
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromatic heterobicycle-fused porphyrins: impact on aromaticity and excited state electron transfer leading to long-lived charge separation†
Austen
Moss‡
,
Youngwoo
Jang‡
,
Jacob
Arvidson
,
Vladimir N.
Nesterov
,
Francis
D'Souza
 * and
Hong
Wang
*
* and
Hong
Wang
*
Department of Chemistry, University of North Texas, 1155 Union Circle, #305070, Denton, TX 76203-5017, USA. E-mail: francis.dsouza@unt.edu; hong.wang@unt.edu
First published on 12th August 2022
Abstract
A new synthetic method to fuse benzo[4,5]imidazo[2,1-a]isoindole to the porphyrin periphery at the β,β-positions has been developed, and its impact on the aromaticity and electronic structures is investigated. Reactivity investigation of the fused benzoimidazo-isoindole component reveals fluorescence quenching of a zinc porphyrin (AMIm-2) upon treatment with a Brønsted acid. The reaction of the zinc porphyrin (AMIm-2) with methyl iodide initiated a new organic transformation, resulting in the ring-opening of isoindole with the formation of an aldehyde and dimethylation of the benzoimidazo component. The fused benzoimidazo-isoindole component acted as a good ligand to bind platinum(II), forming novel homobimetallic and heterobimetallic porphyrin complexes. The fusion of benzoimidazo-isoindole on the porphyrin ring resulted in bathochromically shifted absorptions and emissions, reflecting the extended conjugation of the porphyrin π-system. Time-resolved emission and transient absorption spectroscopy revealed stable excited state species of the benzoimidazo-isoindole fused porphyrins. Zinc porphyrin AMIm-2 promoted excited state electron transfer upon coordinating with an electron acceptor, C60, generating a long-lived charge-separated state, in the order of 37.4 μs. The formation of the exceptionally long-lived charge-separated state is attributed to the involvement of both singlet and triplet excited states of AMIm-2, which is rarely reported in porphyrins.
Introduction
Highly π-extended carbon-rich materials represent fascinating yet challenging frontiers in chemistry and materials science as they offer numerous opportunities in molecular electronics.1–5 Incorporation of heteroatom(s) into highly π-extended structures can provide control of the electronic structure, and has become a critical strategy to manipulate the materials in order to endow them with desirable properties, such as electrochemical activity, optical absorption spectra, photoluminescence, redox behaviour, charge transfer, bandgap tuning, and other characteristics.6–9 The development of new methods to introduce heteroatoms or heterocycles is very important to facilitate the growth of this field. Electron delocalization is an important phenomenon in any highly π-conjugated systems. Understanding how the incorporated heteroatoms/heterocycles affect the conjugated systems is of paramount importance to advancing the field.Porphyrins are aromatic macrocycles, which are nature-selected pigments playing major roles in important biological activities. Due to their unique electronic and photophysical properties, porphyrins have been extensively used in various applications ranging from organic electronics to biomedicines. In recent years, porphyrins have been proven to be effective building blocks in self-assembled materials such as covalent organic frameworks (COFs), metal–organic frameworks (MOFs), and others.10–18 In this regard, highly π-extended systems obtained through fusing porphyrins with aromatic rings such as polycyclic aromatic hydrocarbons (PAHs) are especially attractive as they are reminiscent of “nanographene” doped with heteroatoms.1,2,19–42 Theoretically, the fusion of aromatic moieties to the porphyrin periphery can be realized at the meso, β- and β,β-positions (Fig. 1). Although synthetically challenging, PAH-fused porphyrins have been achieved through fusion at the meso, β-, and β,β-positions.1,2,19–42 In contrast, the π-extension of porphyrins through the incorporation of aromatic heterocycles is rare and is synthetically more challenging. Only two types of aromatic heterocycles have been used to extend the porphyrin π-conjugated system: pyrazine43–50 and imidazole51,52 (Fig. 1). Since the successful development of the synthetic method by Crossley and co-workers in 1991,48 the pyrazine approach has been widely used in constructing many organic materials for various fundamental studies and applications.43–50,53,54 Given the ubiquitous roles aromatic heterocycles play in organic materials science, it is important to develop new strategies to incorporate aromatic heterocycles into highly π-extended structures.
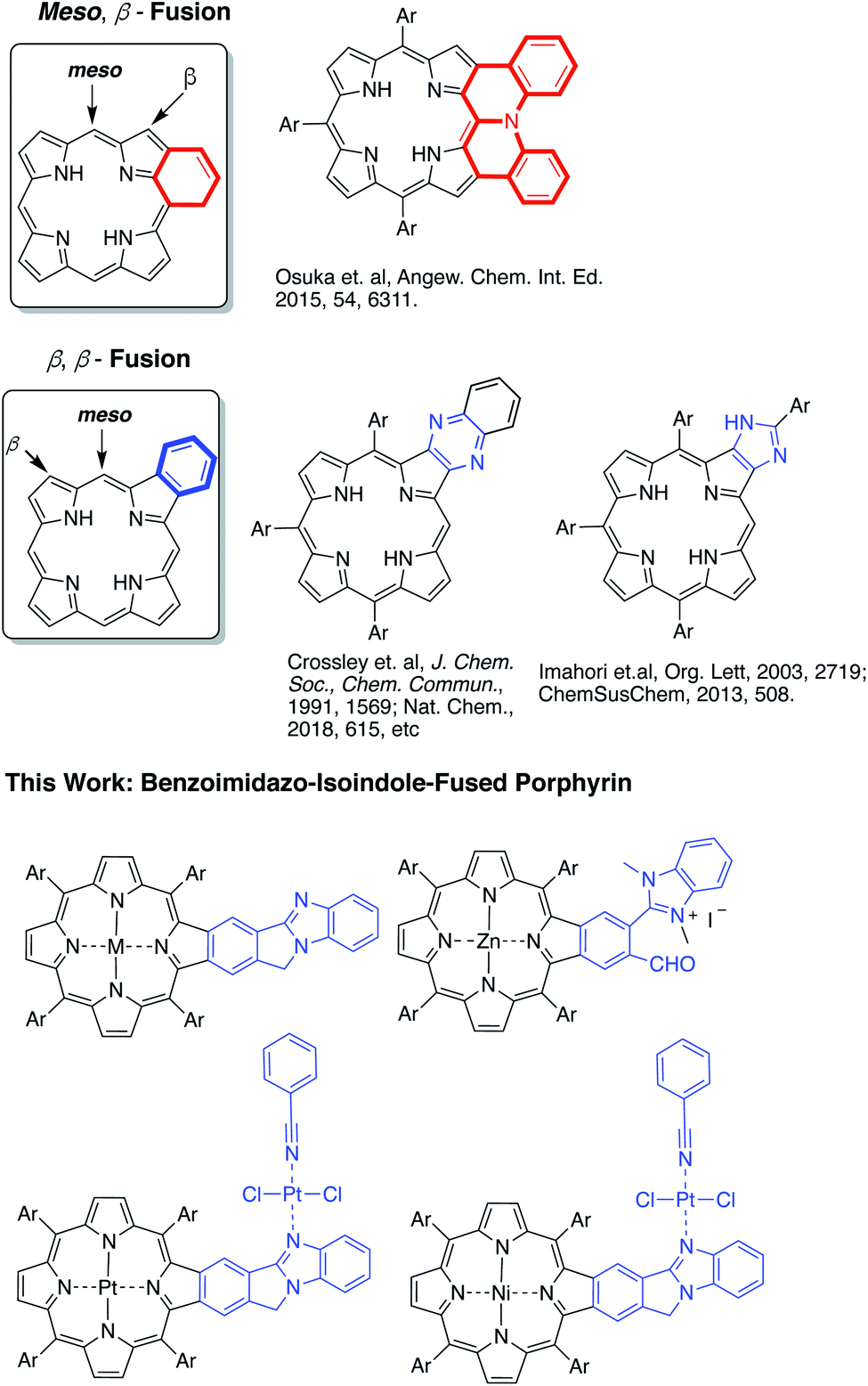 | ||
| Fig. 1 Structures of the benzoimidazo-isoindole-fused porphyrins. | ||
In this work, a new synthetic method was developed to fuse benzo[4,5]imidazo[2,1-a]isoindole to porphyrins at the porphyrin β,β-positions (Fig. 1).55 The benzimidazole moiety has been widely used in drug development.56,57 Benzimidazole is also known to serve as a ligand to metals.58–60 However, the impact of the benzimidazole moiety on aromaticity and electronic structures of π-extended structures remains elusive. Benzo[4,5]imidazo[2,1-a]isoindole induces intrinsic dipole moments in the π-extended molecule owing to the presence of the amidine component (Fig. 1) and is expected to bring in new features. The newly synthesized benzo[4,5]imidazo[2,1-a]isoindole-fused porphyrins have been studied using spectral, electrochemical, DFT, and time-resolved emission and transient absorption spectral methods to probe the effect of benzoimidazo-isoindole fusion on the overall physico-chemical properties. The ability of this class of compounds to promote excited-state electron transfer has also been investigated using zinc porphyrin AMIm-2 as an electron donor and fullerene C60 as an electron acceptor. Our studies show that both the singlet and triplet excited states of AMIm-2 were involved in the electron transfer process, yielding a long-lived charge-separated state of the order of 35 μs.
Results and discussion
Molecular design and synthesis
The synthetic methods developed in our group for π-extended porphyrins open the door to functionalizing π-extended porphyrins at porphyrin β,β-positions.61–65 Our initial plan was to synthesize a phthalaldehyde-fused porphyrin (1, Scheme 1) that would react with o-phenylenediamine to form a 1,4-diazocine-fused porphyrin, which is “non-aromatic” in the ground state, in order to study the interaction between the heteroatom-containing diazocine and porphyrin (Scheme 1). To our surprise, the reaction between the monobenzoporphyrin (1) and o-phenylenediamine under acidic conditions (p-TsOH/dichloromethane (DCM)) did not yield the desired 1,4-diazocine-fused porphyrin. Instead, a complex mixture resulted. After careful analysis of this mixture, we were able to isolate a new porphyrin, which was identified as a benzo[4,5]imidazo[2,1-a]isoindole-fused porphyrin (AMIm-1) (Scheme 1). This exciting result prompted us to scrutinize the literature and we found that Chen, J. et al. reported a similar method to synthesize benzo[4,5]imidazo[2,1-a]isoindoles.55 We then optimized Chen's original procedure (HCOOH/MeOH) to develop suitable conditions for porphyrins. Changing the solvent from MeOH to dichloromethane (DCM) and raising the temperature from 0 °C to room temperature significantly increased the efficacy of this reaction, affording AMIm-1 in 80% yield. Due to the immiscibility of the dichloromethane and the 88% formic acid used in the reaction, the reaction is biphasic, which led to easy separation and purification of the product. This methodology is high-yielding and extremely concise as it does not require lengthy purification protocols using column chromatography. Subsequent insertion of zinc(II) into the porphyrin core afforded AMIm-2 in 92% yield (see the ESI for synthetic details and Fig. S1 and S2† for 1H NMR and FT-IR spectra).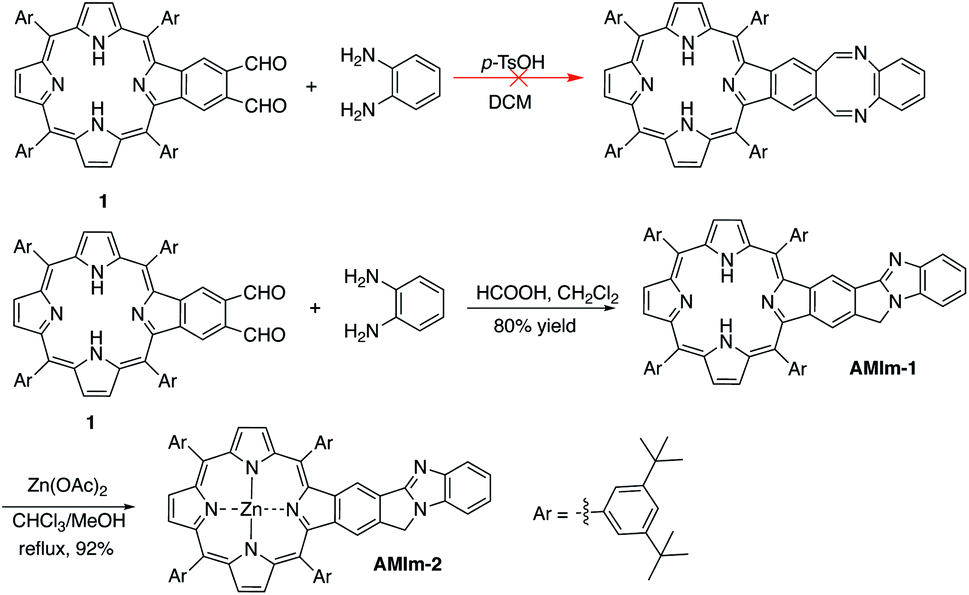 | ||
| Scheme 1 Synthesis of benzo[4,5]imidazo[2,1-a]isoindole-fused porphyrins. | ||
Activity of the fused benzo[4,5]imidazo[2,1-a]isoindole
Benzo[4,5]imidazo[2,1-a]isoindole induces intrinsic dipole moments as shown in Scheme 2. Benzo[4,5]imidazo[2,1-a]isoindole can be treated as isoindole fused with benzimidazole. If the resonance structure A (Scheme 2) can be trapped, it will provide these porphyrins with new features and opportunities. Methylation of the more basic nitrogen of the amidine appeared to be a plausible approach. We decided to treat AMIm-2 with excess MeI (0.2 ml) in DCM (1 ml). The reaction was tracked with UV-vis spectroscopy. Changes started to appear after one day and the reaction was stopped until no further changes were observed after three days. The product was isolated through recrystallization in DCM/MeOH. To our surprise, the 1H NMR spectra of this product displayed a completely different pattern from that of AMIm-2 showing an aldehyde peak at 9.75 ppm, which did not match the expected AMIm-3. The nature of this reaction remained a mystery until the crystal structure of this product was resolved (see ESI Fig. S3 for the structure and ESI Table S1 for the crystallographic data, CCDC: 2172828†). The crystal structure of AMIm-4 reveals that methylation occurred to both nitrogen atoms on the imidazole ring, and the 5-membered ring of the isoindole component was cleaved leaving a hanging methylated imidazole group and an aldehyde group. This organic transformation reaction has not been reported in the literature. The mechanism remains elusive. Cleavage of the C–N bond may be plausible given that the nitrogen carries a positive charge. However, the conversion to a carbonyl group was not understood, but could be related to the minor amount of H2O present in the reaction mixture and needs further investigation.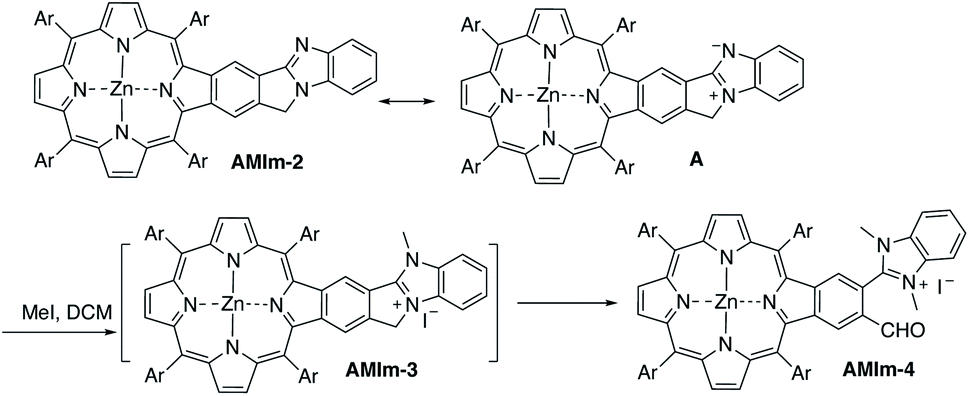 | ||
| Scheme 2 Resonance structure and reactivity of AMIm-2 with methyl iodide. | ||
Benzimidazole is a base. It can also serve as a ligand to metals. However, this benzimidazole component is fused in the system and thus has no rotational flexibility. It will be interesting to explore its binding ability as it can potentially provide two metal-binding sites within the highly π-extended system (Scheme 3). AMIm-1 was treated with Zn(OAc)2 and Ni(OAc)2 under respective metal insertion conditions, and metalation of the porphyrin core was observed for both metals, affording the corresponding metalated products (AMIm-2 and AMIm-5) (Scheme 3). However, binding to the benzimidazole site was not observed in both cases. We then attempted to metalate AMIm-1 with PtCl2 as platinum porphyrins/complexes are of broad interest in various areas.66–70AMIm-1 was treated with PtCl2 in benzonitrile under reflux. To our surprise, Pt(II) was able to bind both at the porphyrin core and at the imidazole site, affording the homo-bimetallic complex AMIm-6 in a 56% yield. Encouraged by this result, AMIm-2 and AMIm-5 were subjected to reactions with platinum chloride in benzonitrile respectively to obtain heterobimetallic complexes. Heterobimetallic AMIm-7 was successfully obtained with AMIm-5. However, the reaction of PtCl2 with AMIm-2 resulted in an inseparable mixture with the homometallic AMIm-6 identified as the major product. Apparently, Zn(II) was kicked out of AMIm-2 and Pt(II) was inserted at both binding sites.
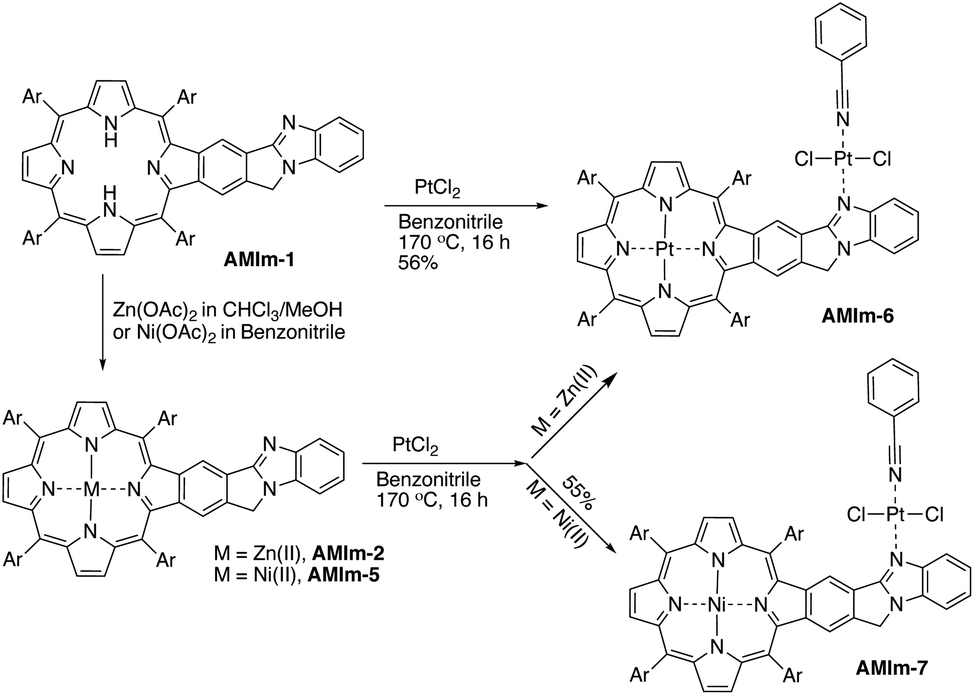 | ||
| Scheme 3 Reactivity of AMIm-1 with different metal ions and the resulting products. | ||
Crystal structures
The single crystal structure of AMIm-6 (CCDC: 2172827†) and AMIm-7 (CCDC: 2172829, see Tables S2 and S3† for structural parameters) was obtained (Fig. 2). In AMIm-6, the Pt(II) porphyrin ring and the fused benzo[4,5]imidazo[2,1-a]isoindole unit stay essentially in the same plane. The second Pt(II) is bonded to two chloride groups, the imidazole nitrogen, and a benzonitrile molecule which came from the solvent for the platinum insertion reaction. The second Pt(II) has a typical square planar geometry and is assumedly locked perpendicular to the porphyrin plane likely due to steric interaction of the neighbouring di-tert-butylphenyl group. In AMIm-7, the coordination sphere of Pt(II) is similar to that of AMIm-6, adopting a square planar geometry, which is perpendicular to the porphyrin ring. The porphyrin ring of AMIm-7 is distorted due to the presence of Ni(II). These structures provide direct evidence for the involvement of the benzimidazole functionality in metal binding.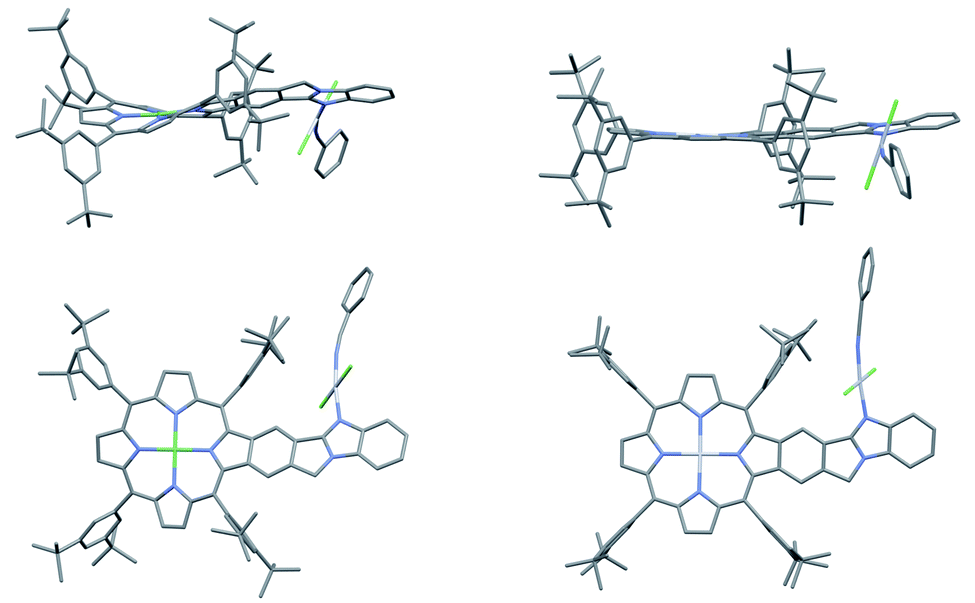 | ||
| Fig. 2 X-ray crystal structures of AMIm-6 (right) and AMIm-7 (left). Top: top view. Bottom: side view. All hydrogen atoms are omitted for clarity. | ||
Spectral, computational and electrochemical studies
Having established the synthesis, structure, and reactivity of the benzoimidazo-isoindole-fused porphyrins, systematic studies were then performed to evaluate the effect of benzoimidazo-isoindole-fusion on the spectral, electrochemical, and photochemical properties of these novel π-extended porphyrins.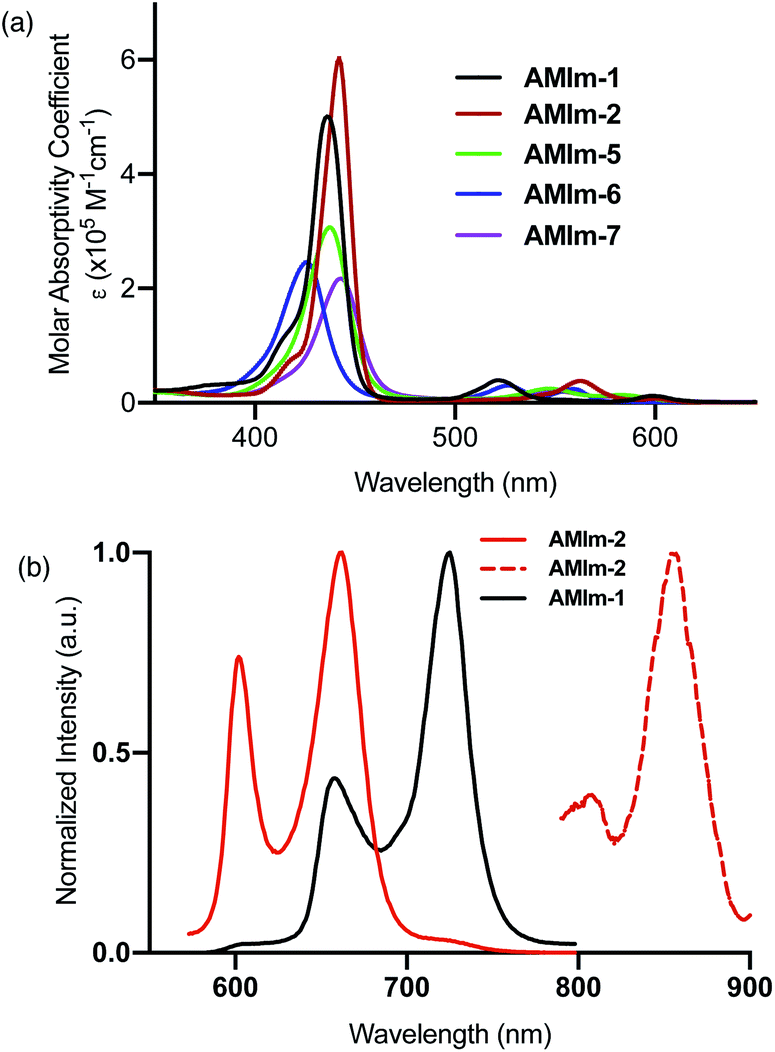 | ||
| Fig. 3 (a) Absorption and (b) normalized fluorescence and phosphorescence spectra of the indicated compounds in dichlorobenzene (DCB). | ||
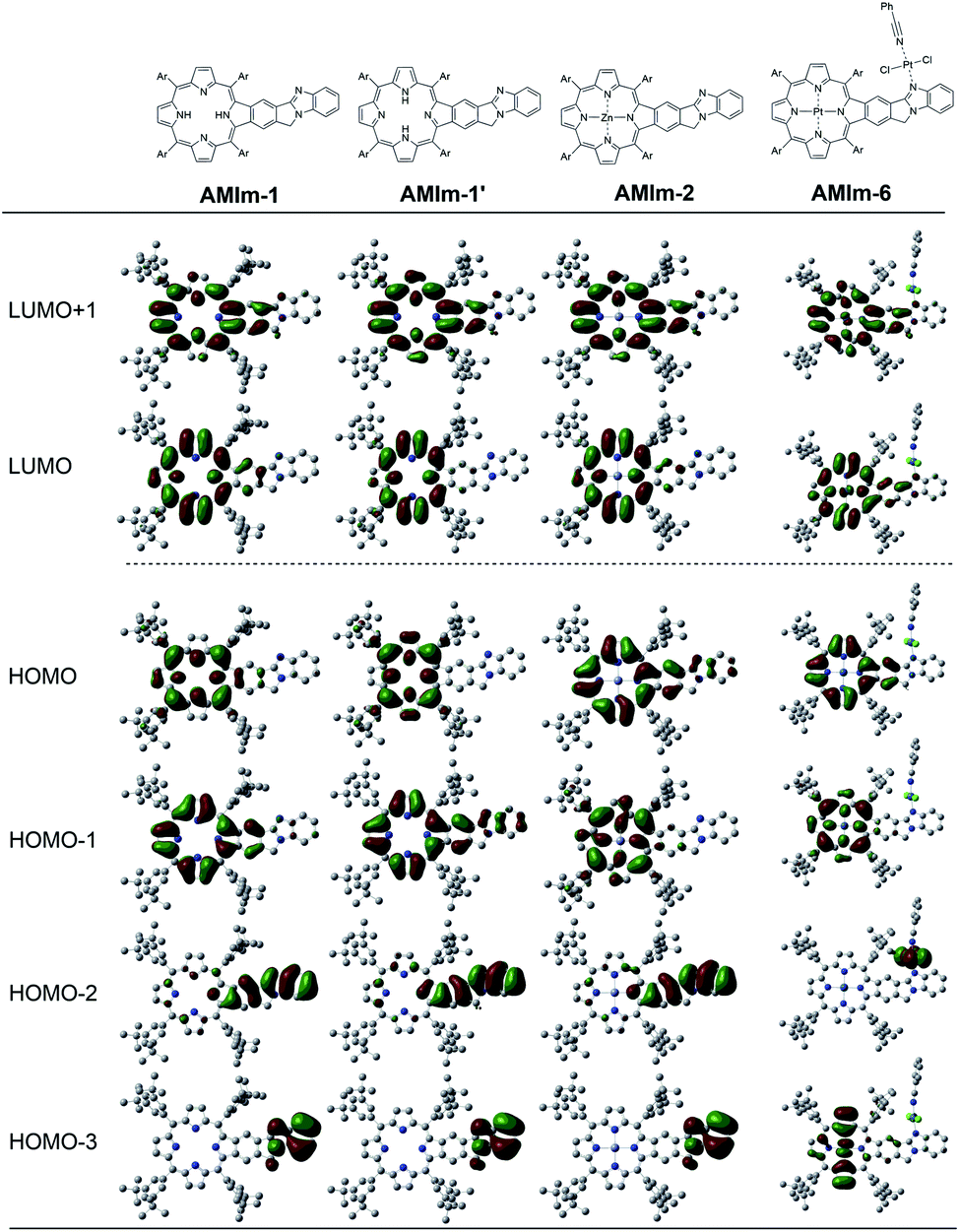 | ||
| Fig. 4 B3LYP/6-31G* optimized structures and frontier orbitals of the indicated molecular systems. | ||
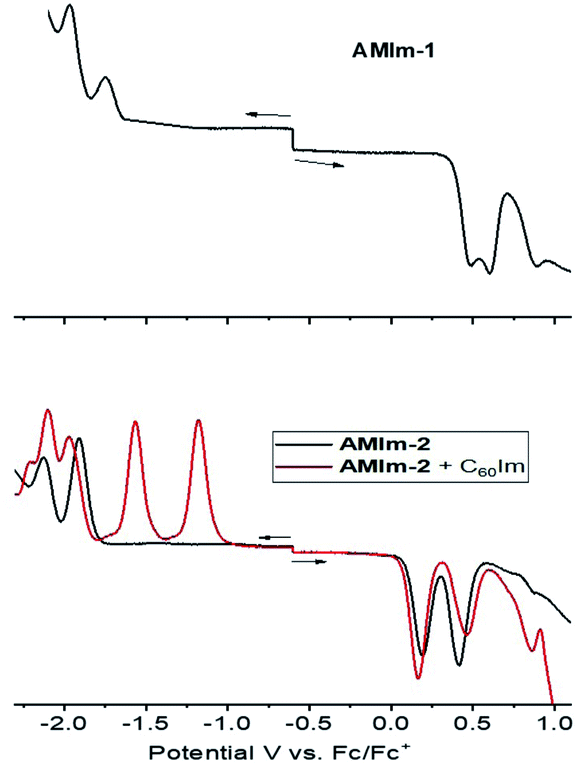 | ||
| Fig. 5 Differential pulse voltammograms of the indicated molecular systems in DCB containing 0.1 M (TBA)ClO4. | ||
Photochemical properties
The excellent spectral and redox properties of AMIm-1 and AMIm-2 further prompted us to investigate their photophysical properties along with their ability to undergo excited-state electron transfer reactions. With this in mind, first, the transient spectral behaviour of both compounds was investigated using femtosecond- (fs-TA) and nanosecond transient absorption (ns-TA) techniques. The former technique probes mainly photo-events originating from the singlet excited state while the latter probes events from the triplet excited state.Fig. 6a shows the fs-TA spectra of AMIm-1 at the indicated delay times. At the Soret band excitation, the excited S2 state populated the S1 state within the first ps via the process of internal conversion (IC). The S1 state revealed excited state absorption peaks at 457, 550, 623, 749, 1125, and 1293 nm. This was accompanied by negative peaks at 518, 595, 660, and 722 nm. By comparison with the earlier discussed absorption and fluorescence, it could be deduced that the first two peaks corresponded to the process of ground-state bleaching (GSB) and the latter two to the stimulated emission (SE) process. The decay of the ESA peaks and the recovery of the SE and GSB peaks were rather slow, consistent with the long lifetime of 1AMIm-1* of 13.6 ns (see the Fig. 6a inset for the decay profile of the 1120 nm peak). Ns-TA spectra were also subsequently recorded to characterize the 3AMIm-1* formed via the process of intersystem crossing (ISC) of 1AMIm-1*. Triplet–triplet absorption peaks of 3AMIm-1* were located at 455, 559, 628, and 918 nm. From the decay profile of the 455 nm band (see the Fig. 6b inset) a lifetime of 34.65 μs was obtained.
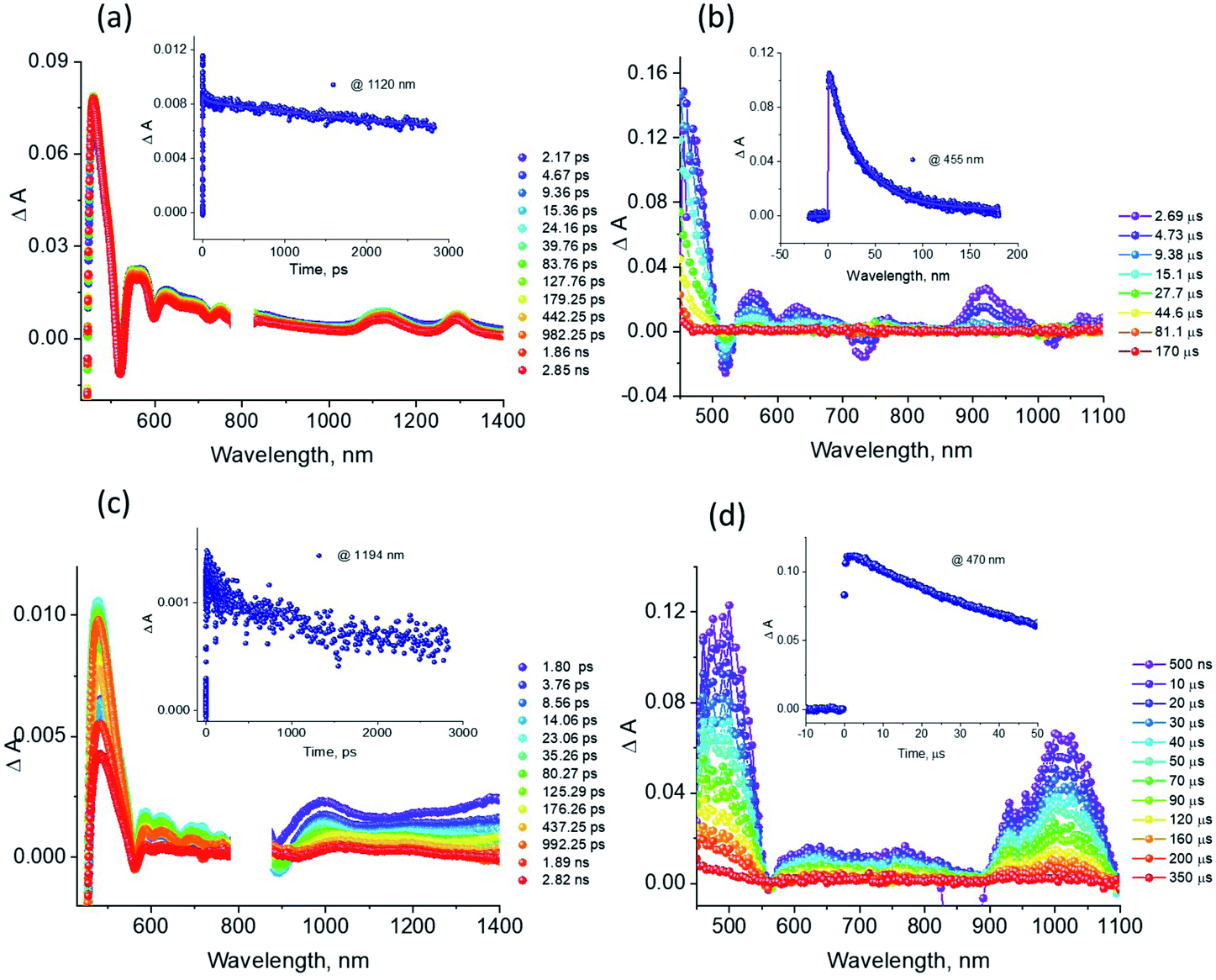 | ||
| Fig. 6 (a and c) Femtosecond transient absorption spectra at the indicated delay times, and (b and d) nanosecond transient absorption spectra of (a and b) AMIm-1 and (c and d) AMIm-2 in DCB. The samples were excited at their corresponding Soret band maxima. The decay profiles at the indicated wavelengths are shown in the figure inset. | ||
The fs-TA spectra of 1AMIm-2* at the indicated delay times are shown in Fig. 6c. The S1 state formed from the rapid internal conversion of the S2 state revealed ESA peaks at 478, 585, 621, 692, 988, and 1194 nm. This was accompanied by negative peaks at 558, 604, and 667 nm. By spectral comparison, it could be deduced that among these peaks, the first two corresponded to GSB and part of the second and third peaks to SE. Decay of the positive peaks and recovery of the negative peaks were relatively fast (see the Fig. 6c inset for the decay profile of the 1194 nm peak), consistent with the short lifetime of 1AMIm-2* of 2 ns. Subsequent ns-TA, shown in Fig. 6d, revealed peaks corresponding to 3AMIm-2* at 440, 624, 769, and 1013 nm. By monitoring the decay profile of the 470 nm peak (see the Fig. 6d inset), a lifetime of 82.42 μs was obtained.
Light-induced electron transfer
The physico-chemical properties along with the excited state features of the benzoimidazo-isoindole-fused porphyrins further prompted us to use them as electron donors in excited state electron transfer reactions. The zinc derivative AMIm-2 was utilized in this case study owing to its facile oxidation, favorable E0,0 and ET values, and well-behaved singlet and triplet transient spectral features. With regard to the electron acceptor, the well-known electron acceptor fullerene C6084 has been employed using a self-assembly approach of metal–ligand axial coordination. For this, C60 is functionalized with an axially coordinated ligand, phenyl imidazole, C60Im.85The coordinatively unsaturated zinc in zinc porphyrins is known to bind nitrogenous ligands with moderate stability.85,86 This also seems to be the case in AMIm-2. As shown in Fig. 7a, the increased addition of C60Im to a solution of AMIm-2 in DCB revealed the diminished intensity of the Soret and visible bands with redshifts. Isosbestic points at 410, 455, and 569 nm were observed indicating the existence of an equilibrium. The equilibrium constant, K, evaluated using the Benesi–Hildebrand method86 was found to be 7.2 × 104 M−1 (see the Fig. 7a inset for the plot) suggesting moderate stability. The linear plot also suggests 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry. The fluorescence spectrum of AMIm-2 was also recorded during the process of titration, as shown in Fig. 7b. Both bands revealed quenching over 80% indicating the occurrence of excited state photo-events such as electron or energy transfer.86 It may be mentioned here that when the AMIm-2:ImC60 complex was excited either at the Soret (Fig. 7b) or visible band maxima, no emission around 715 nm corresponding to C60Im was observed, thus ruling out the possibility of energy transfer as the quenching mechanism or indicating that such a process is a minor one in the donor–acceptor dyad.
:1 stoichiometry. The fluorescence spectrum of AMIm-2 was also recorded during the process of titration, as shown in Fig. 7b. Both bands revealed quenching over 80% indicating the occurrence of excited state photo-events such as electron or energy transfer.86 It may be mentioned here that when the AMIm-2:ImC60 complex was excited either at the Soret (Fig. 7b) or visible band maxima, no emission around 715 nm corresponding to C60Im was observed, thus ruling out the possibility of energy transfer as the quenching mechanism or indicating that such a process is a minor one in the donor–acceptor dyad.
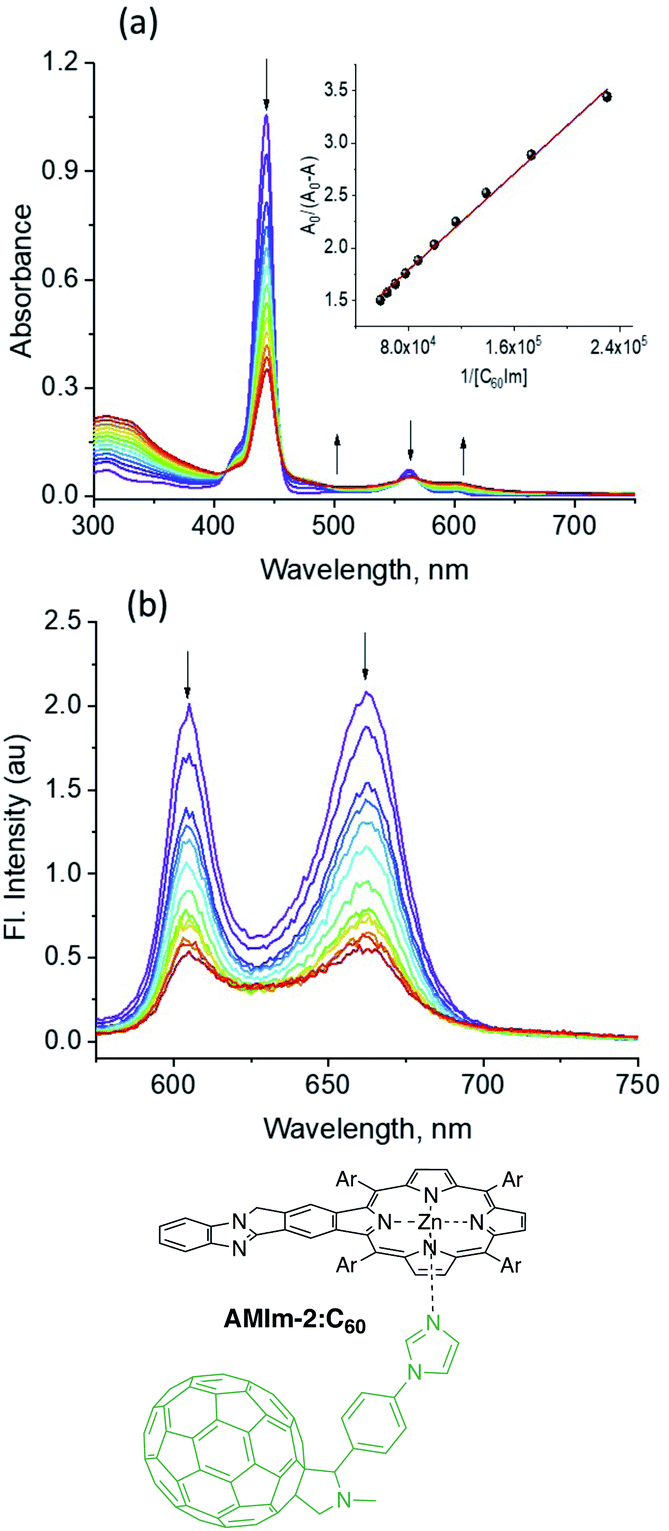 | ||
| Fig. 7 (a) Absorption and (b) fluorescence spectral changes of AMIm-2 on increasing addition of C60Im. The Benesi–Hildebrand plot to evaluate the binding constant is shown in the inset of (a). The sample was excited at the Soret band peak maximum. | ||
The B3LYP/6-31G* optimized structures and frontier orbitals of the AMIm-2:ImC60 complex (see Fig. S11 in the ESI†) clearly reveal the formation of a 1:1 complex without any steric constraints. Interestingly, the LUMO confined to the AMIm-2 macrocycle (see Fig. S11 in the ESI†) is now on C60 establishing the latter's role as an electron acceptor. The majority of the HOMO was on the porphyrin π-structure while HOMO−1 was extended to the benzoimidazole part of the molecule suggesting π-extension also in the oxidized form of AMIm-2.
The electrochemical studies also helped in realizing the metal–ligand binding induced redox changes and facile reduction of C60 compared to that of the zinc porphyrin in the AMIm-2:ImC60 complex, as shown in Fig. 5. Coordination of C60Im cathodically shifted the oxidation peak of AMIm-2 by 30 mV at 0.16 V vs. Fc/Fc+ and the reduction peak by 60 mV at −1.97 V. The first two reductions of C60Im were located at −1.17 and −1.55 V (see the red trace in the lower panel of Fig. 5).
An energy diagram was subsequently established to visualize the electron transfer reaction (Fig. 8). The energy of the excited states was calculated from the earlier discussed spectral data while that of the charge-separated state was calculated using redox data according to the Rehm–Weller approach.87 The thermodynamic feasibility of excited electron transfer from both the singlet and triplet excited states of AMIm-2 is obvious from the energy diagram, with −ΔGSCS = 0.83 eV and −ΔGTCS = 0.22 eV. The energy of the charge-separated state which equals the free-energy change for charge recombination (−ΔGCR) is relatively high, being about 1.23 eV (this also assumes the energy of the singlet and triplet radical ion-pairs to be almost the same). Under such circumstances, the charge recombination could fall in the inverted region of the Marcus parabola88 potentially extending the lifetime of the charge-separated state. In addition, if electron transfer also occurs from the 3AMIm-2* state, then one would expect a long-lived charge-separated state due to different spins of the radical ion-pair. To secure evidence of triplet quenching, the phosphorescence spectrum of AMIm-2 was recorded in the presence of C60Im at liquid nitrogen temperature, as shown in Fig. S12 in the ESI.† Quenching of phosphorescence was observed (over 80%) indicating the occurrence of photochemical events from the triplet excited state. It may also be pointed out here that since the spectrum was recorded at liquid nitrogen temperature, it is safe to conclude that the quenching process is intramolecular involving the AMIm-2:ImC60 complex, and not intermolecular as no diffusion of donor and acceptor entities could be expected in the frozen state. Systematic transient absorption spectral studies were performed using both fs-TA and ns-TA techniques to characterize the electron transfer events from the singlet and triplet excited states and to determine the kinetics of these photo-events.
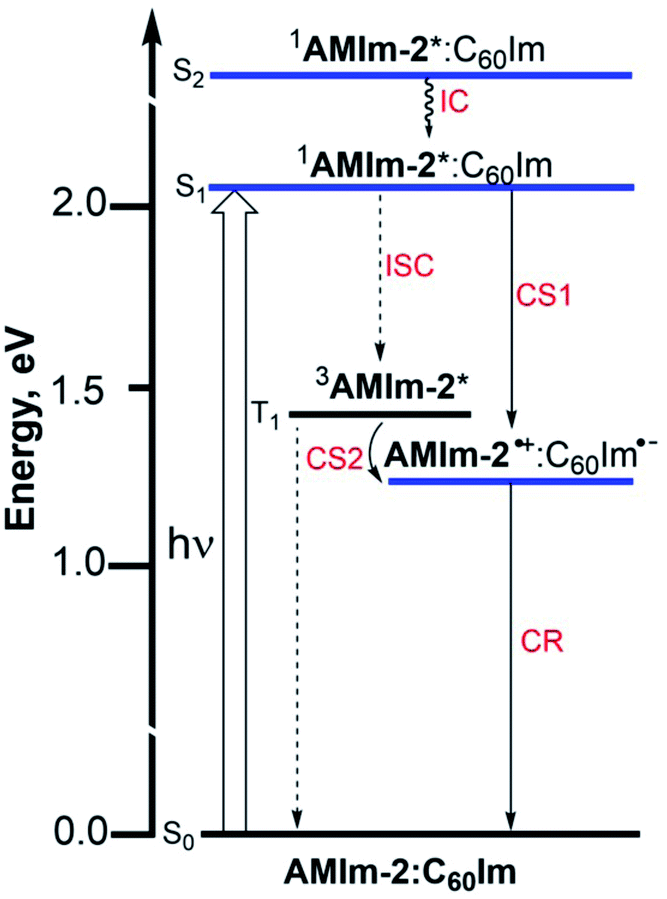 | ||
| Fig. 8 Energy level diagram showing photoinduced electron transfer originating from both singlet and triplet excited states of AMIm-2 in the presence of coordinated C60Im. | ||
Fig. 9a compares the fs-TA spectra of AMIm-2 and AMIm-2:ImC60 complex in dichlorobenzene (DCB) at a delay time of 242 ps. Complete fs-TA spectra of AMIm-2:ImC60 at different delay times are shown in Fig. S13a in the ESI.† The presence of ImC60 deactivated the ESA and GSB/SE peaks of AMIm-2 much more rapidly accompanied by the appearance of new peaks in the visible region at 644 and 690 nm and a near-IR peak in the 1000 nm region.
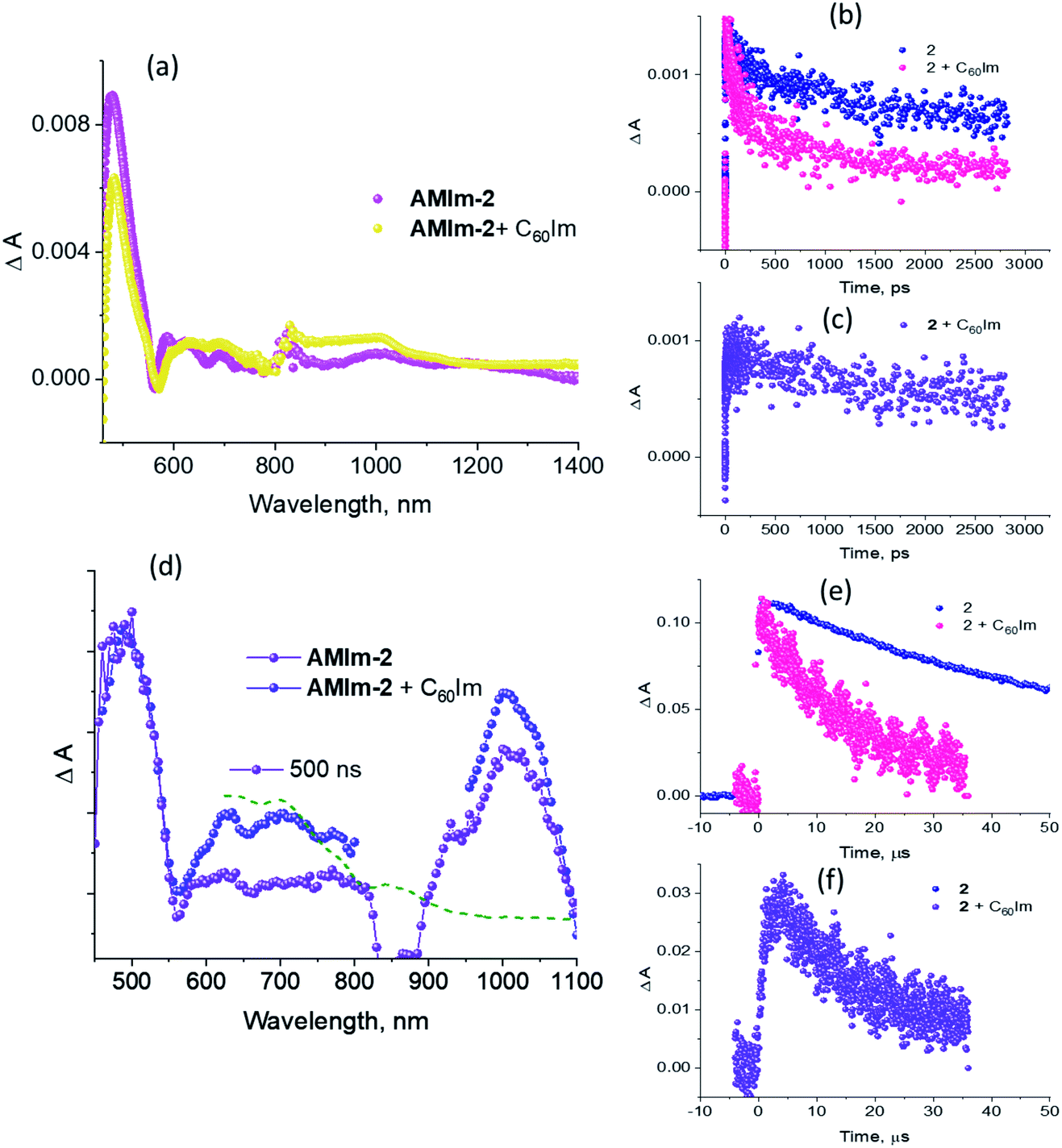 | ||
| Fig. 9 (a) Fs-TA spectra of AMIm-2 and AMIm-2:ImC60 complex in DCB at a delay time of 242 ps and an excitation wavelength of 420 nm. (b) Decay profile of the near-IR ESA peaks. (c) Decay profile of AMIm-2˙+ monitored at 695 nm for the dyad. (d) Nanosecond transient spectra, normalized to the 470 nm peak, of AMIm-2 and AMIm-2:ImC60 complex in DCB at a delay time of 500 ns and an excitation wavelength of 420 nm. (e) Decay profile of the 470 nm peak corresponding to 3AMIm-2*. (f) Decay profile of the AMIm-2˙+ monitored at 690 nm. The dashed line in (d) represents the spectrum of chemically oxidized AMIm-2 (NOBF4 was used as an oxidizing agent). | ||
Chemical oxidation of AMIm-2 confirmed that the visible bands are due to the formation of AMIm-2˙+ while the near-IR peak is due to the formation of 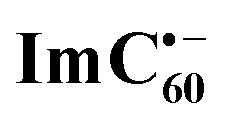 (see the dashed line in Fig. 9d). These spectral results provide direct proof for the occurrence of electron transfer from the S1 state of porphyrin in the AMIm-2:ImC60 complex. For the determination of the rate of charge separation from the singlet excited state, kSCS, the decay time constant of the near-IR ESA peak of AMIm-2 was monitored, as shown in Fig. 9b. Faster decay of this peak in the presence of C60Im can be clearly seen in this plot. From the decay time constants, a value of kSCS = 3.24 × 1010 s−1 was obtained. For the determination of the rate of charge recombination of the singlet radical ion-pair, kSCR, the AMIm-2˙+ peak at 695 nm was monitored, as shown in Fig. 9c. A kSCR value of 3.52 × 108 s−1 was determined; however, the signal persisted beyond the 3 ns monitoring window of the femtosecond transient setup. Under such conditions, the measured kSCR could be treated as the upper limit.
(see the dashed line in Fig. 9d). These spectral results provide direct proof for the occurrence of electron transfer from the S1 state of porphyrin in the AMIm-2:ImC60 complex. For the determination of the rate of charge separation from the singlet excited state, kSCS, the decay time constant of the near-IR ESA peak of AMIm-2 was monitored, as shown in Fig. 9b. Faster decay of this peak in the presence of C60Im can be clearly seen in this plot. From the decay time constants, a value of kSCS = 3.24 × 1010 s−1 was obtained. For the determination of the rate of charge recombination of the singlet radical ion-pair, kSCR, the AMIm-2˙+ peak at 695 nm was monitored, as shown in Fig. 9c. A kSCR value of 3.52 × 108 s−1 was determined; however, the signal persisted beyond the 3 ns monitoring window of the femtosecond transient setup. Under such conditions, the measured kSCR could be treated as the upper limit.
Further, ns-TA spectra were also recorded for the AMIm-2:ImC60 complex. Fig. 9d shows the corresponding spectra, normalized to the 470 nm triplet peak, of AMIm-2 and AMIm-2:ImC60 complex at a delay time of 500 ns in DCB (see Fig. S13b in the ESI† for full spectral features of the complex). Such a spectral comparison was warranted since the spectrum of 3AMIm-2* had ESA peaks both in the visible and near-IR regions where radical cation and radical anion peaks were expected. In the presence of C60Im, 3AMIm-2* revealed faster decay, similar to that observed for the 1AMIm-2* species suggesting its involvement in the electron transfer process (see Fig. 9e for decay curves). Interestingly, the AMIm-2˙+ peaks in the visible region and the 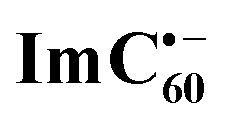 peak in the near-IR region were clearly visible with contributions from 3AMIm-2*. From the decay curves of 3AMIm-2* monitored at 470 nm, a value of kTCS = 3.24 × 104 s−1 was obtained. Similarly, by monitoring the AMIm-2˙+ peak at 690 nm (see Fig. 9f for the decay curve) a value of kTCR = 2.68 × 104 s−1 was obtained. From the kTCR value, a lifetime of the charge-separated state of 37.4 μs was obtained, the highest observed for zinc porphyrin–fullerene dyads assembled via the metal–ligand axial coordination approach.
peak in the near-IR region were clearly visible with contributions from 3AMIm-2*. From the decay curves of 3AMIm-2* monitored at 470 nm, a value of kTCS = 3.24 × 104 s−1 was obtained. Similarly, by monitoring the AMIm-2˙+ peak at 690 nm (see Fig. 9f for the decay curve) a value of kTCR = 2.68 × 104 s−1 was obtained. From the kTCR value, a lifetime of the charge-separated state of 37.4 μs was obtained, the highest observed for zinc porphyrin–fullerene dyads assembled via the metal–ligand axial coordination approach.
Conclusions
This work introduces a novel class of highly π-extended structures with interesting properties through fusing porphyrin with aromatic heterobicycles. A new method has been developed to fuse an aromatic heterobicycle, i.e., benzo[4,5]imidazo[2,1-a]isoindole, to the porphyrin periphery. The π-extended porphyrins fused with benzo[4,5]imidazo[2,1-a]isoindole displayed unprecedented reactivities toward methyl iodide and platinum(II). A new organic transformation was discovered when AMIm-2 was treated with MeI, resulting in ring-opening of the fused isoindole component and formation of an aldehyde functional group. Upon treatment with PtCl2, both homobimetallic and heterobimetallic complexes were obtained (AMIm-6 and AMIm-7), offering a new class of metal complexes.The fusion of heterobicycles to the porphyrin ring results in a bathochromic shift of absorptions for the free base, zinc, and nickel benzoimidazo-isoindole-fused porphyrins (AMIm-1, AMIm-2, and AMIm-5) relative to those of their unfused analogues. On the other hand, while the homobimetallic Pt(II) complex AMIm-6 displayed a blue-shifted Soret band as compared with that of AMIm-1, AMIm-2, and AMIm-5, that of the heterobimetallic AMIm-7 was very red-shifted, reflecting a significant effect on metal ion induced spectral changes. It is notable that zinc porphyrin AMIm-2 demonstrated fluorescence quenching upon treatment with an acid. In contrast to the experimental spectroscopic data which indicate the conjugation of the heterocycles to the porphyrin π-system, the HOMO and LUMO obtained from DFT calculation showed minimal involvement of the heterocycles. These contradicting data suggest that the traditional Gouterman 4-orbital model78,79 cannot interpret the observed spectroscopic data, and a more complex model should be developed.
Photoinduced electron transfer properties of the zinc porphyrin derivative, AMIm-2, in the presence of C60 revealed a long-lived charge-separated state, in the order of 37.4 μs. It is interesting that the intramolecular electron transfer occurred from both the singlet and the triplet excited states. Intramolecular photoinduced electron transfer from the singlet excited state is commonly reported in the literature;84–86 however, such reactions from the triplet excited state are not common.89 The exceptionally long-lived charge-separated state observed in this work was attributed to the involvement of the triplet excited state. Stable excited state species of the benzoimidazo-isoindole-fused porphyrins were also identified through time-resolved emission and transient absorption spectroscopy. The availability of this highly π-extended structure provides new knowledge and an opportunity to design and explore new organic materials for organic electronics and energy harvesting.
Data availability
Crystallographic data for AMIm-4, AMIm-6 and AMIm-7 have been deposited at the Cambridge Crystallographic Data Centre (AMIm-4, CCDC: 2172828; AMIm-6, CCDC: 2172827, AMIm-7, CCDC: 2172829). Experimental procedures, 1H and 13C NMR spectra and computational methods, which support this article, have been uploaded as part of the supplementary material.Author contributions
H. W. and A. M. designed the project. A. M. and J. A. conducted the synthesis and characterization, and initial spectral studies of the compounds. Y. J. and F. D. designed and carried out the experiments for electrochemical measurement and transient spectroscopy. A. M. and Y. J. performed all the computations. V. N. N. collected the X-ray diffraction data and solved the crystal structures. All authors contributed to data analysis and manuscript writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under Award DE-SC0016766. Our gratitude goes out to Dr Guido Verbeck and the Laboratory for Imaging Mass Spectrometry at the University of North Texas for MALDI-Orbitrap Mass Spectrometry data. We acknowledge the National Science Foundation MRI Program (CHE-1726652) and the University of North Texas for supporting the acquisition of the Rigaku XtaLAB Synergy-S X-ray diffractometer.Notes and references
- M. Stepien, E. Gonka, M. Zyla and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef CAS PubMed.
- A. Narita, X. Y. Wang, X. Feng and K. Mullen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- I. Pozo, E. Guitian, D. Perez and D. Pena, Acc. Chem. Res., 2019, 52, 2472–2481 CrossRef CAS PubMed.
- X. Liu, S. Y. Chen, Q. Chen, X. Yao, M. Gelleri, S. Ritz, S. Kumar, C. Cremer, K. Landfester, K. Mullen, S. H. Parekh, A. Narita and M. Bonn, Angew. Chem., Int. Ed. Engl., 2020, 59, 496–502 CrossRef CAS PubMed.
- E. M. Muzammil, D. Halilovic and M. C. Stuparu, Commun. Chem., 2019, 2, 58 CrossRef.
- X. Wang, G. Sun, P. Routh, D. H. Kim, W. Huang and P. Chen, Chem. Soc. Rev., 2014, 43, 7067–7098 RSC.
- S. Kawai, S. Nakatsuka, T. Hatakeyama, R. Pawlak, T. Meier, J. Tracey, E. Meyer and A. S. Foster, Sci. Adv., 2018, 4, eaar7181 CrossRef PubMed.
- X. Y. Wang, X. Yao, A. Narita and K. Mullen, Acc. Chem. Res., 2019, 52, 2491–2505 CrossRef CAS PubMed.
- C. Hu, D. Liu, Y. Xiao and L. Dai, Prog. Nat. Sci., 2018, 28, 121–132 CrossRef CAS.
- H. B. Wu and X. W. D. Lou, Sci. Adv., 2017, 3, eaap9252 CrossRef PubMed.
- J. Chen, Y. Zhu and S. Kaskel, Angew. Chem., Int. Ed. Engl., 2021, 60, 5010–5035 CrossRef CAS PubMed.
- S. De, T. Devic and A. Fateeva, Dalton Trans., 2021, 50, 1166–1188 RSC.
- F. Figueira and F. A. A. Paz, C, 2021, 7, 47 CAS.
- S. S. Rajasree, X. Li and P. Deria, Commun. Chem., 2021, 4, 47 CrossRef CAS.
- X. Zhao, P. Pachfule and A. Thomas, Chem. Soc. Rev., 2021, 50, 6871–6913 RSC.
- M. Lu, M. Zhang, J. Liu, Y. Chen, J. P. Liao, M. Y. Yang, Y. P. Cai, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed. Engl., 2022, 61, e202200003 CAS.
- Z. Liang, H. Y. Wang, H. Zheng, W. Zhang and R. Cao, Chem. Soc. Rev., 2021, 50, 2540–2581 RSC.
- S. Huang, K. Chen and T.-T. Li, Coord. Chem. Rev., 2022, 464, 214563 CrossRef CAS.
- N. Kobayashi, Chem. Phys. Lett., 1993, 205, 51–54 CrossRef CAS.
- T. D. Lash, J. Porphyrins Phthalocyanines, 2001, 5, 267–288 CrossRef CAS.
- S. Hayashi, M. Tanaka, H. Hayashi, S. Eu, T. Umeyama, Y. Matano, Y. Araki and H. Imahori, J. Phys. Chem. C, 2008, 112, 15576–15585 CrossRef.
- A. V. Cheprakov and M. A. Filatov, J. Porphyrins Phthalocyanines, 2009, 13, 291–303 CrossRef CAS.
- N. Ono, H. Yamada and T. Okujima, Synthesis of Porphyrins Fused with Aromatic Rings, World Scientific, Singapore, 2010 Search PubMed.
- L. J. K. Boerner, S. Mazumder, M. Pink, J. M. Zaleski and M.-H. Baik, Chem.–Eur. J., 2011, 17, 14539–14551 CrossRef CAS PubMed.
- A. V. Cheprakov, The Synthesis of pi-Extended Porphyrins, World Scientific Publishing, Singapore, 2011 Search PubMed.
- C.-L. Wang, Y.-C. Chang, C.-M. Lan, C.-F. Lo, E. Wei-Guang Diau and C.-Y. Lin, Energy Environ. Sci., 2011, 4, 1788–1795 RSC.
- J. M. Ball, N. K. S. Davis, J. D. Wilkinson, J. Kirkpatrick, J. Teuscher, R. Gunning, H. L. Anderson and H. J. Snaith, RSC Adv., 2012, 2, 6846–6853 RSC.
- C. M. Carvalho, T. J. Brocksom and K. T. de Oliveira, Chem. Soc. Rev., 2013, 42, 3302–3317 RSC.
- V. V. Roznyatovskiy, C. H. Lee and J. L. Sessler, Chem. Soc. Rev., 2013, 42, 1921–1933 RSC.
- A. Chaudhary, A. Srinivasan and T. K. Chandrashekar, Handbook of Porphyrin Science, 2014 Search PubMed.
- H. Yamada, D. Kuzuhara, T. Takahashi, Y. Shimizu, K. Uota, T. Okujima, H. Uno and N. Ono, Org. Lett., 2008, 10, 2947–2950 CrossRef CAS PubMed.
- N. K. S. Davis, A. L. Thompson and H. L. Anderson, J. Am. Chem. Soc., 2011, 133, 30–31 CrossRef CAS PubMed.
- J. P. Lewtak and D. T. Gryko, Chem. Commun., 2012, 48, 10069–10086 RSC.
- D. Mysliwiec, B. Donnio, P. J. Chmielewski, B. Heinrich and M. Stepien, J. Am. Chem. Soc., 2012, 134, 4822–4833 CrossRef CAS PubMed.
- P. Chen, Y. Fang, K. M. Kadish, J. P. Lewtak, D. Koszelewski, A. Janiga and D. T. Gryko, Inorg. Chem., 2013, 52, 9532–9538 CrossRef CAS PubMed.
- D. Koszelewski, A. Nowak-Król, M. Drobizhev, C. J. Wilson, J. E. Haley, T. M. Cooper, J. Romiszewski, E. Górecka, H. L. Anderson, A. Rebane and D. T. Gryko, J. Mater. Chem. C, 2013, 1, 2044–2053 RSC.
- Y. Fang, D. Koszelewski, K. M. Kadish and D. T. Gryko, Chem. Commun., 2014, 50, 8864–8867 RSC.
- N. Fukui, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed. Engl., 2015, 54, 6311–6314 CrossRef CAS PubMed.
- N. Fukui, S. K. Lee, K. Kato, D. Shimizu, T. Tanaka, S. Lee, H. Yorimitsu, D. Kim and A. Osuka, Chem. Sci., 2016, 7, 4059–4066 RSC.
- N. Fukui and A. Osuka, Angew. Chem., Int. Ed. Engl., 2018, 57, 6304–6308 CrossRef CAS PubMed.
- R. W. Hooper, A. Zhang, D. Koszelewski, J. P. Lewtak, B. Koszarna, C. J. Levy, D. T. Gryko and M. J. Stillman, J. Porphyrins Phthalocyanines, 2018, 22, 1111–1128 CrossRef CAS.
- K. Kato, K. Furukawa, T. Mori and A. Osuka, Chem.–Eur. J., 2018, 24, 572–575 CrossRef CAS PubMed.
- J. R. Reimers, L. E. Hall, M. J. Crossley and N. S. Hush, J. Phys. Chem. A, 1999, 103, 4385–4397 CrossRef CAS.
- J. N. H. Reek, A. E. Rowan, R. d. Gelder, P. T. Beurskens, M. J. Crossley, S. D. Feyter, F. d. Schryver and R. J. M. Nolte, Angew. Chem., Int. Ed. Engl., 1997, 36, 361–363 CrossRef CAS.
- Z. Ou, T. Khoury, Y. Fang, W. Zhu, P. J. Sintic, M. J. Crossley and K. M. Kadish, Inorg. Chem., 2013, 52, 2474–2483 CrossRef CAS PubMed.
- C. B. Dover, J. K. Gallaher, L. Frazer, P. C. Tapping, A. J. Petty II, M. J. Crossley, J. E. Anthony, T. W. Kee and T. W. Schmidt, Nat. Chem., 2018, 10, 305–310 CrossRef CAS PubMed.
- M. J. Crossley and L. A. Johnston, Chem. Commun., 2002, 1122–1123 RSC.
- M. J. Crossley and P. L. Burn, J. Chem. Soc., Chem. Commun., 1991, 1569–1571 RSC.
- P. J. Canfield, I. M. Blake, Z. L. Cai, I. J. Luck, E. Krausz, R. Kobayashi, J. R. Reimers and M. J. Crossley, Nat. Chem., 2018, 10, 615–624 CrossRef CAS PubMed.
- K. P. Birin, A. I. Poddubnaya, I. A. Abdulaeva, Y. G. Gorbunova and A. Y. Tsivadze, Dyes Pigm., 2018, 156, 243–249 CrossRef CAS.
- Y. Kashiwagi, K. Ohkubo, J. A. McDonald, I. M. Blake, M. J. Crossley, Y. Araki, O. Ito, H. Imahori and S. Fukuzumi, Org. Lett., 2003, 5, 2719–2721 CrossRef CAS PubMed.
- H. Hayashi, A. S. Touchy, Y. Kinjo, K. Kurotobi, Y. Toude, S. Ito, H. Saarenpaa, N. V. Tkachenko, H. Lemmetyinen and H. Imahori, ChemSusChem, 2013, 6, 508–517 CrossRef CAS PubMed.
- J. Zhao, J. I. Wong, J. Gao, G. Li, G. Xing, H. Zhang, T. C. Sum, H. Y. Yang, Y. Zhao, S. L. Ake Kjelleberg, W. Huang, S. C. Joachim Loo and Q. Zhang, RSC Adv., 2014, 4, 17822–17831 RSC.
- J. Guo, Y. Xu, S. Jin, L. Chen, T. Kaji, Y. Honsho, M. A. Addicoat, J. Kim, A. Saeki, H. Ihee, S. Seki, S. Irle, M. Hiramoto, J. Gao and D. Jiang, Nat. Commun., 2013, 4, 2736 CrossRef PubMed.
- J. Chen, J. Qu, Y. Zhang, Y. Chen, N. Liu and B. Chen, Tetrahedron, 2013, 69, 316–319 CrossRef CAS.
- J. Velik, V. Baliharova, J. Fink-Gremmels, S. Bull, J. Lamka and L. Skalova, Res. Vet. Sci., 2004, 76, 95–108 CrossRef CAS PubMed.
- S. R. Brishty, M. J. Hossain, M. U. Khandaker, M. R. I. Faruque, H. Osman and S. M. A. Rahman, Front. Pharmacol., 2021, 12, 762807 CrossRef CAS PubMed.
- E. Mishra, J. L. Worlinsky, T. M. Gilbert, C. Bruckner and V. Ryzhov, J. Am. Soc. Mass Spectrom., 2012, 23, 1135–1146 CrossRef CAS PubMed.
- M.-S. Liao, M.-J. Huang and J. D. Watts, J. Phys. Chem. A, 2010, 114, 9554–9569 CrossRef CAS PubMed.
- E. Y. Kaigorodova, G. M. Mamardashvili, O. R. Simonova, N. V. Chizhova and N. Z. Mamardashvili, J. Coord. Chem., 2021, 74, 2443–2462 CrossRef CAS.
- R. Deshpande, L. Jiang, G. Schmidt, J. Rakovan, X. Wang, K. Wheeler and H. Wang, Org. Lett., 2009, 11, 4251–4253 CrossRef CAS PubMed.
- L. Jiang, J. T. Engle, L. Sirk, C. S. Hartley, C. J. Ziegler and H. Wang, Org. Lett., 2011, 13, 3020–3023 CrossRef CAS PubMed.
- L. Jiang, R. A. Zaenglein, J. T. Engle, C. Mittal, C. S. Hartley, C. J. Ziegler and H. Wang, Chem. Commun., 2012, 48, 6927–6929 RSC.
- L. Jiang, J. T. Engle, R. A. Zaenglein, A. Matus, C. J. Ziegler, H. Wang and M. J. Stillman, Chem.–Eur. J., 2014, 20, 13865–13870 CrossRef CAS PubMed.
- Y. Hu, M. B. Thomas, W. A. Webre, A. Moss, R. G. W. Jinadasa, V. N. Nesterov, F. D'Souza and H. Wang, Angew. Chem., Int. Ed., 2020, 132, 20250–20257 CrossRef.
- S. A. Vinogradov and D. F. Wilson, In Designing Dendrimers, Wiley, New York, 2012 Search PubMed.
- M. Quaranta, S. M. Borisov and I. Klimant, Bioanalytical Reviews, 2012, 4, 115–157 CrossRef PubMed.
- D. B. Papkovsky and R. I. Dmitriev, Chem. Soc. Rev., 2013, 42, 8700–8732 RSC.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS PubMed.
- K. S. Kim, J. M. Lim, A. Osuka and D. Kim, J. Photochem. Photobiol., C, 2008, 9, 13–28 CrossRef CAS.
- C. Y. Liu, M. Deb, A. S. Sadhu, R. Karmakar, P. T. Huang, Y. N. Lin, C. S. Chu, B. N. Pal, S. H. Chang and S. Biring, Sensors, 2021, 21, 6940 CrossRef CAS PubMed.
- Y. Zhao, X. Geng, X. Shi, Y. Guo, Y. Sun, L. Qu and Z. Li, J. Mater. Chem. C, 2021, 9, 4300–4306 RSC.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- W. R. Wadt, P. J. Hay and J. Chem, Phys, 1985, 82, 284–298 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- C. Latouche, D. Skouteris, F. Palazzetti and J. Barone, J. Chem. Theory Comput., 2015, 11, 3281–3289 CrossRef CAS PubMed.
- A. Jana, L. McKenzie, A. B. Wragg, M. Ishida, J. P. Hill, J. A. Weinstein, E. Baggaley and M. D. Ward, Chem.–Eur. J., 2016, 22, 4164 CrossRef CAS PubMed.
- M. J. Gouterman, The Porphyrins, Academic Press, New York, 1978 Search PubMed.
- M. J. Gouterman, Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- S. Montanaro, D. G. Congrave, M. K. Etherington and I. A. Wright, J. Mater. Chem. C, 2019, 7, 12886–12890 RSC.
- K. Jana and J. N. Moorthy, New J. Chem., 2022, 46, 1416–1422 RSC.
- K. Baumgartner, M. Hoffmann, F. Rominger, S. M. Elbert, A. Dreuw and M. Mastalerz, J. Org. Chem., 2020, 85, 15256–15272 CrossRef PubMed.
- K. M. Kadish, Prog. Inorg. Chem., 1986, 34, 435–605 CrossRef CAS.
- D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22–36 RSC.
- C. B. KC and F. D'Souza, Coord. Chem. Rev., 2016, 322, 104–141 CrossRef CAS.
- F. D'Souza, O. Ito and O. Coord, Chem. Rev., 2005, 249, 1410–1422 Search PubMed.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 10, 259–271 CrossRef.
- R. A. Marcus, Angew. Chem., Int. Ed. Engl., 1993, 32, 1111–1121 CrossRef.
- C. O. Obondi, G. N. Lim, B. Churchill, P. K. Poddutoori, A. vaa der Est and F. D'Souza, Nanoscale, 2016, 8, 8333–8345 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2172827–2172829. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03238d |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |