
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of a polymer modifier on directing the non-classical crystallization pathway of TS-1 zeolite: accelerating nucleation and enriching active sites†
Jiani
Zhang
a,
Risheng
Bai
 a,
Yida
Zhou
a,
Ziyi
Chen
b,
Peng
Zhang
b,
Jiyang
Li
*a and
Jihong
Yu
*a
a,
Yida
Zhou
a,
Ziyi
Chen
b,
Peng
Zhang
b,
Jiyang
Li
*a and
Jihong
Yu
*a
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130012, China. E-mail: jihong@jlu.edu.cn; lijiyang@jlu.edu.cn
bDepartment of Chemistry, Dalhousie University, Halifax, Nova Scotia B4H4R2, Canada
First published on 30th September 2022
Abstract
The crystallization process directly affects the physicochemical properties and active centers of zeolites; however, controllable tuning of the zeolite crystallization process remains a challenge. Herein, we utilized a polymer (polyacrylamide, PAM) to control the precursor structure evolution of TS-1 zeolite through a two-step crystallization process, so that the crystallization path was switched from a classical to a non-classical mechanism, which greatly accelerated nucleation and enriched active Ti sites. The TS-1 crystallization process was investigated by means of various advanced characterization techniques. It was found that specific interactions between PAM and Si/Ti species promoted the assembly of colloidal precursors containing ordered structural fragments and stabilized Ti species in the precursors, leading to a 1.5-fold shortened crystallization time and enriched Ti content in TS-1 (Si/Ti = 29). The PAM-regulated TS-1 zeolite exhibited enhanced catalytic performance in oxidative reactions compared to conventional samples.
Introduction
As a kind of crystalline microporous material, zeolites have made great contributions to the catalysis applications of the petrochemical industry and energy sustainability due to their unique nanoporous architectures and diverse active sites formed by introducing heteroatoms (e.g., Al, Ti, Sn, Zr, etc.) in the framework.1–7 The structural characteristics of zeolites, including morphology, particle size and porosity, have a great influence on the catalytic activity.8–11 Moreover, the controllable adjustment of the microenvironmental structure of active sites in zeolites is crucial to improve the catalytic performance.12–14 These facts have inspired studies on crystal engineering to control the crystallization process and then to tailor the physicochemical properties and active site structure of zeolite catalysts, which requires in-depth research on the crystal nucleation and growth of zeolites.The crystal growth of zeolites usually proceeds via a classical or non-classical crystallization pathway.15–18 The former one mainly involves spontaneous nucleation and the addition of monomers like ions or molecules to kink, step, and terrace sites on crystals, resulting in smooth surfaces;16,19,20 while the latter one refers to crystallization through the attachment of particles (CPA), leading to the formation of crystals with rough crystalline surfaces and loose hierarchical structures,21–23 which may facilitate the diffusion of substrates and the accessibility of active sites in catalytic reactions.15 Given the involvement of heteroatoms in the crystallization process of zeolite catalysts, it is foreseeable that the changes in crystallization pathways are bound to have an impact on the number and microenvironment of active heteroatoms in zeolites. However, because of the complexity of the zeolite reaction system comprised of multiple species and the lack of a fundamental understanding of the crystallization mechanism, it is challenging to develop feasible methods to effectively control the crystallization process.
Studies on crystal engineering have demonstrated that the crystallization of zeolites can be remarkably influenced by crystal growth modifiers (CGMs) through specific adsorption and interaction.24–26 It has been found that CGMs can adjust the crystal morphology and size, produce a hierarchical structure, and even influence the crystallization process.8,27 For examples, Rimer and co-workers have demonstrated the regulation of non-classical pathways of silicalite-1 by TAA and TBPO,22 and the size tune of SSZ-13 (CHA) from 0.1 μm to 20 μm by modifiers PEIM, CTAB, and PDDA.28 Recently, they further revealed that the suitable amount of PDDA could accelerate the nucleation of SSZ-13 by regulating the evolution of the amorphous precursor and colloidal stability in a non-classical pathway.29 However, such studies mainly focus on several model zeolites, and the specific effects of modifiers on the active species in zeolite catalysts are rarely investigated.
The discovery of titanosilicate-1 (TS-1) zeolite with MFI zeotype is a milestone in the history of zeolite and heterogeneous catalysis, which has triggered the revolution of green oxidation systems with H2O2 as an oxidant. The number and coordination environment of Ti in the framework determine the catalytic performance of TS-1 zeolite.30–34 Some small-molecule additives (e.g., (NH4)2CO3, (NH4)2SO4 and 1,3,5-benzenetricarboxylic acid) and polymers (e.g., PAA) have been utilized to lower the pH of the system or solidify the gel, allowing the rate of Ti entering the framework to match with the rate of nucleation and crystallization to increase the titanium content of TS-1.35–38 With the help of CGMs (e.g., ethanol and amino acids), TS-1 zeolite with high activity mono-nuclear hexacoordinated titanium (TiO6) was prepared.39 In spite of this, the underlying mechanism of TS-1 zeolite from the perspective of the crystallization process is still unclear, which brings great difficulties to the controllable construction of high-content active species.
Herein, we address the challenge of switching the dominant zeolite crystallization pathway from classical to non-classical by virtue of the specific interaction between the polymer (e.g., PAM) and Si/Ti species, achieving accelerated nucleation and enriched active sites in TS-1 zeolites. Systematic studies reveal the synergistic effect of the long chains and functional groups of PAM on the aggregation of amorphous precursors, the evolution of zeolite framework fragments, and stabilization of active sites. This has great advantages for reducing the crystallization time and producing abundant TiO6/TiO4 species, as well as enhancing the catalytic performance in 1-hexene epoxidation and dibenzothiophene (DBT) oxidative desulfurization.
Results and discussion
PAM accelerating nucleation and directing non-classical crystallization
In a wide variety of CGMs, polymers have been found to have the advantages of relatively high potency (that is, they are effective at low concentration), low cost and recyclability.27 Considering that the functional part of polymers can interact with different reactive species to change the formation and evolution of amorphous precursors in the crystallization of zeolite, in this work, a polyacrylamide (PAM) with acyl functional groups (Fig. S1a†) has been utilized as a modifier to impact the crystallization of TS-1 zeolite. Fig. 1a illustrates the synthesis process of TS-1 zeolite in the presence of PAM. A two-step crystallization process with a relatively low temperature compared to the conventional synthesis of TS-1 is preferred to have a more clear insight into the time-resolved process taking into account the large window of time between the onset of nucleation and a fully crystalline product.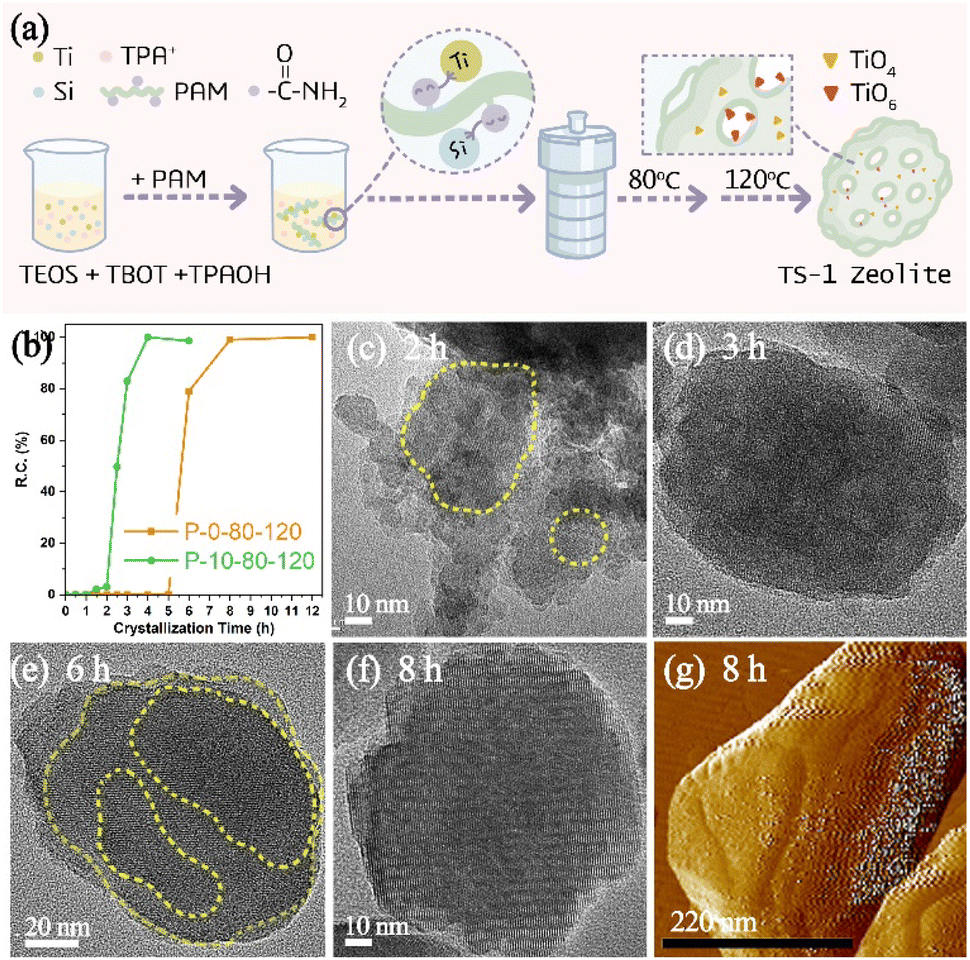 | ||
| Fig. 1 (a) Schematic diagram of the PAM-assisted synthesis of TS-1 zeolite, (b) relative crystallinity of TS-1 samples crystallized at 80 °C for 3 h and then at 120 °C for different time, TEM images of P-10-80-120 crystallized at 120 °C for (c) 2 h and (d) 3 h, and P-0-80-120 crystallized at 120 °C for (e) 6 h and (f) 8 h, and (g) AFM image of P-0-80-120 crystallized at 120 °C for 8 h. | ||
As expected, the introduction of PAM could affect the crystallization process of TS-1 zeolite, which dramatically reduces the crystallization time. As shown in Fig. 1b, the crystallized products of P-10-80-120 begin to form at the crystallization time of 1.5 h (120 °C), and then a rapid crystal growth is observed which finished after ca. 2.5 h with relative crystallinity reaching 100%. In the case of the control sample (P-0-80-120) without PAM, there are no solid products until 6 h of crystallization (120 °C), whereas TS-1 crystals exhibit relatively high crystallinity (ca. 78%) and undergo complete crystallization within 2 h. The induction period for TS-1 nucleation can be estimated from the powder XRD patterns of extracted solids based on the initial emergence of characteristic diffraction peaks (Fig. S2†), which slightly overestimates the time corresponding to the onset of nucleation.29 The comparison reveals that PAM could affect the induction period of TS-1 zeolite, which gives a two-fold decrease (4.5 h vs. 9 h) in induction time, resulting in the total crystallization time decreasing from 11 h to 7 h. In order to further confirm the influence of PAM on the crystallization time of TS-1 zeolite, the syntheses of TS-1 at a single heating temperature of 120 °C and 170 °C have been studied, wherein the nucleation induction time is shortened from 6 h to 2 h (120 °C, Fig. S3†) and from 2 h to 1 h (170 °C, Fig. S4†) in the presence of PAM.
Moreover, the TEM study shows that the morphologies of the samples during the crystallization are quite different in the presence or absence of PAM (Fig. 1c–f). The small aggregated particles in Fig. 1c show that the PAM-assisted samples tend to grow more in a non-classical crystallization path, while the sample without PAM addition shows signs of lamellar growth (Fig. 1e). The results of dynamic light scattering of the supernatant of P-0-80-120 crystallized at 120 °C for 6 h (Fig. S5†) indicate that there are only crystal particles of ∼80 nm, rather than smaller particles (<40 nm), and the 29Si NMR spectrum (Fig. S6†) shows the signal of soluble silicon species, which indicates that the crystallization path without PAM may be more dominated by the classical pathway.40,41
The formation process of P-10-80-120 during the initial crystallization time of 0–3 h (120 °C) has been studied by TEM. When prolonging the crystallization time from 0 h to 2.5 h, the separated solid products are all composed of wormlike particles (WLPs) with small particles (ca. 20 nm) attached to the surface of WLPs. The WLPs with ca. 200 nm in size are rapidly formed before the crystallization stage (0 h) (Fig. 2a). With the increase of heating time (0.5–2.5 h), WLPs grow to ca. 300 nm and then keep unchanged, but the number of WLPs decreases gradually (Fig. 2b–f). This indicates that WLPs go through the early stage of aggregation and fusion, then stop growing and reach the final stage with a stable size and shape. The selected area electron diffraction (SAED) of zone II (Fig. 2j) in Fig. 2h shows that the wormlike particles are amorphous, which mainly consist of amorphous titanosilicate precursors (Si/Ti = 23 determined by ICP) and TPA molecules (Fig. S7†).42,43 In contrast, the changing trend of small particles is opposite to that of WLPs. With the increase of crystallization time, the number of small particles increases gradually, and TS-1 sub-crystals with an obvious electron diffraction pattern appear close to the WLPs after crystallization for 1.5 hours (Fig. 2i), consistent with XRD characterization (Fig. S2†). This indicates that the zeolite nuclei are initially formed on the WLPs and the observed small particles of ca. 20 nm are sub-crystals containing crystal nuclei, which is distinct from spontaneous nucleation. After 3 h of crystallization, WLPs basically disappear, and TS-1 crystals with a size of about ca. 130 nm are obtained (Fig. 2g and S8†). It seems that the transformation of WLPs to TS-1 crystals may be through the direct splitting and transformation of loose WLPs into sub-crystals, instead of molecular exchange between the solid phase and liquid phase. It can be clearly seen from Fig. S9† and 1c that the small particles with obvious lattice diffraction fringes as shown in the area circled in yellow grow into crystals through attachment and aggregation, which is the characteristic of a typical non-classical crystallization pathway, resulting in obvious mesoporous structures (Fig. 1d).
 | ||
| Fig. 2 TEM images of P-10-80-120 samples crystallized at 80 °C for 3 h and then at 120 °C for (a) 0 h, (b) 0.5 h, (c) 1.0 h, (d) and (h) 1.5 h, (e) 2.0 h, (f) 2.5 h, and (g) 3.0 h (scale bar = 200 nm, especially, 100 nm for (g)), and SAED images of (i) area I and (j) area II. | ||
As for PAM-free P-0-80-120 samples, a solid product can only be obtained after 6 h crystallization (120 °C) (Fig. S2d†). The obtained product possesses sharper boundaries and smoother surfaces, in which signs of layer-by-layer growth can be seemingly found as shown in the area circled in yellow (Fig. 1e). As the crystallization time increases (8–12 h), the surface of crystals becomes smoother and the shape becomes much more regular (Fig. 1f and S10†). AFM was also used to monitor the surface of P-0-80-120 samples, showing obvious layers (Fig. 1g).44 Besides, we did not capture the obvious phenomenon of the attachment of small particles or the appearance of amorphous WLPs throughout the crystallization process, which suggests that the dominant growth path is likely to follow the classical crystallization pathway.
To further understand the fundamental reason why PAM changes the crystallization pathways of TS-1 zeolite, the detailed characterization of P-10-80-120 compared with P-0-80-120 has been performed. TG analyses (Fig. 3a) show that the weight loss of the P-10-80-120 sample is higher than that of P-0-80-120 (24 vs. 15 wt%), indicating the encapsulation of PAM in the TS-1 zeolite crystal. Furthermore, the interaction between PAM and silicon species is illustrated by XPS spectra, in which the binding energy (BE) has a close relationship with the estimated charges on the central atoms. As shown in Fig. 3b, the Si 2p peak of the uncalcined sample obviously shifts to lower electron energy levels compared with the calcined P-10-80-120 (103.9 eV vs. 103.1 eV). However, the peak position of P-0-80-120 only shifts slightly before and after calcination (104.2 eV vs. 103.8 eV). This verifies that the amide groups in PAM have interacted with Si species by forming hydrogen bonds with the silanol during the crystallization process, in accordance with the previously published studies.45–4829Si MAS NMR of the synthetic gels of samples P-0-80-120 and P-10-80-120 are shown in Fig. 3c. The peaks around −89.2 ppm and −99.4 ppm are assigned to the hydroxyl-associated silicon species Q2 ((HO)2-Si-(OSi)2) and Q3 (HO-Si-(OSi)3), respectively. Notably, a peak assigned to the Q4 species (Si-(OSi)4) appears in the P-10-80-120 gel (around −110.4 ppm), which suggests a higher degree of cross-linking of Si species in the PAM-containing reaction gel that is the basis for the formation of WLPs.49–53 FT-IR spectra further confirm that more ordered framework fragments already exist in the reaction gel with PAM. As shown in Fig. 3d, the enhancement of band intensity at 1050 cm−1 corresponding to the asymmetric stretching of Si–O–Si bonds in silicate proves the progress of the silicon condensation process in the P-10-80-120 gel. In addition, an intensity-enhanced band at 570 cm−1 is observed in the P-10-80-120-gel, which is specifically assigned to five-ring structures in pentasil zeolites.49
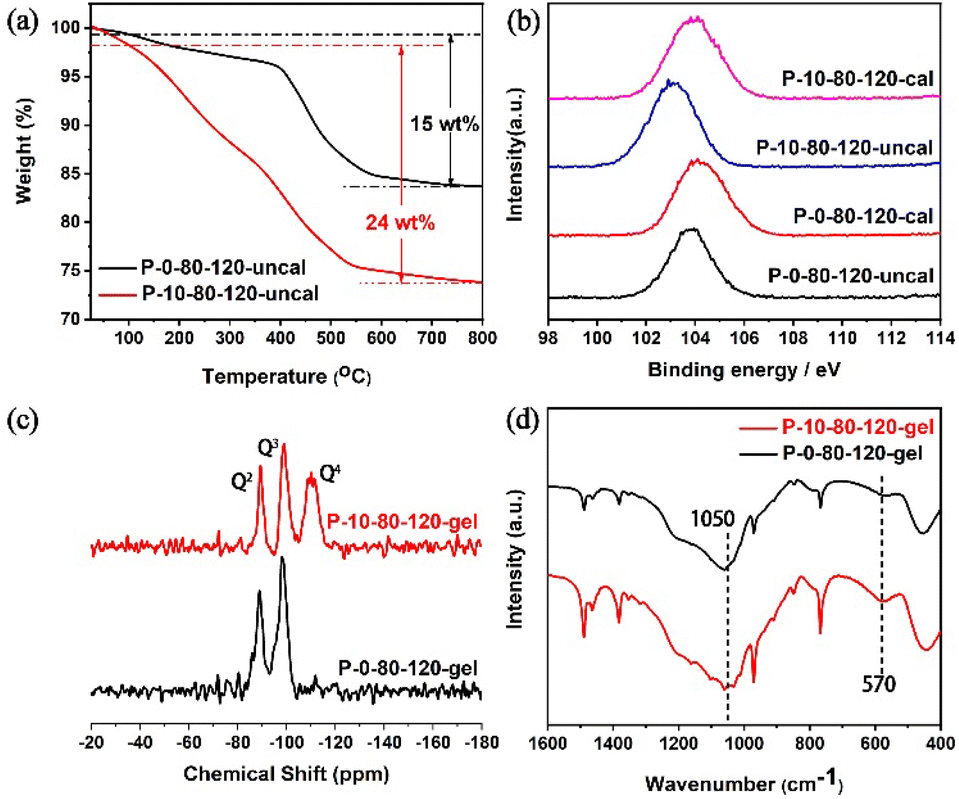 | ||
| Fig. 3 (a) TG curves of P-10-80-120 and P-0-80-120 before calcination, (b) XPS spectra of Si 2p of P-10-80-120 and P-0-80-120 before and after calcination, (c) 29Si MAS NMR spectra and (d) FT-IR spectra of the P-0-80-120 gel and P-10-80-120 gel. | ||
On the other hand, the acceleration effect of PAM on the nucleation and the non-classical crystallization mechanism are also presented on pure silica silicalite-1 zeolite sample (P-S-10-80-120) as shown in Fig. S11 and S12.† Therefore, we speculate that the acyl groups on PAM could interact with TEOS hydrolysate, resulting in the self-assembly and precipitation of amorphous nanoparticles in the form of WLPs with some possible crystalline groups, facilitating the formation of zeolite crystal nuclei. Different from the PAM-free sample which may be more inclined to grow in the classical crystallization path, the formation of highly ordered sub-crystals on WLPs determines the promotion of zeolite crystallization mainly via a non-classical crystallization path.54
PAM enriching active Ti sites in TS-1 zeolite
The active sites in TS-1 zeolite are crucial for selective catalytic oxidation, and thus it is of great significance to study the effect of PAM on the formation of Ti species in the crystallization process of TS-1. As shown in Fig. S13,† FT-IR spectra indicate that both P-0-80-120 and P-10-80-120 contain tetracoordinated framework titanium species (TiO4) with a peak at 960 cm−1 caused by the vibrations of the Si–O–Ti/Si–O–Si bonds closely connected with Si–O–Ti.34 The UV-vis spectra (Fig. 4a) show that these two samples have a main absorption band around 210–220 nm, indicating the existence of framework TiO4 species. Strikingly, after addition of PAM, a new absorption band around 265 nm appears, which is usually attributed to the highly coordinated TiO6 species.32 The coordination environment and local structure of Ti species are further revealed by XAFS. In general, the intensity of the pre-edge peaks in the Ti K-edge XANES spectrum is related to the structural symmetry around the Ti species. As shown in Fig. 4b, a single sharp peak is found at about 4968 eV corresponding to the symmetric structure of TiO4 in the sample of P-0-80-120. Such a peak decreases obviously in P-10-80-120, which should be attributed to the structural distortion caused by the formation of some TiO6 species. The positive shift of the Ti–O peak in FT-EXAFS in P-10-80-120 compared with P-0-80-120 also confirms the existence of highly coordinated Ti species (Fig. 4c).55–58 Similar to the UV-vis analysis, anatase has not been found in these two samples.32,59 Notably, PAM addition is directly related to the noticeable increase of Ti content measured by ICP in P-10-80-120 compared with P-0-80-120 (Si/Ti = 29 vs. 107). The ratio of Si/Ti given by SEM-EDS is consistent with ICP results (Fig. S14†), and the HAADF STEM image and elemental maps show that Ti elements are distributed in the P-10-80-120 sample with good dispersibility (Fig. S15†). These results suggest that PAM may have some interaction with Ti precursors in the crystallization of TS-1, which allows more titanium species to be “fixed” in zeolite in the form of highly coordinated titanium.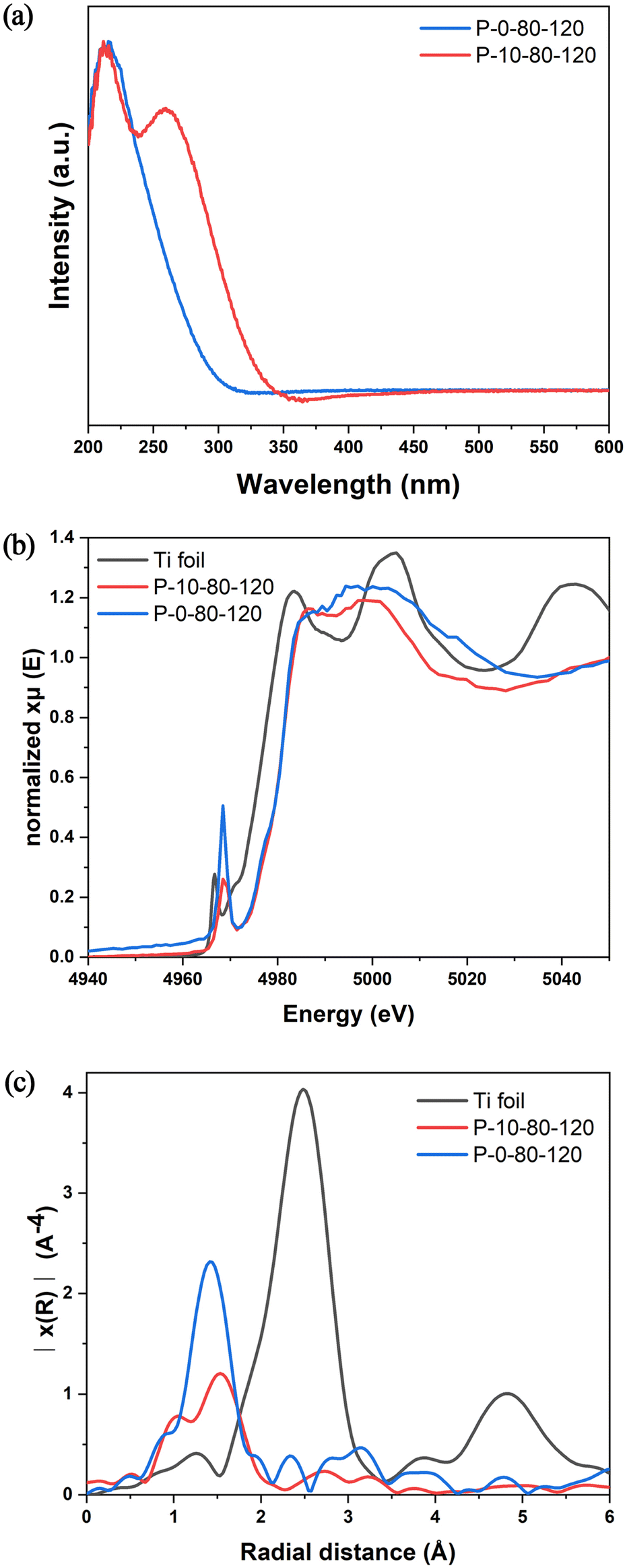 | ||
| Fig. 4 (a) UV-vis spectra, (b) Ti K-edge XANES spectra, and (c) Fourier transform k2-weighted Ti EXAFS spectra in the R spacing of P-0-80-120 and P-10-80-120. | ||
In previous studies, it has been found that the Ti–O bonds could be rapidly formed between the amino acid and Ti IV in aqueous alcohol, and such interactions are beneficial to generate mononuclear TiO6 species in the TS-1 framework.39,60 Hence, in this work, we have performed detailed analyses of XPS spectra to study the specific interaction between PAM and Ti, and to show how this interaction can introduce Ti species into TS-1 in the form of TiO6. As shown in Fig. 5a, the Ti 2p3/2 and Ti 2p1/2 peaks centred at about 460.2 eV and 465.0–465.4 eV are attributed to TiO4 species, and the unique peak at 459.1 eV appearing in the samples prepared involving PAM can be attributed to the TiO6 species, which is consistent with the UV-vis results (Fig. 4a).32 The introduction of PAM reduces the BE of Ti 2p3/2 from 460.2 eV and 459.1 eV of P-0-80-120-uncal to 459.1 eV and 457.8 eV of P-10-80-120-uncal, respectively, which indicates that the charge distribution of the Ti ions in P-10-80-120-uncal becomes more negative due to the interaction with PAM. Further calcination results in BE moving back to 460.2 eV and 459.1 eV, and the peak at 457.8 eV in P-10-80-120-uncal disappears with the decomposition of the organic modifier.61 The XPS of N 1s provides more evidence for the direct coordination between PAM and Ti (Fig. 5b). In PAM-assisted P-10-80-120-uncal samples, a peak at 399.9 eV is observed in addition to the acyl functional group (401.5 eV) on PAM and the N atom (402.6 eV) on the template TPAOH, which might be ascribed to the N–Ti–O bond formed by the coordination of the amino group on PAM with titanium.62–68 Combined with the previous reports and the above results, we hypothesize that some PAM molecules may have coordinated directly to the Ti sites, forming a hexacoordinate Ti species with PAM molecule ligands (TiO6 (PAM)) (Fig. 5c). After calcination, the organic ligands are replaced by water molecules, and the TiO6 (PAM) species are transformed into TiO6 species (Fig. 5d), meanwhile some TiO6 sites are converted into TiO4 sites (Fig. 5e).61
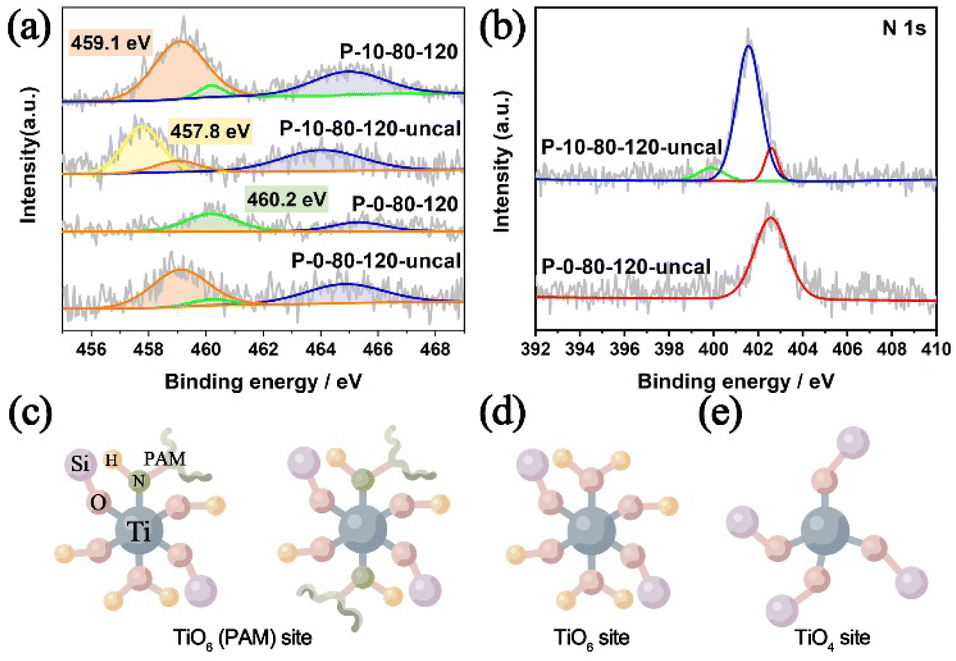 | ||
| Fig. 5 (a) XPS spectra of Ti 2p in P-10-80-120 and P-0-80-120 before and after calcination, (b) XPS spectra of N 1s of P-10-80-120 and P-0-80-120 before calcination and proposed titanium active sites: (c) TiO6 (PAM) site, (d) TiO6 site and (e) TiO4 site. | ||
Furthermore, by tracking the evolution process of Ti species in P-10-80-120 and P-0-80-120 samples, we clarify the reasons for the increased Ti content in PAM-assisted samples. Fig. S16† shows the ICP analysis of solids extracted from the intermediate time-controlled synthesis of P-10-80-120. It can be found that the Si/Ti ratio of the solid obtained after crystallization at 120 °C (0 h) is 23, and the value reaches 19.6 at 120 °C (0.5 h), and then gradually increases to 29 at 120 °C (3 h). SEM-EDS (Fig. S17†) and TEM elemental maps (Fig. S18†) also prove that the amorphous precursor has a relatively high content of Ti species. UV-visible spectra (Fig. S19†) reveal that the relative content of TiO6 is almost unchanged with crystallization time, indicating that titanium species exist stably in the forms of TiO4 and TiO6 in WLPs. Then, during the process of nucleation and the following crystal growth via a non-classical crystallization pathway, these Ti species are directly transformed from WLPs to crystals, without the loss and change of Ti species, that is, Ti participates in the nucleation and the early stage of crystallization of the zeolite from the beginning, rather than inserting into the framework when crystallization is nearly complete in the classical liquid phase crystallization mechanism. In the absence of PAM, the Ti content in the solid product of P-0-80-120 gradually increases (Si/Ti ratio from 107 to 98) with the increase of the crystallization time (6–24 h) (Fig. S20†). The Ti species exist in the form of framework TiO4 at 6 h, and TiO6 species appear at 8 h (Fig. S21†). This demonstrates that the process of crystal formation may occur via the spontaneous nucleation and classical crystallization path, the Ti species gradually enter the crystals with the progress of crystallization, and the existing form of Ti species is not static, but undergoes an unstable dynamic change process. It should be noted that the Ti content in P-0-80-120 (Si/Ti = 107) is relatively lower than that of the conventional TS-1 zeolite (Si/Ti = 31–70) due to the low crystallization temperature (120 °C), which is consistent with the previous study that low temperature (under 140 °C) is not conducive to the entry of titanium species into the TS-1 framework.32,34,69–71 Notably, the Ti content (Si/Ti = 29) could be greatly enhanced at low temperature due to the change of the nucleation and crystallization pathway by modifier PAM, which highlights the advantage of the PAM-assisted synthesis method.
Putative crystallization process of TS-1 zeolite assisted by PAM
As such, we propose the putative crystallization process of TS-1 zeolite assisted by PAM to show the unique effect of PAM on accelerating the nucleation, changing the crystallization pathway, and further controlling the microenvironment of active sites in TS-1 zeolite. As shown in Fig. 6, the acyl groups on PAM can form hydrogen bonds with the monomers and oligomers of Si and Ti, facilitating the reach of the critical aggregation concentration (CAC) in the network region formed by the PAM long-chain entanglement, and promoting the aggregation and participation of Si and Ti precipitates in the form of WLPs.72 Different from the slow spontaneous nucleation of crystals, WLPs containing some ordered structural fragments provide more sites for nucleation, thus crystal nuclei are rapidly generated on the surface of WLPs, and the highly ordered sub-crystals (about 20 nm) are continuously formed by consuming WLPs. Sub-crystals grow through attachment, structural rearrangement and integration, and the final TS-1 crystal is obtained through the non-classical-dominated route. Meanwhile, due to the direct coordination between the amino groups on PAM and the titanium species in the synthesis gel, a large amount of titanium species are stably fixed in WLPs in the form of TiO6(PAM) before nucleation. When the nucleation and crystallization process proceeds through a non-classical path, the TiO6(PAM) species are directly incorporated into TS-1 crystals via CPA. Upon calcination to remove PAM, rich TiO6 sites together with framework TiO4 are formed in TS-1 zeolite. Due to the synergistic effect of the long chains and functional groups of PAM on the zeolite precursor evolution, the crystallization pathway of TS-1 could be switched from a classical-dominated crystallization pathway based on solid–liquid phase conversion to a non-classical-dominated crystallization pathway based on CPA, enabling the rapid synthesis of TS-1 zeolite with high titanium content and rich TiO6 species.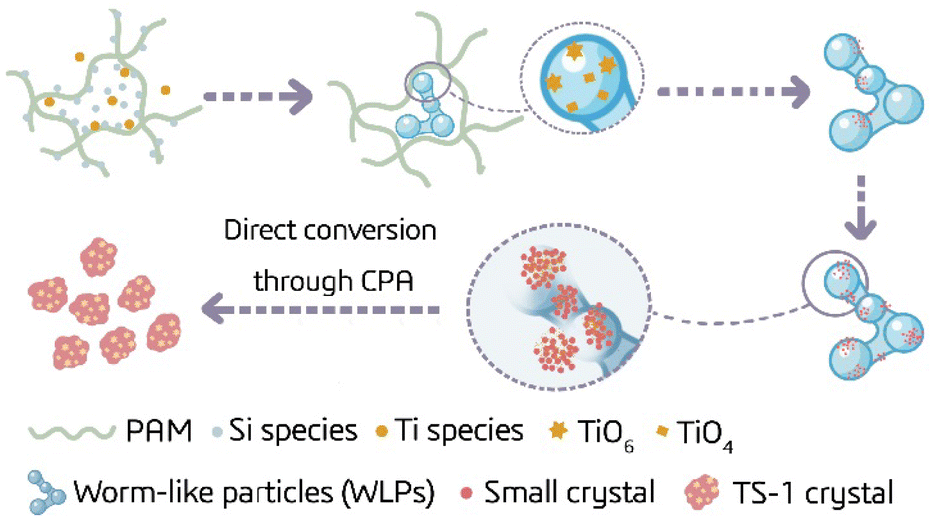 | ||
| Fig. 6 Scheme of the putative crystallization pathway of TS-1 in the presence of PAM. | ||
In order to further study the effect of different CGMs related to PAM, PAM with different molecular weights (MW) and their basic building block acrylamide (AM), as well as the cation-containing polymer polydimethyl diallyl ammonium chloride (PDDA) and the amino-containing polymer polyethylenimine (PEI) have been used as CGMs (see structures in Fig. S1†). The results indicate that AM and PDDA do not show the effect of accelerating the crystallization process in the TS-1 growth system, while PEI and PAM (with a MW of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 and 18000000) have different degrees of acceleration effect (Fig. S22 and S23†). At the same time, except PDDA, other CGMs could introduce Ti species into the TS-1 framework (Table S3†), among which PAM with a MW of 8000000 directs the highest Ti content (Si/Ti = 28) of TS-1 zeolite, while the addition of PAM (18000000) and PEI results in anatase formation in the samples (Fig. S24†). These results demonstrate that the effect of accelerating crystallization and introducing rich titanium requires that the PAM has a MW in a suitable range, and the cation may not be the key factor, and acyl functional groups on the polymer play a greater role than amino groups.
000000 and 18000000) have different degrees of acceleration effect (Fig. S22 and S23†). At the same time, except PDDA, other CGMs could introduce Ti species into the TS-1 framework (Table S3†), among which PAM with a MW of 8000000 directs the highest Ti content (Si/Ti = 28) of TS-1 zeolite, while the addition of PAM (18000000) and PEI results in anatase formation in the samples (Fig. S24†). These results demonstrate that the effect of accelerating crystallization and introducing rich titanium requires that the PAM has a MW in a suitable range, and the cation may not be the key factor, and acyl functional groups on the polymer play a greater role than amino groups.
Optimization of TS-1 zeolites with enhanced catalytic performance
On the basis of the above strategy, TS-1 zeolites with different contents of Ti active species are screened and optimized by adjusting the amount of PAM to achieve excellent catalytic performance. The results indicate that all PAM-assisted samples can crystallize within 6 hours and show the characteristic diffraction peaks of the MFI structure (Fig. S25†). The overall titanium content of the samples can be adjusted from 106 to 26 (Si/Ti ratio) by adding PAM/SiO2 from 0 to 0.2 (Table S2†). These samples all contain framework TiO4 species (Fig. S26† and S27†), and the increase of PAM leads to a significant increase of TiO6 content in the samples (Fig. S27† and S28†). However, it is worth noting that when a large amount of PAM (PAM/SiO2 = 0.2) is used in the synthesis, anatase TiO2 is formed on the sample surface as evidenced by XPS spectra (a peak at 458.75 eV attributed to anatase, Fig. S28†) and UV-vis spectra (a peak at 310 nm attributed to anatase, Fig. S27†). Therefore, in a suitable range, the amount of PAM is positively correlated with the content of overall Ti and TiO6 in TS-1, and the ratio of TiO4/TiO6 can be adjusted by controlling the amount of PAM. Combined with the above analysis, it is found that the TiO4 and TiO6 contents of P-10-80-120 are the highest in the synthesized samples.Besides, the amount of PAM also affects the crystal size and pore structure of TS-1 zeolites. Compared to samples without PAM, a low PAM amount (PAM/SiO2 ≤ 0.10) has little effect on the particle size, while a high content of PAM (PAM/SiO2 = 0.20) leads to an increase in the particle size from 120–130 nm to 180 nm (Fig. S29†). Additionally, the introduction of PAM produces intragranular mesopores in TS-1, and their size gradually increases with increasing PAM amount. Among the synthesized samples, P-10-80-120 has the most uniform intragranular mesopores (ca. 4 nm), while P-5-80-120 has the largest mesopore volume (Vmeso = 0.4 cm3 g−1) (Fig. S30 and Table S2†).
In view of the significantly enhanced titanium content, abundant TiO6 species and mesoporous characteristics of the PAM-assisted samples, the catalytic performance was investigated by the oxidation of DBT with tert-butyl hydroperoxide (TBHP) and the oxidation of 1-hexene with H2O2. As shown in Fig. 7a, the conversion of DBT gradually increases with the increase of Ti content, especially the TiO6 content, in the P-x-80-120 samples (x = 0, 5 and 10). P-10-80-120 exhibits the best catalytic performance in removing 100.0% of DBT molecules within 40 min, much superior to that of conventional microporous TS-1 zeolite (micro-TS-1, Fig. S31†), which is mainly attributed to the significantly increased content of active species in the sample. The conversion of DBT on nano-anatase TiO2 is 8.8%, indicating that the high catalytic activity of P-10-80-120 may originate from the framework Ti species (TiO4 or TiO6) rather than the extra-framework anatase. Further study indicates that after washing off most TiO6 species by acid treatment without destroying the hierarchical structure (see details in Fig. S32†), the Si/Ti ratio increases from 29 to 67 in P-10-80-120-HCl and the catalytic activity decreases significantly, which highlights the high activity of TiO6 in the TS-1 catalyst. The durability tests over the P-10-80-120 catalyst were also conducted. The used catalyst can be regenerated by calcination in air. As presented in Fig. S33,† 100% of the initial catalytic activity remains after 3 runs, and 87% after 5 runs and the structural properties of the catalyst remain unchanged (Fig. S34†).
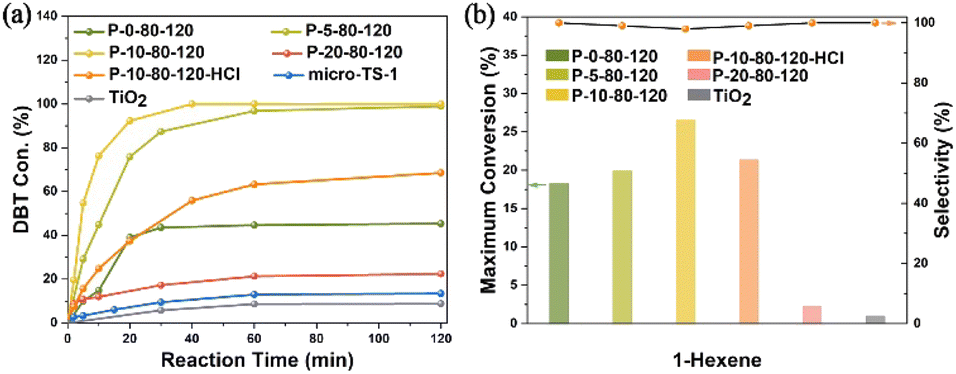 | ||
| Fig. 7 (a) Catalytic oxidation of DBT with TBHP and (b) epoxidation of 1-hexene over synthesized TS-1 samples. | ||
In addition, P-10-80-120 also exhibits the best catalytic performance in the epoxidation of 1-hexene, achieving the highest maximum theoretical conversion of 26.5%, higher than that of P-0-80-120 (18.3%) as shown in Fig. 7b. The conversion of H2O2 and the efficiency are 36.2% and 73.2% respectively (Table S4†). For the P-10-80-120-HCl sample, the catalytic activity drops from 26.5% to 21.4%, although it maintains the mesoporous structure (Fig. S32a†). This also highlights the high activity of TiO6 in the TS-1 catalyst, whereas it is found that the framework TiO4 species seems to exhibit slightly higher product selectivity than the TiO6 species, which may be related to their different catalytic mechanisms and needs further research. As a comparison, the maximum conversion of 1-hexene over the nano-sized anatase only reaches 1.0%, which means TiO2 is almost inert. Durability testing over the P-10-80-120 catalyst shows that 20% of the conversion is maintained after four cycles and the zeolite framework structure is maintained (Fig. S35 and S36†).
Conclusions
In summary, we developed a functional polymer-assisted synthesis method to control nucleation and active sites from the perspective of the crystallization process through transforming the dominant zeolite crystallization pathway from classical to non-classical. Various characterization studies clarified the specific interactions between the functional acyl groups of PAM and Si/Ti species, which promoted the assembly and precipitation of colloidal precursors in the form of wormlike particles, and the evolution of ordered structure fragments, thus accelerating the nucleation of zeolite and leading to the crystallization in a non-classical path. Meanwhile, owing to the coordination with the amino groups of PAM, a large number of Ti species were stabilized in WLPs, participated in the formation of crystal nuclei and directly transformed into the crystal through attachment of particles (CPA), greatly enriching the active sites. By such an approach, we successfully synthesized anatase-free TS-1 zeolite with Si/Ti ratio up to 29, a 1.5-fold reduction in crystallization time, and improved catalytic performance for oxidative desulfurization of dibenzothiophene and epoxidation of 1-hexene. This work sheds light on the rational control of the crystallization process and tailoring the physiochemical properties of zeolite through polymer modifiers, and such a method is expected to be extended to other heteroatom-containing zeolite catalysts.Data availability
All data associated with this article have been included in the main text and ESI.†Author contributions
Jiani Zhang: conceptualization, formal analysis, investigation, writing – original draft. Risheng Bai: formal analysis, investigation. Yida Zhou: formal analysis. Ziyi Chen: formal analysis. Peng Zhang: formal analysis. Jiyang Li: conceptualization, project administration, supervision, writing review & editing, funding acquisition. Jihong Yu: conceptualization, project administration, supervision, writing review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21671075 and 21621001) and the 111 Project (No. B17020).Notes and references
- R. J. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. E. Davies, D. J. Morgan, L. W. Chen, J. Z. Qi, J. Singleton, J. K. Edwards, S. J. Freakley, C. J. Kiely, Y. Yamamoto and G. J. Hutchings, Science, 2022, 376, 615–620 CrossRef CAS PubMed.
- Y. Li and J. Yu, Chem. Rev., 2014, 114, 7268–7316 CrossRef CAS PubMed.
- I. Yarulina, K. De Wispelaere, S. Bailleul, J. Goetze, M. Radersma, E. Abou-Hamad, I. Vollmer, M. Goesten, B. Mezari, E. J. M. Hensen, J. S. Martinez-Espin, M. Morten, S. Mitchell, J. Perez-Ramirez, U. Olsbye, B. M. Weckhuysen, V. Van Speybroeck, F. Kapteijn and J. Gascon, Nat. Chem., 2018, 10, 804–812 CrossRef CAS PubMed.
- Q. Zhang, J. Yu and A. Corma, Adv. Mater., 2020, 32, 2002927 CrossRef PubMed.
- Y. Zhou, J. Zhang, W. Ma, X. Yin, G. Chen, Y. Liu and J. Li, Inorg. Chem. Front., 2022, 9, 1752–1760 RSC.
- X. Zhang, J. Que, Y. Hou, J. Lyu, Z. Liu, K. Lei, S. Yu, X. Li, L. Chen and B. Su, Chem. J. Chin. Univ., 2021, 42, 2529–2539 Search PubMed.
- Y. Wei, M. Chen, X. Ren, Q. Wang, J. Han, W. Wu, X. Yang, S. Wang and J. Yu, CCS Chem., 2022, 4, 1708–1719 CrossRef CAS.
- R. Bai, Y. Song, R. Bai and J. Yu, Adv. Mater. Interfaces, 2020, 8, 2001095 CrossRef.
- J. Xu, Q. Wang and F. Deng, Acc. Chem. Res., 2019, 52, 2179–2189 CrossRef CAS PubMed.
- E. M. Gallego, M. T. Portilla, C. Paris, A. Leon-Escamilla, M. Boronat, M. Moliner and A. Corma, Science, 2017, 355, 1051–1054 CrossRef CAS PubMed.
- W. Schwieger, A. G. Machoke, T. Weissenberger, A. Inayat, T. Selvam, M. Klumpp and A. Inayat, Chem. Soc. Rev., 2016, 45, 3353–3376 RSC.
- M. Shamzhy, M. Opanasenko, P. Concepcion and A. Martinez, Chem. Soc. Rev., 2019, 48, 1095–1149 RSC.
- C. Li, P. Ferri, C. Paris, M. Moliner, M. Boronat and A. Corma, J. Am. Chem. Soc., 2021, 143, 10718–10726 CrossRef CAS PubMed.
- S. Guo, S. Wang, L. Zhang, Z. Qin, P. Wang, M. Dong, J. Wang and W. Fan, Chem. J. Chin. Univ., 2021, 42, 227–238 Search PubMed.
- Y. Zhao, H. B. Zhang, P. C. Wang, F. Q. Xue, Z. Ye, Y. H. Zhang and Y. Tang, Chem. Mater., 2017, 29, 3387–3396 CrossRef CAS.
- S. Karthika, T. K. Radhakrishnan and P. Kalaichelvi, Cryst. Growth Des., 2016, 16, 6663–6681 CrossRef CAS.
- A. I. Lupulescu and J. D. Rimer, Science, 2014, 344, 729–732 CrossRef CAS PubMed.
- K. N. Bozhilov, T. T. Le, Z. X. Qin, T. Terlier, A. Palcic, J. D. Rimer and V. Valtchev, Sci. Adv., 2021, 7, eabg0454 CrossRef CAS PubMed.
- A. I. Lupulescu and J. D. Rimer, Angew. Chem., Int. Ed., 2012, 124, 3401–3405 CrossRef.
- M. Kumar, M. K. Choudhary and J. D. Rimer, Nat. Commun., 2018, 9, 2129 CrossRef PubMed.
- M. Niederberger and H. Colfen, Phys. Chem. Chem. Phys., 2006, 8, 3271–3287 RSC.
- M. K. Choudhary, M. Kumar and J. D. Rimer, Angew. Chem., Int. Ed., 2019, 58, 15712–15716 CrossRef CAS PubMed.
- M. Jehannin, A. Rao and H. Colfen, J. Am. Chem. Soc., 2019, 141, 10120–10136 CrossRef CAS PubMed.
- R. Li, A. Smolyakova, G. Maayan and J. D. Rimer, Chem. Mater., 2017, 29, 9536–9546 CrossRef CAS.
- A. I. Lupulescu, W. Qin and J. D. Rimer, Langmuir, 2016, 32, 11888–11898 CrossRef CAS PubMed.
- C. Sun, Z. Liu, S. Wang, H. Pang, R. Bai, Q. Wang, W. Chen, A. Zheng, W. Yan and J. Yu, CCS Chem., 2021, 3, 189–198 CrossRef CAS.
- A. I. Lupulescu, M. Kumar and J. D. Rimer, J. Am. Chem. Soc., 2013, 135, 6608–6617 CrossRef CAS PubMed.
- M. Kumar, H. Luo, Y. Roman-Leshkov and J. D. Rimer, J. Am. Chem. Soc., 2015, 137, 13007–13017 CrossRef CAS PubMed.
- H. Dai, J. Claret, E. L. Kunkes, V. Vattipalli, N. Linares, C. Huang, M. Fiji, J. Garcia-Martinez, A. Moini and J. D. Rimer, Angew. Chem., Int. Ed., 2022, 61, e202117742 CAS.
- Y. Yu, R. Wang, W. Liu, Z. Chen, H. Liu, X. Huang, Z. Tang, Y. Liu and M. He, Appl. Catal., A, 2021, 610, 117953 CrossRef CAS.
- L. Wu, X. Deng, S. Zhao, H. Yin, Z. Zhuo, X. Fang, Y. Liu and M. He, Chem. Commun., 2016, 52, 8679–8682 RSC.
- W. Xu, T. Zhang, R. Bai, P. Zhang and J. Yu, J. Mater. Chem. A, 2020, 8, 9677–9683 RSC.
- R. Bai, Q. Sun, Y. Song, N. Wang, T. Zhang, F. Wang, Y. Zou, Z. Feng, S. Miao and J. Yu, J. Mater. Chem. A, 2018, 6, 8757–8762 RSC.
- T. J. Zhang, X. X. Chen, G. R. Chen, M. Y. Chen, R. S. Bai, M. J. Jia and J. H. Yu, J. Mater. Chem. A, 2018, 6, 9473–9479 RSC.
- W. B. Fan, R. G. Duan, T. Yokoi, P. Wu, Y. Kubota and T. Tatsumi, J. Am. Chem. Soc., 2008, 130, 10150–10164 CrossRef CAS PubMed.
- J. Zhang, H. Shi, Y. Song, W. Xu, X. Meng and J. Li, Inorg. Chem. Front., 2021, 8, 3077–3084 RSC.
- J. Wang, Y. Zhao, T. Yokoi, J. N. Kondo and T. Tatsumi, ChemCatChem, 2014, 6, 2719–2726 CrossRef CAS.
- W. B. Fan, B. B. Fan, X. H. Shen, J. F. Li, P. Wu, Y. Kubota and T. Tatsumi, Microporous Mesoporous Mater., 2009, 122, 301–308 CrossRef CAS.
- Y. Wang, L. Li, R. Bai, S. Gao, Z. Feng, Q. Zhang and J. Yu, Chin. J. Catal., 2021, 42, 2189–2196 CrossRef CAS.
- N. K. Mal, V. Ramaswamy and A. V. Ramaswamy, Stud. Surf. Sci. Catal., 1998, 113, 743–747 CrossRef CAS.
- L. Ren, C. Li, F. Fan, Q. Guo, D. Liang, Z. Feng, C. Li, S. Li and F. S. Xiao, Chem.–Eur. J., 2011, 17, 6162–6169 CrossRef CAS PubMed.
- Y. Wang and A. Tuel, Microporous Mesoporous Mater., 2008, 113, 286–295 CrossRef CAS.
- V. Pashkova, P. Klein, J. Dedecek, V. Tokarová and B. Wichterlová, Microporous Mesoporous Mater., 2015, 202, 138–146 CrossRef CAS.
- M. K. Choudhary, R. Jain and J. D. Rimer, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 28632–28639 CrossRef CAS PubMed.
- S. P. Chenakin, G. Melaet, R. Szukiewicz and N. Kruse, J. Catal., 2014, 312, 1–11 CrossRef CAS.
- J. M. da Silva, R. C. Sousa, J. C. S. Costa, J. L. Magalhães, G. E. Luz, C. V. R. de Moura and E. M. de Moura, Catal. Lett., 2021, 152, 585–599 CrossRef.
- Y. Sun, G. Li, Y. Gong, Z. Sun, H. Yao and X. Zhou, J. Hazard. Mater., 2021, 403, 124019 CrossRef CAS PubMed.
- S. Long, S. Zhou, F. Yang, K. Lu, T. Xi and Y. Kong, RSC Adv., 2016, 6, 76064–76074 RSC.
- C. E. A. Kirschhock, R. Ravishankar, F. Verspeurt, P. J. Grobet, P. A. Jacobs and J. A. Martens, J. Phys. Chem. B, 1999, 103, 4965–4971 CrossRef CAS.
- V. R. N. K. Mal, P. R. Rajamohanan and A. V. Ramaswam, Microporous Mater., 1997, 12, 331–340 CrossRef.
- C. E. A. Kirschhock, S. P. B. Kremer, P. J. Grobet, P. A. Jacobs and J. A. Martens, J. Phys. Chem. B, 2002, 106, 4897–4900 CrossRef CAS.
- C. J. Y. Houssin, C. E. A. Kirschhock, P. C. M. M. Magusin, B. L. Mojet, P. J. Grobet, P. A. Jacobs, J. A. Martens and R. A. van Santen, Phys. Chem. Chem. Phys., 2003, 5, 3518–3524 RSC.
- Z. Chen, X. Liu, Y. Yu, Z. Tang, J. Wang, D. Liu, N. Fang, Y. Lin, Y. Liu and M. He, Chem. Mater., 2021, 33, 4988–5001 CrossRef CAS.
- Z. Z. Sheng, H. Li, K. Du, L. Gao, J. Ju, Y. H. Zhang and Y. Tang, Angew. Chem., Int. Ed., 2021, 60, 13444–13451 CrossRef CAS PubMed.
- F. B. S. Bordiga, S. Coluccia, F. Genonic, C. Lamberti, G. Leofanti, L. Marchese, G. Petrini, G. Vlaic and A. Zecchina, Catal. Lett., 1994, 26, 195–208 CrossRef.
- Z. Lu, X. Liu, B. Zhang, Z. Gan, S. Tang, L. Ma, T. Wu, G. J. Nelson, Y. Qin, C. H. Turner and Y. Lei, J. Catal., 2019, 377, 419–428 CrossRef CAS.
- M. A. C. T. Blasco, A. Corma and J. Perez-Pariente, J. Am. Chem. Soc., 1993, 115, 11806–11813 CrossRef.
- M. Signorile, L. Braglia, V. Crocella, P. Torelli, E. Groppo, G. Ricchiardi, S. Bordiga and F. Bonino, Angew. Chem., Int. Ed., 2020, 59, 18145–18150 CrossRef CAS PubMed.
- R. Bai, M. T. Navarro, Y. Song, T. Zhang, Y. Zou, Z. Feng, P. Zhang, A. Corma and J. Yu, Chem. Sci., 2020, 11, 12341–12349 RSC.
- X. Song, X. Yang, T. Zhang, H. Zhang, Q. Zhang, D. Hu, X. Chang, Y. Li, Z. Chen, M. Jia, P. Zhang and J. Yu, Inorg. Chem., 2020, 59, 13201–13210 CrossRef CAS PubMed.
- L. Xu, D. D. Huang, C. G. Li, X. Y. Ji, S. Q. Jin, Z. C. Feng, F. Xia, X. H. Li, F. T. Fan, C. Li and P. Wu, Chem. Commun., 2015, 51, 9010–9013 RSC.
- Y. Li, T. Gao, Y. Yao, Z. Liu, Y. Kuang, C. Chen, J. Song, S. Xu, E. M. Hitz, B. Liu, R. J. Jacob, M. R. Zachariah, G. Wang and L. Hu, Adv. Energy Mater., 2018, 8, 1801289 CrossRef.
- F. N. Sayed, O. D. Jayakumar, R. Sasikala, R. M. Kadam, S. R. Bharadwaj, L. Kienle, U. Schürmann, S. Kaps, R. Adelung, J. P. Mittal and A. K. Tyagi, J. Phys. Chem. C, 2012, 116, 12462–12467 CrossRef CAS.
- Y. Song, N. Li, D. Chen, Q. Xu, H. Li, J. He and J. Lu, ACS Sustainable Chem. Eng., 2018, 6, 4000–4007 CrossRef CAS.
- F. Wang, X. He, L. Sun, J. Chen, X. Wang, J. Xu and X. Han, J. Mater. Chem. A, 2018, 6, 2091–2099 RSC.
- X. Zhang, D. Li, J. Wan and X. Yu, RSC Adv., 2016, 6, 17906–17912 RSC.
- R. Fang, W. Xiao, C. Miao, P. Mei, Y. Zhang, X. Yan and Y. Jiang, Electrochim. Acta, 2019, 317, 575–582 CrossRef CAS.
- G.-N. Huo, L.-L. Ma, X.-T. Liu, K.-H. Zhou, Z.-C. Suo, F.-F. Zhang, B.-L. Zhu, S.-M. Zhang and W.-P. Huang, Microchem. J., 2022, 172, 106957 CrossRef CAS.
- Y. Song, R. Bai, Y. Zou, Z. Feng and J. Yu, Inorg. Chem. Front., 2020, 7, 1872–1879 RSC.
- S. Du, H.-M. Chen, H.-X. Shen, J. Chen, C.-P. Li and M. Du, ACS Appl. Nano Mater., 2020, 3, 9393–9400 CrossRef CAS.
- G. Bellussi, R. Millini, Structure and Reactivity of Metals in Zeolite Materials, 2017, vol. 178, pp. 1–52 Search PubMed.
- W. Qin, A. Agarwal, M. K. Choudhary, J. C. Palmer and J. D. Rimer, Chem. Mater., 2019, 31, 3228–3238 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc04544c |
| This journal is © The Royal Society of Chemistry 2022 |