
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron-catalysed alkene and heteroarene H/D exchange by reversible protonation of iron-hydride intermediates†
Luke
Britton
a,
Jamie H.
Docherty
 *a,
Jan
Sklyaruk
a,
Jessica
Cooney
a,
Gary S.
Nichol
a,
Andrew P.
Dominey
b and
Stephen P.
Thomas
*a
*a,
Jan
Sklyaruk
a,
Jessica
Cooney
a,
Gary S.
Nichol
a,
Andrew P.
Dominey
b and
Stephen P.
Thomas
*a
aEaStCHEM School of Chemistry, University of Edinburgh, Edinburgh EH9 3FJ, UK. E-mail: jamie.docherty@ed.ac.uk; stephen.thomas@ed.ac.uk
bGSK Medicines Research Centre, Stevenage SG1 2NY, UK
First published on 11th August 2022
Abstract
C–H functionalisation reactions offer a sustainable method for molecular construction and diversification. These reactions however remain dominated by precious metal catalysis. While significant interest in iron-catalysed C–H activation reactions has emerged, the isolation, characterisation and mechanistic understanding of these processes remain lacking. Herein the iron-catalysed C(sp2)–H bond hydrogen/deuterium exchange reaction using CD3OD is reported for both heterocycles and, for the first time, alkenes (38 examples). Isolation and characterisation, including by single-crystal X-ray diffraction, of the key iron-aryl and iron-alkenyl C–H metallation intermediates provided evidence for a reversible protonation of the active iron hydride catalyst. Good chemoselectivity was observed for both substrate classes. The developed procedure is orthogonal to previous iron-catalysed H/D exchange methods which used C6D6, D2, or D2O as the deuterium source, and uses only bench-stable reagents, including the iron(II) pre-catalyst. Further, a new mechanism of iron-hydride formation is reported in which β-hydride elimination from an alcohol generates the iron hydride. The ability to produce, isolate and characterise the organometallic products arising from C–H activation presents a basis for future discovery and development.
Introduction
Metal-catalysed C–H functionalisation has emerged as a powerful synthetic method for the direct derivatisation of typically inert C–H bonds and obviating the need for pre-functionalised substrates.1 While the application of C–H functionalisation strategies has expanded, these methods generally rely on precious metal catalysts. Examples using Earth-abundant metal,2 and even non-metal,3 catalysts have emerged as potential alternatives, however their use and development has been comparably limited.A number of iron-catalysed C–H functionalisation methods have been developed,2a but the isolation and characterisation of well-defined, catalytically relevant species has remained limited to only a few studies, particularly where C–H metallation, sometimes referred to as C–H activation, has been proposed.4 Holland reported the observation and isolation of an iron-aryl species arising from C–H metallation of benzene using a low oxidation-state iron β-diketiminate complex (Scheme 1a).4a Similarly, Ackermann reported the C–H metallation of pivalophenone using a low oxidation-state iron complex [Fe(PMe3)4] that allowed for the isolation and characterisation of the key iron-aryl species responsible for allene hydroarylation (Scheme 1a).4b,c Additional iron-aryl complexes have been isolated and characterised through independent synthesis, usually from organometallic aryl reagents.4d–g
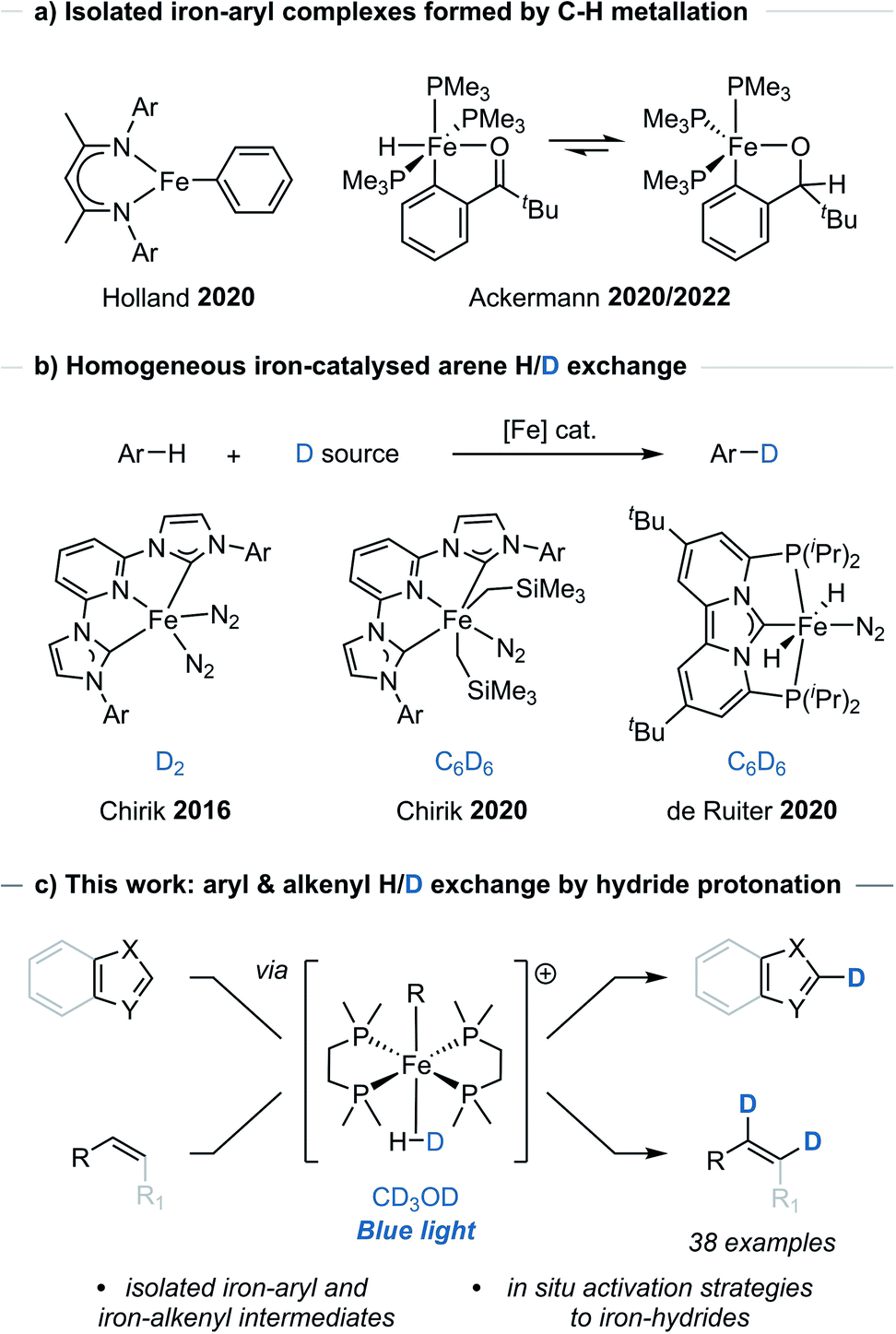 | ||
| Scheme 1 a) Previous examples of isolated iron-aryl complexes by direct C–H metallation. (b) Previous homogeneous iron-catalysed H/D exchange catalysts. (c) This work: arene and alkene C–H metallation and H/D exchange using CD3OD and a bench-stable iron(II) pre-catalyst. | ||
The iron bisdiphosphino complex, [dmpe2FeH2] 2 (dmpe = Me2PCH2CH2PMe2), has been shown to undergo stoichiometric C–H bond metallation of benzene in solution-phase studies.5 It was therefore questioned whether the metallation of C(sp2)–H bonds could be made general and harnessed to generate an iron-aryl species applicable to H/D exchange. In order to achieve H/D exchange, photoirradiation of [dmpe2FeH2] 2 would be used to trigger H2 elimination to give [dmpe2Fe0]. This would undergo C(sp2)–H bond oxidative addition (metallation) to give an aryl(hydrido) iron species, [dmpe2Fe(H)Ar], from which exchange of the hydrido-ligand to the deuteride isotopologue, would give [dmpe2Fe(D)Ar], and leave only a final reductive elimination to complete a potential catalytic cycle.
Commonly, direct H/D exchange reactions have involved the use of precious metal catalysts, with only a few Earth-abundant systems reported. Chirik reported the H/D exchange of arenes using iron-pincer complexes [(MesCNC)Fe(N2)2] and deuterium gas, as well as [(MesCNC)Fe(CH2SiMe3)2(N2)] and C6D6 (Scheme 1b).6 de Ruiter reported the C6D6-mediated H/D exchange reactions of (hetero)arenes using an alternative iron-complex [(PCNHCP)Fe(H)2(N2)] (Scheme 1b).7 Beller and Lei reported the H/D exchange reaction of arenes bearing electron-donating groups using D2O and a heterogeneous iron catalyst.8 Additional examples of Earth-abundant metal-catalysed HIE reactions of arenes have been reported which rely on pre-installed directing groups.9 Of note to this manuscript, Hartwig used a Ag2CO3/JohnPhos catalyst and CH3OD for the H/D exchange reaction of 5-membered heteroarenes.10
Results and discussion
Investigations began by exploring the viability of H/D exchange with a selection of related iron pre-catalysts, deuterium sources and catalyst activation methods (see ESI, Tables S1–S3†). Pre-catalyst activation with alkoxide salts has been shown to generate catalytically active metal-hydride species when combined with commercial silane or borane reagents such as pinacolborane (HBpin).11 This removes the need to generate and handle the highly air- and moisture sensitive iron dihydride. To assess the catalytic feasibility of this strategy in situ generated [dmpe2FeH2] 2 (from [dmpe2FeCl2] 1 + NaOtBu + HBpin) was used in combination with 2-methylfuran 3a, blue light irradiation (450 nm) and a variety of deuterium sources. Deuterium incorporation was achieved using deuterated alcohols as the deuterium source, with CD3OD giving the highest deuterium incorporation and exclusive C-5 regioselectivity. Control reactions showed the necessity for continuous light irradiation, iron complex, phosphine ligand, and the alkoxide activator (see ESI, Table S4†). Notably, good levels of deuterium incorporation were observed even in the absence of HBpin (vida infra). Optimisation of the light source using monochromatic LEDs in the range of 365–525 nm, showed the blue region (450 nm) of the spectrum to be optimal (see ESI, Table S5†). The substitution of CD3OD for CD3OH resulted in no deuterium incorporation, indicating that the most acidic, alcohol, hydrogen was responsible for deuterium-exchange. The state-of-the-art precious metal equivalent for non-directed H/D exchange of arenes using ROD-type deuterium sources resulted in global C–H deuteration and showed no chemoselectivity for alkene versus arene C–H bonds (reaction conditions: Pd(OAc)2 (10 mol%), ligand (15–30 mol%), HFIP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D2O = 3:7, 80–120 °C, 18–48 h).12
:D2O = 3:7, 80–120 °C, 18–48 h).12
Catalytic H/D exchange of heteroarenes
The generality of the method with respect to substrate compatibility and scope was then assessed (Table 1). Furan 3b, furan derivatives bearing a free secondary-amine 3c and imine 3d all underwent successful, chemoselective deuteration to give the monodeuterated furans [D]-3b, [D]-3c and [D]-3d, respectively, with good to excellent deuterium incorporation and complete control of regioselectivity (C-5) in all cases. Pinacol boronic ester protected alcohol 3e reacted to give the deuterated isotopologue [D]-3e, albeit with alcohol deprotection upon work up. Benzofuran 3g underwent site-selective deuteration at the C-2 position, with no detectable deuteration at any of the other five available C(sp2)–H bonds. Unprotected imidazole 3h and methyl-protected imidazole 3i underwent exclusive C-2 selective deuteration to give the deuterated imidazoles [D]-3h and [D]-3i, respectively. This is notable as both previously reported iron-catalysts gave unselective deuteration of N-methyl imidazole with exchange at at all the C(sp2)–H bonds.6,7 Using this system, oxazole 3j, 1,3-thiazole 3k and benzoxazole 3l all underwent deuteration with mild to good deuterium incorporation, again at a single site. 6-Membered heterocycle pyrimidine 3o proved amenable to deuteration at C-2, C-4 and C-6, albeit with only moderate incorporation. When 4-phenylpyrimidine 3p was used, deuteration occurred at both C-2 and C-5 positions with significantly greater inclusion than the unsubstituted analogue.| a Reaction conditions: arene (0.33 mmol), [dmpe2FeCl2] 1 (2.5 mol%), NaOtBu (5 mol%), HBpin (5 mol%), THF (0.2 mL), CD3OD (0.2 mL), blue-light irradiation, 15 h. Reported yields denote quantity of material recovered post-reaction. Deuterium incorporation determined by 1H and/or 13C NMR spectroscopy. b Quantity of recovered product determined using 1,3,5-trimethoxybenzene as an internal standard. c Starting material 3e = 4,4,5,5-tetramethyl-2-(furan-2-ylmethoxy)-1,3,2-dioxaborolane, free alcohol [D]-3e isolated following deprotection. d Performed on a 9.3 mmol scale, 48 h. |
|---|
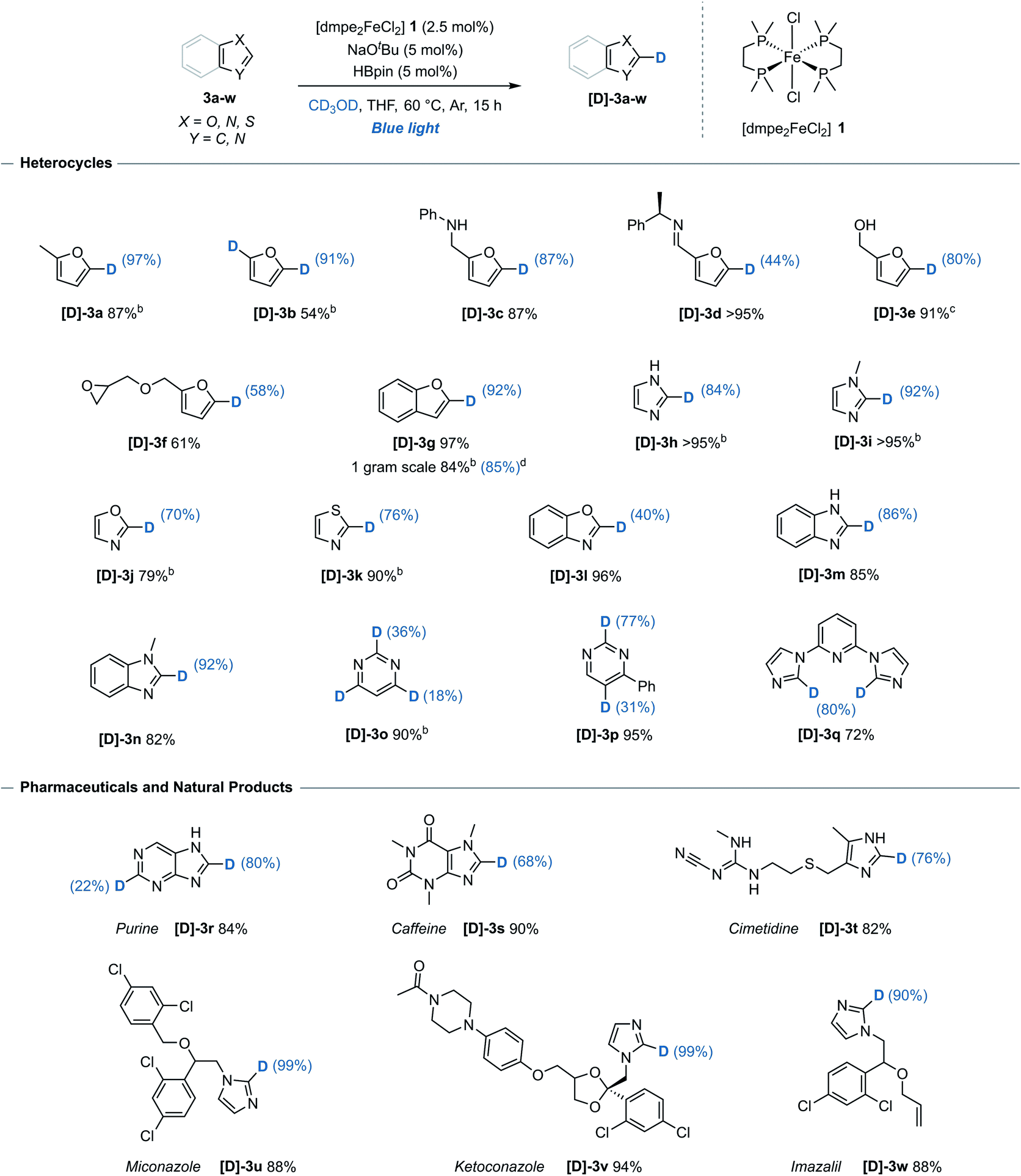
|
To illustrate the utility and tolerance of this system with respect to complex targets, the deuteration procedure was successfully applied to several pharmaceutical and natural product structures (Table 1, lower). Purine 3r, a compound ubiquitous to biological systems and common foodstuffs, reacted efficiently with incorporation at both C-2 and C-8. A further range of biologically active molecules including caffeine 3s, histamine receptor antagonist cimetidine 3t and antifungal compounds; miconazole 3u, ketoconazole 3v and imazalil 3w, all reacted efficiently with predictable deuterium incorporation at the most acidic C(sp2)–H site. While the imidazole unit of 3w underwent H/D exchange under the reaction conditions to give [D]-3w, the allylic ether was not deuterated. The functional group compatibility and regioselectivity shown illustrates the system is suitable for potential late-stage site-selective isotopic exchange of heteroarene units.
While high levels of deuterium incorporation were obtained using this system, it must be noted that an alternative alkoxide-mediated reaction exists for substrates with highly acidic C(sp2)–H bonds. Excluding benzoxazole 3l, which achieved greater levels of deuterium incorporation in the absence of pre-catalyst and HBpin,13 all substrates underwent greater deuterium incorporation and selectivity under the iron-catalysed reaction conditions (see ESI, Table S8†).
Catalytic H/D exchange of alkenes
While H/D exchange reactions of alkenes with non-reductive sources of deuterium, i.e. CD3OD, D2O or C6D6, have been previously reported using precious metals catalysts (iridium,14 rhodium,15 ruthenium,16 and palladium17), Earth-abundant metal-mediated H/D exchange reactions of alkenes have so far been limited to either stoichiometric studies,18 or unwanted side reactions.19 Using this iron-catalysed system, styrene derivatives 4a–f bearing both electron-donating- and electron-withdrawing groups were efficiently deuterated at all alkene positions with no detection of deuterium incorporation at any of the arene C(sp2)–H bonds. This represents the first reported example of an iron-catalysed H/D exchange reaction of alkenes (Table 2). It was observed that for the deuteration of styrene derivatives there was a slight trend for greater deuterium incorporation at the α position relative to both β positions, contrasting with results often seen with precious metal alternatives.14b,15b,17| a Reaction conditions: alkene (0.33 mmol), [dmpe2FeCl2] 1 (2.5 mol%), NaOtBu (5 mol%), HBpin (5 mol%), THF (0.2 mL), CD3OD (0.2 mL), blue-light irradiation, 15 h. Reported yields denote quantity of material recovered post-reaction. Deuterium incorporation determined by 1H and/or 13C NMR spectroscopy. b Quantity of recovered product determined using 1,3,5-trimethoxybenzene as an internal standard. c Performed on a 3.4 mmol scale (1.0 g recovered), 48 h. |
|---|
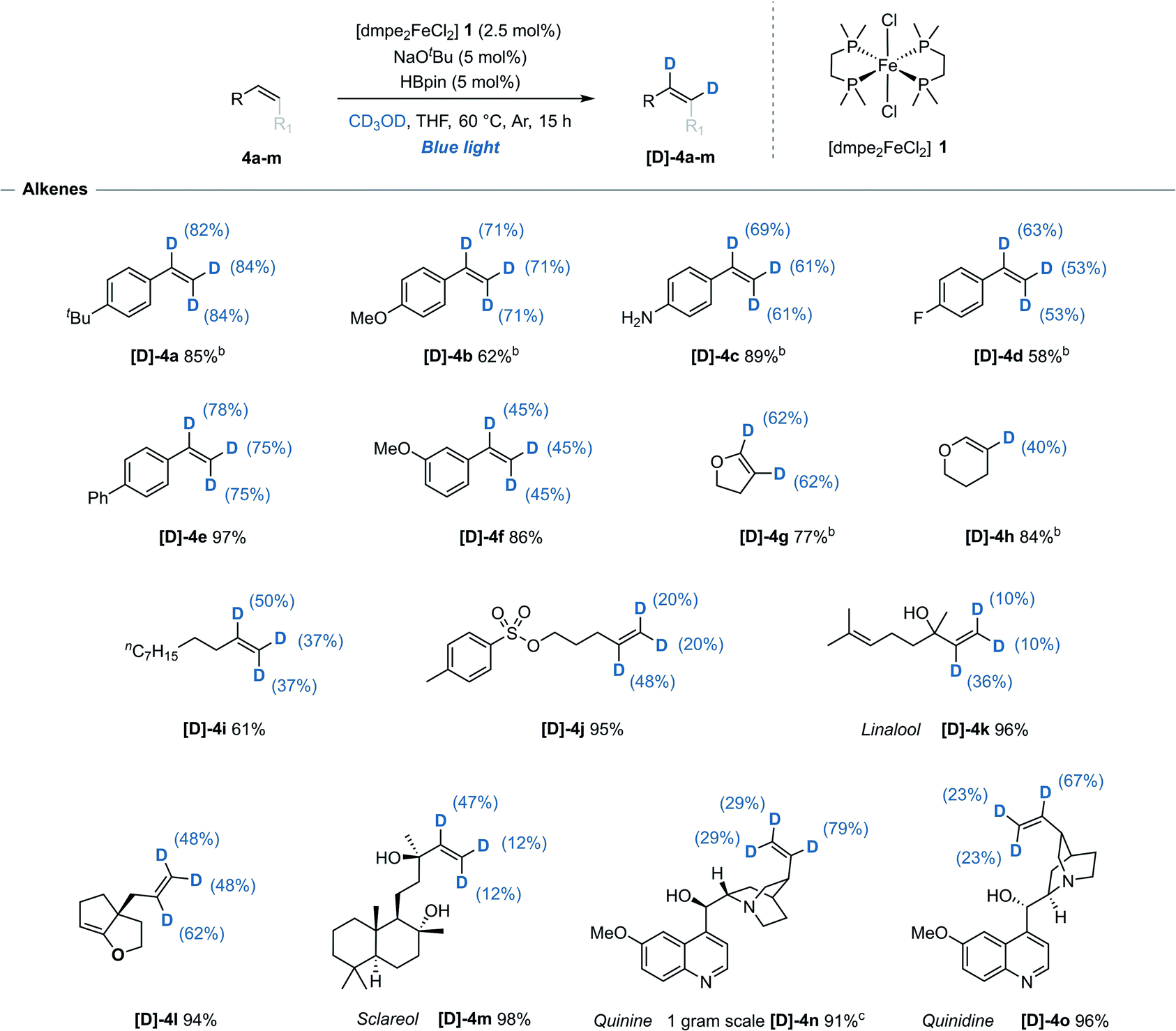
|
2,3-Dihydrofuran 4g underwent deuteration at both alkene C(sp2)–H bonds while the 6-membered 2,3-dihydropyran 4h was selectively deuterated at the 3-position. Monosubstituted acyclic alkenes undecene 4i and O-tosyl alkene 4j both underwent moderate deuterium incorporation. Naturally occurring allylic alcohols Linalool 4k and anti-cancer agent Sclareol 4m, both of which contain free alcohol groups, were deuterated with moderate deuterium incorporation. Cinchona alkaloid quinine 4n and diastereoisomer quinidine 4o were both successfully reacted and with exclusive deuteration of the alkene over the other five available C(sp2)–H sites. Minimal reactivity was displayed towards carbon substituted 1,2-cis alkenes including cyclohexene and cyclooctene, and no reactivity was observed for trans-, 1,1-disubstituted-, trisubstitued- or activated (e.g. acrylates or acrylamides) alkenes. Background, base-mediated H/D exchange was not observed for any alkene substrate (see ESI, Table S8†).20
Mechanistic investigations
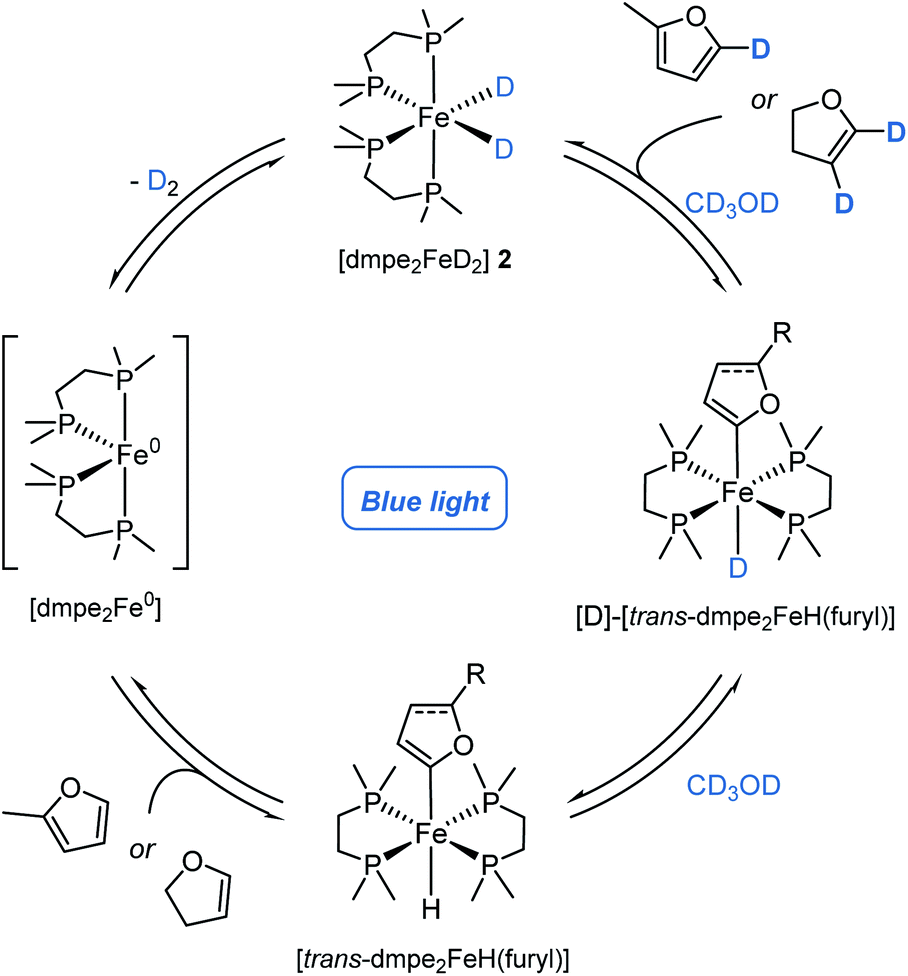
Having demonstrated catalytic H/D exchange across arenes and alkenes, the fundamental catalytic steps were investigated. The generation of the photoactive iron hydride [dmpe2FeH2] 2, through the in situ activation of [dmpe2FeCl2] 1 using an alkoxide salt and HBpin, has been previously reported (Scheme 2a, I),11b but did present a question of how significant deuterium incorporation of 2-methylfuran 3a was achieved in the absence of HBpin (see ESI, Table S4†). Methanol solutions of [dmpe2FeCl2] 1 were observed to exist as a 1:1 electrolyte of [dmpe2Fe(HOMe)Cl]+ and Cl−, as seen by 31P NMR spectroscopy.21 Upon the addition of a stoichiometric amount of sodium tert-butoxide, the immediate generation of iron-hydride species, including [dmpe2FeHCl] and [(dmpe2FeH)2(μ-dmpe)]2+, were observed by 1H and 31P NMR spectroscopy. Following blue light irradiation, [dmpe2FeH2] 2 was also observed in solution alongside an additional unknown iron hydride species. This unknown iron-hydride was not observed under standard activation conditions; sodium tert-butoxide and HBpin (see ESI,† Part 12).
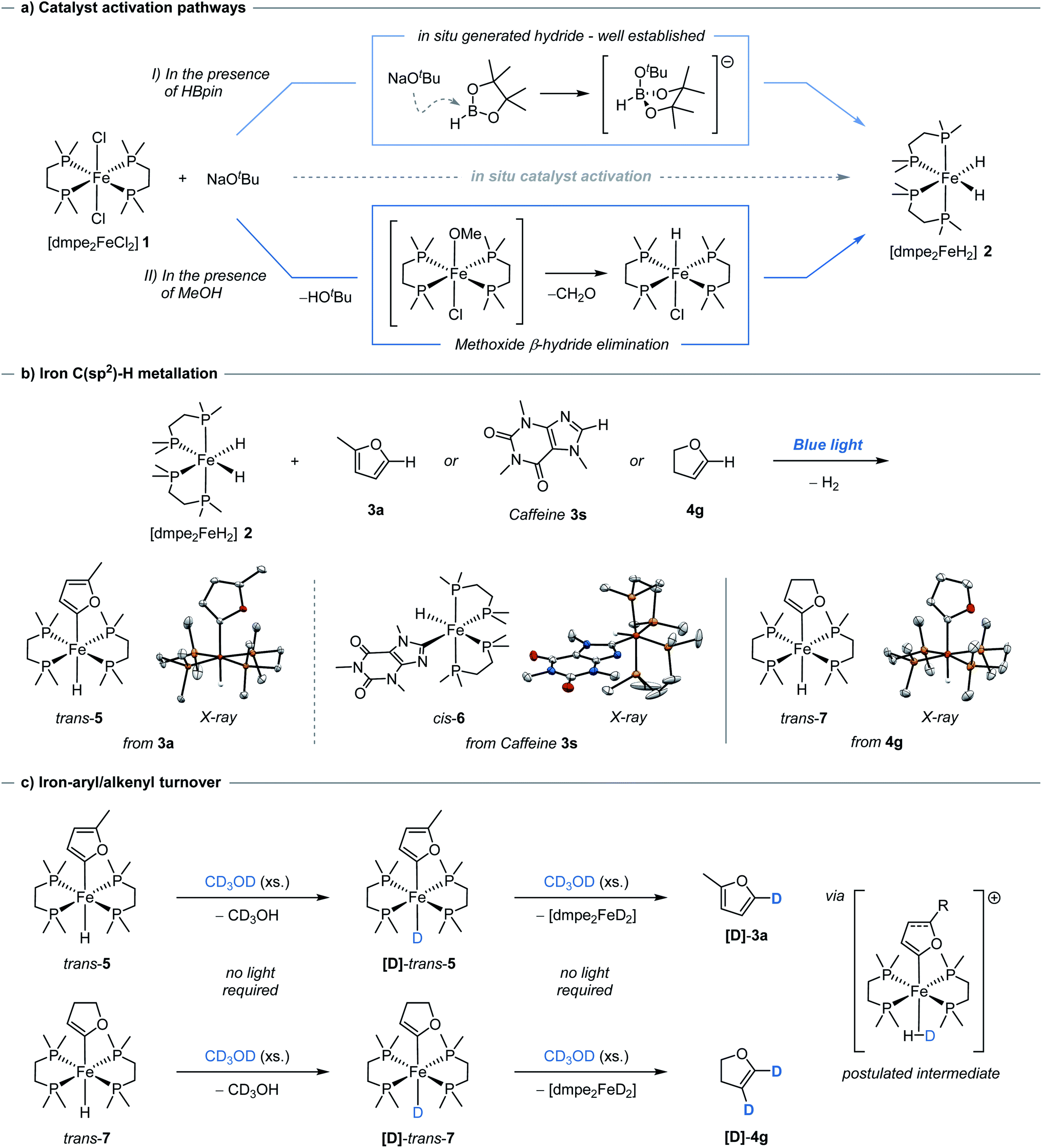 | ||
| Scheme 2 Mechanistic Studies for Iron-catalysed H/D Exchange. (a) in situ Activation pathways of [dmpe2FeCl2] 1 pre-catalyst; (I) in the presence of HBpin; (II) in the presence of CH3OH. (b) C(sp2)–H bond activation to give intermediate trans-5, cis-6, and trans-7. X-ray ellipsoids are shown at 50% probability. Hydrogen atoms, with the exception of Fe-hydrides, have been omitted for clarity. (c) Fe-hydrides (trans-5 and trans-7) to Fe-deuterides ([D]-trans-5 and [D]-trans-7), exchange using CD3OD in the absence of light, postulated intermediate species shown to the right. Generation of [D]-3a and [D]-4g through slow exchange between CD3OD and Fe-deuterides [D]-trans-5 and [D]-trans-7, respectively. | ||
The formation of these catalytically active iron–hydrido complexes was proposed to proceed by tert-butoxide-mediated deprotonation of [dmpe2Fe(HOMe)Cl]+ to generate an unstable iron methoxide complex, [dmpe2Fe(OMe)Cl], which underwent β-hydride elimination releasing formaldehyde and generating [dmpe2FeHCl] (Scheme 2a, II). The formation of [dmpe2FeHCl] was also observed by 1H and 31P NMR spectroscopy when the reaction was performed in ethanol and benzyl alcohol, with benzaldehyde being formed and observed by 13C NMR spectroscopy in the latter case. When the reaction was performed in CD3OH, [dmpe2FeDCl], along with other iron-deuteride complexes, were observed by 31P NMR spectroscopy (see ESI,† Part 12). The use of alcohols lacking available β-hydrogens, tert-butanol and phenol, resulted in no iron-hydride species being observed. Similar reactivity has been proposed for the analogous ruthenium–dmpe complexes.22 It should be noted that although activation to the catalytically active iron-hydride complexes can be achieved in the absence of HBpin, this activation was less controlled in terms of clean formation of [dmpe2FeH2] under catalytic conditions.
[trans-dmpe2Fe(H)(2-methylfuryl)] trans-5, has previously been characterised in solution.11b By careful tuning of the reaction and crystallization conditions, it was characterised in the solid-state by single-crystal X-ray diffraction (Scheme 2b, left). Significantly this reactivity was not limited to furan derivatives and was extended to the reaction of caffeine 3s to give the corresponding complex, [cis-dmpe2Fe(H)(caffeine)] cis-6 (Scheme 2b, center). Although these species were crystallised as the trans- and cis-isomers, respectively, solution phase cis–trans isomerisation was reasonably proposed.23 Rapid and quantitative exchange of hydride for deuteride was observed upon addition of excess CD3OD (approx. 250 eq.) to a THF solution of [trans-dmpe2Fe(H)(2-methylfuryl)] trans-5 to give the deuterated isotopologue [trans-dmpe2Fe(D)(2-methylfuran)] [D]-trans-5 in the absence of blue light irradiation. Exchange of hydride for deuteride occurring by protonation of [trans-dmpe2Fe(H)(2-methylfuryl)] trans-5 with CD3OD,24 to give the cationic complex [dmpe2Fe(HD)(2-methylfuryl)]+ which reversibly released CD3OH (Scheme 2c, upper). Further monitoring of this solution by 1H and 31P NMR spectroscopy showed the slow generation of [dmpe2FeD2] [D]-2 and 2-methyl-5D-furan [D]-3a at room temperature in the absence of blue light irradiation. Additionally, no further intermediary species were observed, and similar reactivity was not observed when using tBuOH. Presumably, turnover proceeded either through a stepwise cationic protonation and release of the arene, or by a concerted sigma bond metathesis reaction with CD3OD, both resulting in an [dmpe2Fe(D)(OMe)] intermediate which collapsed by β-hydride elimination to generate the photoactive complex [dmpe2FeD2] [D]-2 and formaldehyde (vida ante).
The reversible nature of deuterium incorporation led to it being questioned whether this process could be used for selective protodedeuteration to give selectively deuterated products. d4-Furan was regioselectively protodedeuterated at the 2- and 5-positions to give d2-[3D,4D]-furan with >95% in-corporation using 5 mol% of Fe(II) pre-catalyst 2 and MeOH (Scheme 3). The synthesis of d2-[3D,4D]-furan has only been previously prepared using a multistep sequence.25
 | ||
| Scheme 3 Protodedeuteration of d4-furan. Reaction conditions: d4-furan (0.33 mmol), [dmpe2FeCl2] 1 (5.0 mol%), NaOtBu (10 mol%), HBpin (10 mol%), THF (0.2 mL), CH3OH (0.2 mL), blue-light irradiation, 48 h. Deuterium incorporation determined by 1H and 13C NMR spectroscopy. | ||
Considering the H/D exchange of alkenes, blue light irradiation of [dmpe2FeH2] 2 in the presence of 2,3-dihydrofuran, led exclusively to the C-2C(sp2)–H metallation product, [trans-dmpe2Fe(H)(2,3-dihydrofuryl)] trans-7 (Scheme 2b, right). Despite reports of Fe-alkenyl C(sp2)–H metallation products being observed in the solution phase,5b complex trans-7 represents the first example of an Fe-alkenyl complex characterised in the solid-state by single-crystal X-ray diffraction (Fe–C bond distance 1.338(25) Å, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond distance 1.338(2) Å). This was in contrast to previous reports using the analogous bis(diethylphosphino)ethane ligand where C-3 metallation was proposed based on solution phase 1H and 31P NMR spectroscopy.26 In analogy to aryl iron hydride trans-7, the alkenyl iron complex trans-7 displayed similar reactivity in the presence of CD3OD; quantitative conversion to the iron-deuteride isotopologue, [D]-trans-7, was observed upon the addition of excess CD3OD in the absence of blue light irradiation. Slow conversion of [trans-dmpe2Fe(H)(2,3-dihydrofuryl)] trans-7 to [dmpe2FeD2] [D]-2 with the release of 2,3-dihydro-4D,5D-furan [D]-4g was observed by 1H and 31P NMR spectroscopy in the absence of blue light irradiation. The irradiation of [dmpe2FeH2] 2 in the presence of 4-tert-butylstyrene 4a, led to a complex mixture of products observed by 1H and 31P NMR spectroscopy. When performing the deuteration of tert-butylstyrene in the absence of blue light irradiation at 60 or 80 °C, a greater α:β ratio was observed to that under standard reaction conditions (see ESI, Table S6†). When the allylic ethers, 2,5-dihydrofuran and allyl benzyl ether, were reacted under standard conditions, alkene isomerisation to the vinyl ethers was observed producing 2,3-dihydrofuran (62%) and the benzyl propenyl ether (62%, 46:54 Z:E), respectively. Similarly, 4-allylanisole showed small amounts of isomerisation to (Z)-anethole (<10%) (see ESI,† Part 12). Control reactions ruled out any acid- or base-mediated isomerisation. The aforementioned observations, alongside previous work,5b,27 would suggest that the deuteration of alkenes is occurring through a combination of (direct) C–H metallation and hydrometallation mechanisms, with the latter observed to mediate alkene isomerisation alongside exchange (Scheme 4).
C bond distance 1.338(2) Å). This was in contrast to previous reports using the analogous bis(diethylphosphino)ethane ligand where C-3 metallation was proposed based on solution phase 1H and 31P NMR spectroscopy.26 In analogy to aryl iron hydride trans-7, the alkenyl iron complex trans-7 displayed similar reactivity in the presence of CD3OD; quantitative conversion to the iron-deuteride isotopologue, [D]-trans-7, was observed upon the addition of excess CD3OD in the absence of blue light irradiation. Slow conversion of [trans-dmpe2Fe(H)(2,3-dihydrofuryl)] trans-7 to [dmpe2FeD2] [D]-2 with the release of 2,3-dihydro-4D,5D-furan [D]-4g was observed by 1H and 31P NMR spectroscopy in the absence of blue light irradiation. The irradiation of [dmpe2FeH2] 2 in the presence of 4-tert-butylstyrene 4a, led to a complex mixture of products observed by 1H and 31P NMR spectroscopy. When performing the deuteration of tert-butylstyrene in the absence of blue light irradiation at 60 or 80 °C, a greater α:β ratio was observed to that under standard reaction conditions (see ESI, Table S6†). When the allylic ethers, 2,5-dihydrofuran and allyl benzyl ether, were reacted under standard conditions, alkene isomerisation to the vinyl ethers was observed producing 2,3-dihydrofuran (62%) and the benzyl propenyl ether (62%, 46:54 Z:E), respectively. Similarly, 4-allylanisole showed small amounts of isomerisation to (Z)-anethole (<10%) (see ESI,† Part 12). Control reactions ruled out any acid- or base-mediated isomerisation. The aforementioned observations, alongside previous work,5b,27 would suggest that the deuteration of alkenes is occurring through a combination of (direct) C–H metallation and hydrometallation mechanisms, with the latter observed to mediate alkene isomerisation alongside exchange (Scheme 4).
 | ||
| Scheme 4 Proposed reaction mechanism for the iron-catalysed H/D exchange reaction of heteroarenes and alkenes. | ||
Conclusions
In summary, direct C–H metallation of heteroarenes and alkenes using an iron(II) hydride species has enabled the development of an iron-catalysed H/D exchange reaction using a bench-stable iron(II) pre-catalyst, and CD3OD as the deuterium source. This method is mechanistically different from and complementary to previously described iron-catalysed strategies, and allows for precise and predictable formation of C–D bonds without the need for directing groups. Regioselectivity was obtained and controlled through highly selective C–H metallation by the proposed active catalyst. Significantly, the use of a non-reductive deuterium source (CD3OD) has enabled extension to exchange of alkene C–H bonds and to molecules containing reducible functional groups. Isolation and characterisation, including single-crystal X-ray diffraction, of the key iron-aryl and iron-alkenyl intermediates allowed for mechanistic investigations that provided evidence for a reversible protonation of these iron C–H activation products. Further, a new mechanism of iron-hydride formation is presented in alcohol solvents.Data availability
Experimental data is provided in the ESI.†Author contributions
J. H. D. and S. P. T. conceived and discovered the reaction. L. B., J. H. D., J. S. and J. C. conducted the experimental work. G. S. N. conducted the X-ray crystallographic analysis. S. P. T. and A. P. D. advised investigations. All authors contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors thank Dr Andrew Ashley and Lee J. Edwards for useful discussions. S. P. T. thanks The Royal Society for a University Research Fellowship (RF191015). J. H. D. and S. P. T. acknowledge GSK and EPSRC (PIII0002) and The Royal Society (RF191015) for post-doctoral funding. L. B. acknowledges The Royal Society and The University of Edinburgh for a PhD studentship (RF191015).Notes and references
- (a) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173–1193 RSC; (b) M. M. Díaz-Requejo and P. J. Pérez, Chem. Rev., 2008, 108, 3379–3394 CrossRef PubMed; (c) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS; (d) Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Chem. Soc. Rev., 2015, 44, 7764–7786 RSC; (e) F. Roudesly, J. Oble and G. Poli, J. Mol. Catal. A: Chem., 2017, 426, 275–296 CrossRef CAS; (f) D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC; (g) B. Liu, L. Yang, P. Li, F. Wang and X. Li, Org. Chem. Front., 2021, 8, 1085–1101 RSC.
- (a) R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086–9139 CrossRef CAS PubMed; (b) P. Gandeepan, T. Muller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed; (c) L. Ilies, Bull. Chem. Soc. Jpn., 2021, 94, 404–417 CrossRef CAS; (d) J. Loup, U. Dhawa, F. Pesciaioli, J. Wencel-Delord and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 12803–12818 CrossRef CAS PubMed.
- (a) M.-A. Légaré, M.-A. Courtemanche, É. Rochette and F.-G. Fontaine, Science, 2015, 349, 513–516 CrossRef PubMed; (b) K. Chernichenko, M. Lindqvist, B. Kótai, M. Nieger, K. Sorochkina, I. Pápai and T. Repo, J. Am. Chem. Soc., 2016, 138, 4860–4868 CrossRef CAS; (c) J. Lv, X. Chen, X.-S. Xue, B. Zhao, Y. Liang, M. Wang, L. Jin, Y. Yuan, Y. Han, Y. Zhao, Y. Lu, J. Zhao, W.-Y. Sun, K. N. Houk and Z. Shi, Nature, 2019, 575, 336–340 CrossRef CAS PubMed; (d) T. He, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2022, 144, 4734–4738 CrossRef CAS PubMed.
- (a) S. F. McWilliams, D. L. J. Broere, C. J. V. Halliday, S. M. Bhutto, B. Q. Mercado and P. L. Holland, Nature, 2020, 584, 221–226 CrossRef CAS PubMed; (b) A. M. Messinis, L. H. Finger, L. Hu and L. Ackermann, J. Am. Chem. Soc., 2020, 142, 13102–13111 CrossRef CAS PubMed; (c) A. M. Messinis, J. C. A. Oliveira, A. C. Stückl and L. Ackermann, ACS Catal., 2022, 12, 4947–4960 CrossRef CAS; (d) S. L. Daifuku, M. H. Al-Afyouni, B. E. R. Snyder, J. L. Kneebone and M. L. Neidig, J. Am. Chem. Soc., 2014, 136, 9132–9143 CrossRef CAS PubMed; (e) R. B. Bedford, P. B. Brenner, E. Carter, J. Clifton, P. M. Cogswell, N. J. Gower, M. F. Haddow, J. N. Harvey, J. A. Kehl, D. M. Murphy, E. C. Neeve, M. L. Neidig, J. Nunn, B. E. R. Snyder and J. Taylor, Organometallics, 2014, 33, 5767–5780 CrossRef CAS; (f) Y. Liu, M. Shi and L. Deng, Organometallics, 2014, 33, 5660–5669 CrossRef CAS; (g) T. E. Boddie, S. H. Carpenter, T. M. Baker, J. C. DeMuth, G. Cera, W. W. Brennessel, L. Ackermann and M. L. Neidig, J. Am. Chem. Soc., 2019, 141, 12338–12345 CrossRef CAS PubMed.
- (a) S. D. Ittel, C. A. Tolman, A. D. English and J. P. Jesson, J. Am. Chem. Soc., 1976, 98, 6073–6075 CrossRef CAS; (b) M. V. Baker and L. D. Field, J. Am. Chem. Soc., 1986, 108, 7433–7434 CrossRef CAS.
- (a) R. Pony Yu, D. Hesk, N. Rivera, I. Pelczer and P. J. Chirik, Nature, 2016, 529, 195–199 CrossRef CAS PubMed; (b) J. Corpas, P. Viereck and P. J. Chirik, ACS Catal., 2020, 10, 8640–8647 CrossRef CAS.
- S. Garhwal, A. Kaushansky, N. Fridman, L. J. W. Shimon and G. d. Ruiter, J. Am. Chem. Soc., 2020, 142, 17131–17139 CrossRef CAS PubMed.
- W. Li, J. Rabeah, F. Bourriquen, D. Yang, C. Kreyenschulte, N. Rockstroh, H. Lund, S. Bartling, A.-E. Surkus, K. Junge, A. Brückner, A. Lei and M. Beller, Nat. Chem., 2022, 14, 334–341 CrossRef CAS PubMed.
- (a) J. Zhang, S. Zhang, T. Gogula and H. Zou, ACS Catal., 2020, 10, 7486–7494 CrossRef CAS; (b) S. Kopf, H. Neumann and M. Beller, Chem. Commun., 2021, 57, 1137–1140 RSC; (c) F. Bourriquen, N. Rockstroh, S. Bartling, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2022, 61, e202202423 Search PubMed.
- A. Tlahuext-Aca and J. F. Hartwig, ACS Catal., 2021, 11, 1119–1127 CrossRef CAS PubMed.
- (a) J. H. Docherty, J. Peng, A. P. Dominey and S. P. Thomas, Nat. Chem., 2017, 9, 595–600 CrossRef CAS; (b) L. Britton, J. H. Docherty, A. P. Dominey and S. P. Thomas, Molecules, 2020, 25, 905 CrossRef CAS PubMed; (c) L. Britton, M. Skrodzki, G. S. Nichol, A. P. Dominey, P. Pawluć, J. H. Docherty and S. P. Thomas, ACS Catal., 2021, 11, 6857–6864 CrossRef CAS.
- M. Farizyan, A. Mondal, S. Mal, F. Deufel and M. van Gemmeren, J. Am. Chem. Soc., 2021, 143, 16370–16376 CrossRef CAS PubMed.
- Presumed to be a result of a lower concentration of base present in the reaction mixture..
- (a) J. Zhou and J. F. Hartwig, Angew. Chem., Int. Ed., 2008, 47, 5783–5787 CrossRef CAS PubMed; (b) M. Hatano, T. Nishimura and H. Yorimitsu, Org. Lett., 2016, 18, 3674–3677 CrossRef CAS PubMed.
- (a) B. Rybtchinski, R. Cohen, Y. Ben-David, J. M. L. Martin and D. Milstein, J. Am. Chem. Soc., 2003, 125, 11041–11050 CrossRef CAS PubMed; (b) A. Di Giuseppe, R. Castarlenas, J. J. Pérez-Torrente, F. J. Lahoz and L. A. Oro, Chem.–Eur. J., 2014, 20, 8391–8403 CrossRef CAS PubMed.
- (a) G. Erdogan and D. B. Grotjahn, J. Am. Chem. Soc., 2009, 131, 10354–10355 CrossRef CAS PubMed; (b) S. K. S. Tse, P. Xue, Z. Lin and G. Jia, Adv. Synth. Catal., 2010, 352, 1512–1522 CrossRef CAS; (c) A. Bechtoldt and L. Ackermann, ChemCatChem, 2019, 11, 435–438 CrossRef CAS.
- N. Camedda, A. Serafino, R. Maggi, F. Bigi, G. Cera and G. Maestri, Synthesis, 2020, 52, 1762–1772 CrossRef CAS.
- (a) C. P. Lenges, M. Brookhart and B. E. Grant, J. Organomet. Chem., 1997, 528, 199–203 CrossRef CAS; (b) J. Choi, L. Tang and J. R. Norton, J. Am. Chem. Soc., 2007, 129, 234–240 CrossRef CAS PubMed.
- (a) H. Fong, M.-E. Moret, Y. Lee and J. C. Peters, Organometallics, 2013, 32, 3053–3062 CrossRef CAS PubMed; (b) D. Zell, M. Bursch, V. Müller, S. Grimme and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 10378–10382 CrossRef CAS; (c) J.-F. Li, Z.-Z. Wei, Y.-Q. Wang and M. Ye, Green Chem., 2017, 19, 4498–4502 RSC.
- T. R. Puleo, A. J. Strong and J. S. Bandar, J. Am. Chem. Soc., 2019, 141, 1467–1472 CrossRef CAS PubMed.
- J. M. Bellerby, M. J. Mays and P. L. Sears, Dalton Trans., 1976, 1232–1236 RSC.
- (a) J. R. Fulton, S. Sklenak, M. W. Bouwkamp and R. G. Bergman, J. Am. Chem. Soc., 2002, 124, 4722–4737 CrossRef CAS PubMed; (b) X.-J. Dai and C.-J. Li, J. Am. Chem. Soc., 2016, 138, 5433–5440 CrossRef CAS PubMed.
- T. Dombray, C. G. Werncke, S. Jiang, M. Grellier, L. Vendier, S. Bontemps, J. B. Sortais, S. Sabo-Etienne and C. Darcel, J. Am. Chem. Soc., 2015, 137, 4062–4065 CrossRef CAS PubMed.
- M. V. Baker, L. D. Field and D. J. Young, Chem. Commun., 1988, 546–548 RSC.
- R. M. Spycher, L. Hausherr-Primo, G. Grassi and A. Bauder, J. Mol. Struct., 1995, 351, 7–17 CrossRef CAS.
- T. Morikita, M. Hirano, A. Sasaki and S. Komiya, Inorg. Chim. Acta, 1999, 291, 341–354 CrossRef CAS.
- S. Komiya, N. Oyasato and T. Furukawa, Bull. Chem. Soc. Jpn., 1989, 62, 4078–4079 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2073340, 2073339, 2159501. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03802a |
| This journal is © The Royal Society of Chemistry 2022 |