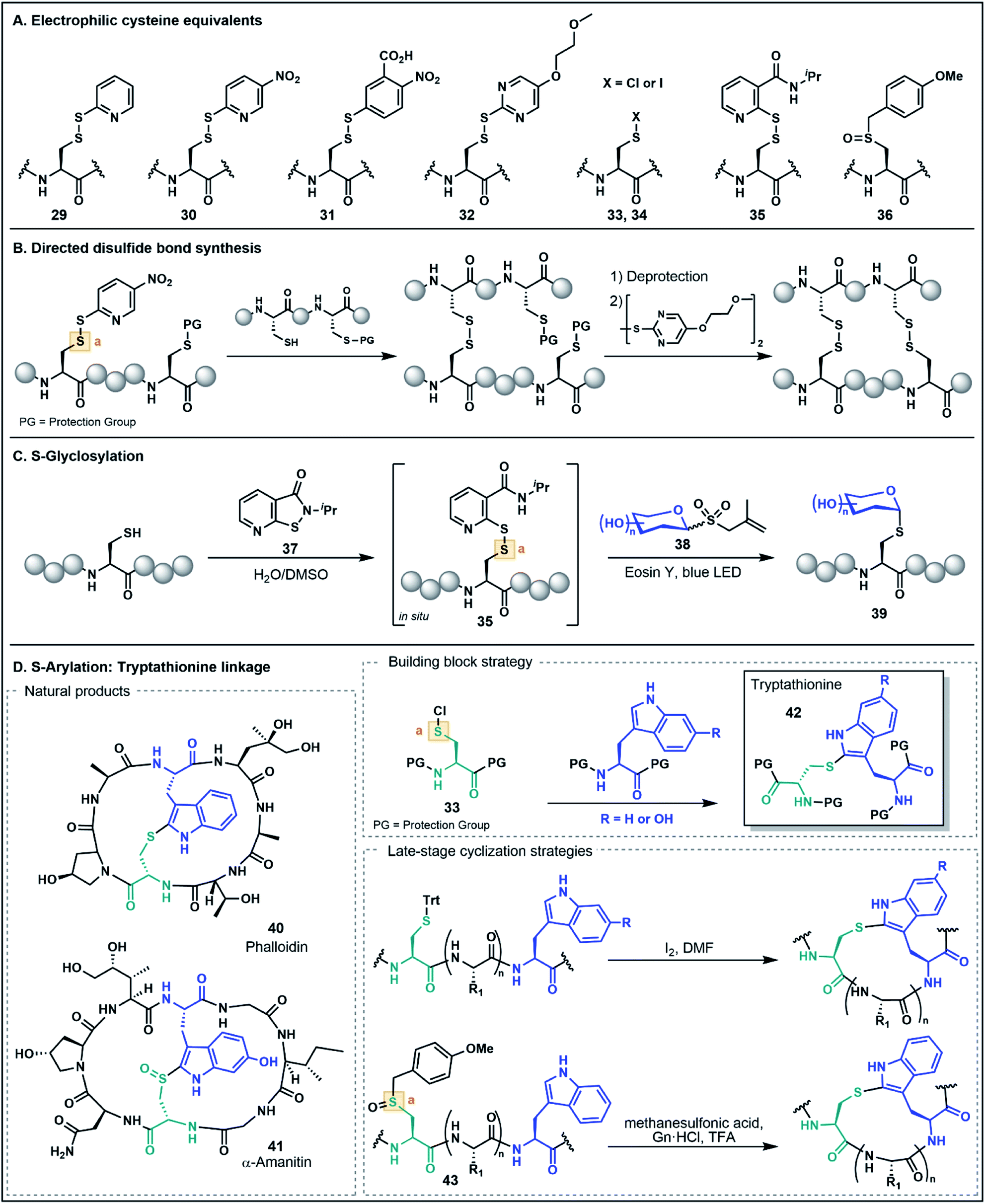 | ||
| Fig. 5 Umpolung strategies using Cys. (A) Electrophilic Cys equivalents implemented in umpolung approaches. (B) An example of a directed disulfide bond synthesis employing electrophilic Cys equivalents for the assembly of a multi-stranded peptide. (C) Late-stage S-glycosylation using an isothiazolone sulfenylating agent. (D) Umpolung preparation of tryptathionine linkages for the synthesis of amatoxins and phallotoxins. | ||
One of the most common approaches for the generation of electrophilic Cys variants is via activation of Cys with a nitropyridine sulfenyl, pyridine sulfenyl, or other electron-deficient thiol-activator (e.g. see 29–32), such as those commonly used in directed disulfide bond synthesis (Fig. 5A and B).34 These electrophilic Cys residues are routinely prepared by reacting the unprotected or protected Cys thiol with an activating symmetrical disulfide (e.g. 2,2′-dithiobis(5-nitropyridine) or 2,2′-dithiodipyridine) which undergoes disulfide exchange to afford an activated cystine under both acidic and basic conditions.34,35 In the case of disulfide synthesis, the thiol moiety of another nucleophilic Cys residue reacts with the activated, electrophilic Cys (inter- or intramolecularly), resulting in the selective formation of a disulfide bond (Fig. 5B). The excellent regioselectivity of this approach has led to its routine use in the synthesis of multi-stranded peptides with several disulfide bonds, and recent examples have utilized these strategies for the efficient preparation of human relaxins and insulins.35,36
The utility of umpolung Cys approaches is not just limited to the formation of disulfide bonds. A recent report by Niu and co-workers described the use of an electrophilic Cys equivalent for the selective late-stage glycosylation of Cys residues (Fig. 5C).37 In this approach, an allyl glycosyl sulfone (38) serves as a precursor to an unprotected, nucleophilic glycosyl radical generated under photocatalytic conditions. Trapping of this radical by the electrophilic Cys furnishes an S-glycosylated Cys residue (39).37 Two iterations of this method were developed, with the first utilizing a classical pyridine sulfenyl electrophilic Cys residue (e.g.29) to generate glycosylated peptides. The second-generation strategy overcomes the need to isolate the activated electrophilic intermediate by generating the activated Cys 35in situ using an isothiazolone sulfenylating agent 37. This strategy provides an operationally simple procedure for stereoselective S-glycosylation under aqueous conditions and in the absence of protecting groups (Fig. 5C)37 and highlights the role of umpolung strategies in the production of peptides bearing valuable post-translational modifications.
Electrophilic Cys has also found application in the assembly of synthetically challenging ribosomally-synthesized and post-translationally modified peptides (RiPPs), such as phalloidin (40) and α-amanitin (41) found in death cap mushrooms from the genus Amanita (Fig. 5D).38 This class of highly toxic peptides is characterized by a unique tryptathionine linkage (42) between the side chains of Cys and tryptophan (Trp) that forms the core bicyclic structural motif of these peptides. One strategy to synthetically access the tryptathionine linkage has been through utilization of electrophilic sulfenyl halides (33–34) that undergo electrophilic aromatic substitution with the nucleophilic 2-position of the Trp indole functionality to forge the side chain thioether linkage.39 This has been employed by both Guy and co-workers39a as well as Süssmuth and co-workers39b in a building block approach whereby a suitably N- and C-protected Cys is treated with sulfuryl chloride to prepare the sulfenyl chloride (33), and subsequent addition of a protected Trp residue yields the tryptathionine dipeptide (e.g.42, Fig. 5D). Tryptathionine dipeptide building blocks have been utilized in the assembly of both a phalloidin derivative and for the total synthesis of α-amanitin.39
In another umpolung approach to the tryptathionine linkage reported by Schuresko and Lokey, iodine was used to both deprotect and activate Cys residues for thioether formation with the Trp indole (Fig. 5D).40 This strategy was inspired by a side reaction observed during the I2-mediated deprotection and oxidation of Cys side chains, namely the identification of a minor Trp–Cys thioether-linked byproduct presumably occurring through a sulfenyl iodide intermediate (e.g.34) that reacts via electrophilic aromatic substitution with the indole ring.40,41 Optimization of this strategy on-resin was accomplished by using a low-loading resin (0.1 g mol−1), which resulted in favorable formation of the Trp–Cys linkage over competing intermolecular disulfide formation, thus enabling the synthesis of a fluorescently labeled phalloidin analogue in good yield.40 This iodine-mediated reaction has also been utilized in the synthesis of amanitins and related analogues, with the efficiency of the approach facilitating systematic SAR studies further advancing the potential of the death cap toxins for medicinal and chemical biology applications.42
Recently, Otaka and co-workers also prepared the tryptathionine linkage using a masked sulfenyl chloride intermediate derived from an S-p-methoxybenzyl Cys sulfoxide 36.43 In this approach, the methoxybenzyl Cys sulfoxide (43, Fig. 5D) is oxidatively activated to a sulfonium cation under the acidic reaction conditions. It is then postulated that the presence of Gn·HCl mediates formation of a Cys-derived sulfenyl chloride which engages the Trp indole side chain to form the tryptathionine linkage. In contrast to the aforementioned sulfenyl chloride building block approaches, the mild electrophilicity of the S-p-methoxybenzyl Cys sulfoxide allows for facile incorporation using SPPS and the ‘on-demand’ formation of the more activated sulfenyl chloride intermediate enables late-stage modification. This has been exemplified in the synthesis of an amanitin derivative with the tryptathionine linkage formed as the last step of the synthesis on the unprotected peptide substrate. Moreover, these conditions were amenable for a stapling strategy in the preparation of a tryptathionine-stapled estrogen receptor-α activity-regulator peptide (ERAP) analogue.43
Selenocysteine (Sec) umpolung strategies
The 21st amino acid, Sec, is a versatile residue for functionalization owing to its nucleophilicity and similarities to its close relative Cys. Accordingly, many approaches used for Cys modification are also mirrored with Sec. However, in contrast to Cys, the selenol functionality has greater acidity (pKa ∼ 5.5) and a lower redox potential, which promotes facile oxidation of the selenol to the corresponding diselenide under mild conditions.44 These characteristics can be advantageous in certain instances, enabling the selective functionalization of Sec in the presence of Cys. Nevertheless, the high polarizability of selenium can lead to dehydroalanine (Dha) formation via elimination, and the use of reducing agents and the stringent exclusion of oxygen to preserve the nucleophilicity of the reduced selenol can be operationally challenging. Accordingly, umpolung based strategies that utilize the electrophilicity of oxidized Sec residues represent attractive alternatives.As with Cys-based umpolung strategies, electrophilic Sec equivalents present a stable but activated functionality that are susceptible to nucleophilic attack. This concept was first explored for late-stage peptide modification by Pentelute, Buchwald and co-workers through chemoselective Se-arylation of 2-thiol-5-nitropyridine activated Sec (44) with aryl boronic acid nucleophiles (Fig. 6).45 The reaction proceeds via a copper-mediated pathway whereby oxidative addition of the electrophilic Sec onto a copper–ligand complex is followed by transmetalation with an aryl boronic acid and reductive elimination to yield an arylated Sec. The strategy is broadly applicable in aqueous conditions and has been demonstrated on a variety of aryl substrates with chemoselectivity for Sec in the context of unprotected peptides.45
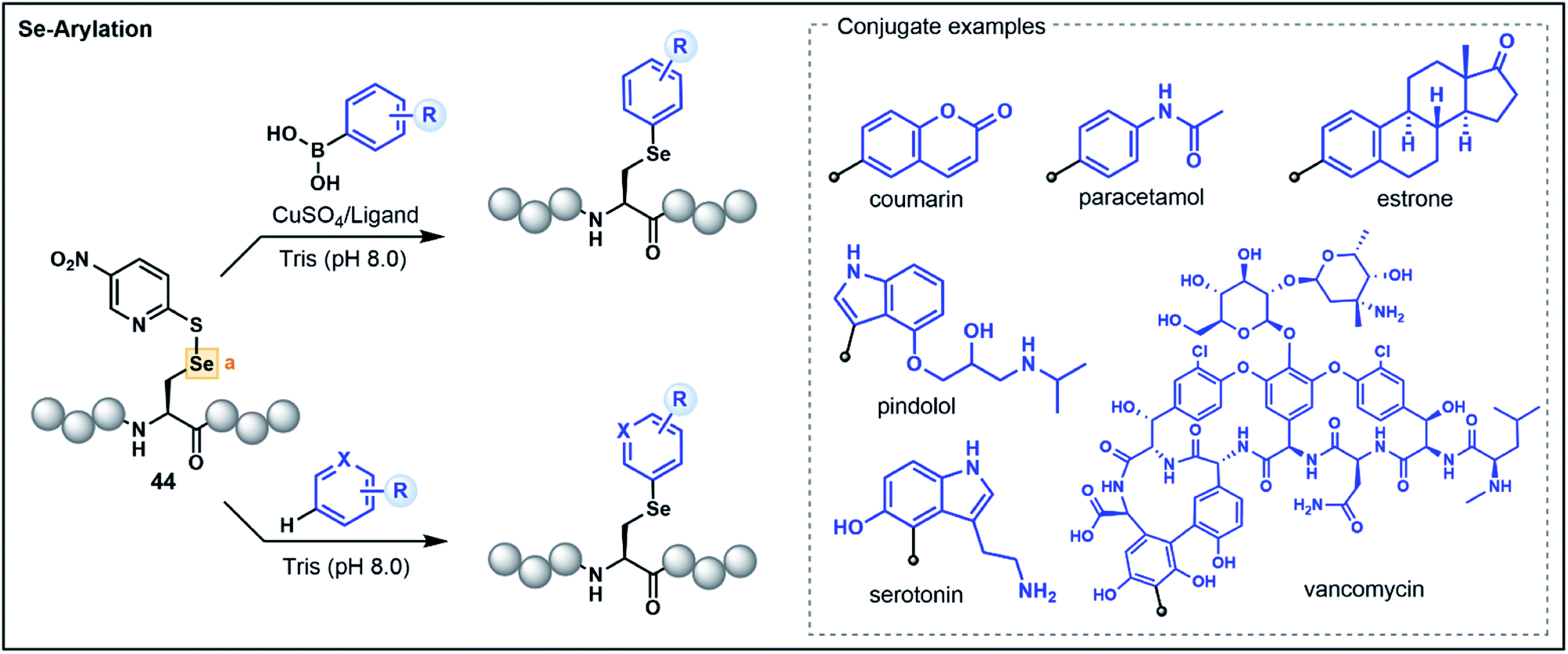 | ||
| Fig. 6 Late-stage umpolung arylation strategies using a 2-thiol-5-nitropyridine activated Sec. | ||
A more recent report extended the strategy to nucleophilic, electron-rich aromatic substrates without the need for boronic acid-functionalized precursors or a copper catalyst (Fig. 6).46 This electrophilic selenium conjugation proceeded in aqueous buffered conditions (pH 8.0) in the absence of additives, and was used to conjugate arenes and heteroarenes, including various natural product and pharmaceutical examples, onto peptide substrates in good yields.46 Where poor yields of the conjugated products were observed (<40%), the addition of a copper bound ligand was shown to enhance the desired yield (>60%), further extending the utility of the approach for challenging substrates. The authors also demonstrated that the strategy could be used to access a variety of conjugated biomolecules, including peptide–vancomycin and protein–vancomycin conjugates by utilizing the embedded electron-rich resorcinol ring in vancomycin as the conjugation site.46 The exceptional versatility of this Sec functionalization has enabled efficient access to challenging peptide conjugates and will be of great value for medicinal chemists for the development of novel drug conjugates.
Alanine (Ala) umpolung strategies
On first inspection the aliphatic Ala side chain stands as a recalcitrant target for the chemoselective functionalization of peptides and proteins. The β-carbon is ostensibly neither a donor nor an acceptor. However, the structurally-related non-proteinogenic derivative dehydroalanine (Dha) has gained considerable attention as a functionalization handle due to its inherent electrophilicity at the β-position and ability to undergo Michael-type conjugate additions with a diverse range of nucleophilic substrates to generate Ala derivatives.47 One drawback of this strategy is the lack of stereochemical control at the α-position leading to a diastereomeric mixture of substituted products, limiting the potential applications of the approach. Nevertheless, reactions at Dha remain the most prominent and versatile strategies to forge new bonds to the Ala β-carbon.In light of the well-explored electrophilicity of Dha, strategies which enable the Ala β-carbon to serve instead as a nucleophile are discussed here as a form of non-classical umpolung reactivity for the synthesis of modified Ala residues. Such strategies have emerged as powerful new methods to build C–C bonds at the β-position via Pd-catalyzed cross-coupling reactions, expanding the repertoire of strategies for Ala functionalization. In particular, metalated Ala (AlaM) building blocks, including carbagermatrane and carbastannatrane derivatives, have emerged as highly stable and versatile nucleophilic handles that can undergo functionalization with various halogenated electrophiles.48 This work was first exemplified by Xiao and co-workers48a through the preparation of various orthogonally-protected Ala building blocks bearing a carbagermatrane group at the β-position (e.g. AlaGe, 45) (Fig. 7A). In the presence of catalytic Pd(dba)2 and a phosphine ligand 46, the AlaGe building blocks were coupled with bromoaryl substrates to generate unnatural β-aryl amino acids without compromising stereochemistry at the α-carbon position.48a This strategy was next extended to the preparation of Fmoc- and Boc-protected AlaGe building blocks which were employed under standard SPPS conditions to assemble an octapeptide (47) containing two AlaGe and two bromophenylalanine residues. Remarkably, the resin bound peptide underwent a dual cross-coupling sequence to generate a bicyclic peptide (48) in good yield (20% overall). Although the requirement for an inert atmosphere and relatively harsh reaction conditions (120 °C for 16 h) might limit the applicability to more sensitive peptide substrates, this example highlights the broad potential for Ala-based umpolung strategies in peptide modifications.48a
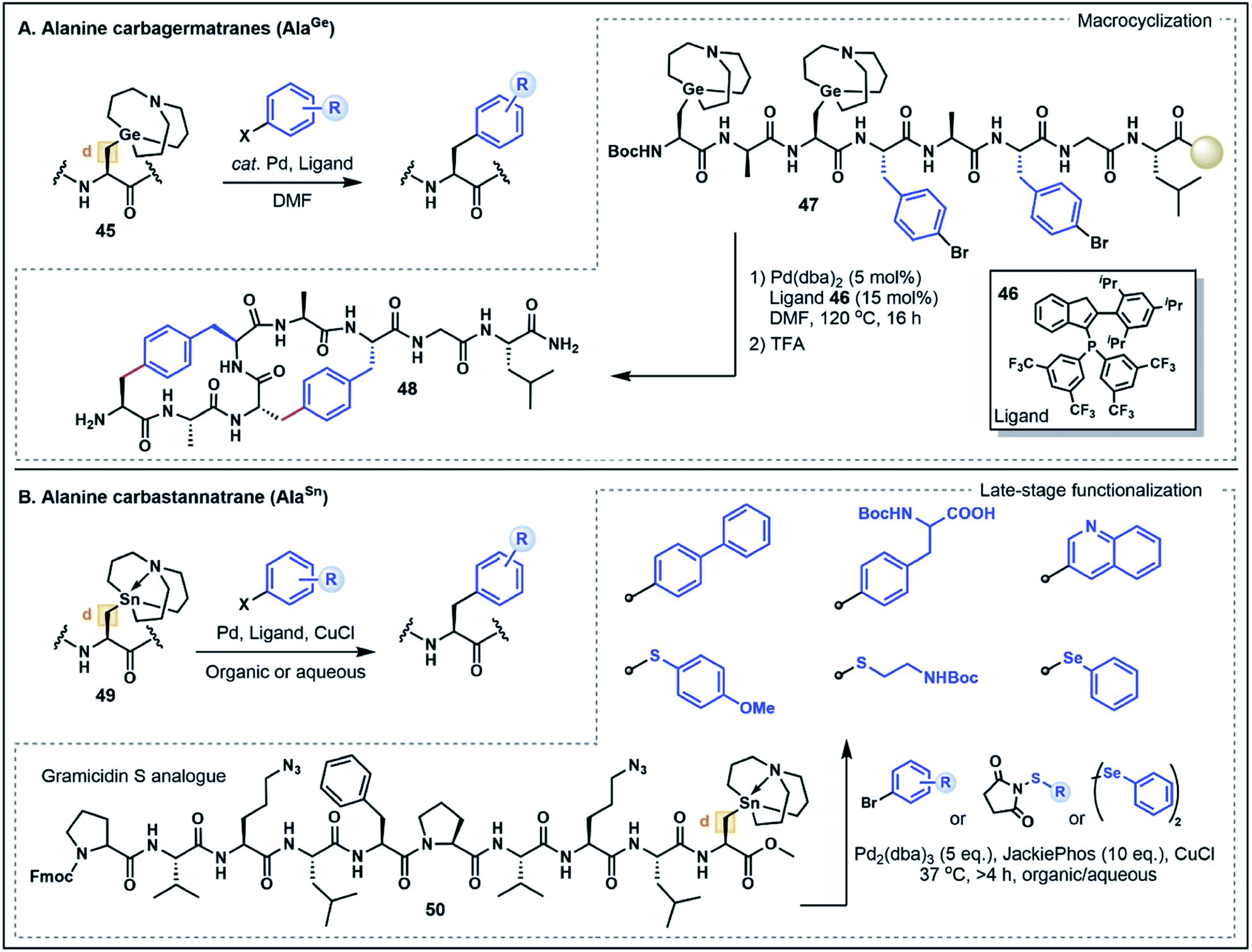 | ||
| Fig. 7 Ala umpolung strategies. (A) Overview of Ala carbagermatrane derivatives and their use in peptide macrocyclization. (B) Overview of Ala carbastannatrane derivatives and their applications in late-stage functionalization. | ||
A more recent AlaM umpolung strategy reported by Walczak and co-workers explored the use of functionalized carbastannatrane Ala residues 49 (AlaSn) that can be transformed under Stille cross-coupling conditions (Fig. 7B).48b This method was first optimized on dipeptide fragments showing that the reaction can be used to generate C–C, C–S and C–Se functionalized Ala residues. In contrast to the AlaGe species, the AlaSn derivatives are stable in aqueous media and reactions were performed in good yield in both organic and biocompatible aqueous buffered solutions (pH 6.5–8.5), often at ambient temperature.48b The broad scope of this strategy prompted further investigation of the method for peptide cyclization to afford small di- and tripeptide cyclic products. The reaction platform was also applied to the late-stage functionalization of a gramicidin analogue 50, where the embedded AlaSn residue was modified with aryl and alkyl electrophiles in moderate to good yields.48b Notably, the assembled gramicidin analogue contained ornithine to azidoornithine mutations and the AlaSn building block was installed via solution peptide phase synthesis techniques. Further exploration of the broader functional group tolerance and compatibility of the AlaSn building blocks with standard SPPS conditions remain to be explored. Nevertheless, both the AlaSn and AlaGe strategies highlight unique umpolung transformations that complement existing Dha-based functionalization approaches. These methods thus serve to greatly expand the chemist's toolbox for broad applications in the assembly of complex peptides.
Histidine (His) umpolung strategies
The His residue is a rather unique amino acid due to its ionizable aromatic side chain (pKa ∼ 6.1)49 that can exist as both the protonated imidazolium or the deprotonated imidazole under physiologically relevant conditions. Although the His side chain is known for its nucleophilic properties (e.g. modification through N-alkylation via substitution or addition reactions),50 in the context of peptides and proteins, the mild nucleophilicity of the nitrogen donors on the imidazole ring is often overshadowed by the nucleophilicity of Cys and Lys which limits the possibility of using His for selective functionalization strategies. This limitation can be overcome by exploiting the inherent electrophilicity of the imidazole C2 position (vide infra) or using unique umpolung strategies that generate alternative electrophilic sites on the His side chain. The latter approach has recently been explored by Sato and co-workers for labeling at the C5 position of the imidazole ring in peptides and proteins.51 This strategy was inspired by prior reports which identified His–His, His–Cys and His–Lys cross-linked side chains in the analytical analyses of monoclonal antibodies that were attributed to a photooxidative cross-linking mechanism.52 Capitalizing on this pathway, the His labeling approach was developed through the photocatalytic generation of singlet oxygen, which subsequently undergoes a Diels–Alder reaction with the His imidazole side chain to afford an electrophilic endoperoxide intermediate 51 (Fig. 8A).51 In the presence of a 1-methyl-4-arylurazole nucleophile 52, the oxidized His intermediate is trapped, leading to a C5 conjugated product 53. This strategy was exemplified by labeling peptides and antibodies with azide handles for fluorescent tagging; modifications occurred rapidly (∼15 min) at the embedded His residues under biocompatible conditions. It is envisaged that the efficiency of this approach may also allow for the extension of the strategy to intracellular applications.51 Notably, this His labeling strategy does not maintain the imidazole structure in the conjugated product 53. This method therefore represents a non-classical umpolung approach as the majority of umpolung strategies preserve the primary structural features of the starting amino acids in their conjugated products.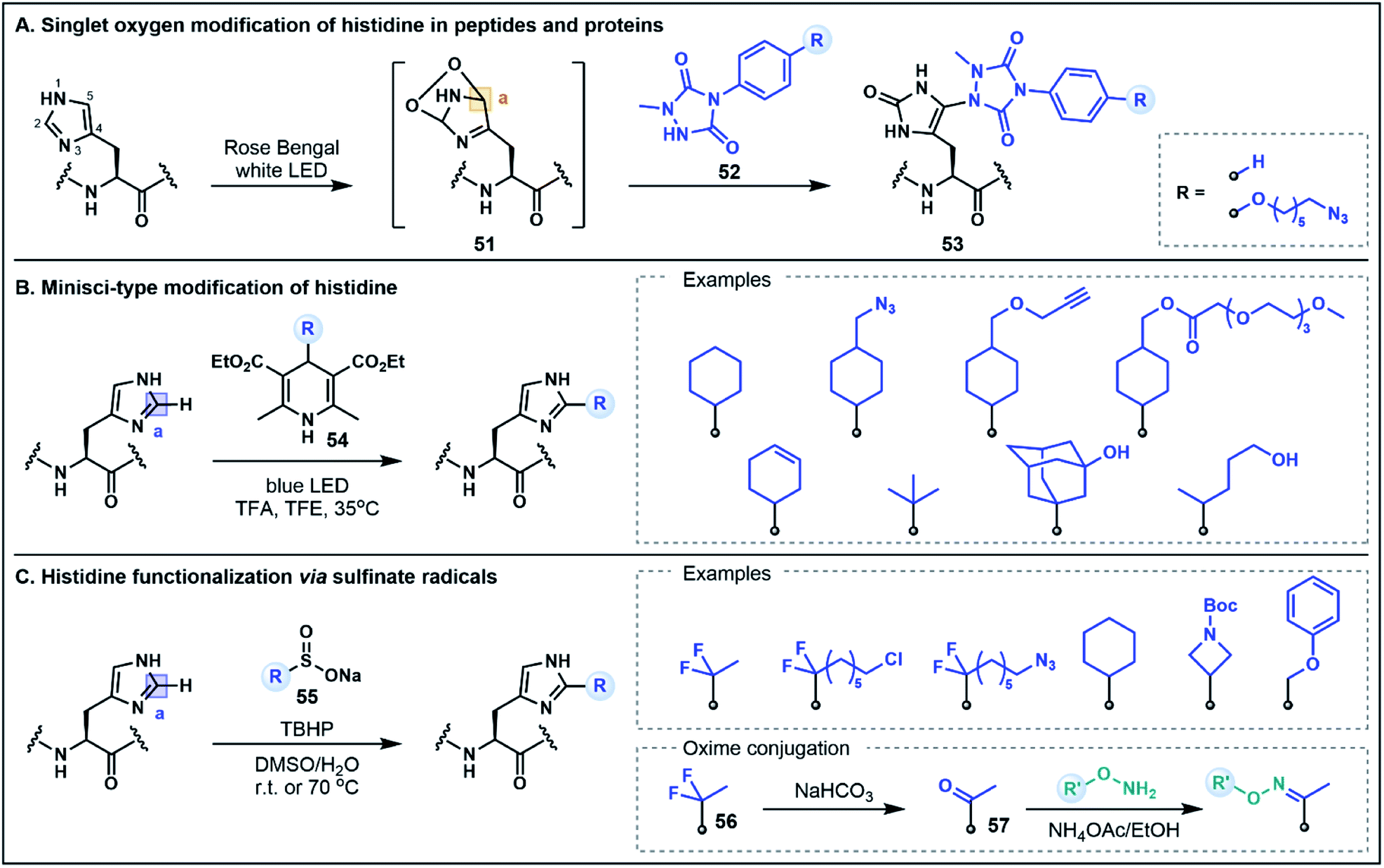 | ||
| Fig. 8 His functionalization using umpolung-like strategies. (A) Singlet oxygen photooxidative functionalization using 1-methyl-4-arylurazole nucleophiles. (B) Minisci-type functionalization of His using photochemically generated alkyl radicals. (C) His functionalization using sulfinate salts as radical precursors. | ||
As evidenced above, harnessing the electrophilic nature of His is an innovative way to devise new methodologies to selectively modify the imidazole side chain. Although not formally defined as umpolung chemistry, there are new methods emerging that capitalize on the inherent electrophilic reactivity of His that is often overlooked in favor of the more conventional nucleophilic His modification strategies. Specifically, in its protonated state, the imidazolium ring contains an electron deficient C2 position that can be modified using nucleophilic radicals in Minisci-type processes. In one such strategy developed by Chen, Wang and co-workers, photochemical activation of 4-alkyl-1,4-dihydropyridine reagents (e.g.54) serves to generate alkyl radicals capable of alkylation at the C2 position of the His side chain (Fig. 8B).53 Using this approach, a range of different peptide scaffolds (e.g. saralasin, bremelanotide, secretin, ubiquitin) were selectively labeled at His with a variety of different alkyl substituents, including azide and alkyne handles, generally in moderate to good yields.53 Similarly, Noisier, Gopalakrishnan and co-workers demonstrated that His can be selectively labeled using aliphatic sulfinate salts (55) as radical precursors (Fig. 8C).54 This alkylation strategy was applied to a wide variety of bioactive peptide substrates, leading to good residue specificity with a number of alkyl sulfinates. Although electrophilic radicals derived from sulfinate salts (e.g. ˙CF3) have also been shown to engage Trp and Tyr residues,55 the selectivity for His in this instance stems from the favorable pairing of nucleophilic alkyl radicals with the enhanced electrophilicity of the protonated imidazolium formed under the acidic reaction conditions. Furthermore, His alkylation using a gem-difluoro handle (56) could be followed by quantitative hydrolysis to a substituted ketone (57), which was then utilized for oxime ligations, generating a range of peptide conjugates that included a fluorescent tag, a hydrophilic tag, a lipid, a small molecule drug and even an oligonucleotide conjugate.54,56 Both of these C2 His tagging strategies are excellent examples of novel methodologies that utilize underexplored reactivity pathways for diverse applications in organic and medicinal chemistry.53,54
Tyrosine (Tyr) umpolung strategies
The electron-rich side chain of Tyr is known for its nucleophilic character and the vast majority of Tyr functionalization strategies rely on the donor properties of the phenolic oxygen and the adjacent ortho carbons at C3 and C5 (Fig. 9).5b Expanding the breadth of Tyr functionalization, various strategies are emerging that depart from the conventional nucleophilic pathways by utilizing the phenolic motif instead as an acceptor. The oxidative coupling of phenols is one approach in which phenolic motifs serve as both the nucleophile (donor) and electrophile (acceptor). This strategy has been elegantly used in the synthesis of cross-linked peptide natural products featuring Tyr and 4-hydroxyphenylglycine (e.g. the arylomycins).57 In this perspective we will focus on recent strategies for which Tyr serves unambiguously as the acceptor. Many such approaches employ an initial dearomatization to enable secondary reactions at various sites within the Tyr side chain. A recent enzymatic approach by Fellmann, Doudna, Francis and co-workers utilizes tyrosinase to selectively and efficiently (<1 h) oxidize Tyr to the o-quinone electrophilic intermediate 58, under mild aerobic conditions (Fig. 9A).58 The o-quinone intermediate can undergo nucleophilic attack at the C3 position with Cys thiols to generate substituted diol products (e.g.59). This strategy was utilized as a selective protein–protein and protein–peptide conjugation tool, providing a rapid approach (proceeding in <2 h) to link large biomolecules without the need for unnatural amino acids.58 It is worth noting that the core phenolic structure of Tyr is not regenerated following this functionalization strategy, and thus, the approach would not satisfy a strict definition of umpolung chemistry. However, as the site selective functionalization strategy relies on the inverted donor/acceptor properties of the Tyr ring, it bears considerable resemblance to other umpolung strategies discussed. Moreover, the underlying concept illustrates the value of looking beyond conventional side chain reactivities to deliver powerful new tools for protein bioconjugation.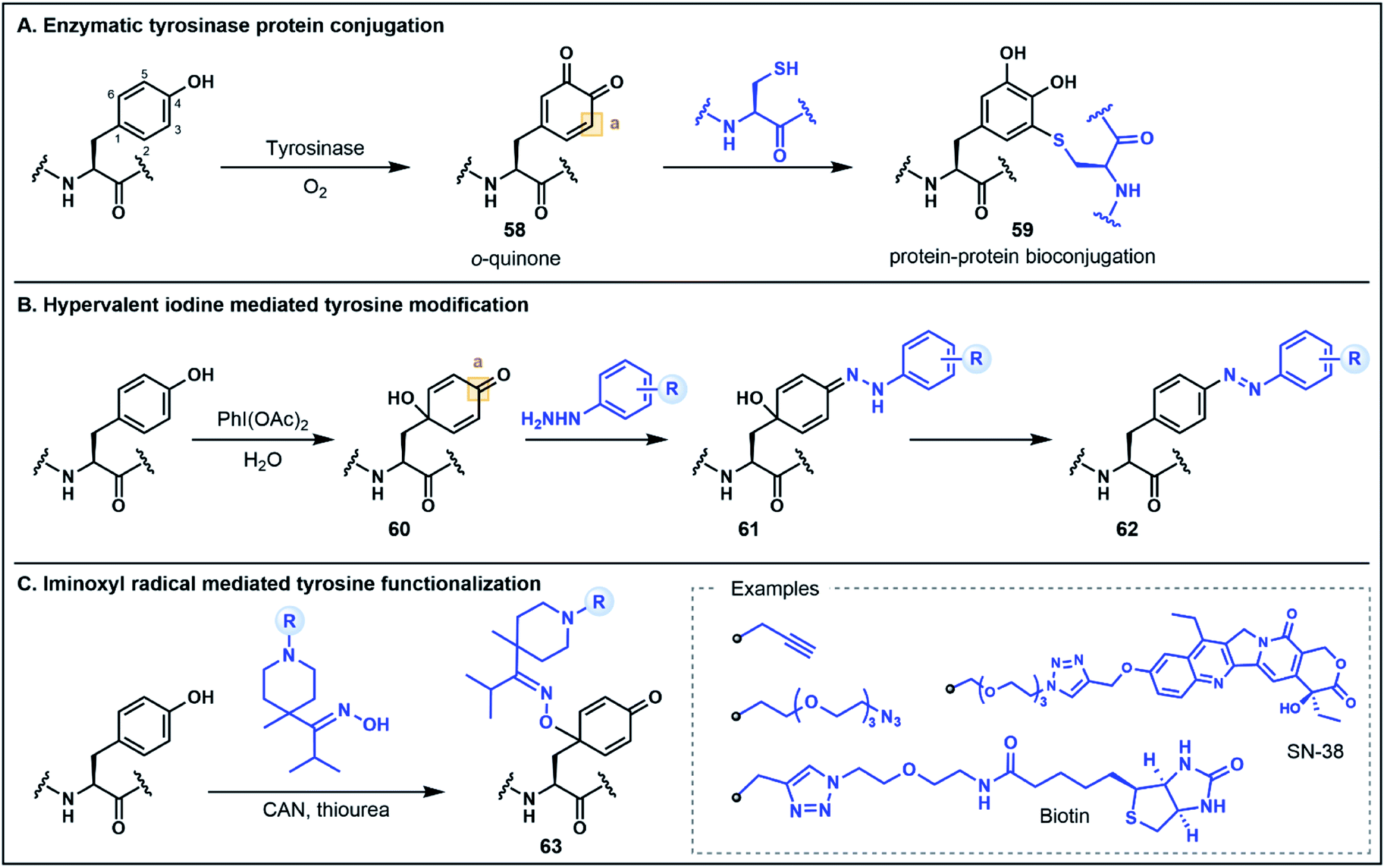 | ||
| Fig. 9 Tyr functionalization using non-conventional umpolung strategies. (A) Tyr–Cys bioconjugation strategy using an enzymatically activated o-quinone intermediate. (B) Tyr dearomatization–rearomatization functionalization using phenyl hydrazine nucleophiles. (C) Tyr functionalization using iminoxyl radicals; CAN = ceric ammonium nitrate. | ||
Tyr functionalization can also be mediated via chemical dearomatization strategies which have led to the development of additional conjugation methods that exploit underutilized electrophilic Tyr derivatives. In one such strategy developed by Wang, Su, Fang and co-workers, hypervalent iodine(III) was used to oxidatively dearomatize Tyr to afford a 4-hydroxylcyclohexadienone intermediate 60 (Fig. 9B).59 The addition of phenyl hydrazine leads to the formation of a hydrazone intermediate 61 which undergoes rearomatization to generate a C4 substituted azobenzene residue 62. This one-pot strategy was used to modify various peptide substrates, including through the direct conjugation of two peptides, as well as through attachment of alkyne functionalized hydrazides for secondary conjugation reactions.59 In a similar dearomatization approach reported by Oisaki, Kanai and co-workers, iminoxyl radicals were used to modify Tyr at the C1 carbon to afford functionalized oximes (Fig. 9C).60 The iminoxyl radicals were prepared via single electron oxidation with ceric ammonium nitrate—conditions which lead to dearomatization of the Tyr side chain and conjugation of the iminoxyl to the C1 position. Unlike the aforementioned approach, there is no rearomatization of the phenol ring. However, the conjugated oxime product 63 was shown to be stable or reversible depending on the steric and electronic properties of the oxime substituents. This approach was used to generate a number of different protein conjugates including through the installation of azide and alkyne handles as well as through conjugation with various small molecules.60 Collectively, these oxidation strategies highlight innovative methods for Tyr modification that extend the conventional utility of the phenolic side chain for peptide and protein functionalization.
Perspective
Umpolung based side chain modification strategies are an exciting and rapidly expanding area, with many new peptide and protein functionalization methodologies having emerged in the last five years alone. These strategies have laid the critical groundwork to establish umpolung chemistry as a common approach for peptide and protein modification. Nevertheless, with the field still very much in its infancy, there is a wealth of opportunity for further growth and development.One area in which we envisage umpolung strategies will be particularly impactful is in the chemical synthesis of RiPPs and nonribosomal peptides (NRPs). As highlighted above, Cys umpolung strategies have enabled the synthesis of multi-stranded disulfide-linked peptides34,35b,36 and the tryptathionine linkages of the amatoxins and phallotoxins.39,40,42 However, there remains a multitude of other peptides (e.g. lanthipeptides, thiopeptides, lasso peptides) that are synthetically challenging or inaccessible using current methodologies.61 These peptides have valuable medicinal applications which provide strong impetus for the continued pursuit of efficient strategies for chemical synthesis. Umpolung reactivity may hold the key to accessing challenging targets by amplifying approaches to retrosynthetic planning and unlocking new levels of orthogonality. The use of umpolung strategies for the late-stage functionalization of peptides and proteins likewise warrants additional investigation. This perspective has highlighted a number of exciting strategies that have emerged at Cys, Sec, Ala, His and Tyr residues, many of which proceed under biocompatible conditions for the selective modification of both proteins and peptides. While it remains to be seen which of these nascent strategies will be widely utilized by the broader community, these approaches undoubtedly diversify the available methods for late-stage functionalization and complement the array of existing strategies for peptide/protein conjugation and protein labeling, ultimately advancing their use in medicinal and biological applications.
With umpolung reactivity pathways highlighted herein for just five distinct amino acid side chains, the remaining 16 amino acids comprise a wealth of untapped functionality that could serve as additional sites for novel umpolung modifications. Indeed, we might expect that new strategies will emerge first at amino acids bearing polar functionalities (e.g. Lys, Met, Ser, Thr, Trp, Asp, Glu, Gln, Asn, Arg) owing to their plasticity toward chemical modification. In particular, residues that may be oxidatively activated (cf. His, Tyr, vide supra) may facilitate inversion of innate nucleophilicity patterns. Nonetheless, emerging Ala cross-coupling approaches48a,b exemplify the value of aliphatic residues as pliable sites for innovative modifications, particularly when paired with cutting-edge advances in catalysis. It is also worth noting that novel peptide and protein methodologies may judiciously exploit the logic of polarity reversal by merging small molecule umpolung approaches with the inherent reactivity of amino acid side chains, for example in the generation of valuable electrophiles.62 This approach highlights the opportunity to build unprecedented connectivity through the modification of peptide side chains using new synthetic technologies based broadly on the principles of umpolung chemistry.
Conclusions
Almost 50 years after the term umpolung was first coined by Seebach,10 we continue to witness the value of polarity reversal as an elegant and enabling platform for the design of new synthetic methods. Beyond established applications in small molecule synthesis and catalysis, the emerging umpolung strategies for peptide and protein modification outlined in this perspective underscore the enormous potential of these principles as a guiding force for the functionalization of biomolecules. We anticipate that the development of new strategies which capitalize on reactivity umpolung and exploit other underutilized modes of reactivity will continue to complement the growing repertoire of residue-specific modifications and bioconjugation approaches and will drive innovation in organic synthesis, medicinal chemistry, and chemical biology well into the future.Author contributions
A. M. W. and L. R. M. conceived of the work. I. R. P. developed and assembled the section on amide umpolung strategies. A. M. W. developed and assembled the section on side chain modifications. A. M. W., I. R. P., and L. R. M. wrote and edited the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We would like to acknowledge the Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science (CE200100012), the Australian Government Research Training Program PhD Scholarship scheme (I. R. P.) and the Westpac Research Fellowship (L. R. M.) for financial support.References
- (a) M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed; (b) D. J. Drucker, Nat. Rev. Drug Discovery, 2020, 19, 277–289 CrossRef CAS PubMed; (c) D. S. Dimitrov, in Therapeutic Proteins, ed. V. Voynov and J. A. Caravella, Humana Press, Totowa, NJ, 2012, vol. 899, pp. 1–26 Search PubMed; (d) B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed.
- (a) S. Shabani, Y. Wu, H. G. Ryan and C. A. Hutton, Chem. Soc. Rev., 2021, 50, 9278–9343 RSC; (b) J. Rodríguez and M. Martínez-Calvo, Chem.–Eur. J., 2020, 26, 9792–9813 CrossRef PubMed; (c) P. G. Isenegger and B. G. Davis, J. Am. Chem. Soc., 2019, 141, 8005–8013 CrossRef CAS PubMed; (d) L. R. Malins, Curr. Opin. Chem. Biol., 2018, 46, 25–32 CrossRef CAS PubMed.
- (a) V. M. Lechner, M. Nappi, P. J. Deneny, S. Folliet, J. C. K. Chu and M. J. Gaunt, Chem. Rev., 2021 DOI:10.1021/acs.chemrev.1c00357; (b) L. Raynal, N. C. Rose, J. R. Donald and C. D. Spicer, Chem.–Eur. J., 2021, 27, 69–88 CrossRef CAS PubMed; (c) C. Bottecchia and T. Noël, Chem.–Eur. J., 2019, 25, 26–42 CrossRef CAS PubMed.
- (a) Y. Weng, C. Song, C.-W. Chiang and A. Lei, Commun. Chem., 2020, 3, 171, DOI:10.1038/s42004-020-00413-x; (b) A. S. Mackay, R. J. Payne and L. R. Malins, J. Am. Chem. Soc., 2021 DOI:10.1021/jacs.1c11185.
- (a) E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Nat. Rev. Chem., 2019, 3, 147–171 CrossRef CAS; (b) J. N. deGruyter, L. R. Malins and P. S. Baran, Biochemistry, 2017, 56, 3863–3873 CrossRef CAS PubMed.
- (a) S. B. Gunnoo and A. Madder, Chem.–Eur. J., 2016, 17, 529–553 CAS; (b) P. Ochtrop and C. P. R. Hackenberger, Curr. Opin. Chem. Biol., 2020, 58, 28–36 CrossRef CAS PubMed.
- E. J. Corey, Pure Appl. Chem., 1967, 14, 19–38 CAS.
- (a) D. Seebach and E. J. Corey, J. Org. Chem., 1975, 40, 231–237 CrossRef CAS; (b) B.-T. Gröbel and D. Seebach, Synthesis, 1977, 1977, 357–402 CrossRef.
- D. A. Evans and G. C. Andrews, Acc. Chem. Res., 1974, 7, 147–155 CrossRef CAS.
- Used in a lecture at the University of Hamburg on July 7, 1972.
- D. Seebach, Angew. Chem., Int. Ed., 1979, 18, 239–258 CrossRef.
- IUPAC, Compendium of Chemical Terminology, (the “Gold Book”), Compiled by A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 2nd edn, 1997, Online version (2019) created by S. J. Chalk, DOI:10.1351/goldbook.
- (a) P. Ertl, E. Altmann and J. M. McKenna, J. Med. Chem., 2020, 63, 8408–8418 CrossRef CAS PubMed; (b) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef CAS.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed.
- B. Shen, D. M. Makley and J. N. Johnston, Nature, 2010, 465, 1027–1032 CrossRef CAS PubMed.
- For a review of non-classical amidation routes, see: R. M. de Figueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS PubMed.
- J. P. Shackleford, B. Shen and J. N. Johnston, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 44–46 CrossRef CAS PubMed.
- M. S. Crocker, H. Foy, K. Tokumaru, T. Dudding, M. Pink and J. N. Johnston, Chem, 2019, 5, 1248–1264 CAS.
- Z. Ke and Y.-Y. Yeung, Chem, 2019, 5, 1014–1016 CAS.
- K. E. Schwieter, B. Shen, J. P. Shackleford, M. W. Leighty and J. N. Johnston, Org. Lett., 2014, 16, 4714–4717 CrossRef CAS PubMed.
- K. E. Schwieter and J. N. Johnston, Chem. Commun., 2016, 52, 152–155 RSC.
- J. Li, M. J. Lear, Y. Kawamoto, S. Umemiya, A. R. Wong, E. Kwon, I. Sato and Y. Hayashi, Angew. Chem., Int. Ed., 2015, 54, 12986–12990 CrossRef CAS PubMed.
- J. Li, M. J. Lear, E. Kwon and Y. Hayashi, Chem.–Eur. J., 2016, 22, 5538–5542 CrossRef CAS PubMed.
- (a) D. M. Makley and J. N. Johnston, Org. Lett., 2014, 16, 3146–3149 CrossRef CAS PubMed; (b) M. Vishe and J. N. Johnston, Chem. Sci., 2019, 10, 1138–1143 RSC; (c) K. E. Schwieter and J. N. Johnston, Chem. Sci., 2015, 6, 2590–2595 RSC; (d) K. E. Schwieter and J. N. Johnston, ACS Catal., 2015, 5, 6559–6562 CrossRef CAS PubMed; (e) B. M. Nugent, R. A. Yoder and J. N. Johnston, J. Am. Chem. Soc., 2004, 126, 3418–3419 CrossRef CAS PubMed.
- M. W. Leighty, B. Shen and J. N. Johnston, J. Am. Chem. Soc., 2012, 134, 15233–15236 CrossRef CAS PubMed.
- A. H. Fauq, K. Simpson, G. M. Maharvi, T. Golde and P. Das, Bioorg. Med. Chem. Lett., 2007, 17, 6392–6395 CrossRef CAS PubMed.
- Y. Ning, S. Wang, M. Li, J. Han, C. Zhu and J. Xie, Nat. Commun., 2021, 12, 4637 CrossRef CAS PubMed.
- (a) M. Lecomte, J. M. Lipshultz, S.-H. Kim-Lee, G. Li and A. T. Radosevich, J. Am. Chem. Soc., 2019, 141, 12507–12512 CrossRef CAS PubMed; (b) G. Li, T. V. Nykaza, J. C. Cooper, A. Ramirez, M. R. Luzung and A. T. Radosevich, J. Am. Chem. Soc., 2020, 142, 6786–6799 CrossRef CAS PubMed.
- (a) Y.-P. Zhu, S. Sergeyev, P. Franck, R. V. A. Orru and B. U. W. Maes, Org. Lett., 2016, 18, 4602–4605 CrossRef CAS PubMed; (b) Y.-P. Zhu, P. Mampuys, S. Sergeyev, S. Ballet and B. U. W. Maes, Adv. Synth. Catal., 2017, 359, 2481–2498 CrossRef CAS; (c) H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714–2742 RSC; (d) X. Wang, Nat. Catal., 2019, 2, 98–102 CrossRef; (e) J.-S. Suppo, G. Subra, M. Bergès, R. Marciade Figueiredo and J.-M. Campagne, Angew. Chem., Int. Ed., 2014, 53, 5389–5393 CrossRef CAS PubMed.
- Z. Zhao, Z. Peng, Y. Zhao, H. Liu, C. Li and J. Zhao, J. Org. Chem., 2017, 82, 11848–11853 CrossRef CAS PubMed.
- J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chem.–Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- F. Liu, A. N. Zaykov, J. J. Levy, R. D. DiMarchi and J. P. Mayer, J. Pept. Sci., 2016, 22, 260–270 CrossRef CAS PubMed.
- (a) A. N. Zaykov, V. M. Gelfanov, F. Liu and R. D. DiMarchi, Org. Lett., 2018, 20, 3695–3699 CrossRef CAS PubMed; (b) F. Liu, E. Y. Luo, D. B. Flora and A. R. Mezo, Angew. Chem., Int. Ed., 2014, 53, 3983–3987 CrossRef CAS PubMed.
- (a) X. Yang, V. Gelfanov, F. Liu and R. DiMarchi, Org. Lett., 2016, 18, 5516–5519 CrossRef CAS PubMed; (b) F. Liu, Q. Liu and A. R. Mezo, Org. Lett., 2014, 16, 3126–3129 CrossRef CAS PubMed; (c) J. A. Karas, D. B. Scanlon, B. E. Forbes, I. Vetter, R. J. Lewis, J. Gardiner, F. Separovic, J. D. Wade and M. A. Hossain, Chem.–Eur. J., 2014, 20, 9549–9552 CrossRef CAS PubMed.
- L.-Q. Wan, X. Zhang, Y. Zou, R. Shi, J.-G. Cao, S.-Y. Xu, L.-F. Deng, L. Zhou, Y. Gong, X. Shu, G. Y. Lee, H. Ren, L. Dai, S. Qi, K. N. Houk and D. Niu, J. Am. Chem. Soc., 2021, 143, 11919–11926 CrossRef CAS PubMed.
- J. Vetter, Toxicon, 1998, 36, 13–24 CrossRef CAS PubMed.
- (a) M. O. Anderson, A. A. Shelat and R. K. Guy, J. Org. Chem., 2005, 70, 4578–4584 CrossRef CAS PubMed; (b) M.-A. J. Siegert, C. H. Knittel and R. D. Süssmuth, Angew. Chem., Int. Ed., 2020, 59, 5500–5504 CrossRef CAS PubMed.
- L. A. Schuresko and R. S. Lokey, Angew. Chem., Int. Ed., 2007, 46, 3547–3549 CrossRef CAS PubMed.
- P. Sieber, B. Kamber, B. Riniker and W. Rittel, Helv. Chim. Acta, 1980, 63, 2358–2363 CrossRef CAS.
- (a) G. Yao, C. H. Knittel, S. Kosol, M. T. Wenz, B. G. Keller, H. Gruß, A. C. Braun, C. Lutz, T. Hechler, A. Pahl and R. D. Süssmuth, J. Am. Chem. Soc., 2021, 143, 14322–14331 CrossRef CAS PubMed; (b) G. Yao, J.-O. Joswig, B. G. Keller and R. D. Süssmuth, Chem.–Eur. J., 2019, 25, 8030–8034 CrossRef CAS PubMed.
- D. Kobayashi, Y. Kohmura, T. Sugiki, E. Kuraoka, M. Denda, T. Fujiwara and A. Otaka, Chem.–Eur. J., 2021, 27, 14092–14099 CrossRef CAS PubMed.
- (a) M. Muttenthaler and P. F. Alewood, J. Pept. Sci., 2008, 14, 1223–1239 CrossRef CAS PubMed; (b) L. R. Malins, N. J. Mitchell and R. J. Payne, J. Pept. Sci., 2014, 20, 64–77 CrossRef CAS PubMed.
- D. T. Cohen, C. Zhang, B. L. Pentelute and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 9784–9787 CrossRef CAS PubMed.
- D. T. Cohen, C. Zhang, C. M. Fadzen, A. J. Mijalis, L. Hie, K. D. Johnson, Z. Shriver, O. Plante, S. J. Miller, S. L. Buchwald and B. L. Pentelute, Nat. Chem., 2019, 11, 78–85 CrossRef CAS PubMed.
- (a) T. H. Wright, B. J. Bower, J. M. Chalker, G. J. L. Bernardes, R. Wiewiora, W.-L. Ng, R. Raj, S. Faulkner, M. R. J. Vallée, A. Phanumartwiwath, O. D. Coleman, M.-L. Thézénas, M. Khan, S. R. G. Galan, L. Lercher, M. W. Schombs, S. Gerstberger, M. E. Palm-Espling, A. J. Baldwin, B. M. Kessler, T. D. W. Claridge, S. Mohammed and B. G. Davis, Science, 2016, 354, aag1465 CrossRef PubMed; (b) J. Dadová, S. R. G. Galan and B. G. Davis, Curr. Opin. Chem. Biol., 2018, 46, 71–81 CrossRef PubMed.
- (a) M.-Y. Xu, W.-T. Jiang, Y. Li, Q.-H. Xu, Q.-L. Zhou, S. Yang and B. Xiao, J. Am. Chem. Soc., 2019, 141, 7582–7588 CrossRef CAS PubMed; (b) F. Zhu, W. C. Powell, R. Jing and M. A. Walczak, Chem Catalysis, 2021, 1, 870–884 CrossRef PubMed; (c) C. Elgindy and L. R. Malins, Chem Catalysis, 2021, 1, 758–760 CrossRef.
- R. J. Sundberg and R. B. J. C. r. Martin, Chem. Rev., 1974, 74, 471–517 CrossRef CAS.
- (a) P. N. Joshi and V. Rai, Chem. Commun., 2019, 55, 1100–1103 RSC; (b) K. Peciak, E. Laurine, R. Tommasi, J.-w. Choi and S. Brocchini, Chem. Sci., 2019, 10, 427–439 RSC; (c) T. C. Bruice and G. L. Schmir, J. Am. Chem. Soc., 1958, 80, 148–156 CrossRef CAS.
- K. Nakane, S. Sato, T. Niwa, M. Tsushima, S. Tomoshige, H. Taguchi, M. Ishikawa and H. Nakamura, J. Am. Chem. Soc., 2021, 143, 7726–7731 CrossRef CAS PubMed.
- (a) M. Liu, Z. Zhang, J. Cheetham, D. Ren and Z. S. Zhou, Anal. Chem., 2014, 86, 4940–4948 CrossRef CAS PubMed; (b) C.-F. Xu, Y. Chen, L. Yi, T. Brantley, B. Stanley, Z. Sosic and L. Zang, Anal. Chem., 2017, 89, 7915–7923 CrossRef CAS PubMed.
- X. Chen, F. Ye, X. Luo, X. Liu, J. Zhao, S. Wang, Q. Zhou, G. Chen and P. Wang, J. Am. Chem. Soc., 2019, 141, 18230–18237 CrossRef CAS PubMed.
- A. F. M. Noisier, M. J. Johansson, L. Knerr, M. A. Hayes, W. J. Drury III, E. Valeur, L. R. Malins and R. Gopalakrishnan, Angew. Chem., Int. Ed., 2019, 58, 19096–19102 CrossRef CAS PubMed.
- (a) C. W. Kee, O. Tack, F. Guibbal, T. C. Wilson, P. G. Isenegger, M. Imiołek, S. Verhoog, M. Tilby, G. Boscutti, S. Ashworth, J. Chupin, R. Kashani, A. W. J. Poh, J. K. Sosabowski, S. Macholl, C. Plisson, B. Cornelissen, M. C. Willis, J. Passchier, B. G. Davis and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 1180–1185 CrossRef CAS PubMed; (b) N. Ichiishi, J. P. Caldwell, M. Lin, W. Zhong, X. Zhu, E. Streckfuss, H.-Y. Kim, C. A. Parish and S. W. Krska, Chem. Sci., 2018, 9, 4168–4175 RSC.
- A. F. M. Noisier and R. Gopalakrishnan in Oxime/hydrazone conjugation at histidine: late-stage functionalization approach of unprotected peptides, ed. W. M. Hussein, R. J. Stephenson and I. Toth, Springer US, New York, NY, 2021, pp. 35–48 Search PubMed.
- D. S. Peters, F. E. Romesberg and P. S. Baran, J. Am. Chem. Soc., 2018, 140, 2072–2075 CrossRef CAS PubMed.
- M. J. Lobba, C. Fellmann, A. M. Marmelstein, J. C. Maza, E. N. Kissman, S. A. Robinson, B. T. Staahl, C. Urnes, R. J. Lew, C. S. Mogilevsky, J. A. Doudna and M. B. Francis, ACS Cent. Sci., 2020, 6, 1564–1571 CrossRef CAS PubMed.
- P. Wang, Y. Cheng, C. Wu, Y. Zhou, Z. Cheng, H. Li, R. Wang, W. Su and L. Fang, Org. Lett., 2021, 23, 4137–4141 CrossRef CAS PubMed.
- K. Maruyama, T. Ishiyama, Y. Seki, K. Sakai, T. Togo, K. Oisaki and M. Kanai, J. Am. Chem. Soc., 2021, 143, 19844–19855 CrossRef CAS PubMed.
- M. Montalbán-López, T. A. Scott, S. Ramesh, I. R. Rahman, A. J. van Heel, J. H. Viel, V. Bandarian, E. Dittmann, O. Genilloud, Y. Goto, M. J. Grande Burgos, C. Hill, S. Kim, J. Koehnke, J. A. Latham, A. J. Link, B. Martínez, S. K. Nair, Y. Nicolet, S. Rebuffat, H.-G. Sahl, D. Sareen, E. W. Schmidt, L. Schmitt, K. Severinov, R. D. Süssmuth, A. W. Truman, H. Wang, J.-K. Weng, G. P. van Wezel, Q. Zhang, J. Zhong, J. Piel, D. A. Mitchell, O. P. Kuipers and W. A. van der Donk, Nat. Prod. Rep., 2021, 38, 130–239 RSC.
- Recent examples have utilized the innate nucleophilicity of Cys in combination with small molecule umpolung approaches for the generation of electrophiles, e.g., see: (a) D. P. Hari, P. Caramenti and J. Waser, Acc. Chem. Res., 2018, 51, 3212–3225 CrossRef CAS PubMed; (b) J. Blom, G. J. Reyes-Rodríguez, H. N. Tobiesen, J. N. Lamhauge, M. V. Iversen, C. L. Barløse, N. Hammer, M. Rusbjerg and K. A. Jørgensen, Angew. Chem., Int. Ed., 2019, 58, 17856–17862 CrossRef CAS PubMed.
Footnote |
| † A. M. W. and I. R. P. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |