
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA covalent cobalt diimine-dioxime – fullerene assembly for photoelectrochemical hydrogen production from near-neutral aqueous media†
Dongyue
Sun
,
Adina
Morozan
,
Matthieu
Koepf
 * and
Vincent
Artero
*
* and
Vincent
Artero
*
Univ. Grenoble Alpes, CNRS, CEA, IRIG, Laboratoire de Chimie et Biologie des Métaux, 17 rue des Martyrs, F-38054 Grenoble, France. E-mail: matthieu.koepf@cea.fr; vincent.artero@cea.fr
First published on 7th March 2022
Abstract
The covalent assembly between a cobalt diimine-dioxime complex and a fullerenic moiety results in enhanced catalytic properties in terms of overpotential requirement for H2 evolution. The interaction between the fullerene moiety and PCBM heterojunction further allows for the easy integration of the cobalt diimine-dioxime – fullerene catalyst with a poly-3-hexylthiophene (P3HT):[6,6]-phenyl-C61-butyric acid methyl ester (PCBM) bulk heterojunction, yielding hybrid photoelectrodes for H2 evolution from near-neutral aqueous solutions.
The shift towards more sustainable economic models for the production of fuels and commodity chemicals requires the emergence of radically new industrial schemes utilizing renewable energy resources as well as abundant raw materials. In that context, the idea of artificial photosynthesis is particularly appealing.1–3 This emerging field of research aims to harvest the largely abundant solar energy to produce fuels and commodity chemicals from widely available feedstocks from the atmosphere. An archetypal reaction targeted in artificial photosynthesis is the solar-driven splitting of water for hydrogen production.4 Various strategies are currently under investigation, including the combination of photovoltaic cells and electrolyzers (PV + electrolysis) or the development of photoelectrochemical systems in which light-absorbing materials and catalysts are brought in intimate contact within photoelectrodes. The latter strategy usually implies the final assembly of a photoanode and a photocathode in a tandem configuration to obtain fully functional self-standing photoelectrochemical cells (PEC).4 Importantly, for photoelectrochemical cells to take over PV + electrolysis systems requires them to show advantages in terms of sustainability as far as their constitutive materials are concerned.4 In that prospect, the direct integration of well-defined molecular electrocatalysts on organic bulk heterojunctions (BHJ) is particularly appealing. Not only BHJs performances have greatly improved during the last decade demonstrating their relevance for energy applications, but also their intrinsic design possesses several technical advantages. Firstly, they mostly rely on abundant materials compatible with the emergence of sustainable production schemes with minimal environmental costs. Secondly, based on all-organic semiconductors and solution processing, BHJs can be deposited under extremely mild conditions, which greatly reduces the constraint on the technological implementation. Furthermore, their compatibility with the use of roll-to-roll processing may allow in fine a rapid transfer towards large-scale production of any functional system. It is therefore appealing to move towards the development of PEC based on organic semiconductors, and efforts towards this endeavor boomed.5–15
Regarding photocathodes based on BHJ, several architectures have been proposed integrating various additional layered materials as charge selective carriers and protecting layers. Such architectures easily sustain current densities of several mA cm−2 under 1-sun simulated solar irradiation and do offer promises for developing functional devices. However, thus far these systems rely on the use of efficient yet limited classes of catalysts (Pt, MoSx),5–7,10–13 which only work under strongly acidic conditions. This limits the long-term stability of the systems. A solution could be found via the integration of molecular-based catalysts operating under near-neutral aqueous conditions.16 In particular, cobaloximes and cobalt diimine-dioxime complexes are popular hydrogen-evolving catalysts known to operate in near-neutral aqueous solution with moderate overpotential requirements.17–19 Over the years these complexes have been successfully used to obtain efficient noble-metal free homogeneous photocatalytic systems20 as well as integrated in photocathodes based on inorganic semiconductors21,22 and dye-sensitized mesoporous oxides.23–28 Such catalysts, active at or close to neutral pHs, allow for the design of devices working under particularly mild conditions which are more compatible with the use of sensitive organic light-harvesting components. This calls for increased efforts towards the integration of molecular catalysts at the BHJ-interface.
We report here on the integration of cobalt diimine-dioxime ([Co(DO)(DOH)pnBr2], (DOH)2pn = N2,N2′-propanediylbis-butan-2-imine 3-oxime) proton reduction catalysts29 within poly-3-hexylthiophene (P3HT):[6,6]-phenyl-C61-butyric acid methyl ester (PCBM) bulk heterojunctions to assemble functional photocathodes for photocatalytic hydrogen production (Fig. 1). In such BHJ photoelectrode constructs, light is harvested by the P3HT component to form excitons that dissociate at the interface between P3HT and PCBM. Photogenerated holes are then transferred to the supporting NiO electrode while photogenerated electrons accumulate in the PCBM phase and then transferred to the catalyst. We reasoned that conjugating the catalyst with a C60 moiety in a structure such as 1 (Scheme 1) should allow to develop favorable interactions between this fullerene moiety with PCBM in P3HT:PCBM BHJ, thus ensuring the optimal interfacing of the catalyst with the n-type domain of the junction.
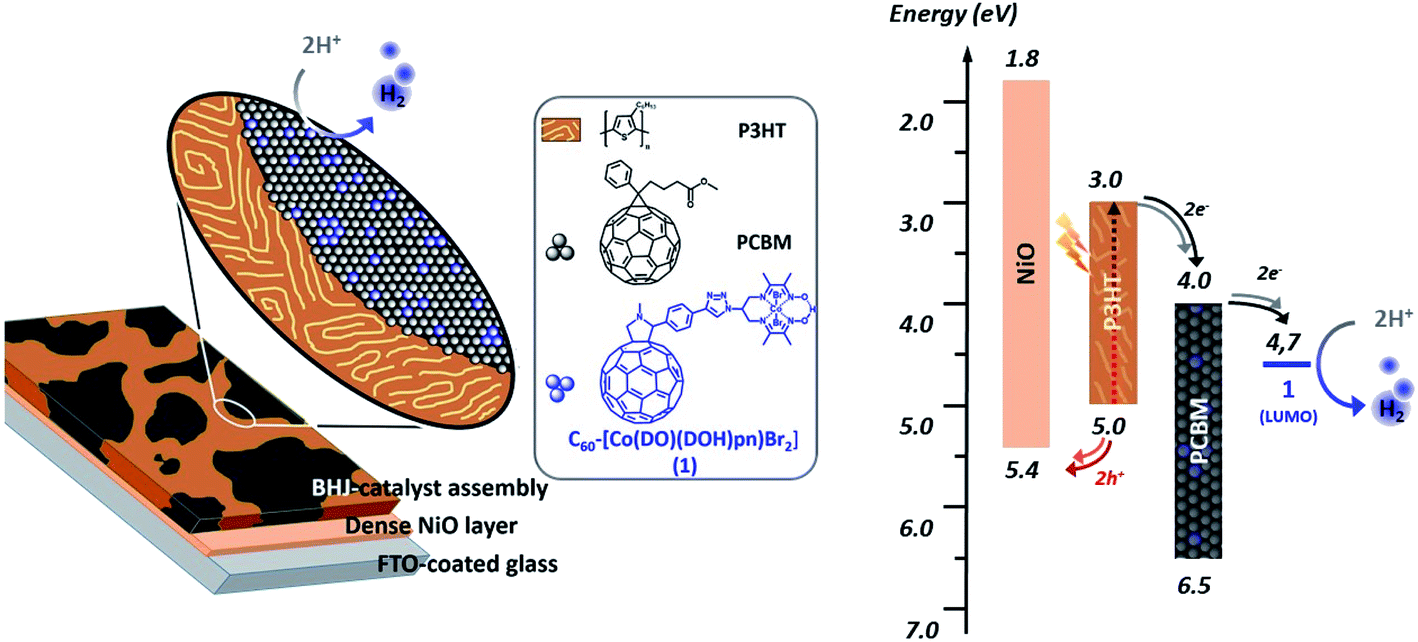 | ||
| Fig. 1 Representation (left) and energy diagram (right) of a photocathode for H2 evolution based on a PCBM:P3HT bulk heterojunction BHJ combined with the cobalt diimine-dioxime – fullerene assembly 1 as H2-evolving catalyst (see ESI† and ref. 30 for energy values). | ||
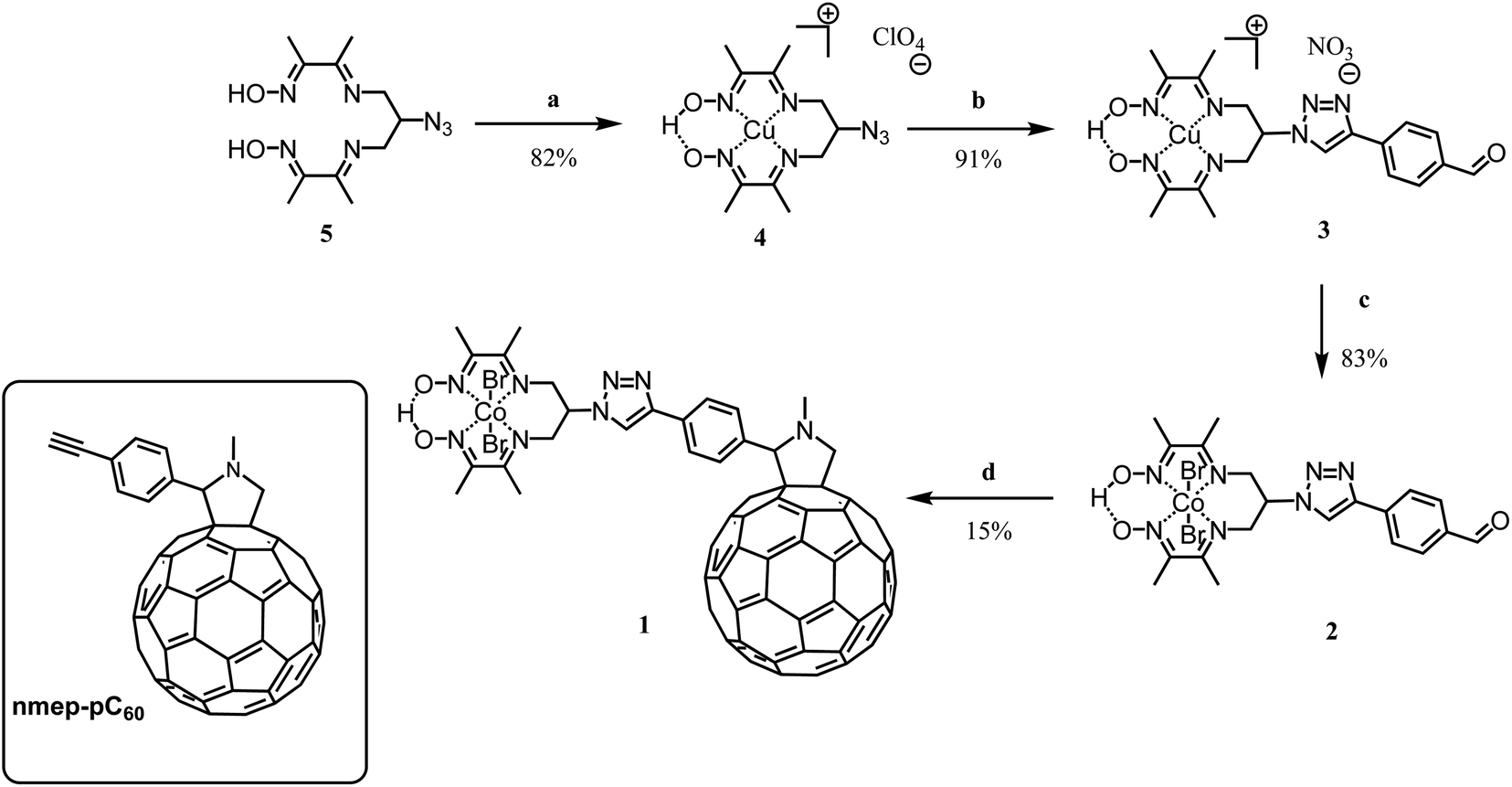 | ||
| Scheme 1 Synthesis of the cobalt diimine-dioxime – fullerene assembly 1; (a) Cu(ClO4)2·6H2O, acetone, RT; (b) 4-ethynylbenzaldehyde, N,N′,N′′,N′′-pentamethyldiethylenetriamine, CuBr, MeOH, Ar, 50 °C, 3.5 h; (c) CoBr2, O2, RT, 2 h; (d) C60, sarcosine, toluene:1,2-dichloroethane 2:1, Ar, reflux, 5 h; inset: structure of nmep-pC60. | ||
Furthermore the first reduction potential of fullerenopyrrolidines anchor (pC60/pC60− = −1.05 V vs. Fc+/Fc in CH3CN/toluene mixtures)31 is properly aligned with the Co(II)/Co(I) reduction potential of the cobalt diimine-dioxime complex (−1.11 V vs. Fc+/Fc in CH3CN for [Co(DO)(DOH)pnBr2] in CH3CN),32 thus allowing for a directional electron transfer from the PCBM/C60 domains toward the active catalyst.
1 was prepared through a Prato condensation between the fullerene and an appropriately substituted cobalt diimine-dioxime complex. The critical aspect of this synthesis was to use a proper high boiling point solvent or combination of solvents, able to solubilize simultaneously both the strongly apolar fullerene and polar diimine-dioxime metal complex moieties while allowing reaching the temperature needed to drive the Prato condensation. 1 is obtained straightforwardly in 4 steps form the previously described azido derivative 533 with an overall yield of 9%. With the exception of compound 4 that was recrystallized,34 the purification of the different compounds was done using flash chromatography, on silica, as described in details in the experimental part. The overall low yield obtained for this synthesis is likely due to the sluggish condensation between the fullerene moiety and the cobalt complex. In brief, following the previously established procedure for azido-diimine-dioxime derivative functionalization, the copper complex 4 was prepared before being engaged in the copper catalyzed Huisgen 1,3-dipolar cycloaddition to introduce the 4-formylphenyl substituent.34–36 The latter derivative 3 was then transmetallated with cobalt in presence of oxygen to obtain the cobalt derivative 2, which was engaged in a Prato reaction to couple the catalyst on the fullerene moiety. In addition to the 1H NMR and MALDI-TOF mass spectra, the FTIR spectrum of the product obtained from the condensation confirmed the formation of the desired covalent diimine-dioxime cobalt – fullerene assembly 1. As shown on Fig. S1a,† the absorption band corresponding to the stretching mode of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1688 cm−1) of the precursor 2 is lost while two new absorption bands (1461 cm−1 and 1430 cm−1) appear around the triazole NN stretching band (1451 cm−1). These two additional bands can be assigned to the C–H bending modes of the newly formed pyrrolidine ring,37 which are also observed in the FTIR spectrum of the reference N-methyl-2-(4′-ethynyl)phenyl-3,4-fulleropyrrolidine compound (nmep-pC60). Importantly the strong absorption band corresponding to the stretching mode of the CN bonds of the Co-coordinated diimine-dioxime ligand is clearly visible on the FTIR of 1 and 2 at 1513 cm−1 and 1507 cm−1, respectively indicating that the latter is preserved during the Prato condensation.22 Other strategies including the direct copper catalyzed Huisgen 1,3-dipolar cycloaddition between copper- or cobalt-diimine dioxime azide derivatives and an acetylene functionalized fullerene, or the stepwise building of diimine-dioxime ligand on the fullerene did not succeed in our hands, likely due to the very limited range of solvents able to solubilize the fullerene derivatives. Alternatively, our attempts to react the free ligand 5 on 4-ethynylbenzaldehyde via a thermal (microwave assisted) Huisgen 1,3-dipolar cycloaddition systematically resulted in the degradation of the dioxime-diimine moiety under the high temperature required to drive the condensation.38
O (1688 cm−1) of the precursor 2 is lost while two new absorption bands (1461 cm−1 and 1430 cm−1) appear around the triazole NN stretching band (1451 cm−1). These two additional bands can be assigned to the C–H bending modes of the newly formed pyrrolidine ring,37 which are also observed in the FTIR spectrum of the reference N-methyl-2-(4′-ethynyl)phenyl-3,4-fulleropyrrolidine compound (nmep-pC60). Importantly the strong absorption band corresponding to the stretching mode of the CN bonds of the Co-coordinated diimine-dioxime ligand is clearly visible on the FTIR of 1 and 2 at 1513 cm−1 and 1507 cm−1, respectively indicating that the latter is preserved during the Prato condensation.22 Other strategies including the direct copper catalyzed Huisgen 1,3-dipolar cycloaddition between copper- or cobalt-diimine dioxime azide derivatives and an acetylene functionalized fullerene, or the stepwise building of diimine-dioxime ligand on the fullerene did not succeed in our hands, likely due to the very limited range of solvents able to solubilize the fullerene derivatives. Alternatively, our attempts to react the free ligand 5 on 4-ethynylbenzaldehyde via a thermal (microwave assisted) Huisgen 1,3-dipolar cycloaddition systematically resulted in the degradation of the dioxime-diimine moiety under the high temperature required to drive the condensation.38
Cyclic voltammograms of the covalent cobalt diimine-dioxime – fullerene assembly 1, the cobalt diimine-dioxime derivative 2 and nmep-pC6039 are depicted in Fig. 2 and corresponding differential pulse voltammetry (DPV) measurements for 1 and 2 are shown in Fig. S4.† Obviously, the covalent assembly of the two moieties results in a modification of the electrochemical responses of both the cobalt diimine-dioxime and the fullerene redox centers.
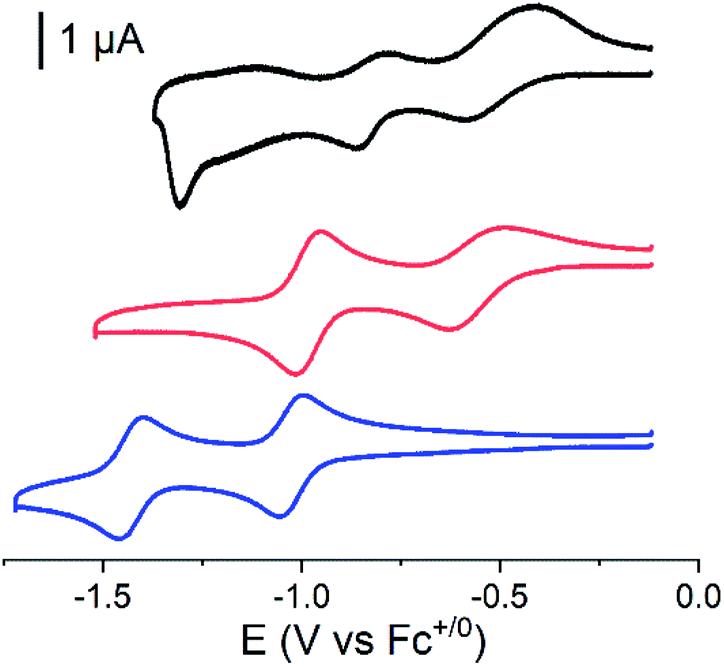 | ||
Fig. 2 Cyclic voltammograms for nmep-pC60 (blue), 2 (red) and 1 (black), recorded in a 0.3 mM CH3CN/1,2-dichlorobenzene 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) solution in the presence of 0.1 M of n-Bu4NBF4, on glassy carbon (ø 1.6 mm); scan rate: 100 mV s−1. All measurements were started after equilibration at the open-circuit potential around −0.1 V vs. Fc+/Fc with initial scan towards more negative potentials. :1 (v/v) solution in the presence of 0.1 M of n-Bu4NBF4, on glassy carbon (ø 1.6 mm); scan rate: 100 mV s−1. All measurements were started after equilibration at the open-circuit potential around −0.1 V vs. Fc+/Fc with initial scan towards more negative potentials. | ||
The quasi-reversible Co(III)/Co(II) couple can unambiguously be identified at −0.62 V (CV) or −0.67 V (DPV) and −0.57 V (CV) or −0.59 V (DPV) vs. Fc+/0 in both the cobalt diimine-dioxime complex 2 and the cobalt diimine-dioxime – fullerene assembly 1, respectively. Coupling with the fullerene unit therefore entails a 50 mV anodic shift for the standard potential of the Co(III)/Co(II) redox couple. The second reversible redox event occurs for 1 at −0.85 V (CV) or −0.86 V (DPV) vs. Fc+/0, which we tentatively assign to the Co(II)/Co(I) couple, the standard potential of which would be anodically shifted by 160 mV compared to that measured in 2 (−1.01 V (CV) or −1.03 V (DPV) vs. Fc+/0). By comparison the reversible nmep-pC60/nmep-pC60˙− couple (−1.05 V vs. Fc+/0) is observed at 40 mV more cathodic potential and could correspond to the third broad redox event observed for 1 at −1.15 V (CV) or −1.07 V (DPV) vs. Fc+/0. A peculiar irreversible reduction process is observed at Epc = −1.3 V (CV) or −1.2 V (DPV) vs. Fc+/0. Based on its shape, it could be associated with desorption of some compound that has adsorbed onto the electrode upon the preceding processes.40 We unfortunately failed in confirming these assignments spectroscopically. Indeed spectroelectrochemical measurements could not help determining the nature of the redox center involved in this second reduction wave due to the insoluble nature of the reduced species and its immediate precipitations in all conditions tested.
To investigate the H2 evolution properties of 1, we turned to CH3CN electrolyte in which the thermodynamical parameters are tabulated41 and parent cobalt diimine-dioxime compounds have been assessed.32,41 Compound 1 was deposited on glassy carbon electrode at a surface concentration of 52 ± 1 nmol cm−2. The cyclic voltammogram measured at such a modified electrode in CH3CN displays three successive quasi-reversible redox processes at −0.71 V, −1.11 V and −1.55 V vs. Fc+/0 (Fig. 3). These waves reproduce the signals measured for 1 in solution, with a ∼20–40 mV cathodic shift likely associated with the change of electrolyte composition.
Addition of 10 mM para-cyanoanilinium tetrafluoroborate as a proton source in the medium triggers a catalytic wave with half-wave potential of −0.62 V vs. Fc+/0 (or −4.66 eV vs. vacuum in Fig. 1, see ESI†). This process is significantly anodically shifted with regards to the Co(II)/Co(I), reproducing the behavior observed for the unfunctionalized [Co(DO)(DOH)pnBr2] complex under similar conditions. For this complex,32 as well as for 2, the catalytic potential at the mid wave has been measured at −0.82 V vs. Fc+/0. Thus, the presence of the fullerene moiety in close proximity to the cobalt center lowers the overpotential requirement for H2 evolution by 200 mV. The latter is estimated to 160 mV based on the equilibrium potential of the H+/H2 couple (−0.46 V vs. Fc+/0)41 in CH3CN with 10 mM para-cyanoanilinium tetrafluoroborate as proton source, and ranks amongst the lowest values for Earth-abundant molecular catalysts for H2 evolution.
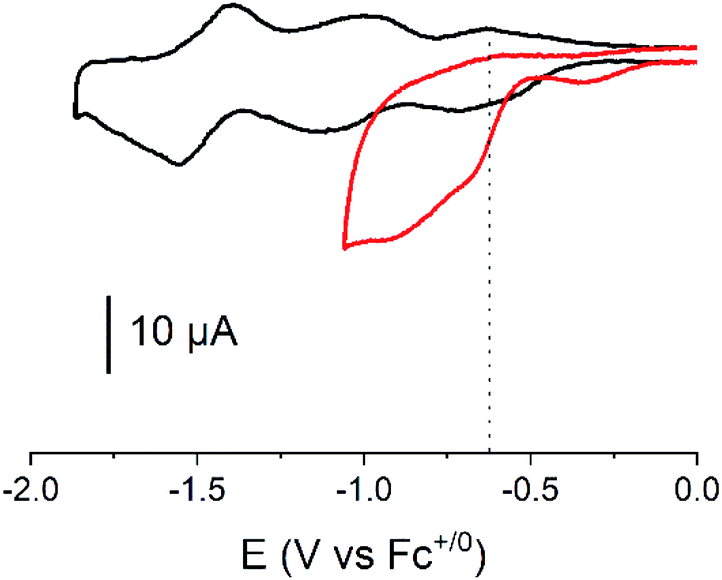 | ||
| Fig. 3 Cyclic voltammograms for compound 1 deposited on a GC electrode (ø 3 mm) recorded at 100 mV s−1 in CH3CN (with 0.1 M of n-Bu4NBF4) in the absence (black) and in the presence (red) of p-cyanoanilinium tetrafluoroborate (10 mM); the vertical dotted line indicates the mid-wave potential of the catalytic process. Measurements were started after equilibration at the open-circuit potential around 0 V vs. Fc+/Fc with initial scan towards more negative potentials. | ||
To confirm the catalytic performance of 1 for H2 evolution, we measured the same modified electrode in 0.1 M aqueous acetate buffer (pH 4.5) (Fig. 4 and S5†). Under such conditions, H2 production could be quantified with 88% faradaic yield during a 4 h chronoamperometric experiment performed at −0.63 V vs. RHE (see the ESI† and ref. 42 for correspondence between potential measured in non-aqueous and aqueous solvents). Considering the surface density of the electrochemically active complexes on the GC electrodes (∼0.10 (±0.02) nmol cm−2) obtained from the integration of the redox process associated to the Co(III)/Co(II) couple, we can estimate the maximum turnover number (TON) and turnover frequency (TOF) of the electrochemically active centers for H2 evolution. We find TONmax = 4.57 × 106 and TOFmax = 317 s−1 which are clearly higher than the values reported for a related material based on covalently modified carbon nanotubes where the observed TON and TOF reached 33 × 103 et TOF 2.2 s−1 respectively.33 Taking a more conservative standpoint and calculating the minimum TON and TOF values based on the total quantity of catalyst deposited (52 ± 1 nmol cm−2) we still find significant TONmin = 8.79 × 103 and TOFmin = 0.6 s−1 values, that demonstrate the stability and good activity of our catalytic assembly.
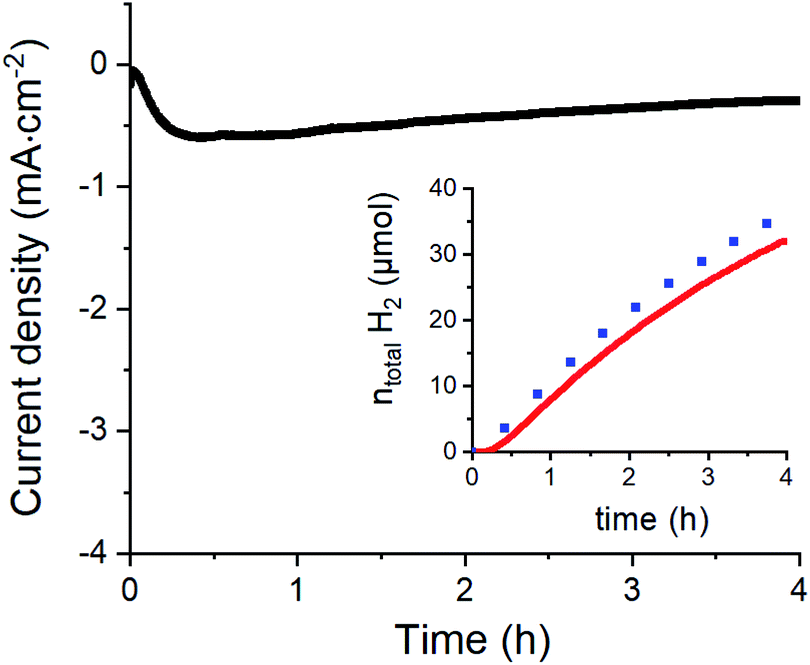 | ||
| Fig. 4 Long-term chronoamperometric experiment at −0.63 V vs. RHE in acetate buffer (0.1 M, pH 4.5) with concomitant H2 quantification (red trace in inset); the blue dots indicate the expected H2 amount for a 100% faradaic yield. | ||
These promising results prompted us to interface 1 to a P3HT:PCBM BHJ so as to prepare a hybrid photocathode for H2 evolution. We tested various architectures: first, 1 was simply spin-coated onto a preassembled P3HT:PCBM BHJ. This approach was not fully satisfactory since the dichlorobenzene used to solubilize 1 resulted in the partial dissolution and degradation of the BHJ in the FTO/NiO/P3HT:PCBM/1 architecture. Attempts to protect the BHJ with a spin-coated layer of 2,9-dimethyl-4,7-diphenyl-1,10-phenantroline (BCP) did not prove successful. We then took advantage of the structural similarities between 1 and PCBM and directly added 1 in the P3HT:PCBM solution used to fabricate the BHJ. This yield the FTO/NiO/[P3HT:PCBM+1] architecture in which the BHJ contains fullerene domains including the cobalt catalytic moiety. In all cases, the BHJ was deposited on a compact NiO layer that proved superior to alternate PEDOT:PSS layers.7 This photocathode was then tested for photoelectrochemical hydrogen production in half-cell configuration in pH 4.5 acetate buffer (0.1 M) with back irradiation with visible light (400 < λ < 800 nm, 65 mW cm−2). A photocurrent is observed from ca. 0.9 V vs. RHE throughout the potential range (Fig. 5) and plateaus from 0.6 V vs. RHE. The Incident Photon to Current Efficiencies (IPCE) values26 measured for both FTO/NiO/[P3HT:PCBM] and FTO/NiO/[P3HT:PCBM+1] assemblies (Fig. S6†) closely follow the absorption spectra of each film, with maximum values at the maximum absorption (λabs = 550 nm) of P3HT:PCBM BHJ.
 | ||
| Fig. 5 LSVs at FTO/NiO/[P3HT:PCBM+1] electrode with (red line), without (black line) or with chopped (blue line) visible light (400 < λ < 800 nm, 65 mW cm−2) recorded at a scan rate of 5 mV s−1 in 0.1 M acetate buffer (pH 4.5). | ||
Chronoamperometry was performed to ascertain the magnitude and stability of the photocurrent. Photocurrents of 35 μA cm−2 are measured at 0.065 V vs. RHE (Fig. 6) that proved stable over time. By comparison, a catalyst-free P3HT:PCBM BHJ displays much lower photocurrents (∼5 μA cm−2). Chronoamperometry was then performed under continuous irradiation for 2 and 3.5 hours (Fig. S7†), corresponding to charges of 195 and 385 mC cm−2 and faradaic efficiencies of 10.2 ± 0.5 and 10.3 ± 0.5 for H2 evolution, respectively. Such values are comparable to other photocathodes constructions involving similar cobalt diimine-dioxime catalysts.23–25 Although the photocurrents measured here remain modest, they are 20-fold higher than those measured for dye-sensitized photocathodes based on the same catalytic moiety.23–25 In terms of photocurrent density, our construction is also 10 times more efficient than a P3HT/nmep-pC60 planar heterojunction functionalized by a cobalt diimine-dioxime complex via in situ copper catalyzed 1,3-dipolar Huisgen cycloaddition.43,44 The latter photocathode yielded maximum current densities of 3 μA cm−2. Hydrogen evolution was not characterized as the product of the light-driven process in that case, but it is also reasonable to grant this construction with a ∼10% faradaic efficiency, as observed for all the other photocathode constructions involving similar cobalt diimine-dioxime catalysts.23–25
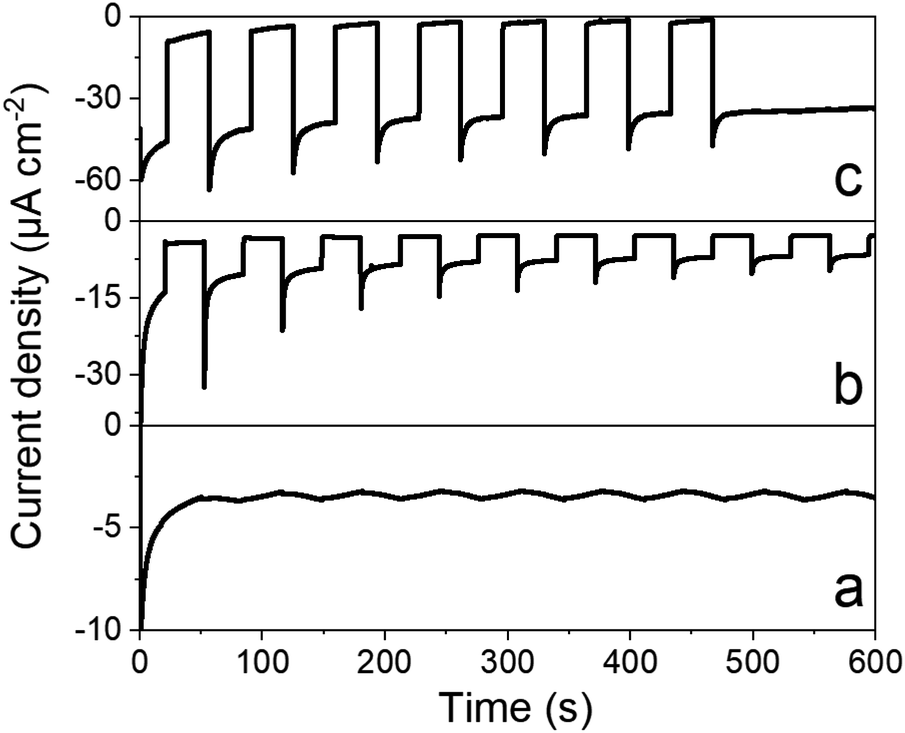 | ||
| Fig. 6 Photocurrent densities measured under chopped visible light (400 < λ < 800 nm, 65 mW cm−2) at an applied potential of 0.065 V vs. RHE in 0.1 M acetate buffer (pH 4.5) for the following electrodes: FTO/NiO (a), FTO/NiO/P3HT:PCBM (b) and FTO/NiO/[P3HT:PCBM+1] (c). | ||
Conclusion
The field of hybrid photoelectrodes combining molecular catalysts and inorganic light-harvesting materials or vice versa is rapidly expanding.22,45–48 Indeed, molecular chemistry offers an unmatched tunability in the design of highly selective and active catalysts or light harvesting units, therefore opening new horizons and versatility for photoelectrochemical cells. Organic semi-conductors gathers advantages from both words, being highly tunable and displaying high current densities when formulated as planar8,14 or bulk5–7,12,13 heterojunctions. Here we show for the first time that such junction of organic semi-conductors can be interfaced with molecular catalysts to yield operating photoelectrodes. We could evidence a clear influence of the strategy used to incorporate the catalyst into the devices, which paves the way towards a rational approach for the integration of molecular catalysts into (or interfacing with) organic BHJ, therefore opening the field to a large variety of chemical reactions and more diverse working conditions.Author contributions
D. S., M. K. and V. A. designed the study. D. S., A. M. and M. K. carried out the experiments. D. S., A. M., M. K. and V. A. analyzed data. The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the French National Research Agency (ANR-17-EURE-0003 and Labex ARCANE and CBH-EUR-GS), the European Research Council and European Commission's Seventh Framework Program (FP7/2007–2013) under grant agreement no 306398 (project PhotocatH2ode).References
- M. Koepf, A.-L. Teillout and M. J. Llansola-Portoles, in Handbook of Ecomaterials, ed. L. M. T. Martínez, O. V. Kharissova and B. I. Kharisov, Springer International Publishing, Cham, 2017, pp. 1–25 Search PubMed.
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946–964 CrossRef CAS PubMed.
- A. Thapper, S. Styring, G. Saracco, A. W. Rutherford, B. Robert, A. Magnuson, W. Lubitz, A. Llobet, P. Kurz, A. Holzwarth, S. Fiechter, H. de Groot, S. Campagna, A. Braun, H. Bercegol and V. Artero, Green, 2013, 3, 43–57 Search PubMed.
- S. Ardo, D. F. Rivas, M. A. Modestino, V. S. Greiving, F. F. Abdi, E. A. Llado, V. Artero, K. Ayers, C. Battaglia, J. P. Becker, D. Bederak, A. Berger, F. Buda, E. Chinello, B. Dam, V. Di Palma, T. Edvinsson, K. Fujii, H. Gardeniers, H. Geerlings, S. M. H. Hashemi, S. Haussener, F. Houle, J. Huskens, B. D. James, K. Konrad, A. Kudo, P. P. Kunturu, D. Lohse, B. Mei, E. L. Miller, G. F. Moore, J. Muller, K. L. Orchard, T. E. Rosser, F. H. Saadi, J. W. Schuttauf, B. Seger, S. W. Sheehan, W. A. Smith, J. Spurgeon, M. H. Tang, R. van de Krol, P. C. K. Vesborg and P. Westerik, Energy Environ. Sci., 2018, 11, 2768–2783 RSC.
- T. Bourgeteau, D. Tondelier, B. Geffroy, R. Brisse, C. Laberty-Robert, S. Campidelli, R. de Bettignies, V. Artero, S. Palacin and B. Jousselme, Energy Environ. Sci., 2013, 6, 2706–2713 RSC.
- T. Bourgeteau, D. Tondelier, B. Geffroy, R. Brisse, R. Cornut, V. Artero and B. Jousselme, ACS Appl. Mater. Interfaces, 2015, 7, 16395–16403 CrossRef CAS PubMed.
- T. Bourgeteau, D. Tondelier, B. Geffroy, R. Brisse, S. Campidelli, R. Cornut and B. Jousselme, J. Mater. Chem. A, 2016, 4, 4831–4839 RSC.
- A. Morozan, T. Bourgeteau, D. Tondelier, B. Geffroy, B. Jousselme and V. Artero, Nanotechnology, 2016, 27, 355401 CrossRef CAS PubMed.
- N. Queyriaux, N. Kaeffer, A. Morozan, M. Chavarot-Kerlidou and V. Artero, J. Photochem. Photobiol., C, 2015, 25, 90–105 CrossRef CAS.
- L. Francas, E. Burns, L. Steier, H. Cha, L. Sola-Hernandez, X. Li, P. Shakya Tuladhar, R. Bofill, J. Garcia-Anton, X. Sala and J. R. Durrant, Chem. Commun., 2018, 54, 5732–5735 RSC.
- L. Steier, S. Bellani, H. C. Rojas, L. Pan, M. Laitinen, T. Sajavaara, F. Di Fonzo, M. Gratzel, M. R. Antognazza and M. T. Mayer, Sustainable Energy Fuels, 2017, 1, 1915–1920 RSC.
- M. Haro, C. Solis, G. Molina, L. Otero, J. Bisquert, S. Gimenez and A. Guerrero, J. Phys. Chem. C, 2015, 119, 6488–6494 CrossRef CAS.
- A. Guerrero, M. Haro, S. Bellani, M. R. Antognazza, L. Meda, S. Gimenez and J. Bisquert, Energy Environ. Sci., 2014, 7, 3666–3673 RSC.
- T. Abe, S. Tobinai, N. Taira, J. Chiba, T. Itoh and K. Nagai, J. Phys. Chem. C, 2011, 115, 7701–7705 CrossRef CAS.
- T. Abe, K. Nagai, S. Kabutomori, M. Kaneko, A. Tajiri and T. Noritnatsit, Angew. Chem., Int. Ed., 2006, 45, 2778–2781 CrossRef CAS PubMed.
- N. Coutard, N. Kaeffer and V. Artero, Chem. Commun., 2016, 52, 13728–13748 RSC.
- S. Donck, J. Fize, E. Gravel, E. Doris and V. Artero, Chem. Commun., 2016, 52, 11783–11786 RSC.
- N. Kaeffer, M. Chavarot-Kerlidou and V. Artero, Acc. Chem. Res., 2015, 48, 1286–1295 CrossRef CAS PubMed.
- D. Sun, A. K. Harshan, J. Pecaut, S. Hammes-Schiffer, C. Costentin and V. Artero, ChemElectroChem, 2021, 8, 2671–2679 CrossRef CAS.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- D. Cedeno, A. Krawicz and G. F. Moore, Interface Focus, 2015, 5, 20140085 CrossRef PubMed.
- S. Chandrasekaran, N. Kaeffer, L. Cagnon, D. Aldakov, J. Fize, G. Nonglaton, F. Baleras, P. Mailley and V. Artero, Chem. Sci., 2019, 10, 4469–4475 RSC.
- C. D. Windle, J. Massin, M. Chavarot-Kerlidou and V. Artero, Dalton Trans., 2018, 47, 10509–10516 RSC.
- N. Kaeffer, C. D. Windle, R. Brisse, C. Gablin, D. Léonard, B. Jousselme, M. Chavarot-Kerlidou and V. Artero, Chem. Sci., 2018, 9, 6721–6738 RSC.
- N. Kaeffer, J. Massin, C. Lebrun, O. Renault, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2016, 138, 12308–12311 CrossRef CAS PubMed.
- C. Windle, H. Kumagai, M. Higashi, R. Brisse, S. Bold, B. Jousselme, M. Chavarot-Kerlidou, K. Maeda, R. Abe, O. Ishitani and V. Artero, J. Am. Chem. Soc., 2019, 141, 9593–9602 CrossRef CAS PubMed.
- E. Giannoudis, S. Bold, C. Müller, A. Schwab, J. Bruhnke, N. Queyriaux, C. Gablin, D. Leonard, C. Saint-Pierre, D. Gasparutto, D. Aldakov, S. Kupfer, V. Artero, B. Dietzek and M. Chavarot-Kerlidou, ACS Appl. Mater. Interfaces, 2021, 13, 49802–49815 CrossRef CAS PubMed.
- A. Charisiadis, E. Giannoudis, Z. Pournara, A. Kosma, V. Nikolaou, G. Charalambidis, V. Artero, M. Chavarot-Kerlidou and A. G. Coutsolelos, Eur. J. Inorg. Chem., 2021, 2021, 1122–1129 CrossRef CAS.
- N. Kaeffer, M. Chavarot-Kerlidou and V. Artero, Acc. Chem. Res., 2015, 48, 1286–1295 CrossRef CAS PubMed.
- M. D. Irwin, D. B. Buchholz, A. W. Hains, R. P. H. Chang and T. J. Marks, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2783–2787 CrossRef CAS.
- M. Prato, M. Maggini, C. Giacometti, G. Scorrano, G. Sandona and G. Farnia, Tetrahedron, 1996, 52, 5221–5234 CrossRef CAS.
- P.-A. Jacques, V. Artero, J. Pécaut and M. Fontecave, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20627–20632 CrossRef CAS PubMed.
- E. S. Andreiadis, P. A. Jacques, P. D. Tran, A. Leyris, M. Chavarot-Kerlidou, B. Jousselme, M. Matheron, J. Pecaut, S. Palacin, M. Fontecave and V. Artero, Nat. Chem., 2013, 5, 48–53 CrossRef CAS PubMed.
- N. Queyriaux, E. S. Andreiadis, S. Torelli, J. Pecaut, B. S. Veldkamp, E. A. Margulies, M. R. Wasielewski, M. Chavarot-Kerlidou and V. Artero, Faraday Discuss., 2017, 198, 251–261 RSC.
- N. Kaeffer, C. D. Windle, R. Brisse, C. Gablin, D. Leonard, B. Jousselme, M. Chavarot-Kerlidou and V. Artero, Chem. Sci., 2018, 9, 6721–6738 RSC.
- N. Kaeffer, J. Massin, C. Lebrun, O. Renault, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2016, 138, 12308–12311 CrossRef CAS PubMed.
- J. C. Evans and J. C. Wahr, J. Chem. Phys., 1959, 31, 655–662 CrossRef CAS.
- In an earlier study, Tian and al discuss the interesting possibility of obtaining 1via the direct copper catalyzed Huisgen 1,3-dipolar cycloaddition between cobalt-diimine dioxime azide derivatives and surface-assembled nmep-pC60, thus, avoiding the solubility issues we faced in the current work.40 However, we note that the IR spectra of the product given as 1 by Tian and al. lacks the characteristic CN stretching band of [Co(DO)(DOH)pnBr2] moiety observed here and in earlier work (cf. Fig. S1b†) R. Blinc and D. Hadži, J. Chem. Soc., 1958, 4536–4540 RSC.
- A. Lembo, P. Tagliatesta and D. M. Guldi, J. Phys. Chem. A, 2006, 110, 11424–11434 CrossRef CAS PubMed.
- M. F. Suárez, F. Marken, R. G. Compton, A. M. Bond, W. Miao and C. L. Raston, J. Phys. Chem. B, 1999, 103, 5637–5644 CrossRef.
- V. Fourmond, P. A. Jacques, M. Fontecave and V. Artero, Inorg. Chem., 2010, 49, 10338–10347 CrossRef CAS PubMed.
- T. N. Huan, P. Simon, G. Rousse, I. Genois, V. Artero and M. Fontecave, Chem. Sci., 2017, 8, 742–747 RSC.
- Y. Chen, H. Chen and H. Tian, Chem. Commun., 2015, 51, 11508–11511 RSC.
- In our hand, copper-catalyzed 1,3 dipolar Huisgen cycloaddition between nmep-pC60 and the azido derivative of the cobalt or copper diimine-dioxime complex failed to produce the desired product..
- D. H. Nam, J. Z. Zhang, V. Andrei, N. Kornienko, N. Heidary, A. Wagner, K. Nakanishi, K. P. Sokol, B. Slater, I. Zebger, S. Hofmann, J. C. Fontecilla-Camps, C. B. Park and E. Reisner, Angew. Chem., Int. Ed., 2018, 57, 10595–10599 CrossRef CAS PubMed.
- D. Khusnutdinova, A. M. Beiler, B. L. Wadsworth, S. I. Jacob and G. F. Moore, Chem. Sci., 2017, 8, 253–259 RSC.
- B. Shan, A. Nayak, R. N. Sampaio, M. S. Eberhart, L. Troian-Gautier, M. K. Brennaman, G. J. Meyer and T. J. Meyer, Energy Environ. Sci., 2018, 11, 447–455 RSC.
- M. Wang, Y. Yang, J. Shen, J. Jiang and L. Sun, Sustainable Energy Fuels, 2017, 1, 1641–1663 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc06335a |
| This journal is © The Royal Society of Chemistry 2022 |