
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Open-air green-light-driven ATRP enabled by dual photoredox/copper catalysis†
Grzegorz
Szczepaniak
 *ab,
Jaepil
Jeong‡
a,
Kriti
Kapil‡
a,
Sajjad
Dadashi-Silab
a,
Saigopalakrishna S.
Yerneni
c,
Paulina
Ratajczyk
ad,
Sushil
Lathwal
a,
Dirk J.
Schild
a,
Subha R.
Das
ae and
Krzysztof
Matyjaszewski
*a
*ab,
Jaepil
Jeong‡
a,
Kriti
Kapil‡
a,
Sajjad
Dadashi-Silab
a,
Saigopalakrishna S.
Yerneni
c,
Paulina
Ratajczyk
ad,
Sushil
Lathwal
a,
Dirk J.
Schild
a,
Subha R.
Das
ae and
Krzysztof
Matyjaszewski
*a
aDepartment of Chemistry, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, USA. E-mail: gszczepa@andrew.cmu.edu; matyjaszewski@cmu.edu
bFaculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland
cDepartment of Biomedical Engineering, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, USA
dFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland
eCenter for Nucleic Acids Science & Technology, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, USA
First published on 20th September 2022
Abstract
Photoinduced atom transfer radical polymerization (photo-ATRP) has risen to the forefront of modern polymer chemistry as a powerful tool giving access to well-defined materials with complex architecture. However, most photo-ATRP systems can only generate radicals under biocidal UV light and are oxygen-sensitive, hindering their practical use in the synthesis of polymer biohybrids. Herein, inspired by the photoinduced electron transfer-reversible addition–fragmentation chain transfer (PET-RAFT) polymerization, we demonstrate a dual photoredox/copper catalysis that allows open-air ATRP under green light irradiation. Eosin Y was used as an organic photoredox catalyst (PC) in combination with a copper complex (X–CuII/L). The role of PC was to trigger and drive the polymerization, while X–CuII/L acted as a deactivator, providing a well-controlled polymerization. The excited PC was oxidatively quenched by X–CuII/L, generating CuI/L activator and PC˙+. The ATRP ligand (L) used in excess then reduced the PC˙+, closing the photocatalytic cycle. The continuous reduction of X–CuII/L back to CuI/L by excited PC provided high oxygen tolerance. As a result, a well-controlled and rapid ATRP could proceed even in an open vessel despite continuous oxygen diffusion. This method allowed the synthesis of polymers with narrow molecular weight distributions and controlled molecular weights using Cu catalyst and PC at ppm levels in both aqueous and organic media. A detailed comparison of photo-ATRP with PET-RAFT polymerization revealed the superiority of dual photoredox/copper catalysis under biologically relevant conditions. The kinetic studies and fluorescence measurements indicated that in the absence of the X–CuII/L complex, green light irradiation caused faster photobleaching of eosin Y, leading to inhibition of PET-RAFT polymerization. Importantly, PET-RAFT polymerizations showed significantly higher dispersity values (1.14 ≤ Đ ≤ 4.01) in contrast to photo-ATRP (1.15 ≤ Đ ≤ 1.22) under identical conditions.
Introduction
Reversible-deactivation radical polymerization (RDRP) has been recognized by IUPAC as one of the most important emerging technologies in chemistry that could change our world.1 The key RDRP techniques are reversible addition fragmentation chain-transfer (RAFT) polymerization and atom transfer radical polymerization (ATRP).2–4 Both of these methods allow the polymerization of various vinyl monomers under mild conditions, giving unprecedented control over polymer architecture.5–7ATRP is a reversible redox process, typically catalyzed by copper complexes.8,9 The control over radical propagation in Cu-catalyzed ATRP is provided via a reversible redox reaction (Fig. 1). First, the CuI/L catalyst (L is typically a polydentate amine ligand) reacts with the dormant C(sp3)–X polymer chain end through a concerted inner sphere electron transfer to form two species: a X–CuII/L deactivator and a propagating carbon-based radical. Then, in the reverse reaction, the propagating radical reacts with the X–CuII/L, recovering the active form of the catalyst (CuI/L) and the dormant chain end (C(sp3)–X).10–12 A limitation of ATRP, like any other RDRP technique is its sensitivity to oxygen, as both initiating and propagating radicals are quenched by oxygen. Furthermore, molecular oxygen can oxidize an ATRP catalyst to its inactive form, inhibiting polymerization. Therefore, normal ATRP requires strictly anaerobic conditions.13 The sensitivity of ATRP techniques to oxygen hinders their use under ambient conditions and necessitates deoxygenation by inert gas purging, freeze–pump–thaw cycles, or using a glovebox. As a result, ATRP methods are time-consuming and can be challenging for non-experts.
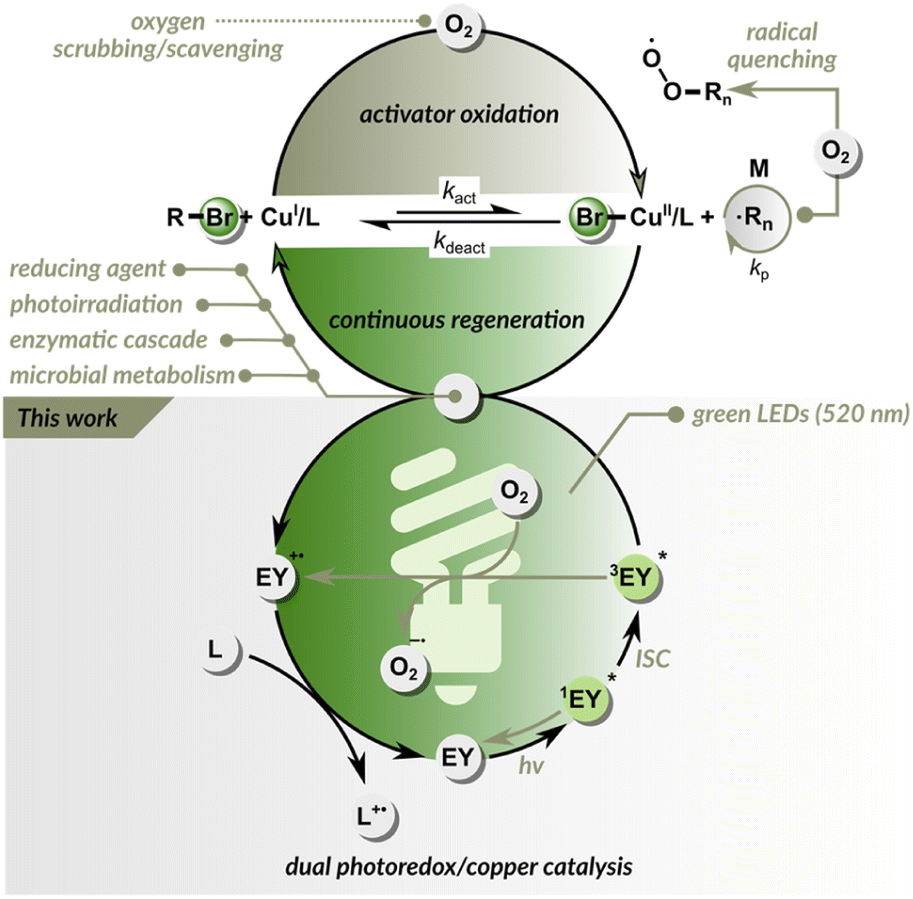 | ||
| Fig. 1 Approaches for attaining oxygen tolerance in ATRP. | ||
To address these challenges, many efforts have been made to increase oxygen tolerance in ATRP.14–16 Since, at the ATRP equilibrium, the concentration of CuI/L is much higher than the concentration of propagating radicals, oxygen predominantly oxidizes CuI/L rather than reacting with radicals. This inspired researchers to harness CuI/CuII catalysis to function also as an oxygen scavenger.17–22 The continuous reduction of CuII/L back to CuI/L makes the ATRP inherently resistant to oxygen (Fig. 1).14 The alternative approach is based on adding glucose oxidase enzyme that removes oxygen without affecting the polymerization process, as in enzyme-assisted ATRP techniques.23–26 Despite these great improvements, most modern ATRP techniques tolerate only a limited amount of oxygen and can be performed in sealed vessels.22,27–31 When an open reaction vessel is used, the regeneration of the catalyst is usually slower than oxygen diffusion into the reaction site. To date, only a few ATRP systems are fully oxygen-tolerant and can be run in open-air reaction vessels.32–37
Photoinduced ATRP (photo-ATRP) techniques use light energy to generate activator CuI/L species, thus initiating polymerization.38–44 They typically require the use of biocidal UV light (<400 nm), which can degrade proteins, damage DNA, or initiate unwanted side reactions.45 Organocatalyzed ATRP (O-ATRP) relies on direct activation of the dormant polymer chain end by electron transfer from the photocatalyst in an excited state.46–52 It allows polymerization over a broad visible light spectrum,53–56 but moderate control and limited scope of monomers hinder its practical application, particularly under biologically relevant conditions.57 Dual catalytic ATRP systems use ppm-level copper catalysts to attain a controlled polymerization process in the ground state and photoredox catalysts (PCs) to reduce deactivators via photoinduced electron transfer, maintaining radical propagation.58–65 Moreover, when a PC in the excited state has sufficient redox potential, it can react directly with a dormant C(sp3)–X polymer chain end, generating radicals and thus offering an additional activation pathway. These methods are highly efficient under long-wavelength light, but have limited oxygen tolerance.
Another powerful photochemically driven RDRP technique is the photoinduced electron transfer RAFT (PET-RAFT) polymerization method developed by Boyer and co-workers.66 In the PET-RAFT, an excited PC can transfer energy/electrons to a chain transfer agent (CTA), generating propagating radicals.67–69 As in the conventional RAFT process, control over polymerization is provided by a CTA via a degenerative chain transfer mechanism.3 PET-RAFT can be used to polymerize a wide range of monomers, is oxygen tolerant, and exhibits perfect temporal control. Initially, PET-RAFT was performed using tris(phenylpyridinium) iridium(III) catalyst.66 The method was later expanded to several other photoredox systems, allowing polymerization under longer wavelength light.70–76 Eosin Y is one of the most widely used photocatalysts,77–79 especially for biological applications, due to its solubility in water, low toxicity, and cost. PET-RAFT catalyzed by eosin Y has recently been used to engineer protein and cell surfaces.45,80
Here, we demonstrate a dual photoredox/copper catalysis that enables ATRP under green light irradiation (Fig. 1). Inspired by PET-RAFT polymerization, we used eosin Y as the PC. In this dual catalytic system, control over radical propagation is provided by ATRP equilibrium, while eosin Y is essential for triggering and maintaining polymerization. In addition, the dual photoredox/copper catalysis removes oxygen, enabling the rapid open-to-air synthesis of well-defined polymers in both aqueous and organic media, outperforming PET-RAFT under identical conditions.
Results and discussion
EY/Cu dual catalysis: optimization of ATRP conditions
A set of polymerizations was performed to evaluate the influence of the dual photoredox/copper catalysis on the ATRP process. Oligo(ethylene glycol) methyl ether methacrylate (average Mn = 500, OEOMA500) monomer was polymerized under green LEDs (520 nm, 9.0 mW cm−2), using 2-hydroxyethyl 2-bromoisobutyrate (HOBiB) as the initiator, eosin Y in neutral form (EYH2) as the organic photoredox catalyst and CuBr2/TPMA (TPMA = tris(2-pyridylmethyl)amine) as the precatalyst (Fig. 2). TPMA was used as a ligand since it forms a stable CuI/TPMA complex in water, enabling a well-controlled polymerization of methacrylates.81 The polymerizations were conducted in phosphate-buffered saline (PBS) with DMSO (10% v/v) in open vials (Fig. S1†).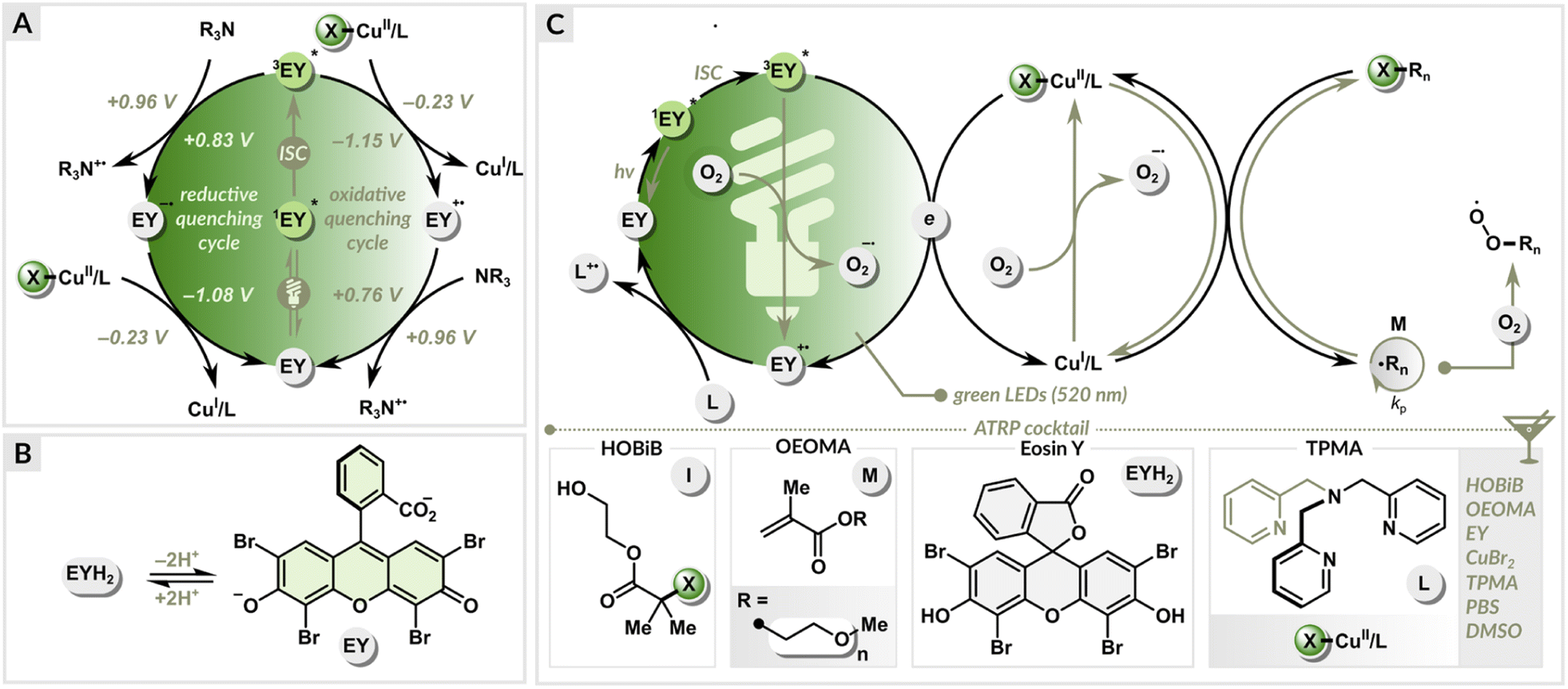 | ||
| Fig. 2 (A) Reductive quenching vs. oxidative quenching cycle, (B) formation of EY in PBS solution, (C) proposed mechanism. | ||
Photo-ATRP was first attempted using molar ratios of [OEOMA500]/[HOBiB]/[CuBr2]/[TPMA] = 200/1/0.2/0.6. As expected, no OEOMA500 conversion, as measured by 1H NMR, was observed in the absence of the photoredox catalyst (entry 1, Table 1). O-ATRP catalyzed with the EY/TPMA (without CuBr2) enabled rapid polymerization, reaching 89% monomer conversion within 30 min (entry 2, Table 1),56 but size exclusion chromatography (SEC) analysis showed that the polymer had a high dispersity (Đ) of 4.30. On the other hand, when EYH2 was used in combination with CuBr2 and excess TPMA ligand, high monomer conversion (88%) and well-controlled polymerization (Đ = 1.19) were achieved (entry 3, Table 1). These experiments confirmed the critical role of dual catalysis in ensuring a well-controlled polymerization under green light irradiation.
| No. | EYH2 (equiv) | CuBr2 (equiv) | TPMA (equiv) | rpmb | Conv.c (%) | M n,th | M n,abs | M n,app | Đ |
|---|---|---|---|---|---|---|---|---|---|
| a Reactions conditions: [OEOMA500]/[HOBiB]/[EYH2]/[CuBr2]/[TPMA] = 200/1/x/x/x, [OEOMA500] = 300 mM, in PBS with DMSO (10% v/v), irradiated for 30 min under green LEDs (520 nm, 9.0 mW cm−2) in an open vial with stirring at 0–1000 rpm. b Revolutions per minute (rpm). c Monomer conversion was determined by using 1H NMR spectroscopy. d Molecular weight (Mn,abs) was determined by Mark–Houwink calibration. e Apparent molecular weight (Mn,app) and dispersity (Đ) were determined by GPC analysis (DMF as eluent) calibrated to poly(methyl methacrylate) standards. | |||||||||
| 1 | — | 0.2 | 0.6 | 0 | 0 | — | — | — | — |
| 2 | 0.05 | — | 0.6 | 0 | 89 | 89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
187500 |
126000 |
4.30 |
| 3 | 0.05 | 0.2 | 0.6 | 0 | 88 | 88000 |
84000 |
65000 |
1.19 |
| 4 | 0.05 | 0.2 | 0.6 | 250 | 86 | 86000 |
82500 |
64000 |
1.18 |
| 5 | 0.05 | 0.2 | 0.6 | 500 | 86 | 86000 |
86000 |
66000 |
1.15 |
| 6 | 0.05 | 0.2 | 0.6 | 1000 | 86 | 86000 |
86000 |
66000 |
1.18 |
| 7 | 0.1 | 0.2 | 0.6 | 500 | 89 | 89000 |
89500 |
68500 |
1.20 |
| 8 | 0.01 | 0.2 | 0.6 | 500 | 84 | 84000 |
79500 |
62000 |
1.15 |
| 9 | 0.005 | 0.2 | 0.6 | 500 | 80 | 80000 |
73000 |
58000 |
1.16 |
| 10 | 0.01 | 0.3 | 0.6 | 500 | 74 | 74000 |
70000 |
56000 |
1.14 |
| 11 | 0.01 | 0.1 | 0.6 | 500 | 92 | 92000 |
90500 |
69000 |
1.21 |
| 12 | 0.01 | 0.2 | 0.4 | 500 | 77 | 77000 |
61000 |
50000 |
1.19 |
| 13 | 0.01 | 0.2 | 1.2 | 500 | 91 | 91000 |
90500 |
69000 |
1.16 |
To evaluate oxygen tolerance, the stirring rate was increased from 0 to 250, 500, and 1000 rpm (entries 4–6, Table 1). All polymerizations were successful, yielding well-defined polymers with low dispersity (Đ < 1.18) and controlled molecular weights, indicating that the EY/Cu system is highly tolerant to increased oxygen mass transfer.
The amount of EYH2 was then varied to explore the performance of the dual catalytic system (entries 7–9, Table 1). Increasing the EYH2 concentration from 75 μM to 150 μM resulted in 89% monomer conversion and slightly higher dispersity (Đ = 1.20; entry 7, Table 1). In contrast, a 5-fold decrease in EYH2 concentration to 15 μM caused only a slight decrease in conversion (84%) while improving control over the polymerization (Mn,th = 84000, Mn,abs = 79500, Đ = 1.15; entry 8, Table 1). After further reduction of EYH2 to 7.5 μM (25 ppm relative to the monomer), the dual EY/Cu catalysis still provided a well-controlled polymerization (Đ = 1.16) with predetermined molecular weight (Mn,th = 80000, Mn,abs = 73000) and high monomer conversion of 80% (entry 9, Table 1).
Finally, the effect of CuBr2 and TPMA ligand concentrations was investigated (entries 10–13, Table 1). Increasing the amount of copper diminished the polymerization rate while improving its control (conv. = 74%, Đ = 1.14; entry 10, Table 1). Increasing the ligand concentration led to higher monomer conversion (conv. = 91%) and dispersity of 1.16 (entry 13, Table 1).
Proposed mechanism
The neutral form of eosin Y with spirocyclic structure (EYH2) exhibits low absorbance in the visible region.82,83 In PBS solution, EYH2 undergoes sequential deprotonation leading to the photoactive ring-opened form (EY) (Fig. 2), which exhibits an absorption maximum at ∼520 nm. Under green light irradiation, EY in the excited triplet state (3EY*) can both accept (E1/2(3EY*/EY˙−) = +0.83 V vs. SCE) or donate an electron (E1/2(EY˙+/3EY*) = −1.15 V vs. SCE) (Fig. 2A).83 In the reductive quenching cycle, 3EY* is quenched by accepting an electron from an ATRP ligand (with a tertiary nitrogen atom), which acts as a sacrificial electron donor. This results in the formation of the EY radical anion (EY˙−) and an amine radical cation (L˙+). The formed EY˙− (E1/2(EY/EY˙−) = −1.08 V vs. SCE) then donates an electron to X–CuII/L, generating CuI/L activator and EY in the ground state, completing the photocatalytic cycle. In the oxidative quenching cycle, 3EY* is quenched by donating an electron to CuII/L, leading to the formation of EY˙+ and CuI/L. Finally, the photocatalytic cycle is closed by reducing the oxidized EY (E1/2(EY˙+/EY) = +0.76 V vs. SCE) with an alkylamine.The free energy of a photoinduced electron transfer can be determined using the Gibbs energy of photoinduced electron transfer equation:
| ΔGet (eV) = −[E1/2(A/A˙−) − E1/2(D˙+/D)] − EPC* + ΔE |
| ΔGet (eV) = −[E1/2(CuIIL/CuIL) − E1/2(EY˙+/EY)] − EEY* |
| ΔGet (eV) = −[E1/2(EY/EY˙−) − E1/2(L˙+/L)] − EEY* |
In the EY/Cu dual catalysis, both CuI/L and 3EY* can react with molecular oxygen (E1/2(O2/O2˙−) = −0.87 V vs. SCE). The continuous regeneration of the CuI/L activator by EY provides high oxygen tolerance (Fig. 2C). The control over radical propagation is achieved through a reversible redox equilibrium between Cu(I) and Cu(II) complexes, where they act as intermittent activators of dormant species and deactivators of radicals. EY and the ligand used in excess are essential to initiate and sustain green light-induced polymerization in the presence of oxygen.
Comparison of EY/Cu-catalyzed ATRP with PET-RAFT
Since EY/Cu-catalyzed ATRP is based on the same photoredox catalysis as PET-RAFT polymerization triggered by EY, we were interested in directly comparing the two methods. Such comparisons are rare, and to our knowledge have been reported only for conventional RAFT.2,85,86 Moreover, PET-RAFT polymerization is considered one of the most efficient biocompatible oxygen-tolerant polymerization techniques.4,45 Thus, a detailed comparison of the two methods is warranted.First, photo-ATRP was performed under optimized conditions ([OEOMA500]/[HOBiB]/[EYH2]/[CuBr2]/[TPMA] = 200/1/0.01/0.2/0.6) at a stirring rate of 500 rpm in an open vial. Kinetic analysis revealed a linear relationship between ln([M]0/[M]t) and time with a short induction period of 5 min, followed by a rapid polymerization that reached 91% monomer conversion within 40 min (Fig. 3A). In addition, the molecular weights increased as a function of monomer conversion, and molecular weight distribution values remained low (Đ ≤ 1.16) during the reaction (Fig. 3B and C).
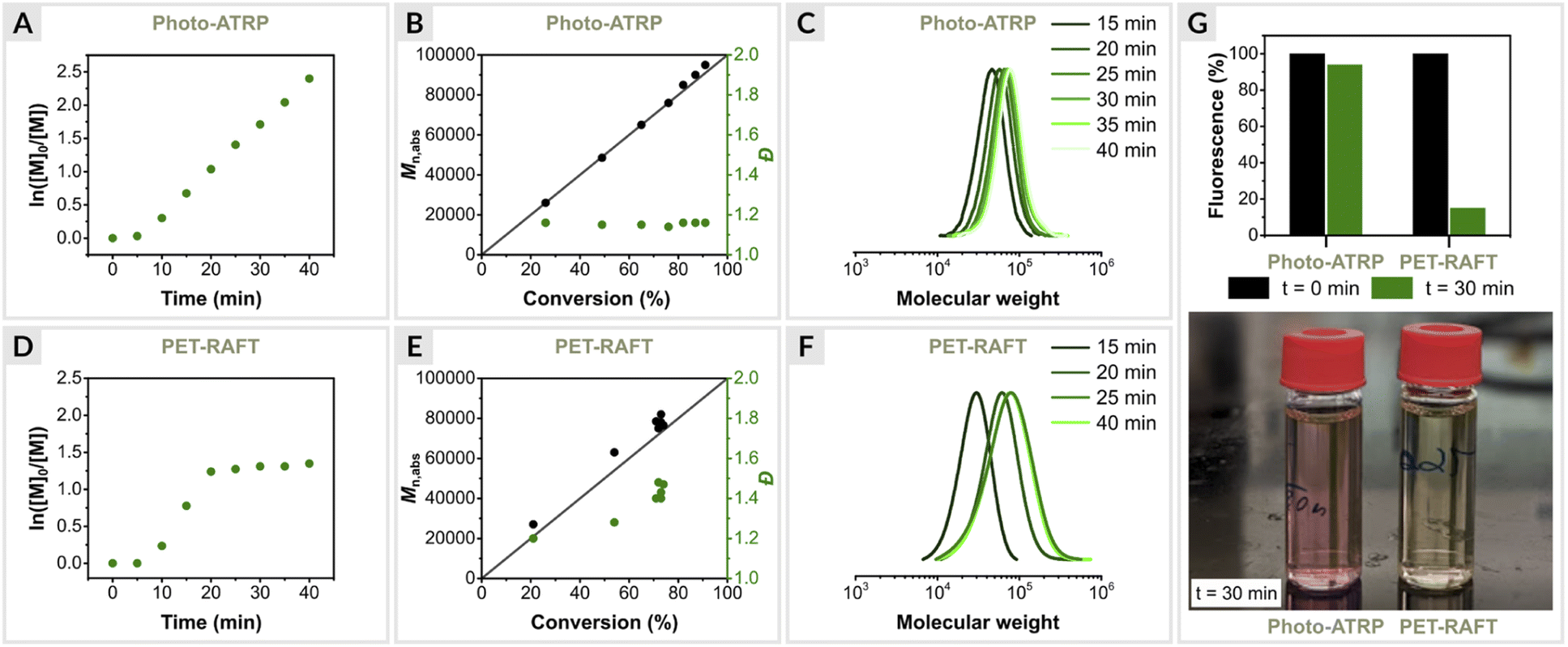 | ||
| Fig. 3 Polymerization kinetics of OEOMA500, (A–C) EY/Cu mediated photo-ATRP and (D–F) EY-catalyzed PET-RAFT polymerization. (G) EY fluorescence intensity measurements. | ||
Next, PET-RAFT polymerization of OEOMA500 was performed under identical conditions using 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPADB) as the CTA, EY as the photoredox catalyst and TEOA (TEOA = triethanolamine) as the sacrificial electron donor with [OEOMA500]/[CPADB]/[EYH2]/[TEOA] molar ratios of 200/1/0.01/0.6. Similar to photo-ATRP, a short induction period of 5 min was observed, followed by rapid polymerization, which reached 71% monomer conversion within 20 min (Fig. 3D). However, no further increase in conversion was observed over time. Fluorescence intensity measurements showed an 85% decrease in EY fluorescence after 30 min of PET-RAFT polymerization, while only 6% of the initial amount of EY was photobleached in photo-ATRP (Fig. 3G). The better performance of the EY/Cu dual catalysis can be attributed to the rapid electron transfer from 3EY* to CuII/L complex, increasing the long-term stability of EY under green light irradiation. In the absence of a CuII/L, EY degrades faster, leading to the inhibition of PET-RAFT polymerization. In addition, PET-RAFT polymerizations generated polymers with significantly higher dispersity values (Đ ≤ 1.45) (Fig. 3E and F), in contrast to photo-ATRP (Đ ≤ 1.16).
Varying targeted degrees of photo-ATRP and PET RAFT polymerization
The EY/Cu system was further evaluated for the synthesis of polymers with different molecular weights (Table 2). The target degrees of polymerization (DPT) were varied by adjusting the initiator concentration, while the concentrations of the other polymerization components were fixed for each reaction. The results showed a high degree of control for a wide targeted DP range (50–1000) (entries 1–7, Table 2 and Fig. 4A). The monomer conversions reached 69–84% within 30 min, and dispersities remained very low (1.15 ≤ Đ ≤ 1.22). However, deviations from the theoretical molecular weights were observed for DPT > 600 (entries 6 and 7, Table 2), which can be attributed to the presence of oxygen. For DPT = 800 and 1000, the initiator concentration (∼0.3 mM) was close to the oxygen concentration in the reaction mixture (∼0.2 mM). The kinetic experiments (Fig. 3A) and oxygen concentration measurements (see later section) suggest that the initiator reacts with oxygen in the initial phase of the reaction when the oxygen concentration in the reaction mixture is highest. This explains the increase in the molecular weight of the polymers obtained (entries 4–7, Table 2). Nevertheless, the polymerizations proceeded with excellent control (Đ ≤ 1.16), indicating that the loss of the initiator occurs mainly during the inhibition period, while during polymerization, oxygen is removed by EY/Cu catalysis.| No. | [OEOMA500]/[HOBiB]/[EYH2]/[CuBr2]/[TPMA] | [HOBiB] (mM) | Conv.b (%) | M n,th | M n,abs | M n,app | Đ |
|---|---|---|---|---|---|---|---|
| a Reaction conditions [OEOMA500] = 300 mM, [HOBiB] = 6.0–0.3 mM, [EYH2] = 15 μM, [CuBr2] = 0.3 mM, [TPMA] = 0.9 mM in PBS with DMSO (10% v/v), irradiated for 30 min under green LEDs (520 nm, 9.0 mW cm−2) in an open vial with stirring at 500 rpm. b Monomer conversion was determined by using 1H NMR spectroscopy. c Molecular weight (Mn,abs) was determined by Mark–Houwink calibration (see ESI). d Apparent molecular weight (Mn,app) and dispersity (Đ) were determined by GPC analysis (DMF as eluent) calibrated to poly(methyl methacrylate) standards. | |||||||
| 1 | 50/1/0.0025/0.05/0.15 | 6.0 | 74 | 18500 |
16500 |
17000 |
1.22 |
| 2 | 100/1/0.005/0.1/0.3 | 3.0 | 81 | 40500 |
38000 |
34000 |
1.16 |
| 3 | 200/1/0.01/0.2/0.6 | 1.5 | 84 | 84000 |
79500 |
62000 |
1.15 |
| 4 | 400/1/0.02/0.4/1.2 | 0.75 | 79 | 158000 |
179000 |
121000 |
1.16 |
| 5 | 600/1/0.03/0.6/1.8 | 0.5 | 73 | 219000 |
305000 |
188000 |
1.16 |
| 6 | 800/1/0.04/0.8/2.4 | 0.325 | 68 | 272000 |
483000 |
275000 |
1.15 |
| 7 | 1000/1/0.05/1.0/3.0 | 0.3 | 69 | 345000 |
607000 |
332000 |
1.15 |
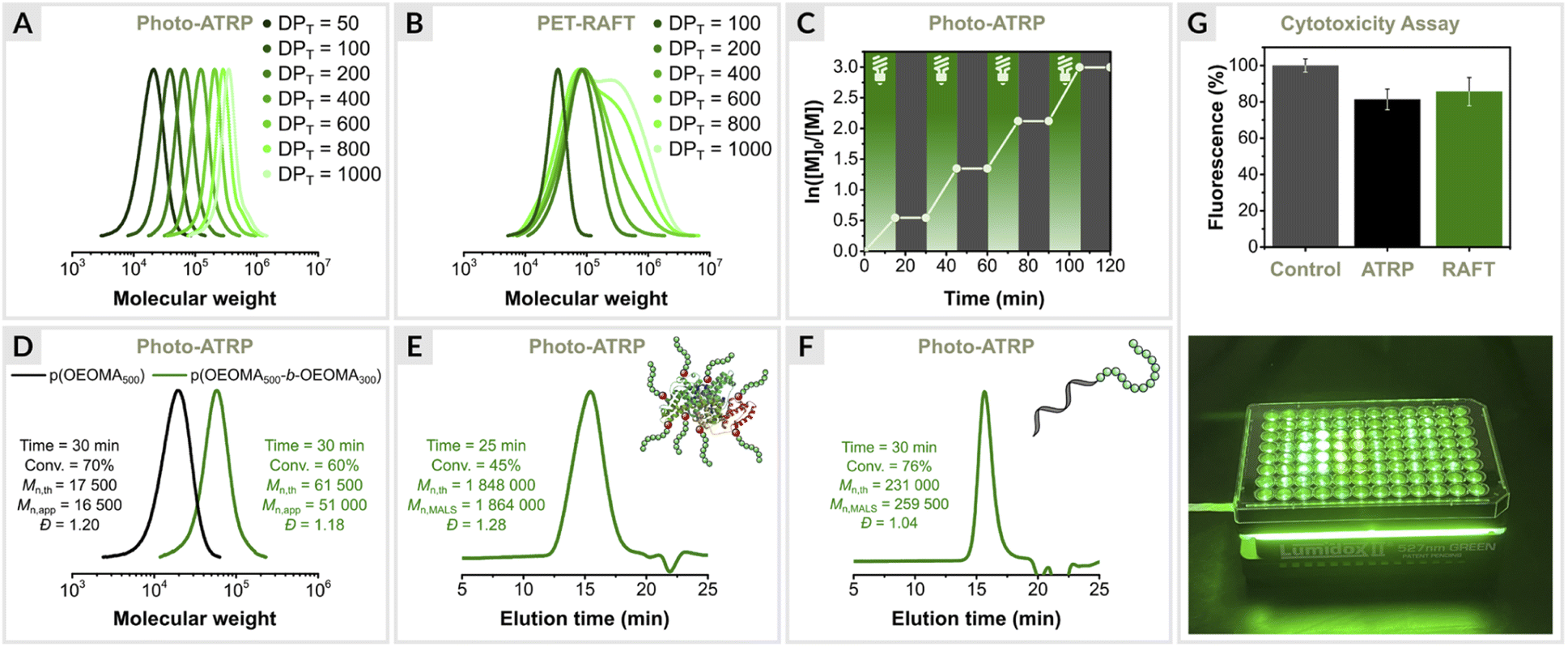 | ||
| Fig. 4 Varying targeted degrees of polymerization, (A) EY/Cu mediated photo-ATRP, (B) EY-catalyzed PET-RAFT polymerization. (C) Temporal control. (D) Chain extension. Synthesis of biohybrids, (E) BSA-p(OEOMA500), (F) DNA-p(OEOMA500). (G) Comparison of the cytocompatibility of photo-ATRP with PET-RAFT polymerization. | ||
PET-RAFT polymerizations were then performed under identical conditions (Table 3). The target DP was set by adjusting the CTA concentration, while the concentrations of the other reagents were fixed for all experiments. The results showed an increase in molecular weight distribution values with increasing DPT (entries 1–7, Table 3 and Fig. 4B). In addition, significant deviations from the theoretical molecular weights were observed. Only for DPT = 100, the polymerization was well controlled (Mn,abs = 32000, Đ = 1.14). The loss of control over polymerization could be attributed to a very low CTA concentration. Similar to photo-ATRP, a CTA can react with oxygen in the initial phase of the reaction, resulting in its degradation. In the case of RAFT, a CTA not only determines the initial target polymer chain length but is also responsible for the degenerative chain transfer process. In ATRP, control over polymerization is provided by a CuII/L deactivator, which is resistant to oxygen. In addition, Cu catalysis increases the tolerance of ATRP to oxygen since CuI/L species can scavenge oxygen. Therefore, in ATRP, deviations from theoretical molecular weights are observed for higher targeted DTT (entries 5–7, Table 2). At the same time, control over polymerization is not affected, demonstrating the superiority of EY/Cu dual catalysis. Interestingly, for the highest concentration of the CTA (6.0 mM, DPT = 50), no OEOMA500 conversion was observed (entry 1, Table 3). This could be explained by the insufficient loading of the photoredox catalyst. In contrast, under the same conditions, photo-ATRP reached 74% monomer conversion and low dispersity (Mn,abs = 16500, Đ = 1.22, entry 1, Table 1).
| No. | [OEOMA500]/[CPADB]/[EYH2]/[TEOA] | [CPADB] (mM) | Conv.b (%) | M n,th | M n,abs | M n,app | Đ |
|---|---|---|---|---|---|---|---|
| a Reaction conditions [OEOMA500] = 300 mM, [CPADB] = 6.0–0.3 mM, [EYH2] = 15 μM, [TEOA] = 0.9 mM in PBS with DMSO (10% v/v), irradiated for 30 min under green LEDs (520 nm, 9.0 mW cm−2) in an open vial with stirring at 500 rpm. b Monomer conversion was determined by using 1H NMR spectroscopy. c Molecular weight (Mn,abs) was determined by Mark–Houwink calibration (see ESI). d Apparent molecular weight (Mn,app) and dispersity (Đ) were determined by GPC analysis (DMF as eluent) calibrated to poly(methyl methacrylate) standard. | |||||||
| 1 | 50/1/0.0025/0.15 | 6.0 | 0 | — | — | — | — |
| 2 | 100/1/0.005/0.3 | 3.0 | 53 | 28000 |
32000 |
29500 |
1.14 |
| 3 | 200/1/0.01/0.6 | 1.5 | 75 | 75000 |
82500 |
64000 |
1.42 |
| 4 | 400/1/0.02/1.2 | 0.75 | 58 | 116000 |
98500 |
74000 |
1.68 |
| 5 | 600/1/0.03/1.8 | 0.5 | 50 | 150000 |
108000 |
80000 |
2.52 |
| 6 | 800/1/0.04/2.4 | 0.325 | 51 | 204000 |
98500 |
74000 |
3.63 |
| 7 | 1000/1/0.05/3.0 | 0.3 | 50 | 250000 |
115000 |
84000 |
4.01 |
Temporal control of green-light-driven ATRP
The EY/Cu dual catalytic system exhibited a high degree of temporal control, as demonstrated by switching the light on/off (Fig. 4C).87 Polymerization proceeded only under green light irradiation. No monomer conversion was observed after the light was turned off. Stirring accelerated the diffusion of oxygen into the reaction mixture, facilitating the oxidation of the activator (CuI/L) to its inactive form (CuII/L), while turning off the light prevented its regeneration. This explains the immediate inhibition of open-to-air ATRP when the light is turned off as the Cu(I) species react with oxygen, halting the activation and subsequent propagation process. A high degree of temporal control is usually achieved by using a copper catalyst at a very low concentration, but this results in much broader molar mass distributions.88,89Block copolymerization
To confirm chain-end fidelity, a chain extension experiment was performed (Fig. 4D). The macroinitiator p(OEOMA500) was synthesized with [OEOMA500]/[HOBiB]/[EYH2]/[CuBr2]/[TPMA] molar ratios of 50/1/0.0025/0.05/0.15 (conv. = 70%, Mn,app = 16500, Đ = 1.20). A sample was then taken from the post-polymerization mixture and used without further purification to prepare an ATRP “cocktail” with OEOMA300 monomer (DPT = 250). After 30 min of green light irradiation, the monomer conversion was 70%. SEC analysis showed a clear shift toward higher molecular weights without any shoulder or tailing at lower molecular weights (Mn,app = 51000, Đ = 1.18), indicating well-controlled polymerization and high retention of chain-end fidelity.
Low volume polymerization
Conducting RDRP in low volume opens avenues for many important applications,90–92 such as high-throughput combinatorial synthesis.93–97 However, the use of external deoxygenation methods on a small scale can lead to the loss of volatile substrates. Oxygen tolerance eliminates the need for degassing before polymerization, facilitating the synthesis of polymers in a small volume.To investigate the low volume performance of the EY/Cu system, a series of reactions at volumes of 250, 150, and 50 μL were performed in open reaction vessels (Table S1†). Polymerizations were carried out with [OEOMA500]/[HOBiB]/[EYH2]/[CuBr2]/[TPMA] molar ratios of 1000/1/0.05/1/3. Despite the reduced volume and target DP of 1000, high monomer conversions (72–80%) were achieved in all reactions, and only for a volume of 50 μL the polymerization was less controlled (Đ = 1.46, entry 4, Table S1†).
These results indicate the great potential of this technique for high-throughput screening applications.
Synthesis of biohybrids
Functional proteins are often inherently unstable and prone to aggregation, which significantly hinders their practical applications. Anchoring polymers to proteins protects protein–polymer hybrids from denaturation, delays their clearance from the body, and reduces the immunological response toward them.98 ATRP is very useful for the preparation of polymer–protein conjugates,45,99–106 but degassing prior to polymerization can trigger aggregation.107The dual EY/Cu system was applied in protein modifications. First, ATRP initiators were covalently attached to the accessible lysine residues in bovine serum albumin (BSA) to yield the protein macroinitiator BSA-[iBBr]22. The model protein–polymer bioconjugate was then prepared by grafting polymer chains from the surface of BSA using molar ratios of [OEOMA500]/[BSA-iBBr22]/[EYH2]/[CuBr2]/[TPMA] = 400/0.045/0.02/0.4/0.8 (Fig. 4E). Within 25 minutes, 45% monomer conversion was reached. SEC with multi-angle light scattering (MALS) analysis confirmed the well-controlled synthesis of the protein–polymer hybrid (Mn,th = 1848000, Mn,MALS = 1864000, Đ = 1.28, Fig. S3†).
Nucleic acid–polymer conjugates represent another important class of biohybrids used as multifunctional biomaterials in nanoscience and biomedicine.91,104,108–110 Therefore, we decided to use the EY/Cu technique to graft polymers from DNA. A 23-mer DNA-based macroinitiator (DNA-iBBr) with α-bromoisobutyrate group at the 5′-end was prepared in a DNA synthesizer111 and then extended with OEOMA500, and molar ratios of [OEOMA500]/[DNA-iBBr]/[EYH2]/[CuBr2]/[TPMA] = 600/1/0.03/0.6/1.8 at a low reaction volume of 250 μL (Fig. 4F). After 30 min of green light irradiation, 76% monomer conversion was achieved. SEC-MALS analysis showed that the DNA-polymer biohybrid had a dispersity of 1.04, and absolute molecular weight close to the theoretical value (Mn,th = 231000, Mn,MALS = 259500, Fig. S4†), indicating a well-controlled polymerization.
ATRP in the presence of cells
Engineering cell surfaces with synthetic polymers enables the modulation of physicochemical and biological properties of cells.112 Both ATRP and RAFT techniques have been used to initiate polymers from the surface of living cells.80,113 We wanted to investigate whether our method could also be used for grafting polymers from living cells. For this purpose, the method must allow rapid polymerization in water at low temperature to minimize cell exposure to a potentially harmful polymerization environment and tolerate oxygen since degassing procedures such as freeze–pump–thaw cycles cannot be applied to living cells. In addition, the polymerization process must not cause cell death.It is a common belief that Cu-catalyzed ATRP methods are more cytotoxic than metal-free RAFT techniques. The cytocompatibility of EY/Cu dual catalysis was investigated and compared with EY-catalyzed PET-RAFT (Fig. 4G). Polymerizations were performed in the presence of human embryonic kidney 293 (HEK293) cells at a low volume of 250 μL using a 96-Well LED array (520 nm, 25 mW cm−2). The cells tolerated both the ATRP and RAFT polymerization in vitro at a similar level. Compared to the control (untreated cells), 81% of the cells exposed to photo-ATRP for 10 min remained viable vs. 85% in PET-RAFT, indicating that the EY/Cu-catalyzed ATRP technique can be utilized in live cell surface engineering.
Green light-induced ATRP in organic solvents
Next, we expanded the scope of the EY/Cu system to the hydrophobic methyl acrylate (MA) monomer. Polymerizations of MA with a target DP of 200 were performed using EYH2, the CuBr2/Me6TREN complex (Me6TREN = tris[2-(dimethylamino)ethyl]amine) and methyl α-bromoisobutyrate (MBiB) initiator in DMSO in open vials without stirring (Table 3). The initial conditions used [MA]/[MBiB]/[EYH2]/[CuBr2]/[Me6TREN] molar ratios of 200/1/0.01/0.05/0.3 and MA monomer concentration of 5.5 M. After 60 min of green light irradiation (520 nm, 9.0 mW cm−2), the monomer conversion was 48% (entry 1, Table 4) and SEC analysis showed that the polymer had a bimodal molecular weight distribution (Fig. S6A†). This could be attributed to the over-reduction of the Br–CuII/L deactivator by EY, leading to termination by radical–radical coupling. To counter this problem, the EYH2 loading was reduced 2-fold (entry 2, Table 4). As a result, a much higher monomer conversion was reached (81%), yielding a well-defined polymer with monomodal, narrow molecular weight distribution (Đ = 1.05) and controlled molecular weight (Mn,th = 14100, Mn,abs = 14300). Further reduction of the amount of EYH2 improved monomer conversion (84%), but formed polymer with the dispersity of 1.07 and deviation from the theoretical molecular weight value (Mn,th = 14600, Mn,abs = 12200). Despite reducing CuBr2 to just 100 ppm (relative to monomer), the ATRP was well-controlled (Đ = 1.08). Importantly, reducing EYH2 5-fold to just 1 ppm still allowed polymerization of MA (entry 5, Table 4). However, the monomer conversion dropped to 44%, and the dispersity increased to 1.23.
| No. | [EYH2] (equiv) | CuBr2 (equiv) | Me6TREN (equiv) | Conv.b (%) | M n,th | M n,abs | M n,app | Đ |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: [MA]/[MBiB]/[EYH2]/[CuBr2]/[Me6TREN] = 200/1/x/x/x, [MA] = 5.5 M, in DMSO, irradiated for 60 min under green LEDs (520 nm, 9.0 mW cm−2) in an open vial with no stirring. b Monomer conversion was determined by using 1H NMR spectroscopy. c Molecular weight (Mn,abs) was determined by Mark–Houwink calibration. d Apparent molecular weight (Mn,app) and dispersity (Đ) were determined by GPC analysis (THF as eluent) calibrated to poly(methyl methacrylate) standards. | ||||||||
| 1 | 0.01 | 0.05 | 0.3 | 48 | 8500 | 17200 |
21000 |
1.12 |
| 2 | 0.005 | 0.05 | 0.3 | 81 | 14100 |
14300 |
17300 |
1.05 |
| 3 | 0.001 | 0.05 | 0.3 | 84 | 14600 |
12200 |
14600 |
1.07 |
| 4 | 0.001 | 0.02 | 0.12 | 79 | 13800 |
11500 |
13700 |
1.08 |
| 5 | 0.0002 | 0.02 | 0.12 | 44 | 7800 | 8300 | 9800 | 1.23 |
Oxygen concentration measurements
Finally, the effect of EY/Cu catalysis on oxygen consumption rate was investigated in situ using an oxygen probe (Fig. 5).114 Measurements were carried out without monomer in PBS with DMSO (10% v/v) in open vials at a stirring rate of 500 rpm. The initial conditions used [HOBiB]/[EYH2]/[CuBr2]/[TPMA] molar ratios of 1/0.01/0.2/0.6. After ∼7 min of green light irradiation, all dissolved oxygen was scavenged, which correlates well with the induction period observed during kinetic studies (Fig. 3A). Despite the continuous diffusion of oxygen into the reaction mixture, no oxygen re-saturation was observed. As expected, oxygen depletion did not occur in the absence of EY, whereas exclusion of the HOBiB initiator resulted in slower oxygen removal. The Cu-free system initially showed faster oxygen consumption, but after a short time, the oxygen concentration started to increase again. This could be attributed to the photobleaching of EY caused by reactive oxygen species. The Cu/EY catalysis without TPMA ligand showed an even faster initial drop of oxygen concentration followed by a rapid increase. Presumably, in the absence of the ligand, the oxidized EY could not be regenerated, closing the catalytic cycle. The control experiments showed that all components of EY/Cu catalysis are critical to achieving high oxygen tolerance.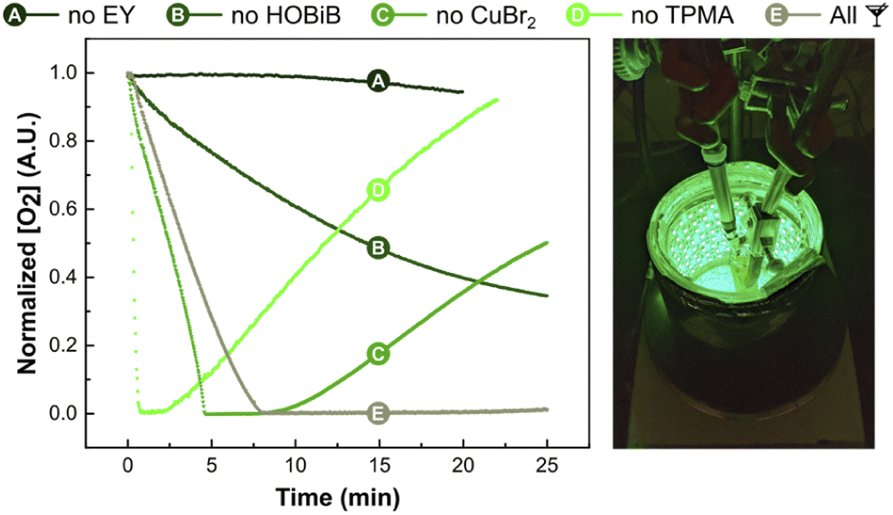 | ||
| Fig. 5 Oxygen concentration measurements. Reactions conditions: [HOBiB]/[EYH2]/[CuBr2]/[TPMA] = 1/0.01/0.2/0.6, [HOBiB] = 1.5 mM, in PBS with DMSO (10% v/v), irradiated for 30 min under green LEDs (520 nm, 9.0 mW cm−2) in an open vial with stirring at 500 rpm. | ||
Conclusions
We have developed a highly efficient, biocompatible dual photoredox/copper catalytic system enabling ATRP under low-energy green light irradiation. Eosin Y was used as an organic photocatalyst to trigger and drive the polymerization, while the copper catalyst provided control over the radical propagation via ATRP equilibrium. Excited EY was oxidatively quenched by X–CuII/L, generating CuI/L activator and EY˙+. The ATRP ligand used in excess then reduced the EY˙+, closing the photocatalytic cycle. The technique showed high oxygen tolerance, allowing ATRP in an open vessel at stirring rates up to 1000 rpm. Well-defined polymers (1.15 < Đ < 1.22) were synthesized with high OEOMA500 monomer conversions for a wide targeted DP range (50–1000) within 30 min under biologically relevant conditions. This contrasts with PET-RAFT polymerization, which under the same conditions led to poorer molecular weight control and higher dispersity values (1.14 < Đ < 4.01). Moreover, the photo-ATRP could be stopped immediately and then resumed by turning the green light off and on. The method proved to be highly efficient in the synthesis of protein–polymer and DNA-polymer hybrids without the need for deoxygenation showing similar cytocompatibility to PET-RAFT polymerization. It also enabled the successful open-to-air polymerization of MA monomer in DMSO using Cu and eosin Y catalyst at ppm levels, which extends the scope of this method toward hydrophobic monomers. We envision that this method will provide non-experts with easy access to advanced materials and polymer biohybrids.Data availability
Additional data and detailed experimental details are available in the ESI.†Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSF (CHE-2000391). G.S. gratefully acknowledges the Polish Ministry of Science and Higher Education (“Mobilnosc Plus” grant no. 1646/MOB/V/2017/0) for financial support. P.R. gratefully acknowledges The Kosciuszko Foundation for financial support. The authors would like to thank Prof. L. Peteanu for help with the fluorescence quenching measurements.References
- F. Gomollón-Bel, Chem. Int., 2019, 41, 12–17 Search PubMed.
- N. P. Truong, G. R. Jones, K. G. E. Bradford, D. Konkolewicz and A. Anastasaki, Nat. Rev. Chem., 2021, 5, 859–869 CrossRef CAS.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- K. Parkatzidis, H. S. Wang, N. P. Truong and A. Anastasaki, Chem, 2020, 6, 1575–1588 CAS.
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS PubMed.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 CrossRef CAS PubMed.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS PubMed.
- T. G. Ribelli, F. Lorandi, M. Fantin and K. Matyjaszewski, Macromol. Rapid Commun., 2019, 40, 1800616 CrossRef.
- A. E. Enciso, F. Lorandi, A. Mehmood, M. Fantin, G. Szczepaniak, B. G. Janesko and K. Matyjaszewski, Angew. Chem., Int. Ed., 2020, 59, 14910–14920 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- F. Lorandi, M. Fantin and K. Matyjaszewski, J. Am. Chem. Soc., 2022, 144, 15413–15430 CrossRef CAS PubMed.
- A. K. K. Fung and M. L. Coote, Polym. Int., 2021, 70, 918–926 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- G. Szczepaniak, L. Fu, H. Jafari, K. Kapil and K. Matyjaszewski, Acc. Chem. Res., 2021, 54, 1779–1790 CrossRef CAS.
- M. Fromel, E. M. Benetti and C. W. Pester, ACS Macro Lett., 2022, 11, 415–421 CrossRef CAS PubMed.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC.
- K. Matyjaszewski, S. Coca, S. G. Gaynor, M. Wei and B. E. Woodworth, Macromolecules, 1998, 31, 5967–5969 CrossRef CAS.
- K. Min, W. Jakubowski and K. Matyjaszewski, Macromol. Rapid Commun., 2006, 27, 594–598 CrossRef CAS.
- K. Matyjaszewski, H. Dong, W. Jakubowski, J. Pietrasik and A. Kusumo, Langmuir, 2007, 23, 4528–4531 CrossRef CAS PubMed.
- F. De Bon, R. G. Fonseca, F. Lorandi, A. C. Serra, A. A. Isse, K. Matyjaszewski and J. F. J. Coelho, Chem. Eng. J., 2022, 445, 136690 CrossRef CAS.
- W. Li, W. Sheng, B. Li and R. Jordan, Angew. Chem., Int. Ed., 2021, 60, 13621–13625 CrossRef CAS PubMed.
- E. Liarou, R. Whitfield, A. Anastasaki, N. G. Engelis, G. R. Jones, K. Velonia and D. M. Haddleton, Angew. Chem., Int. Ed., 2018, 57, 8998–9002 CrossRef CAS PubMed.
- R. Li, W. Kong and Z. An, Angew. Chem., Int. Ed., 2022, 61, e202202033 CAS.
- L. A. Navarro, A. E. Enciso, K. Matyjaszewski and S. Zauscher, J. Am. Chem. Soc., 2019, 141, 3100–3109 CrossRef CAS PubMed.
- A. E. Enciso, L. Fu, A. J. Russell and K. Matyjaszewski, Angew. Chem., Int. Ed., 2018, 57, 933–936 CrossRef CAS.
- A. E. Enciso, L. Fu, S. Lathwal, M. Olszewski, Z. Wang, S. R. Das, A. J. Russell and K. Matyjaszewski, Angew. Chem., Int. Ed., 2018, 57, 16157–16161 CrossRef CAS PubMed.
- Q. Wei, M. Sun, F. Lorandi, R. Yin, J. Yan, T. Liu, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2021, 54, 1631–1638 CrossRef CAS.
- W. Yan, S. Dadashi-Silab, K. Matyjaszewski, N. D. Spencer and E. M. Benetti, Macromolecules, 2020, 53, 2801–2810 CrossRef CAS.
- S. Dadashi-Silab, G. Szczepaniak, S. Lathwal and K. Matyjaszewski, Polym. Chem., 2020, 11, 843–848 RSC.
- S. Dadashi-Silab, X. Pan and K. Matyjaszewski, Macromolecules, 2017, 50, 7967–7977 CrossRef CAS.
- J. Mosnáček, A. Eckstein-Andicsová and K. Borská, Polym. Chem., 2015, 6, 2523–2530 RSC.
- G. Szczepaniak, M. Łagodzińska, S. Dadashi-Silab, A. Gorczyński and K. Matyjaszewski, Chem. Sci., 2020, 11, 8809–8816 RSC.
- L. Qiao, M. Zhou, G. Shi, Z. Cui, X. Zhang, P. Fu, M. Liu, X. Qiao, Y. He and X. Pang, J. Am. Chem. Soc., 2022, 144, 9817–9826 CrossRef CAS PubMed.
- E. Liarou, Y. Han, A. M. Sanchez, M. Walker and D. M. Haddleton, Chem. Sci., 2020, 11, 5257–5266 RSC.
- G. Fan, A. J. Graham, J. Kolli, N. A. Lynd and B. K. Keitz, Nat. Chem., 2020, 12, 638–646 CrossRef CAS PubMed.
- H. Kang, W. Jeong and D. Hong, Langmuir, 2019, 35, 7744–7750 CrossRef CAS PubMed.
- G. J. Dunderdale, C. Urata, D. F. Miranda and A. Hozumi, ACS Appl. Mater. Interfaces, 2014, 6, 11864–11868 CrossRef CAS PubMed.
- C. Aydogan, G. Yilmaz, A. Shegiwal, D. M. Haddleton and Y. Yagci, Angew. Chem., Int. Ed., 2022, 61, e202117377 CrossRef CAS PubMed.
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef CAS.
- S. Dadashi-Silab, S. Doran and Y. Yagci, Chem. Rev., 2016, 116, 10212–10275 CrossRef CAS.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- T. G. Ribelli, D. Konkolewicz, S. Bernhard and K. Matyjaszewski, J. Am. Chem. Soc., 2014, 136, 13303–13312 CrossRef CAS.
- J. Mosnáček and M. Ilčíková, Macromolecules, 2012, 45, 5859–5865 CrossRef.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Rapid Commun., 2011, 32, 58–62 CrossRef CAS PubMed.
- R. A. Olson, A. B. Korpusik and B. S. Sumerlin, Chem. Sci., 2020, 11, 5142–5156 RSC.
- D. A. Corbin and G. M. Miyake, Chem. Rev., 2022, 122, 1830–1874 CrossRef CAS PubMed.
- E. H. Discekici, A. Anastasaki, J. Read de Alaniz and C. J. Hawker, Macromolecules, 2018, 51, 7421–7434 CrossRef CAS.
- B. McCarthy and G. M. Miyake, ACS Macro Lett., 2018, 7, 1016–1021 CrossRef CAS PubMed.
- B. Narupai, Z. A. Page, N. J. Treat, A. J. McGrath, C. W. Pester, E. H. Discekici, N. D. Dolinski, G. F. Meyers, J. Read de Alaniz and C. J. Hawker, Angew. Chem., Int. Ed., 2018, 57, 13433–13438 CrossRef CAS PubMed.
- C. Theriot Jordan, C.-H. Lim, H. Yang, D. Ryan Matthew, B. Musgrave Charles and M. Miyake Garret, Science, 2016, 352, 1082–1086 CrossRef CAS.
- X. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 2411–2425 CrossRef CAS PubMed.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed.
- L. Zhou, Z. Zhang, M. Li, Q. Wang, J. Gao, K. Li and L. Lei, Green Chem., 2021, 23, 9617–9624 RSC.
- X. Xu, X. Xu, Y. Zeng and F. Zhang, J. Photochem. Photobiol. A, 2021, 411, 113191 CrossRef CAS.
- B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343–346 CrossRef CAS PubMed.
- C. Kutahya, F. S. Aykac, G. Yilmaz and Y. Yagci, Polym. Chem., 2016, 7, 6094–6098 RSC.
- S. Averick, A. Simakova, S. Park, D. Konkolewicz, A. J. D. Magenau, R. A. Mehl and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 6–10 CrossRef CAS PubMed.
- S. Dadashi-Silab, K. Kim, F. Lorandi, G. Szczepaniak, S. Kramer, L. Peteanu and K. Matyjaszewski, ACS Macro Lett., 2022, 11, 376–381 CrossRef CAS PubMed.
- S. Dadashi-Silab, F. Lorandi, M. J. DiTucci, M. Sun, G. Szczepaniak, T. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2021, 143, 9630–9638 CrossRef CAS PubMed.
- M. Sun, F. Lorandi, R. Yuan, S. Dadashi-Silab, T. Kowalewski and K. Matyjaszewski, Front. Chem., 2021, 9, 734076 CrossRef CAS PubMed.
- C. Kütahya, Y. Zhai, S. Li, S. Liu, J. Li, V. Strehmel, Z. Chen and B. Strehmel, Angew. Chem., Int. Ed., 2021, 60, 10983–10991 CrossRef PubMed.
- C. Kütahya, N. Meckbach, V. Strehmel and B. Strehmel, J. Polym. Sci., 2021, 59, 2023–2035 CrossRef.
- W. Zhang, J. He, C. Lv, Q. Wang, X. Pang, K. Matyjaszewski and X. Pan, Macromolecules, 2020, 53, 4678–4684 CrossRef CAS.
- C. Kütahya, C. Schmitz, V. Strehmel, Y. Yagci and B. Strehmel, Angew. Chem., Int. Ed., 2018, 57, 7898–7902 CrossRef PubMed.
- M. A. Tasdelen, M. Ciftci and Y. Yagci, Macromol. Chem. Phys., 2012, 213, 1391–1396 CrossRef CAS.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS.
- C. Wu, N. Corrigan, C.-H. Lim, W. Liu, G. Miyake and C. Boyer, Chem. Rev., 2022, 122, 5476–5518 CrossRef CAS.
- C. Wu, K. Jung, Y. Ma, W. Liu and C. Boyer, Nat. Commun., 2021, 12, 478 CrossRef CAS PubMed.
- M. L. Allegrezza and D. Konkolewicz, ACS Macro Lett., 2021, 10, 433–446 CrossRef CAS PubMed.
- S. Shanmugam, J. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef CAS PubMed.
- S. Shanmugam, J. Xu and C. Boyer, Chem. Sci., 2015, 6, 1341–1349 RSC.
- S. Shanmugam, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2016, 55, 1036–1040 CrossRef CAS PubMed.
- J. Xu, K. Jung, N. A. Corrigan and C. Boyer, Chem. Sci., 2014, 5, 3568–3575 RSC.
- J. C. Theriot, G. M. Miyake and C. A. Boyer, ACS Macro Lett., 2018, 7, 662–666 CrossRef CAS.
- Z. Wu, W. Fang, C. Wu, N. Corrigan, T. Zhang, S. Xu and C. Boyer, Chem. Sci., 2022 10.1039/D2SC03952D.
- J. Yeow, R. Chapman, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 5012–5022 RSC.
- T. Lueckerath, T. Strauch, K. Koynov, C. Barner-Kowollik, D. Y. W. Ng and T. Weil, Biomacromolecules, 2019, 20, 212–221 CrossRef CAS.
- C. A. Figg, J. D. Hickman, G. M. Scheutz, S. Shanmugam, R. N. Carmean, B. S. Tucker, C. Boyer and B. S. Sumerlin, Macromolecules, 2018, 51, 1370–1376 CrossRef CAS.
- J. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6, 5615–5624 RSC.
- J. Niu, D. J. Lunn, A. Pusuluri, J. I. Yoo, M. A. O’Malley, S. Mitragotri, H. T. Soh and C. J. Hawker, Nat. Chem., 2017, 9, 537–545 CrossRef CAS.
- M. Fantin, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2015, 48, 6862–6875 CrossRef CAS.
- M. Majek and A. Jacobi von Wangelin, Acc. Chem. Res., 2016, 49, 2316–2327 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- R. Martínez-Haya, A. A. Heredia, W. D. Castro-Godoy, L. C. Schmidt, M. L. Marin and J. E. Argüello, J. Org. Chem., 2021, 86, 5832–5844 CrossRef.
- K. Parkatzidis, S. Boner, H. S. Wang and A. Anastasaki, ACS Macro Lett., 2022, 11, 841–846 CrossRef CAS PubMed.
- J. Cuthbert, S. V. Wanasinghe, K. Matyjaszewski and D. Konkolewicz, Macromolecules, 2021, 54, 8331–8340 CrossRef CAS.
- S. Dadashi-Silab, I.-H. Lee, A. Anastasaki, F. Lorandi, B. Narupai, N. D. Dolinski, M. L. Allegrezza, M. Fantin, D. Konkolewicz, C. J. Hawker and K. Matyjaszewski, Macromolecules, 2020, 53, 5280–5288 CrossRef CAS.
- R. Whitfield, K. Parkatzidis, M. Rolland, N. P. Truong and A. Anastasaki, Angew. Chem., Int. Ed., 2019, 58, 13323–13328 CrossRef CAS PubMed.
- H. S. Wang, K. Parkatzidis, S. Harrisson, N. P. Truong and A. Anastasaki, Chem. Sci., 2021, 12, 14376–14382 RSC.
- K. Ślusarczyk, M. Flejszar and P. Chmielarz, Polymer, 2021, 233, 124212 CrossRef.
- Y. Sun, S. Lathwal, Y. Wang, L. Fu, M. Olszewski, M. Fantin, A. E. Enciso, G. Szczepaniak, S. Das and K. Matyjaszewski, ACS Macro Lett., 2019, 8, 603–609 CrossRef CAS.
- E. Liarou, A. Anastasaki, R. Whitfield, C. E. Iacono, G. Patias, N. G. Engelis, A. Marathianos, G. R. Jones and D. M. Haddleton, Polym. Chem., 2019, 10, 963–971 RSC.
- A. J. Gormley and M. A. Webb, Nat. Rev. Mater., 2021, 6, 642–644 CrossRef CAS PubMed.
- Z. Li, Z. Han, M. H. Stenzel and R. Chapman, Nano Lett., 2022, 22, 2660–2666 CrossRef CAS PubMed.
- F. Soheilmoghaddam, M. Rumble and J. Cooper-White, Chem. Rev., 2021, 121, 10792–10864 CrossRef CAS PubMed.
- P. R. Judzewitsch, N. Corrigan, F. Trujillo, J. Xu, G. Moad, C. J. Hawker, E. H. H. Wong and C. Boyer, Macromolecules, 2020, 53, 631–639 CrossRef CAS.
- M. Tamasi, S. Kosuri, J. DiStefano, R. Chapman and A. J. Gormley, Adv. Intell. Syst., 2020, 2, 1900126 CrossRef PubMed.
- E. M. Pelegri-O’Day, E.-W. Lin and H. D. Maynard, J. Am. Chem. Soc., 2014, 136, 14323–14332 CrossRef.
- M. Olszewski, J. Jeong, G. Szczepaniak, S. Li, A. Enciso, H. Murata, S. Averick, K. Kapil, S. R. Das and K. Matyjaszewski, ACS Macro Lett., 2022, 11, 1091–1096 CrossRef CAS PubMed.
- A. Theodorou, P. Mandriotis, A. Anastasaki and K. Velonia, Polym. Chem., 2021, 12, 2228–2235 RSC.
- M. S. Messina, K. M. M. Messina, A. Bhattacharya, H. R. Montgomery and H. D. Maynard, Prog. Polym. Sci., 2020, 100, 101186 CrossRef CAS PubMed.
- A. Theodorou, E. Liarou, D. M. Haddleton, I. G. Stavrakaki, P. Skordalidis, R. Whitfield, A. Anastasaki and K. Velonia, Nat. Commun., 2020, 11, 1486 CrossRef CAS PubMed.
- C. Chen, D. Y. W. Ng and T. Weil, Prog. Polym. Sci., 2020, 105, 101241 CrossRef CAS.
- S. L. Baker, B. Kaupbayeva, S. Lathwal, S. R. Das, A. J. Russell and K. Matyjaszewski, Biomacromolecules, 2019, 20, 4272–4298 CrossRef CAS PubMed.
- H. Murata, S. Carmali, S. L. Baker, K. Matyjaszewski and A. J. Russell, Nat. Commun., 2018, 9, 845 CrossRef PubMed.
- A. J. Russell, S. L. Baker, C. M. Colina, C. A. Figg, J. L. Kaar, K. Matyjaszewski, A. Simakova and B. S. Sumerlin, AIChE J., 2018, 64, 3230–3245 CrossRef CAS.
- L. Zhang, Y. Zhang, J. Cheng, L. Wang, X. Wang, M. Zhang, Y. Gao, J. Hu, X. Zhang, J. Lü, G. Li, R. Tai and H. Fang, Sci. Rep., 2017, 7, 10176 CrossRef PubMed.
- C. J. Whitfield, M. Zhang, P. Winterwerber, Y. Wu, D. Y. W. Ng and T. Weil, Chem. Rev., 2021, 121, 11030–11084 CrossRef CAS PubMed.
- S. S. Yerneni, S. Lathwal, J. Cuthbert, K. Kapil, G. Szczepaniak, J. Jeong, S. R. Das, P. G. Campbell and K. Matyjaszewski, Biomacromolecules, 2022, 23, 1713–1722 CrossRef CAS PubMed.
- S. Lathwal, S. S. Yerneni, S. Boye, U. L. Muza, S. Takahashi, N. Sugimoto, A. Lederer, S. R. Das, P. G. Campbell and K. Matyjaszewski, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2020241118 CrossRef CAS PubMed.
- X. Pan, S. Lathwal, S. Mack, J. Yan, S. R. Das and K. Matyjaszewski, Angew. Chem., Int. Ed., 2017, 56, 2740–2743 CrossRef CAS.
- J. Liu and B. Liu, Prog. Polym. Sci., 2022, 129, 101545 CrossRef CAS.
- J. Y. Kim, B. S. Lee, J. Choi, B. J. Kim, J. Y. Choi, S. M. Kang, S. H. Yang and I. S. Choi, Angew. Chem., Int. Ed., 2016, 55, 15306–15309 CrossRef CAS PubMed.
- M. Rolland, R. Whitfield, D. Messmer, K. Parkatzidis, N. P. Truong and A. Anastasaki, ACS Macro Lett., 2019, 8, 1546–1551 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc04210j |
| ‡ J.J. and K.K. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |