DOI:
10.1039/D2SC05621F
(Perspective)
Chem. Sci., 2022,
13, 12625-12633
Historical perspective on ruthenium-catalyzed hydrogen transfer and survey of enantioselective hydrogen auto-transfer processes for the conversion of lower alcohols to higher alcohols
Received
10th October 2022
, Accepted 20th October 2022
First published on 26th October 2022
Abstract
Ruthenium-catalyzed hydrogen auto-transfer reactions for the direct enantioselective conversion of lower alcohols to higher alcohols are surveyed. These processes enable completely atom-efficient carbonyl addition from alcohol proelectrophiles in the absence of premetalated reagents or metallic reductants. Applications in target-oriented synthesis are highlighted, and a brief historical perspective on ruthenium-catalyzed hydrogen transfer processes is given.
I. Historical perspective
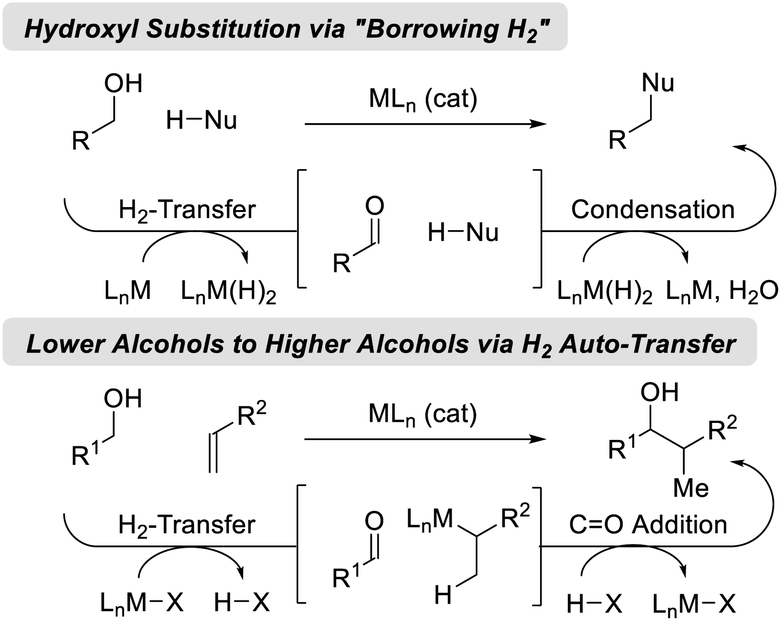
Catalytic hydrogenation and transfer hydrogenation account for roughly 14% of reactions used to prepare small-molecule clinical candidates (GMP reactions),1 and it is estimated that hydrogenation catalysis will continue to expand.2 Beyond hydrogenation, a survey of >9 million patents demonstrate that carbonyl additions (alongside Suzuki couplings) are among the most widely utilized methods for C–C bond formation in the pharmaceutical industry.3 These data have inspired our laboratory to develop a family of metal-catalyzed hydrogen auto-transfer reactions4 that merge the characteristics of catalytic hydrogenation and carbonyl addition by exploiting the native reducing power of alcohols for the concomitant generation of organometallic nucleophiles (from π-unsaturated pronucleophiles) and carbonyl electrophiles. In this way, carbonyl addition is achieved from the alcohol oxidation level in the absence of stoichiometric organometallic reagents. Processes of this type convert lower alcohols to higher alcohols, and are distinct from so-called “borrowing hydrogen” reactions,5 which affect formal hydroxyl substitution (Fig. 1).
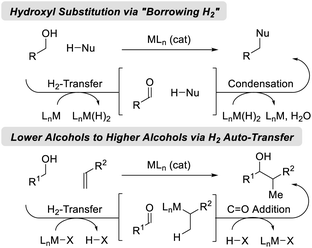 |
| | Fig. 1 Two major classes of hydrogen auto-transfer processes. | |
Ruthenium(II) complexes are octahedral d6 metal ions with unoccupied dx2−y2 orbitals, making them well-suited for alkoxide β-hydride elimination (Fig. 2). Indeed, beyond borrowing hydrogen processes,5 diverse transformations based on ruthenium(II)-catalyzed alcohol dehydrogenation have been developed.6 Important milestones in ruthenium-catalyzed hydrogen transfer include the very first examples of alkene homogenous hydrogenation developed by Halpern (1961),7 ruthenium-catalyzed transfer hydrogenations reported by Blum (1971),8 so-called “acceptorless” alcohol dehydrogenations described by Robinson (1977),9 oxidative esterifications devised by Shvo and Murahashi (1981)10a,b and related oxidative amidations (Murahashi 1991),10c respectively, as well as borrowing hydrogen alcohol aminations described by Watanabe (1981).11 In a major advance, Noyori developed highly enantioselective ruthenium-catalyzed hydrogenations and transfer hydrogenations (1986 and 1995, respectively).12 Finally, ruthenium-catalyzed hydrogen auto-transfer reactions that transform lower alcohols to higher alcohols were developed in the present authors' laboratory (2008).13,14 Corresponding enantioselective ruthenium-catalyzed processes soon followed (2011) and, in recent years, have expanded to encompass diverse reaction types. In this monograph, enantioselective methods for conversion of lower alcohols to higher alcohols via ruthenium-catalyzed hydrogen auto-transfer are cataloged.15 For surveys on the broader topic of metal-catalyzed carbonyl reductive couplings, the reader is referred to the review literature.16
 |
| | Fig. 2 Selected milestones in homogeneous ruthenium-catalyzed hydrogen transfer. | |
II. Enantioselective reactions of alcohols with diene and allene pronucleophiles
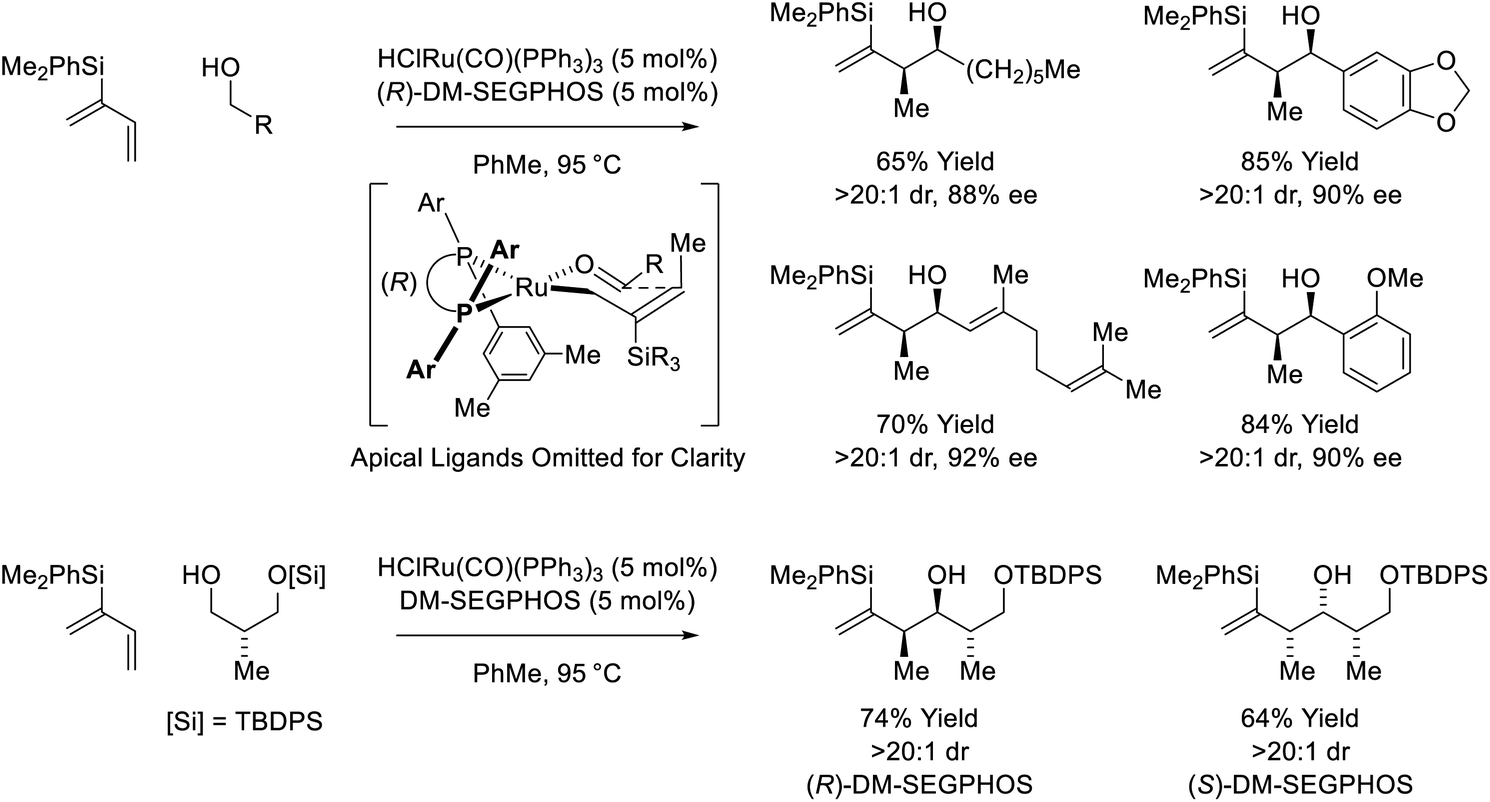
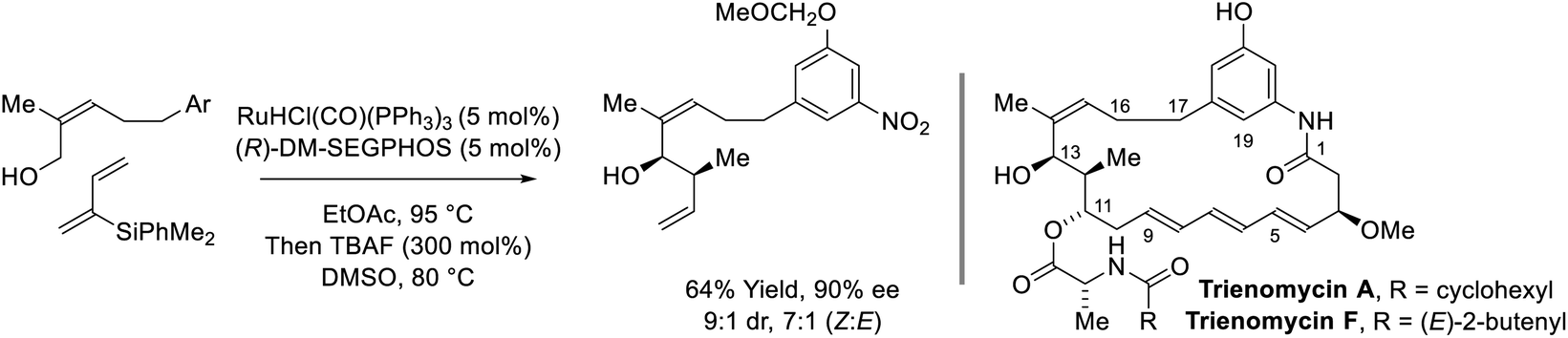
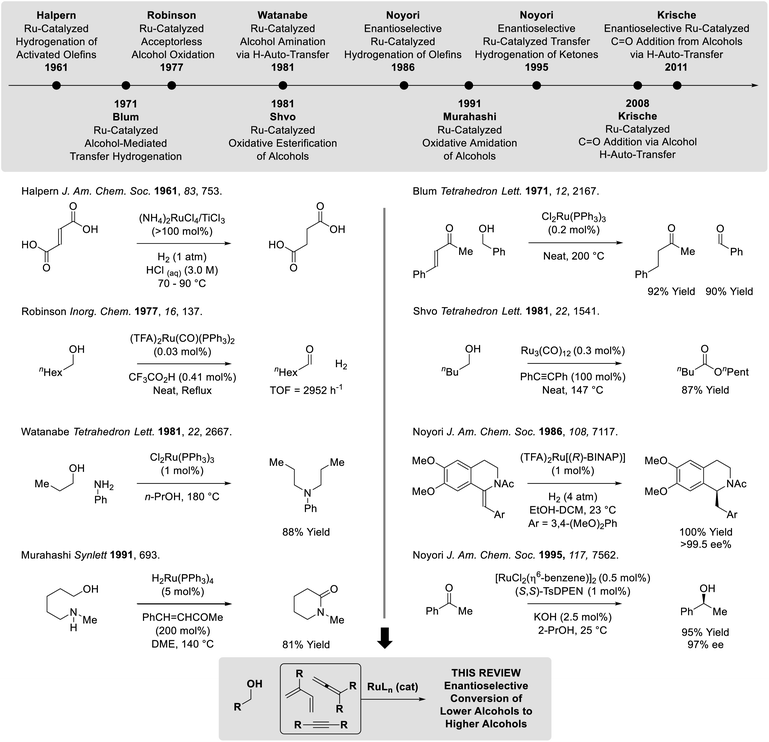
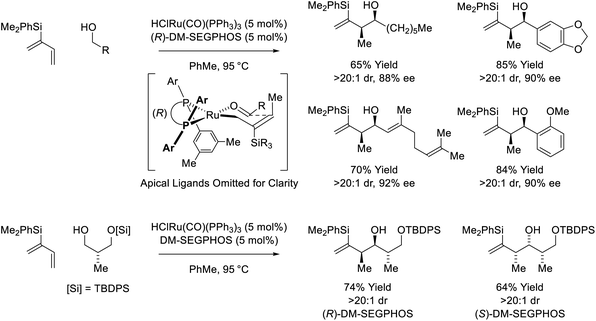
Following reports of racemic diene-alcohol C–C couplings via hydrogen auto-transfer,13,17 the first enantioselective ruthenium-catalyzed transformations of this type were developed (Scheme 1).18a Using a chiral ruthenium catalyst modified by (R)-DM-SEGPHOS, hydrogen transfer from primary alcohols to 2-trialkylsilyl-butadienes (“silaprenes”) generates crotylruthenium–aldehyde pairs that combine to form branched secondary homoallylic alcohols with high levels of enantiomeric enrichment. Allylic 1,2-strain imposed by the trialkylsilyl moiety defines alkene geometry of the transient σ-crotylruthenium nucleophile, which participates in stereospecific aldehyde addition through a closed transition structure to deliver the syn-diastereomers. Brimble exploited enantiomeric ruthenium catalysts in couplings of silaprene with the indicated chiral α-stereogenic alcohol derived from the Roche ester, enabling access to the syn,anti- or syn,syn-diketide stereotriads with good levels of catalyst-directed diastereocontrol.19 The silaprene-mediated conversion of lower alcohols to higher branched alcohols was utilized in total syntheses of trienomycins A and F (Scheme 2)21a and soraphen A (not shown).21b
 |
| | Scheme 1
syn-Diastereo- and enantioselective silaprene-mediated crotylation of primary alcohols. | |
 |
| | Scheme 2
syn-Diastereo- and enantioselective silaprene-mediated crotylation of primary alcohols for the total synthesis of trienomycins A and F. | |
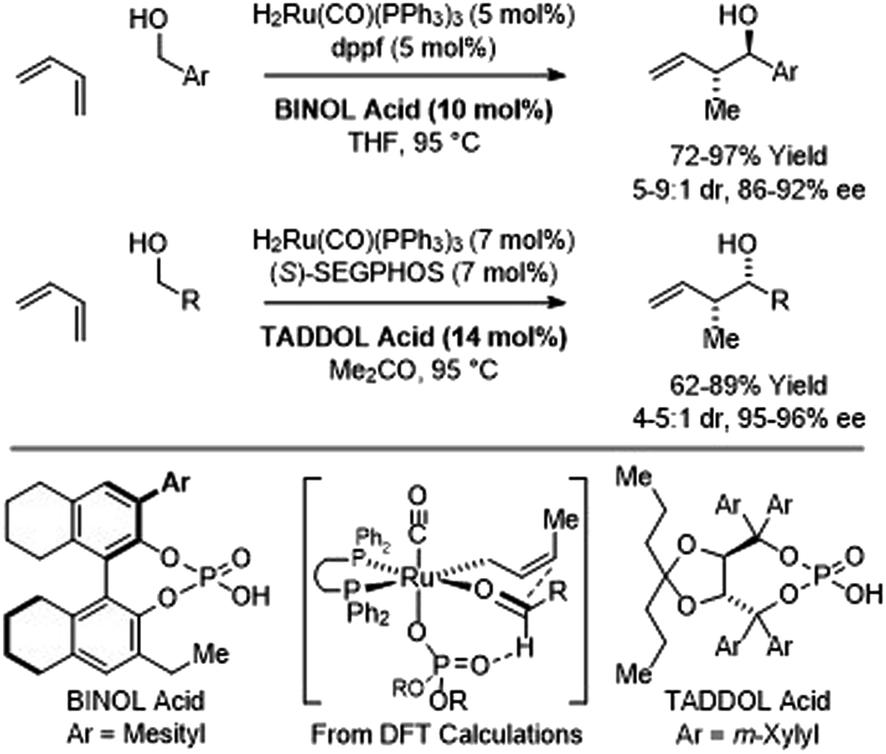
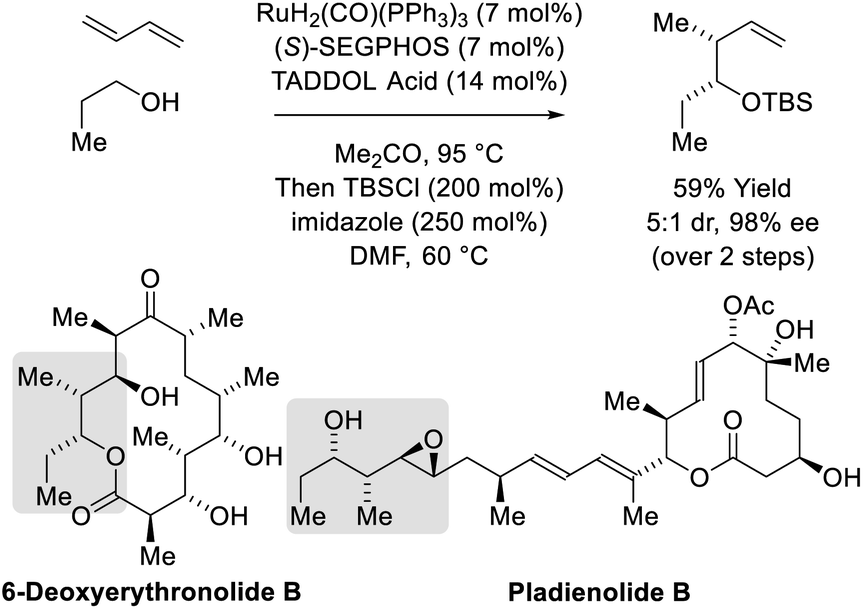
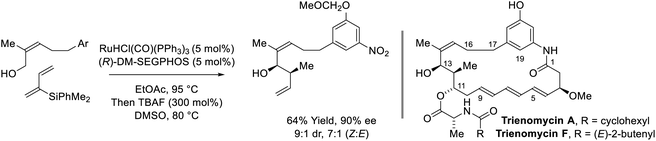
In C–C couplings of primary alcohols with the parent diene, butadiene (an abundant petrochemical feedstock), high levels of stereoselectivity are more difficult to achieve due to the challenge of controlling the geometry of the intervening σ-crotylruthenium nucleophile. Eventually, it was found that good levels of anti-diastereoselectivity and enantioselectivity could be obtained using ruthenium catalysts bound by chiral BINOL-derived phosphate counterions (Scheme 3).18b However, these processes were restricted to benzylic alcohols. Interestingly, using the corresponding chiral tartaric acid-derived phosphate counterion in combination with (S)-SEGPHOS, syn-diastereo- and enantioselective crotylations could be achieved (Scheme 3).18c It was postulated that the more Lewis basic tartaric acid-derived phosphate counterion forms a contact ion pair with ruthenium, retarding the rate of isomerization of the kinetic (Z)-σ-crotylruthenium haptomer (formed by hydroruthenation of the s-cis conformer of butadiene) with respect to carbonyl addition. Computational studies suggest the transition state for aldehyde addition leading to the syn-diastereomer is stabilized by a formyl hydrogen bond.20,22 The syn-diastereoselective butadiene-mediated conversion of lower alcohols to higher branched alcohols was utilized in total syntheses of 6-deoxyerythronolide B23 and pladienolide B (Scheme 4).24
 |
| | Scheme 3 Chiral counterion-directed anti- and syn-diastereo- and enantioselective butadiene-mediated crotylations of primary alcohols. | |
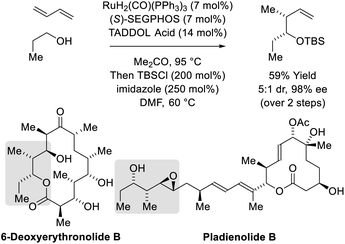 |
| | Scheme 4
syn-Diastereo- and enantioselective butadiene-mediated crotylation of primary alcohols in the total synthesis of 6-deoxyerythronolide B and pladienolide B. | |
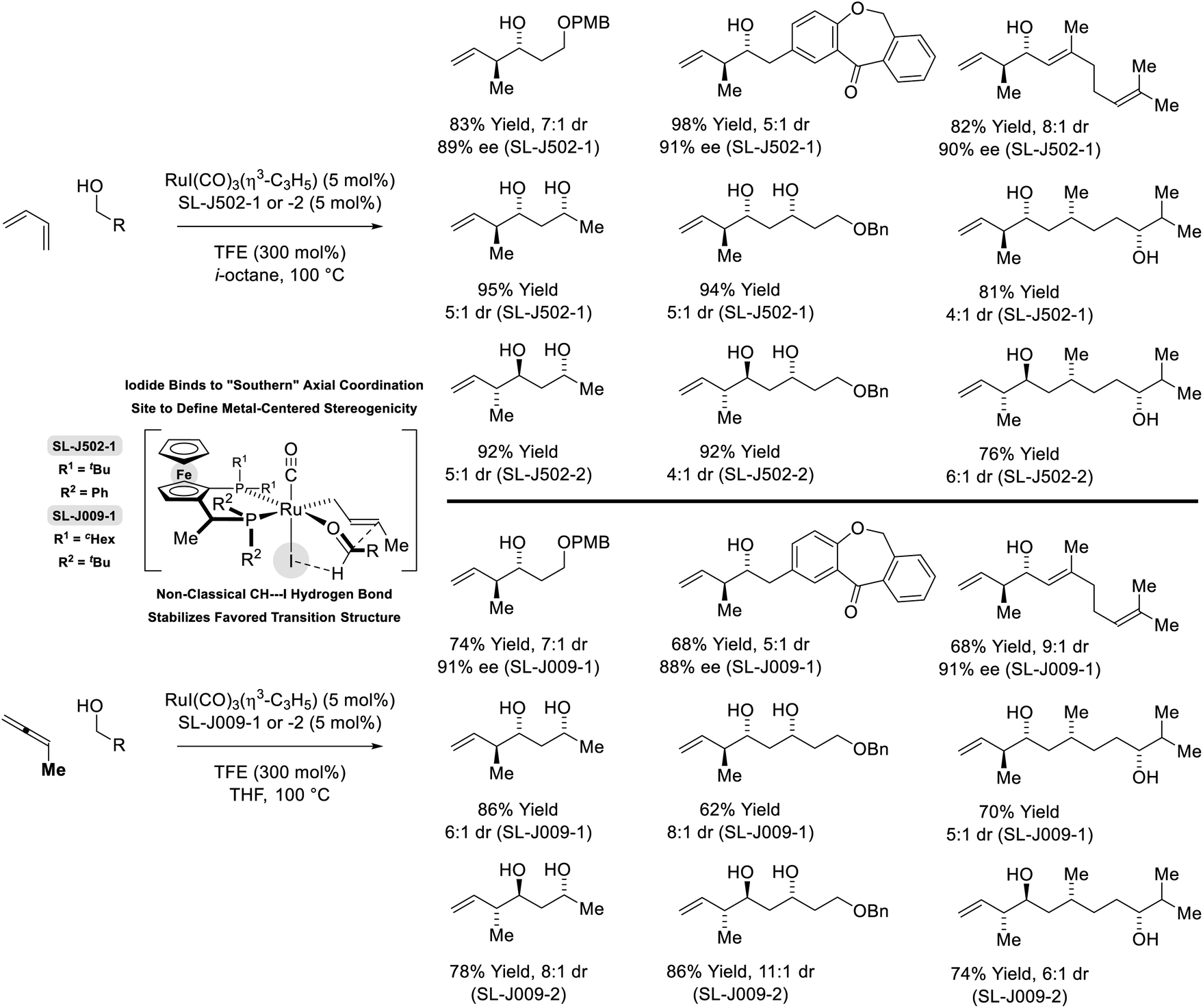
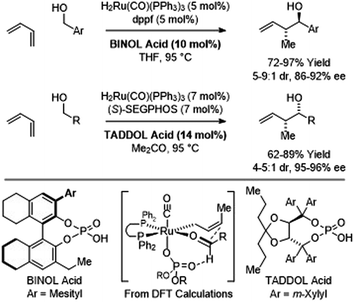
In more recent work, it was found that iodide-bound ruthenium-JOSIPHOS complexes catalyze the coupling of primary alcohols with butadiene to generate diastereo- and enantiomerically enriched products of anti-crotylation (Scheme 5).18d This catalytic system displays broad scope and is of greater practicality, as it does not require chiral counterions and the JOSIPHOS ligand is commercially available. One remarkable feature of the ruthenium-JOSIPHOS-catalyzed primary alcohol-butadiene C–C coupling relates to the ability to affect C–C coupling of primary alcohols in the presence of unprotected secondary alcohols without the need for hydroxyl protecting groups, which is attributed to the relatively rapid kinetics of primary alcohol dehydrogenation. As supported by experimental and computational studies, iodide-bound ruthenium-JOSIPHOS complexes (unlike the corresponding chloride or bromide complexes) enforce high levels of enantioselectivity by defining the stereogenic center at ruthenium and through formation of a formyl CH⋯I hydrogen-bond that stabilizes the favored transition state for carbonyl addition.22 Nearly identical reaction conditions were effective in couplings of primary alcohols with methylallene to form a duplicate set of diastereo- and enantiomerically enriched products. These processes represent an alternative to the longstanding use of discrete allylmetal reagents in enantioselective carbonyl crotylation.25
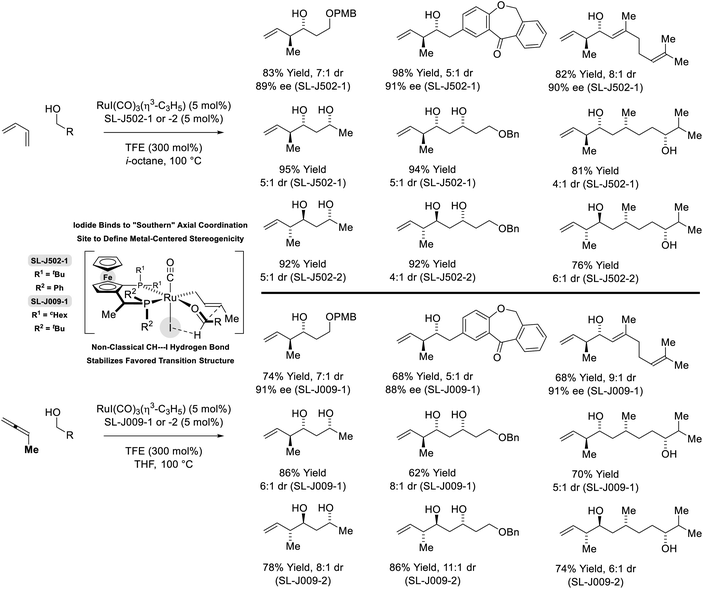 |
| | Scheme 5
anti-Diastereo- and enantioselective crotylation of primary alcohols by the C4 feedstocks 1,2- and 1,3-butadiene. | |
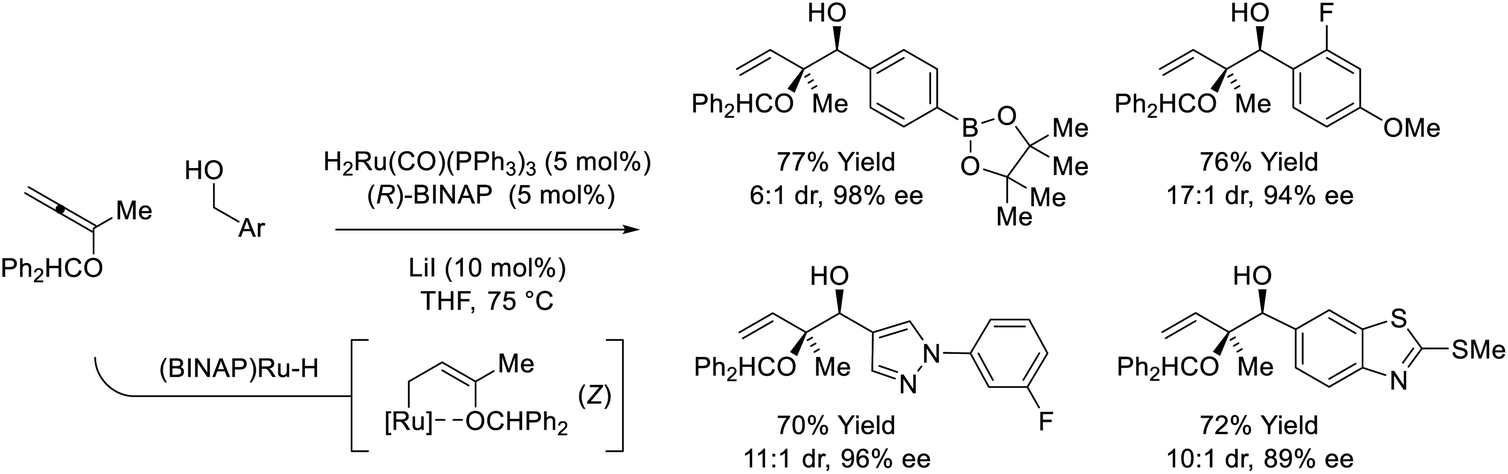
Ruthenium-BINAP-catalyzed couplings of primary benzylic alcohols with the indicated 1,1-disubstituted alkoxyallene enables formation of enantiomerically enriched syn–sec,tert-diols (Scheme 6).26 Unusual syn-diastereoselectivity is attributed to internal chelation of the benzhydryl oxygen to ruthenium, which directs formation of (Z)-σ-(alkoxy)allylruthenium isomers that participate in stereospecific carbonyl addition through chair-like transition structures. Low conversion was observed in attempted reactions of aliphatic alcohols, however, 2-propanol-mediated reductive couplings of alkyl-substituted aldehydes were achieved in good yield (not shown).
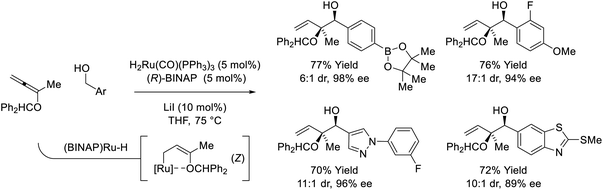 |
| | Scheme 6
syn-Diastereo- and enantioselective alkoxyallene-mediated conversion of primary alcohols to syn–sec,tert-homoallylic diols. | |
III. Enantioselective reactions of alcohols with alkyne pronucleophiles
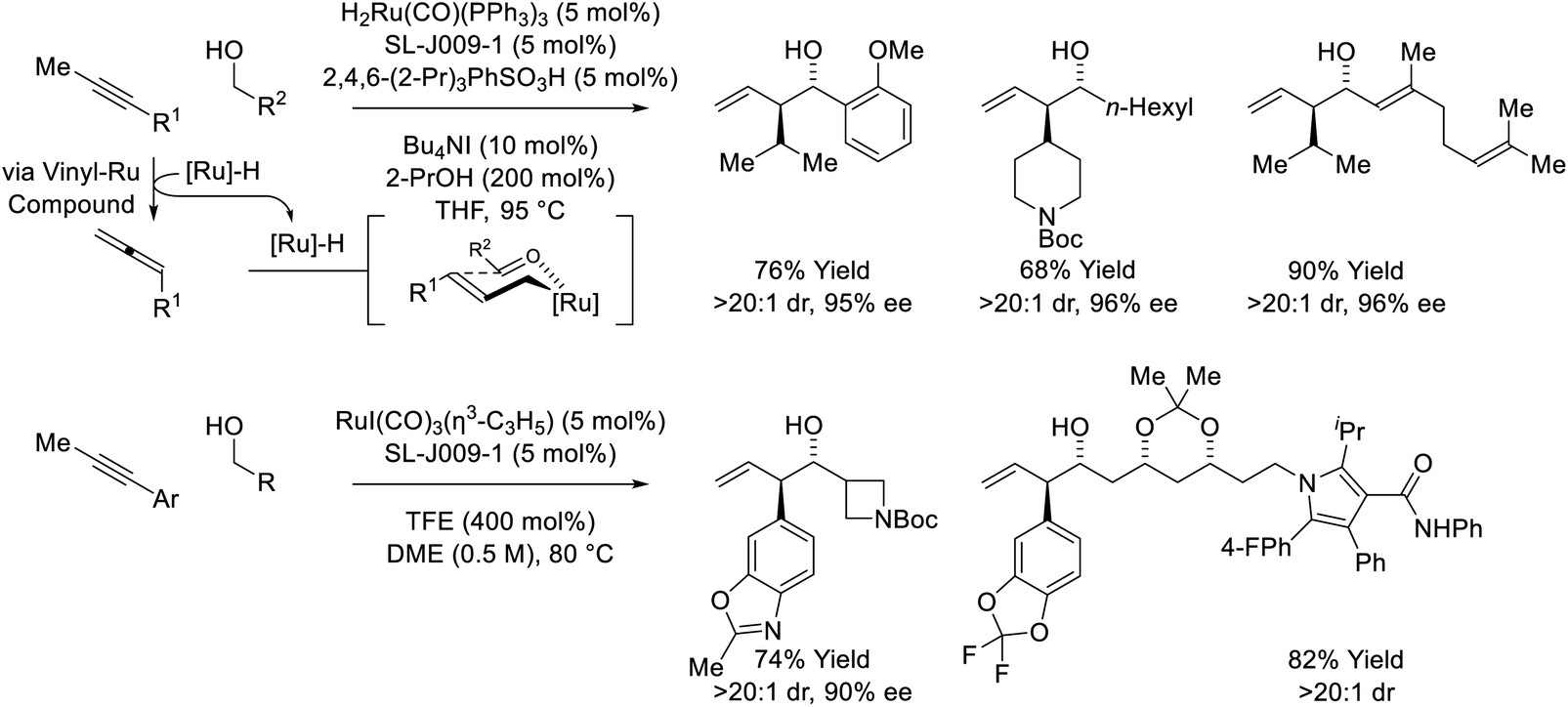
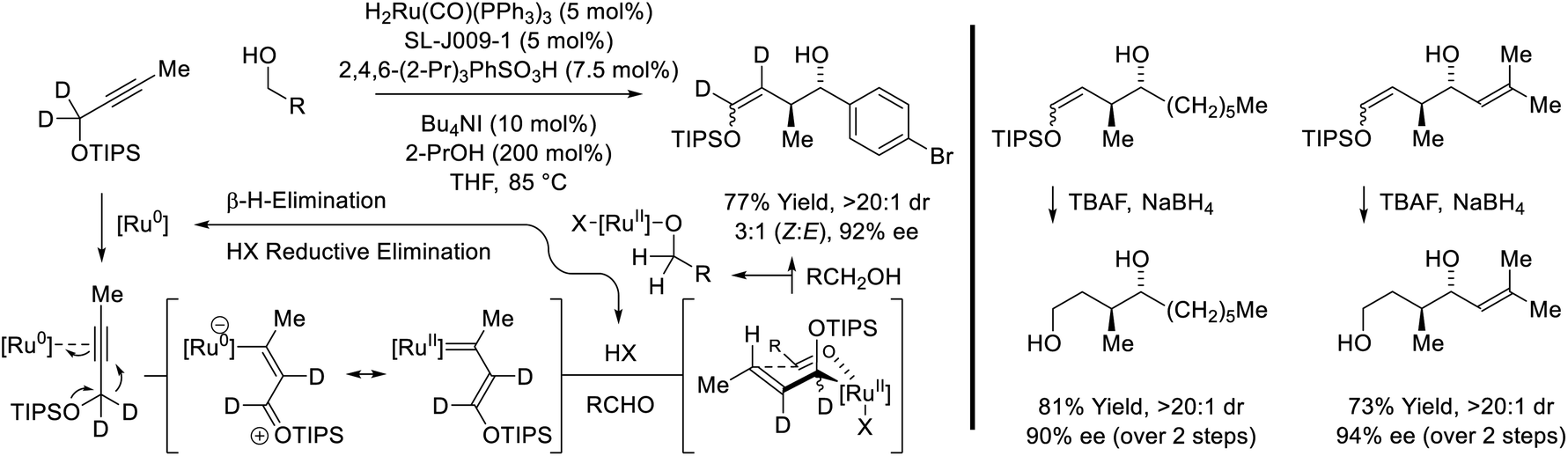
The first reported use of alkyne pronucleophiles in the enantioselective ruthenium-catalyzed conversion of lower alcohols to higher alcohols takes advantage of a dual catalytic cycle in which alkyne-to-allene isomerization (via hydrometallation-β-hydride elimination) is followed by allene-mediated carbonyl allylation to deliver branched homoallylic secondary alcohols (Scheme 7).27a In these processes, iodide-bound ruthenium-JOSIPHOS catalysts are generated in situ through the acid–base reaction of H2Ru(CO)(PPh3)3 with 2,4,6-(2-Pr)3PhSO3H, followed by substitution of the sulfonate counterion with iodide. Chloride and bromide-containing catalysts were inferior. As previously indicated (vide supra, Scheme 5), iodide defines the stereogenic center at ruthenium and stabilizes the favored transition state for carbonyl addition through formyl CH⋯I hydrogen-bonding with the transient aldehyde. Several improvements to this catalytic system were subsequently made. Replacement of 2,4,6-(iPr)3PhSO3H with 4-NO2PhSO3H resulted in >50% increase in isolated yield in reactions of 1-aryl-1-propynes, although catalyst loadings of 10 mol% remained necessary (not shown).27b The use of the iodide-containing precatalyst RuI(CO)3(η3-C3H5) in combination with trifluoroethanol (TFE) provided the most effective catalytic system for the reaction of primary alcohols with 1-aryl-1-propynes to form products of carbonyl anti-(α-aryl)allylation.27c In this process, TFE catalyses exchange of the homoallylic ruthenium alkoxide with reactant alcohol, mitigating issues of product inhibition.
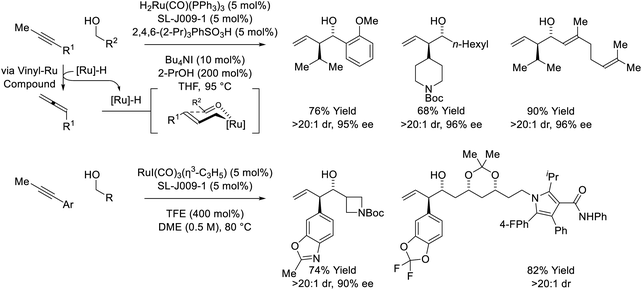 |
| | Scheme 7
anti-Diastereo- and enantioselective alkyne-mediated conversion of primary alcohols to branched homoallylic secondary alcohols. | |
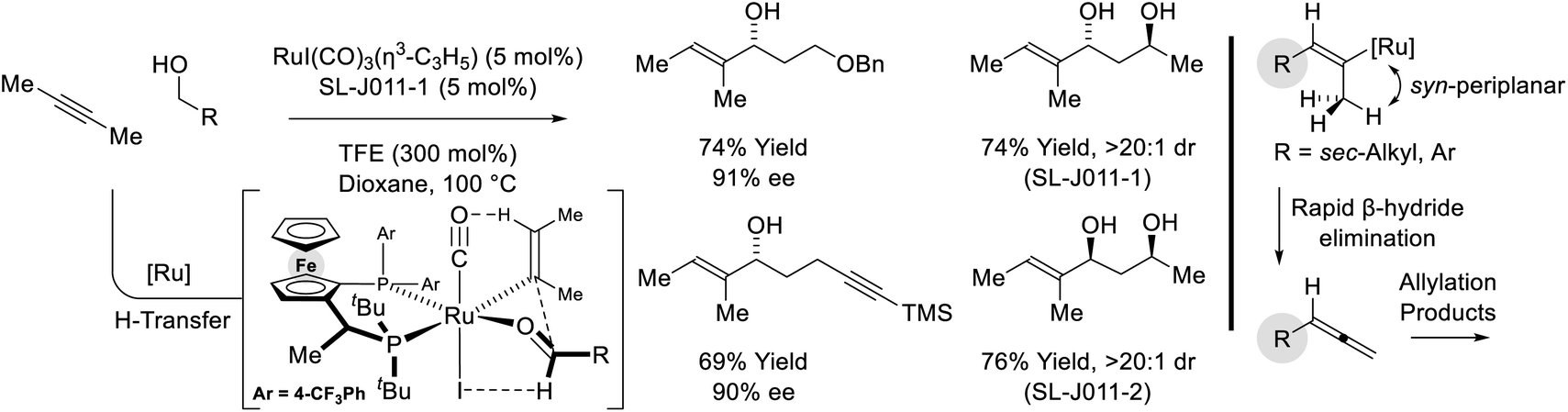
Remarkably, when the latter conditions are applied to 2-butyne, alkyne-to-allene isomerization is interrupted at the stage of intermediate vinylruthenium species via carbonyl addition to form products of alkenylation (Scheme 8).28,29 This divergent reactivity suggests acetylenic substituents larger than methyl (i.e. aryl or sec-alkyl) stabilize a syn-periplanar conformation of the carbon–ruthenium and propargylic C–H bonds, reducing the entropy of activation for β-hydride elimination to accelerate allene formation. The conversion of 2-butyne and primary alcohols to secondary allylic alcohols is highly chemoselective and proceeds in the presence of silyl-substituted alkynes and unprotected secondary alcohols. The indicated stereochemical model invokes a formyl CH⋯I hydrogen-bond22,27c and a ruthenium CH···O![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C[Ru] hydrogen-bond30 in the favored transition state for carbonyl addition.
C[Ru] hydrogen-bond30 in the favored transition state for carbonyl addition.
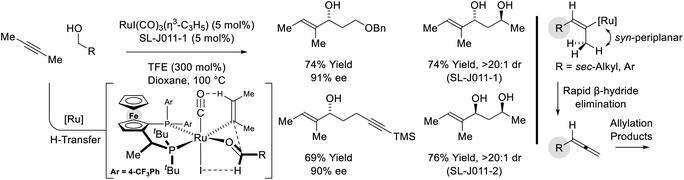 |
| | Scheme 8 Enantioselective conversion of primary alcohols and 2-butyne to secondary allylic alcohols. | |
Yet another mode of reactivity is observed in connection with the couplings of trialkylsilyl propargyl ethers and primary alcohols (Scheme 9).31 Under conditions essentially identical to those used in the coupling sec-alkyl propynes (vide supra, Scheme 7),27a alkyne-to-allene isomerization is not observed. Rather, as corroborated by deuterium labeling studies, alkyne coordination by zero-valent ruthenium triggers a 1,2-hydride shift to form a vinyl carbene, which upon protonation, generates a siloxy-π-allylruthenium nucleophile. Aldehyde addition by way of the indicated σ-allylruthenium haptomer delivers branched homoallylic alcohols that incorporate enol silyl ethers. Silyl deprotection–reduction of the crude reaction products provides the corresponding 1,4-diols with excellent control of regio-, diastereo- and enantioselectivity.
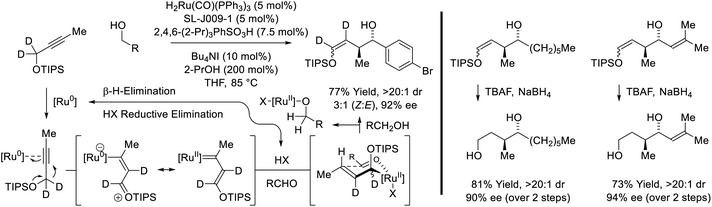 |
| | Scheme 9
anti-Diastereo- and enantioselective conversion of primary alcohols and trialkylsilyl propargyl ethers to branched secondary alcohols. | |
IV. Enantioselective reactions of alcohols with enyne pronucleophiles
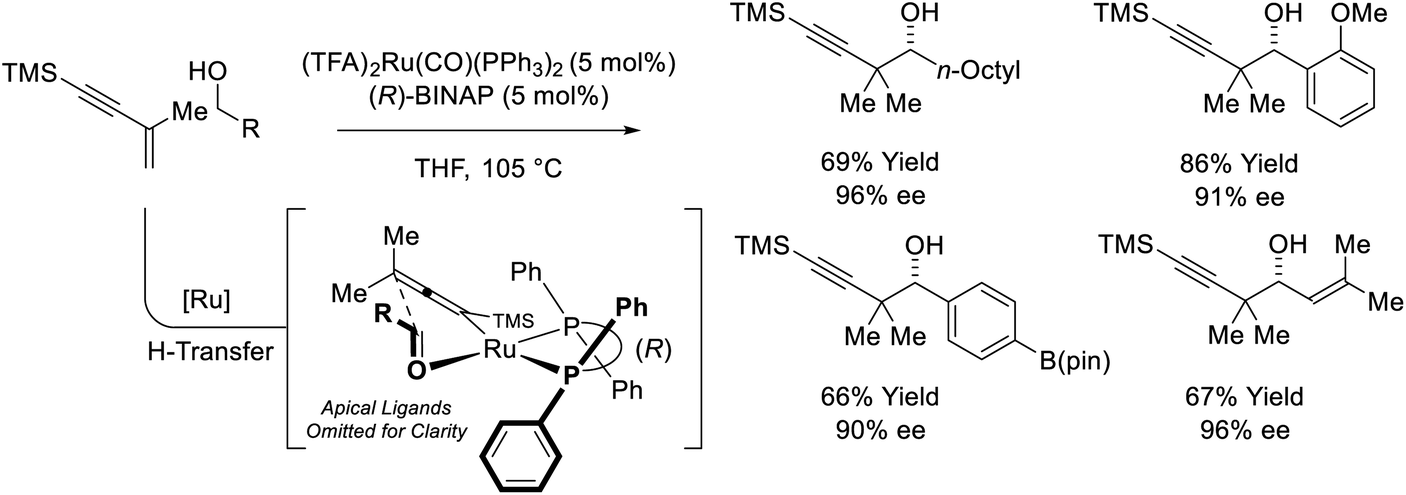
The first examples of enyne-mediated carbonyl propargylation were reported from the present authors laboratory.32–34 The first enantioselective ruthenium-catalyzed conversion of 1,3-enynes and primary alcohols to form homopropargylic secondary alcohol was achieved using the chiral ruthenium complex assembled from (TFA)2Ru(CO)(PPh3)2 and (R)-BINAP (Scheme 10).32c The resulting secondary homopropargyl alcohols bearing gem-dimethyl groups are formed with excellent levels of enantiomeric enrichment. In this process, hydrogen transfer from the primary alcohol to the 1,3-enyne generates a prochiral allenylruthenium nucleophile, which engages the transient aldehyde in carbonyl addition by way of the indicated closed transition structure. In support of this mechanism, the reaction of HClRu(CO)(PPh3)3 with 1,3-enynes to form an isolable σ-allenylruthenium complex characterized by single crystal X-ray diffraction has been described.35 This protocol represents an alternative to the longstanding use of discrete allenylmetal reagents in enantioselective carbonyl propargylation.36
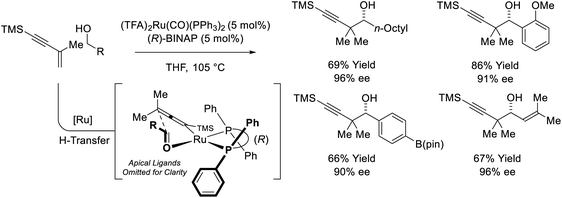 |
| | Scheme 10 Enantioselective conversion of primary alcohols and 2-butyne to secondary allylic alcohols. | |
V. Conclusion and future outlook
Ruthenium complexes are the prototypical hydrogen transfer catalysts and they continue to facilitate an ever-increasing array of chemical processes. As described in this perspective article, a new reactivity mode in the field of ruthenium catalysis is represented by the emerging class of chemical transformations in which alcohol dehydrogenation triggers reductive generation of aldehyde-organometal pairs that combine to affect byproduct-free carbonyl addition. Such reactions enable direct conversion of lower alcohols to higher alcohols and allow abundant π-unsaturated feedstocks to serve as surrogates to stoichiometric organometallic reagents. Discovered and developed wholly in the present authors laboratory, processes of this type raise numerous other possibilities. Beyond continued efforts to adapt this concept to other types of carbonyl additions (alkylations,37 vinylations,38 alkynylations, and arylation), methods for the direct conversion of lower amine to higher amines (hydroaminoalkylation)39 represents an important challenge that merits further attention. It is the authors' hope that the present state of the art summary will expedite progress toward this and other challenges in this growing area of research.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM069445) are acknowledged for partial support of this research.
References
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC.
- M. A. Stoffels, F. J. R. Klauck, T. Hamadi, F. Glorius and J. Leker, Adv. Synth. Catal., 2020, 362, 1258–1274 CrossRef CAS PubMed.
- N. Schneider, D. M. Lowe, R. A. Sayle, M. A. Tarselli and G. A. Landrum, J. Med. Chem., 2016, 59, 4385–4402 CrossRef CAS PubMed.
- For recent reviews on hydrogen auto-transfer for the conversion of lower alcohols to higher alcohols, see:
(a) S. W. Kim, W. Zhang and M. J. Krische, Acc. Chem. Res., 2017, 50, 2371–2380 CrossRef CAS PubMed;
(b) R. S. Doerksen, C. C. Meyer and M. J. Krische, Angew. Chem., Int. Ed., 2019, 58, 14055–14064 CrossRef CAS PubMed;
(c) C. G. Santana and M. J. Krische, ACS Catal., 2021, 11, 5572–5585 CrossRef CAS PubMed.
- For selected reviews on “borrowing hydrogen” processes that result in hydroxyl substitution, see:
(a) M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS;
(b) G. Guillena, D. J. Ramón and M. Yus, Angew. Chem., Int. Ed., 2007, 46, 2358–2364 CrossRef CAS PubMed;
(c) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed;
(d) S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1853–1864 CrossRef;
(e) Q. Yang, Q. Wang and Z. Yu, Chem. Soc. Rev., 2015, 44, 2305–2329 RSC;
(f) H. Aitchison, R. L. Wingad and D. F. Wass, ACS Catal., 2016, 6, 7125–7132 CrossRef CAS;
(g) A. Quintard and J. Rodriguez, Chem. Commun., 2016, 52, 10456–10473 RSC;
(h) B. G. Reed-Berendt, K. Polidano and L. C. Morrill, Org. Biomol. Chem., 2019, 17, 1595–1607 RSC;
(i) T. Kwok, O. Hoff, R. J. Armstrong and T. J. Donohoe, Chem.–Eur. J., 2020, 26, 12912–12926 CrossRef CAS PubMed.
- For selected general reviews on ruthenium-catalyzed hydrogen transfer processes, see:
(a) U. Matteoli, P. Frediani, M. Bianchi, C. Botteghi and S. Gladiali, J. Mol. Catal., 1981, 12, 265–319 CrossRef CAS;
(b) S. I. Murahashi and T. Naota, Adv. Met.-Org. Chem., 1994, 3, 225–254 CAS;
(c) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS;
(d) S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS;
(e) Y. Ahn, S.-B. Ko, M.-J. Kim and J. Park, Coord. Chem. Rev., 2008, 252, 647–658 CrossRef CAS;
(f) M. C. Warner, C. P. Casey and J.-E. Bäeckvall, Top. Organomet. Chem., 2011, 37, 85–125 CrossRef CAS;
(g) C. Gunanathan and D. Milstein, Science, 2013, 341, 249–260 CrossRef CAS PubMed;
(h) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed;
(i) J. Zhang, B. Guo, D. J. Young and H.-X. Li, Dalton Trans., 2020, 49, 15527–15547 RSC;
(j) V. Ritleng and J. G. de Vries, Molecules, 2021, 26, 4076–4121 CrossRef CAS PubMed.
- J. Halpern, J. F. Harrod and B. R. James, J. Am. Chem. Soc., 1961, 83, 753–754 CrossRef CAS.
- Y. Sasson and J. Blum, Tetrahedron Lett., 1971, 12, 2167–2170 CrossRef.
- A. Dobson and S. D. Robinson, Inorg. Chem., 1977, 16, 137–142 CrossRef CAS.
- For oxidative esterification and oxidative amidation of primary alcohols, see:
(a) Y. Blum, D. Reshef and Y. Shvo, Tetrahedron Lett., 1981, 22, 1541–1544 CrossRef CAS;
(b) S.-I. Murahashi, K.-I. Ito, T. Naota and Y. Maeda, Tetrahedron Lett., 1981, 22, 5327–5330 CrossRef CAS;
(c) T. Naoto and S.-I. Murahashi, Synlett, 1991, 10, 693–694 CrossRef . Also see,;
(d) C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS PubMed.
- Alcohol amination via metal-catalyzed hydrogen auto-transfer was reported contemporaneously by Wantanabe and Grigg using ruthenium and rhodium catalysts, respectively:
(a) Y. Watanabe, Y. Tsuji and Y. Ohsugi, Tetrahedron Lett., 1981, 22, 2667–2670 CrossRef CAS;
(b) R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit and N. Tongpenyai, J. Chem. Soc., Chem. Commun., 1981, 611–612 RSC.
-
(a) R. Noyori, M. Ohta, Y. Hsiao, M. Kitamura, T. Ohta and H. Takaya, J. Am. Chem. Soc., 1986, 108, 7117–7119 CrossRef CAS;
(b) S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562–7563 CrossRef CAS.
- The first metal-catalyzed hydrogen auto-transfer reactions to convert lower alcohols to higher alcohols employed catalysts based on iridium and ruthenium:
(a) J. F. Bower, E. Skucas, R. L. Patman and M. J. Krische, J. Am. Chem. Soc., 2007, 129, 15134–15135 CrossRef CAS PubMed;
(b) F. Shibahara, J. F. Bower and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 6338–6339 CrossRef CAS PubMed.
- This reactivity pattern appears quite unique. To our knowledge, the closest precedent is the ruthenium-catalyzed conversion of primary alcohols and allyl acetate to α,β-unsaturated enones: T. Kondo, T. Mukai and Y. Watanabe, J. Org. Chem., 1991, 56, 487 CrossRef CAS.
- For earlier reviews on ruthenium-catalyzed hydrogen auto-transfer for the conversion of lower alcohols to higher alcohols, see:
(a) F. Shibahara and M. J. Krische, Chem. Lett., 2008, 37, 1102–1107 CrossRef CAS PubMed;
(b) J. Moran and M. J. Krische, Pure Appl. Chem., 2012, 84, 1729–1739 CrossRef CAS PubMed;
(c) F. Perez, S. Oda, L. M. Geary and M. J. Krische, Top. Curr. Chem., 2016, 374, 365–387 Search PubMed.
- For selected reviews on the general topic of metal-catalyzed carbonyl reductive coupling, see:
(a) J. Montgomery, Angew. Chem. Int. Ed., 2004, 43, 3890–3908 CrossRef CAS PubMed;
(b)
M. J. Krische and H.-Y. Jang, in Comprehensive Organometallic Chemistry III, ed. P. Mingos, and R. Crabtree, Elsevier, Oxford, 3rd edn, 2006, vol. 10, ch. 4, pp. 493–536 Search PubMed;
(c) M. J. Krische, Top. Curr. Chem., 2007, 279, 77–104 CrossRef;
(d) M. Kimura and Y. Tamaru, Top. Curr. Chem., 2007, 279, 173–207 CrossRef CAS;
(e) T. Moragas, A. Correa and R. Martin, Chem.–Eur. J., 2014, 20, 8242–8258 CrossRef CAS PubMed;
(f) K. D. Nguyen, B. Y. Park, T. Luong, H. Sato, V. J. Garza and M. J. Krische, Science, 2016, 354, aah5133 CrossRef PubMed;
(g) M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026–6052 CrossRef CAS PubMed;
(h) E. Ortiz, J. Z. Shezaf, Y.-H. Chang and M. J. Krische, ACS Catal., 2022, 12, 8164–8174 CrossRef CAS.
- For ruthenium-catalyzed diene-alcohol C–C couplings to form secondary homoallylic alcohols, see ref. 13 and following studies:
(a) H. Han and M. J. Krische, Org. Lett., 2010, 12, 2844–2846 CrossRef CAS PubMed;
(b) J. C. Leung, L. M. Geary, T.-Y. Chen, J. R. Zbieg and M. J. Krische, J. Am. Chem. Soc., 2012, 134, 15700–15703 CrossRef CAS PubMed;
(c) T.-Y. Chen and M. J. Krische, Org. Lett., 2013, 15, 2994–2997 CrossRef CAS PubMed;
(d) B. Y. Park, T. P. Montgomery, V. J. Garza and M. J. Krische, J. Am. Chem. Soc., 2013, 135, 16320–16323 CrossRef CAS PubMed.
- For enantioselective ruthenium-catalyzed diene-alcohol C–C couplings to form higher homoallylic alcohols, see:
(a) J. R. Zbieg, J. Moran and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 10582–10586 CrossRef CAS PubMed;
(b) J. R. Zbieg, E. Yamaguchi, E. L. McInturff and M. J. Krische, Science, 2012, 336, 324–327 CrossRef CAS PubMed;
(c) E. L. McInturff, E. Yamaguchi and M. J. Krische, J. Am. Chem. Soc., 2012, 134, 20628–20631 CrossRef CAS PubMed;
(d) E. Ortiz, B. J. Spinello, Y. Cho, J. Wu and M. J. Krische, Angew. Chem., Int. Ed., 2022, 61, e202212814 Search PubMed.
- M. Pantin, J. G. Hubert, T. Söhnel, M. A. Brimble and D. P. Furkert, J. Org. Chem., 2017, 82, 11225–11229 CrossRef CAS PubMed.
- For computational studies of chiral counterion-directed enantioselective ruthenium-catalyzed diene-alcohol C–C couplings to provide higher homoallylic alcohols, see: M. N. Grayson, M. J. Krische and K. N. Houk, J. Am. Chem. Soc., 2015, 137, 8838–8850 CrossRef CAS PubMed.
-
(a) D. J. Del Valle and M. J. Krische, J. Am. Chem. Soc., 2013, 135, 10986–10989 CrossRef CAS PubMed;
(b) T. T. Schempp and M. J. Krische, J. Am. Chem. Soc., 2022, 144, 1016–1022 CrossRef CAS PubMed.
- For selected studies of formyl CH hydrogen bonds, see ref. 20 and following reports:
(a) E. J. Corey and T. W. Lee, Chem. Commun., 2001, 1321–1329 RSC;
(b) T. S. Thakur, M. T. Kirchner, D. Bläser, R. Boese and G. R. Desiraju, Phys. Chem. Chem. Phys., 2011, 13, 14076–14091 RSC.
- X. Gao, S. K. Woo and M. J. Krische, J. Am. Chem. Soc., 2013, 135, 4223–4226 CrossRef CAS PubMed.
- M. Yoo and M. J. Krische, Angew. Chem., Int. Ed., 2021, 60, 13923–13928 CrossRef CAS PubMed.
- For selected reviews on the use of allylmetal reagents in carbonyl allylation and crotylation, see:
(a) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763–2794 CrossRef CAS PubMed;
(b) D. G. Hall, Synlett, 2007, 11, 1644–1655 CrossRef;
(c) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2011, 111, 7774–7854 CrossRef CAS;
(d) H.-X. Huo, J. R. Duvall, M.-Y. Huang and R. Hong, Org. Chem. Front., 2014, 1, 303–320 RSC;
(e) K. Spielmann, G. Niel, R. M. de Figueiredo and J.-M. Campagne, Chem. Soc. Rev., 2018, 47, 1159–1173 RSC.
- M. Xiang, D. Pfaffinger, E. Ortiz, G. A. Brito and M. J. Krische, J. Am. Chem. Soc., 2021, 143, 8849–8854 CrossRef CAS PubMed.
-
(a) T. Liang, K. D. Nguyen, W. Zhang and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 3161–3164 CrossRef CAS PubMed;
(b) M. Xiang, A. Ghosh and M. J. Krische, J. Am. Chem. Soc., 2021, 143, 2838–2845 CrossRef CAS PubMed;
(c) E. Ortiz, J. Z. Shezaf, Y.-H. Chang, T. P. Gonçalves, K.-W. Huang and M. J. Krische, J. Am. Chem. Soc., 2021, 143, 16709–16717 CrossRef CAS PubMed.
- E. Ortiz, Y.-H. Chang, J. Z. Shezaf, W. Shen and M. J. Krische, J. Am. Chem. Soc., 2022, 144, 8861–8869 CrossRef CAS PubMed.
- The coupling of 2-butyne with primary alcohols to form racemic products of carbonyl alkenylation was previously described: R. L. Patman, M. R. Chaulagain, V. M. Williams and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 2066–2067 CrossRef CAS PubMed.
- An exhaustive survey of X-ray diffraction data reveal CH⋯OC[Ru] hydrogen-bonds are conserved across diverse vinylruthenium carbonyl complexes. These hydrogen bonds are < 2.96 Å in length, which is significantly shorter than the <3.2 Å distance usually observed for CH⋯O hydrogen bonds. See ref. 27 for primary literature references.
- T. Liang, W. Zhang, T.-Y. Chen, K. D. Nguyen and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 13066–13071 CrossRef CAS PubMed.
- For ruthenium-catalyzed coupling of 1,3-enynes with primary alcohols to form secondary homopropargylic alcohols, see:
(a) R. L. Patman, V. M. Williams, J. F. Bower and M. J. Krische, Angew. Chem., Int. Ed., 2008, 47, 5220–5223 CrossRef CAS PubMed;
(b) L. M. Geary, J. C. Leung and M. J. Krische, Chem. - Eur. J., 2012, 18, 16823–16827 CrossRef CAS PubMed;
(c) K. D. Nguyen, D. Herkommer and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 5238–5241 CrossRef CAS PubMed.
- For iridium- and rhodium-catalyzed coupling of 1,3-enynes or propargyl chlorides with primary alcohols to form secondary homopropargylic alcohols, see:
(a) L. M. Geary, S. K. Woo, J. C. Leung and M. J. Krische, Angew. Chem., Int. Ed., 2012, 51, 2972–2976 CrossRef CAS PubMed;
(b) S. K. Woo, L. M. Geary and M. J. Krische, Angew. Chem., Int. Ed., 2012, 51, 7830–7834 CrossRef CAS PubMed;
(c) T. Liang, S. K. Woo and M. J. Krische, Angew. Chem., Int. Ed., 2016, 55, 9207–9211 CrossRef CAS PubMed.
- For a review on carbonyl propargylation via alcohol-mediated hydrogen auto-transfer, see: B. R. Ambler, S. K. Woo and M. J. Krische, ChemCatChem, 2019, 11, 324–332 CrossRef CAS PubMed.
- Y. Wakatsuki, H. Yamazaki, Y. Maruyama and I. Shimizu, J. Chem. Soc., Chem. Commun., 1991, 261–263 RSC.
- For selected reviews on the use of allenylmetal reagents in carbonyl propargylation, see:
(a) J. A. Marshall, Chem. Rev., 1996, 96, 31–48 CrossRef CAS PubMed;
(b) C.-H. Ding and X.-L. Hou, Chem. Rev., 2011, 111, 1914–1937 CrossRef CAS PubMed;
(c) H. M. Wisniewska and E. R. Jarvo, J. Org. Chem., 2013, 78, 11629–11636 CrossRef CAS PubMed;
(d) T. Thaima, F. Zamani, C. J. T. Hyland and S. G. Pyne, Synthesis, 2017, 49, 1461–1480 CrossRef CAS.
- For use of ethylene and α-olefins as alkylmetal equivalents in carbonyl addition from alcohol proelectrophiles, see:
(a) E. Yamaguchi, J. Mowat, T. Luong and M. J. Krische, Angew. Chem., Int. Ed., 2013, 52, 8428–8431 CrossRef CAS PubMed;
(b) B. Y. Park, T. Luong, H. Sato and M. J. Krische, J. Org. Chem., 2016, 81, 8585–8594 CrossRef CAS PubMed.
- For vinyl transfer beyond use of alkyne pronucleophiles, see: B. Y. Park, T. Luong, H. Sato and M. J. Krische, J. Am.
Chem. Soc., 2015, 137, 7652–7655 CrossRef CAS PubMed.
- For late transition metal-catalyzed hydroaminoalkylation beyond directing groups, see: T.-Y. Chen, R. Tsutsumi, T. P. Montgomery, I. Volchkov and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 1798–1801 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Jonathan Z.
Shezaf
,
Jonathan Z.
Shezaf