
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA switchable redox annulation of 2-nitroarylethanols affording N-heterocycles: photoexcited nitro as a multifunctional handle†
Bin
Wang‡
a,
Hongyuan
Ren‡
 a,
Hou-Ji
Cao
*b,
Changsheng
Lu
*a and
Hong
Yan
*a
a,
Hou-Ji
Cao
*b,
Changsheng
Lu
*a and
Hong
Yan
*a
aState Key Laboratory of Coordination Chemistry, Jiangsu Key Laboratory of Advanced Organic Materials, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: hyan1965@nju.edu.cn
bSchool of Chemistry and Chemical Engineering, Henan Normal University, XinXiang, Henan 453007, China. E-mail: houjicao@sina.cn
First published on 31st August 2022
Abstract
The efficient transformation of nitroaromatics to functional molecules such as N-heterocycles has been an attractive and significant topic in synthesis chemistry. Herein, a photoexcited nitro-induced strategy for switchable annulations of 2-nitroarylethanols was developed to construct N-heterocycles including indoles, N-hydroxyl oxindoles and N–H oxindoles. The metal- and photocatalyst-free reaction proceeds through intramolecular redox C–N coupling of branched hydroxyalkyl and nitro units, which is initiated by a double hydrogen atom abstraction (d-HAA) process. The key to the switchable reaction outcomes is the mediation of a diboron reagent by its favorable oxy-transfer reactivity to in situ generated nitroso species. The utility of this protocol was well demonstrated by broad substrate scope, excellent yields, functional group tolerance and wide applications. Finally, detailed mechanistic studies were performed, and kinetic isotope effect (KIE) experiments indicate that the homolysis of the C–H bond is involved in the rate-determining step.
Introduction
N-Heterocycles are ubiquitous skeletons of various biologically active and pharmaceutical substances.1 Among them, indole2 and oxindole2a,3 are privileged skeletons, which have attracted chemists for over a century and remained sustained topics in synthetic chemistry with high value-added features. Although a considerable collection of synthetic methods for these skeleton molecules has been developed, there is still a continuous thirst for highly efficient strategies to create these functional substructures while maximizing atom economy.4 Besides, new application scenarios also issue new challenges to the development of these methodologies, such as the modularity for pharmaceutical synthesis5 and facile conditions for biocompatible synthesis.6 Given the widespread availability and easy preparation of nitroarenes, the most straightforward protocol to construct N-heterocycles is using nitro as the nitrogen source.4c,7 A few renowned reactions8 for the synthesis of indoles have witnessed the potential of nitroarenes, especially for ortho-branched ones which possess robust features for reductive cyclization. More recently, switchable transformations of ortho-branched nitroarenes to indoles or oxindoles by metal-free methods were reported by Radosevich’s group9 and Driver’s group,10 which provided new perspectives for the construction of these N-heterocycles (Scheme 1A). Compared to these strategies starting from ortho-vinyl or -carbonyl nitroarenes, the redox cyclization of ortho-hydroxyalkyl nitroarenes could offer a more facile and practical choice for the synthesis of the corresponding N-heterocycles.11 However, the tactic using an ortho-branched hydroxyalkyl moiety as both a C-precursor and a hydrogen donor under metal-free conditions is still undeveloped. This tactic could circumvent the use of problematic reductants such as sensitive Grignard reagents,8c,12 pressured CO gas,13 hazardous organophosphine reagents7c,9,14 and stoichiometric metal reductants.7a,b,15 Moreover, exogenetic hydrogen donors for reductive cyclization of nitroarenes are no longer needed.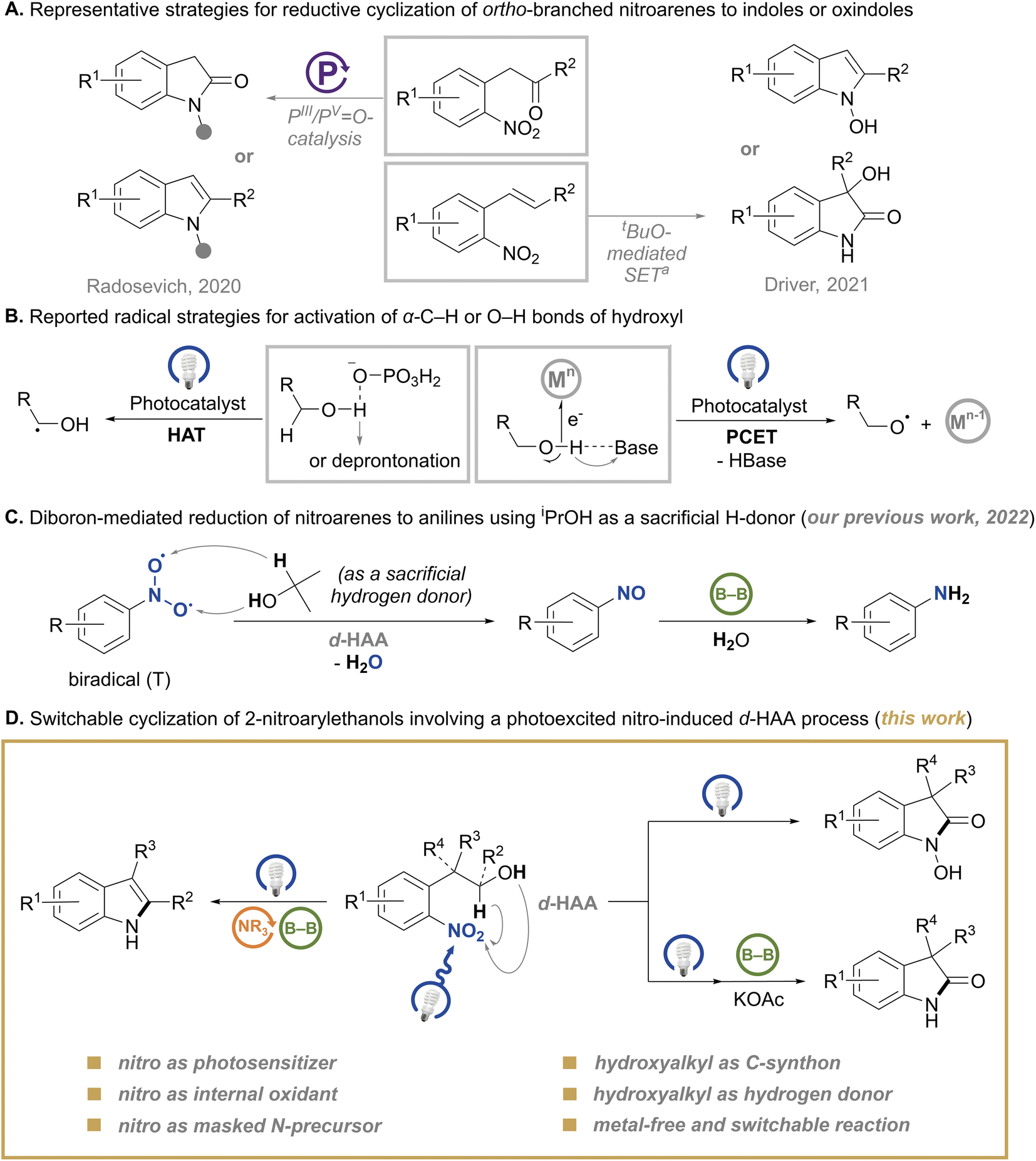 | ||
| Scheme 1 Research backgrounds and this work. a SET: single electron transfer. | ||
The recent development of the activation of alcohols into α-carbon-centered or oxygen-centered radical fragments by photoredox catalysis contributed to valuable transformations of alcohols,16 which were otherwise unattainable. The photocatalytic hydrogen atom transfer (HAT) strategy for activating an electron-rich α-C–H bond of alcohols is commonly facilitated via hydrogen-bond interaction or deprotonation of the hydroxyl group17 (Scheme 1B). Besides, the proton-coupled electron transfer (PCET) strategy for activating the O–H bond of alcohols is heavily dependent on the combination of a Brønsted base with an oxidant to remove a proton and an electron from the O–H bond, respectively18 (Scheme 1B). Inspired by these profound strategies for radical generation, we envisaged that the photoexcited nitro might act as a potential dual HAT-functionality for the activation of both α-C–H and O–H bonds of hydroxyl owing to its nature as a biradical in the triplet state,19 and we describe this process as double hydrogen atom abstraction (d-HAA). As a preliminary attempt for this hypothesis, our previous work achieved a tandem reduction of nitroarenes to anilines triggered by a d-HAA process with isopropanol20 (Scheme 1C). However, in the reaction, alcohol serves a sacrificial and disposable reductant, which limits further evolution of the nitro-triggered radical process. In order to provide more reaction possibilities for the d-HAA process and construct a diversity of functionalized molecules by using this reaction platform, we tried new designs in an intramolecular setting. Herein, we disclose a redox annulation of 2-nitroarylethanols via a photoexcited nitro-triggered d-HAA process under blue-light irradiation, which delivers diboron-switched indoles, N-hydroxyl oxindoles or N–H oxindoles (Scheme 1D) in good to excellent yields. This reaction does not need any metal catalyst or photocatalyst, and three characteristics stand out as given below: (1) nitro serves as a masked nitrogen precursor, as well as an internal photosensitive and oxidative functionality; (2) the branched hydroxyalkyl serves as an internal carbon precursor and a double hydrogen donor (α-C–H and O–H); (3) the in situ generated nitrosoarene and N-hydroxylaniline intermediates may undergo diboron-based deoxygenation,20,21 thus avoiding the addition of extra activators.22 The different usages of a diboron reagent could lead to different products (Scheme 1D).
Results and discussion
We started our study with the reaction of 2-(2-nitrophenyl)ethan-1-ol (1a) in the presence of a base catalyst and a diboron reagent under the irradiation of 400 nm blue LEDs. A broad screening of conditions gave the optimal conditions containing N,N-diisopropylethylamine (DIPEA, 20 mol%) as the catalyst, bis(neopentyl glycolato)diboron (B2nep2, 2.2 equiv.) as the reductant, THF/MeOH with an 8/1 ratio as co-solvents, under a nitrogen atmosphere and at room temperature for 12 h, delivering the product 2a in 82% isolated yield (Table 1, entry 1). Deviations from the standard conditions were manipulated by varying the reaction parameters to validate the superiority of the optimal conditions. In this regard, other solvents have been screened including oxygenated and chlorinated hydrocarbons, such as 1,4-dioxane, toluene, dichloroethane, and acetonitrile instead of THF/MeOH, but led to lower yields (Table 1, entry 2 and Table S1†). Altering the ratio of mixed THF/MeOH or using THF as the solvent diminished the yield of 2a (Table S1†). A decreased yield of 2a was observed if B2nep2 was replaced by B2(OH)4 due to the competitive formation of N-hydroxyoxindole (Table 1, entry 3 and Table S1†). A detrimental effect on the yield of 2a was also observed while other diboron reagents such as bis(pinacolato)diboron (B2pin2) and bis(catecholato)diboron (B2cat2) were examined (Table 1, entry 4 and Table S1†). When the amount of B2nep2 was reduced instead of 2.2 equiv., a diminished yield of 2a was observed (Table 1, entries 5 and 6 and Table S1†). To check the effect of other catalytic bases, Et3N, K2CO3, and KOtBu were examined, which led to lower yields (Table 1, entry 7 and Table S1†). Notably, the yield of 2a dropped without the addition of DIPEA (Table 1, entry 8). The control experiments further demonstrated that the diboron reagent and light are indispensable for the redox cyclization process (Table 1, entries 9 and 10), and air is a detrimental factor (Table 1, entry 11).|
|
||
|---|---|---|
| Entry | Deviation from the standard conditions | Yieldb/% |
| a Standard reaction conditions: 1a (0.2 mmol), DIPEA (0.04 mmol), B2nep2 (0.44 mol), THF/MeOH (8/1, 0.45 mL), N2, rt, under the irradiation of blue LEDs (400 nm), 12 h. b Isolated yields of 2a. c B2(OH)4: tetrahydroxydiboron; B2nep2: bis(neopentylglycolato)diboron. | ||
| 1 | None | 82 |
| 2 | Other solvents instead of the mixed solvents | <70 |
| 3c | B2(OH)4 instead of B2nep2 | 62 |
| 4 | Other diboron reagents instead of B2(OH)4 or B2nep2 | <30 |
| 5 | 0.5 equiv. instead of 2.2 equiv. B2nep2 | 35 |
| 6 | 1.0 equiv. instead of 2.2 equiv. B2nep2 | 52 |
| 7 | Other catalytic bases instead of DIPEA | <60 |
| 8 | Without DIPEA | 40 |
| 9 | Without B2nep2 | Trace |
| 10 | In the dark | 0 |
| 11 | In air | 48 |
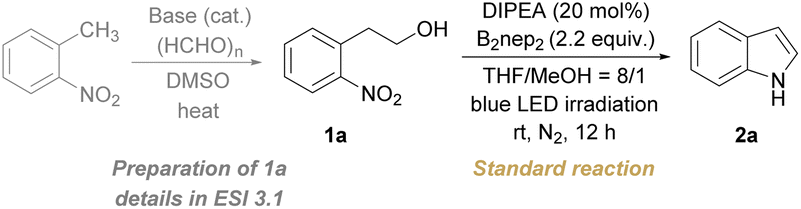
The substrate scope of the metal-free and photoinduced cyclization of 2-nitroarylethanols for the preparation of indoles was explored under the above optimal reaction conditions (Scheme 2). Most of our substrates of 2-nitroarylethanols were either commercially accessible or easily prepared in one step (ESI 3.1†), which differentiate from those substrates used in some reported methods such as o-nitrostyrenes7c,10,23 and o-nitrobenzyl ketone analogues9,24 that were prepared by noble metal-catalysis. A broad range of substrates bearing electron-donating and electron-withdrawing substituents installed at the phenyl group were evaluated. These substrates exhibited good to excellent reactivity, affording functionalized indoles 2a–2k in 62–92% isolated yields. It is noteworthy that halogen substituents, especially for iodo, which could be utilized as coupling handles for late-stage modification, were intact (2b, 2d, 2e, 2h, and 2m), in contrast to the reported dehalogenation.25 This can presumably be attributed to the nitro-triggered direct intramolecular 1,2-double-hydrogen atom abstraction (1,2-d-HAA) of the hydroxyalkyl moiety, which could avoid reactive hydride dehalogenation. In addition, the alkynyl-substituted substrate was also converted to the corresponding C4-substituted indole in 78% yield (2i), which is difficult to directly prepare by traditional methods.26 C7-functionalized indoles were also produced in satisfactory yields (2j and 2aa in Scheme 4), whereas scarcely reported synthetic methods were qualified for this target.8c,12 Fortunately, this annulative method was applicable to the challenging 6-azaindoles (2l and 2m), notwithstanding moderate yields were obtained due to the low reactivity of these electron-deficient pyridines.27 To assess the generality toward C2-functionalized indoles, panels of substituents at the α-position of hydroxyl were inspected under slightly modified conditions. It turned out that C2-alkyl or aryl functionalized indoles (2n–2w) were obtained in quantitative yields, exhibiting insensitivity toward sterically hindered tertiary alkyl (2n), cyclopropyl (2o) and polycyclic (2q) substituents. This system also demonstrates remarkable compatibility with sensitive groups including aldehyde (2t), cyano (2u), alkene (2p, 2q), and ether (2r). To further validate the practicality of this method, we extrapolated this strategy to the synthesis of the aggregation-induced emission (AIE)28 molecule (2y) and medicinal skeletons (2x, 2z and 2aa in Scheme 4) in moderate to excellent yields. In particular, the AIE molecule 2y can be employed as a specific lipid droplet (LD) probe as it could colocalize perfectly with the commercial LD dye of Nile Red and Pearson’s correlation coefficient can reach 90.49% (Fig. S10†). Therefore, various indole derivatives including interesting functionalities can be facilely synthesized by this methodology, demonstrating its practicability. To our surprise, the β-aryl-substituted substrates release the reactivity of the β-C–H bond to form a benzylic radical, which delivers α,β-dihydroxyl-substituted amines in good yields (2ab–2ae in Scheme 2).
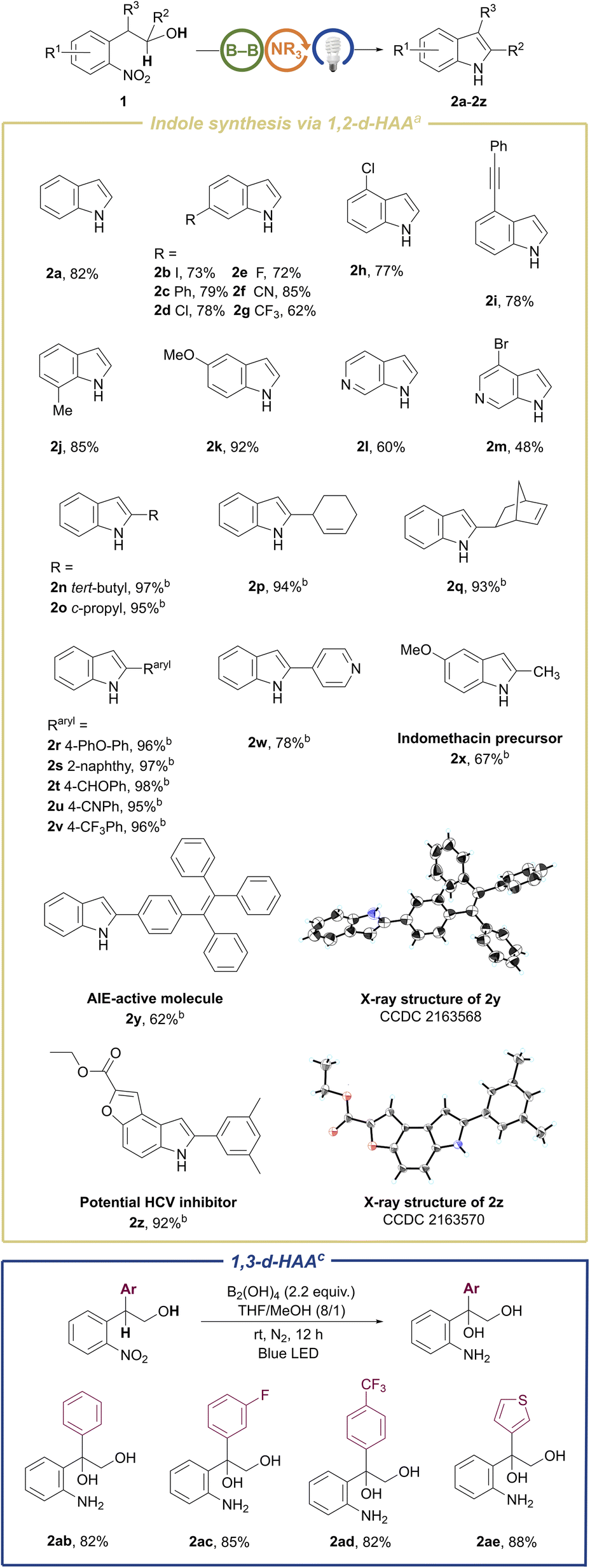 | ||
| Scheme 2 Substrate scope for the synthesis of indoles and other products. aStandard conditions: 0.2 mmol scale reaction in THF/MeOH (8/1, 0.45 mL), DIPEA (0.04 mmol, 20 mol%), B2nep2 (0.44 mmol, 2.2 equiv.), blue LED, N2, rt, 12 h; isolated yields. bB2(OH)4 (0.44 mmol) instead of B2nep2. c Without the addition of DIPEA. | ||
To our delight, the aforementioned competitive N-hydroxyl oxindoles could become the sole products just by removal of the diboron reagent from the standard conditions (Table S2† and Scheme 3, left). This reaction system does not need any additive and exhibits admirable functional group compatibility toward group-abundant N-hydroxyl oxindole molecules, including halogen-(3b, 3d–3f, 3m, and 3o), cyano-(3g), thienyl-(3i), alkynyl-(3l) and ester-(3r) bearing products. Spiro-N-hydroxyoxindoles with a five-membered ring (3p) and even a strained four-membered ring (3q) were readily afforded in 85% and 82% isolated yields, respectively. Substrates with either electron-withdrawing or electron-donating groups performed well regardless of substituents embedded either on the phenyl ring or the benzylic site. Potentially bio-active hetero-tricyclic molecule 3r was isolated in 90% yield, which shows the practicality of this method. Unexpectedly, the substrate with a cyclohexyl substituent in the benzylic position underwent incredible oxy-transfer and intramolecular rearrangement, finally leading to spiro-[cyclohexane-1, 2′-indolin]-3′-one (3s) in an isolated yield of 92%. To test the scalability of our N-hydroxyl oxindole synthesis, we conducted the preparation of 3a, 3b, and 3f on the gram-scale and obtained 70–85% isolated yields. In our system, with exclusive conversion of the substrates, the products could be easily isolated and purified by using a pad of silica gel or by quick recrystallization in chloroform. Although this class of skeletons is widely present in natural products and pharmaceuticals, for example, as calcium channel blockers or agents possessing anti-angiogenic and analgesic effects, only few synthetic methods of N-hydroxyl oxindoles have been described.29 Thus, these results imply the utility of the protocol developed here. Moreover, hydroxyl attached at the nitrogen site of N–OH oxindoles can obviously serve as a diversified handle for post-functionalization of O–H or N–O bonds. However, it has not yet been reported as a leaving group to access N–H oxindoles.
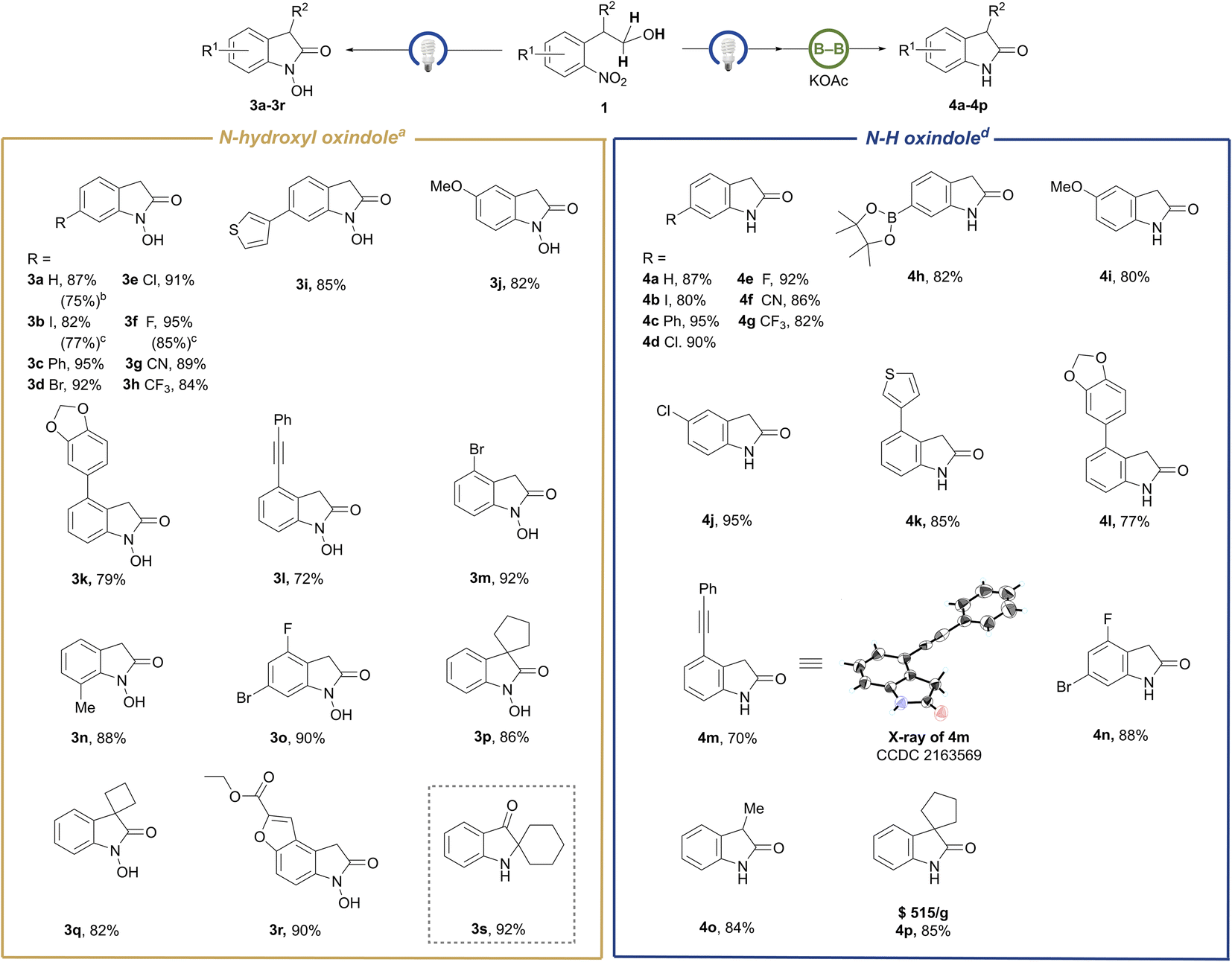 | ||
| Scheme 3 Substrate scope for the synthesis of N-hydroxyl oxindoles and N-free oxindoles. a0.2 mmol scale reaction in THF/MeOH (8/1, 0.45 mL), a blue LED, N2, rt, 6 h; isolated yields. b10 mmol scale. c5 mmol scale. dThe reaction was performed via a one-pot, two-step procedure (ESI 4.1†): 0.2 mmol scale reaction in THF/MeOH (8/1, 0.45 mL), blue LED, N2, rt, 6 h; after 6 h, the solvent was evaporated and the subsequent reaction was performed in the same pot, B2(OH)4 (0.3 mmol, 1.5 equiv.), KOAc (0.4 mmol, 2.0 equiv.), MeOH (0.2 M), 50 °C, 2 h, isolated yields. | ||
Next, we demonstrated the switchability of our reaction with respect to the formation of N–H oxindoles via a one-pot two-step procedure (Table S3† and Scheme 3, right). A range of N–H oxindole products were achieved through a subsequent transborylative deoxygenation of in situ generated N–OH oxindoles under a weakly basic condition. Substrates containing sensitive groups, including iodo (4b), cyano (4f), oxaboron (4h), and alkynyl (4m) were accommodated perfectly, yielding N–H oxindoles exclusively with the reducible functionalities intact upon borylation. It is noteworthy that the method could provide value-added 3,3-spiro-oxindole (4p) at a lower cost compared to the precedent methods.1b
Then we provide representative synthetic applications to signify the practicality of our protocols. To show the unique features of the N–OH oxindoles, the post-functionalization of the O–H bond of compound 3f led to the N-esterification product 5a in 91% yield (Scheme 4A). Besides, starting from the drug Flutamide, our methodology can transform it into the indole product 2aa (Scheme 4B). This may provide a useful strategy to access potential lead compounds for medicinal chemistry and structure–activity relationship studies without the need of de novo synthesis.30 To further demonstrate the synthetic utility of our methods, we exampled the synthesis of Sunitnib, a multitargeted receptor of tyrosine kinase for the treatment of cancer.31 Using our protocol first to prepare 5-fluoroindolin-2-one (4q), and then one more step could lead to Sunitnib (Scheme 4C), in contrast to the 5 steps required for the synthesis of Sunitnib in the literature.31 Of note, our reaction could be scaled up to 2.3 mmol without loss of efficiency for 4q (78% yield).
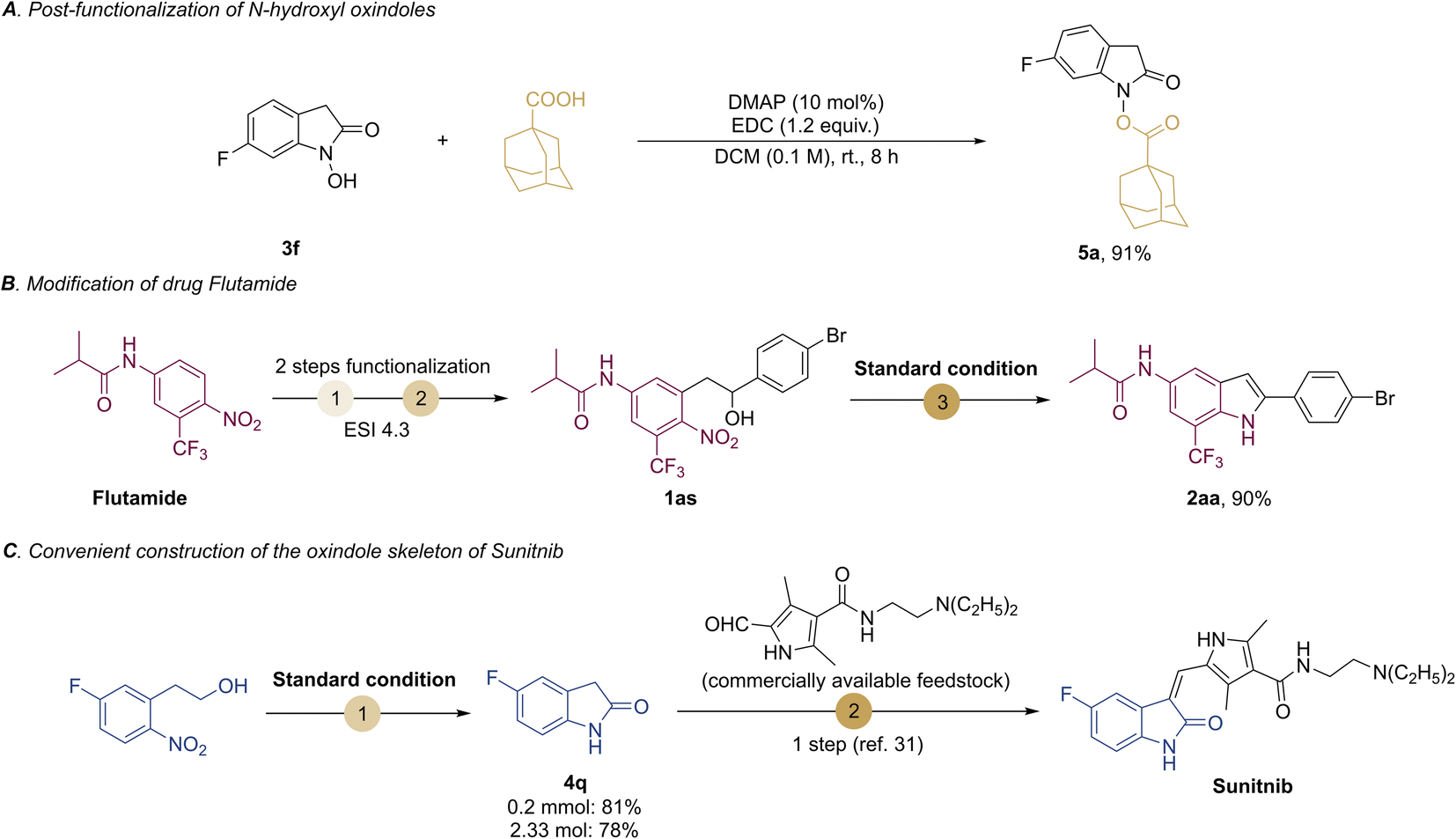 | ||
| Scheme 4 Synthetic applications. | ||
To shed insight into the mechanisms of the switchable annulation reactions of 2-nitroarylethanols, a series of mechanistic investigations were conducted (Scheme 5 and ESI†). First, the UV-Vis absorption spectra of 1a, B2nep2, and a mixture of 1a and B2nep2 (molar ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.2) were recorded (Fig. S12†). No spectral shift was observed for the mixture versus the individual species, which indicates that no electron-donor acceptor (EDA) was involved between 1a and B2nep2. Then we turned our attention to investigate the radical nature of the reactions. The addition of 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) as a radical scavenger under standard conditions resulted in no production of the target product 2a, but the formation of TEMPO-H, as detected by HRMS (Scheme 5A, ESI 5.1†). This indicates that the radical reaction is quenched. The same phenomenon was observed in the synthesis of N–OH oxindole, as only a trace amount of 3a was obtained if TEMPO was added (ESI 5.1†). Combined with the control experiment in the dark (Table 1, entry 10), we assume that the photoinduced hydrogen atom abstraction (HAA) process is involved in the annulation reaction. Moreover, the light on–off experiments showed that no transformation could occur in the dark and constant irradiation is required for completion of the reaction (Fig. 1 and ESI 5.3†).
:2.2) were recorded (Fig. S12†). No spectral shift was observed for the mixture versus the individual species, which indicates that no electron-donor acceptor (EDA) was involved between 1a and B2nep2. Then we turned our attention to investigate the radical nature of the reactions. The addition of 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) as a radical scavenger under standard conditions resulted in no production of the target product 2a, but the formation of TEMPO-H, as detected by HRMS (Scheme 5A, ESI 5.1†). This indicates that the radical reaction is quenched. The same phenomenon was observed in the synthesis of N–OH oxindole, as only a trace amount of 3a was obtained if TEMPO was added (ESI 5.1†). Combined with the control experiment in the dark (Table 1, entry 10), we assume that the photoinduced hydrogen atom abstraction (HAA) process is involved in the annulation reaction. Moreover, the light on–off experiments showed that no transformation could occur in the dark and constant irradiation is required for completion of the reaction (Fig. 1 and ESI 5.3†).
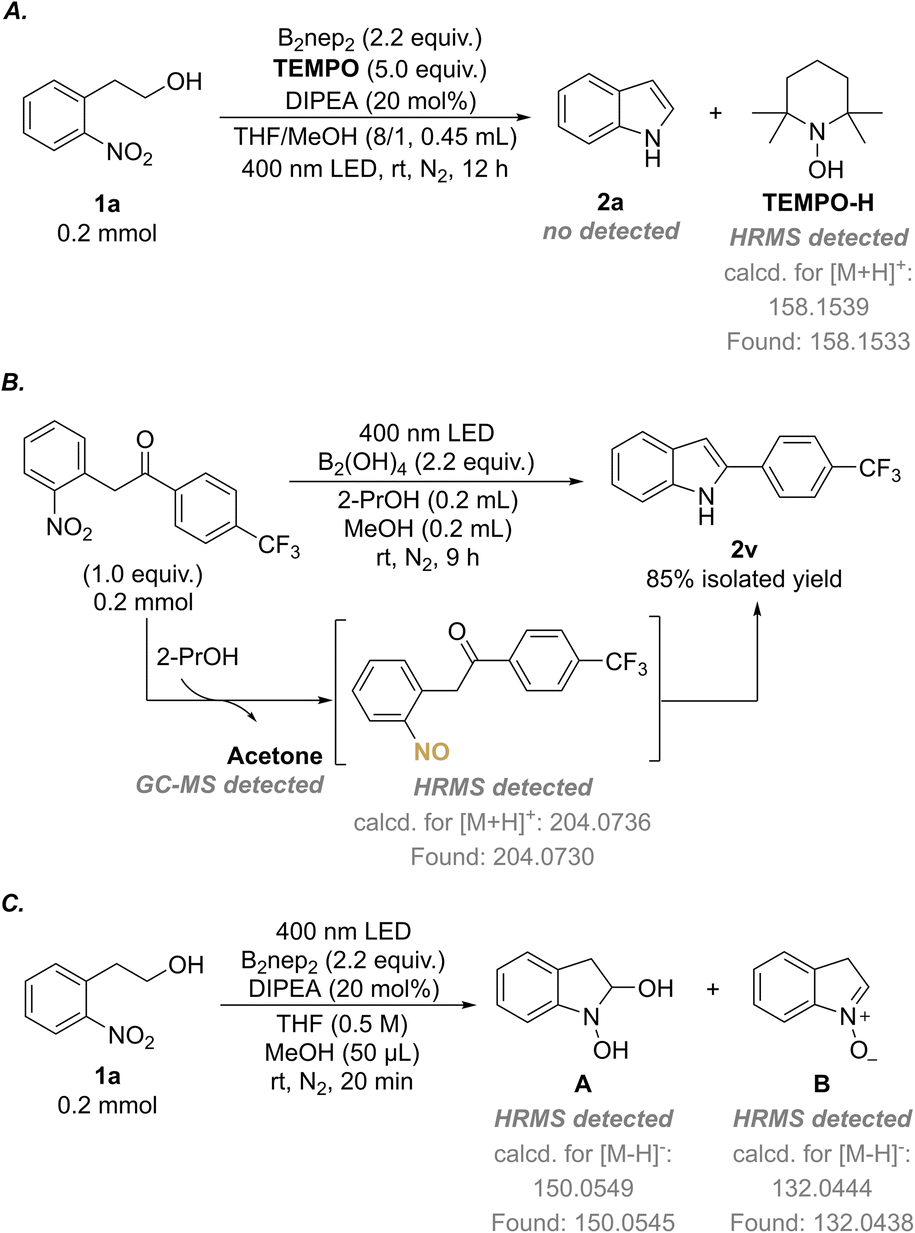 | ||
| Scheme 5 Mechanistic studies. (A) Radical-quenching experiment for the synthesis of indole. (B) Annulation of o-nitrobenzylic ketone with an exogenetic H-donor. (C) HRMS analysis of the proposed intermediates. | ||
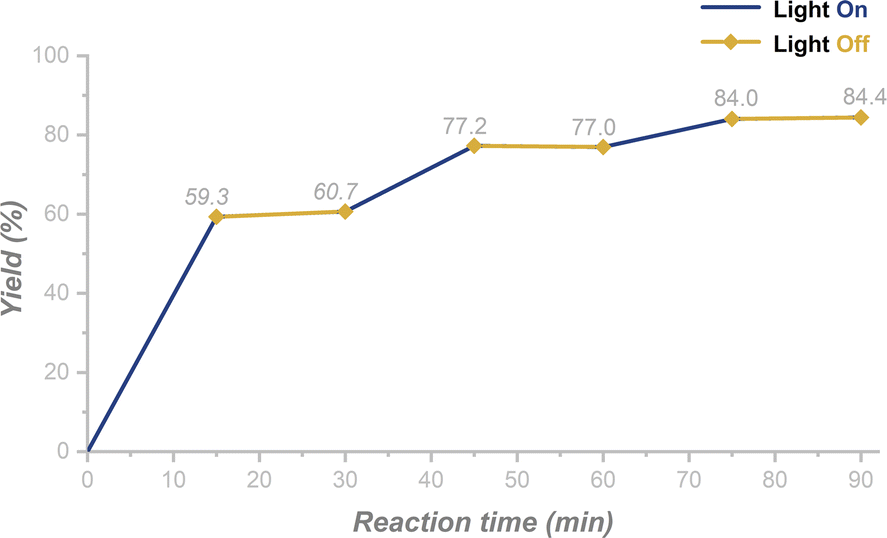 | ||
| Fig. 1 Light on–off experiments for the synthesis of N–OH oxindole (the reaction was performed in a sealed NMR tube and NMR yields were shown, ESI 5.3†). | ||
According to the previous reports concerning the reduction of nitroarenes to nitroso compounds,19,19a,20 we assume that nitroso and carbonyl might be the outcomes of the d-HAA process. Therefore, we designed the less fast intermolecular HAA reaction to verify this hypothesis and try to detect the active nitroso species. In doing so, the reductive cyclization of o-nitrobenzylic ketone using 2-PrOH as a hydrogen donor was examined. As a result, not only the indole product 2v was obtained in 85% isolated yield, accompanied by the generation of acetone as detected by GC-MS (Scheme 5B, Fig. S23†), but the nitroso intermediate was also detected in the reaction mixture by HRMS (Scheme 5B, Fig. S22†). Moreover, we also attempted to experimentally detect more possible intermediates involved in the redox–cyclization reaction and we were successful to catch the proposed intermediates A and B32 by HRMS analysis (Scheme 5C, Fig. S21†).
To gain more solid data concerning the reaction mechanism, the intermolecular kinetic isotope effect (KIE) experiments were performed by using the reactions of equivalent molar 1a and the deuterium-labeled substrate [D]-1a under the standard conditions (Scheme 6 and ESI 5.5†). Two kH/kD values were obtained as kH/kD = 5.0 for the N–OH oxindole formation, and kH/kD = 3.8 for the indole formation. The KIE studies suggest that the homolytic cleavage of the α-C–H bond is the rate-determining step for the formation of both N–OH oxindole and indole, which conforms to the primary kinetic isotope effect (PKIE). The difference of the two KIE values also suggest that the formation of N-hydroxyl oxindoles may be related to dual homolysis of α-C–H bonds.33
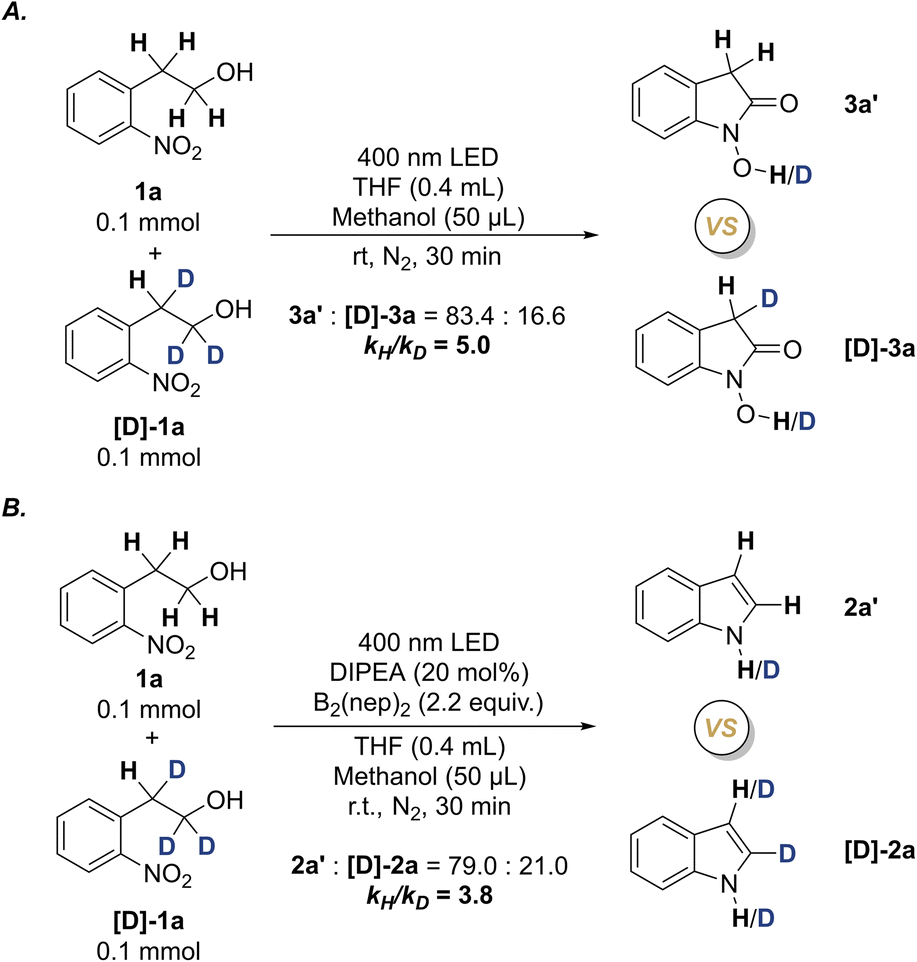 | ||
| Scheme 6 Kinetic isotope effect (KIE) experiments. | ||
Based on the above mechanistic experiments and previous reports,19,20,34 the plausible mechanisms were proposed (Scheme 7). If ground-state nitroarene (1a) is exposed to appropriate irradiation, it could be photoexcited to the long-lived triplet state (T), which has a biradical character.19 Then the active biradical participates in an intramolecular double hydrogen atom abstraction (d-HAA) from the adjacent hydroxyalkyl, affording the nitroso intermediate 6 (as illustrated in Scheme 7, top). Thereafter the key species 6 could switch two paths dependent on a diboron reagent. In Path I, the quick diborylation of the nitroso intermediate results in 7 which gives rise to 8 upon hydrolysis.21 Then intramolecular nucleophilic addition occurs to yield 9, whose subsequent proton transfer would produce species A (detected by HRMS, ESI 5.6†). A undergoes sequential deprotonation and dehydration under the catalysis of tertiary amine to deliver 10 and species B (detected by HRMS, ESI 5.6†).32 Next, the N-oxide B undergoes the oxygen-transfer process assisted by a diboron reagent to release 11 which undergoes a fast tautomerization to give the indole product 2a. In Path II, the nitroso species 6 could undergo a photoinduced intramolecular 1,6-HAT process to yield 12, which subsequently gives rise to the N–OH oxindole 3avia an intramolecular radical coupling. After sequential deprotonation, deoxygenation by a diboron reagent and protonation, 3a delivers the N–H oxindole 4a (Scheme 7). Additionally, the mechanisms for the rearrangement reactions of β-cyclohexyl and β-aryl-substituted substrates via 1,3-double hydrogen atom abstraction (1,3-d-HAA) were rationally explained which leads to 2ab–2ae and 3s (ESI 5.8, Fig. S24†).
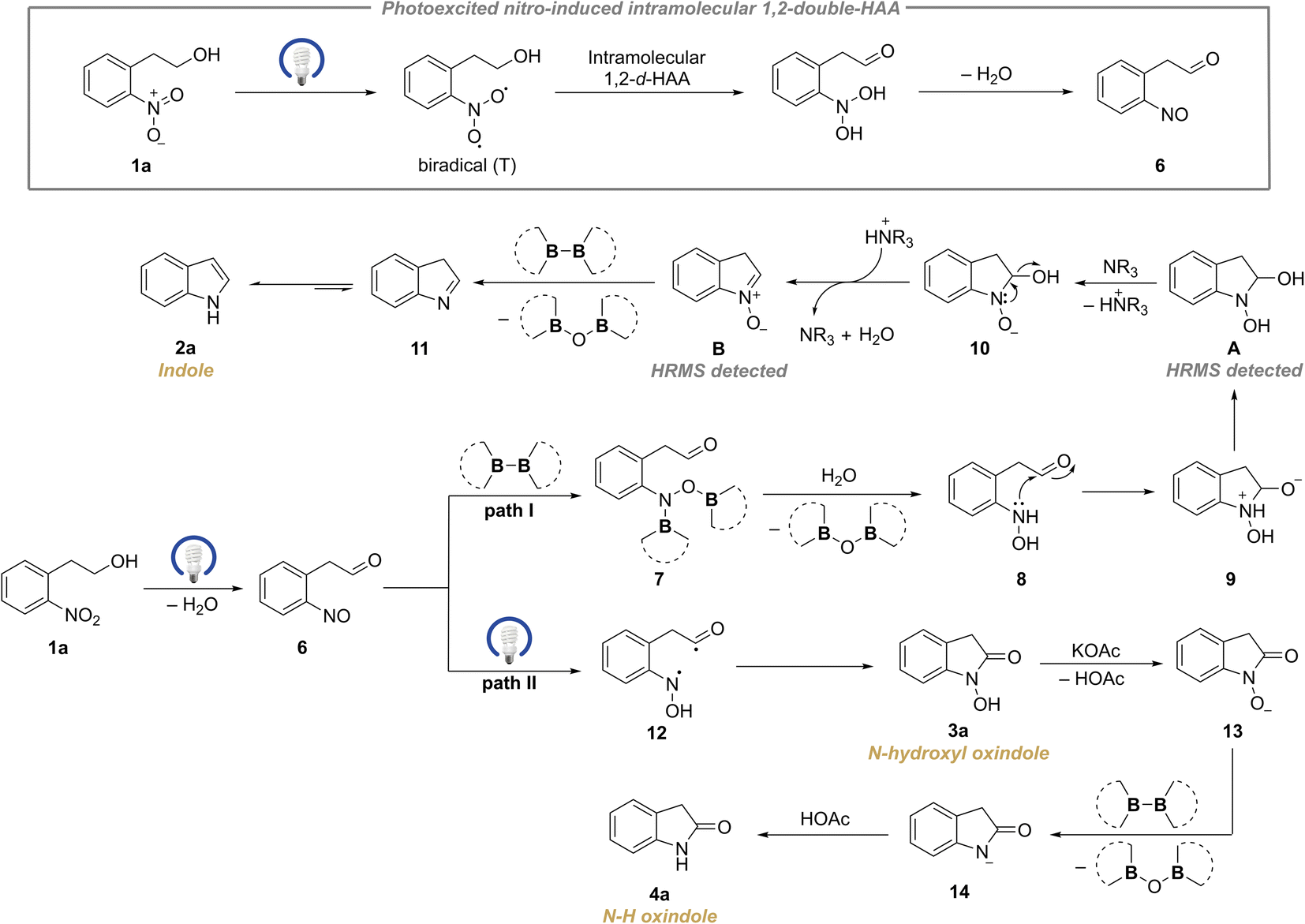 | ||
| Scheme 7 Proposed mechanisms for the generation of indoles, N–OH oxindoles and N–H oxindoles. | ||
Conclusions
In conclusion, we developed a metal- and photocatalyst-free toolbox for intramolecular annulation of 2-nitroarylethanols under blue-light irradiation, to deliver a diversity of indoles, N-hydroxyl oxindoles and N–H oxindoles in good to excellent yields. A notable feature of the strategy is a photoexcited nitro-triggered double hydrogen atom abstraction (d-HAA) process, which initiates the intramolecular redox C–N coupling of the nitro unit and the ortho-hydroxyalkyl moiety. Using the concept of nitro-triggered d-HAA for the synthesis of N-heterocycles is novel and unprecedented. This work also demonstrates the practical applications as an efficient methodology in the synthesis of important molecules of indoles and oxindoles. Moreover, abundant nitro-triggered radical chemistry is involved in this protocol. This prompts us to extend the concept to other robust synthetic studies.Data availability
The data that support the findings of this study are available in the ESI† or on request from the corresponding author.Author contributions
B. Wang discovered the reductive cyclization reaction of 1a and guided the synthesis. On this basis, B. Wang and H. Ren conducted the experiments. B. Wang wrote the draft. H. Ren analysed the experimental data and wrote the ESI.† H. Yan, C. Lu and H.-J. Cao designed this project, directed the study and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21820102004 and 91961104) and the Ministry of Science and Technology (2021YFE0114800). We are also grateful to Prof. Zhuangzhi Shi, Prof. Shaolin Zhu, Prof. Jin Xie and Prof. Shouyun Yu for their helpful suggestions.Notes and references
- (a) T. Janosik, A. Rannug, U. Rannug, N. Wahlström, J. Slätt and J. Bergman, Chem. Rev., 2018, 118, 9058–9128 CrossRef CAS PubMed; (b) G. S. Cockerill, R. M. Angell, A. Bedernjak, I. Chuckowree, I. Fraser, J. Gascon-Simorte, M. S. A. Gilman, J. A. D. Good, R. Harland, S. M. Johnson, J. H. Ludes-Meyers, E. Littler, J. Lumley, G. Lunn, N. Mathews, J. S. McLellan, M. Paradowski, M. E. Peeples, C. Scott, D. Tait, G. Taylor, M. Thom, E. Thomas, C. Villalonga Barber, S. E. Ward, D. Watterson, G. Williams, P. Young and K. Powell, J. Med. Chem., 2021, 64, 3658–3676 CrossRef CAS PubMed.
- (a) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed; (b) T. V. Sravanthi and S. L. Manju, Eur. J. Pharm. Sci., 2016, 91, 1–10 CrossRef CAS PubMed.
- (a) E. R. Wood, L. Kuyper, K. G. Petrov, R. N. Hunter, P. A. Harris and K. Lackey, Bioorg. Med. Chem. Lett., 2004, 14, 953–957 CrossRef CAS PubMed; (b) A. D. Jagtap, P.-T. Chang, J.-R. Liu, H.-C. Wang, N. B. Kondekar, L.-J. Shen, H.-W. Tseng, G. S. Chen and J.-W. Chern, Eur. J. Med. Chem., 2014, 85, 268–288 CrossRef CAS PubMed.
- (a) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875–2911 CrossRef CAS PubMed; (b) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873–2920 CrossRef CAS PubMed; (c) A. D. Marchese, E. M. Larin, B. Mirabi and M. Lautens, Acc. Chem. Res., 2020, 53, 1605–1619 CrossRef CAS PubMed.
- A. W. Sun, S. Lackner and B. M. Stoltz, Trends Chem., 2019, 1, 630–643 CrossRef CAS.
- (a) A. S. Mackay, R. J. Payne and L. R. Malins, J. Am. Chem. Soc., 2022, 144(1), 23–41 CrossRef CAS PubMed; (b) P. N. Daniels, H. Lee, R. A. Splain, C. P. Ting, L. Zhu, X. Zhao, B. S. Moore and W. A. van der Donk, Nat. Chem., 2022, 14, 71–77 CrossRef CAS PubMed.
- (a) S. Tong, Z. Xu, M. Mamboury, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2015, 54, 11809–11812 CrossRef CAS; (b) B. Özkaya, C. L. Bub and F. W. Patureau, Chem. Commun., 2020, 56, 13185–13188 RSC; (c) T. V. Nykaza, A. Ramirez, T. S. Harrison, M. R. Luzung and A. T. Radosevich, J. Am. Chem. Soc., 2018, 140, 3103–3113 CrossRef CAS PubMed.
- (a) A. Reissert, Ber. Dtsch. Chem. Ges., 1897, 30, 1030–1053 CrossRef CAS; (b) A. D. Batcho and W. Leimgruber, Org. Synth., 1985, 63, 214–220 CrossRef CAS; (c) G. Bartoli, R. Dalpozzo and M. Nardi, Chem. Soc. Rev., 2014, 43, 4728–4750 RSC.
- T. V. Nykaza, G. Li, J. Yang, M. R. Luzung and A. T. Radosevich, Angew. Chem., Int. Ed., 2020, 59, 4505–4510 CrossRef CAS.
- Y. Zhao, H. Zhu, S. Sung, D. J. Wink, J. M. Zadrozny and T. G. Driver, Angew. Chem., Int. Ed., 2021, 60, 19207–19213 CrossRef CAS PubMed.
- K.-i. Fujita, K. Yamanoto and R. Yamaguchi, Org. Lett., 2002, 4, 2691–2694 CrossRef CAS PubMed.
- G. Bartoli, G. Palmieri, M. Bosco and R. Dalpozzo, Tetrahedron Lett., 1989, 30, 2129–2132 CrossRef CAS.
- (a) A. Penoni and K. M. Nicholas, Chem. Commun., 2002, 5, 484–485 RSC; (b) M. Akazome, T. Kondo and Y. Watanabe, Chem. Lett., 1992, 21, 769–772 CrossRefK. Okuro, J. Gurnham and H. Alper, J. Org. Chem., 2011, 76, 4715–4720 CrossRef CAS PubMed.
- R. J. Sundberg, J. Am. Chem. Soc., 1966, 88, 3781–3789 CrossRef CAS.
- B. Delayre, C. Piemontesi, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2020, 59, 13990–13997 CrossRef CAS PubMed.
- (a) L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS PubMed; (b) Y. Chen, X. Wang, X. He, Q. An and Z. Zuo, J. Am. Chem. Soc., 2021, 143, 4896–4902 CrossRef CAS PubMed; (c) K. Zhang, L. Chang, Q. An, X. Wang and Z. Zuo, J. Am. Chem. Soc., 2019, 141, 10556–10564 CrossRef CAS PubMed; (d) A. Hu, J. J. Guo, H. Pan, H. Tang, Z. Gao and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 1612–1616 CrossRef PubMed; (e) A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS.
- (a) A. C. Colgan, R. S. J. Proctor, D. C. Gibson, P. Chuentragool, A. S. K. Lahdenperä, K. Ermanis and R. J. Phipps, Angew. Chem., Int. Ed., 2022, 61, e202200266 CrossRef CAS PubMed; (b) H. Fuse, H. Mitsunuma and M. Kanai, J. Am. Chem. Soc., 2020, 142, 4493–4499 CrossRef CAS PubMed; (c) L. Jeffrey Jenna, A. Terrett Jack and W. C. MacMillan David, Science, 2015, 349, 1532–1536 CrossRef CAS PubMed; (d) J. Twilton, M. Christensen, D. A. DiRocco, R. T. Ruck, I. W. Davies and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 5369–5373 CrossRef CAS PubMed.
- (a) K. Zhao, G. Seidler and R. R. Knowles, Angew. Chem., Int. Ed., 2021, 60, 20190–20195 CrossRef CAS; (b) L. Huang, T. Ji and M. Rueping, J. Am. Chem. Soc., 2020, 142, 3532–3539 CrossRef CAS PubMed; (c) D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed; (d) Y. Zhu, Z. Zhang, R. Jin, J. Liu, G. Liu, B. Han and N. Jiao, Angew. Chem., Int. Ed., 2020, 59, 19851–19856 CrossRef CAS.
- (a) R. Hurley and A. C. Testa, J. Am. Chem. Soc., 1966, 88, 4330–4332 CrossRef CAS; (b) A. Ruffoni, C. Hampton, M. Simonetti and D. Leonori, Nature, 2022 DOI:10.1038/s41586-022-05211-0; (c) D. E. Wise, E. S. Gogarnoiu, A. D. Duke, J. M. Paolillo, T. L. Vacala, W. A. Hussain and M. Parasram, J. Am. Chem. Soc., 2022, 144, 15437–15442 CrossRef CAS PubMed.
- B. Wang, J. Ma, H. Ren, S. Lu, J. Xu, Y. Liang, C. Lu and H. Yan, Chin. Chem. Lett., 2022, 33, 2420–2424 CrossRef CAS.
- J.-Q. Qi and L. Jiao, J. Org. Chem., 2020, 85, 13877–13885 CrossRef CAS PubMed.
- (a) H. Zhao, Z. Lin and T. B. Marder, J. Am. Chem. Soc., 2006, 128, 15637–15643 CrossRef CAS PubMed; (b) W. Kong, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 3987–3991 CrossRef CAS PubMed; (c) S. P. Cummings, T.-N. Le, G. E. Fernandez, L. G. Quiambao and B. J. Stokes, J. Am. Chem. Soc., 2016, 138, 6107–6110 CrossRef CAS PubMed; (d) M. Flinker, H. Yin, R. W. Juhl, E. Z. Eikeland, J. Overgaard, D. U. Nielsen and T. Skrydstrup, Angew. Chem., Int. Ed., 2017, 56, 15910–15915 CrossRef CAS PubMed; (e) M. Rauser, R. Eckert, M. Gerbershagen and M. Niggemann, Angew. Chem., Int. Ed., 2019, 58, 6713–6717 CrossRef CAS; (f) J. Kim and C. R. Bertozzi, Angew. Chem., Int. Ed., 2015, 54, 15777–15781 CrossRef CAS PubMed; (g) L. Wang, T. Zhang, W. Sun, Z. He, C. Xia, Y. Lan and C. Liu, J. Am. Chem. Soc., 2017, 139, 5257–5264 CrossRef PubMed; (h) L. Tao, X. Guo, J. Li, R. Li, Z. Lin and W. Zhao, J. Am. Chem. Soc., 2020, 142, 18118–18127 CrossRef CAS PubMed.
- J. T. Kuethe, A. Wong and I. W. Davies, Org. Lett., 2003, 5, 3721–3723 CrossRef CAS PubMed.
- J. L. Rutherford, M. P. Rainka and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 15168–15169 CrossRef CAS PubMed.
- (a) M. Rauser, C. Ascheberg and M. Niggemann, Angew. Chem., Int. Ed., 2017, 56, 11570–11574 CrossRef CAS PubMed; (b) H. Chung, J. Kim, G. A. González-Montiel, P. Ha-Yeon Cheong and H. G. Lee, Org. Lett., 2021, 23, 1096–1102 CrossRef CAS PubMed; (c) G. E. Bell, J. W. B. Fyfe, E. M. Israel, A. M. Z. Slawin, M. Campbell and A. J. B. Watson, Org. Lett., 2022, 24, 3024–3027 CrossRef CAS; (d) R. C. Larock and E. K. Yum, J. Am. Chem. Soc., 1991, 113, 6689–6690 CrossRef CAS; (e) B. Witulski, C. Alayrac and L. Tevzadze-Saeftel, Angew. Chem., Int. Ed., 2003, 42, 4257–4260 CrossRef CAS PubMed.
- (a) Y.-Q. Fang and M. Lautens, Org. Lett., 2005, 7, 3549–3552 CrossRef CAS PubMed; (b) R. Sanz, M. P. Castroviejo, V. Guilarte, A. Pérez and F. J. Fañanás, J. Org. Chem., 2007, 72, 5113–5118 CrossRef CAS.
- D. R. Motati, R. Amaradhi and T. Ganesh, Org. Chem. Front., 2021, 8, 466–513 RSC.
- D. Yan, Q. Wu, D. Wang and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 15724–15742 CrossRef CAS PubMed.
- (a) L. Musso, R. Cincinelli, V. Zuco, M. De Cesare, F. Zunino, A. L. Fallacara, M. Botta and S. Dallavalle, ChemMedChem, 2016, 11, 1700–1704 CrossRef CAS PubMed; (b) R. Singh, K. Nagesh, D. Yugandhar and A. V. G. Prasanthi, Org. Lett., 2018, 20, 4848–4853 CrossRef CAS PubMed; (c) W. Ji, Y. A. Liu and X. Liao, Angew. Chem., Int. Ed., 2016, 55, 13286–13289 CrossRef CAS PubMed.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- C. J. Matheson, K. A. Casalvieri, D. S. Backos, M. Minhajuddin, C. T. Jordan and P. Reigan, Eur. J. Med. Chem., 2020, 197, 112316 CrossRef CAS PubMed.
- Although intermediate B is an isomeride of indolin-2-one, we identify the molecular peak detected by HRMS as belonging to B rather than indolin-2-one by analyzing the experimental data. Please see ESI5.6.†.
- H.-X. Zheng, X.-H. Shan, J.-P. Qu and Y.-B. Kang, Org. Lett., 2017, 19, 5114–5117 CrossRef CAS PubMed.
- K. Manna, T. Ganguly, S. Baitalik and R. Jana, Org. Lett., 2021, 23, 8634–8639 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2163566, 2163568, 2163569 and 2163570. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03590a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |