
Recent advances in non-precious group metal-based catalysts for water electrolysis and beyond
Hee Jin
Kim†
 a,
Ho Young
Kim†
b,
Jinwhan
Joo†
c,
Sang Hoon
Joo
*d,
June Sung
Lim
e,
Jinwoo
Lee
*f,
Huawei
Huang
f,
Minhua
Shao
*g,
Jue
Hu
*h,
Jin Young
Kim
*bi,
Byeong Jo
Min
bi,
Seung Woo
Lee
*j,
Minsoo
Kang
j,
Kwangyeol
Lee
*c,
Songa
Choi
c,
Yeji
Park
c,
Yao
Wang
k,
Junjun
Li
l,
Zhicheng
Zhang
*l,
Jianmin
Ma
m and
Sang-Il
Choi
*an
a,
Ho Young
Kim†
b,
Jinwhan
Joo†
c,
Sang Hoon
Joo
*d,
June Sung
Lim
e,
Jinwoo
Lee
*f,
Huawei
Huang
f,
Minhua
Shao
*g,
Jue
Hu
*h,
Jin Young
Kim
*bi,
Byeong Jo
Min
bi,
Seung Woo
Lee
*j,
Minsoo
Kang
j,
Kwangyeol
Lee
*c,
Songa
Choi
c,
Yeji
Park
c,
Yao
Wang
k,
Junjun
Li
l,
Zhicheng
Zhang
*l,
Jianmin
Ma
m and
Sang-Il
Choi
*an
aDepartment of Chemistry and Green-Nano Materials Research Center, Kyungpook National University, Daegu, 41566, Republic of Korea. E-mail: sichoi@knu.ac.kr
bCenter for Hydrogen and Fuel Cell Research, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea. E-mail: jinykim@kist.re.kr
cDepartment of Chemistry and Research Institute for Natural Sciences, Korea University, Seoul, 02841, Republic of Korea. E-mail: kylee1@korea.ac.kr
dDepartment of Chemistry, Ulsan National Institute of Science & Technology (UNIST), Ulsan 44919, Republic of Korea. E-mail: shjoo@unist.ac.kr
eSchool of Energy and Chemical Engineering, Ulsan National Institute of Science & Technology (UNIST), Ulsan 44919, Republic of Korea
fDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: jwlee1@kaist.ac.kr
gDepartment of Chemical and Biological Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: kemshao@ust.hk
hFaculty of Metallurgical and Energy Engineering, Kunming University of Science and Technology, Kunming 650093, China. E-mail: hujue@kust.edu.cn
iDivision of Energy-Environment Engineering, KIST School, Korea University of Science and Technology (UST), Daejeon, Republic of Korea
jWoodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332, USA. E-mail: seung.lee@me.gatech.edu
kDepartment of Chemistry, Tsinghua University, Beijing 100084, China
lTianjin Key Laboratory of Molecular Optoelectronic Sciences, Department of Chemistry, School of Science, Tianjin University, Tianjin 300072, China. E-mail: zczhang19@tju.edu.cn
mSchool of Materials and Energy, University of Electronic Science and Technology of China, Chengdu 610054, China
nDepartment of Hydrogen & Renewable Energy, Kyungpook National University, Daegu, 41566, Republic of Korea
First published on 20th October 2021
Abstract
As the demand for green hydrogen (H2) rapidly increases, the development of water electrolysis technology has been receiving great attention. Indeed, recent remarkable advances in catalyst materials increased the feasibility of water electrolysis for a future H2 economy and technology. In this review, we summarize representative non-precious group metal-based materials for achieving active and stable water electrolysis performances. Our comprehensive range of the state-of-the-art catalysts includes doped carbon catalysts, metal borides, metal carbides, metal oxides, metal phosphides, metal sulfides, and single-atom catalysts. For each class of materials, we focus on the synthesis and catalytic performances of the state-of-the-art materials toward water electrolysis and present the current challenges and outlooks of such materials, along with prospective insights to develop and realize practical systems.
 Jin Young Kim | Jin Young Kim is a principal research scientist at the Center for Hydrogen and Fuel Cell Research at the Korea Institute of Science and Technology (KIST). He received a PhD degree in Materials Science and Engineering from the Massachusetts Institute of Technology under the supervision of Prof. Francesco Stellacci. Prior to joining KIST in 2013, he was a postdoctoral associate with Prof. Edward Sargent at the University of Toronto and was a research staff member at the Samsung Advanced Institute of Technology. His research area is the development of functional nanomaterials for electrochemical-based energy and environmental applications. |
 Kwangyeol Lee | Professor Kwangyeol Lee obtained his PhD degree (1997) in chemistry from the University of Illinois at Urbana-Champaign. After fulfilling his military obligation, he joined Korea University in 2003 as a chemistry faculty member, before being appointed as a professor. He is the recipient of the 2009 Wiley-KCS Young Scholar Award and 2019 Excellent Research Award (KCS Inorganic Chemistry Division). His current interests include the development of synthetic methodologies for nanoscale materials, the application of nanomaterials in biomedical fields, and the development of nanotechnologies to support the environment by creating sustainable energy sources. |
 Sang-Il Choi | Professor Sang-Il Choi received his PhD degree in inorganic chemistry from the KAIST (2011). After his postdoctoral research in the Xia group at the Georgia Institute of Technology, USA, he joined Kyungpook National University in 2015 as an assistant professor. He is the recipient of the 2018 POSCO TJ Park Science Fellowship. His research interests include the design and synthesis of nanomaterials and exploration of their applications in electro-catalysis. |
1. Introduction
Hydrogen (H2) has been considered as one of the promising alternative energy sources to fossil fuels. However, since the steam reforming process is the major process for H2 production, significant CO2 emission, separation of the gases produced, and environmental impacts are the main drawbacks of the relevant technology.1,2 Therefore, water electrolysis, a carbon-free H2 production method, has been reconsidered significantly over the past decade, although technical limitations such as low power-to-gas generation efficiency and poor long-term durability have been revealed. Indeed, the remarkable advances in catalyst materials in recent years have enabled the re-evaluation of water electrolysis for future H2 economy and technology.3–8Since water electrolysis occurs by supplying electric power to overcome the thermodynamic barrier, both the anode and cathode must adapt suitable electrocatalysts to minimize the overpotential (η, the potential difference between the theoretical and experimental values for a redox reaction).9 For the hydrogen evolution reaction (HER) at the cathode, Pt-based catalysts have been utilized as the most efficient electrode materials thanks to their optimal free energy of atomic H adsorption (ΔGH), achieving H2 production with a high exchange current density (j0) and a small Tafel slope value.7,8 Meanwhile, Ir- and Ru-based catalysts form active surface oxide layers during the anode reaction, providing optimal catalytic sites towards the oxygen evolution reaction (OER).10,11 However, owing to the high price and scarcity of Pt, Ir, and Ru, the practical application of water electrolysis on a global scale has been limited. Given this situation, tremendous effort has been made to develop highly active, durable, and non-precious group metal (PGM) catalysts. Notable examples of the state-of-the-art catalysts include doped carbon catalysts, metal borides, metal carbides, metal oxides, metal phosphides, metal sulfides, and single-atom catalysts.12–17
Although there have been extensive interest and research development of catalyst materials for water electrolysis, most of the review articles on the relevant topics have a limited coverage for half-cell reactions and material classes. Therefore, to provide a comprehensive overview of electrocatalysts for the anode and cathode reactions of water electrolysis, this review paper has compiled information from technical levels on the performance of the HER/OER in eight different classes of the state-of-the-art non-PGM-based catalysts. We first introduce the fundamental understanding of the HER and OER and the performance of representative catalysts. Then, we categorize eight different classes and introduce the current status and challenges of synthesis and catalysis with a critical view on the performance and stability. The scope of this review excludes the applications of bifunctional catalysts. Finally, we provide outlooks and future directions of model catalysts to realize highly active and durable catalysts. With the explosive increase in the demand for current technological developments, we believe that this review will provide promising and very interesting prospects for future research related to water electrolysis.
2. Water electrolysis
Water electrolysis, or electrochemical water splitting to produce H2 (and O2), has been considered to be a clean and sustainable system to replace fossil fuels. Water electrolysis includes two half-cell reactions, namely the HER and OER at the cathode and anode, respectively, and the electrolysis can be performed at various pHs. Fig. 1 shows a scheme of a water electrolyser system and the corresponding chemical equations for the electrode reactions in acidic or alkaline electrolytes are presented below.18–20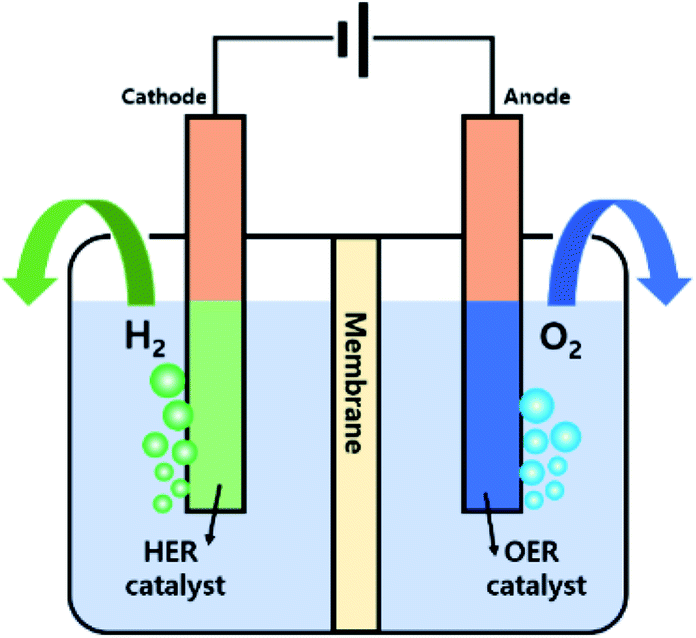 | ||
| Fig. 1 A scheme of a two-electrode system for overall water electrolysis. | ||
In an acidic electrolyte:
| Cathode: 2H+ + 2e− → H2, E0 = 0 V |
| Anode: 2H2O → O2 + 4H+ + 4e−, E0 = 1.23 V |
In an alkaline electrolyte:
| Cathode: 2H2O+ 2e− → H2 + 2OH−, E0 = −0.83 V |
| Anode: 4OH− → O2 + 2H2O+ 4e−, E0 = 0.40 V |
A voltage of 1.23 V is a theoretical value required for water electrolysis. However, additional η must be applied practically to address the potential losses because of the kinetic constraints in the electrode reactions. Developing efficient electrocatalysts can therefore reduce the η of the HER and OER to kinetically accelerate water electrolysis.21
Water electrolysis can take place in different pathways at different pHs. In the case of the HER in acidic electrolytes, H2 is generated by the reduction of H+ (2H+ + e− → H2) and for this, two mechanisms of Volmer–Heyrovsky and Volmer–Tafel mechanisms have been widely accepted. Both the mechanisms firstly involve the adsorption of H+ on the surface of the electrode (Volmer step: H+ + e− → Hads). The second step occurs in different ways; when Hads is stable, the Tafel step (2Hads → H2) is predominant, and vice versa, the Heyrovsky step (H+ + Hads + e− → H2) can occur. Therefore, a catalyst with ΔGH close to zero shows the most optimal conditions for the HER process.22,23 Meanwhile, the HER in alkaline electrolytes also proceeds through the Volmer–Heyrovsky and Volmer–Tafel mechanisms, but their reaction paths are totally different from those under acidic conditions. As water dissociation occurs in the alkaline HER, the reaction kinetics is about two to three orders of magnitude lower than that in acidic media. Based on the Tafel slope of the polarization curve in linear sweep voltammetry (LSV), it is possible to assume whether the Volmer or the Tafel/Heyrovsky step is rate determining.8,24,25
The reaction pathway of the OER also depends on the pH of the electrolyte. In acidic media, O2 is evolved by the oxidation of two water molecules. However, in alkaline and neutral solutions, the OER involves the oxidation of four OH− to water and O2. Many research groups have proposed possible mechanisms for the OER under acidic and alkaline conditions. Most of the proposed mechanisms include the same intermediates such as MOH, MO, and MOOH (M = electrode material surface). The major difference is the circumstances surrounding the O2 evolution site as shown with blue and red arrows in Fig. 2. In addition, there is another O2 evolution pathway from a MO intermediate under acidic conditions. As marked with a yellow route, the direct combination of 2MO produces O2, which is different from the red route involving the formation of the MOOH intermediate, which subsequently decomposes to O2. Despite these differences, the common consensus is that the M–O bond interactions within the intermediates (M–OH, M–O and M–OOH) are crucial for the overall OER performance.19,21,26
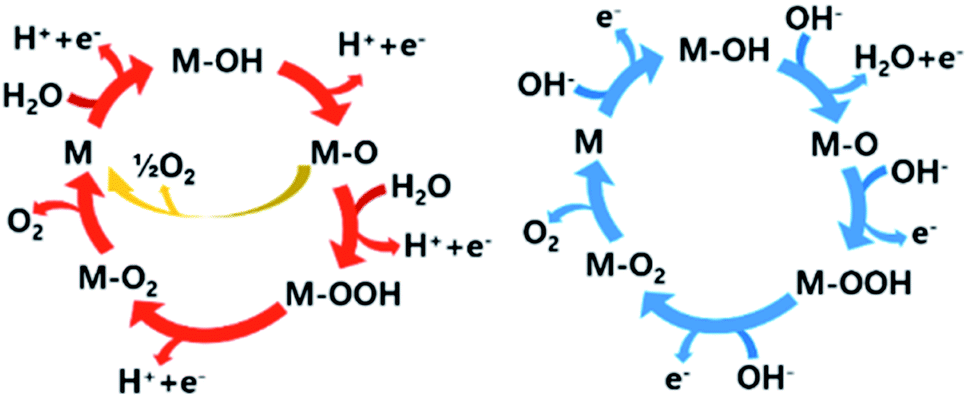 | ||
| Fig. 2 The OER mechanisms under acidic (red route) and alkaline (blue route) conditions. Under acidic conditions, the red route indicates that the oxygen evolution involves the formation of a peroxide (M–OOH) intermediate, while the yellow route shows the direct reaction of two adjacent M–O intermediates to produce O2. | ||
Although PGM-based electrocatalysts have demonstrated the most optimal activities toward the HER/OER, their scarcity and high cost impede practical applications on a large scale. Thus, research has been focused on the development of earth-abundant and cheap, yet efficient catalysts. Thereafter, the effects of different factors and different ways of improvement are thoroughly studied for alternative catalysts. An electrocatalyst should possess moderate adsorption energy to reaction intermediates on the surface. In addition, faster electron transfer during the HER/OER process in acidic/alkaline media is considered a desirable feature.7,27,28 The surface phase stability is also an important factor since the elemental dissolution and surface structure degradation are easily observed under elevated potentials.29,30 To date, the development of new designs and viable model electrocatalysts has represented a breakthrough in replacing PGMs. To improve the electrocatalytic responses, numerous experimental and theoretical results of doped carbon catalysts, transition metal borides (TMBs), transition metal carbides (TMCs), transition metal oxides (TMOs), transition metal phosphides (TMPs), transition metal sulfides (TMSs), and single-atom catalysts have made significant advances. Therefore, we here summarize and review the recent developments in non-PGM HER/OER electrocatalysts with particular attention paid to the discussion of the structural design, relationship between the structure/composition and specific/intrinsic activities, and various tactics to improve the performance.
3. Doped carbon catalysts
As described in the preceding section, PGM catalysts have been the mainstay for water electrolysis reactions (Pt for the HER and Ir oxide or Ru oxide for the OER). To replace these PGM-based catalysts, various non-metallic catalysts such as doped carbon, carbon nitride, boron nitride, phosphorus carbide, and black phosphorous are being studied for water electrolysis reactions. Among them, doped carbon electrocatalysts have received considerable attention due to their inexpensiveness, earth-abundance, and high conductivity.12,31–34 Since pristine carbon materials have poor activity due to the unfavourable binding energies of reaction intermediates (H*, O*, OH*, and OOH*) on carbon surfaces, the introduction of dopants is imperative to endow carbon materials with appropriate binding energy and therefore improved performances for the HER and OER.Doped carbon catalysts can be classified into two major classes: metal-free, heteroatom-doped carbon (heteroatom-doped carbon) catalysts and transition-metal and nitrogen co-doped carbon (M–N/C) catalysts. In the heteroatom-doped catalysts, the heteroatoms of p-block elements (O, N, P, and S) are doped in carbon host materials, including graphene, carbon nanotubes (CNTs), and porous carbons. By introducing a heteroatom having different electronegativity and electron affinity of carbon, the electronic structure, chemical state, and electrocatalytic activity of carbon adjacent heteroatoms can be modulated.12,31,33 In M–N/C catalysts, a coordinated moiety of a transition metal (Fe, Co, and Ni) and nitrogen (M–Nx) is introduced into carbon. In this structure, nitrogen can be replaced with other heteroatoms or their multiple combinations. M–N/C catalysts are inspired from natural enzymes or homogeneous metal-complexes. The metal centre has been proposed as the active site and its electronic structure depends on the type of transition metal and its coordination environment.32,34
In this section, we introduce the key strategies for rationally designing highly active doped carbon electrocatalysts for the HER and OER from both experimental and theoretical viewpoints. Also, we discuss the kinetics, mechanism, and critical factors for enhancing the catalytic activities of doped carbon catalysts. Finally, based on this understanding, we summarize the challenges of doped carbon catalysts.
3.1 The state-of-the-art catalysts and their HER/OER performances
In the early stage of doped carbon-based OER catalyst research, various dopants were investigated to enhance the OER performance of doped carbon catalysts. N-doped carbon showed the highest performance;35,36 however its performance was far inferior to that of other types of non-PGM-based catalysts. As a means of further boosting the OER activity, a dual-doping strategy was exploited. Most dual-doped carbon catalysts were synthesized by introducing secondary heteroatoms (S and P) along with N.37–39 As a notable example, Zhu et al. developed efficient dual-doped carbon nanofiber catalysts with the distribution of N and P on carbon paper (NPC-CP) (Fig. 3a).38 To reveal the effect of dual-doping, N-doped carbon paper (NC-CP) was also prepared, and pristine CP and IrO2 catalysts served as the standards. The LSV curve in Fig. 3b indicates that NPC-CP exhibited the highest activity among the compared catalysts. NPC-CP required the lowest η of 310 mV to drive 10 mA cm−2 (η10), followed by NC-CP (370 mV) and CP (560 mV). This result clearly suggests a role of the additional P dopant in enhancing the catalytic activity over mono-doped NC-CP. The Tafel slope of NPC-CP was 87.4 mV dec−1, which is lower than those of IrO2 (92.1 mV dec−1), NC-CP (105.3 mV dec−1), and CP (131.8 mV dec−1), implying much more favourable electrochemical OER kinetics on NPC-CP. Density functional theory (DFT) calculations suggested that the pyridinic N and P dopants located at the edges in the NPC-CP catalyst could activate the adjacent edge C atoms, and the edge C atoms were proposed as the active sites of the OER. When the P atoms are doped, the adsorption energy of OH*, O* and OOH* intermediates on the edge C atoms adjacent to the P atoms could be appropriately modulated, thereby enhancing the OER activity. In another example, Hu et al. developed a conductive and porous hydrogel catalyst composed of phytic-acid-doped polypyrrole (P, N-doped) on carbon cloth (PA-PPy/CC), which showed efficient and stable catalytic performances. The PA-PPy/CC catalysts exhibited the highest activity (η10 = 340 mV) among the CC (pristine), PPy/CC (N-doped), and PA/CC (P-doped) catalysts, demonstrating the efficacy of dual-doping.39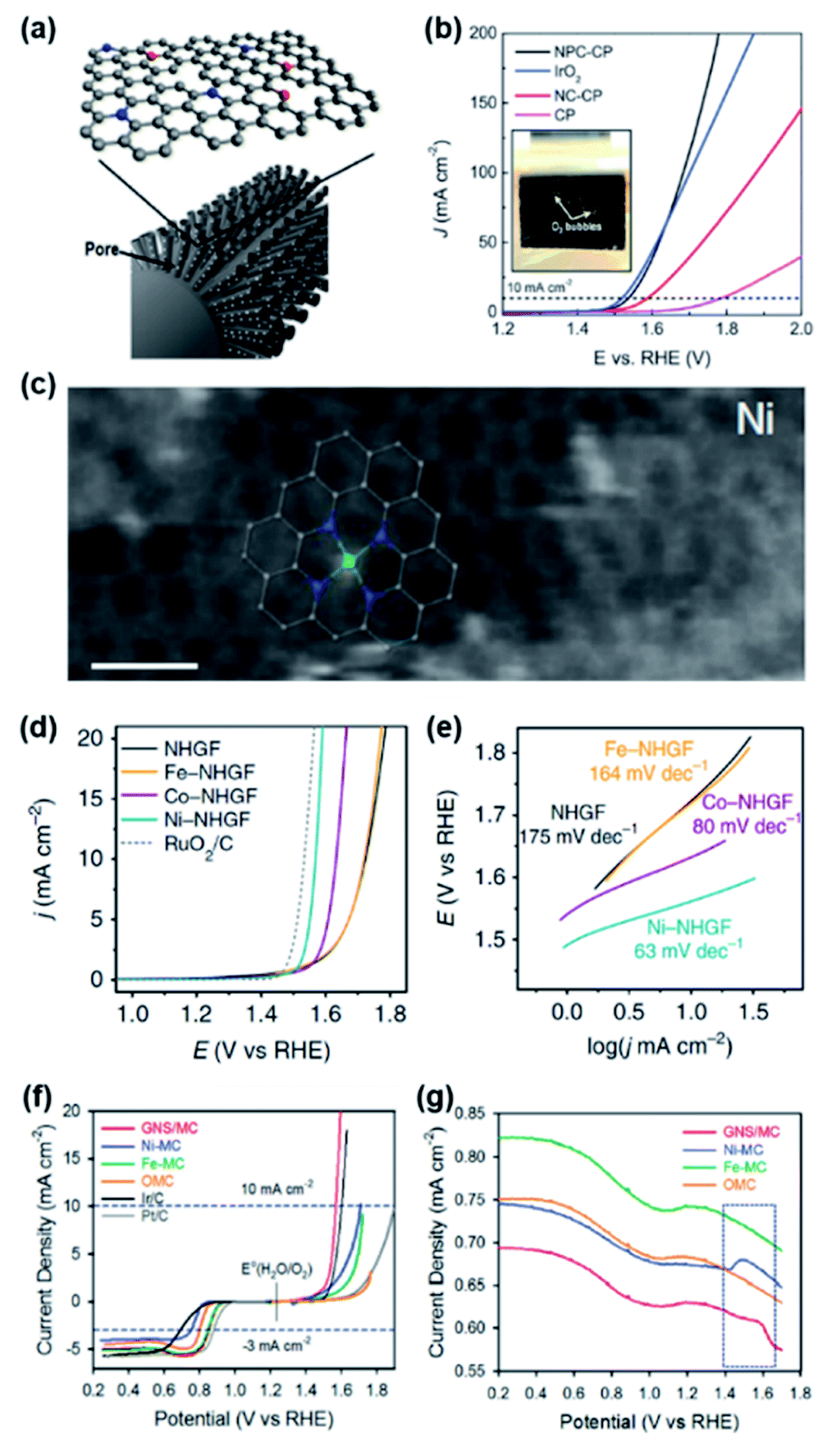 | ||
| Fig. 3 (a) Schematic illustration of N and P-doped carbon nanofibers on carbon paper. (b) OER LSV curves of NPC-CP, NC-CP, IrO2 and pristine carbon catalysts in 1 M KOH. Reproduced with permission,38 Copyright 2017, Wiley-VCH. (c) High-resolution TEM image of Ni-NHGF. (d) OER LSV curves and (e) Tafel plots of NHGF, Fe-NHGF, Co-NHGF, Ni-NHGF, and RuO2/C catalysts in 1 M KOH. Reproduced with permission,40 Copyright 2018, Springer Nature. (f) OER and ORR polarization curves of GNS/MC, Ni-MC, Fe-MC, OMC, Ir/C, and Pt/C catalysts in 0.1 M KOH. (g) SWV profiles of GNS/MC, Ni-MC, Fe-MC, and OMC catalysts collected in 0.1 M KOH. Reproduced with permission,44 Copyright 2016, Wiley-VCH. | ||
While heteroatom-doped carbons can serve as non-PGM catalysts for the OER, their intrinsic OER activity is unsatisfactory compared to their rival PGM-based catalysts. Instead, M–N/C catalysts emerged as more promising OER catalysts with enhanced activity.40–42 A DFT calculation study predicted that Ni coordinated with pyridinic N (Ni-N/C) and Co coordinated with pyrrolic N (Co-N/C) can function as active sites for the OER.43 Fei et al. validated this conjecture with a combined experimental and computational study.40 A series of 3d transition metals embedded in N-doped graphene frameworks (M-NHGFs, M = Fe, Co, or Ni) were prepared and exploited as model catalysts. The combination of spectroscopic analyses of M-NHGFs clearly revealed that the structure of all catalysts comprised single metal centres with four adjacent N atoms and four C atoms in the second coordination sphere (MN4C4 structure). As shown in Fig. 3c, the annular dark-field scanning transmission electron microscopy (ADF-STEM) image of Ni-NHGF clearly visualised the atomic structure within single-layer graphene, which was further confirmed by the spectroscopic results. The LSV curves of the catalysts revealed that dopant-free NHGF exhibited poor OER activity (η10 = 494 mV) and with the addition of a metal, the activity increased in the order of Fe (η10 = 488 mV) < Co (η10 = 402 mV) < Ni (η10 = 330 mV) (Fig. 3d). The Tafel slope of Ni-NHGFs (63 mV dec−1) was smaller than those of Co-NHGFs (80 mV dec−1) and Fe-NHGFs (164 mV dec−1). This work established the trend of OER activity in M–N/C catalysts and suggested an important role of the metal in enhancing reaction kinetics for the OER (Fig. 3e).
Toward achieving dual catalytic functionality, two different types of transition metals (M1 and M2) were exploited in the design of M–N/C catalysts. In this structure, each of the two metal-N species catalyses the OER and oxygen reduction reaction (ORR), respectively. The resulting OER–ORR bifunctional catalysts can be used as electrode materials in metal–air batteries and electrode catalysts in unitised regenerative fuel cells.44–46 As representative work, Cheon et al. synthesized a graphitic nanoshell/mesoporous carbon comprising Fe and Ni-N species (GNS/MC) and used as a bifunctional OER–ORR catalyst. To experimentally identify the reason for high catalytic performance, mono-metallic Fe-N doped (Fe-MC) and Ni-N doped (Ni-MC) catalysts and N-doped carbon (OMC) were also compared.44 The OER polarization curve indicated that GNS/MC showed the best activity with η10 = 340 mV, which was followed by Ni-MC (η10 = 480 mV), Fe-MC (η10 = 500 mV), and OMC (Fig. 3f). In situ X-ray absorption spectroscopy (XAS) suggested that the Ni centre is the active site of GNS/MC and both Ni and Fe species are associated with the superior OER activity. Also, square wave voltammetry (SWV) measurement revealed that the Ni centre in GNS/MC is more amenable to the adsorption of the OH group than Ni-NC catalysts, which means that Fe species induce an appropriate binding energy between Ni and OH groups (Fig. 3g).
Turning to HER catalysts, most of the developed catalysts have been based on M–N/C catalysts, whereas only a limited number of studies were carried out for doped carbon catalysts due to their much lower activity than that of other benchmarked non-PGM-based catalysts. Hence, the main focus was directed towards identifying the origin of HER activity in doped carbons and establishing the catalyst design principle to enhance the HER performance.37,47–49 Representatively, Jiao et al. carefully investigated a series of heteroatom-doped graphene materials (a-G; a = B, N, O, S, and P) as efficient HER electrocatalysts by combining spectroscopic characterization, electrochemical measurement, and DFT calculation.48 Fifteen different doping configurations in the graphene matrix were identified by near edge X-ray absorption fine spectroscopy (NEXAFS) analyses (Fig. 4a). Next, the most favourable sites for the HER which include heteroatoms themselves and several carbon sites around them on the 15 doping models were assessed by DFT calculations. The results indicated that, for each dopant, the graphitic doping sites exhibited higher HER activity than edge type sites with lower ΔGH* (Fig. 4b). Examining the reaction mechanism for each heteroatom doping model with the lowest ΔGH*, it was found that all the doping models followed the Volmer–Heyrovsky mechanism, with the Volmer step being the rate determining step (Fig. 4c). The plot of j0 and ΔGH* showed the volcano-shaped plot of a-G catalysts and indicated that all a-G catalysts showed lower HER activity than the state-of-the-art HER catalysts (Pt and MoS2) (Fig. 4d). Calculations were then performed to predict a new catalyst with higher HER performance, and it was found that dual doping and structural engineering can enhance the HER activity. On the basis of these findings, dual doped graphene materials (a,b-G/a = N, b = B, S, and P) were synthesised. As shown in the HER polarization curves, N,S-G exhibited a lower η than N-G and G, agreeing well with the calculation results (Fig. 4e). The general volcano plot including pure (G), a-G, a,b-G samples also supported the above results (Fig. 4f). Finally, it was predicted that the tuning dopant level and surface area of heteroatom-doped carbon catalysts can lead to improved HER activity.
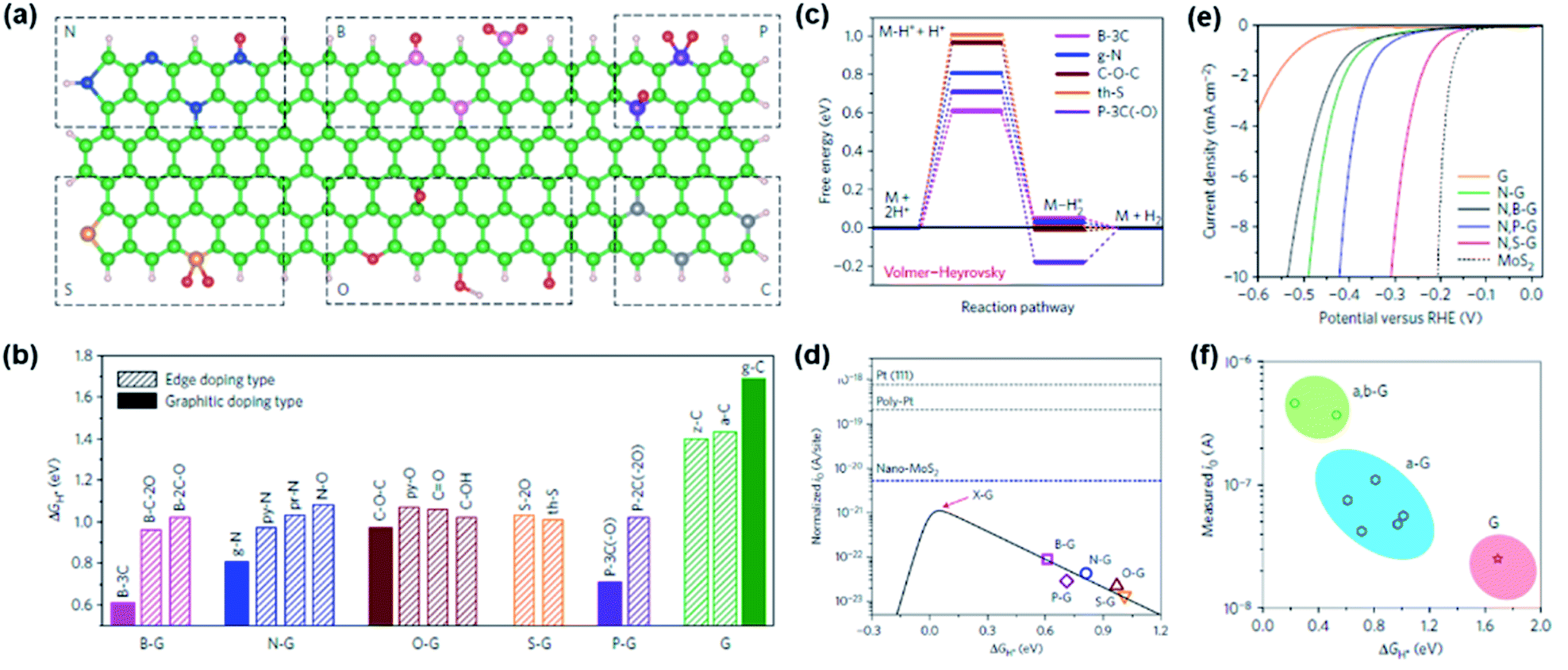 | ||
Fig. 4 (a) Schematic summary of the heteroatom doping configurations: (top row, from left to right) pr-N, py-N, g-N, N–O, B-2C-O, B-3C, B-C-2O, P-3C(–O) and P-2C(–2O); (bottom row, from left to right) th-S, S-2O, py-O, C–O–C, C–OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, g-C, z-C and a-C. Green/grey, pink, blue, red, gold, purple, and white represent C, B, N, O, S, P and H atoms, respectively. (b) The computed lowest ΔGH* for different models. The ΔGH* values on graphitic type doping models are labelled with solid bars, and those on edge doping models are labelled with shaded bars. (c) Free energy diagram for the HER following the Volmer–Heyrovsky pathway on various graphene models. (d) Volcano plot between io and ΔGH* with the charge-transfer coefficient, α = 0.125 (black solid line). The open symbols represent io obtained from Tafel plots and DFT-derived ΔGH* for each graphene sample/model. (e) Electrochemical HER measurements on various graphene-based materials and MoS2 in 0.5 M H2SO4. (f) A volcano plot that includes pure (G), single-doped (a-G) and dual-doped (a,b-G) graphene samples. The io values are those measured without normalization. Reproduced with permission,48 Copyright 2016, Springer Nature. O, g-C, z-C and a-C. Green/grey, pink, blue, red, gold, purple, and white represent C, B, N, O, S, P and H atoms, respectively. (b) The computed lowest ΔGH* for different models. The ΔGH* values on graphitic type doping models are labelled with solid bars, and those on edge doping models are labelled with shaded bars. (c) Free energy diagram for the HER following the Volmer–Heyrovsky pathway on various graphene models. (d) Volcano plot between io and ΔGH* with the charge-transfer coefficient, α = 0.125 (black solid line). The open symbols represent io obtained from Tafel plots and DFT-derived ΔGH* for each graphene sample/model. (e) Electrochemical HER measurements on various graphene-based materials and MoS2 in 0.5 M H2SO4. (f) A volcano plot that includes pure (G), single-doped (a-G) and dual-doped (a,b-G) graphene samples. The io values are those measured without normalization. Reproduced with permission,48 Copyright 2016, Springer Nature. | ||
M–N/C catalysts for use in the HER were derived from the studies of hydrogenase enzymes.50 By mimicking these enzymes, several homogeneous metal complexes such as cobaloxime and cobalt diamine-dioxime were synthesized and studied for the HER.51,52 Through these efforts, Co-Nx sites were proposed as the active sites of the HER. Inspired by these early studies, various heterogeneous Co-N/C catalysts are being investigated as potent non-PGM catalysts. Morozan et al. attempted to determine the HER activity trend of various M–N/C catalysts and suggested that Co-N/C showed the highest HER activity in both acidic and alkaline electrolytes.53 However, their catalysts contained both Co-Nx and Co@C sites, which made the determination of exact active sites difficult. These problems could be resolved by subsequent efforts. Liang et al. successfully synthesized Co-N/C catalysts that contained molecular CoNx centres by pyrolysis of several types of cobalt complex molecules (Fig. 5a).54 Then, the HER performance of the prepared CoNx/C, Co/C, and N/C catalysts was analysed. Interestingly, CoNx/C exhibited the highest HER activity under all electrolyte pH conditions. The HER activity trend of the above catalysts under acidic conditions was in the order of CoNx/C (η10 = 133 mV) > Co/C (η10 = 310 mV) > N/C (η10 = 460 mV) (Fig. 5b). The Tafel slopes of the CoNx/C, Co/C, and N/C catalysts were 57, 106, and 98 mV dec−1, respectively, suggesting that CoNx/C catalysts show faster kinetics than Co/C and N/C catalysts (Fig. 5c). An acid-leaching experiment clearly reconfirmed that CoNx sites contribute to HER performance predominantly compared to Co nanoparticle sites (Fig. 5d). In order to further clarify the active sites in Co-N/C catalysts, Sa et al. carried out a systematic study by preparing Co-N/C model catalysts with controlled Co-Nx and Co@C site densities to clarify their contribution to HER activity (Fig. 5e).55 Through extended X-ray absorption fine structure (EXAFS) analysis, the coordination numbers (CNs) of Co-N/C and Co@C were quantified to determine the correlation between the CN ratio of Co-N/C (CNCo-N/C) and HER mass activity in both acidic and alkaline electrolytes. In both electrolytes, linear relationships between CNCo-N/C and mass activities were established, suggesting that the Co-N/C sites play a crucial role in the acidic and alkaline HER, whereas Co@C sites exhibit negligible catalytic performance (Fig. 5f and g). According to DFT calculations, the CoN4C10 structure was considered as the best active site and the Co@C structure is inactive for the HER (Fig. 5h).
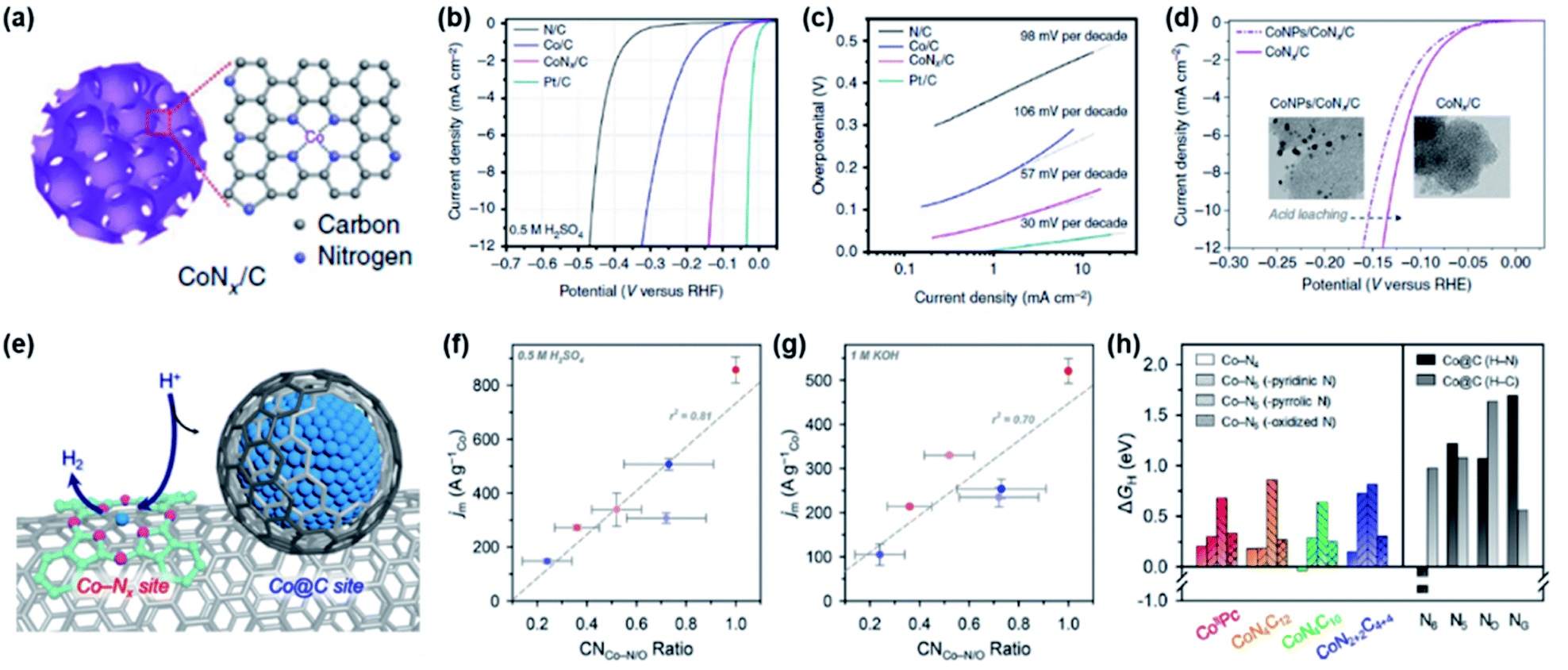 | ||
| Fig. 5 (a) Schematic illustration of the porous CoNx/C electrocatalysts. (b) HER polarization curves and (c) Tafel plots of the CoNx/C, N/C, Co/C and Pt/C catalysts in 0.5 M H2SO4. (d) Comparison of the HER activity of the CoNPs/CoNx/C and CoNx/C catalysts showing the influence of acid leaching. Reproduced with permission,54 Copyright 2015, Springer Nature. (e) Schematic illustration of CNT/Co-PcC catalysts with controlled Co-Nx and Co@C site densities. Correlation between the Co-N/O coordination number (CNCo-N/O) and the HER mass activity measured in (f) 0.5 M H2SO4 and (g) 1 M KOH. (h) Comparison of calculated ΔGH values for Co-Nx and Co@C sites. For Co-Nx sites, Co-N4 clusters and Co-N5 clusters composed of additional axial ligands were considered. For Co@C models, N dopants (denoted as H–N, black bars) and adjacent carbon sites (denoted as H–C, gray bars) in the graphene models (i.e., pyridinic (N6), pyrrolic (N5), graphitic (NG), and oxidized (NO) species) were considered as different hydrogen adsorption sites. Reproduced with permission,55 Copyright 2019, American Chemical Society. | ||
3.2 Challenges in synthesis and catalysis
During the last decade, there have been continuous efforts to develop doped carbon catalysts for use in the OER and HER. Although considerable advances have been made, further improvements in their synthesis and catalysis are still needed. First, it is important to develop synthetic methods that can create active sites in a high density. Currently developed doped carbon catalysts commonly require a pyrolysis step, which induces the decomposition of some heteroatom and transition metal precursors. Moreover, during the synthesis of M–N/C catalysts, the transition metal atoms aggregate together in the pyrolysis step. For these reasons, the active sites of doped carbon catalysts are generated in a low density. Next, it is also important to synthesize a catalyst which has only desirable sites. In the case of doped carbon catalysts, various sites are generated simultaneously. This can lead to ambiguity on the true active sites for the OER or HER. Finally, it is necessary to widen the pH range of the OER and HER in which doped carbon catalysts can effectively operate. Doped carbon catalysts are generally studied under alkaline conditions for the OER and acidic conditions for the HER. These research trends are due to the low stability of the acidic OER due to the corrosive environment, and low alkaline HER activity due to the additional water dissociation step. OER and HER studies in wider pH regions facilitate the universal use of doped carbon catalysts in water electrolysis devices.4. Metal borides
As a class of intermetallic inorganics, TMBs have special chemical and physical properties, including high conductivity, superior hardness, and excellent corrosion resistance in both acidic and alkaline media. Recently, with the increase in research interest in electrochemical water splitting, TMBs began to gain more attention and demonstrated tuneable catalytic activity for the HER and OER. The catalytic activity of TMBs is affected by the electron transfer between the metal and boron, which modulates the electronic structure and changes the surface properties, thereby regulating the reaction energy barrier involved in the catalytic process. So far, a variety of strategies have been developed to optimize the catalytic activity of TMBs, such as component regulation (including the boron content and metal species/contents), crystallinity regulation, and nanostructure construction.In this part, we review and compare the catalytic activities of different TMBs and analyse the important parameters that are related to the intrinsic catalytic activity. In addition, some effective methods that can regulate catalytic activity are summarized and discussed. Furthermore, we also analysed the structure and component changes of TMBs during the OER process.
4.1 The state-of-the-art catalysts and their HER/OER performances
Understanding the trend and mechanism of the catalytic activity of TMBs is helpful to get design guidance for the development of advanced catalysts. In order to study the difference in the catalytic activity of TMBs based on different metal elements, Mazánek et al. evaluated the electrochemical HER catalytic behaviour of different TMBs with an MB2 structure, including AlB2, CrB2, HfB2, MgB2, NbB2, TaB2, TiB2, VB2, and ZrB2.13 These borides were purchased from reagent companies without any treatment, which are all bulk materials on the microscale and are helpful to compare the intrinsic catalytic activity and eliminate the influence of factors like the porosity and defective structure. Notably, the test results suggested that TiB2, ZrB2 and HfB2 exhibit better HER performance than bare glassy carbon (Fig. 6a). ZrB2 exhibited the best catalytic activity and showed an η10 of 970 mV. Their corresponding Tafel slopes were all larger than 120 mV dec−1, suggesting that they all showed sluggish reaction kinetics (Fig. 6b). Recently, Li et al. conducted an elaborate study to unravel the relationship between the d-band centre of TMBs and catalytic activity.56 They investigated 12 MB2 that included 9 AlB2-type borides (TiB2, ZrB2, HfB2, VB2, NbB2, TaB2, CrB2, MoB2, and WB2), 2 RuB2-type borides (RuB2 and OsB2), and ReB2. Their theoretical calculations suggested that the ΔGH* absolute value generally decreases from Group IV TMBs to Group VII TMBs, namely increasing their HER catalytic activity (Fig. 6c). Remarkably, they found that the d-band centres of these TMBs show similar trends, which are almost linearly related to the ΔGH* absolute value. The reason for this is that if the d-band centre is more negative with respect to the Fermi level, the antibonding states will shift to lower energy and become more occupied, which helps to reduce the binding energy of H* on the surface. The predicted catalytic performance by theoretical calculations was further verified by their experimental results. As shown in Fig. 6d, the Group VIII TMBs (RuB2 and OsB2) exhibited much better HER performance than other synthesized borides under both acidic and alkaline conditions, and RuB2 with the best catalytic activity only needed an ultra-low η10 of 18 and 28 mV in 0.5 M H2SO4 and 1 M KOH, respectively. Their study demonstrated that the d-band centre of TMBs can be used as an important indicator to predict and guide the synthesis of advanced TMB catalysts. Ai et al. reported that the H* adsorption energy on metal-terminated MB type borides is weaker than their corresponding pure metal phase, which is caused by the strong hybridization between the d-orbitals of metal atoms and the sp orbitals of B atoms.57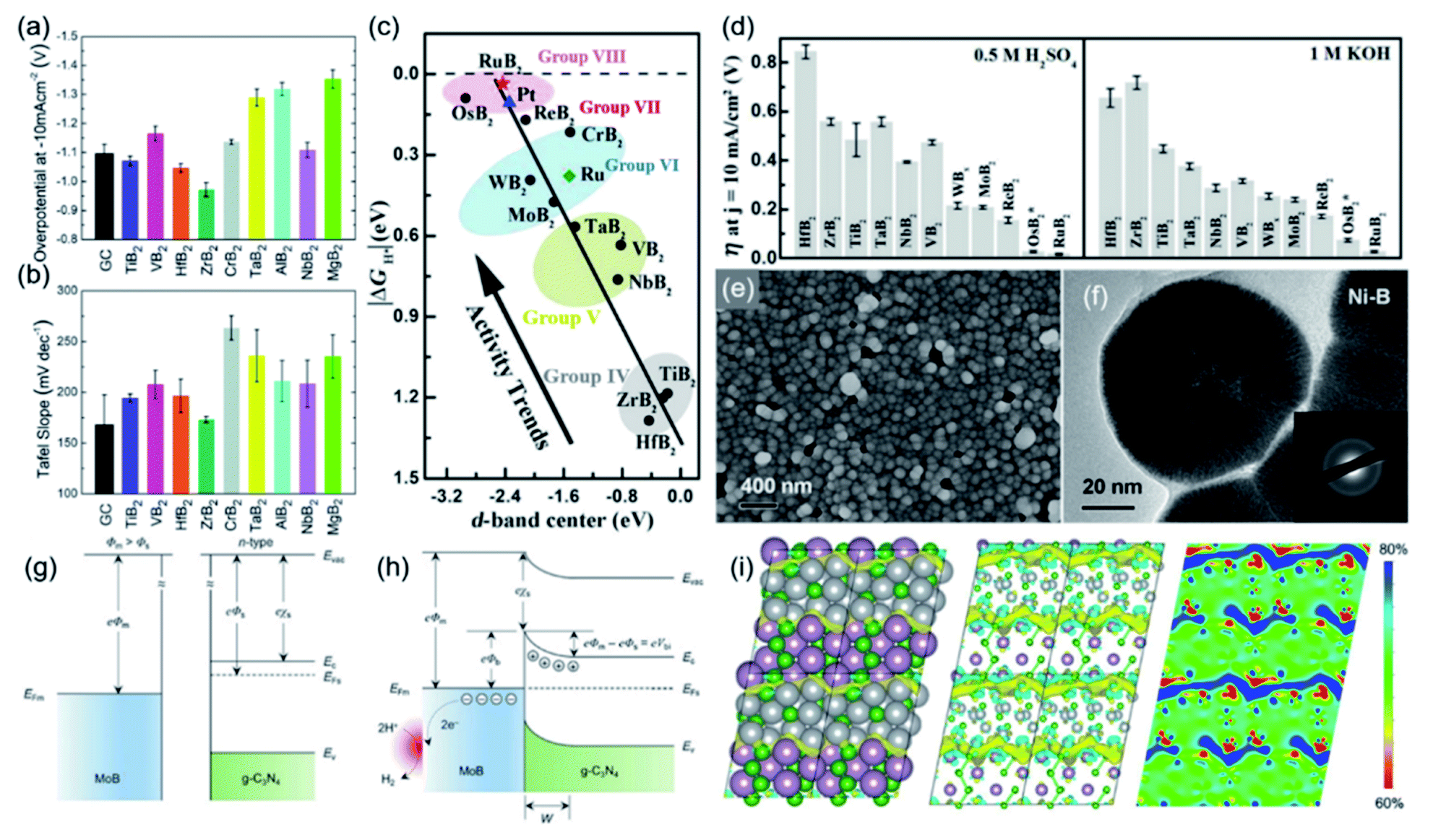 | ||
| Fig. 6 (a) The η10 in 0.5 M H2SO4 and (b) corresponding Tafel slopes of AlB2, CrB2, HfB2, MgB2, NbB2, TaB2, TiB2, VB2 and ZrB2. Reproduced with permission,13 Copyright 2018, Royal Society of Chemistry. (c) The relationship between the d-band center of metal borides and ΔGH* absolute value and (d) the η10 of different metal borides in 0.5 M H2SO4 and 1 M KOH. Reproduced with permission,56 Copyright 2018, Wiley-VCH. (e) SEM image and (f) TEM image of amorphous Ni-B; the inset shows the corresponding SAED pattern. Reproduced with permission,59 Copyright 2018, Wiley-VCH. Energy band schematic diagram of (g) pure MoB and (h) MoB/g-C3N4. Reproduced with permission,61 Copyright 2018, Wiley-VCH. (i) DFT-optimized model of the Ni3B/MoB heterostructure, and the charge density distribution and electronic local function in the corresponding structure. Reproduced with permission,62 Copyright 2021, Elsevier. | ||
Among the reported borides, PGM-based borides showed excellent HER catalytic performance that exceeds that of benchmark Pt/C, such as Pd2B, PdB, RuB, ReB, RuB2 and OsB2.56–58 However, the catalytic activity of non-PGM-based TMBs has been limited. Researchers have developed some effective strategies to improve the intrinsic HER catalytic activity of non-PGM-based TMBs, like crystallinity engineering, component regulation, nanostructure control, interface constructing, etc. For example, Zeng et al. synthesized amorphous Ni–B nanoparticles with an average diameter of ca. 80 nm (Fig. 6e and f) by the electroless plating method.59 The X-ray photoelectron spectroscopy (XPS) results suggested that the Ni/B ratio is 2.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and electrons are transferred from B to Ni. The synthesized Ni-B showed a low η20 of 123 mV in a 1 M HClO4 electrolyte. Xu et al. constructed Ni-ZIF/Ni-B ultra-thin nanosheet arrays with numerous crystalline–amorphous phase boundaries.60 DFT calculation results for the HER process demonstrated that compared with Ni-ZIF and Ni-B, the Ni-ZIF/Ni-B catalyst shows a more optimal ΔGH* that is closer to zero. Benefiting from the structural advantage, Ni-ZIF/Ni-B required an η10 of 67 mV for the HER. Zhuang et al. developed a type of Schottky catalyst by constructing an interface between metallic MoB and g-C3N4 so that a Schottky junction can be formed.61 Since the Fermi energy levels of metals and semiconductors are very different, it causes the charge flow propagated by electrons or holes at the interface to reach equilibrium, which eventually lead to band bending and the formation of a Schottky barrier (Fig. 6g and h). As a result, the electron density on the MoB surface was increased and the kinetic barriers for the HER process were dramatically lowered. Huang et al. developed a method for synthesizing a Ni3B/MoB heterostructure with rich grain boundaries, which has an enhanced intrinsic catalytic activity.62 Ni3B/MoB exhibited much better HER performance than pure Ni3B and pure MoB and showed a low η10 of 75 mV. Their DFT calculations revealed that the Ni atoms near the interface donate more electrons to the B atoms, and electrons are transferred from Ni3B to MoB at the grain boundary, which results in a high local density of electrons at the grain boundary and a low density of electrons distributed on both sides (Fig. 6i). Such interface electric dipole formation strengthened the electronic interactions between MoB and Ni3B so that the ΔGH* of Ni3B/MoB was closer to zero than that of pure Ni3B and MoB.
:1 and electrons are transferred from B to Ni. The synthesized Ni-B showed a low η20 of 123 mV in a 1 M HClO4 electrolyte. Xu et al. constructed Ni-ZIF/Ni-B ultra-thin nanosheet arrays with numerous crystalline–amorphous phase boundaries.60 DFT calculation results for the HER process demonstrated that compared with Ni-ZIF and Ni-B, the Ni-ZIF/Ni-B catalyst shows a more optimal ΔGH* that is closer to zero. Benefiting from the structural advantage, Ni-ZIF/Ni-B required an η10 of 67 mV for the HER. Zhuang et al. developed a type of Schottky catalyst by constructing an interface between metallic MoB and g-C3N4 so that a Schottky junction can be formed.61 Since the Fermi energy levels of metals and semiconductors are very different, it causes the charge flow propagated by electrons or holes at the interface to reach equilibrium, which eventually lead to band bending and the formation of a Schottky barrier (Fig. 6g and h). As a result, the electron density on the MoB surface was increased and the kinetic barriers for the HER process were dramatically lowered. Huang et al. developed a method for synthesizing a Ni3B/MoB heterostructure with rich grain boundaries, which has an enhanced intrinsic catalytic activity.62 Ni3B/MoB exhibited much better HER performance than pure Ni3B and pure MoB and showed a low η10 of 75 mV. Their DFT calculations revealed that the Ni atoms near the interface donate more electrons to the B atoms, and electrons are transferred from Ni3B to MoB at the grain boundary, which results in a high local density of electrons at the grain boundary and a low density of electrons distributed on both sides (Fig. 6i). Such interface electric dipole formation strengthened the electronic interactions between MoB and Ni3B so that the ΔGH* of Ni3B/MoB was closer to zero than that of pure Ni3B and MoB.
The atomic ratio of B/M in TMBs also significantly affects their morphology, structure and surface properties, which ultimately affects their intrinsic catalytic activity. In 2017, Park et al. used the arc melting method to synthesize molybdenum boride with different B contents and crystal phases, including Mo2B, α-MoB, β-MoB, and MoB2.63 As shown by the model structure in Fig. 7a, with the increase of the B content, the B–B connectivity changed from 0-D isolated B atoms (Mo2B) to 1-D zigzag B chains (α-MoB and β-MoB) and further to 2-D graphene-like boron layers (MoB2). For electrocatalysis, MoB2 with the highest boron content showed the best HER catalytic activity, followed by β-MoB, α-MoB, and Mo2B (Fig. 7b). Chen et al. revealed the reason for the high activity of MoB2 with a subunit B layer (borophene) by experimental results and theoretical calculations.64 They found that the density and activity of HER active sites on the surface of α-MoB2 are even higher than that of the Pt(111) surface. Similarly, the phenomenon of changes in catalytic performance caused by the B content difference of TMBs has also been found in iron boride. Li et al. synthesized and compared the catalytic activity of Fe2B and FeB2.65 Their nanostructure and crystal phases were analysed by TEM and XRD (Fig. 7c, d), and the results showed that Fe2B and FeB2 have similar structures and even specific surface areas. For the HER, FeB2 had much better performance than Fe2B (Fig. 7e). They analysed the adsorption energy of H* at different sites of FeB2 through DFT calculations (Fig. 7f) and compared that with that of Fe2B. The results showed that the absolute ΔGH* values on different sites of FeB2 are all much closer to zero than that of Fe2B. From the above results, it can be inferred that increasing the boron contents in TMBs may be an effective strategy to enhance their HER activity.
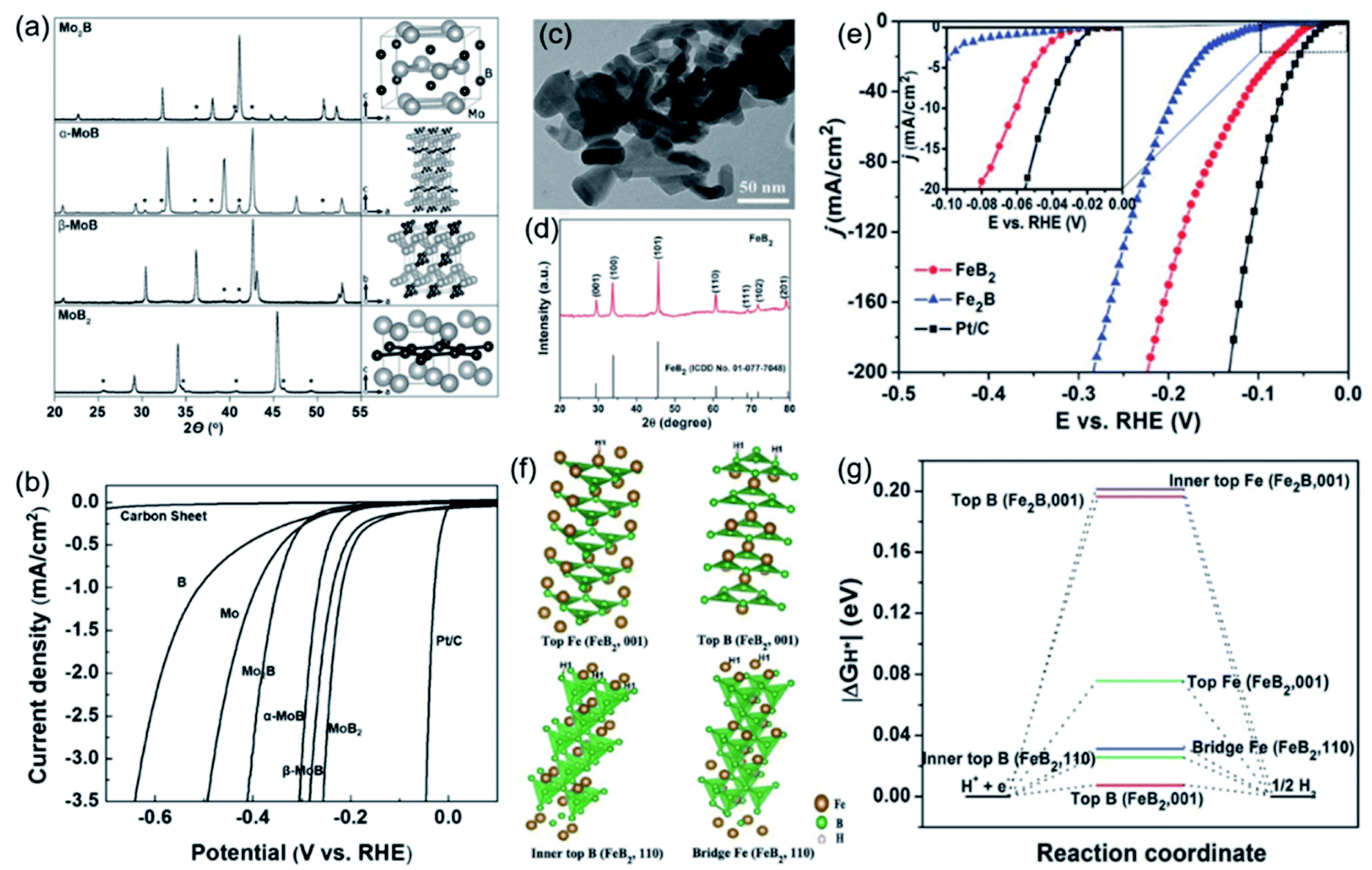 | ||
| Fig. 7 (a) XRD patterns and model structures of Mo2B, α-MoB, β-MoB, and MoB2 and (b) their HER polarization curves obtained in 0.5 M H2SO4. Reproduced with permission,63 Copyright 2017, Wiley-VCH. (c) TEM image and (d) XRD pattern of FeB2; (e) HER polarization curves of Fe2B, FeB2 and benchmark Pt/C; (f) several stable structure models of H* absorbed on different sites of FeB2; (g) calculated free–energy diagram of the HER on FeB2 and Fe2B. Reproduced with permission,65 Copyright 2017, Wiley-VCH. | ||
Recently, TMBs have also been reported to be used as efficient catalysts for the OER. As is well known, the OER is a half-reaction that occurs at the anode in the process of electrochemical water splitting, which involves oxygen generation and electrochemical oxidation. Similar to TMSs and TMPs, TMBs are also a class of compounds that are easily oxidized, so their composition and electronic structure change during OER processes. For example, Guo et al. boronized a series of metal foils (Ni, Co, Fe, NiFe alloy and SUS 304 steel) by high temperature treatment with amorphous boron powder as the boron source (Fig. 8a).66 The electrochemical OER performance of boronized metal foils was evaluated and compared with that of their corresponding metal phases in 1 M KOH. As shown in Fig. 8b, all the boronized mono-metal foils possessed better OER performance than their metal foils, and the boronized Ni foil exhibited the best catalytic activity. These results proved that boronization is an effective method to improve the OER catalytic activity of metals. They further analysed the composition and structural changes of the boronized Ni foil after catalyzing the OER for 10 h at 10 mA cm−2. Raman analysis confirmed that a B doped γ-NiOOH film is generated on the surface of boronized Ni foil after the OER process. XPS results (Fig. 8c) revealed that the metaborate species exists in the surface oxidized layer, which reduces the oxidation state of Ni. The high resolution TEM image (Fig. 8d) suggested that the thickness of the oxidized layer is about 2–5 nm. Therefore, it can be determined that TMBs will generate an oxidized layer containing metaborate in the process of catalysing the OER. Han et al. reported a V-doped Ni-Co boride hollow nanoprism through a self-templated ion exchange strategy followed by atomic layer deposition (Fig. 8e and f).67 The doped V in the hollow layered Ni-Co boride structure induced more unsaturated atoms that can serve as active sites, which exhibited a better catalytic performance than Ni-Co boride (Fig. 8g). DFT calculations suggested that the enhanced OER catalytic activity comes from the synergistic catalysis effect of different elements: Co and B served as the active sites, Ni acted as the regulator of surface electronic structures, and V facilitated the charge transfer. The analysis of the samples after the stability test also showed that the boride catalyst surface is partially oxidized.
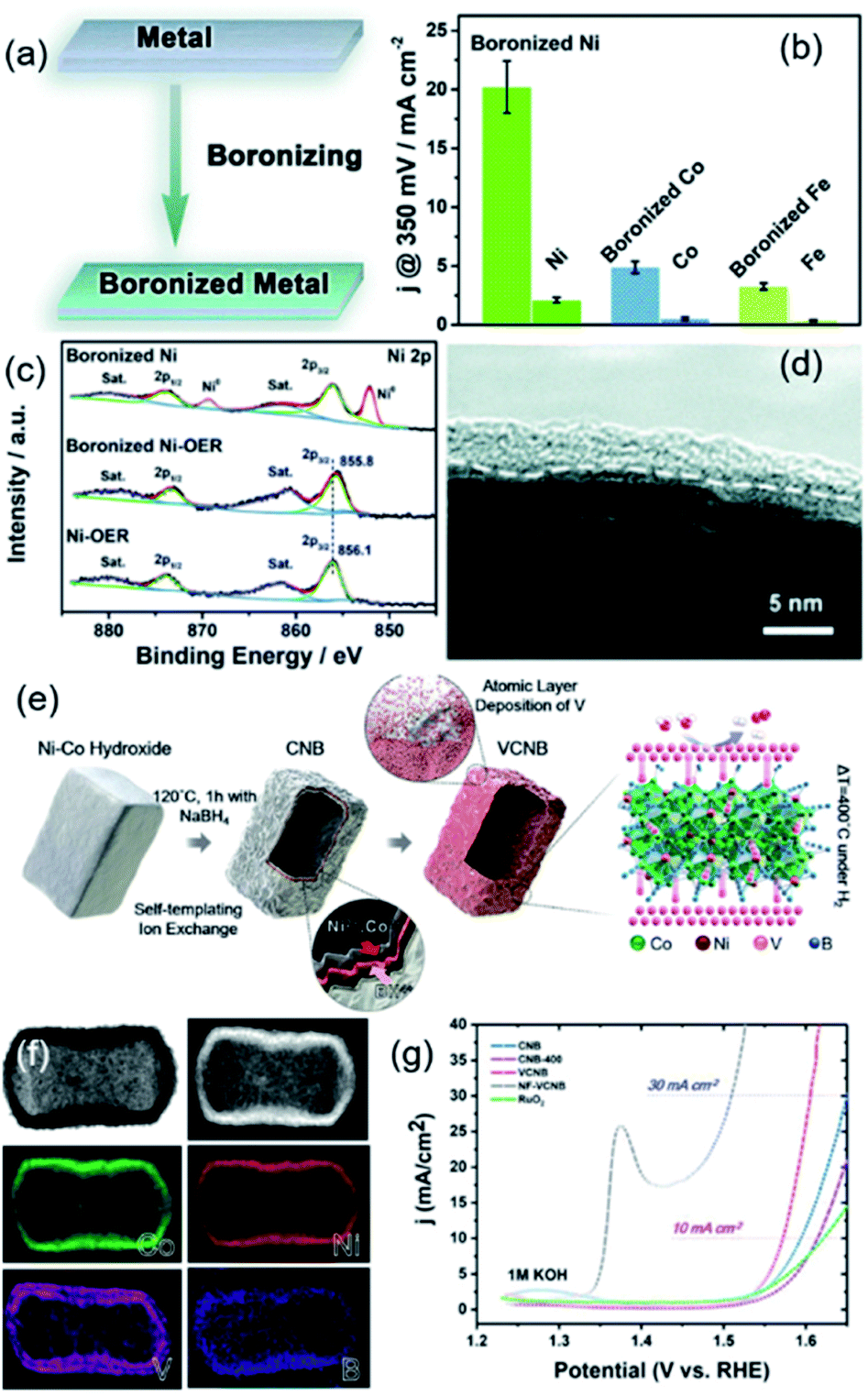 | ||
| Fig. 8 (a) Schematic for the preparation of metal borides from metal foil; (b) The η of different metal foils and boronized metal foils; (c) XPS analysis of metal foils and boronized metal foils before and after the OER; (d) TEM image of boronized metal foils after the OER. Reproduced with permission,66 Copyright 2019, Royal Society of Chemistry. (e) Schematic for the preparation of hollow V doped cobalt nickel boride; (f) EDX mapping of the synthesized hollow V doped cobalt nickel boride; (g) OER polarization curves of different samples. Reproduced with permission,67 Copyright 2019, Wiley-VCH. | ||
4.2 Challenges in synthesis and catalysis
So far, various TMBs have been synthesized by CVD, electrodeposition, solid-state reactions, liquid-phase methods, and ball milling.68 Although lots of achievements have been made regarding developing efficient TMBs for the HER and OER, there are still some challenges that need to be addressed. (i) The morphologies and structures of the currently reported borides are mostly uncontrollable together with a low specific surface area, which is not conducive to the exposure of active sites and the progress of mass transfer. Therefore, more research work is needed in the future to develop porous TMBs with controllable nanostructures to enhance the catalytic activity and application areas. (ii) At present, the research of TMBs is mainly focused on regulating the metal species in TMBs. It is necessary to study the influence of the doping of non-metallic elements (N, P, S, etc.) on the catalytic activity of TMBs. (iii) In the case of the OER, the active components of TMBs are mainly surface-generated hydroxides or oxyhydroxides, but their activity is far superior to that of their corresponding metals or metal hydroxides used in the OER. More research work is needed to reveal the mechanism and the role of B in the OER process. In situ characterization is also needed to explore the real active species of TMBs during the catalytic process.5. Metal carbides
The HER performance of TMCs is still restricted by the strong Hads on their surfaces. Optimizing the electronic properties of TMCs to tune the Hads energy is crucial to further improve their HER activity. Therefore, recent studies on TMCs mainly focused on designing MC-based materials with heterostructures (e.g., hybridizing with transition-metal compounds).69 TMCs have also been regarded as promising OER catalysts due to their high chemical stability at high potentials. However, compared to the acceptable HER activity, the OER activity of TMCs is poor which seriously limited their practical application. This is mainly attributed to the strong adsorption of the OER intermediates on the surfaces of catalysts leading to the sluggish process of removing reaction intermediates and products. In this section, the recent progress in TMC development for the HER and OER is summarized. In addition, the synthesis method and mechanism of the enhanced catalysis are also discussed.5.1 The state-of-the-art catalysts and their HER/OER performances
MoC and Mo2C materials have been explored as promising HER electrocatalysts owing to their low cost and high chemical stability. The carbon atoms in molybdenum carbides are filled in the Mo lattice, leading to the increase of lattice spacing and contraction of its d-band.70 The synthesis of MoC and Mo2C with a highly crystalline structure was usually carried out by pyrolysis of compounds containing Mo and organic precursors at high temperatures.14 Unfortunately, this process leads to a low surface area and poor mass transport. Therefore, great effort was devoted to improving the HER performance by synthesizing novel structures.71,72 Hu et al. synthesized β-Mo2C for the first time and achieved a good HER performance with an η20 of 225 and 210 mV under acidic and alkaline conditions, respectively.73 Recently, Asefa et al. reported nanoporous η-MoC nanosheets on N doped graphene (np-η-MoC NSs).14 In the synthesis, the precursor Mo-CN preserved a tetragonal structure during the pyrolysis at 750 °C (Fig. 9a). The np-η-MoC NSs showed a low onset potential of 28 mV, a small η10 of 112 mV, and a small Tafel slope of 53 mV dec−1 in a 1 M KOH electrolyte, as well as a more favorable ΔGH* (Fig. 9b).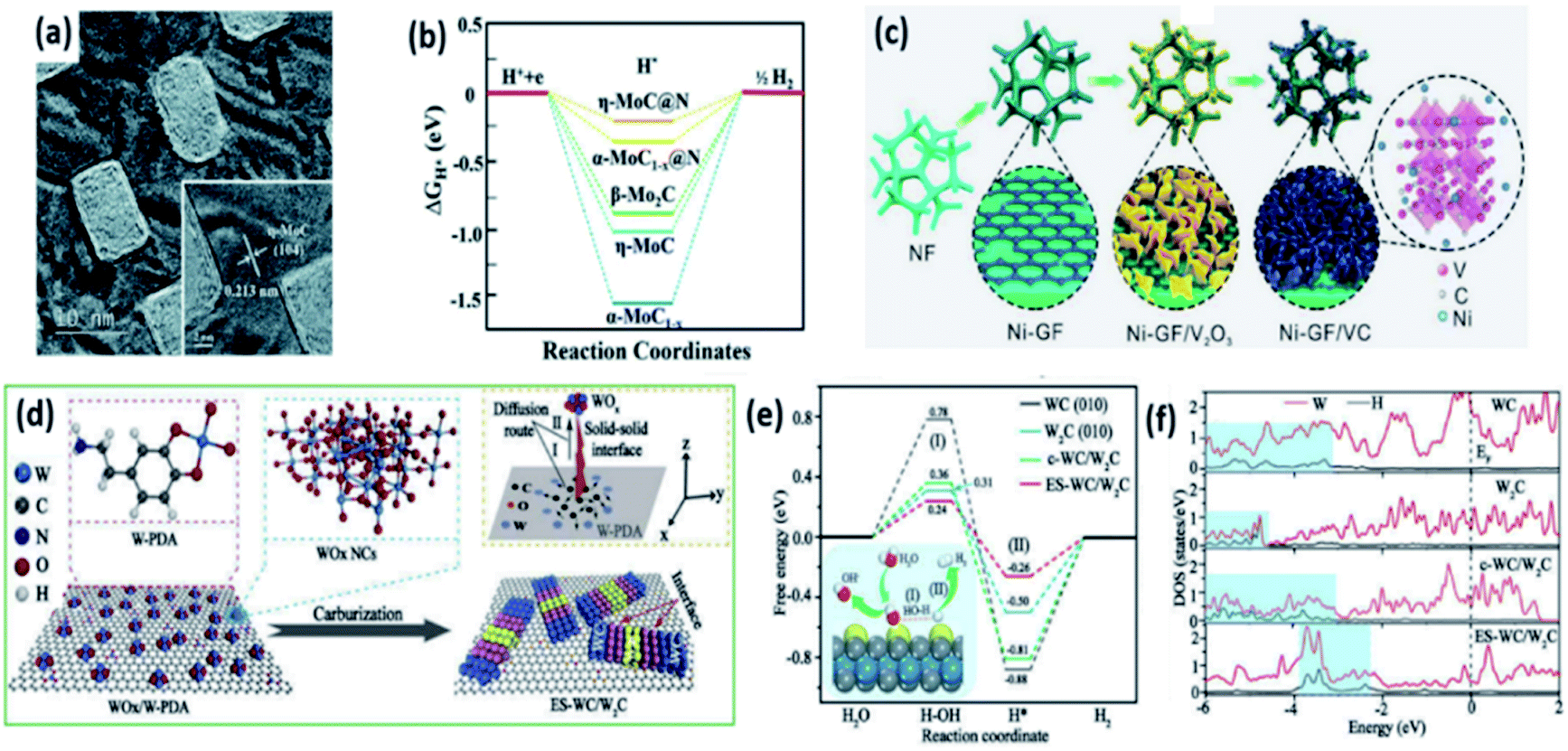 | ||
| Fig. 9 (a) High-resolution TEM images of np-η-MoC NSs synthesized by pyrolysis of the Mo-CN precursor at 750 °C. (b) ΔGH* diagram of Hads on different electrocatalysts. Reproduced with permission,14 Copyright 2019, Royal Society of Chemistry. (c) Schematic illustration of the synthesis process of the Ni-GF/VC catalyst. Reproduced with permission,75 Copyright 2020, Wiley-VCH. (d) Schematic representation of the fabrication of ES-WC/W2C from organic–inorganic tungsten precursors. (e) The free energy diagram for the alkaline HER process, with the schematic illustration of water activation and H* formation (I) and hydrogen generation (II) processes in inset. (f) Local state densities of W and H on WC, W2C, c-WC/W2C and ES-WC/W2C catalysts. Reproduced with permission,81 Copyright 2020, Elsevier. | ||
Doping foreign atoms into TMCs can further improve the HER performance by tuning the ΔGH*. Yu et al. prepared a series of Mo2C particles doped with Ni, Co, Fe, and Cr. Among them, Ni-Mo2C@C showed the lowest η10 of 72 mV in 0.5 M H2SO4. DFT calculations suggested that the ΔGH* of these transition metal doped Mo2C materials follows the trend: Ni-Mo2C > Co-Mo2C > Fe-Mo2C > Cr-Mo2C.74 Li et al. doped Ni atoms into VC and Fe3C nanosheets via a hydrothermal method and the subsequent magnesium thermal reaction (Fig. 9c).75 The ΔGH* values for the (100) and (111) planes of Niads site were 0.161 and 0.254 eV, respectively, which were much lower than that of C and V sites, indicating an active Ni site in Ni-VC. Recently, Mu et al. fabricated Ni-Ni3C heterostructures to adjust the adsorption of H*.76 The Ni-Ni3C/CC catalyst exhibited a low η10 of 98 mV in a 1 M KOH electrolyte. Chen et al. reported N, B co-doped Co3C spheres for multifunctional electrocatalysis. The N, B co-doped Co3C catalyst exhibited superior HER performance with an η10 of 154 mV in 1 M KOH, which was 70 mV lower than that of the Co3C catalyst.77 Fan et al. integrated transition metal carbides M3C (M = Ni, Co, Fe) with graphitic shells supported on vertically aligned graphene nanoribbons (M3C-GNRs) and found that the catalysts possessed good HER activities with an η10 of 49, 91 and 48 mV for Fe3C-GNRs, Co3C-GNRs and Ni3C-GNRs in 0.5 M H2SO4, respectively, due to the small size of M3C and the microporous structure of M3C-GNRs.78 Mullins et al. demonstrated a more favourable hydrogen adsorption free energy and better conductivity of Co2C than those of Co3C, indicating superior HER activity of Co2C.79 Co2C nanoparticles were synthesized by Ma et al. and exhibited high HER activity with an η10 of 181 mV in 0.1 M KOH.80
Tungsten carbides exhibited similar HER performance to molybdenum carbides.81 Because of the higher electronic density of states (DOS) at the Fermi level and more favourable H* adsorption, W2C exhibits better HER activity than WC.82,83 However, the chemical stability of W2C is worse than that of WC. Li et al. synthesized carbon covered ultrafine WC/W2C nanowires (WC/W2C@C NWs) to combine the advantages of these two components.82 The WC/W2C@C NW catalyst showed a low η10 of 69 and 56 mV in 0.5 M H2SO4 and 1 M KOH electrolytes, respectively, as well as good long-term stability. Chen et al. fabricated a eutectoid WC/W2C heterostructure (ES-WC/W2C) with defect-rich WC/W2C interfaces (Fig. 9d).81 The ES-WC/W2C catalyst exhibited excellent HER activity in an alkaline electrolyte with a low onset potential of 17 mV, η10 of 75 mV. DFT calculations presented a lower water dissociation barrier at the defect-rich WC/W2C interface, indicating the easier cleavage of HO–H bonds for ES-WC/W2C (Fig. 9e). As shown in Fig. 9f, hybridization peaks were only observed at higher energies (about −3.4 and −3.6 eV) for ES-WC/W2C, implying a weakened W–H interaction.
Metal carbides, such as Fe3C, Ni3C, Co3C, Mo2C, and W2C, are also promising for the OER due to their superior chemical stability. Xiao et al. prepared an Fe3C-based electrocatalyst by electrochemical metallurgy using Fe2O3 and CO2 as the feedstock (Fig. 10a).85 The carbon deposition (CO32− + 4e− = C + 3O2−) and iron formation (Fe2O3 + 6e− = 2Fe + 3O2−) on cathode resulted in highly dispersed Fe/Fe3C (Fe/Fe3C-MC) generation. The η10 of the Fe/Fe3C-MC catalyst was 320 mV which is lower than that of benchmark RuO2 (353 mV). Chemical structure analyses confirmed the transition of Fe/Fe3C into Fe3C–FeOOH during the OER process. The *OOH intermediates were linearly adsorbed on the Fe3C–FeOOH surface, which would be conducive to the formation of O2 (Fig. 10b). The energy barrier for the rate determining step on the Fe3C–FeOOH model was 1.37 eV, which is lower than that of Fe3C–Fe (5.43 eV) and Fe3C (2.73 eV), indicating that the in situ produced FeOOH improves the catalytic OER kinetics (Fig. 10c). To further clarify the structural changes, Mullins et al. characterized the elemental composition and microstructure of Co3C particles at various times during the OER process. They found that the Co3C particles are firstly converted to a transitory Co3C core-amorphous Co oxide shell and then to an amorphous Co oxide particle.84 Gou et al. reported the preparation of Co-doped Fe3C@carbon nano-onions (FCC@CNOs) via a high-pressure annealing method.86 The high resolution TEM images showed the lattice fringes of 0.24 nm, which are indexed to the (210) crystal plane of Fe3C. The OER η10 of FCC@CNOs was 271 mV, which is much better than that of the benchmark RuO2. DFT results showed that Co tends to form weaker *O adsorption than Fe do. Coupling Ni with Ni3C can optimize the adsorption energies of water and OER intermediates, benefiting the OER catalytic kinetics. A Ni-Ni3C heterostructure composed of Ni NPs and Ni3C NSs on carbon cloth (Ni-Ni3C/CC) was obtained by annealing Ni(OH)2 and melamine.76 The OER η20 of the Ni-Ni3C/CC catalyst was 299 mV, which is lower than that of Ni3C/CC and RuO2. DFT calculations showed that the ΔG value of the rate determining step on Ni-Ni3C is 1.815 eV, which is significantly lower than that of Ni3C (3.701 eV) and Ni (3.877 eV), confirming more efficient OER kinetics on the Ni-Ni3C surface.
 | ||
| Fig. 10 (a) Schematic of electrochemical co-reduction to produce cathodic Fe/Fe3C-MC. (b) Geometric structures for the rate-determining *OOH step on Fe3C–FeOOH models. (c) The calculated energy profile for the OER on several models at pH = 14 and U = 1.23 eV vs. SHE. Reproduced with permission,85 Copyright 2021, Wiley-VCH. (d) The synthesis illustration of Co SAs/Mo2C. Reproduced with permission,87 Copyright 2020, Royal Society of Chemistry. (e) LSV curves of Co-Mo2C toward the alkaline OER. Reproduced with permission,69 Copyright 2020, American Chemical Society. | ||
Bimetallic TMCs were found to be more efficient for the OER. For instance, Kou et al. synthesized metal single atom (e.g., Co, Ni, and Cu) doped Mo2C nanosheets (M SAs/Mo2C) by metallic cation exchange and carbonization using MoZn bimetallic imidazolate frameworks as the precursors (Fig. 10d).87 Metal single atoms were coordinated with three Mo atoms on the surface of Mo2C to form M-Mo3 sites. Co SAs/Mo2C provided an ultralow η10 of 270 mV and high turnover frequency (TOF). Partial Mo K-edge XANES plots suggested a decrease in the average electron density of Mo after the decoration of Co atoms on Mo2C, leading to a favourable OH* adsorption strength for an efficient OER. Wang et al. synthesized a Co-Mo2C heterostructure in a similar way.69 Due to the long-range ordered Co-O–Mo connectivity in the Co-ZIF-L-MoO4 precursor, the in situ generated Co and Mo2C further formed heterostructure nanoparticles with uniform distribution of Co and Mo2C after pyrolysis. The η10 of Co-Mo2C was only 190 mV that is much lower than that of RuO2 (Fig. 10e). After OER cycling, a γ-phase cobalt oxyhydroxide (γ-CoOOH) was formed. The newly created γ-CoOOH would boost the electron flow from Mo to Co through the bridging oxygen, benefiting the electrostatic adsorption of OH−, thus increasing the catalytic activity of the OER. Bimetallic cobalt tungsten carbide nanosheets embedded in N-doped carbon (Co6W6C@NC) were prepared for efficient OER catalysis with an η10 of 286 mV.88 Pan et al. synthesized trimetallic CoCuW-based carbide hybrids by pyrolysis of a core–shell CuWO4@ZIF-67 precursor.89 The catalyst was comprised of metallic Cu, hexagonal WC, and Cu-doped cubic Co3W3C, and exhibited a low η10 of 238 mV for the OER. The introduction of Cu into Co3Wo3C could prevent the reduction of Co to a metallic state, maximize the use of the active Co sites, and modulate the electronic structure of catalysts for an efficient OER.
5.2 Challenges in synthesis and catalysis
Although TMC-based catalysts achieved enhanced HER and OER activities, it is still a great challenge to replace PGM catalysts. Mo2C is considered an ideal substitute for Pt in the HER. DFT calculations suggest that the synergistic effect between N-doped carbon and Mo2C can significantly improve the HER activity of Mo2C. Accordingly, N-rich carbon precursors are usually used for preparing such materials. However, the Mo2C particles synthesized by this method are bulky and usually encapsulated in carbon. Therefore, building Mo2C/NC composites with a porous structure and low-dimensional morphology is expected to further improve the HER activity. Although tungsten carbide-based catalysts exhibit a good HER activity in acidic solutions, their activities in alkaline solutions are unsatisfactory. W2C has a strong affinity for OH* intermediates in alkaline solutions, which will lead to the poisoning of active W sites into inert WxOy species. The doping of other metals, such as Co and Ni, is a common strategy to improve the HER activity of W2C.90,91 However, due to the low chemical stability of the doped metals, this strategy may have adverse effects on the durability of W2C-based catalysts.92 Therefore, it is interesting to develop W2C with a unique structure to obtain excellent HER activity and stability in alkaline solutions. For instance, Chen et al. introduced a eutectoid structure into the field of electrocatalysis and the eutectoid WC/W2C heterostructure showed good HER catalytic performances under alkaline conditions.81In the case of the OER, the component and structural changes of TMC-based catalysts during the OER process become crucial factors limiting the improvement of electrocatalysis. Therefore, it is of great significance for the rational design of TMC-based catalysts by considering the surface reconstruction during the OER process. A computational thermodynamic model can be used to predict the catalyst structure reconstruction in different reaction environments, which will be constructive for building efficient TMC-based OER catalysts.
6. Metal oxides
TMOs based on first-row 3d metals (Mn, Fe, Co, and Ni) have been commercially used as anode catalysts in mature alkaline electrolysers because they feature high alkaline stability, flexibility in oxygen vacancies, and distinctive electron distribution.15,21 The high intrinsic activity and stability of TMOs in alkaline media allow them to be applied even in technically advanced and well-defined anion exchange membrane water electrolyser (AEMWE) systems. The high OER activity of TMOs originates from the overlap of the d-band of 3d metals and the p-band of oxygen.21 The electronic interaction among these band structures tunes the binding energies between each intermediate species and the oxide surface and determines the OER rate. In particular, the binding energy difference between O* and OH* (ΔGO − ΔGOH) has been considered a useful OER activity descriptor of the oxide surfaces.21 Compositional and structural optimization enabled producing various crystal structures (e.g. rock salt, spinel, layer-structured oxides, and perovskites) having optimal ΔGO − ΔGOH and excellent OER performances.21,93–95 These advanced TMO catalysts have often demonstrated superior OER performances to PGM-based catalysts.Recently, TMOs have also emerged as promising HER catalysts.96 Since TMOs can possess diverse crystal structures, their electronic configuration is flexible. TMOs, which are considered inactive materials for the HER, can be activated by structural engineering and modifying electronic structures.97–100 These strategies, including oxygen-vacancy tuning, phase transformation, and developing multimetallic composition, generated HER-reactive TMO catalysts with various crystal structures via boosting the low electric conductivity and optimizing the ΔGH of oxide surfaces to promote the HER.
In this section, we review remarkable examples the TMO catalysts promoting the OER and HER. The underpinning mechanisms of enhanced catalysis are also discussed. We also summarize the remaining challenges in the TMO field.
6.1 The state-of-the-art catalysts and their HER/OER performances
During the past few decades, the advances of TMO-based OER catalysts have been accelerated by inspiration from the cubane-like CaMn4Ox structure in the nature's oxygen evolution complex.101 An enormous number of TMO catalysts have been developed with diverse strategies. Because the class of TMOs is too broad, advances in perovskites and pyrochlores are introduced in the next section (Section 7). Among the developed catalysts, (oxy)hydroxide (MOxHy) materials represent the most promising OER electrocatalysts.15,21 In particular, NiFeOxHy catalysts have demonstrated remarkable OER activity.102–104 Interestingly, pure NiOxHy, which shows low OER activity, shows a drastic activity increase when Fe is intercalated into the structure, even the Fe amount is impurity-level.105,106 Systematic studies using XAS or electrochemical scanning microscopy have suggested that the Fe moiety in the NiFeOxHy matrices is the OER active site.107,108 Recently, Chung et al. revealed that Fe–MOxHy systems generate dynamically stable Fe active sites on the MOxHy clusters (Fig. 11).109 A model experiment combined with isotopic labeling and inductively coupled plasma mass spectroscopy (ICP-MS) analyses exhibited that the dissolution and redeposition of Fe species take place continuously over the MOxHy host materials during the OER process with Fe–MOxHy catalysts. Balancing the rates of Fe dissolution/redeposition is pivotal for an active and stable OER. Markovic et al. also suggested two routes for boosting catalytic activity: (i) increasing the surface area of the host MOxHy cluster to achieve the maximized number of Fe species, and (ii) tuning Fe adsorption energy on the host materials (ΔGFe–M) to boost the number of effective Fe active sites.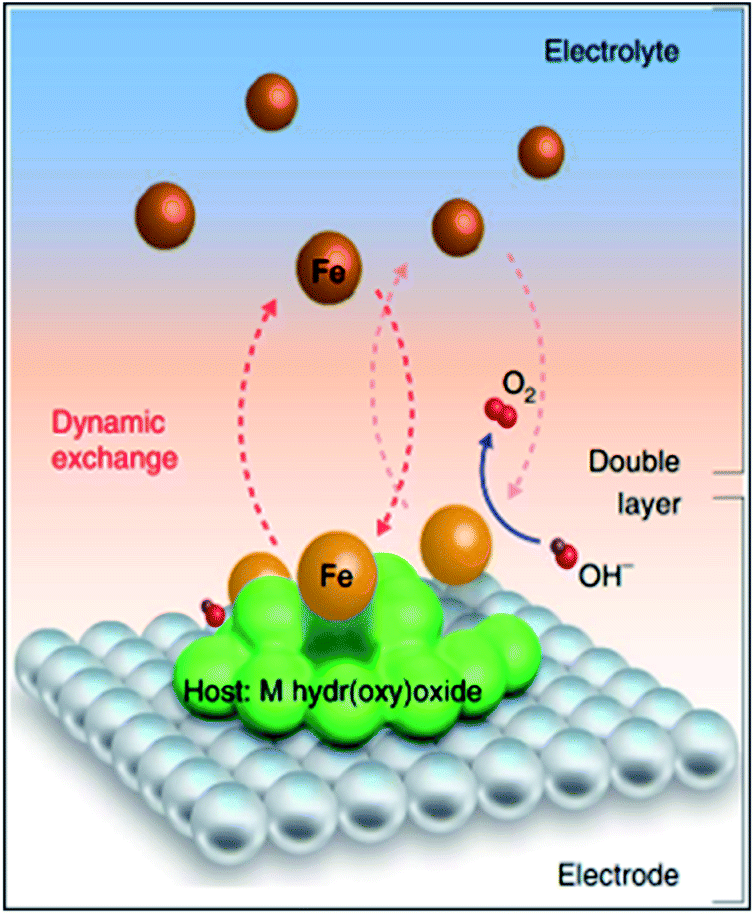 | ||
| Fig. 11 Schematic diagram of the dynamically stable active-site/host pair at the electrode/electrolyte interface, highlighting the role of M oxyhydroxide as a suitable host for Fe species to stay at the interface long enough to catalyse the conversion of OH− into O2 molecules, with the presence of Fe in the electrolyte ensuring that Fe species can return to the interface and redeposit at oxyhydroxide sites. Reproduced with permission,109 Copyright 2020, Springer Nature. | ||
Adopting multimetallic M1M2OxHy host materials can further boost the OER performance by tuning the ΔGFe–M of Fe-MOxHy. For example, Zhang et al. developed highly efficient ternary oxyhydroxide catalysts with a homogeneous atomic distribution.95 They adopted FeCoW composition based on DFT calculations. Computational prediction estimated that the OER activity of bimetallic TMOs based on first-row 3d metals can be greatly improved by W doping. On the basis of DFT calculations, gelled FeCo (G-FeCo) and gelled FeCoW (G-FeCoW) oxyhydroxides with a homogeneous atomic distribution were prepared via a room-temperature sol–gel process (Fig. 12a). To elucidate the impact of the structural homogeneity of the G-FeCoW catalyst on the OER performance, an annealed G-FeCoW (A-FeCoW) sample was synthesized by heating G-FeCoW oxyhydroxide at 500 °C. The prepared A-FeCoW contains Co3O4 and CoWO4 phases, exhibiting the heterogeneous distribution of elements. An FeCo layer double hydroxide (LDH) was also prepared for the activity benchmark. The OER activity of catalysts was measured on gold-plated Ni foam. The OER polarization curves (Fig. 12b) and η10 of the catalysts clearly suggest an OER activity in the following order: G-FeCoW (191 mV) > G-FeCo (215 mV) > A-FeCoW (232 mV) > LDH FeCo (279 mV). The intrinsic activity of catalysts was further compared using TOFs. The TOF of G-FeCoW (0.46 s−1) also surpassed those of G-FeCo (0.043 s−1), A-FeCoW (0.17 s−1), and LDH FeCo (0.0085 s−1). In addition, G-FeCoW catalysts exhibited high stability preserving the initial η30 for 550 h (Fig. 12c). Notably, the OER faradaic efficiency for G-FeCoW was well preserved during the continuous OER process. The electrochemical quartz crystal microbalance technique and ICP-atomic emission spectroscopy (ICP-AES) analysis also confirmed the excellent stability of G-FeCoW catalysts. This synthetic method has recently been extended to the preparation of ternary NiFeM and FeCoM oxyhydroxides (M = W, Mo, Nb, Ta, Re) and quaternary NiFeMoW and FeCoMoW by the same group.110 Among those catalysts, the best-performing NiFeMo oxyhydroxide catalysts demonstrated excellent activity and durability in an industrial alkaline electrolyser system (Fig. 12d). Under various applied cell potentials, the obtained current densities of NiFeMo electrode-base cells are 17 times higher than those of the commercial RANEY® Ni electrode-based one. Furthermore, the NiFeMo catalyst demonstrated high stability, exhibiting no appreciable increase in the cell voltage during the continuous operation of an alkaline electrolyser at 300 mA cm−2 for 12 h (Fig. 12e).110
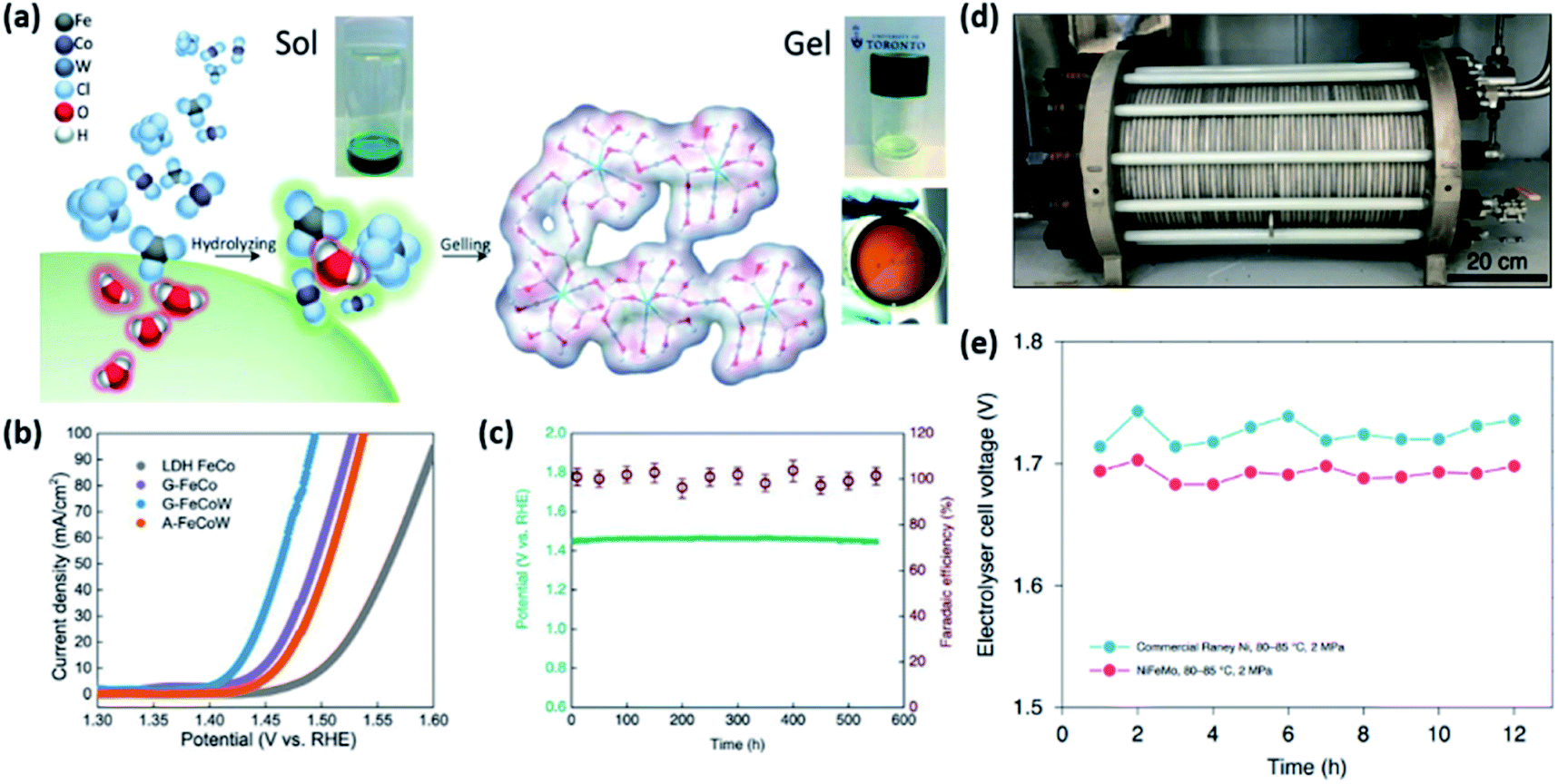 | ||
| Fig. 12 (a) Schematic illustration of the preparation process for the gelled structure and pictures of the corresponding sol, gel, and gelled film. (b) The OER polarization curve of catalysts loaded on two different substrates with a 1 mV s−1 scan rate, without iR correction. (c) Chronopotentiometric curves obtained with the G-FeCoW oxyhydroxides with a constant current density of 30 mA cm−2, and the corresponding faradaic efficiency from gas chromatography measurement of evolved O2. Reproduced with permission,95 Copyright 2016, AAAS. (d) Photograph of an industrial electrolyser device. (e) The cell voltage of the electrolyser held at 300mA cm−2 for 12 h at 80–85 °C and 2 MPa. Reproduced with permission,110 Copyright 2020, Springer Nature. | ||
In the case of the HER, various structures of active catalysts have been explored and evaluated. The most studied compositions for the acidic HER are MoO2, MoO3, WO3, and TiO2, perhaps owing to their acidic stability. However, it is well known that these pristine oxides with well-defined stoichiometry are usually inactive for the HER. In this vein, the creation of oxygen vacancies has been regarded as an efficient way to activate these materials. Oxygen vacancy tuning can modulate the electronic structure, electrical conductivity, and ΔGH leading to enhanced intrinsic HER activity. For example, Li et al. showed that the activity of WO3 was improved by facile structural engineering to form oxygen-defect WO2.9 (Fig. 13a).97 As shown in the HER polarization curves (Fig. 13b), the η10 of WO2.9 is much lower than that of pristine WO3 (637 mV). DFT calculations also suggested that the vacancy engineering tuned the ΔGH toward an optimal value to promote the HER. The vacancy engineering of TMOs is also applicable to improve alkaline HER performance. Ling et al. reported the activity enhancement of CoO nanorods (NRs) via surface strain engineering.98 They demonstrated that the electronic structures of CoO can be optimized by the tensile strain effect induced by generating a large number of oxygen vacancies. These oxygen vacancies can facilitate the dissociation of water molecules and lowered the ΔGH. Consequently, the surface-strained CoO NRs showed excellent alkaline HER activity, which is comparable to that of the benchmark Pt/C catalyst. Hu et al. also reported that vacancy engineering is effective in improving the HER activity.111 They developed metal oxide nanofibers, which are composed of interconnected Co-Ni oxide NPs possessing a plethora of various lattice defects and unsaturated metal sites (D-CoNiOx-P-NFs). D-CoNiOx-P-NFs demonstrated enhanced HER performance compared to defect-free CoNiOx-P-NFs. It originates from the promoted water dissociation by the defective structure, consistent with the above CoO NR catalysts.
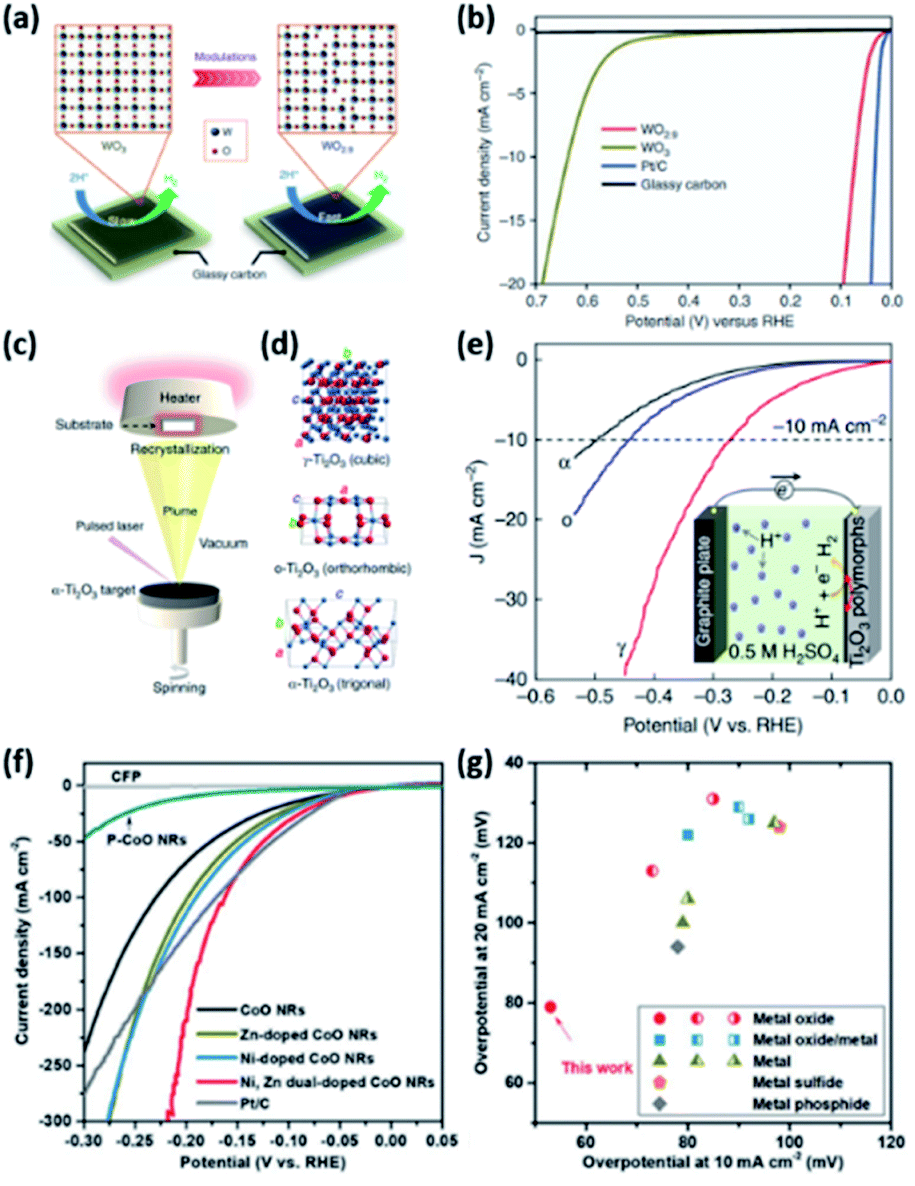 | ||
| Fig. 13 (a) Schematic illustration of the plausible reaction mechanism of the electrocatalytic HER for WO3 and WO2.9. (b) HER polarization curves of catalysts in 0.5 M H2SO4. Reproduced with permission,97 Copyright 2015, Springer Nature. (c) Schematic of the PLD chamber where Ti2O3 polymorphs were fabricated using the same (α-Ti2O3) target. (d) Unit cells for γ-Ti2O3, o-Ti2O3, and α-Ti2O3 polymorphs from top to bottom, respectively. (e) HER polarization curves of catalysts in 0.5 M H2SO4. Reproduced with permission,99 Copyright 2019, Springer Nature. (f) HER polarization curves of P-CoO, pure CoO, Zn-doped, Ni-doped, Ni, Zn dual-doped CoO NRs, and the benchmark Pt/C catalyst recorded in 1 M KOH. (g) Alkaline HER performance of Ni, Zn dual-doped CoO NRs compared to recently reported highly active alkaline HER catalysts. Reproduced with permission,100 Copyright 2019, Wiley-VCH. | ||
The phase transformation of TMOs is also a vital strategy for boosting catalytic activity via tuning the electronic structure. Li et al. highlighted the importance of phase engineering to achieve efficient HER catalysts. They prepared three different bulk-absent Ti2O3 polymorphs (trigonal α-Ti2O, orthorhombic o-Ti2O3, and cubic γ-Ti2O3) using the pulsed laser deposition (PLD) technique (Fig. 13c). By recrystallization on substrates during epitaxial growth, electronic reconstruction was classified into three types (Fig. 13d). The prepared Ti2O3 thin films were utilized as the model catalysts for investigating the correlation between the electronic structure and HER activity. As shown in Fig. 13e, the HER activity of cubic γ-Ti2O3 was much higher than those of trigonal α-Ti2O and orthorhombic o-Ti2O3. The higher activity of γ-Ti2O3 is attributed to the strongest hybridization of Ti 3d and O 2p orbitals.
As illustrated in OER catalysts, adopting a multimetallic composition is also an effective and easy way to activate TMOs towards the HER. Ling et al. elaborately tailored the electronic structure and electrical conductivity of inactive CoO NRs by dual doping of Ni and Zn elements, resulting in activity enhancement.110 In this work, the Ni dopant modulated the electronic environment of host CoO NRs, tuning the binding energy of intermediates. The Zn dopant also improves the electrical conductivity of the CoO matrix by tuning the bulk electronic structure. As shown in the HER polarization curves (Fig. 13f), the Ni, Zn dual-doped CoO NRs outperformed the pure CoO NRs, polycrystalline CoO NRs (P-CoO NRs), and M-doped CoO NRs (M = Ni, Zn). Notably, the η of Ni, Zn dual-doped CoO NRs was even smaller than that of the benchmark Pt/C catalyst at high-current density. More importantly, the comparison of η10 and η20 demonstrated that the activity of Ni, Zn-doped CoO NRs surpasses those of present state-of-the-art alkaline HER catalysts (Fig. 13g).
Recently, high-performance multimetallic (oxy)hydroxide catalysts for the HER have also been developed. Monometallic (oxy)hydroxide materials are considered to have low HER activity due to their inappropriate ΔGH. However, they can dissociate water efficiently and achieve high HER activity by adopting multimetallic composition. For example, Fe-substituted VOOH hollow spheres composed of 2D flakes exhibited an η10 of 93 mV.112 DFT calculations suggested that the Fe site of the Fe-doped VOOH tunes the ΔGH of V sites to an optimal value. In another example, the dynamically self-optimized NiFe LDH nanosheets reported by Edvinsson et al. showed an η10 of 59 mV in 1 M KOH.113 The in situ Raman spectra revealed that the dynamically generated FeOOH species promote water dissociation and induce Had–NiO bonds, resulting in HER activation.
6.2 Challenges in synthesis and catalysis
Despite the great promise of TMO catalysts for water electrolysis, several critical issues remain toward widespread application. For OER catalysis, the use of TMO-based OER catalysts is only available in AEMWEs due to their unstable nature in acidic media. Developing robust TMO catalysts with excellent acid stability and corrosion resistance is crucial for accomplishing low-cost catalysts, which are alternatives to Ir- and Ru-based catalysts for proton exchange membrane water electrolysers (PEMWEs). As reported by Simonov et al., CoFePb multimetallic oxide shows excellent acid stability, and further advances in TMO catalysts for PEMWEs are also highly expected.For HER catalysis, the catalytic activity of oxide materials is still not comparable to that of Pt catalysts. There are huge opportunities for exploring new HER catalysts for optimal ΔGH, given a myriad of TMO compositions that are not yet realized. Further improvements in structural engineering strategies for modulating the electronic structure are also demanding. Combining systematic compositional screening with structural optimization can produce highly conductive, efficient, and robust TMO-based HER catalysts. Intensive research along this direction should be conducted in the near future.
Overall, single-cell studies should be performed to accelerate the growth of this field. Currently, the fabrication of TMO-based PEMWE or AEMWE cells has been rarely reported. In addition, the standard measurement conditions for the performance evaluation of TMO catalysts in a single-cell configuration are not established firmly. Furthermore, developing in situ techniques for the structural analyses of TMO catalysts under single-cell driving conditions is an urgent task to glean insights into practically useable catalysts.
7. Perovskite and pyrochlore
Functional oxides with perovskite and pyrochlore structures have been intensively studied as electrocatalysts for the ORR and OER due to their low cost, compositional flexibility, and tuneable properties. The general formula of perovskites is ABO3, where the A-site is an alkaline, alkaline-earth or rare-earth cation in 12-fold coordination with O and the B-site represents a smaller transition or p-block metal ion in corner-sharing BO6 octahedral units (Fig. 14a).27 Pyrochlore is a complex oxide with a general formula of A2B2O7−δ where A is an alkaline-earth or rare-earth cation and the B-site represents a transition metal cation.114 The formula of pyrochlore can also be described as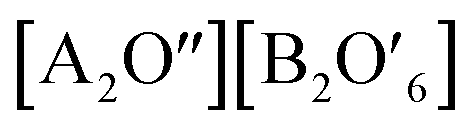 with two types of oxygen, consisting of a network of corner-sharing
with two types of oxygen, consisting of a network of corner-sharing 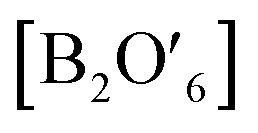 octahedra with A and O′′ atoms occupying interstitial sites (A2O′′) (Fig. 14b).114 The compositional flexibility of the A and B sites of these perovskite and pyrochlore oxides provide numerous opportunities to manipulate their electronic structures and corresponding catalytic properties.27,114,115 Furthermore, the ability to substitute other ions for the A- and B-site elements while maintaining the crystal structure provides more opportunities for adjusting the electronic structure and oxygen vacancy concentration, and thus their catalytic activity and stability.114 In this section, we first discuss the recent advances in understanding the OER mechanism based on perovskites, and then introduce representative examples of perovskite and pyrochlore catalysts for the OER and HER. We also discuss the remaining challenges and emerging opportunities in these oxide materials.
octahedra with A and O′′ atoms occupying interstitial sites (A2O′′) (Fig. 14b).114 The compositional flexibility of the A and B sites of these perovskite and pyrochlore oxides provide numerous opportunities to manipulate their electronic structures and corresponding catalytic properties.27,114,115 Furthermore, the ability to substitute other ions for the A- and B-site elements while maintaining the crystal structure provides more opportunities for adjusting the electronic structure and oxygen vacancy concentration, and thus their catalytic activity and stability.114 In this section, we first discuss the recent advances in understanding the OER mechanism based on perovskites, and then introduce representative examples of perovskite and pyrochlore catalysts for the OER and HER. We also discuss the remaining challenges and emerging opportunities in these oxide materials.
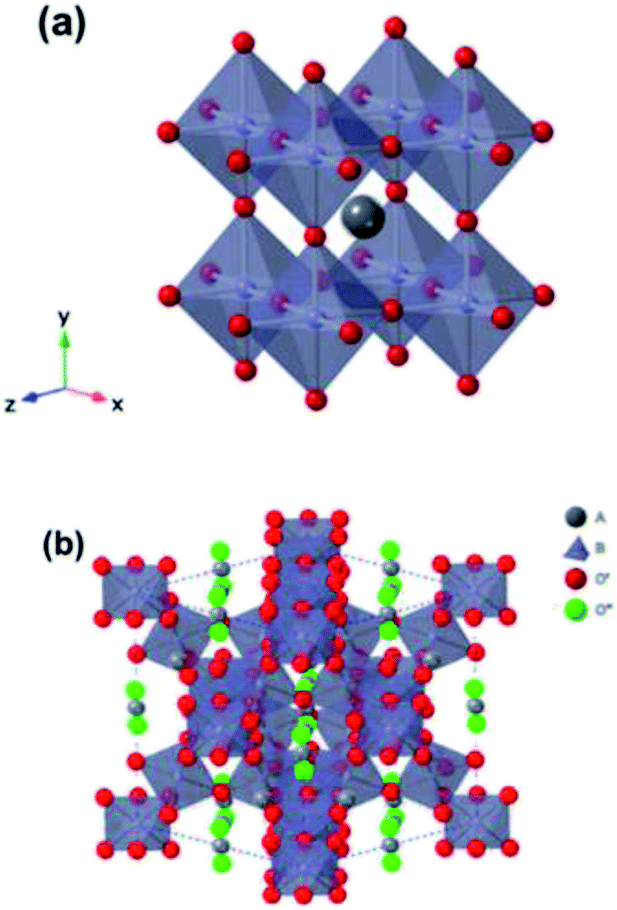 | ||
| Fig. 14 Structure of (a) perovskite, A2B2O7 and (b) pyrochlore, A2B2O7. | ||
7.1 The state-of-the-art catalysts and their HER/OER performances
The conventional OER mechanism for metal oxides stems from the basis that the oxygen evolves only around the metal centre, where the main parameter controlling the reaction η is the binding strength of oxygenated adsorbates (Fig. 15a).116,117 However, recent studies on perovskite oxide catalysts have revealed a new OER mechanism based on the involvement of lattice oxygens in perovskite, the so-called lattice oxygen evolution reaction (LOER) mechanism (Fig. 15a).117–119 Particularly under alkaline conditions, perovskite oxide catalysts have recently been central to the study of the LOER mechanism to improve the OER activity. Suntivich et al. systematically investigated the OER activity of perovskite oxides and suggested that the eg occupancy of the 3d electron can be employed as an effective descriptor.94 Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) showed the highest OER activity in 0.1 M KOH due to its optimal eg occupancy close to unity (Fig. 15b).94 However, the eg occupancy descriptor was established based on the ionic model and could not capture the electron sharing between the metal and oxygen atoms.115 Therefore, the metal–oxygen covalency was proposed as a more effective descriptor for predicting both OER activity and stability.115 The metal–oxygen covalency can be quantified by charge-transfer energy (energy difference between unoccupied metal 3d- and occupied O 2p-band centres) or the O 2p-band center.115 For example, Grimaud et al. used the O p-band centre to screen the OER activity and stability of perovskites (Fig. 15c).120 Bringing the O p-band centre closer to the Fermi level can increase the OER activities under alkaline conditions, and moving the O p-band centre too close to the Fermi level reduces stability.120 It should also be noted that the metal–oxygen covalency can provide the basis for adjusting the electronic structure of metal oxides to control the LOER mechanism.121 In a highly covalent network in which the metal d band penetrates the O p-band, the electrons from the p band can move to the d band, generating ligand holes.121 The generated ligand holes can facilitate the formation of oxygenated (O2)n− species, which tend to oxidize and release gaseous oxygen.121 Therefore, increasing the metal–oxygen covalency is expected to promote the LOER mechanism.27 | ||
| Fig. 15 (a) Conventional and lattice oxygen participating mechanisms of perovskite oxides for the OER. Reproduced with permission,116 Copyright 2007, Elsevier. (b) The relationship between the OER catalytic activity and the occupancy of the eg-symmetry electron of perovskites. Reproduced with permission,94 Copyright 2011, AAAS. (c) Schematic representation of the O p-band for transition metal oxides and the OER activity versus the O p-band center relative to the Fermi level for perovskite oxides. Reproduced with permission,120 Copyright 2013, Springer Nature. (d) Tafel plot comparing the specific activity of IrOx/SrIrO3 in 0.5 M H2SO4. Reproduced with permission,121 Copyright 2016, AAAS. (e) Crystal structure of 6H-SrIrO3, including face-sharing IrO6 octahedral dimers and a corner-sharing, isolated IrO6 octahedron. Reproduced with permission,123 Copyright 2018, Springer Nature. (f) High-resolution TEM images of cycled SrCo0.9Ir0.1O3−δ, where the white curve indicates the interfaces between the crystallized region and the reconstructed region.124 Copyright 2019, Springer Nature. (g) TEM image of nanoscale Bi2Ir2O7. (h) Stability measurements for various pyrochlore samples in 0.1 M HClO4. Reproduced with permission,128 Copyright 2017, American Chemical Society. (i) OER performance of Y2Ru2O7−δ and reference RuO2 catalysts in 0.1 M HClO4. Reproduced with permission,129 Copyright 2017, American Chemical Society. | ||
Under acidic OER conditions, research efforts have been focused on Ir- and Ru-based oxides due to their balanced activity and stability. Seitz et al. first reported that IrOx/SrIrO3 can be formed via strontium (Sr) leaching from the surface layer of SrIrO3 thin films under acidic OER conditions.122 The in situ formed IrOx/SrIrO3 catalyst delivered high OER activity with an η10 of 270 mV in 0.5 M H2SO4 (Fig. 15d).113 In addition, Yang et al. showed that a 6H-SrIrO3 perovskite catalyst exhibited a high OER activity with a low η10 of 248 mV in 0.5 M H2SO4. This perovskite catalyst also showed excellent stability with only ∼1% Sr leaching over a 30 h-long OER test. These improved activity and stability of 6H-SrIrO3 were attributed to its unique face-sharing IrO6 octahedral subunits (Fig. 15e).123 Chen et al. also introduced a pseudo-cubic SrCo0.9Ir0.1O3−δ perovskite, containing corner-sharing IrO6 octahedrons.124 The Ir in SrCo0.9Ir0.1O3−δ exhibited a significantly higher TOF than that of benchmark IrO2 in 0.1 M HClO4.124 The enhanced activity was explained by surface reconstruction caused by Sr and Co leaching, which can lead to the formation of active IrOx layers under acidic OER conditions (Fig. 15f).124 Moreover, Retuerto et al. showed that A-site Na doping into SrRuO3 can improve both the activity and stability of the catalyst under acidic conditions.125 Specifically, Sr0.95Na0.05RuO3 and S0.90Na0.10RuO3 delivered high OER activity with a low η0.5 of 120mV in 0.1 M HClO4.125 For Na-doped SrRuO3, the increase in activity was attributed to a weakened adsorption energy of the OER intermediates, and the improved stability was due to lower surface energy and higher dissolution potentials.125
Ir and Ru-based pyrochlore oxides have also been actively investigated as acidic OER catalysts due to their low Ir/Ru content, high activity, and stability in acidic environments.114 For example, Sardar et al. synthesized nanoscale Bi2Ir2O7 with an average crystal size of 10 nm as an acidic OER catalyst by a one-step hydrothermal reaction (Fig. 15g).126 Later, the same group first synthesized mixed Ru/Ir pyrochlores, (Na0.33Ce0.67)2(Ir1−xRux)2O7, and showed that Ru is more active toward the OER than Ir.127 Lebedev et al. also synthesized a series of Ir pyrochlores, (A, A′)2Ir2O6.5+x (A, A′ = Bi, Pb, Y), with a high surface area of 40 m2 g−1 using the Adams fusion method at moderate temperatures (500–575 °C).128 Electrochemical tests in 0.1 M HClO4 showed that yttrium pyrochlore catalysts exhibited high OER activity and reasonable stability (Fig. 15h), which is due to the formation of an active IrOx surface layer driven by the leaching of Y3+.128 Moreover, Kim et al. reported that a pyrochlore yttrium ruthenate (Y2Ru2O7−δ) could provide significantly enhanced OER activity and stability over RuO2 in 0.1 M HClO4 (Fig. 15i).129 The improved activity and stability of Y2Ru2O7−δ were attributed to its favourable electronic structures with a low valence state and a lower band centre energy for the overlap between Ru 4d and O 2p orbitals.129 Motivated by this study, Kuznetsov et al. synthesized a series of A-site substituted yttrium ruthenium pyrochlores, Y1.8M0.2Ru2O7−δ (M = Cu, Co, Ni, Fe, Y), and correlated their OER activity with the concentration of surface oxygen vacancies.130 Electrochemical tests in 1 N H2SO4 showed that Y1.8Cu0.2Ru2O7−δ had the highest OER activity due to the upshift of the O 2p band centre closer to the Fermi level.130
In addition to their application to OER catalysts, researchers have recently explored perovskite oxides as HER catalysts under alkaline conditions. Xu et al. demonstrated for the first time that A-site praseodymium (Pr)-doped Pr0.5(Ba0.5Sr0.5)0.5Co0.8Fe0.2O3−δ (Pr0.5BSCF) perovskite delivers a high HER activity with an η10 of 237 mV and a Tafel slope of 45 mV dec−1, which is significantly superior to Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) in 1 M KOH (Fig. 16a).131 The enhanced HER activity of Pr0.5BSCF was attributed to the combination of increased concentration of lattice oxygen and partially oxidized cobalt induced by Pr-doping, an increased electrochemical surface area (ECSA), and promoted electron transfer.131 Hua et al. also reported that a perovskite oxyfluoride catalyst, La0.5Ba0.25Sr0.25CoO2.9−δF0.1 (LBSCOF), exhibits an enhanced HER activity compared to La0.5Ba0.25Sr0.25CoO3−δ (LBSCO) in 1 M KOH.132 Specifically, LBSCOF showed a decreased Tafel slope of 44 mV dec−1 compared to that of LBSCO (51 mV dec−1) in a low current density region (0.3–8 mA cm−2) (Fig. 16b).132 The improved HER activity of LBSCOF was explained by fluorine (F)-anion doping that can uplift the O p band centre and activate the redox capability of lattice O.132 Therefore, LBSCOF has a preferred ΔGH* of −0.279 eV, which is close to zero compared to that of LBSCO (−0.634 eV) (Fig. 16c).132
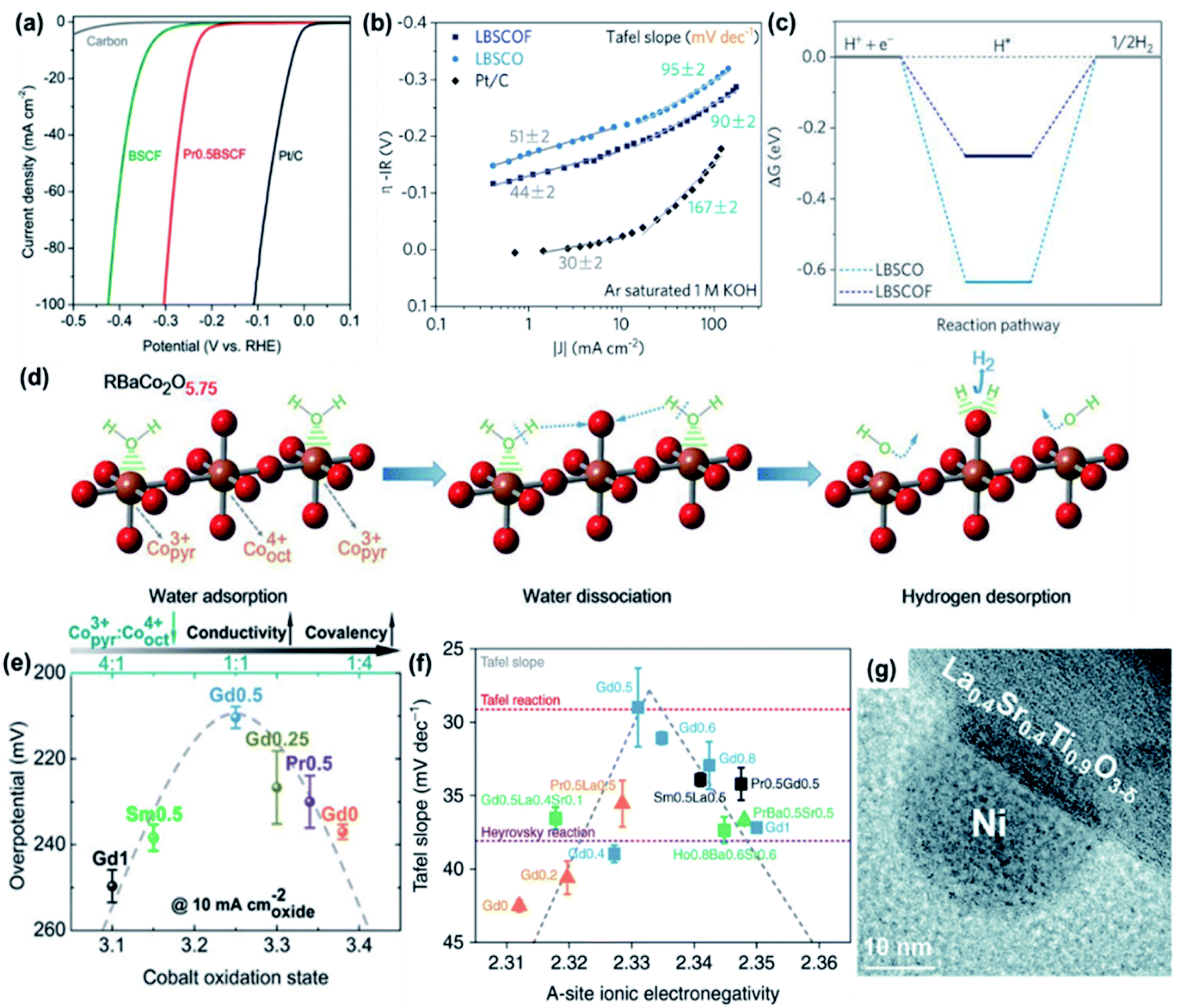 | ||
| Fig. 16 (a) HER Polarization curves of BSCF, Pr0.5BSCF and benchmark Pt/C catalysts in 1 M KOH. Reproduced with permission,131 Copyright 2016, Wiley-VCH. (b) Tafel plots and (c) computed Gibbs free energies of LBSCO and LBSCOF for the HER. Reproduced with permission,132 Copyright 2018, Elsevier. (d) Computational illustration for the alkaline HER processes on the RBaCo2O5.75 surface, including H2O adsorption, H2O dissociation, H* intermediate formation, and subsequent recombination of H* to form molecular H2. Reproduced with permission,133 Copyright 2019, Wiley-VCH. (e) Volcano plot of the η10 as a function of the Co valence state showing the highest HER activity for Gd0.5. Reproduced with permission,124 Copyright 2019, Wiley-VCH. (f) Volcano plot of the Tafel slope as a function of A-site ionic electronegativity. Reproduced with permission,134 Copyright 2019, Springer Nature. (g) High-magnification TEM image of Ni nanoparticle-decorated La0.4Sr0.4Ti0.9O3−δ perovskite. Reproduced with permission,133 Copyright 2019, Wiley-VCH. | ||
Double perovskite oxides with a general formula of AA′B2O6−δ, where A sites are alkaline-earth metal ions and A′ are lanthanide elements, have also been studied as alkaline HER electrocatalysts. Based on computational research, Guan et al. predicted that synergistic interactions between the ordered oxygen vacancies (at pyramidal high-spin Co3+ sites) and the O 2p ligand holes (at metallic octahedral intermediate-spin Co4+ sites) in RBaCo2O5.5+δ (δ = 0.25; R = lanthanides) can provide a near-optimal HER reaction pathway to adsorb H2O and release H2, respectively (Fig. 16d).133 They also experimentally showed that (Gd0.5La0.5)BaCo2O5.75 (δ = 0.25; Gd0.5) had the highest HER activity among the RBaCo2O5.5+δ catalysts in 1 M KOH (Fig. 16e).133 In addition, the same group reported that A-site ionic electronegativity (AIE) can be employed as an efficient descriptor to predict the HER activities of Co-based perovskites via a volcano-type activity trend (Fig. 16f).134 Based on this prediction, (Gd0.5La0.5)BaCo2O5.5+δ(Gd0.5) with an AIE value of ∼2.33 exhibited superior HER activity with a small Tafel slope of 27.6mVdec−1 in 1 M KOH.134 These studies represent a descriptor-based design that can accelerate the screening of highly active electrocatalysts towards the HER.
Perovskite and pyrochlore oxides have also been used as supporting materials for metal nanoparticle deposition in the design of hybrid catalysts for the alkaline HER. For example, metal nanoparticles can be deposited on a perovskite or pyrochlore oxide host through an in situ exsolution process.135–137 Zhu et al. synthesized a Ni nanoparticle-decorated La0.4Sr0.4Ti0.9O3−δ perovskite through the exsolution process as an alkaline HER catalyst (Fig. 16g).135 Similarly, Kim et al. designed NiRu alloy nanoparticle-supported Pb2Ru2−xNixO6.5 pyrochlore through a temperature-controlled exsolution process.137 The NiRu alloy nanoparticle supported pyrochlore oxide delivered a high HER activity with a small Tafel slope of 30 mV dec−1 in 0.1 M KOH.137 The enhanced HER activity of these hybrid catalysts under alkaline media can be attributed to the bifunctional mechanism where the oxide substrate can promote the dissociation of water molecules and provide Had to the neighbouring active site of metal nanoparticles for the recombination of Had into molecular hydrogen.135,137 Hu et al. modified the surface composition and structure of K0.469La0.531TiO3 perovskite (KLTO) by a hydrothermal reaction with a RuCl3 solution.138 During the hydrothermal treatment, ion exchange caused the incorporation of Ru into the surface of KLTO, and nucleation growth generated Ti-doped RuO2 (TRO) nanoparticles on the oxide surface.138 These hybrid catalysts exhibited a very high HER activity with a low η10 of 20 mV in 1 M KOH.138 DFT computation results showed that TRO nanoparticles can promote water dissociation and the oxide surface facilitates hydrogen evolution.138
7.2 Challenges in synthesis and catalysis
Considering the numerous combinations of A- and B-site compositions of perovskite and pyrochlore oxides, a computer-based screening process is necessary to identify the desired material structure for the best catalytic activity and stability.114 Recently, a computation-based approach has been used to identify a series of pyrochlore oxides as OER catalysts by calculating their phase diagrams, Pourbaix diagrams, projected density of states, and band energy diagram.139 In particular, DFT-based Pourbaix diagrams are found to be effective to predict the stability of oxide catalysts under acidic OER conditions.139,140 Additionally, a recent study has shown that a machine-learning model can be used to screen the covalency competition in spinel oxides to predict the composition of a highly active OER catalyst.141 This machine-learning based computational approach is also essential for identifying the ideal composition of perovskite and pyrochlore oxides for the HER and OER.In addition to the composition, oxide size control is also important because electrocatalytic reactions occur on or near the surface. The conventional synthesis of perovskite and pyrochlore oxides typically involves a high-temperature calcination process (>800 °C), resulting in particle sintering and surface area reduction.114 Therefore, it is highly desirable to synthesize nanoscale perovskites and pyrochlore oxides under low-temperature conditions. Recent studies have used a porogen method to synthesize nanostructured porous pyrochlore oxides142 and a polymer entrapment flash pyrolysis (PEFP) method to demonstrate the synthesis of phase-pure pyrochlore oxides at low temperatures (500 °C).143 Such efforts represent an effective strategy for improving the active sites of oxides for electrocatalytic reactions.
Surface reconstruction has been shown to occur for perovskite and pyrochlore oxides during the OER.140,144,145 The most recent studies have shown that understanding the dissolution of the A-site and the associated exposure of the B-site metal in perovskite and pyrochlore oxides is crucial for improving both the activity and stability of the OER.140,145 For example, Sr-doped LaCoO3 (La1−xSrxCoO3) perovskite showed enhanced activity during the alkaline OER with surface evolution due to A-site Sr dissolution.145 The enhanced OER activity was attributed to the formation of a Co hydr(oxy)oxide layer that interacts with trace amounts of Fe (aq.) in the alkaline electrolyte to generate dynamically active sites.145 It has also been reported that the dissolution of the A-site element in A2Ru2O7 pyrochlore oxides during the acidic OER can expose highly oxidized Ru sites that exhibit enhanced activity.140 Therefore, it is very important to understand the actual active sites and associated reaction mechanisms for mixed metal oxide catalysts during the OER process.
Finally, the in situ growth of metal nanoparticles on perovskite or pyrochlore oxide hosts through the exsolution process can be a powerful strategy to design high-performance hybrid catalysts for both the HER and OER.136,137 In detail, the transition metals incorporated into the B-site of perovskite or pyrochlore oxides can be exsolved from the oxide backbone as highly dispersed nanoparticles under a reducing environment. In particular, a recent study has revealed that the generated vacancies in the pyrochlore oxide support during the exsolution process can facilitate charge transfer between the exsolved metal nanoparticles and oxide support for enhanced catalytic reactions.137 These results open up new opportunities to design a variety of hybrid catalysts by carefully controlling the compositions and reduction conditions of perovskite and pyrochlore oxides.
8. Metal phosphides
TMPs are increasingly being developed through various technologies owing to the synergistic effect of metals and phosphides realizing a high efficiency of water electrolysis. Most of these materials involved Fe-, Co-, and Ni-based phosphides, and their alloy forms have been shown to efficiently improve the performance of electrolysis by altering the composition. In the case of the OER, TMOs are well known to promote the performance when the interaction of M2+ with OH* intermediates is weakened.21 This is mainly attributed to the increased repulsion between the metal d-band and the coordinated oxygen p-band (3d–2p). However, TMPs are not promising as OER catalysts because electronegative P atoms located near the metal atoms result in the obstruction of reaction intermediate OH* coordination on TMPs and thus the OER.16 Instead, TMPs have been applied as pre-catalysts to form OER active metal-oxo/hydroxo species under anodic conditions. In contrast, TMPs have been proven as excellent catalysts for the HER. The negatively charged P atoms and the positively charged metal atoms serve as the proton and anion acceptors, respectively. Therefore, both influence the ensemble effect of cooperatively promoting the HER under alkaline/acidic conditions. In addition, increasing the P atomic content within an appropriate range was found to be more effective in enhancing HER activity. Thereafter, much effort has been made to understand the role of P as the catalyst for the HER. Recently, Shin et al. designed a synthetic process to obtain FexNi2−xP nanocatalysts with various Fe-to-Ni compositions while maintaining the crystal structure.22 This work demonstrated the alloying effect of TMPs, excluding the structural effect, and that Fe0.5Ni1.5P nanocatalysts required the lowest η50 of 163 mV and a Tafel slope value of 65 mV dec−1 among the various FexNi2−xP nanocatalysts (Fig. 17a). According to XPS analysis, the P atoms in Fe0.5Ni1.5P showed the most electron-deficient character among the P atoms in FexNi2−xP (Fig. 17b). By EXAFS, it was found that the highly ordered M–M (Ni–M, Fe–M) bonds and disordered Ni–P bonds at Fe0.5Ni1.5P led to reduced electronegativity of P and thus to significantly improved HER kinetics (Fig. 17c). As a result, the optimal electron distribution of P is important for improving HER performance.146 However, the HER performance of currently developed TMP electrocatalysts has not reached the industry level, and much effort has been made to design new catalyst models to further improve HER performances.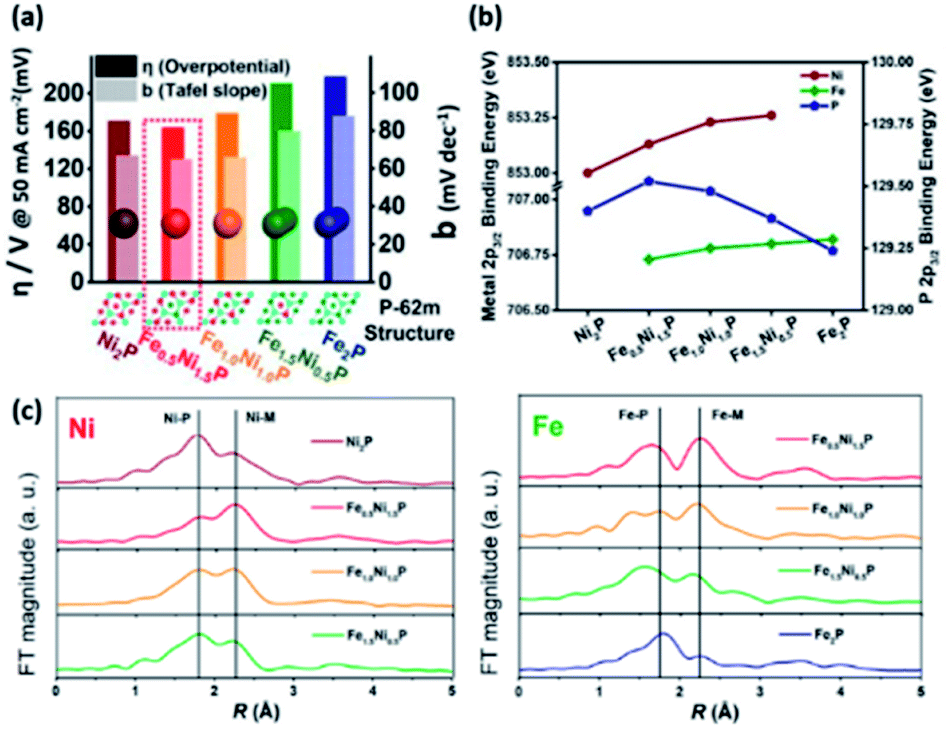 | ||
| Fig. 17 (a) The histogram of the b and the η at j = 50 mA cm−2 of Ni2P (wine), Fe0.5Ni1.5P (red), Fe1.0Ni1.0P (orange), Fe1.5Ni0.5P (green), and Fe2P (blue) nanocatalysts. (b) XANES spectra of the FexNi2−xP nanocatalysts: Ni 2p, Fe 2p, and P 2p binding energies of FexNi2−xP nanocatalysts. Fourier transforms of the (c) Ni K-edge and Fe K-edge EXAFS spectra for FexNi2−xP nanocatalysts. Reproduced with permission,22 Copyright 2020, American Chemical Society. | ||
8.1 The state-of-the-art catalysts and their HER performances
To significantly improve the HER performance, TMP development strategies, such as N doping to induce lattice contraction, heterostructure formation, and vacancy/defect engineering, have been the primary topics. Firstly, the N doping effect is introduced to improve the HER performance. As discussed, TMPs with abundant P sites have been predicted to be more favorable for HER catalysis. However, the actual activities of P-rich TMPs did not work as expected, and the fundamental understanding of P-rich conditions was unclear at the atomic level. Therefore, from a structural perspective, modulating the nonpolar P–P interaction to tailor the overlap of atomic wave functions has been considered to further boost the intrinsic HER activity of TMPs. Cai et al. developed N-doped CoP2 nanowires (N-CoP2 NWs) to increase the overlap of atomic wave functions via N-induced lattice contraction.147 XRD patterns showed similar diffraction patterns of CoP2 and N-CoP2 NWs. The diffraction peaks were slightly shifted to the higher angle region with the N incorporation (Fig. 18a), suggesting that N doping does not change the crystal structure of CoP2 but leads to slightly compressed interplanar distances. The HAADF-STEM and the corresponding EDX elemental mapping images indicated that Co, P, and N are homogeneously distributed over the NW (Fig. 18b). The broad XPS N 1s spectrum of N-CoP2 can be deconvoluted into the chemical states of N–Co and N–P, suggesting that N is successfully doped into the CoP2 lattices (Fig. 18c). Together, material analyses further demonstrated that N is substitutionally doped into the structure of CoP2, which can essentially manipulate the electronic and coordination structures of CoP2. The N-CoP2 NWs showed high catalytic performance with an η10 of 38 mV and a Tafel slope of 46 mV dec−1, which are substantially smaller than those of CoP2 NWs (125 mV and 73 mV dec−1) and even close to those of benchmark Pt/C catalysts (28 mV and 29 mV dec−1) (Fig. 18d). This catalyst also showed superior stability up to 30 h (Fig. 18e). Characterization and DFT calculations consistently revealed that the created strong N–P interaction in CoP2 could increase the atomic wave function overlap via lattice contraction and consequently broaden the local energy bandwidth with the downshift of the valence band centre. Furthermore, the free energy of ΔGH on N-CoP2 NWs was consistently much closer to neutral than those on CoP2 (Fig. 18f). As a results, N-CoP2 NWs facilitated HER catalysis by weakening the electron coupling between CoP2 and Hads. | ||
| Fig. 18 (a) XRD patterns of CoP2 and N-CoP2 NWs and the corresponding magnification of the diffraction peaks of the (200) facet. (b) HAADF-STEM image and EDX elemental mapping of Co, P, and N for a single N-CoP2 NW. (c) XPS N 1s spectrum of N-CoP2. (d) Polarization curves for CC, CoP2, N-CoP2, and Pt/C. (e) Chronopotentiometric curve for N-CoP2 at a current density of 20 mA cm−2. (f) Calculated ΔGH on CoP2 and N-CoP2 with various facets. Reproduced with permission,147 Copyright 2020, AAAS. (g) High-resolution TEM image of Ni12P5–Ni4Nb5P4/PCC. (h) The SEM images of CC treated with DBD plasma. (i) The XRD pattern of Ni4Nb2O9/PCC and Ni12P5–Ni4Nb5P4/PCC. (j) Polarization curves of PCC, CC, Pt/C/PCC, Ni4Nb2O9/PCC and Ni12P5–Ni4Nb5P4/PCC. (k) Polarization curves of Ni12P5–Ni4Nb5P4/PCC before and after 3000 cycles, and its chronoamperometric curves at a current density of 15 mA cm−2 for 100 h in the inset of (k). (l) Gibbs energy of different catalysts for the HER (dotted lines represent bonds facing the plane). Reproduced with permission,148 Copyright 2020, Wiley-VCH. | ||
Heterointerface engineering can be used to develop excellent catalysts through electronic coupling effects between different components or phases. In addition, carbon defects can be used to trap and stabilize reaction products, further promoting electrocatalytic performances. Based on this, Chen et al. reported that bi-phase Ni12P5–Ni4Nb5P4 nanocrystals with rich heterointerfaces and phase edges are successfully fabricated on carbon cloth (PCC), which is utilized by intentional defect creation by atmospheric pressure dielectric barrier discharge (DBD) plasma.148Fig. 18g exhibits the high-resolution TEM image of Ni12P5–Ni4Nb5P4 featuring the crystal planes of Ni12P5 and Ni4Nb5P4 are characterized. After the DBD plasma treatment, a large number of circular pits were formed on the carbon fiber surface (Fig. 18h). Fig. 18i shows the XRD patterns of Ni4Nb2O9/PCC and Ni12P5–Ni4Nb5P4/PCC. According to these results, a hetero-structured Ni12P5–Ni4Nb5P4 compound was formed on the PCC. The Ni12P5–Ni4Nb5P4/PCC electrocatalyst exhibited excellent HER performance, showing a low η10 and η50 of 81 and 287 mV, respectively, which outperforms the industry-relevant benchmark Pt/C/PCC catalyst (Fig. 18j). As shown in Fig. 18k, the polarization curves after 3000 cycles were close to the initial one. A long-term chronoamperometric measurement at a current density of j15 (the inset of Fig. 18k) for 100 h was performed that the obtained Ni12P5–Ni4Nb5P4/PCC catalyst presents an excellent stability. Therefore, it can be demonstrated that Ni12P5–Ni4Nb5P4 tightly coated on the surface of PCC results in a stable electrocatalyst. As shown in Fig. 18l, the ΔGH at the interface of Ni12P5–Ni4Nb5P4 was about 0.16 eV, which is much lower than that at the surfaces of Ni12P5 (0.23 eV) and Ni4Nb5P4 (0.35 eV), indicating favorable H* adsorption kinetics on the Ni12P5–Ni4Nb5P4 heterostructure during the HER process. They claimed that the superior HER performances of the Ni12P5–Ni4Nb5P4/PCC electrocatalyst are attributed to the well-defined heterointerface between the Ni12P5 and Ni4Nb5P4 phases and strong bonding between the composite and PCC. This feature reduced the loss of current, facilitated the adsorption or activation of active species, and caused a faster electron transfer and kinetic process for the HER.
In view of vacancy/defect engineering, Duan et al. succeeded in creating P vacancies (Pv) in Ni12P5 through a thermal-annealing process.149 As shown in Fig. 19a, the Ni12P5 with Pv (v-Ni12P5) showed a porous nanosheet texture with homogeneous dispersion of Ni and P atoms. Synchrotron-based XAS indicated that the higher intensity of the Ni K-edge in Ni12P5 than that in metal Ni suggests more probable 1s → 3d transition, while it was much lower than that of NiO, indicating that Ni in Ni12P5 is close to the metallic state with a slightly smaller number of electrons in Ni 3d (Fig. 19b). A slightly lower intensity of v-Ni12P5 than that of pristine Ni12P5 (p-Ni12P5) suggested a higher electron density in the Ni 3d band because of Pv. The v-Ni12P5 catalyst required a small η10 of 27.7 mV and a Tafel slope of 30.88 mV dec−1 in comparison with p-Ni12P5 (120.1 mV and 83.60 mV dec−1), and even outperformed the benchmark Pt/C (32.7 mV and 30.90 mV dec−1) (Fig. 19c). DFT calculations performed that conducted that a significant electron redistribution in the Ni12P5 is induced by Pv with considerable electron accumulation and depletion parts inside v-Ni12P5 (Fig. 19d). As shown in Fig. 19e, the electrons depleted in the neighboring area (colored blue) of Ni 3d and P 2p, while accumulated on the Ni and P atoms (colored red). This electron redistribution at the Pv site may have a trivial effect on the adsorption/desorption of reaction intermediates during the HER process. The free energy is proven to be a key descriptor to characterize the HER activity of the electrocatalyst. The ΔGH values were −0.36 eV for v-Ni12P5 and −0.43 eV for p-Ni12P5, demonstrating that the H* desorption step can be boosted by Pv, while the OH* desorption step was unaffected owing to the unchanged energies (Fig. 19f). According to XAS characterization together with DFT calculations, the Pv can weaken the hybridization of Ni 3d and P 2p orbitals, enrich the electron density of Ni and P atoms nearby Pv, and facilitate the H* desorption process, contributing to outstanding HER activity and facile kinetics.
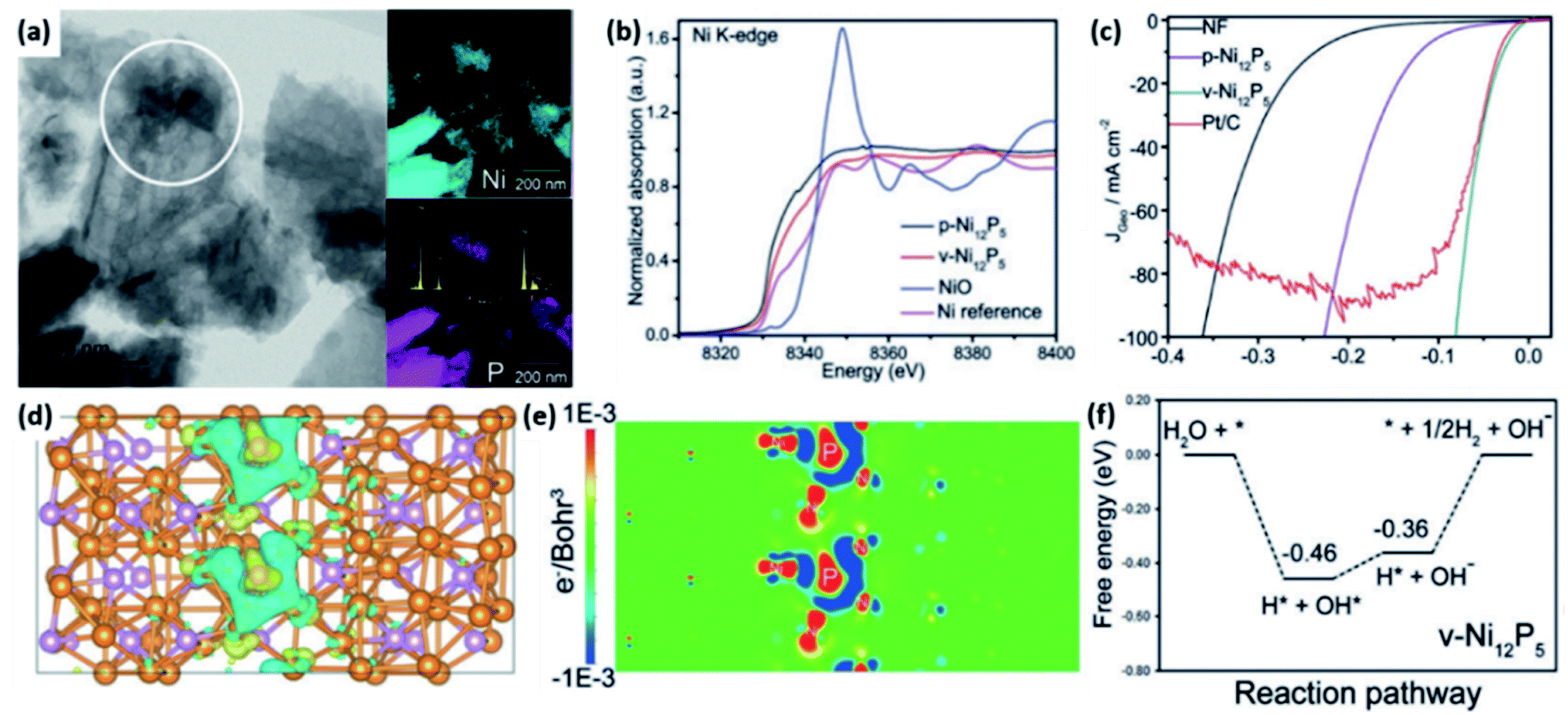 | ||
| Fig. 19 (a) TEM image and TEM EDS elemental mapping images of Ni and P of v-Ni12P5 (inset: EDS spectrum). (b) XAS spectra (fluorescence mode) of nanocataylsts in energy and R-spaces. (c) Polarization curves of NF, p-Ni12P5, v-Ni12P2, and Pt/C. (d) Electron distribution; yellow area indicates electron accumulation while cyan means electron depletion. (e) Two-dimensional charge difference isosurface; red is the electron-rich area while blue is the deficient area. (f) DFT calculated reaction pathways of v-Ni12P5. Reproduced with permission,149 Copyright 2020, American Chemical Society. | ||
8.2 Challenges in synthesis and catalysis
Although many TMP-based catalysts have been developed to enhance HER performance, the scalable and cost-effective synthesis of catalysts still remain challenging. Fundamental methods to enhance the electrocatalytic HER performance of TMPs have been developed to increase the number of active sites and the intrinsic activity of active sites. Accordingly, the development of facet-controlled and hollow TMP nanostructures has received great interest because these material structures can achieve highly active and large surface areas due to their morphological characteristics. The facet effect on the HER has been reported by several researchers. As an example, Wu et al. reported that CoPS single crystals with exposed (100) and (111) facets exhibited different HER behaviors as a function of η.150 In the low potential region (0–0.35 V), the HER catalytic activity of the (100) facet was superior to that of the (111) facet, and the trend was reversed in the high-potential region (>0.35 V). At low potentials, rate limiting H adsorption was faster on the (100) facet, yielding an enhanced HER performance. However, at high potentials, H2 recombination/desorption became the rate determining step and thus the (111) facet, with lower Hads barriers, showed better HER activity. Therefore, hollow TMP nanostructures enclosed with a specific facet may possess a larger density of active sites than solid ones, contributing to the improvement in HER electrocatalysis. However, morphology control of catalysts, including hollow structures, is much less explored for these TMP systems because of difficulties in the synthesis, which requires a deep understanding of the nanocrystal growth process. Therefore, by further developing morphology and facet-controlled nanostructures, we can expect a high-density of low-coordinated metals and abundant P atoms as catalytic sites, which can prove to be better water splitting catalysts.9. Metal sulfides
TMSs have been widely developed as potential electrocatalyst candidates for water splitting because of their unique physical and chemical properties.16,151,152 Compared to TMOs, TMSs usually have high electrical conductivities stemming from the small bandgap between their valence and conduction bands. More impressively, 2D layered MS2 (M = Mo, W) have abundant exposed active sites and moderate adsorption energy for atomic H, leading to remarkable electrocatalytic performance for the HER. Another class of TMSs, namely, nonlayered MxSy (M = Fe, Ni, Co, etc.), have also emerged as economical HER catalysts due to their high electrical conductivity, fast charge transfer kinetics, and low cost. The catalytic performance of these catalysts can be enhanced by various strategies, including morphology control, doping heteroatoms, phase control, defect engineering, and composite engineering.Compared to the electrocatalytic mechanism of the HER, the OER shows a different mechanism and thus different strategies to improve catalytic performance. TMSs are characterized by lower thermodynamic stabilities than metal oxides, indicating that TMSs can be easily oxidized to the corresponding metal oxides under strongly oxidative conditions of the OER.153 Therefore, TMSs act as a pre-catalyst in forming the oxide/hydroxide surface as actual catalytic active sites. In this section, the recent progress in different classes of TMS for the HER and OER is summarized. Moreover, the mechanism to enhance the catalytic activities of TMSs is discussed in detail.
9.1 The state-of-the-art catalysts and their HER/OER performances
The study of TMSs for the electrochemical HER can be traced back to 2005, which was inspired by the composition and structure of nitrogenases and hydrogenases.154 The electrocatalytic performance results showed that (1010) Mo-edged MoS2 is highly active toward the HER. Furthermore, the computational result of the ΔGH on a Mo-edge was close to that of Pt, indicating the great potential of MoS2 as a promising HER electrocatalyst. This innovative material opened up a new door for economical PGM free electrocatalysts. To further study the active sites of MoS2, Jaramillo et al. synthesized different sizes of MoS2 with a systematically varied number of active sites.155 They reported that the catalytic performance of MoS2 is directly proportional to the edge length, regardless of the particle size. After numerous studies about MoS2, the nonlayered first-row transition TMSs were studied as PGM-free HER electrocatalyst candidates. Kong et al. reported that pyrite FeS2, NiS2, and CoS2 exhibit attractive activity and stability toward the HER in an acidic environment.156 Moreover, Fe0.43Co0.57S2 had enhanced catalytic activity compared to FeS2 and CoS2.The strategies to enhance the HER performance of TMSs were classified into two distinct groups: increasing active sites and electronic structure modification. The most general method to increase the number of active sites was designing new kinds of morphologies with a large surface area.157–160 Kibsgaard et al. synthesized mesoporous MoS2 with a double-gyroid structure, exposing more Mo-edges.157 This unique structure of MoS2 exhibited an increased surface area with a longer electrodeposition time. After optimal deposition time, double-gyroid MoS2 showed a 2.2-fold larger surface area compared to MoO3–MoS2 nanowires. Furthermore, various shapes of TMSs such as nanorods, nanosheets, nanowires, and hollow structures were synthesized.158–163 These morphology control studies have recently expanded to hetero-interface structures.164–167 Solomon et al. reported Ag2S/MoS2/RGO with a ternary composite by using a simple one-pot polyol method.164Fig. 20a shows the high-resolution TEM image of the Ag2S/MoS2/RGO composite composed of some large Ag2S nanoparticles (10–50 nm) embedded in MoS2. In addition, Fig. 20b exhibits the fast Fourier transform (FFT) pattern, and extracted inverse FFT images from the spots in the FFT pattern (red and yellow, [0,0,1] and [1,1,0] zone axes of Ag2S; green, [0,0,2] zone axis of MoS2). Moreover, the HAADF-STEM and the corresponding EDX elemental mapping results indicated that Ag and S are homogeneously distributed over the nanoparticles, while Mo is located within the outer part (Fig. 20c). The XRD pattern and Raman spectrum of the Ag2S/MoS2/RGO composite exhibited distinct phases of Ag2S and RGO signals (Fig. 20d and e). According to these results, the Ag2S/MoS2/RGO composite has a large number of hetero-interfaces between two phases. The η10 and η50 of the Ag2S/MoS2/RGO composite required to drive the HER were 190 and 300 mV, while the η50 of MoS2 and MoS2/RGO was 460 and 400 mV, respectively (Fig. 20f). The smaller required η of the Ag2S/MoS2/RGO composite compared to those of MoS2 and binary composites indicated that the hetero-interfaces in the composite promotes HER performance. In addition, the double-layer capacitance (Cdl) results could evaluate the ECSA of each catalyst based on the linear relationship between the Cdl and ECSA. The Cdl of the Ag2S/MoS2/RGO composite (23.7 mF cm−2) was larger than those of MoS2 (8.9 mF cm−2) and MoS2/RGO (16 mF cm−2), indicating a larger ECSA (Fig. 20g). This could imply that the larger ECSA of the Ag2S/MoS2/RGO composite contributed to an enhanced electrocatalytic HER. As shown in Fig. 20h, to demonstrate the improved HER activity, they performed electrochemical impedance spectroscopy and data were fitted using a two-series of constant phase elements (the inset of Fig. 20h). The charge transfer resistance of the Ag2S/MoS2/RGO composite was 96 Ω, which is much smaller than those of MoS2 (495 Ω) and MoS2/RGO (259 Ω). This dramatically reduced charge transfer resistance clearly indicated that fast electron transfer occurs at the hetero-interfaces, leading to superior HER performances of the Ag2S/MoS2/RGO composite.
 | ||
| Fig. 20 (a and b) High-resolution TEM image of the Ag2S/MoS2/RGO composite, and the corresponding FFT patterns and extracted inverse FFT images from the spots in the FFT pattern (red and yellow, [0,0,1] and [1,1,0] zone axes of Ag2S; green, [0,0,2] zone axis of MoS2). (c) HAADF-STEM image of the Ag2S/MoS2/RGO composite and the corresponding EDX elemental maps. (d) XRD pattern of the Ag2S/MoS2/RGO composite. (e) Raman spectra of the Ag2S/MoS2/RGO composite. (f) Polarization curves of the Ag2S/MoS2/RGO composite and other counter groups. (g) Linear fit for double-layer capacitance from cyclic voltammetry results at different scan rates. (h) Nyquist plots and equivalent circuit (inset) of MoS2, MoS2/RGO, and Ag2S/MoS2/RGO composites. Reproduced with permission,164 Copyright 2019 American Chemical Society. | ||
In view of electronic structure modification, alloying or doping with other transition metals has been widely used.101,158,168–172 Yin et al. synthesized NiS2 nanosheets with various transition metal dopants (Co, Cu, and Fe) to modify the electronic structure.172 This work demonstrated the incorporating transition metal effect by the projected partial DOS (PDOS) of surface metal sites (Fig. 21a). The surface Ni sites adjacent to the dopants serve as electron-depleting centres and promote electron transfer. Especially, Co doped NiS2 (Co-NiS2) shows a smaller eg–t2g splitting gap compared to Fe and Cu doped samples, indicating a higher efficiency of electron transfer across the Fermi level. In addition, DFT calculations were performed to understand the correlation between the doping and overall energetic pathway of the HER. As shown in Fig. 21b, the transition state barrier of water splitting at Co-NiS2 was 0.80 eV, which is much smaller compared to that of NiS2 (1.38 eV), indicating an energetically favourable HER. Based on these computational results, Co-NiS2 required the lowest η10 of 80 mV and the smallest Tafel slope of 43 mV dec−1, which is much smaller than that of Fe doped, Cu doped, and undoped NiS2 (Fig. 21c). In addition, vacancy engineering has been used as a strategy to modify the electronic structure of catalysts. Jia et al. reported that sulfur vacancies (Vs) could be generated by Ar plasma etching.168 As shown in Fig. 21d and e, Ni3S2 with Vs (Vs-Ni3S2) synthesized by Ar plasma treatment for 300 seconds shows a Ni3S2 crystal structure with a porous morphology, indicating that the Ar plasma does not affect the crystal structure. To further analyse the Vs, they performed electron paramagnetic resonance (EPR) and XPS measurements (Fig. 21f and g). Vs-Ni3S2 exhibited an approximately two times larger EPR signal compared to Ni3S2, corresponding to Vs. Moreover, Ni 2p spectra shifted negatively due to Vs. Vs-Ni3S2 showed much lower η10 for the HER (88 mV) than Ni3S2 (274 mV) (Fig. 21h). As shown in Fig. 21i, Vs in Ni3S2 change the electron density of the Ni site and the d-band centre shifts positively toward the Fermi level, demonstrating stronger binding between adsorbate and active sites. Therefore, the calculated ΔGH at the Ni and S sites of Vs-Ni3S2 was lower than that of Ni3S2 and these results back up the excellent HER performance of Vs-Ni3S2 (Fig. 21j).
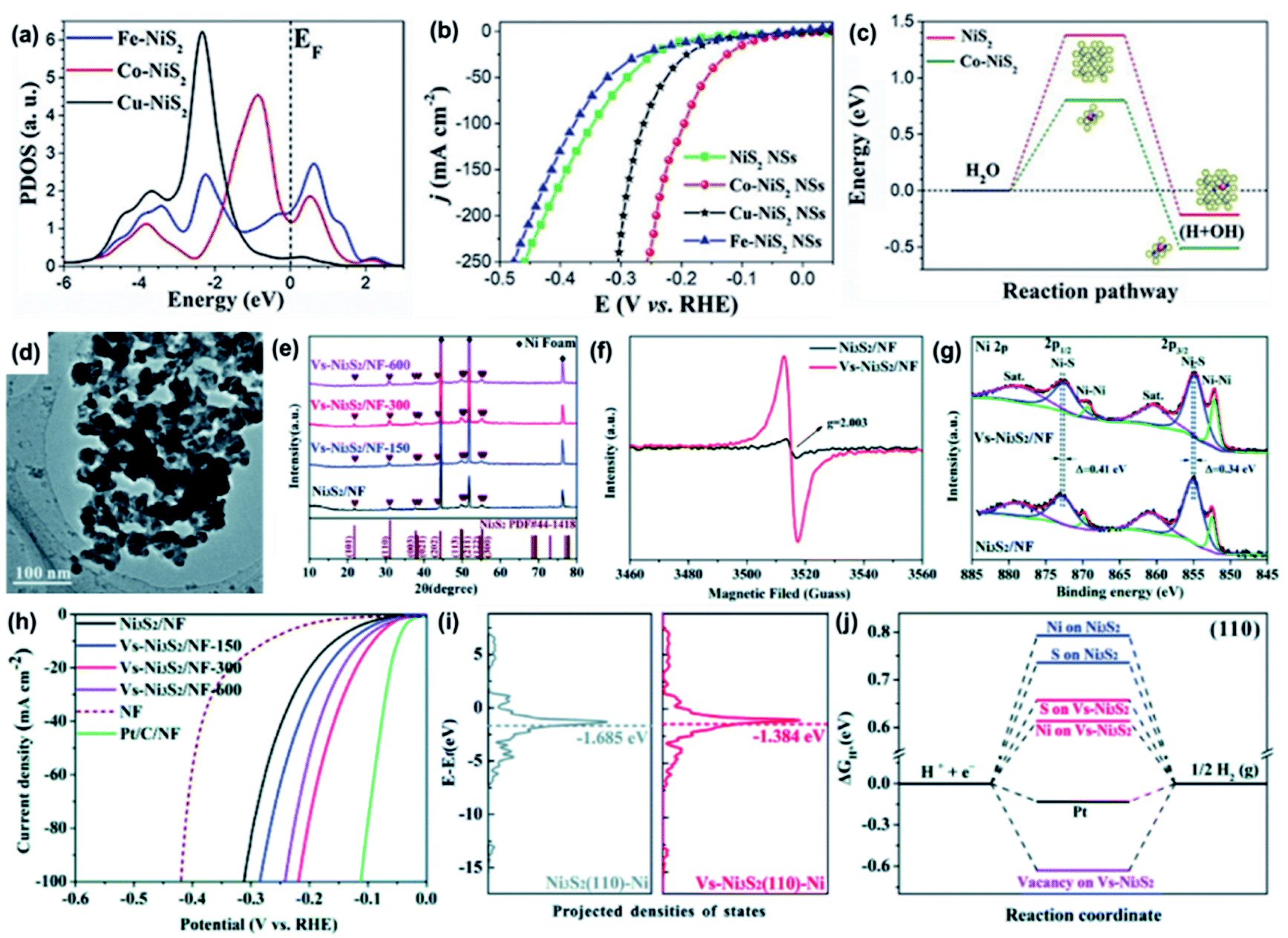 | ||
| Fig. 21 (a) PDOS of the 3d-bands of Co, Fe, and Cu doped NiS2. (b) Polarization curves of undoped NiS2, Co, Fe, and Cu doped NiS2. (c) Transition state barrier for H2O splitting on the surface of NiS2 and Co-NiS2. Reproduced with permission,172 Copyright 2019 Wiley-VCH. (d) TEM image of Vs-Ni3S2. (e) XRD pattern of Vs-Ni3S2 treated for different time durations and Ni3S2. (f) EPR spectra of Ni3S2 and Vs-Ni3S2. (g) XPS spectra of Ni3S2 and Vs-Ni3S2. (h) Polarization curves of Vs-Ni3S2 treated for different time durations and other counter groups. (i) PDOS of the d-band for the (110) Ni surfaces of Ni3S2 and Vs-Ni3S2. (j) Gibbs free energies of H adsorption on the S and Ni sites of the (110) surface for Ni3S2 and Vs-Ni3S2. Reproduced with permission,166 Copyright 2020 Royal Society of Chemistry. | ||
The scientific approach to enhancing catalytic activity toward the OER is quite different from that of the HER. As mentioned earlier, the surface of sulfides is converted into oxides/hydroxides at harsh OER potentials, indicating that the sulfide surface cannot act as an active site. Thus, TMS electrocatalysts were considered a pre-catalyst with a hetero-structure of metal sulfides and metal oxides/hydroxides.173 Recently, researchers have introduced the concept of a high entropy alloy to implement stable metal sulfides at OER potentials. Cui et al. reported high entropy metal sulfide nanoparticles through a pulse thermal decomposition method.174 They utilized Cr, Mn, Fe, Co, and Ni metal elements to prepare various compositions of metal sulfides ranging from unary to quinary. As shown in Fig. 22a, the HAADF-STEM and the corresponding EDX elemental mapping images indicated that all metal elements and sulfur are homogeneously distributed. Quinary high entropy metal sulfide nanoparticles (i.e., (CrMnFeCoNi)Sx) showed enhanced OER activity and stability compared to binary, ternary, and quaternary nanoparticles (Fig. 22b and c). Moreover, even after an OER stability test, the (CrMnFeCoNi)Sx nanoparticles maintained their pristine crystal structure (Fig. 22d). This enhanced OER performance was consistent with DFT calculation, which reveals that (CrMnFeCoNi)Sx has optimal adsorption energy ΔEo* (Fig. 22e). As a result, high entropy metal sulfides offer new insights into utilizing TMSs toward OER electrocatalysts.
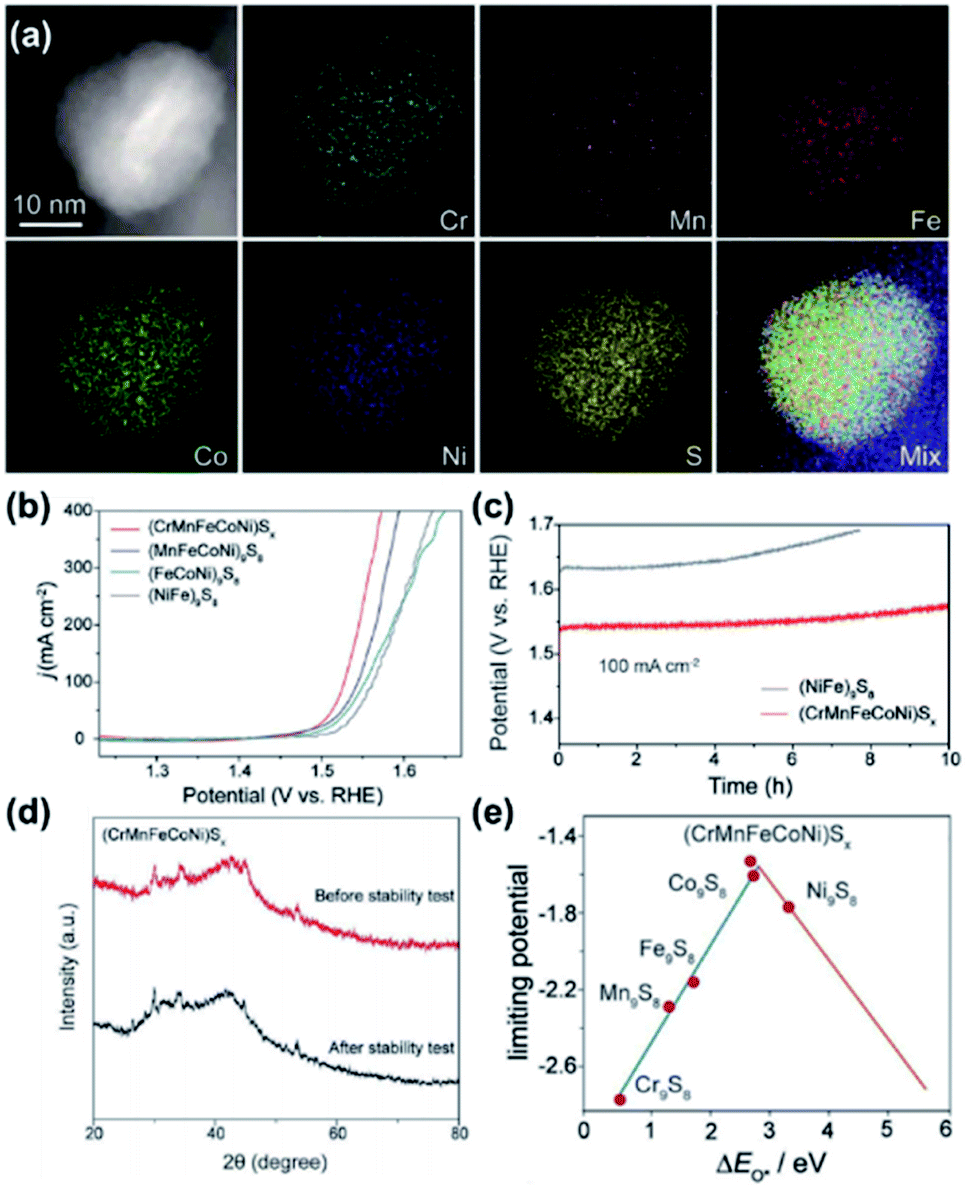 | ||
| Fig. 22 (a) HAADF-STEM image of (CrMnFeCoNi)Sx and the corresponding EDX elemental maps. (b) Polarization curves of (CrMnFeCoNi)Sx and other counter groups. (c) Chronopotentiometry test of (CrMnFeCoNi)Sx at a constant current density of 100 mA cm−2. (d) XRD patterns of (CrMnFeCoNi)Sx before (red) and after (black) the OER stability test. (e) Volcano plot of metal sulfides for the OER. Reproduced with permission,174 Copyright 2021 Wiley-VCH. | ||
9.2 Challenges in synthesis and catalysis
TMS based electrocatalysts have been widely studied to enhance HER and OER activity through the traditional strategies of increasing active sites and electronic structure modification. However, advanced and recent strategies, such as forming hetero-interfaces, vacancies, and high entropy alloys, are very underdeveloped. Especially, hetero-structured TMSs show great potential toward HER and OER nanocatalysts. Hetero-interfaces are able to modulate the electronic structure of the both sides of materials and also provide a synergistic effect between two materials, such as the spill-over effect of adsorbates.167,175,176 These unique advantages of hetero-interfaces simultaneously improve the electrocatalytic performance toward the HER and OER. However, understanding of hetero-interfaces, vacancies, and high entropy alloys is still at their early stages in boosting electrocatalytic performance. Therefore, continuous effort on understanding the relationship between the aforementioned strategies and the catalytic performance is crucial for the further development of TMS-based electrocatalysts.In addition to a poor understanding of these state-of-the-art strategies, the lack of single-cell performance evaluations shows a threshold for the practical utilization of TMS electrocatalysts toward water electrolysers. According to Kim et al., electrocatalytic performance in a half-cell test and single-cell test is similar in the low current density region, where the electrocatalytic performance is determined by the reaction kinetics.177 On the other hand, in a high current density region, the mass transfer at the electrode surface becomes a significant factor affecting the electrocatalytic performance, which gives a disjunction between the half-cell performance and the single-cell performance. Therefore, in the field of TMSs, where most of the research is concentrated on half-cell evaluation, it is believed that sufficient research on whether there is practical applicability will be needed.
10. Single-atom catalysts
Water splitting under an electrochemical force is a promising strategy to convert electricity and water into a high-purity hydrogen fuel.15,178 The key point of electrochemical water splitting is high-performance catalysts to catalyze the half reaction of the HER and OER. Overcoming the limitation of high reaction kinetic barriers needs to be given a priority for the rational design of target catalysts. Single-atom catalysts (SACs) show great application prospects in the fields of the HER and OER, and many breakthroughs have been made recently.10.1 The state-of-the-art catalysts and their HER/OER performances
Recently, SACs showed comparable HER performance with maximal utilization efficiency. Therefore, many researchers show interest in the fields of SACs to improve the HER performance. Using carbon materials as the support to anchor isolated metal atoms to fabricate Pt-based SACs has attracted enormous attention due to their high electrical conductivity, stability in a harsh liquid-phase, structural diversity, and tailored surface chemistry. Although Pt is a PGM, Pt-based SACs dramatically reduce the utilization of Pt, resulting in cost savings comparable to non-PGM-based catalysts. Implanting foreign metal atoms into the defects of the carbon support not only stabilizes isolated metal atoms, but also modifies their electronic structures via metal–support interactions. For example, Qu et al. developed a facile route to access active and well-defined single atom site catalysts.17 In general, a thermal emitting method using Pt bulk as the precursor under ammonia derived from dicyandiamide pyrolysis was developed. Because of the strong chelating action between ammonia and Pt atoms, the atomically dispersed Pt atoms could be caught by ammonia to form Pt(NH3)x and anchored on the defective graphene surface, as illustrated in Fig. 23a. In this case, the zero-valent Pt was oxidized to Ptδ (0 < δ < 4), and at the same time, the most oxygen-containing functional group on GO was removed through thermal treatment, generating defective graphene (DG). Fig. 23b and c show the HAADF-STEM images of the as-obtained Pt SAs/DG catalyst. As can be seen, the light spots in Fig. 23c are the atomically dispersed Pt atoms. The coordination environment of isolated Pt atoms was further demonstrated by EXAFS characterization (Fig. 23d). The resultant Pt SAs/DG catalyst has exhibited superior performance for the HER as shown in Fig. 23e. As seen from Fig. 23f, the Pt SAs/DG catalyst shows the lowest ΔGH* in comparison with other catalysts. Yin et al. demonstrated a new-type Pt-based SAC via supporting two Pt atoms on graphdiyne (donated as Pt-GDY2) to tune the electronic effect of Pt.179 From the HER performance results, the activity of Pt-GDY2 was 26.9 times higher than that of benchmark Pt/C. A deeper study revealed that the five-coordinated Pt metal centre possessing zero-value ΔGH* showed an enhanced HER performance. Zhang et al. documented a novel strategy to anchor isolated Pt atoms in N-doped mesoporous carbon via high-temperature pyrolysis. The obtained SACs exhibited 25 times higher mass activity than benchmark Pt/C.180 In addition, the electroplating deposition method was also adapted to prepare atomically dispersed metal atoms on carbon materials. Zhang et al. demonstrated a facile and general electrochemical method to construct Pt-based SACs by using CoP nanoarrays as the support,181 which show an excellent HER performance in neutral media.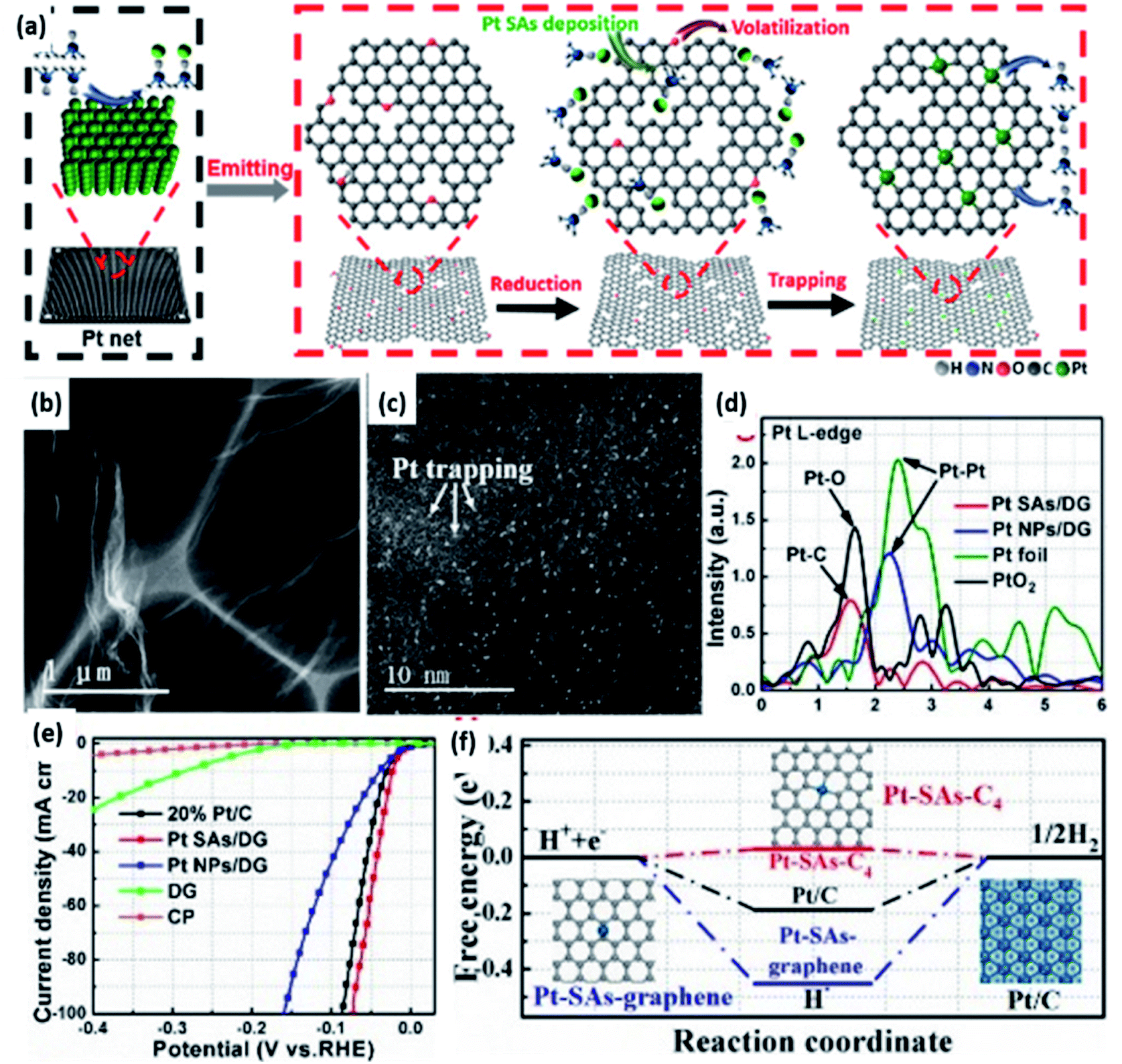 | ||
| Fig. 23 (a) Proposed reaction mechanism for the preparation of Pt SAs/DG. (b and c) HAADF STEM image, (d) R-space spectra from EXAFS, and (e) HER LSV curves of the catalysts. (f) Calculated Gibbs free energy diagram of the HER on Pt/C, Pt-SAs-graphene and Pt-SAs-C4 at the equilibrium potential. Reproduced with permission,17 Copyright 2019, American Chemical Society. | ||
Besides Pt-based SACs, other non-Pt based SACs were also designed and prepared. Earth-abundant catalysts, especially M–N/C, have been widely studied as appealing non-PGM catalysts for HER application.182 Isolated Mo atoms anchored on N-doped carbon materials were designed and fabricated using a versatile template and high-temperature pyrolysis route.183 AC-STEM and EXAFS characterization revealed that the single Mo atom is successfully anchored. Benefiting from the unique structural features, Mo-NC SACs showed extraordinary HER performance compared with Mo2C, MoN, and benchmark Pt/C. DFT calculations disclosed that Mo-NC SACs possess a low ΔGH* and a large density of states. In addition, metal oxide/carbide/nitride and carbon encapsulated metal atoms showed enhanced HER performance. However, the synergistic effects among these active species make it difficult to uncover the real active site and electrocatalytic mechanism.
As the other half reaction of water dissociation, the OER is kinetically sluggish in comparison with the HER, and SACs showed promising potential for OER catalysis. Zhang et al. provided an atomically dispersed Ir and Ir cluster supported on Co(OH)2 nanosheets via a facile NaBH4 reduction strategy.184 The atomic Ir/Co(OH)2 catalyst showed an overpotential of 373 mV at 10 mA cm−2 in 1.0 M PBS. After activation, the valence state of Ir species increased derived from the strong peak of Ir–O coordination. On the other hand, the Co K-edge showed an evident shift to higher energy, indicating that the Co species are oxidized to a higher valence. XRD results revealed that the α-Co(OH)2 phase is transformed into β-phase Co oxyhydroxide (β-CoOOH). Deep characterization revealed that high-valence unique β-CoOOH with a low-coordination structure and Ir species under oxidation potential are the main active sites for the OER.
Hu et al. developed a hybrid amorphous/crystalline FeCoNi LDH-supported and -stabilized single Ru atom catalyst (Ru SAs/AC-FeCoNi) through facile self-templating cation-exchange tactics,185 as shown in Fig. 24a. The combined characterization of AC-STEM and XANES disclosed that the Ru atoms are evenly dispersed over the whole FeCoNi LDH support (Fig. 24b–f). Under the synergistic effect of abundant defect sites and unsaturated coordination sites, as well as a highly symmetric rigid structure, the Ru SAs/AC-FeCoNi catalysts showed enhanced OER performance (Fig. 24g). The intrinsic strong metal–support interaction between Ru and FeCoNi LDH was the main reason for promoting the OER performance via triggering the local electronic configuration and internal electron rearrangement. DFT calculations revealed that Ru SAs/AC-FeCoNi could optimize the adsorption energies for catalytic intermediates, subsequently enhancing the OER activity (Fig. 24h and i).
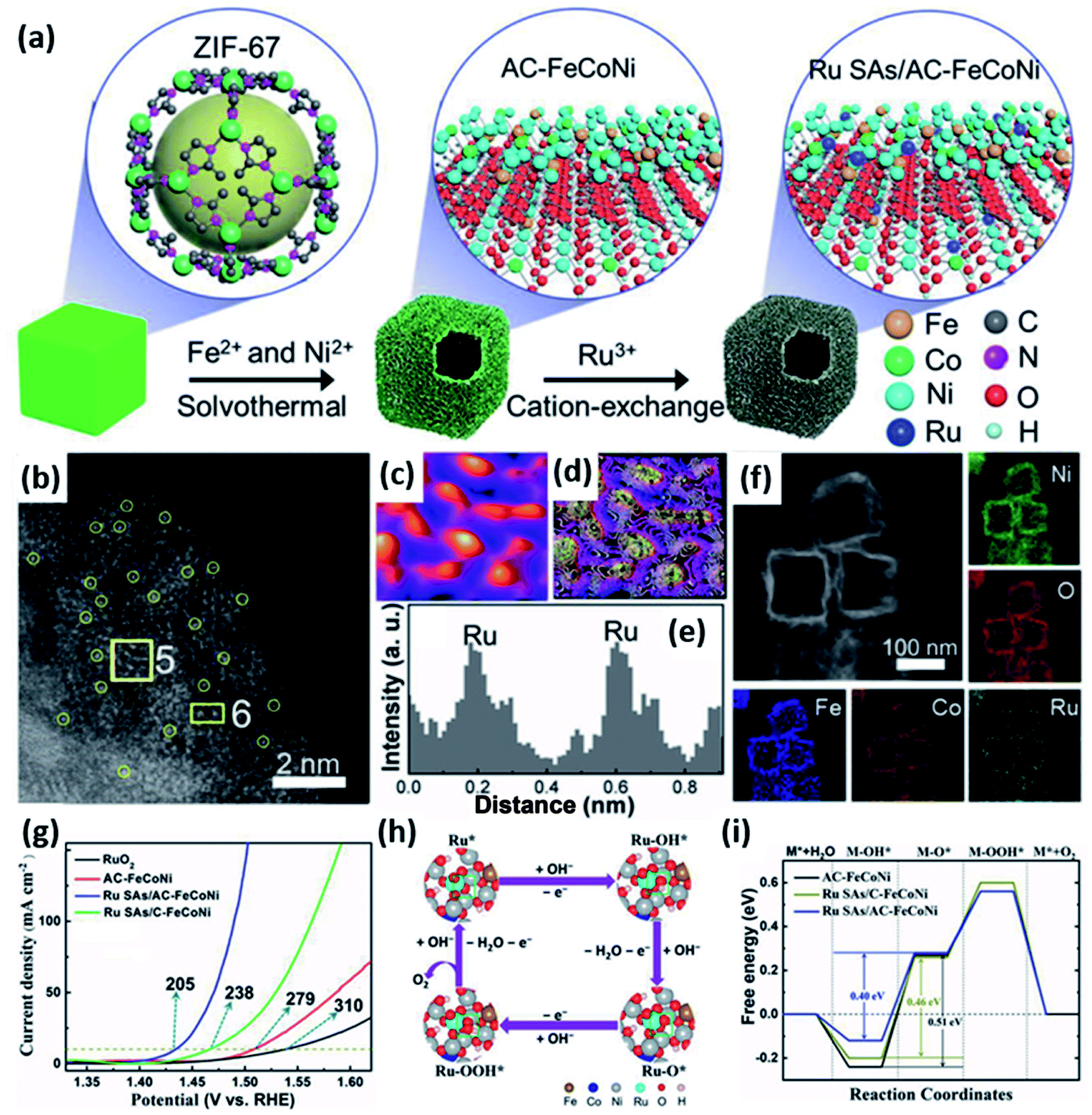 | ||
| Fig. 24 (a) Illustration for the synthesis of Ru SAs/AC-FeCoNi. (b) AC HAADF-STEM image of Ru SAs/AC-FeCoNi. (c) 3D AOGF mapping and (d) 3D isolines of 5 shown in (b). (e) Intensity profile along the middle of 6 from (b). (f) HAADF-STEM image and corresponding elemental mappings of Ru SAs/AC-FeCoNi. (g) LSV curves of RuO2, AC-FeCoNi, Ru SAs/AC-FeCoNi, and Ru SAs/C-FeCoNi. (h) Proposed OER pathway on Ru SAs/AC-FeCoNi for DFT + U calculation. (i) Free energy on AC-FeCoNi, Ru SAs/C-FeCoNi, and Ru SAs/AC-FeCoNi at the equilibrium potential. Reproduced with permission,186 Copyright 2017, American Chemical Society. | ||
Similarly, compared with their PGM counterparts, theoretical and experimental studies on non-PGM based SACs for the OER have sparked much research interest because of their distinctive OER activity. Besides the verification of the single metal atom and related active moieties, the metal species evolution is also important. For the OER, due to high potential, the microenvironment of monatomic catalysts is complex and changeable, and the electronic structure and coordination environment of the metal active centre are also dynamically evolving in the process of catalysis, which leads to the variability of the catalyst structure and the complexity of the catalytic reaction mechanism. In this case, given the complex profuse pyridine-N in the C3N4 matrix, Zheng et al. designed a molecule-level Co-C3N4/CNT catalyst accompanied by a CoN3C2 ring via a direct grafting strategy.186 The well-defined Co-C3N4/CNT showed an excellent OER performance.
10.2 Challenges in synthesis and catalysis
A great impact of SACs on energy generation is foreseeing, such as, providing catalysts for water splitting, whose realization depends on highly efficient materials for both the HER and OER, greatly influencing economically and technologically our society. However, seeking high-efficiency, low cost, and sustainable SACs is promising for their large-scale practical applications. In fact, advanced characterization has been used to reveal the underlying relationships between the single atom and support to give a fundamental understanding of rational design and construction of novel SACs.Among the numerous substrates for SACs, carbon materials (such as, graphitic materials, CNTs, graphene and derivatives) show huge application prospects for electrocatalytic water dissociation, involving the HER and OER due to excellent mechanical and chemical properties. Their intrinsic catalytic performance could be further improved by implanting nitrogen to form N-doped carbon materials. Moreover, most HER or OER catalysts were prepared by combining metal atoms with a suitable support with improved electronic interactions and a unique coordination environment. Therefore, in the future, constructing different atomic interfaces and modulating the surrounding coordination structure of active sites of SACs will be the main subject to improve the performance of water splitting.
Compared with SACs, dual-atom catalysts have also received increasing interest owing to their higher metal loading, more versatile active sites and unique reactivity.187 For example, Yang et al. prepared a dual-atom catalyst consisting of an O-coordinated W-Mo heterodimer embedded in N-doped graphene (W1Mo1-NG). Impressively, the obtained W1Mo1-NG catalysts exhibited Pt-like activity and excellent stability for the HER in a pH-universal electrolyte. DFT calculations demonstrated that electron delocalization of W1Mo1-NG could show desirable ΔGH and good HER kinetics, thereby enhancing the HER activity.188
11. Conclusion and outlooks
This review article has summarized recent research advances in the development of HER/OER electrocatalysts for water electrolysis. The major goal of this research field is to replace expensive and rare PGM-based electrocatalysts with low-cost and highly active non-PGM bifunctional ones or greatly reduce PGMs by their single atom dispersion. By far, the state-of-the-art catalysts including carbon-based catalysts, TMBs, TMCs, TMOs, TMPs, TMSs, and single-atom catalysts have been investigated toward both the HER and OER, and they showed comparable activities to PGM catalysts as listed in Tables 1 and 2. In addition, catalyst modeling has been proposed to advance electrocatalysts for HER/OER performances and their stability.| Class | Catalysts | Method | Electrolytes | η @ 10 mA cm−2 (mV) | Tafel slope (mV dec−1) | Ref. |
|---|---|---|---|---|---|---|
| a N/A: not available. | ||||||
| Carbon | N/C | Pyrolysis + etching | KOH | 380 | N/A | 35 |
| NPMC foam | Polymerization + pyrolysis | 6 M KOH | 300 | 193 | 36 | |
| GNS/MC | Templating + pyrolysis + etching | 0.1 M KOH | 340 | 80 | 43 | |
| SHG | Hybridization + pyrolysis + etching | 0.1 M KOH | 260 | 71 | 37 | |
| NPC-CP | Polymerization + carbonization | 1 M KOH | 310 | 87.4 | 38 | |
| Ni-NHGF | Hydrothermal + pyrolysis | 1 M KOH | 331 | 63 | 40 | |
| A-Ni@DG | Annealing + etching | 1 M KOH | 270 | 47 | 41 | |
| CoNi-SAs/NC | MOF + pyrolysis | 0.1 M KOH | 340 | 58.7 | 45 | |
| Ni-N4/GHSs/Fe-N4 | Deposition + annealing + etching | 0.1 M KOH | 390 | 81 | 46 | |
| PHI-Co-0.5 | Hydrothermal | 1 M KOH | 324 | 44 | 42 | |
| TMBs | Co2B/N-doped graphene | Chemical reduction | 1 M KOH | 360 | 45 | 192 |
| NixB | Chemical reduction | 1 M KOH | 380 | 89 | 193 | |
| FeB2 | Chemical reduction | 1 M KOH | 296 | 52.4 | 65 | |
| Fe–Co-2.3Ni–B | Chemical reduction | 1 M KOH | 274 | 38 | 194 | |
| CoBx@h-BN | Chemical reduction + annealing | 1 M KOH | 290 | 81.9 | 195 | |
| Ni3B/Ni foam | Annealing | 1 M KOH | 300 | 43 | 196 | |
| V doped Ni–Co boride | Chemical reduction + CVD | 1 M KOH | 340 | 58 | 67 | |
| boronized NiFe foil | Annealing | 1 M KOH | 270 | N/A | 66 | |
| FeCoNiBOx/PPy/rGO | Chemical reduction | 1 M KOH | 290 | 47 | 197 | |
| W, P-FeB | Chemical reduction | 1 M KOH | 209 | 40 | 198 | |
| TMC | Fe/Fe3C-MC | Electrochemical reduction | 1 M KOH | 320 | 51 | 91 |
| FCC@CNOs/NF | High pressure annealing | 1 M KOH | 271 | 48.9 | 92 | |
| PB@Met-700-acid | MOF derived method | 1 M KOH | 390 | N/A | 199 | |
| Co SAs/Mo2C | MOF derived method | 1 M KOH | 270 | 74.9 | 87 | |
| Co-Mo2C | MOF derived method | 1 M KOH | 190 | 94.3 | 69 | |
| Co/Mo2C@NC-800 | MOF derived method | 1 M KOH | 311 | 131.50 | 200 | |
| Co6W6C@NC | MOF derived method | 1 M KOH | 286 | 53.96 | 88 | |
| NC@CuCoW-C (S-2) | MOF derived method | 1 M KOH | 238 | 59 | 89 | |
| Ni/WCx-CNFs-3 | Electrospinning + carbonization | 1 M KOH | 350 | 93 | 201 | |
| a-MnOx/TiC | Hydrothermal | 0.1 M KOH | 330 | 110 | 202 | |
| Ni-Ni3C/CC | Annealing | 1 M KOH | 299 (@ 20 mA cm−2) | 43.8 | 76 | |
| Co3C | Wet-chemistry | 1 M NaOH | 455 | N/A | 84 | |
| CoNi–C/rGO-450 | Annealing | 1 M KOH | 364 | 101.6 | 203 | |
| TMO | Ni0.9Fe0.1Ox/ITO | Solution casting + annealing | 1 M KOH | 336 | 30 | 103 |
| NiFe-LDH nanosheets | Hydrothermal + anion exchange | 1 M KOH | 300 | 40 | 204 | |
| Co3O4/Ni foam | Hydrothermal + plasma | 1 M KOH | 339 | 108 | 205 | |
| G-FeCoW/Au foam | Sol–gel | 1 M KOH | 191 | 37 | 95 | |
| CoNi(OH)x/Cu foil | Electrodeposition + annealing + etching | 1 M KOH | 280 | 77 | 206 | |
| NiCoFe LDH NA/carbon cloth | Electrodeposition | 1 M KOH | 239 | 32 | 207 | |
| Fe2+–NiFe LDH NA/carbon fiber paper | Co-precipitation | 1 M KOH | 195 | 40.3 | 208 | |
| NiFeV LDH NA/Ni foam | Hydrothermal | 1 M KOH | 192 | 42 | 209 | |
| Pv/Py | Ba0.5Sr0.5Co0.8Fe0.2O3−δ | Co-precipitation | 0.1 M KOH | ∼360 (@ 10 mA cmoxide−2) | ∼60 | 94 |
| (Pr0.5Ba0.5)CoO3 | Solid-state | 0.1 M KOH | ∼60 | 120 | ||
| IrOx/SrIrO3 | Pulsed laser deposition + electrochemical leaching | 0.5 M H2SO4 | 270 (@ 10 mA cmoxide−2) | N/A | 122 | |
| 6H-SrIrO3 | Wet-chemistry | 0.5 M H2SO4 | 248 | N/A | 123 | |
| SrCo0.9Ir0.1O3−δ | Solid-state method | 0.1 M KOH | ∼320 (@ 10 mA cmoxide−2) | ∼50 | 124 | |
| Sr1−xNaxRuO3 | Wet-chemistry | 0.1 M KOH | 170 (@ 10 mA cmoxide−2) | N/A | 125 | |
| Bi2Ir2O7 | Hydrothermal | 1 M H2SO4 | ∼350 | 45 | 126 | |
| (Na0.33Ce0.67)2(Ir1−xRux)2O7 | Hydrothermal | 0.5 M H2SO4 | N/A | 48.6–85.6 | 127 | |
| (A, A′)2Ir2O6.5+x, (A, A′ = Bi, Pb, Y) | Adams fusion method | 0.1 M HClO4 | N/A | 45–46 | 128 | |
| Y2Ru2O7−δ | Sol–gel | 0.1 M HClO4 | 270 (@ 1 mA cmoxide−2) | 46–55 | 129 | |
| Y1.8M0.2Ru2O7−δ (M = Fe, Co, Ni, Cu, Y) | Sol–gel | 1 N H2SO4 | 325–395 (@ 1 mA cmoxide−2) | 52–63 | 130 | |
| TMS | Ni(Fe)S2@Ni(Fe)OOH | Hydrothermal | 1 M KOH | 230 | 42.6 | 173 |
| Co1−xNixS2/N-doped rGO aerogel | Hydrothermal | 1 M KOH | 330 | 47 | 210 | |
| FeNiS2 nanosheets/rGO | Solvothermal + pyrolysis under NH3 | 1 M KOH | 200 | 40 | 211 | |
| N–(Ni,Fe)3S2 nanosheets/Ni–Fe alloy foam | Pyrolysis with sulfur | 1 M KOH | 167 | 33 | 212 | |
| Hollow nanostructured CoxNi1−xS2/rGO | Solvothermal | 1 M KOH | 290 | 46 | 213 | |
| Ni3S2 | Solvothermal | 1 M KOH | 330 | 52 | 214 | |
| FeSx/CoSx heterophase | Solvothermal | 1 M KOH | 304 | 48.7 | 215 | |
| Fe–CoMoS | Hydrothermal + pyrolysis | 1 M KOH | 282 | 58 | 216 | |
| (CrMnFeCoNi)Sx high-entropy metal sulfide nanoparticles | Pulse thermal decomposition | 1 M KOH | 295 (@ 100 mA cm−2) | 66 | 174 | |
| Class | Catalyst | Method | Electrolytes | η @ 10 mA cm−2 (mV) | Tafel slope (mV dec−1) | Ref. |
|---|---|---|---|---|---|---|
| a N/A: not available. | ||||||
| Carbon | C3N4@NG | Polymerization + annealing | 0.5 M H2SO4 | 240 | 51.5 | 47 |
| CoNx/C | Pyrolysis + etching | 1 M KOH | 170 | 75 | 54 | |
| 1 M PBS | 247 | N/A | ||||
| 0.5 M H2SO4 | 130 | 57 | ||||
| N,S-G | Templating + etching | 0.5 M H2SO4 | 310 | 120 | 48 | |
| SHG | Hybridization + pyrolysis + etching | 0.1 M KOH | 310 | 112 | 37 | |
| Hierarchical NS500 | CVD + etching | 0.5 M H2SO4 | 230 | 72 | 49 | |
| A-Ni@DG | Annealing + etching | 0.5 M H2SO4 | 70 | 31 | 41 | |
| CNT/Co-PcC-1 | Pyrolysis + etching | 1 M KOH | 219 | 78 | 55 | |
| 0.5 M H2SO4 | 202 | 82 | ||||
| Co1/PCN | Polymerization + annealing | 1 M KOH | 89 | 52 | 217 | |
| TMBs | FeB2 | Chemical reduction | 1 M KOH | 61 | 87.5 | 65 |
| Co-Ni–B@NF | Electroless plating | 1 M KOH | 205 | N/A | 218 | |
| Mo3B film | CVD | 0.5 M H2SO4 | 249 (@ 20 mA cm−2) | 52 | 219 | |
| Etched MoAlB | Etching of MXenes | 0.5 M H2SO4 | 301 | 68 | 220 | |
| Co-B/Ni electrode | Electroless plating | 1 M KOH | 70 | 68 | 221 | |
| MoB/g-C3N4 | Physical grinding | 0.5 M H2SO4 | 133 | 46 | 61 | |
| RuB2 | Quasi solid-state metathesis | 1 M KOH | 28 | 28.7 | 56 | |
| 0.5 M H2SO4 | 18 | 38.9 | ||||
| Ni doped WB | Molten salt-assisted reaction | 0.5 M H2SO4 | 144 | 63 | 222 | |
| Pd2B | Hydrothermal | 0.5 M H2SO4 | 15.3 | 22.5 | 58 | |
| RuB | Annealing | 0.5 M H2SO4 | 22 | 30.7 | 57 | |
| Ni3B/MoB | Annealing | 0.5 M H2SO4 | 75 | 61 | 62 | |
| TMC | Np-η-MoC NSs | Hydrothermal | 0.5 M H2SO4 | 112 | 53 | 14 |
| 1 M KOH | 119 | 39 | ||||
| Np-α-MoC1−x NSs | Hydrothermal | 0.5 M H2SO4 | 158 | 54 | ||
| 1 M KOH | 147 | 43.6 | ||||
| Ni-Mo2C@C | Annealing | 1 M KOH | 72 | 65.6 | ||
| Co-Mo2C@C | Annealing | 1 M KOH | 122 | 80.9 | 74 | |
| Fe-Mo2C@C | Annealing | 1 M KOH | 129 | 102.4 | ||
| Cr-Mo2C@C | Annealing | 1 M KOH | 147 | 114.2 | 74 | |
| WC/W2C@C NWs | CVD | 0.5 M H2SO4 | 69 | 52 | 82 | |
| 1 M KOH | 56 | 59 | ||||
| Ni/WC@NC | Hydrothermal + annealing | 0.5 M H2SO4 | 53 | 43.5 | 223 | |
| Cu@WC | Wet chemical oxidation + in situ electroreduction | 0.5 M H2SO4 | 92 | 50.5 | 224 | |
| 1 M KOH | 119 | 88.7 | ||||
| 1 M PBS | 173 | 118.3 | ||||
| W-W2C/CNT | Say-drying process + carbonization | 0.5 M H2SO4 | 155 | 56 | 90 | |
| 1 M KOH | 147 | 51 | ||||
| CoW/CN | Annealing | 1 M KOH | 98 | 125 | 91 | |
| ES-WC/W2C | CVD | 1 M KOH | 75 | 59 | 81 | |
| Ni/VC | CVD + hydrothermal | 0.5 M H2SO4 | 111 | 86 | 75 | |
| 1 M KOH | 239 | 80 | ||||
| Ni/Fe3C | CVD + hydrothermal | 0.5 M H2SO4 | 112 | N/A | ||
| 1 M KOH | 93 | N/A | ||||
| Ni–Ni3C/CC | CVD + hydrothermal | 1 M KOH | 98 | 88.5 | 76 | |
| Co2C | Bromide-induced wet-chemistry | 0.1 M KOH | 181 | 89 | 80 | |
| N, B co-doped Co3C | Ball-milling | 1 M KOH | 154 | 56 | 77 | |
| Fe3C-GNRs | Hot filament chemical vapor deposition | 0.5 M H2SO4 | 49 | 46 | ||
| Co3C-GNRs | 0.5 M H2SO4 | 91 | 57 | 78 | ||
| Ni3C-GNRs | 0.5 M H2SO4 | 48 | 54 | |||
| TMO | WO2.9 | Annealing | 0.5 M H2SO4 | 70 | 50 | 97 |
| Porous MoO2 NSs/Ni foam | Hydrothermal + annealing | 1 M KOH | 25 | N/A | 225 | |
| 3D urchin-like Mo–W18O49 | Hydrothermal | 0.5 M H2SO4 | 45 | 54 | 226 | |
| S-CoO NRs/carbon fiber paper | Hydrothermal + cation exchange | 1 M KOH | 73 | 82 | 98 | |
| Porous WO2 HNs/Ni foam | Hydrothermal + annealing | 1 M KOH | 48 | 43 | 227 | |
| Ni, Zn-dual doped CoO NRs/carbon fiber paper | Hydrothermal + cation exchange | 1 M KOH | 53 | 47 | 100 | |
| Ni0.35Mo0.65O2/carbon paper | Hydrothermal + annealing | 0.5 M H2SO4 | 43 | 37 | 228 | |
| F-CoO NWs/carbon cloth | Hydrothermal + annealing | 1 M KOH | 53 | 65 | 229 | |
| Pv/Py | Pr0.5(Ba0.5Sr0.5)0.5Co0.8Fe0.2O3−δ | Sol–gel | 1 M KOH | 237 | 45 | 131 |
| La0.5Ba0.25Sr0.25CoO2.9−δF0.1 | Solid-state reaction | 1 M KOH | 256 (@ 100 mA cm−2) | 44 | 12 | |
| (Gd0.5La0.5)BaCo2O5.75 | Sol–gel | 1 M KOH | 185 | 27.6 | 133 | |
| Ni NP/La0.4Sr0.4Ti0.9O3−δ | Sol–gel + exsolution | 0.1 M KOH | ∼430 | 97 | 135 | |
| NiRu NP/Pb2Ru2−xNixO6.5 | Sol–gel + exsolution | 0.1 M KOH | 35 | 30 | 137 | |
| Ti-doped RuO2/Ru-doped K0.469La0.531TiO3 | Hydrothermal | 1 M KOH | 20 | 30 | 138 | |
| TMP | 3D urchin-like CoP NCs | Hydrothermal + phosphidation | 0.5 M H2SO4 | 180 | 46 | 230 |
| NiCoPx holey nanosheet/Ni foam | Calcination + plasma CVD | 1 M KOH | 58 | 57 | 231 | |
| Ni-doped FeP/carbon hollow nanorods | Hydrothermal + etching + annealing | 1 M KOH | 95 | 72 | 180 | |
| 1 M PBS | 117 | 70 | ||||
| 0.5 M H2SO4 | 72 | 54 | ||||
| Ni2P/Ni foam | Hydrothermal | 1 M KOH | 37 | 76 | 232 | |
| Co0.6Fe0.4P nanoframe | Template growth + etching + phosphidation | 1 M KOH | 133 | 61 | 233 | |
| N-CoP2 NWs/carbon cloth | Hydrothermal + phosphidation + N doping | 0.5 M H2SO4 | 38 | 46 | 147 | |
| Pv-modified Ni12P5/Ni foam | Annealing | 1 M KOH | 27.7 | 30.88 | 149 | |
| CoPx@carbon nanosheets | Carbonization + phosphidation | 1 M KOH | 91 | 129 | 234 | |
| 0.5 M H2SO4 | 98 | N/A | ||||
| Fe0.5Ni1.5P | Colloidal synthesis | 0.5 M H2SO4 | 163 (@ 50 mA cm−2) | 65 | 22 | |
| CoP–InNC@CNT | Carbonization + phosphidation | 1 M KOH | 159 | 56 | 235 | |
| 0.5 M H2SO4 | 153 | 62 | ||||
| Ni12P5–Ni4Nb5P4/PCC | Plasma + hydrothermal + phosphidation | 1 M KOH | 81 | 76.4 | 148 | |
| P-NM-CF HNRs/Ni foam | Hydrothermal + phosphidation | 1 M KOH | 100 | 54.3 | 236 | |
| 0.5 M H2SO4 | 130 | 51.3 | ||||
| 2D Ni–CoP ultrathin nanosheets | Phosphidation | 1 M KOH | 88 | 41 | 237 | |
| 2D porous MoP/Mo2N heterojunction/Ni foam | Polyethylene glycol-mediated assembly | 1 M KOH | 89 | 78 | 238 | |
| TMS | Fe@FeOxSy/Fe foam | Solvothermal | 1 M KOH | 300 | 77 | 239 |
| a-CoSx/FTO | Electrochemical activation | 1 M PBS | 168 | 76 | 240 | |
| NiWS/carbon fiber | Electrodeposition + hydrothermal | 1 M KOH | 38 | 98 | 241 | |
| 0.2 M PBS | 120 | 244 | ||||
| 0.5 M H2SO4 | 56 | 128 | ||||
| Co-NiS2/Ni foam | Electrodeposition + annealing | 1 M KOH | 80 | 43 | 172 | |
| F-Ni3S2/Ni foam | Solvothermal + pyrolysis | 1 M KOH | 38 | 78 | 242 | |
| Ag2S/MoS2/rGO | Hydrothermal | 0.5 M H2SO4 | 190 | 56 | 164 | |
| N-NiS/MoS2/Ni foam | Hydrothermal + pyrolysis with thiourea | 1 M KOH | 71 | 79 | 166 | |
| V-MoS2 | Solvothermal | 1 M KOH | 206 | 89 | 243 | |
| 0.5 M H2SO4 | 194 | 59 | ||||
| Fe-CoMoS | Hydrothermal + pyrolysis | 1 M KOH | 137 | 98 | 216 | |
| Vs-Ni3S2/Ni foam | Hydrothermal + plasma | 1 M KOH | 88 | 87 | 168 | |
Although a large number of electrocatalysts have been developed with promising performances for water electrolysis, their catalytic mechanism is not yet fully understood. In general, ex situ experiments for catalysts in pre- and post-reaction states have been commonly carried out to explain the behaviors of active species. However, these results do not represent the real-time action of catalysts due to the short-lived intermediates during the reaction. Motivated by these challenges, in situ characterization techniques, such as XAS, Raman spectroscopy, FT-IR spectroscopy, electron microscopy, ambient pressure XPS, ICP-MS, and synchrotron radiation Fourier transform infrared spectroscopy, have been extensively developed to monitor the structural evolution of catalysts, the changes of electronic configurations, and key intermediates in real-time during HER/OER processes.189 For examples, Wei et al. unambiguously probed the catalytically active sites and the stability of Co SACs during the HER by in situ XAS.190 Using an in situ TEM, Yu et al. captured the morphology evolution of an amorphous CoSx catalyst during the OER process.191 The results from these in situ characterization will provide in-depth understandings of the catalytic mechanism and help for designing next-generation catalysts. However, there are still limitations to the development, standardization, and wide application of these techniques. In addition, current reports focused on single in situ characterization. Therefore, combining multiple in situ experiments is highly desirable to synergistically provide comprehensive insights into electrocatalysts.
The next step should be the practical application of the prepared non-PGM-based electrocatalysts and SACs for water electrolysis technology, providing potential directions. To date, limited work has been carried out on electrolysers including PEMWEs and AEMWEs. Especially, issues such as electrode stability, power efficiency, membrane handling, and gas treatment have to be solved. In addition, the use of chemically stable and cheap PEMs/AEMs with PGM-free catalysts will be suitable for reducing the overall cost of H2 production and fuel cell operation. Finally, solving the durability issue of PEMWEs and the efficiency issue of AEMWEs will accelerate the widespread application of water electrolysis technologies.
Author contributions
H. J. K., H. Y. K., J. J., J. Y. K., K. L., and S.-I. C. conceived and designed the review article. H. J. K. and S.-I. C. contributed to chapters 1, 2, 8, and 11. H. Y. K., B. Y. M., and J. Y. K. contributed to chapters 1, 6, and 11. J. J., S. C., Y. P., and K. L. contributed to chapters 1, 9, and 11. J. S. L. and S. H. J. contributed to chapters 3 and 11. H. H. and J. L. contributed to chapters 4 and 11. J. H. and M. S. contributed to chapters 5 and 11. M. K. and S. W. L. contributed to chapters 7 and 11. Y. W., J. L., Z. Z., and J. M. contributed to chapters 10 and 11. All authors contributed to the finalization of the text and figures of the manuscript and commented on the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors at Kyungpook National University acknowledge support from the National Research Foundation (NRF) of Korea funded by the Ministry of Science and ICT (NRF-2021R1A2C4001411 and 2020R1A4A1018393). S. H. J. and J. S. L. were supported by the NRF of Korea (NRF-2021R1A2C200749511). M. Shao acknowledges the support from the Research Grant Council (16308420 and HKUST C6011-20G) of the Hong Kong Special Administrative Region, China. J. Hu acknowledges the support from the National Nature Science Foundation of China (No. 21862011). The authors at the Georgia Institute of Technology acknowledge support from the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korea government (MOTIE) (No. 20188550000440). The authors at Tianjin University acknowledge support from the National Natural Science Foundation of China (22071172).Notes and references
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- J. K. Nørskov and C. H. Christensen, Science, 2006, 312, 1322–1323 CrossRef PubMed.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- J. Yu, Q. He, G. Yang, W. Zhou, Z. Shao and M. Ni, ACS Catal., 2019, 11, 9973–10011 CrossRef.
- S. M. Alia, S. Shulda, C. Ngo, S. Pylypenko and B. S. Pivovar, ACS Catal., 2018, 8, 2111–2120 CrossRef CAS.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- N. Mahmood, Y. Yao, J.-W. Zhang, L. Pan, X. Zhang and J.-J. Zou, Adv. Sci., 2017, 5, 1700464 CrossRef PubMed.
- W. Sheng, M. Myint, J. G. Chen and Y. Yan, Energy Environ. Sci., 2013, 6, 1509–1512 RSC.
- F. Lu, M. Zhou, Y. Zhou and X. Zeng, Small, 2017, 13, 1701931 CrossRef PubMed.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- S. Cherevko, A. R. Zeradjanin, A. A. Topalov, N. Kulyk, I. Katsounaros and K. J. J. Mayrhofer, ChemCatChem, 2014, 6, 2219–2223 CrossRef CAS.
- X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 16064 CrossRef CAS.
- V. Mazánek, H. Nahdi, J. Luxa, Z. Sofer and M. Pumera, Nanoscale, 2018, 10, 11544–11552 RSC.
- C. Tang, H. Zhang, K. Xu, Q. Zhang, J. Liu, C. He, L. Fan and T. Asefa, J. Mater. Chem. A, 2019, 7, 18030–18038 RSC.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 13, eaad4998 CrossRef PubMed.
- J. Joo, T. Kim, J. Lee, S.-I. Choi and K. Lee, Adv. Mater., 2019, 31, 1806682 CrossRef PubMed.
- Y. Qu, B. Chen, Z. Li, X. Duan, L. Wang, Y. Lin, T. Yuan, F. Zhou, Y. Hu, Z. Yang, C. Zhao, J. Wang, C. Zhao, Y. Hu, G. Wu, Q. Zhang, Q. Xu, B. Liu, P. Gao, R. You, W. Huang, L. Zheng, L. Gu, Y. Wu and Y. Li, J. Am. Chem. Soc., 2019, 141, 4505–4509 CrossRef CAS PubMed.
- C. Spöri, J. T. H. Kwan, A. Bonakdapour, D. P. Wilkinson and P. Strasser, Angew. Chem., Int. Ed., 2017, 56, 5994–6021 CrossRef PubMed.
- Z. Yan, H. Liu, Z. Hao, M. Yu, X. Chen and J. Chen, Chem. Sci., 2020, 11, 10614–10625 RSC.
- J. Peng, W. Dong, Z. Wang, Y. Meng, W. Liu, P. Song and Z. Liu, Mater. Today Adv., 2020, 8, 100081 CrossRef.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- D. Shin, H. J. Kim, M. Kim, D. Shin, H. Kim, H. Song and S.-I. Choi, ACS Catal., 2020, 10, 11665–11673 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef.
- B. Ruqia and S.-I. Choi, ChemSusChem, 2018, 11, 2643–2653 CrossRef CAS PubMed.
- J. Mahmood, F. Li, S.-M. Jung, M. S. Okyay, I. Ahmad, S.-J. Kim, N. Park, H. Y. Jeong and J.-B. Baek, Nat. Nanotechnol., 2017, 12, 441–446 CrossRef CAS PubMed.
- H. Jin, B. Ruqia, Y. Park, H. J. Kim, H.-S. Oh, S.-I. Choi and K. Lee, Adv. Energy Mater., 2020, 11, 2003188 CrossRef.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- M. A. Ahsan, T. He, K. Eid, A. M. Abdullah, M. L. Curry, A. Du, A. R. P. Santiago, L. Echegoyen and J. C. Noveron, J. Am. Chem. Soc., 2021, 143, 1203–1215 CrossRef CAS PubMed.
- B. Kim, A. Oh, M. K. Kabiraz, Y. Hong, J. Joo, H. Baik, S.-I. Choi and K. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 10115–10122 CrossRef CAS PubMed.
- S. Choi, J. Park, M. K. Kabiraz, Y. Hong, T. Kwon, T. Kim, A. Oh, H. Baik, M. Lee, S.-M. Paek, S.-I. Choi and K. Lee, Adv. Funct. Mater., 2020, 30, 2003935 CrossRef CAS.
- C. Hu and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 11736 CrossRef CAS PubMed.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- K. Gao, B. Wang, L. Tao, B. V. Cunning, Z. Zhang, S. Wang, R. S. Ruoff and L. Qu, Adv. Mater., 2019, 31, 1805121 CrossRef PubMed.
- Z. Shi, W. Yang, Y. Gu, T. Liao and Z. Sun, Adv. Sci., 2020, 7, 2001069 CrossRef CAS.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 CrossRef PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- C. Hu and L. Dai, Adv. Mater., 2017, 29, 1604942 CrossRef PubMed.
- Y. P. Zhu, Y. Jing, A. Vasileff, T. Heine and S.-Z. Qiao, Adv. Energy Mater., 2017, 7, 1602928 CrossRef.
- Q. Hu, G. Li, X. Liu, B. Zhu, X. Chai, Q. Zhang, J. Liu and C. He, Adv. Energy Mater., 2017, 7, 1602928 CrossRef.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, Nat. Catal., 2018, 1, 63–72 CrossRef CAS.
- L. Zhang, Y. Jia, G. Gao, X. Yan, N. Chen, J. Chen, M. T. Soo, B. Wood, D. Yang, A. Du and X. Yao, Chem, 2018, 4, 285–297 CAS.
- M.-Y. Ye, S. Li, X. Zhao, N. V. Tarakina, C. Teutloff, W. Y. Chow, R. Bittl and A. Thomas, Adv. Mater., 2020, 32, 1903942 CrossRef CAS PubMed.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- J. Y. Cheon, K. Kim, Y. J. Sa, S. H. Sahgong, Y. Hong, J. Woo, S.-D. Yim, H. Y. Jeong, Y. Kim and S. H. Joo, Adv. Energy Mater., 2016, 6, 1501794 CrossRef.
- X. Han, X. Ling, D. Yu, D. Xie, L. Li, S. Peng, C. Zhong, N. Zhao, Y. Deng and W. Hu, Adv. Mater., 2019, 31, 1905622 CrossRef CAS PubMed.
- J. Chen, H. Li, C. Fan, Q. Meng, Y. Tang, X. Qiu, G. Fu and T. Ma, Adv. Mater., 2020, 32, 2003134 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 CrossRef PubMed.
- Y. Jiao, Y. Zheng, K. Davey and S.-Z. Qiao, Nat. Energy, 2016, 12, 16130 CrossRef.
- L. Chen, J. Han, Y. Ito, T. Fujita, G. Huang, K. Hu, A. Hirata, K. Watanabe and M. Chen, Angew. Chem., Int. Ed., 2018, 57, 13302–13307 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 8, 4081–4148 CrossRef PubMed.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed.
- P.-A. Jacques, Y. Artero, J. Pécaut and M. Fontecave, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20627–20632 CrossRef CAS PubMed.
- A. Morozan, V. Goellner, Y. Nedellec, J. Hannauer and F. Jaouen, J. Electrochem. Soc., 2015, 162, H719 CrossRef CAS.
- H.-W. Liang, S. Brüller, R. Dong, J. Zhang, X. Feng and K. Müllen, Nat. Commun., 2015, 6, 7992 CrossRef CAS PubMed.
- Y. J. Sa, S. O. Park, G. Y. Jung, T. J. Shin, H. Y. Jeong, S. K. Kwak and S. H. Joo, ACS Catal., 2019, 9, 83–97 CrossRef CAS.
- Q. Li, X. Zou, X. Ai, H. Chen, L. Sun and X. Zou, Adv. Energy Mater., 2018, 9, 1803369 CrossRef.
- X. Ai, X. Zou, H. Chen, Y. Su, X. Feng, Q. Li, Y. Liu, Y. Zhang and X. Zou, Angew. Chem., Int. Ed., 2020, 59, 3961–3965 CrossRef CAS PubMed.
- L. Chen, L.-P. Zhang, L.-Y. Yao, Y.-H. Fang, L. He, G.-F. Wei and Z.-P. Liu, Energy Environ. Sci., 2019, 12, 3099–3105 RSC.
- M. Zeng, H. Wang, C. Zhao, J. Wei, K. Qi, W. Wang and X. Bai, ChemCatChem, 2016, 8, 708–712 CrossRef CAS.
- H. Xu, B. Fei, G. Cai, Y. Ha, J. Liu, H. Jia, J. Zhang, M. Liu and R. Wu, Adv. Energy Mater., 2020, 10, 1902714 CrossRef CAS.
- Z. Zhuang, Y. Li, Z. Li, F. Lv, Z. Lang, K. Lang, K. Zhao, L. Zhou, L. Moskaleva, S. Guo and L. Mai, Angew. Chem., Int. Ed., 2018, 57, 496–500 CrossRef CAS PubMed.
- H. Huang, H. Jung, H. Jun, D. Y. Woo, J. W. Han and J. Lee, Chem. Eng. Sci., 2021, 405, 126977 CrossRef CAS.
- H. Park, A. Encinas, J. P. Scheifers, Y. Zhang and B. P. T. Fokwa, Angew. Chem., Int. Ed., 2017, 56, 5575–5578 CrossRef CAS PubMed.
- Y. Chen, G. Yu, W. Chen, Y. Liu, G.-D. Li, P. Zhu, Q. Tao, Q. Li, J. Liu, X. Shen, H. Li, X. Huang, D. Wang, T. Asefa and X. Zou, J. Am. Chem. Soc., 2017, 139, 12370–12373 CrossRef CAS PubMed.
- H. Li, P. Wen, Q. Li, C. Dun, J. Xing, C. Lu, S. Adhikari, L. Jiang, D. L. Carroll and S. M. Geyer, Adv. Energy Mater., 2017, 7, 1700513 CrossRef.
- F. Guo, Y. Wu, H. Chen, Y. Liu, L. Yang, X. Ai and X. Zou, Energy Environ. Sci., 2019, 12, 684–692 RSC.
- H. Han, Y.-R. Hong, J. Woo, S. Mhin, K. M. Kim, J. Kwon, H. Choi, Y.-C. Chung and T. Song, Adv. Energy Mater., 2019, 9, 1803799 CrossRef.
- S. Carenco, D. Portehault, C. Boissière, N. Mézailles and C. Sanchez, Chem. Rev., 2013, 113, 7981–8065 CrossRef CAS PubMed.
- Z. Kou, Y. Yu, X. Liu, X. Gao, L. Zheng, H. Zou, Y. Pang, Z. Wang, Z. Pan, J. He, S. J. Pennycook and J. Wang, ACS Catal., 2020, 10, 4411–4419 CrossRef CAS.
- A.-M. Alexander and J. S. J. Hargreaves, Chem. Soc. Rev., 2010, 39, 4388–4401 RSC.
- H. Lin, N. Liu, Z. Shi, Y. Guo, Y. Tang and Q. Gao, Adv. Funct. Mater., 2016, 26, 5590–5598 CrossRef CAS.
- Y. Huang, Q. Gong, X. Song, K. Feng, K. Nie, F. Zhao, Y. Wang, M. Zeng, J. Zhong and Y. Li, ACS Nano, 2016, 10, 11337–11343 CrossRef CAS PubMed.
- H. Vrubel and X. Hu, Angew. Chem., Int. Ed., 2012, 51, 12703–12706 CrossRef CAS PubMed.
- F. Yu, Y. Gao, Z. Lang, Y. Ma, L. Yin, J. Du, H. Tan, Y. Wang and Y. Li, Nanoscale, 2018, 10, 6080–6087 RSC.
- C. Yang, R. Zhao, H. Xiang, J. Wu, W. Zhong, W. Li, Q. Zhang, N. Yang and X. Li, Adv. Energy Mater., 2020, 10, 2002260 CrossRef CAS.
- P. Wang, R. Qin, P. Ji, Z. Pu, J. Zhu, C. Lin, Y. Zhao, H. Tang, W. Li and S. Mu, Small, 2020, 16, 2001642 CrossRef CAS PubMed.
- X. Ma, K. Li, X. Zhang, B. Wei, H. Yang, L. Liu, M. Zhang, X. Zhang and Y. Chen, J. Mater. Chem. A, 2019, 7, 14904–14915 RSC.
- X. Fan, Z. Peng, R. Ye, H. Zhou and X. Guo, ACS Nano, 2015, 9, 7407–7418 CrossRef CAS PubMed.
- K. Kawashima, K. Shin, B. R. Wygant, J.-H. Kim, C. L. Cao, J. Lin, Y. J. Son, Y. Liu, G. Henkelman and C. B. Mullins, ACS Appl. Energy Mater., 2020, 3, 3909–3918 CrossRef CAS.
- S. Li, C. Yang, X. Yin, H. Yang, Y. Chen, L. Lin, M. Li, W. Li, G. Hu and D. Ma, Nano Res., 2017, 10, 1322–1328 CrossRef CAS.
- Z. Chen, W. Gong, S. Cong, Z. Wang, G. Song, T. Pan, X. Tang, J. Chen, W. Lu and Z. Zhao, Nano Energy, 2020, 68, 104335 CrossRef CAS.
- L.-N. Zhang, Y.-Y. Ma, Z.-L. Lang, Y.-H. Wang, S. U. Khan, G. Yan, H.-Q. Tan, H.-Y. Zang and Y. Li, J. Mater. Chem. A, 2018, 6, 15395–15403 RSC.
- N. Takei, C. Sommer, C. Genes, G. Pupillo, H. Goto, K. Koyasu, H. Chiba, M. Weidemüller and K. Ohmori, Nat. Commun., 2016, 7, 13449 CrossRef CAS PubMed.
- J.-H. Kim, K. Kawashima, B. R. Wygant, O. Mabayoje, Y. Liu, J. H. Wang and C. B. Mullins, ACS Appl. Energy Mater., 2018, 1, 5145–5150 CAS.
- X. Liang, J. Xiao, W. Weng and W. Xiao, Angew. Chem., Int. Ed., 2021, 60, 2120–2124 CrossRef CAS PubMed.
- S. Xu, M. Wang, G. Saranya, N. Chen, L. Zhang, Y. He, L. Wu, Y. Gong, Z. Yao, G. Wang, Z. Wang, S. Zhao, H. Tang, M. Chen and H. Gou, Appl. Catal., B, 2020, 268, 118385 CrossRef CAS.
- Z. Kou, W. Zang, W. Pei, L. Zheng, S. Zhou, S. Zhang, L. Zhang and J. Wang, J. Mater. Chem. A, 2020, 8, 3071–3082 RSC.
- J. Chen, B. Ren, H. Cui and C. Wang, Small, 2020, 16, 1907556 CrossRef CAS PubMed.
- L. Qiao, A. Zhu, W. Zeng, R. Dong, P. Tan, Z. Ding, P. Gao, S. Wang and J. Pan, J. Mater. Chem. A, 2020, 8, 2453–2462 RSC.
- Y. Hu, B. Yu, M. Ramadoss, W. Li, D. Yang, B. Wang and Y. Chen, ACS Sustainable Chem. Eng., 2019, 7, 10016–10024 CrossRef CAS.
- H. Jin, J. Chen, S. Mao and Y. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 22094–22101 CrossRef CAS PubMed.
- Y.-J. Ko, J.-M. Cho, I. Kim, D. S. Jeong, K.-S. Lee, J.-K. Park, Y.-J. Baik, H.-J. Choi and W.-S. Lee, Appl. Catal., B, 2017, 203, 684–691 CrossRef CAS.
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612–13614 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. G. Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. G. de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. D. Luna, A. Janmohamed, H. L. Xin, H. Yamg, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- Y. Zhu, Q. Lin, Y. Zhong, H. A. Tahini, Z. Shao and H. Wang, Energy Environ. Sci., 2020, 13, 3361–3392 RSC.
- Y. H. Li, P. F. Liu, L. F. Pan, H. F. Wang, Z. Z. Yang, L. R. Zheng, P. Hu, H. J. Zhao, L. Gu and H. G. Yang, Nat. Commun., 2015, 6, 8064 CrossRef CAS PubMed.
- T. Ling, D.-Y. Yan, H. Wang, Y. Jiao, Z. Hu, Y. Zheng, L. Zheng, J. Mao, H. Liu, X.-W. Du, M. Jaroniec and S.-Z. Qiao, Nat. Commun., 2017, 8, 1509 CrossRef PubMed.
- Y. Li, Z. G. Yu, L. Wang, Y. Weng, C. S. Tang, X. Yin, K. Han, H. Wu, X. Yu, L. M. Wong, D. Wan, X. R. Wang, J. Chai, Y.-W. Zhang, S. Wang, J. Wang, A. T. S. Wee, M. B. H. Breese, S. J. Pennycook, Y. Venkatesan, S. Dong, J. M. Xue and J. Chen, Nat. Commun., 2019, 10, 3149 CrossRef PubMed.
- T. Ling, T. Zhang, B. Ge, L. Han, L. Zheng, F. Lin, Z. Xu, W.-B. Hu, X.-W. Du, K. Davey and S. Z. Qiao, Adv. Mater., 2019, 31, 1807771 CrossRef PubMed.
- K. N. Ferreira, Y. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
- D. A. Corrigan, J. Electrochem. Soc., 1987, 134, 377–384 CrossRef CAS.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettecher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- C. R. B. Sebok, S. B. Scott, E. M. Fiordaliso, J. E. Sørensen, A. Bodin, D. B. Trimarco, C. D. Damsgaard, P. C. K. Vesborg, O. Hansen, I. E. L. Stephens, J. Kibsgaard and I. Chorkendorff, Nat. Catal., 2018, 1, 820–829 CrossRef.
- M. S. Burke, S. Zou, L. J. Enman, J. E. Kellon, C. A. Gabor, E. Pledger and S. W. Boettcher, J. Phys. Chem. Lett., 2015, 6, 3737–3742 CrossRef CAS PubMed.
- M. B. Stevens, C. D. M. Trang, L. J. Enman, J. Deng and S. W. Boettcher, J. Am. Chem. Soc., 2017, 139, 11361–11364 CrossRef CAS PubMed.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, Y.-C. Weng, R. A. Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- H. S. Ahn and A. J. Bard, J. Am. Chem. Soc., 2016, 138, 313–318 CrossRef CAS PubMed.
- D. Y. Chung, P. P. Lopes, P. F. B. D. Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik, Y. Zhu, S. Seifert, S. Lee, V. R. Stamenkovic and N. M. Markovic, Nat. Energy, 2020, 5, 222–230 CrossRef.
- B. Zhang, L. Wang, Z. Cao, S. M. Kozlov, F. P. G. de Arquer, C. T. Dinh, J. Li, Z. Wang, X. Zheng, L. Zhang, Y. Wen, O. Voznyy, R. Comin, P. D. Luna, T. Regier, W. Bi, E. E. Alp, C.-W. Pao, L. Zheng, Y. Hu, Y. Ji, Y. Li, Y. Zhang, L. Cavallo, H. Peng and E. H. Sargent, Nat. Catal., 2020, 3, 985–992 CrossRef CAS.
- Q. Hu, Z. Wang, X. Huang, Y. Qin, H. Yang, X. Ren, Q. Zhang, J. Liu and C. He, Energy Environ. Sci., 2020, 13, 5097–5103 RSC.
- J. Zhang, R. Cui, C. Gao, L. Bian, Y. Pu, X. Zhu, X. Li and W. Huang, Small, 2019, 15, 1904688 CrossRef CAS PubMed.
- Z. Qiu, C.-W. Tai, G. Z. Niklasson and T. Edvinsson, Energy Environ. Sci., 2019, 12, 572–581 RSC.
- M. Kim, J. Park, M. Kang, J. Y. Kim and S. W. Lee, ACS Cent. Sci., 2020, 6, 880–891 CrossRef CAS PubMed.
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Science, 2017, 358, 751–756 CrossRef CAS PubMed.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- B.-J. Kim, E. Fabbri, M. Borlaf, D. F. Abbott, I. E. Castelli, M. Nachtegaal, T. Graule and T. J. Schmidt, Mater. Adv., 2021, 2, 345–355 RSC.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- E. Fabbri and T. J. Schmidt, ACS Catal., 2018, 8, 9765–9774 CrossRef CAS.
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 CrossRef PubMed.
- A. Grimaud, W. T. Hong, Y. Shao-Horn and J.-M. Tarascon, Nat. Mater., 2016, 15, 121–126 CrossRef CAS PubMed.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Nørskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- L. Yang, G. Yu, X. Ai, W. Yan, H. Duan, W. Chen, X. Li, T. Wang, C. Zhang, X. Huang, J.-S. Chen and X. Zou, Nat. Commun., 2018, 9, 5236 CrossRef CAS PubMed.
- Y. Chen, H. Li, J. Wang, Y. Du, S. Xi, Y. Sun, M. Sherburne, J. W. Ager, A. C. Fisher and Z. J. Xu, Nat. Commun., 2019, 10, 572 CrossRef CAS PubMed.
- M. Retuerto, L. Pascual, F. Calle-Vallejo, P. Ferrer, D. Gianolio, A. G. Pereira, Á. García, J. Torrero, M. T. Fernández-Díaz, P. Bencok, M. A. Peña, J. L. G. Fierro and S. Rojas, Nat. Commun., 2019, 10, 2041 CrossRef PubMed.
- K. Sardar, S. C. Ball, J. D. B. Sharman, D. Thompsett, J. M. Fisher, R. A. P. Smith, P. K. Biswas, M. R. Lees, R. J. Kashtiban, J. Sloan and R. I. Walton, Chem. Mater., 2012, 24, 4192–4200 CrossRef CAS.
- K. Sardar, E. Petrucco, C. I. Hiley, J. D. B. Sharman, P. P. Wells, A. E. Russell, R. J. Kashtiban, J. Sloan and R. I. Walton, Angew. Chem., Int. Ed., 2014, 53, 10960–10964 CrossRef CAS PubMed.
- D. Lebedev, M. Povia, K. Waltar, P. M. Abdala, I. E. Castelli, E. Fabbri, M. V. Blanco, A. Fedorov, C. Copéret, N. Marzari and T. J. Schmidt, Chem. Mater., 2017, 29, 5182–5191 CrossRef CAS.
- J. Kim, P.-C. Shih, K.-C. Tsao, Y.-T. Pan, X. Yin, C.-J. Sun and H. Yang, J. Am. Chem. Soc., 2017, 139, 12076–12083 CrossRef CAS PubMed.
- D. A. Kuznetsov, M. A. Naeem, P. V. Kumar, P. M. Abdala, A. Fedorov and C. R. Müller, J. Am. Chem. Soc., 2020, 142, 7883–7888 CrossRef CAS PubMed.
- X. Xu, Y. Chen, W. Zhou, Z. Zhu, C. Su, M. Liu and Z. Shao, Adv. Mater., 2016, 28, 6442–6448 CrossRef CAS PubMed.
- B. Hua, M. Li, W. Pang, W. Tang, S. Zhao, Z. Jin, Y. Zeng, B. S. Amirkhiz and J.-L. Luo, Chem, 2018, 4, 2902–2916 CAS.
- D. Guan, J. Zhou, Z. Hu, W. Zhou, X. Xu, Y. Zhong, B. Liu, Y. Chen, M. Xu, H.-J. Lin, C.-T. Chen, J.-Q. Wang and Z. Shao, Adv. Funct. Mater., 2019, 29, 1900704 CrossRef.
- D. Guan, J. Zhou, Y.-C. Huang, C.-L. Dong, J.-Q. Wang, W. Zhou and Z. Shao, Nat. Commun., 2019, 10, 3755 CrossRef PubMed.
- Y. Zhu, J. Dai, W. Zhou, Y. Zhong, H. Wang and Z. Shao, J. Mater. Chem. A, 2018, 6, 13582–13587 RSC.
- M. Kim, B. Lee, H. Ju, S. W. Lee and J. Kim, Adv. Mater., 2019, 31, 1901977 CrossRef PubMed.
- M. Kim, J. Park, H. Ju, J. Y. Kim, H.-S. Cho, C.-H. Kim, B.-H. Kim and S. W. Lee, Energy Environ. Sci., 2021, 14, 3053–3063 RSC.
- C. Hu, J. Hong, J. Huang, W. Chen, C. U. Segre, K. Suenaga, W. Zhao, F. Huang and J. Wang, Energy Environ. Sci., 2020, 13, 4249–4257 RSC.
- D. F. Abbott, R. K. Pittkowski, K. Macounová, R. Nebel, E. Marelli, E. Fabbri, I. E. Castelli, P. Krtil and T. J. Schmidt, ACS Appl. Mater. Interfaces, 2019, 11, 37748–37760 CrossRef CAS PubMed.
- M. A. Hubert, A. M. Patel, A. Gallo, Y. Liu, E. Valle, M. Ben-Naim, J. Sanchez, D. Sokaras, R. Sinclair, J. K. Nørskov, L. A. King, M. Bajdich and T. F. Jaramillo, ACS Catal., 2020, 10, 12182–12196 CrossRef CAS.
- Y. Sun, H. Liao, J. Wang, B. Chen, S. Sun, S. J. H. Ong, S. Xi, C. Diao, Y. Du, J.-O. Wang, M. B. H. Breese, S. Li, H. Zhang and Z. J. Xu, Nat. Catal., 2020, 3, 554–563 CrossRef CAS.
- J. Kim, P.-C. Shih, Y. Qin, Z. A. Bardan, C.-J. Sun and H. Yang, Angew. Chem., Int. Ed., 2018, 57, 13877–13881 CrossRef CAS PubMed.
- P.-C. Shih, C. Zhang, H. Raheja, C.-J. Sun and H. Yang, ChemNanoMat, 2020, 6, 930–936 CrossRef CAS.
- B. Han, K. A. Stoerzinger, V. Tileli, A. D. Gamalski, E. A. Stach and Y. Shao-Horn, Nat. Mater., 2017, 16, 121–126 CrossRef CAS PubMed.
- P. P. Lopes, D. Y. Chung, X. Rui, H. Zheng, H. He, P. Farinazzo Bergamo Dias Martins, D. Strmcnik, V. R. Stamenkovic, P. Zapol, J. F. Mitchell, R. F. Klie and N. M. Markovic, J. Am. Chem. Soc., 2021, 143, 2741–2750 CrossRef CAS PubMed.
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC.
- J. Cai, Y. Song, Y. Zang, S. Niu, Y. Wu, Y. Xie, X. Zheng, Y. Liu, Y. Lin, X. Liu, G. Wang and Y. Qian, Sci. Adv., 2020, 6, eaaw8113 CrossRef CAS PubMed.
- D. Chen, Z. Xu, W. Chen, G. Chen, J. Huang, C. Song, K. Zheng, Z. Zhang, X. Hu, H.-S. Choi and K. Ostrikov, Small, 2020, 16, 2004843 CrossRef CAS PubMed.
- J. Duan, S. Chen, C. A. O. Ledón, M. Jaroniec and S.-Z. Qiao, Angew. Chem., Int. Ed., 2020, 59, 8181–8186 CrossRef CAS PubMed.
- T. Wu, M. L. Stone, M. J. Shearer, M. J. Stolt, I. A. Guzei, R. J. Hamers, R. Lu, K. Deng, S. Jin and J. R. Schmidt, ACS Catal., 2018, 8, 1143–1152 CrossRef CAS.
- Y. Yan, B. Xia, Z. Xu and X. Wang, ACS Catal., 2014, 4, 1693–1705 CrossRef CAS.
- Y. Guo, T. Park, J. W. Yi, J. Henzie, J. Kim, Z. Wang, B. Jiang, Y. Bando, Y. Sugahara, J. Tang and Y. Yamauchi, Adv. Mater., 2019, 31, 1807134 CrossRef PubMed.
- Z. Grzesik and K. Przybylski, in Developments in High Temperature Corrosion and Protection of Materials, ed. W. Gao and Z. Li, Woodhead Publishing, Cambridge, England, 2008, pp. 599–638 Search PubMed.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- D. Kong, J. J. Cha, H. Wang, H. R. Lee and Y. Cui, Energy Environ. Sci., 2013, 6, 3553–3558 RSC.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963–969 CrossRef CAS PubMed.
- S. Dai, X. Wu, D. Liu, Y. Chu, K. Wang, B. Yang and J. Huang, ACS Appl. Mater. Interfaces, 2018, 10, 9379–9389 CrossRef PubMed.
- B. Guo, K. Yu, H. Li, H. Song, Y. Zhang, X. Lei, H. Fu, Y. Tan and Z. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 5517–5525 CrossRef CAS PubMed.
- J. Kim, H. Jin, A. Oh, H. Baik, S. H. Joo and K. Lee, Nanoscale, 2017, 9, 15397–15406 RSC.
- W. Zhou, X.-J. Wu, X. Cao, X. Huang, C. Tan, J. Tian, H. Liu, J. Wang and H. Zhang, Energy Environ. Sci., 2013, 6, 2921–2924 RSC.
- X. Long, G. Li, Z. Wang, H. Zhu, T. Zhang, S. Xiao, W. Guo and S. Yang, J. Am. Chem. Soc., 2015, 137, 11900–11903 CrossRef CAS PubMed.
- D. Liu, Q. Lu, Y. Luo, X. Sun and A. M. Asiri, Nanoscale, 2015, 7, 15122–15126 RSC.
- G. Solomon, R. Mazzaro, S. You, M. M. Natile, V. Morandi, I. Concina and A. Vomiero, ACS Appl. Mater. Interfaces, 2019, 11, 22380–22389 CrossRef CAS PubMed.
- J.-X. Feng, J.-Q. Wu, Y.-X. Tong and G.-R. Li, J. Am. Chem. Soc., 2018, 140, 610–617 CrossRef CAS PubMed.
- M. Yang, Y. Jiang, S. Liu, M. Zhang, Q. Guo, W. Shen, R. He, W. Su and M. Li, Nanoscale, 2019, 11, 14016–14023 RSC.
- X. Zhang and Y. Liang, Adv. Sci., 2018, 5, 1700644 CrossRef PubMed.
- D. Jia, L. Han, Y. Li, W. He, C. Liu, J. Zhang, C. Chen, H. Liu and H. L. Xin, J. Mater. Chem. A, 2020, 8, 18207–18214 RSC.
- B. Zhang, G. Yang, C. Li, K. Huang, J. Wu, S. Hao, J. Feng, D. Peng and Y. Huang, Nanoscale, 2018, 10, 1774–1778 RSC.
- Y. Li, J. Liu, C. Chen, X. Zhang and J. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 5982–5991 CrossRef CAS PubMed.
- Z.-F. Huang, J. Song, K. Li, M. Tahir, Y.-T. Wang, L. Pan, L. Wang, X. Zhang and J.-J. Zou, J. Am. Chem. Soc., 2016, 138, 1359–1365 CrossRef CAS PubMed.
- J. Yin, J. Jin, H. Zhang, M. Lu, Y. Peng, B. Huang, P. Xi and C.-H. Yan, Angew. Chem., Int. Ed., 2019, 58, 18676–18682 CrossRef CAS PubMed.
- M. Zhou, Q. Weng, X. Zhang, X. Wang, Y. Xue, X. Zeng, Y. Bando and D. Golberg, J. Mater. Chem. A, 2017, 5, 4335–4342 RSC.
- M. Cui, C. Yang, B. Li, Q. Dong, M. Wu, S. Hwang, H. Xie, X. Wang, G. Wang and L. Hu, Adv. Energy Mater., 2021, 11, 2002887 CrossRef CAS.
- Y. Cheng, S. Lu, F. Liao, L. Liu, Y. Li and M. Shao, Adv. Funct. Mater., 2017, 27, 1700359 CrossRef.
- Y. Kwon, M. Jun, J. Joo and K. Lee, J. Mater. Chem. A, 2019, 7, 5090–5110 RSC.
- H. Kim, J. Kim, W. Guo, G. H. Han, S. Hong, S. Y. Kim and S. H. Ahn, ACS Sustainable Chem. Eng., 2020, 8, 15815–15821 CrossRef CAS.
- D. Zhao, K. Sun, W.-C. Cheong, L. Zheng, C. Zhang, S. Liu, X. Cao, K. Wu, Y. Pan, Z. Zhuang, B. Hu, D. Wang, Q. Peng, C. Chen and Y. Li, Angew. Chem., Int. Ed., 2019, 59, 8982–8990 CrossRef PubMed.
- X.-P. Yin, H.-J. Wang, S.-F. Tang, X.-L. Lu, M. Shu, R. Si and T.-B. Lu, Angew. Chem., Int. Ed., 2018, 57, 9382–9386 CrossRef CAS PubMed.
- H. Zhang, P. An, W. Zhou, B. Y. Guan, P. Zhang, J. Dong and X. W. Lou, Sci. Adv., 2018, 4, eaao6657 CrossRef PubMed.
- L. Zhang, L. Han, H. Liu, X. Liu and J. Luo, Angew. Chem., Int. Ed., 2017, 56, 13694–13698 CrossRef CAS PubMed.
- F. Meng, H. Zhong, D. Bao, J. Yan and X. Zhang, J. Am. Chem. Soc., 2016, 138, 10226–10231 CrossRef CAS PubMed.
- W. Chen, J. Pei, C.-T. He, J. Wan, H. Ren, Y. Zhu, Y. Wang, J. Dong, S. Tian, W.-C. Cheong, S. Lu, L. Zheng, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 16086–16090 CrossRef CAS PubMed.
- Y. Zhang, C. Wu, H. Jiang, Y. Lin, H. Liu, Q. He, S. Chen, T. Duan and L. Song, Adv. Mater., 2018, 30, 1707522 CrossRef PubMed.
- Y. Hu, G. Luo, L. Wang, X. Liu, Y. Qu, Y. Zhou, F. Zhou, Z. Li, Y. Li, T. Yao, C. Xiong, B. Yang, Z. Yu and Y. Wu, Adv. Energy Mater., 2021, 11, 2002816 CrossRef CAS.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336–3339 CrossRef CAS PubMed.
- S. Zhang, Y. Wu, Y.-X. Zhang and Z. Niu, Sci. China: Chem., 2021, 64, 1908–1922 CrossRef CAS.
- Y. Yang, Y. Qian, H. Li, Z. Zhang, Y. Mu, D. Do, B. Zhou, J. Dong, W. Yan, Y. Qin, L. Fang, R. Feng, J. Zhou, P. Zhang, J. Dong, G. Yu, Y. Liu, X. Zhang and X. Fan, Sci. Adv., 2021, 6, eaba6586 CrossRef PubMed.
- T. Zhao, Y. Wang, S. Karuturi, K. Catchpole, Q. Zhang and C. Zhao, Carbon Energy, 2020, 2, 582–613 CrossRef CAS.
- L. Cao, Q. Luo, W. Liu, Y. Lin, X. Liu, Y. Cao, W. Zhang, Y. Wu, J. Yang, T. Yao and S. Wei, Nat. Catal., 2019, 2, 134–141 CrossRef CAS.
- K. Fan, H. Zou, Y. Lu, H. Chen, F. Li, J. Liu, L. Sun, L. Tong, M. F. Toney, M. Sui and J. Yu, ACS Nano, 2018, 12, 12369–12379 CrossRef CAS PubMed.
- J. Masa, P. Weide, D. Peeters, I. Sinev, W. Xia, Z. Sun, C. Somsen, M. Muhler and W. Schuhmann, Adv. Energy Mater., 2016, 6, 1502313 CrossRef.
- J. Masa, I. Sinev, H. Mistry, E. Ventosa, M. de la Mata, J. Arbiol, M. Muhler, B. R. Cuenya and W. Schuhmann, Adv. Energy Mater., 2017, 7, 1700381 CrossRef.
- J. M. V. Nsanzimana, Y. Peng, Y. Y. Xu, L. Thia, C. Wang, B. Y. Xia and X. Wang, Adv. Energy Mater., 2018, 8, 1701475 CrossRef.
- S. Chen, Y. Li, Z. Zhang, Q. Fu and X. Bao, J. Mater. Chem. A, 2018, 6, 10644–10648 RSC.
- J. Li, H. Chen, Y. Liu, R. Gao and X. Zou, J. Mater. Chem. A, 2019, 7, 5288–5294 RSC.
- H. Mao, X. Guo, Y. Fu, H. Yang, Y. Zhang, R. Zhang and X.-M. Song, J. Mater. Chem. A, 2020, 8, 1821–1828 RSC.
- Z. Chen, R. Zheng, M. Graś, W. Wei, G. Lota, H. Chen and B.-J. Nia, Appl. Catal., B, 2021, 288, 120037 CrossRef CAS.
- Y. Lian, K. Shi, H. Yang, H. Sun, P. Qi, J. Ye, W. Wu, Z. Deng and Y. Deng, Small, 2020, 16, 1907368 CrossRef CAS PubMed.
- G. Liu, K. Wang, L. Wang, B. Wang, Z. Lim, X. Chen, Y. Hua, W. Zhu, H. Li and J. Xia, J. Colloid Interface Sci., 2021, 583, 614–625 CrossRef CAS PubMed.
- S. Lu, J. Wu, H. Hu, X. Pan, Z. Hu, H. Li, H. Zhu, F. Duan and M. Du, J. Colloid Interface Sci., 2021, 585, 258–266 CrossRef CAS PubMed.
- S. Song, W. Li, Y.-P. Deng, Y. Ruan, Y. Zhang, X. Qin and Z. Chen, Nano Energy, 2020, 67, 104208 CrossRef CAS.
- M. B. Zakaria, D. Zheng, U. P. Apfel, T. Nagata, E.-R. S. Kenawy and J. Lin, ACS Appl. Mater. Interfaces, 2020, 12, 40186–40193 CrossRef CAS PubMed.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS PubMed.
- Y. Zhang, B. Ouyang, J. Xu, G. Jia, S. Chen, R. S. Rawat and H. J. Fan, Angew. Chem., Int. Ed., 2016, 55, 8670–8674 CrossRef CAS PubMed.
- S. Li, Y. Wang, S. Peng, L. Zhang, A. M. Al-Enizi, H. Zhang, X. Sun and G. Zheng, Adv. Energy Mater., 2016, 6, 1501661 CrossRef.
- A.-L. Wang, H. Xu and G.-R. Li, ACS Energy Lett., 2016, 1, 445–453 CrossRef CAS.
- Z. Cai, D. Zhou, M. Wang, S.-M. Bak, Y. Wu, Z. Wu, Y. Tian, X. Xiong, Y. Li, W. Liu, S. Siahrostami, Y. Kuang, X.-Q. Yang, H. Duan, Z. Feng, H. Wang and X. Sun, Angew. Chem., Int. Ed., 2018, 57, 9392–9396 CrossRef CAS PubMed.
- P. Li, X. Duan, Y. Kuang, Y. Li, G. Zhang, W. Liu and X. Sun, Adv. Energy Mater., 2018, 8, 1703341 CrossRef.
- H. Han, K. M. Kim, H. Choi, G. Ali, K. Y. Chung, Y.-R. Hong, J. Choi, J. Kwon, S. W. Lee, J. W. Lee, J. H. Ryu, T. Song and S. Mhin, ACS Catal., 2018, 8, 4091–4102 CrossRef CAS.
- J. Jiang, S. Lu, W.-K. Wang, G.-X. Huang, B.-C. Huang, F. Zhang, Y.-J. Zhang and H.-Q. Yu, Nano Energy, 2018, 43, 300–309 CrossRef CAS.
- Y. Jin, X. Yue, H. Du, K. Wang, S. Huang and P. K. Shen, J. Mater. Chem. A, 2018, 6, 5592–5597 RSC.
- Y.-R. Hong, S. Mhin, K.-M. Kim, W.-S. Han, H. Choi, G. Ali, K. Y. Chung, H. J. Lee, S.-I. Moon, S. Dutta, S. Sun, Y.-G. Jung, T. Song and H. Han, J. Mater. Chem. A, 2019, 7, 3592–3602 RSC.
- X. Zheng, X. Han, Y. Zhang, J. Wang, C. Zhong, Y. Deng and W. Hu, Nanoscale, 2019, 11, 5646–5654 RSC.
- M. Wang, C.-L. Dong, Y.-C. Huang and S. Shen, ACS Catal., 2020, 10, 1855–1864 CrossRef.
- Y. Guo, X. Zhou, J. Tang, S. Tanaka, Y. V. Kaneti, J. Na, B. Jiang, Y. Yamauchi, Y. Bando and Y. Sugahara, Nano Energy, 2020, 75, 104913 CrossRef CAS.
- L. Cao, Q. Luo, W. Liu, Y. Lin, X. Liu, Y. Cao, W. Zhang, Y. Wu, J. Yang, T. Yao and S. Wei, Nat. Catal., 2019, 2, 134–141 CrossRef CAS.
- N. Xu, G. Cao, Z. Chen, Q. Kang, H. Dai and P. Wang, J. Mater. Chem. A, 2017, 5, 12379–12384 RSC.
- X. Wang, G. Tai, Z. Wu, T. Hu and R. Wang, J. Mater. Chem. A, 2017, 5, 23471–23475 RSC.
- B. Zhang, H. Soleimaninejad, D. J. Jones, J. M. White, K. P. Ghiggino, T. A. Smith and W. H. Wong, Chem. Mater., 2017, 29, 8953–8395 CrossRef.
- W. Hao, R. Wu, R. Zhang, Y. Ha, Z. Chen, L. Wang, Y. Yang, X. Ma, D. Sun, F. Fang and Y. Guo, Adv. Energy Mater., 2018, 8, 1801372 CrossRef.
- F. Guo, Y. Wu, X. Ai, H. Chen, G.-D. Li, W. Chen and X. Zou, Chem. Commun., 2019, 55, 8627–8630 RSC.
- Y.-Y. Ma, Z.-L. Lang, L.-K. Yan, Y.-H. Wang, H.-Q. Tan, K. Feng, Y.-J. Xia, J. Zhong, Y. Liu, Z.-H. Kang and Y.-G. Li, Energy Environ. Sci., 2018, 11, 2114–2123 RSC.
- M. Yao, B. Wang, B. Sun, L. Luo, Y. Chen, J. Wang, N. Wang, S. Komarneni, X. Niu and W. Hu, Appl. Catal., B, 2021, 280, 119451 CrossRef CAS.
- Y. Jin, H. Wang, J. Li, X. Yue, Y. Han, P. K. Shen and Y. Cui, Adv. Mater., 2016, 28, 3785–3790 CrossRef CAS PubMed.
- X. Zhong, Y. Sun, X. Chen, G. Zhuang, X. Li and J.-G. Wang, Adv. Funct. Mater., 2016, 26, 5778–5786 CrossRef CAS.
- C. Shu, S. Kang, Y. Jin, X. Yue and P. K. Shen, J. Mater. Chem. A, 2017, 5, 9655–9660 RSC.
- H. Zeng, S. Chen, Y. Q. Jin, J. Li, J. Song, Z. Le, G. Liang, H. Zhang, F. Xie, J. Chen, Y. Jin, X. Chen and H. Meng, ACS Energy Lett., 2020, 5, 1908–1915 CrossRef CAS.
- C. Zhong, Z. Han, T. Wang, Q. Wang, Z. Shen, Q. Zhou, J. Wang, S. Zhang, X. Jin, S. Li, P. Wang, D. Gao, Y. Zhou and H. Zhang, J. Mater. Chem. A, 2020, 8, 10831–10838 RSC.
- H. Yang, Y. Zhang, F. Hu and Q. Wang, Nano Lett., 2015, 15, 7616–7620 CrossRef CAS PubMed.
- Z. Fang, L. Peng, Y. Qian, X. Zhang, Y. Xie, J. J. Cha and G. Yu, J. Am. Chem. Soc., 2018, 140, 5241–5247 CrossRef CAS PubMed.
- X. Yu, Z.-Y. Yu, X.-L. Zhang, Y.-R. Zheng, Y. Duan, Q. Gao, R. Wu, B. Sun, M.-R. Gao, G. Wang and S.-H. Yu, J. Am. Chem. Soc., 2019, 141, 7537–7543 CrossRef CAS PubMed.
- Y. Lian, H. Sun, X. Wang, P. Qi, Q. Mu, Y. Chen, J. Ye, X. Zhao, Z. Deng and Y. Peng, Chem. Sci., 2019, 10, 464–474 RSC.
- C.-C. Hou, L. Zou, Y. Wang and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 21360–21366 CrossRef CAS PubMed.
- L. Chai, Z. Hu, X. Wang, Y. Xu, L. Zhang, T.-T. Li, Y. Hu, J. Qian and S. Huang, Adv. Sci., 2020, 7, 1903195 CrossRef CAS PubMed.
- R. Li, J. Zang, W. Li, J. Li, Q. Zou, S. Zhou, J. Su and Y. Wang, ChemSusChem, 2020, 13, 3718–3725 CrossRef CAS PubMed.
- Y. Zhao, J. Zhang, Y. Xie, B. Sun, J. Jiang, W.-J. Jiang, S. Xi, H. Y. Yang, K. Yan, S. Wang, X. Guo, P. Li, Z. Han, X. Lu, H. Liu and G. Wang, Nano Lett., 2021, 21, 823–832 CrossRef CAS PubMed.
- Y. Gu, A. Wu, Y. Jiao, H. Zheng, X. Wang, Y. Xie, L. Wang, C. Tian and H. Fu, Angew. Chem., Int. Ed., 2021, 133, 6747–6755 CrossRef.
- X. Zou, Y. Wu, Y. Liu, D. Liu, W. Li, L. Gu, H. Liu, P. Wang, L. Sun and Y. Zhang, Chem, 2018, 4, 1139–1152 CAS.
- W. He, R. Ifraemov, A. Raslin and I. Hod, Adv. Funct. Mater., 2018, 28, 1707244 CrossRef.
- S.-S. Lu, X. Shang, L.-M. Zhang, B. Dong, W.-K. Gao, F.-N. Dai, B. Liu, Y.-M. Chai and C.-G. Liu, Appl. Surf. Sci., 2018, 445, 445–453 CrossRef CAS.
- W. He, L. Han, Q. Hao, X. Zheng, Y. Li, J. Zhang, C. Liu, H. Liu and H. L. Xin, ACS Energy Lett., 2019, 4, 2905–2912 CrossRef CAS.
- S. Bolar, S. Shit, J. S. Kumar, N. C. Murmu, R. S. Ganesh, H. Inokawa and T. Kuila, Appl. Catal., B, 2019, 254, 432–442 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |