
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface differences of oxide nanocrystals determined by geometry and exogenously coordinated water molecules†
Junchao
Chen‡
ab,
Xin-Ping
Wu‡
 c,
Michael A.
Hope‡
d,
Zhiye
Lin
a,
Lei
Zhu
e,
Yujie
Wen
a,
Yixiao
Zhang
b,
Tian
Qin
b,
Jia
Wang
c,
Tao
Liu
f,
Xifeng
Xia
g,
Di
Wu
h,
Xue-Qing
Gong
c,
Weiping
Tang
e,
Weiping
Ding
a,
Xi
Liu
b,
Liwei
Chen
bi,
Clare P.
Grey
d and
Luming
Peng
*ajk
c,
Michael A.
Hope‡
d,
Zhiye
Lin
a,
Lei
Zhu
e,
Yujie
Wen
a,
Yixiao
Zhang
b,
Tian
Qin
b,
Jia
Wang
c,
Tao
Liu
f,
Xifeng
Xia
g,
Di
Wu
h,
Xue-Qing
Gong
c,
Weiping
Tang
e,
Weiping
Ding
a,
Xi
Liu
b,
Liwei
Chen
bi,
Clare P.
Grey
d and
Luming
Peng
*ajk
aKey Laboratory of Mesoscopic Chemistry of MOE, Collaborative Innovation Center of Chemistry for Life Sciences, School of Chemistry and Chemical Engineering, Nanjing University, 163 Xianlin Road, Nanjing 210023, China. E-mail: luming@nju.edu.cn
bSchool of Chemistry and Chemical Engineering, Frontiers Science Center for Transformative Molecules, In situ Center for Physical Sciences, Shanghai Jiao Tong University, Shanghai 200240, China
cKey Laboratory for Advanced Materials, Centre for Computational Chemistry, Research Institute of Industrial Catalysis, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China
dDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK
eState Key Laboratory of Space Power Technology, Shanghai Institute of Space Power-Sources (SISP), Shanghai Academy of Spaceflight Technology, Shanghai 200245, China
fShanghai Key Laboratory of Chemical Assessment and Sustainability, Department of Chemistry, Tongji University, Shanghai 200092, China
gAnalysis and Testing Center, Nanjing University of Science and Technology, Nanjing 210094, China
hCollege of Food Science and Engineering/Collaborative Innovation Center for Modern Grain Circulation and Safety/Key Laboratory of Grains and Oils Quality Control and Processing, Nanjing University of Finance and Economics, Nanjing 210023, China
ii-Lab, CAS Center for Excellence in Nanoscience, Suzhou Institute of Nano-Tech and Nano-Bionics (SINANO), Chinese Academy of Sciences, Suzhou 215123, China
jJiangsu Key Laboratory of Vehicle Emissions Control, Nanjing University, Nanjing 210093, China
kFrontiers Science Center for Critical Earth Material Cycling (FSC-CEMaC), Nanjing University, Nanjing, Jiangsu 210023, China
First published on 14th September 2022
Abstract
Determining the different surfaces of oxide nanocrystals is key in developing structure–property relations. In many cases, only surface geometry is considered while ignoring the influence of surroundings, such as ubiquitous water on the surface. Here we apply 17O solid-state NMR spectroscopy to explore the facet differences of morphology-controlled ceria nanocrystals considering both geometry and water adsorption. Tri-coordinated oxygen ions at the 1st layer of ceria (111), (110), and (100) facets exhibit distinct 17O NMR shifts at dry surfaces while these 17O NMR parameters vary in the presence of water, indicating its non-negligible effects on the oxide surface. Thus, the interaction between water and oxide surfaces and its impact on the chemical environment should be considered in future studies, and solid-state NMR spectroscopy is a sensitive approach for obtaining such information. The work provides new insights into elucidating the surface chemistry of oxide nanomaterials.
Introduction
Different surfaces of oxides1,2 usually exhibit diverse properties in heterogeneous catalysis,3–5 photocatalysis,6–8 gas sensors,9 energy storage,10 and biological applications.11 Identification of the chemical environment of different surfaces is, therefore a prerequisite for investigating structure–property relations for oxides. The most popular conventional techniques for determining surface geometry are microscopy-based methods, including Scanning Electron Microscopy (SEM),12,13 High-Resolution Transmission Electron Microscopy (HRTEM)14,15 and Scanning Tunneling Microscopy (STM),16,17 with which the surfaces can be visualized at controlled temperature and pressure conditions, and even in gas or liquid environments with specialized setups.14,16 However, disadvantages remain in the application of such techniques, due to the weak signatures of light elements (e.g., H and O) for oxide and oxide-based materials.18 Complementary spectroscopy-based techniques, such as infrared19,20 and solid-state nuclear magnetic resonance (NMR) spectroscopy,21–23 have been applied to overcome these problems, however, the use of probe molecules (e.g., CO and trimethylphosphine) is often required. In addition to the geometry information obtained from these methods, the chemical environment which may also include the influence of surroundings, is still challenging to probe. For instance, water is ubiquitous in nature and is often involved in surface processes,24,25 yet the impact of water on oxide surfaces26,27 is still not well studied.Recently, we showed that 17O solid-state NMR in combination with 17O surface-selective labeling and density functional theory (DFT) calculations can probe the oxygen anions of oxide surfaces with a very high sensitivity.18,28–30 In particular, we have applied this strategy to study the dominant surfaces of some morphology-controlled ceria nanostructures, which are technologically important in CO oxidation,31 automotive three-way catalysis32 and the water–gas shift (WGS) reaction.33 We found that the 1st, 2nd and 3rd layer oxygen ions as well as surface hydroxyl groups at ceria (111) surface can be distinguished,29 while the reconstruction of ceria (100) surfaces18 and the hydrous ceria (111) surfaces can be elucidated.30 Here we adopt a strategy based on 17O NMR spectroscopy combined with an improved17O surface-selective labelling method (increased rounds of labelling that for the first time, enables us to probe not only the dominant surfaces but also secondary exposed ones) to ceria nanorods containing mixed facets, and reveal the differences of ceria (111), (110) and (100) surfaces under dry and hydrated conditions. We found both the surface geometry and exogenously coordinated water molecules are non-ignorable for determining the chemical environment of oxide surfaces.
Results and discussion
Morphology of the ceria nanorods
Faceted ceria nanorods were synthesized via a solvothermal approach.34 Two samples were prepared with different annealing temperatures (see Methods): NRs700 (nanorods annealed at 700 °C) and NRs300 (nanorods annealed at 300 °C). Scanning transmission electron microscopy (STEM) and HRTEM images in Fig. 1 depict the exposed surfaces of NRs700 and NRs300. Fig. 1a and c exhibit several nanorods measuring around 15–20 nm in width, for NRs700 and NRs300, respectively. Fig. 1b shows that NRs700 is mainly dominated by (111) surfaces along the sides of the nanorods, with a small fraction of (110) and (100) secondary surfaces at the ends. Two images of NRs300 are presented in Fig. 1d and e along with the corresponding Fourier transform (FT) patterns. The strongest electron diffraction peaks in Fig. 1d are assigned to the (111) and (100) peaks, while those in Fig. 1e only correspond to the (100) peak, suggesting the NRs300 predominantly expose (110) and (100) surfaces. All the microscopy signatures are consistent with previous results by Gao et al.34 Synchrotron powder X-ray diffraction (XRD) (Fig. S1†) confirms that both samples crystallize with the fluorite structure (space group: Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m); the broad diffraction peaks denote that they are nano-sized crystals. The BET surface areas of NRs700 and NRs300 (Fig. S2 and Table S1†) are determined as 110.9 and 132.7 m2 g−1, respectively. Na+ or NO3− impurities from the NaOH and Ce(NO3)3·6H2O precursors are not detectable by X-ray photoelectron spectra (XPS) (Fig. S3†), inductively coupled plasma mass spectrometry (ICP-MS) or elemental analysis (Table S1†); therefore, possible surface impurities are not considered.
m); the broad diffraction peaks denote that they are nano-sized crystals. The BET surface areas of NRs700 and NRs300 (Fig. S2 and Table S1†) are determined as 110.9 and 132.7 m2 g−1, respectively. Na+ or NO3− impurities from the NaOH and Ce(NO3)3·6H2O precursors are not detectable by X-ray photoelectron spectra (XPS) (Fig. S3†), inductively coupled plasma mass spectrometry (ICP-MS) or elemental analysis (Table S1†); therefore, possible surface impurities are not considered.
 | ||
| Fig. 1 STEM images and morphology models (top right corner) of NRs700 and NRs300. (a) Secondary electron (SE) and (b) HRTEM images of NRs700. (c) SE and (d and e) HRTEM images of NRs300 (the insets show the corresponding FT patterns that allow assignment of the planes perpendicular to the observation axes of (d) [110] and (e) [100], respectively). | ||
Conventional 17O NMR spectroscopy
Following four rounds of isotopic labeling with 90 atom% 17O2, resulting in a surface with highly enriched oxygen ions at the 1st and 2nd layers (see Methods), the NRs700 and NRs300 were subjected to 17O solid-state NMR measurements. Since dry 17O2 was used in the enrichment, the samples should represent dry surfaces (vide infra). The most intense resonance at 877 ppm in the 17O NMR spectra of both samples (Fig. 2a and the untruncated spectra in Fig. S4†) arises from the tetra-coordinated oxygen ion (O4C) in the bulk.29 The signal at a higher frequency of 1032 ppm for NRs700 is due to tri-coordinated oxygen ions (O3C) at the first layer of the major (111) facet, while the relatively sharp signal at 970 ppm in both samples comes from O3Cs at the 1st layer of ceria (100) surfaces (Fig. 2b).18,30 The broader signal at ∼1020 ppm for both samples, which has not previously been assigned, may be tentatively attributed to the oxygen ions at the 1st layer of other low-index ceria surfaces, most likely the (110) surface given that there is a significant proportion of (110) facets in the NRs300 sample. Other 17O resonances at 920, 905 and 830 ppm are associated with O4Cs at the sub-surface layers of (111) and (100) facets.18,30 Notably, the 17O NMR signals of the 1st layer O3Cs for different dry ceria surfaces are distinct, proving that the chemical environment of the exposed low-coordinated oxygen ions depends critically on the surface geometry.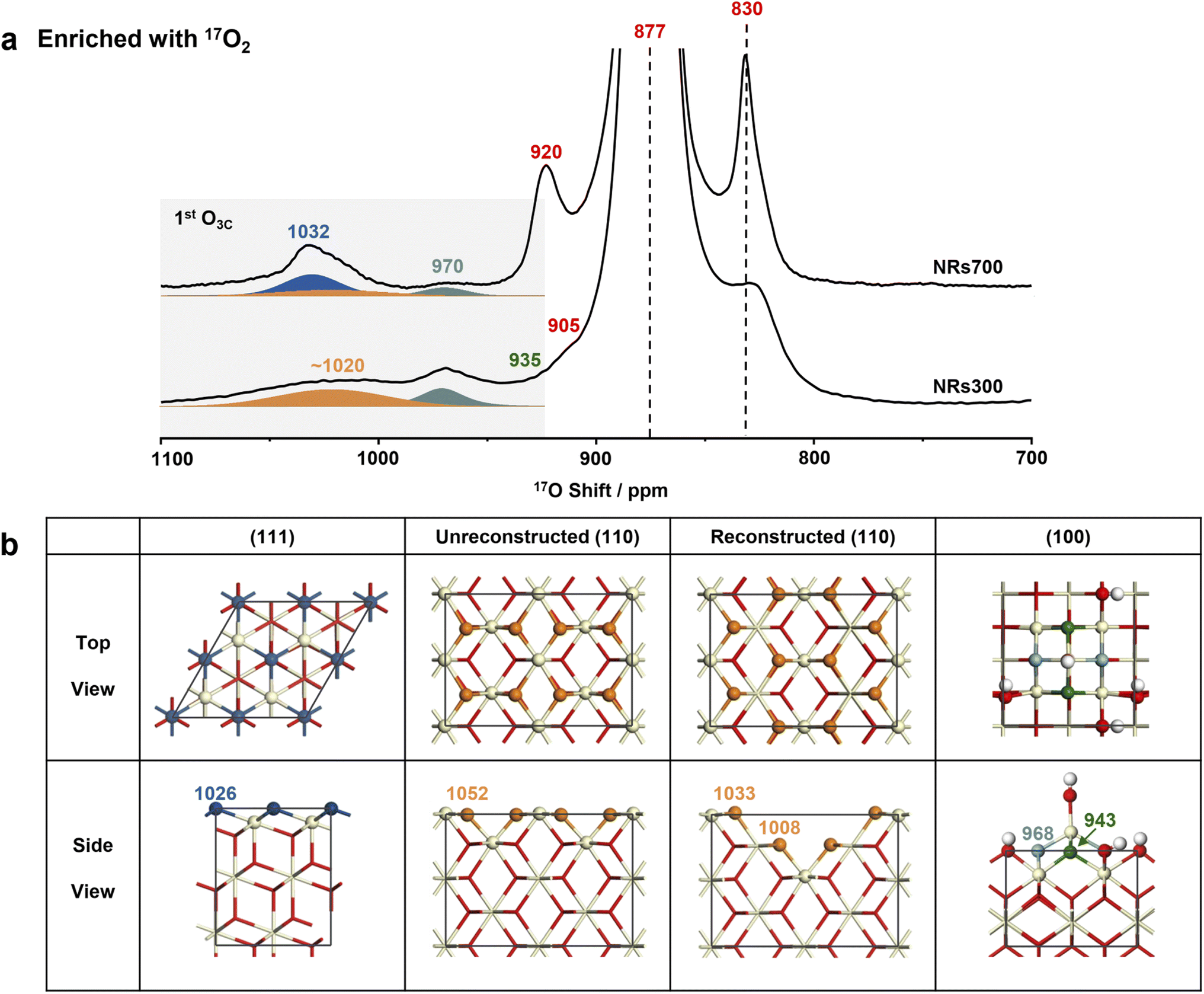 | ||
| Fig. 2 17O NMR spectra (9.4 T), morphology diagrams and the dry structure model of ceria NRs700 and NRs300. (a) 17O solid-state NMR spectra of the 17O2-enriched ceria NRs700 and NRs300. The spectra were recorded at a spinning speed of 20 kHz and a 2.0 s recycle delay using a rotor synchronized Hahn-echo pulse sequence (π/2–τ–π–τ-acquisition) with 1H decoupling. (b) The calculated structures of ceria (111), (110) and (100) surfaces. Red, off-white and white spheres denote bulk oxygen, hydrogen and cerium ions, respectively. 1st-layer oxygen ions from different surfaces are shown in different colors. | ||
Recent investigations have shown that a fraction of dry ceria (110) surfaces, which have a relatively high surface energy (1.00 J m−2), can reconstruct to yield indent (111) nanofacets with a lower surface energy (0.88 J m−2).19,35 To investigate the presence of (111) nanofacets at the (110) surfaces of the nanorod samples, DFT simulations were conducted on two models with and without reconstruction (Fig. 2b, S5–S9, Tables S2 and S3†). For the dry (110) surface, a single shift (δCG) of 1052 ppm for 1st layer O3C is predicted (Table S2 and Fig. S9†); although a non-zero 17O spectral intensity is observed at this frequency, the dry (110) surface alone is not sufficient to explain the broad resonance centered at ∼1020 ppm. Including the reconstructed (110) surface yields further O3CδCGs at 1033 and 1008 ppm (Fig. S9† and 3a), explaining the broad experimental signal, especially considering that the presence of any surface defects and/or vacancies will cause further broadening.29 This provides further experimental evidence for the reconstruction of the (110) surface. In addition, the absence of signal at 920 ppm in both the experimental spectrum of NRs300 (Fig. 2) and the simulated spectrum based on DFT calculations (Fig. S9†) shows that the (111) nanofacets reconstructed from (110) facets can be distinguished from the common (111) surfaces. The simulated δCGs for O4Cs in the sub-surface layers and the bulk of both models are between 898 and 800 ppm, in accordance with the remaining intensity observed in this region (Fig. 2).
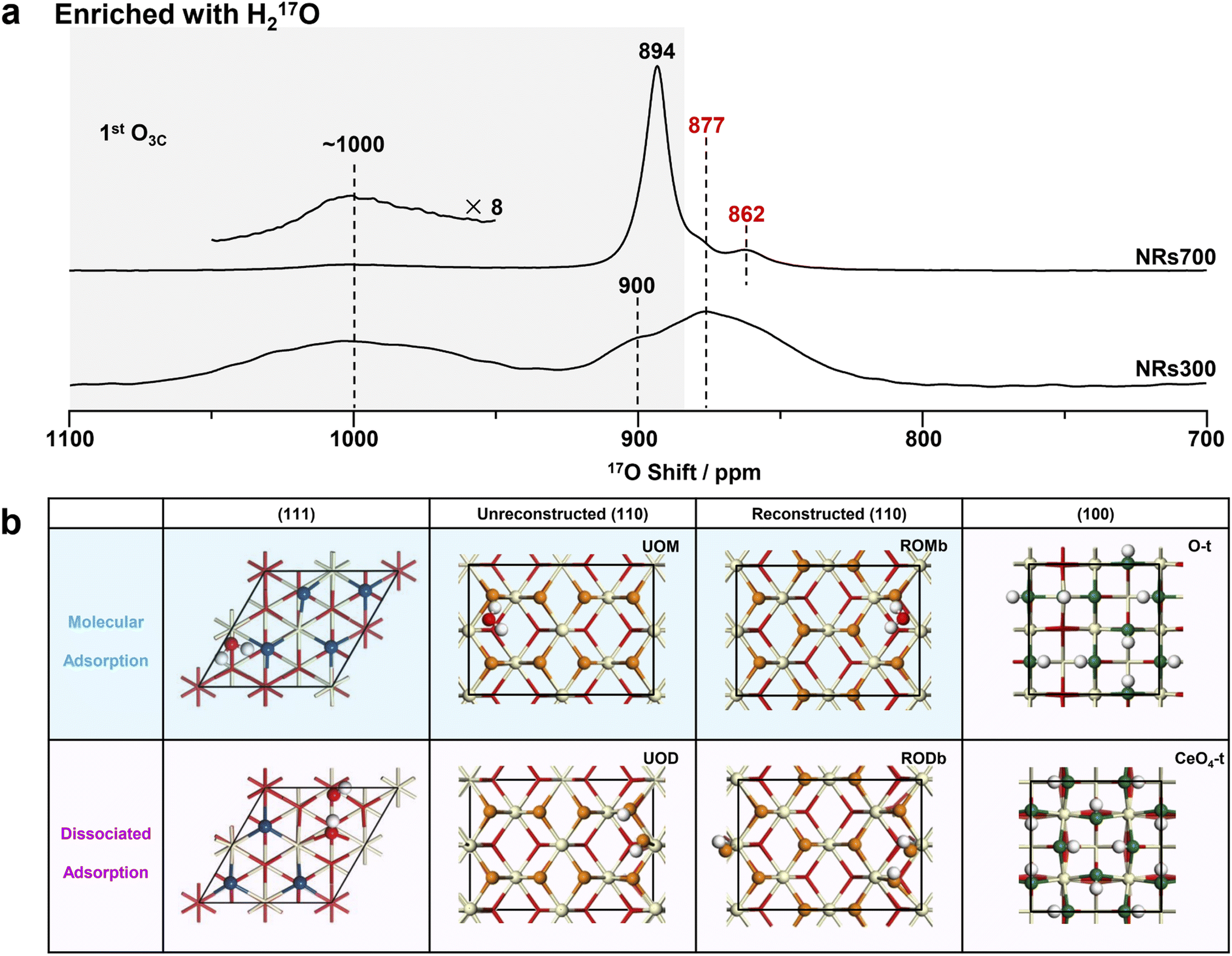 | ||
| Fig. 3 17O NMR spectra of NRs700 and NRs300 and the hydrated structure models of ceria (111), (110) and (100) surfaces. (a) 17O solid-state NMR spectra of the H217O-enriched ceria NRs700 and NRs300 (with hydrated surfaces). The conditions under which these spectra were collected were consistent with the two in Fig. 2a. (b) The calculated structures of hydrated ceria (111), (110) and (100) surfaces. Red, off-white and white spheres denote bulk oxygen, hydrogen and cerium ions, respectively. 1st layer oxygen ions with variable chemical shifts are marked in different colors. O-t and CeO4-t are O-terminated and CeO4-t ceria (100) surfaces, respectively.18 | ||
17O DNP NMR spectroscopy
Direct dynamic nuclear polarization (DNP) is another advanced approach to selectively observe the surface 17O NMR signals of oxides.36–39 The sample is wetted with a solution of organic radicals, cooled to 100 K, and irradiated with microwaves in order to transfer polarization from the unpaired electrons and enhance the 17O NMR signals of oxygen ions at or near the surface. The direct DNP 17O NMR spectra of 17O2-labeled NRs700 and NRs300 (Fig. S10†) exhibit an order-of-magnitude signal enhancement and similar signals to the conventional spectra of surface-selectively labelled samples, but suffer from poor resolution due to increased signal broadening at 100 K and the overlap of spinning sidebands at the higher field and lower magic angle spinning (MAS) rate. On the other hand, the conventional 17O NMR spectra of surface-selectively enriched ceria nanocrystals show much improved resolution with relatively good signal-to-noise ratios, which should represent a better approach for obtaining surface structural information. These results are in agreement with our previous studies on the optimal strategy for surface investigations.30Spectral assignments from DFT calculations
Compared to 17O-enrichment with 17O2 gas, enrichment with H217O is more surface-selective and results in highly hydrated surfaces.29,30 The 17O NMR spectra of ceria NRs700 and NRs300 enriched with H217O were therefore recorded to study the structure of hydrous ceria surfaces (Fig. 3a). The resonance at 877 ppm arising from bulk O4Cs exhibits a low intensity in both spectra, indicating a low degree of labeling for bulk oxygen ions. Other resonances exhibit notable distinctions from those in Fig. 2a. The major signal for NRs700 occurs at 894 ppm, which is due to the 1st and 2nd layer oxygen ions of hydrated ceria (111) surfaces, as previously determined from DFT calculations (Fig. 3b).30 Another previously unobserved resonance at 862 ppm is ascribed to the 3rd layer O4Cs in hydrated (111) surfaces on the basis of DFT calculations, which may not have been effectively labelled with shorter thermal treatment times in the previous study.30 The broad peak at ∼1000 ppm in the spectrum of NRs300, and to a lesser extent NRs700, may be assigned to the surface oxygen ions in hydrated (110) surfaces according to previous studies on ceria (100)18 and (111)30 surfaces, given that NRs300 is dominated by (110) and (100) surfaces, and also overlaps with the ∼1012 ppm resonance arising from hydrous (100) surfaces.18The structures of hydrated (111) and (100) surfaces have previously been revealed by combining DFT calculations and NMR,18,30 however hydrated ceria (110) surfaces have not been analyzed; DFT simulations were therefore performed here. Quantitative 1H NMR (Fig. S11, Table S4 and Note S1†) allows the concentration of hydrogen at the surfaces to be calculated (as surface hydroxyl groups and/or molecularly adsorbed water molecules), giving an estimated coverage of 1.5 water molecules per surface unit for the (110) facets (83.9 Å2, shown in Fig. 2b and Table S4†). We then performed DFT simulations of the 17O shifts (δCGs) and adsorption energy (Eads) with one water molecule per surface unit of hydrated ceria (110) surfaces.
Ten different models were considered: unreconstructed O-terminated models with dissociative and molecular water adsorption (UOD and UOM, respectively), four different reconstructed models with molecular adsorption (ROMa–ROMd), and four reconstructed models with dissociative adsorption (RODa–RODd). The water adsorption energies, Eads, are summarized in Fig. S12.† On the basis of energetic favorability (more negative Eads), models UOD, UOM, RODb and ROMb are the best representations of the structure of the hydrated (110) surface, therefore the 17O shifts were calculated (Fig. S13–S24 and Tables S5–S8†). The broadness of the resonance at ∼1000 ppm for H217O labeled NRs300 arises from the overlap of the different environments for O3Cs in ceria (110) surfaces represented in these models (Fig. 4, S25 and Tables S5–S8†), and further includes the shift at 996 ppm due to the 1st layer O3Cs in hydrated (100) surfaces.18 The shoulder at 900 ppm in the experimental spectrum also arises from 1st layer O3C ions in unreconstructed ceria (110) O-t surfaces with molecularly adsorbed water (UOM) (Fig. 4, S17, S18 and Table S6†). Fig. 3b summarizes the models for the (110) surface and the previously determined models for (111)30 and (100)18 hydrated surfaces. The calculated 17O shifts (δCGs) of the 1st layer O3Cs in all these models are summarized in Fig. 4.
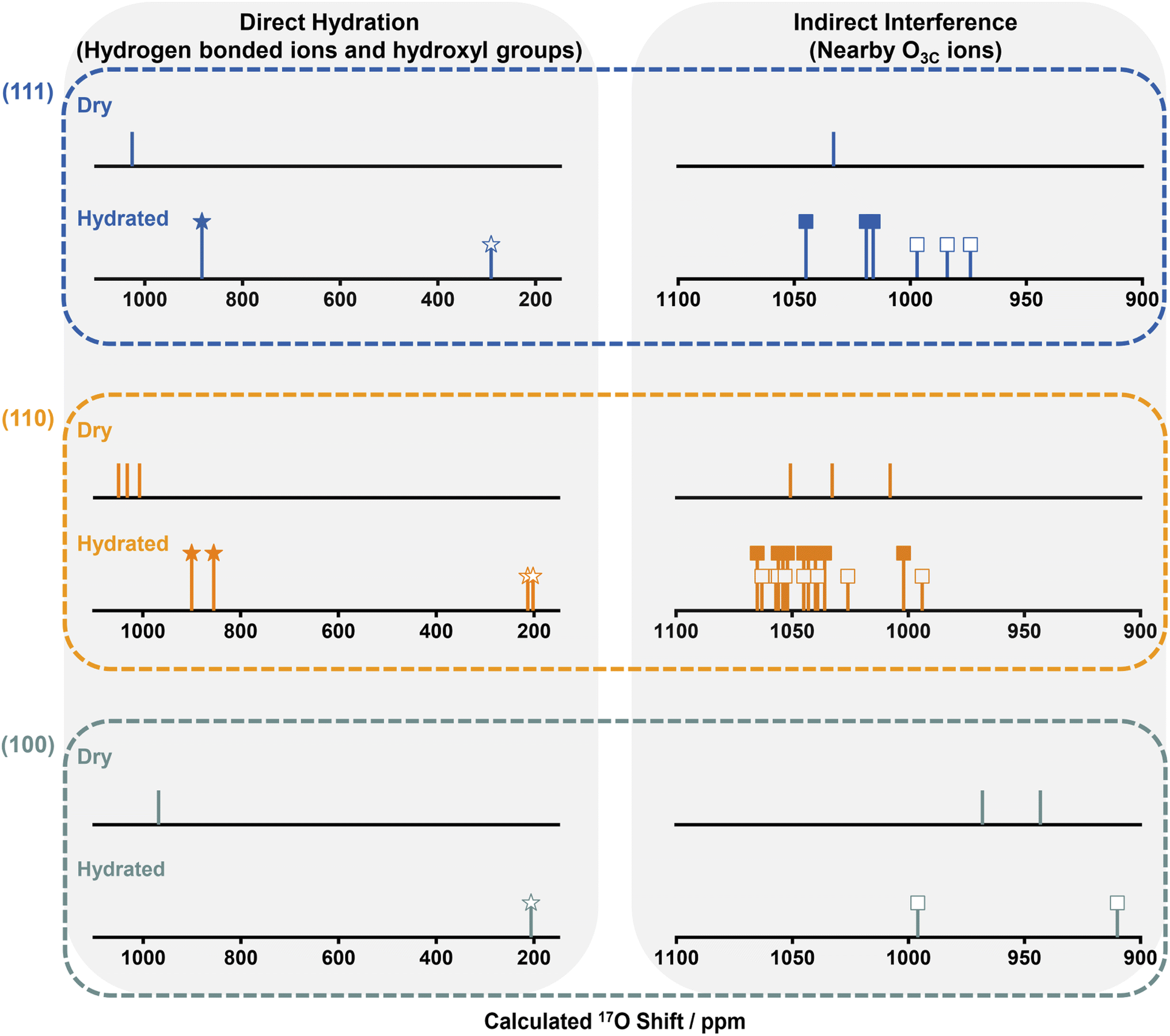 | ||
| Fig. 4 Summarized 17O NMR shifts (δCGs) predicted for the 1st layer oxygen ions in dry and hydrated ceria (111), (110) and (100) surfaces. At hydrous conditions, 17O shifts of 1st layer oxygen ions interacting directly and indirectly with water molecules are denoted by stars and squares, respectively; while hollow and solid shapes represent the 17O shifts of the 1st layer oxygen ions associated with dissociative and molecular water molecules, respectively. | ||
We now compare the surface structures of ceria low-index surfaces ((111), (110) and (100)) at dry and hydrated conditions (Fig. 4 and Table S9†). For all three facets, the molecular or dissociative adsorption of water results in distinctive changes to the 17O shifts for oxygen species on the surface. The (111) surface comprises O3Cs in the 1st layer and O4Cs in the sub-surface and bulk; the (110) surface is partially reconstructed into indent (111) nanofacets, with both reconstructed and unreconstructed surfaces comprising exposed O3Cs and inner O4Cs; the (100) surface consists of reconstructed oxygen-terminated (O-t) and CeO4-terminated (CeO4-t) surfaces.18 Water molecules are adsorbed molecularly and dissociatively on the (111) and (110) surfaces, whereas only dissociative adsorption is observed for (100) surfaces, due to the presence of lower-coordinated oxygen ions.18 The adsorbed water molecules in turn affect the coordinated 1st layer oxygen ions, i.e., transform 1st layer O3Cs into surface hydroxyl groups by dissociation and coordinate with 1st layer O3Cs via hydrogen bonding on molecular adsorption; the 17O shifts of these surface oxygen ions that are affected directly by the water molecule are shown on the left of Fig. 4. More interestingly, hydration also slightly perturbs the chemical environment of the adjacent O3Cs (all of the exposed 1st layer O3Cs shown in Fig. 3b) that are not directly bonded to water molecules, generating 17O NMR shifts in between those of dry surfaces and the directly hydrated surface oxygens; these are shown on the right of Fig. 4. The observation that interactions between exogenously coordinated water and the surfaces can modify the chemical environment of both the immediately coordinated and surrounding ions suggests that not only the surface geometry but the impact of exogenous water should be carefully considered when investigating the “true” chemical environment of oxide surfaces.
CO adsorption on the pristine and hydrated (1/4 ML) ceria (111) surfaces were modeled and the adsorption energies were calculated to explore the effects of adsorbed H2O (Fig. S26†). It is clear that a hydrous surface can lead to a slightly stronger CO adsorption on the bare oxygen sites, and the change might affect reactions involving CO with the presence of H2O (Fig. S27 and Table S10†), which has been observed previously.40,41 Comparing the three bare oxygen sites, the calculated CO adsorption energies suggest that the oxygen ions associated with a smaller 17O chemical shift are inclined to have stronger adsorption (Fig. S28 and Table S10†). Therefore, the 17O NMR shift might be correlated to gas adsorption capability and thus the catalytic activity of a specific oxygen site.
Conclusions
The ceria (111), (110) and (100) facet structures, considering both geometry and the influence of external coordinated water molecules, were investigated with 17O solid-state NMR in combination with DFT calculations. Tri-coordinated oxygen ions in the 1st layer of different dry ceria facets exhibit notable differences in their 17O NMR shifts, resulting in a characteristic “spectral fingerprint” corresponding to the different surface geometries. The spectral signatures change under hydrous conditions, indicating that the chemical environment of the oxygen ions in hydrated oxide surfaces depends not only on the original surface geometry, but also on the interaction with adsorbed water molecules. Thus, the exogenously coordinated water is important and its interaction with the surface is a non-negligible factor when studying oxide surfaces, especially for oxide surfaces with complex structures (e.g., ceria (110) and (100) surfaces exhibiting reconstructions) which may result in multiple adsorption sites and modes. The extension of this work to study the ‘true’ surface chemical environment and identify which oxygen species are involved under specific conditions (e.g., temperature, pressure, and adsorption of guest species) can be envisaged. The insights obtained from this work are expected to help to further the understanding of chemical processes at oxide surfaces.Experimental section
Preparation of ceria NRs700 and NRs300
0.98 g Ce(NO3)3·6H2O (Sigma-Aldrich, 99.9%) was dissolved in 20 mL distilled water. Then 15 mL NaOH (pH = 14) solution was quickly added and stirred for 30 minutes. The prepared solution was transferred to two 50 mL polytetrafluoroethylene hydrothermal reactors. After heating at 100 °C for one day, yellow precipitates were obtained. After washing with distilled water six times, the precipitates were dried at 80 °C overnight. The yellow powders were calcined at 700 and 300 °C for 5 hours, respectively. The two samples are denoted NRs700 (nanorods and calcined at 700 °C) and NRs300 (nanorods and calcined at 300 °C).17O isotopic labeling
150 mg sample was added into a glass tube and evacuated at 300 °C for 2 hours. After cooling down to room temperature, 90% 17O-enriched O2 gas/H2O vapor was introduced into the tube and then heated at 300 °C for 48 hours for 17O2 labeling and room temperature for 3 hours for H217O enrichment. The sample was re-evacuated and this process repeated (filling and evacuation) for four times to obtain highly 17O labeled samples. After the final enrichment, the sample was re-evacuated at room temperature to remove residual 17O2/H217O.17O solid-state NMR spectroscopy
17O magic angle spinning nuclear magnetic resonance (MAS NMR) spectra were measured on a 9.4 T Bruker Avance III 400 spectrometer, corresponding to a 17O Larmor frequency of 54.2 MHz. Samples were packed into 4 mm outer diameter ZrO2 rotors in a N2-filled glove box. 17O shifts were referenced to H2O at 0.0 ppm.17O direct DNP NMR experiments were performed on a 14.1 T Bruker Avance III HD 600 spectrometer equipped with a 395 GHz gyrotron microwave source and a 3.2 mm MAS probe at a Larmor frequency of 81.3 MHz. Samples were mixed with radical solution (16 mM TEKPol42 in dried tetrachloroethane, TCE) in an Ar-filled glove box. 17O chemical shifts of the DNP NMR spectra were referenced to bulk ceria at 875 ppm at 100 K.
Data availability
Requests for materials and data can be addressed to Luming Peng.Author contributions
J. Chen, X.-P. Wu, M. A. Hope, C. P. Grey and L. Peng conceived and developed the idea of the project. J. Chen and Z. Lin carried out the synthesis of ceria nanorods. J. Chen, L. Zhu, Y. Wen, Y. Zhang, T. Qin, T. Liu, X. Xia, D. Wu, and X. Liu carried out XRD, HRTEM, XPS, Raman, Na and N element analysis, and surface area measurement. J. Chen, Z. Lin, Y. Wen, and L. Peng performed 17O isotope enrichment, collected and analysed the NMR spectra. J. Chen, M. A. Hope, and C. P. Grey collected and analysed the high field 17O DNP NMR spectra; X.-P. Wu, J. Wang, and X.-Q. Gong conducted the DFT calculations. J. Chen, X.-P. Wu, M. A. Hope, W. Tang, W. Ding, X.-Q. Gong, L. Chen, and L. Peng wrote the manuscript and all authors discussed the experiments and final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Key R&D Program of China (2021YFA1502803), the National Natural Science Foundation of China (NSFC) (21972066, 91745202, 21573103, 22003016 and 21421004), NSFC – Royal Society Joint Program (21661130149), the Fundamental Research Funds for the Central Universities (1124020512) and National Science Fund for Talent Training in Basic Science (J1103310). Junchao Chen would like to thank Dr Xingmin Zhang at Shanghai Institute of Applied Physics for his invaluable discussions and help in this work. The ECUST group thanks the Programme of Introducing Talents of Discipline to Universities (B16017). Luming Peng thanks the Royal Society and Newton Fund for a Royal Society – Newton Advanced Fellowship. Clare P. Grey thanks the European Research Council for an Advanced Fellowship. This work was also supported by the Research Funds for the Frontiers Science Center for Critical Earth Material Cycling, Nanjing University, and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.References
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zhou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- W. Huang, Acc. Chem. Res., 2016, 49, 520–527 CrossRef CAS.
- X. Xie, Y. Li, Z.-Q. Liu, M. Haruta and W. Shen, Nature, 2009, 458, 746–749 CrossRef CAS PubMed.
- Y. Li and W. Shen, Chem. Soc. Rev., 2014, 43, 1543–1574 RSC.
- Z. Zhang, S.-S. Wang, R. Song, T. Cao, L. Luo, X. Chen, Y. Gao, J. Lu, W.-X. Li and W. Huang, Nat. Commun., 2017, 8, 488 CrossRef PubMed.
- W.-C. Huang, L.-M. Lyu, Y.-C. Yang and M. H. Huang, J. Am. Chem. Soc., 2012, 134, 1261–1267 CrossRef CAS.
- T. Tachikawa, S. Yamashita and T. Majima, J. Am. Chem. Soc., 2011, 133, 7197–7204 CrossRef CAS PubMed.
- Y. Bi, S. Ouyang, N. Umezawa, J. Cao and J. Ye, J. Am. Chem. Soc., 2011, 133, 6490–6492 CrossRef CAS PubMed.
- H. Wang, K. Dou, W. Y. Teoh, Y. Zhan, T. F. Hung, F. Zhang, J. Xu, R. Zhang and A. L. Rogach, Adv. Funct. Mater., 2013, 23, 4847–4853 CAS.
- F. Wang, X. Wang, Z. Chang, Y. Zhu, L. Fu, X. Liu and Y. Wu, Nanoscale Horiz., 2016, 1, 272–289 RSC.
- B. Ren, Y. Wang and J. Z. Ou, J. Mater. Chem. B, 2020, 8, 1108–1127 RSC.
- M. Liu, L. Piao, L. Zhao, S. Ju, Z. Yan, T. He, C. Zhou and W. Wang, Chem. Commun., 2010, 46, 1664–1666 RSC.
- X.-L. Cheng, J.-S. Jiang, D.-M. Jiang and Z. J. Zhao, J. Phys. Chem. C, 2014, 118, 12588–12598 CrossRef CAS.
- P. L. Hansen, J. B. Wagner, S. Helveg, J. R. Rostrup-Nielsen, B. S. Clausen and H. Topsøe, Science, 2002, 295, 2053–2055 CrossRef CAS PubMed.
- T. W. Hansen, J. B. Wagner, P. L. Hansen, S. Dahl, H. Topsøe and C. J. H. Jacobsen, Science, 2001, 294, 1508–1510 CrossRef CAS.
- L. A. Kibler, A. Cuesta, M. Kleinert and D. M. Kolb, J. Electroanal. Chem., 2000, 484, 73–82 CrossRef CAS.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Science, 2005, 309, 752–755 CrossRef CAS.
- J. Chen, X.-P. Wu, M. A. Hope, K. Qian, D. M. Halat, T. Liu, Y. Li, L. Shen, X. Ke, Y. Wen, J.-H. Du, P. C. M. M. Magusin, S. Paul, W. Ding, X.-Q. Gong, C. P. Grey and L. Peng, Nat. Commun., 2019, 10, 5420 CrossRef.
- C. Yang, X. Yu, S. Heißler, A. Nefedov, S. Colussi, J. Llorca, A. Trovarelli, Y. Wang and C. Wöll, Angew. Chem., Int. Ed., 2017, 56, 375–379 CrossRef CAS PubMed.
- A. Vimont, F. Thibault-Starzyk and M. Daturi, Chem. Soc. Rev., 2010, 39, 4928–4950 RSC.
- W. Zhang, Z. Lin, H. Li, F. Wang, Y. Wen, M. Xu, Y. Wang, X. Ke, X. Xia, J. Chen and L. Peng, RSC Adv., 2021, 11, 25004–25009 RSC.
- Y. Hu, B. Guo, Y. Fu, Y. Ren, G. Tang, X. Chen, B. Yue and H. He, Chem. Commun., 2015, 51, 14219–14222 RSC.
- Y.-K. Peng, L. Ye, J. Qu, L. Zhang, Y. Fu, I. F. Teixeira, I. J. McPherson, H. He and S. C. E. Tsang, J. Am. Chem. Soc., 2016, 138, 2225–2234 CrossRef CAS PubMed.
- M. A. Henderson, Surf. Sci. Rep., 2002, 46, 1–308 CrossRef CAS.
- G. E. Brown, V. E. Henrich, W. H. Casey, D. L. Clark, C. Eggleston, A. Felmy, D. W. Goodman, M. Grätzel, G. Maciel, M. I. McCarthy, K. H. Nealson, D. A. Sverjensky, M. F. Toney and J. M. Zachara, Chem. Rev., 1999, 99, 77–174 CrossRef CAS PubMed.
- R. Mu, Z.-J. Zhao, Z. Dohnálek and J. Gong, Chem. Soc. Rev., 2017, 46, 1785–1806 RSC.
- A. Verdaguer, G. M. Sacha, H. Bluhm and M. Salmeron, Chem. Rev., 2006, 106, 1478–1510 CrossRef CAS PubMed.
- Y. Li, X.-P. Wu, N. Jiang, M. Lin, L. Shen, H. Sun, Y. Wang, M. Wang, X. Ke, Z. Yu, F. Gao, L. Dong, X. Guo, W. Hou, W. Ding, X.-Q. Gong, C. P. Grey and L. Peng, Nat. Commun., 2017, 8, 581 CrossRef PubMed.
- M. Wang, X.-P. Wu, S. Zheng, L. Zhao, L. Li, L. Shen, Y. Gao, N. Xue, X. Guo, W. Huang, Z. Gan, F. Blanc, Z. Yu, X. Ke, W. Ding, X.-Q. Gong, C. P. Grey and L. Peng, Sci. Adv., 2015, 1, e1400133 CrossRef PubMed.
- J. Chen, M. A. Hope, Z. Lin, M. Wang, T. Liu, D. M. Halat, Y. Wen, T. Chen, X. Ke, P. C. M. M. Magusin, W. Ding, X. Xia, X.-P. Wu, X.-Q. Gong, C. P. Grey and L. Peng, J. Am. Chem. Soc., 2020, 142, 11173–11182 CrossRef CAS.
- S. Carrettin, P. Concepción, A. Corma, J. M. L. Nieto and V. F. Puntes, Angew. Chem., Int. Ed., 2004, 43, 2538–2540 CrossRef CAS PubMed.
- J. Kašpar, P. Fornasiero and M. Graziani, Catal. Today, 1999, 50, 285–298 CrossRef.
- J. A. Rodriguez, S. Ma, P. Liu, J. Hrbek, J. Evans and M. Pérez, Science, 2007, 318, 1757–1760 CrossRef CAS PubMed.
- Y. Gao, R. Li, S. Chen, L. Luo, T. Cao and W. Huang, Phys. Chem. Chem. Phys., 2015, 17, 31862–31871 RSC.
- J. Zhang, X.-Q. Gong and G. Lu, Surf. Sci., 2015, 632, 164–173 CrossRef CAS.
- M. A. Hope, D. M. Halat, P. C. M. M. Magusin, S. Paul, L. Peng and C. P. Grey, Chem. Commun., 2017, 53, 2142–2145 RSC.
- Q. Z. Ni, E. Daviso, T. V. Can, E. Markhasin, S. K. Jawla, T. M. Swager, R. J. Temkin, J. Herzfeld and R. G. Griffin, Acc. Chem. Res., 2013, 46, 1933–1941 CrossRef CAS PubMed.
- F. Blanc, L. Sperrin, D. A. Jefferson, S. Pawsey, M. Rosay and C. P. Grey, J. Am. Chem. Soc., 2013, 135, 2975–2978 CrossRef CAS PubMed.
- P. Berruyer, L. Emsley and A. Lesage, eMagRes, 2018, 7, 93–104 CAS.
- T. Yan, J. Gong, D. W. Flaherty and C. B. Mullins, J. Phys. Chem. C, 2011, 115, 2057–2065 CrossRef CAS.
- J. T. Calla and R. J. Davis, Ind. Eng. Chem. Res., 2005, 44, 5403–5410 CrossRef CAS.
- A. Zagdoun, G. Casano, O. Ouari, M. Schwarzwälder, A. J. Rossini, F. Aussenac, M. Yulikov, G. Jeschke, C. Copéret, A. Lesage, P. Tordo and L. Emsley, J. Am. Chem. Soc., 2013, 135, 12790–12797 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03885d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |